DOI:
10.1039/D1NA00429H
(Paper)
Nanoscale Adv., 2021,
3, 6992-7001
Water dissociation and association on mirror twin boundaries in two-dimensional MoSe2: insights from density functional theory calculations†
Received
8th June 2021
, Accepted 21st October 2021
First published on 21st October 2021
Abstract
The adsorption and dissociation of water molecules on two-dimensional transition metal dichalcogenides (TMDs) is expected to be dominated by point defects, such as vacancies, and edges. At the same time, the role of grain boundaries, and particularly, mirror twinboundaries (MTBs), whose concentration in TMDs can be quite high, is not fully understood. Using density functional theory calculations, we investigate the interaction of water, hydroxyl groups, as well as oxygen and hydrogen molecules with MoSe2 monolayers when MTBs of various types are present. We show that the adsorption of all species on MTBs is energetically favorable as compared to that on the basal plane of pristine MoSe2, but the interaction with Se vacancies is stronger. We further assess the energetics of various surface chemical reactions involving oxygen and hydrogen atoms. Our results indicate that water dissociation on the basal plane should be dominated by vacancies even when MTBs are present, but they facilitate water clustering through hydroxyl groups at MTBs, which can anchor water molecules and give rise to the decoration of MTBs with water clusters. Also, the presence of MTBs affects oxygen reduction reaction (ORR) on the MoSe2 monolayer. Unlike Se vacancies which inhibit ORR due to a high overpotential, it is found that the ORR process on MTBs is more efficient, indicating their important role in the catalytic activity of MoSe2 monolayer and likely other TMDs.
1 Introduction
Due to their diverse electronic,1–4 optical,4–7 magnetic,8,9 and catalytic properties,3,10–12 transition metal dichalcogenides (TMDs) are currently among the most intensively studied two-dimensional (2D) materials. The diversity in their properties stems not only from various possible chemical compositions of TMDs,13 but also from the possibility to exist in different phases13,14 and strong dependence of the electronic structure on the number of layers.1 Moreover, the 2D nature of these systems enables modification of their properties by defects which can easily be introduced during or after the growth by chemical treatment or irradiation, see ref. 15 and 16 for an overview.
Not only point, but also line defects have been reported to exist in TMDs.17–21 Among them, mirror twin boundaries (MTBs) of various types22 with relatively low formation energies have been found. Moreover, contrary to serpentine or low-angle grain boundaries,17,18 which appear during the growth when domains with arbitrary crystal orientation coalesce, the formation of straight MTBs is energetically favorable in non-stoichiometric chalcogen-deficient TMDs.23 Indeed, their appearance in originally pristine TMD samples has been observed under electron irradiation, which created chalcogen vacancies23–25 or Mo deposition26 at moderate temperatures. Most important, their concentration can be very high,20 and to some extent, controlled using the post-synthesis approaches described above.
TMDs, which are considered as an alternative to expensive Pt-based materials,27,28 have received a considerable amount of attention in the context of catalysis,3,10–12,29e.g., for the oxygen reduction reaction (ORR), which is one of the most important processes in proton exchange membrane fuel cells. Edges of TMDs have been shown to be catalytically active29–33 but unless flakes of small sizes are used,34 their concentration is naturally relatively low. Thus possible activation of the inert basal planes of TMDs has been studied. In particular, substitutional doping with P, N, O, Cu was proposed to be a method for activating the basal plane for ORR via enhancement of oxygen molecule adsorption.35–37 However, this approach still requires the incorporation of impurities into the 2D materials during the growth or via a post-growth treatment. In addition, point defects like vacancies can create active sites for ORR electrocatalytic activity as shown for WTe2.38 Due to their metallic properties,22,39–41 MTBs can potentially be active sites for the ORR process. However, the catalytic performance of ORR on these defects, which is critical for catalysis, has not been investigated so far. Moreover, the dissociation/association of water molecules and the behavior of their derivatives (hydroxyl groups and isolated O/H atoms) on TMDs with MTBs has not been addressed, contrary to that on basal planes and edges,42–44 while knowing the energetics of these processes is important not only for assessing the catalytic properties but also for understanding the stability of TMDs at ambient conditions and clustering of water molecules at the surface.45
Here, using first-principles calculations, we theoretically study the interaction of water, hydroxyl groups, as well as hydrogen and oxygen atoms with MTBs of various types in MoSe2 monolayer, a typical TMD. As a reference, we also study the interaction of these species with vacancies and pristine basal plane. The efficiency of ORR on the MTBs is investigated as well.
2 Methods
Spin-polarized density functional theory (DFT) calculations were performed using the generalized gradient approximation (GGA) with PBE exchange-correlation functional,46 as implemented in the VASP code.47,48 A plane-wave cut-off of 450 eV was used in all the calculations. A vacuum space of around 20 Å was considered in the confinement directions to avoid the spurious inter-layer interaction. The full geometry optimizations were carried out with the force tolerance being set to 0.01 eV Å−1. The Brillouin zone of the supercells was sampled using gamma point approximation. The calculations include a dispersion correction scheme (DFT-D2)49 for the effects of long-range vdW interactions. Calculations with a more accurate vdW exchange-correlation functional based on the Tkatchenko and Scheffler (DFT-TS) method50 were also carried out for test systems. The structural models include nanoribbons of MoSe2 with maximum number of 590 atoms, corresponding to 5 × 20 unit cells of an ideal lattice in a rectangular representation. The adsorption energy (Eads) of the adsorbate on the surface is defined as:| | | Eads = Eslab+adsorbate − (Eslab + Eadsorbate) | (1) |
where the first term denotes the total energy of the slab with adsorbate, and the second and third terms represent the energy of the isolated slab and the isolated adsorbate, respectively.
The ORR reaction involves the trapping of adsorbate on a surface immersed in an electrolyte solution followed by a series of intermediate reactions,51,52 eventually resulting in the reduction of O2 to H2O. Here, we employ the computational hydrogen electrode model (CHE) proposed in ref. 53 to describe the Gibbs free energy difference ΔG of each ORR elementary steps. ΔG is expressed as:
| | | ΔG = ΔE + ΔZPE − TΔS + ΔGU + ΔGpH | (2) |
where Δ
E represents the change in the internal energy,
ΔZPE is the change in the vibrational zero-point energy,
T is the temperature (298.15 K), Δ
S is the entropy difference, Δ
GU and Δ
GpH are the contribution from electrode potential and pH of the solution. The reaction energy, ZPE and entropy change of the intermediate adorbate stages can be directly calculated from the corresponding DFT and vibrational calculations. The entropy is derived by calculating the vibrational entropy using harmonic approximation on the vibrational frequencies as input. However, the ZPE energies and entropies of liquid H
2O and gaseous H
2 are more complex, including the contributions from vibrations, rotations, and translations of molecules; fortunately these can be obtained from the thermodynamic NIST-JANAF tables.
54 The last two energy terms in
eqn (2), for solution acidity (Δ
GpH =
kBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
ln
10 × pH) and electrode potential (Δ
GU = −
qU,
q =
e| here) needs to be considered only for the protonation steps.
The ORR process in acidic involves a 4e− associate or a 2e− dissociative mechanism to reduce oxygen molecule to water. The major reaction pathway for 4e− process employs:53
| | | *OOH + H+ + e– ↔ *O + H2O | (4) |
where * implies the substrate with the respective adsorbate. In principle, the adsorption of molecular O
2 on the surface should also be included, but this is viewed as a chemical step and the steady-state adsorption of such neutral species does not contribute to the potential dependence of the faradaic current. The 2e
− dissociative mechanism involves adsorption of oxygen onto the substrate followed by step 5 and step 6. The presence of a finite electrode potential in the calculation of FED is due to the spontaneous charging of the surface during the ORR process. Thus, in this study we use
U = 1.23 V at pH = 0 (acid solution). The detailed ORR equations are provided in the ESI.
†
3 Result and discussion
3.1 Adsorption of water
To get a microscopic insight into the interaction of adsorbents on TMDs, the reactions on the pristine MoSe2 sheets and those with MTBs were investigated. We considered three possible Mo-rich grain boundaries which are always lower in energy than Se-rich grain boundaries in the wide range of the chemical potential in TMDs.23 In the 44| P and 44| E MTBs, the structure includes 4-fold rings that share a point at the Se site or edge (Mo–Se bond). The structure of the 55| 8 MTB contains two 5-fold rings adjacent to the 8-fold ring.
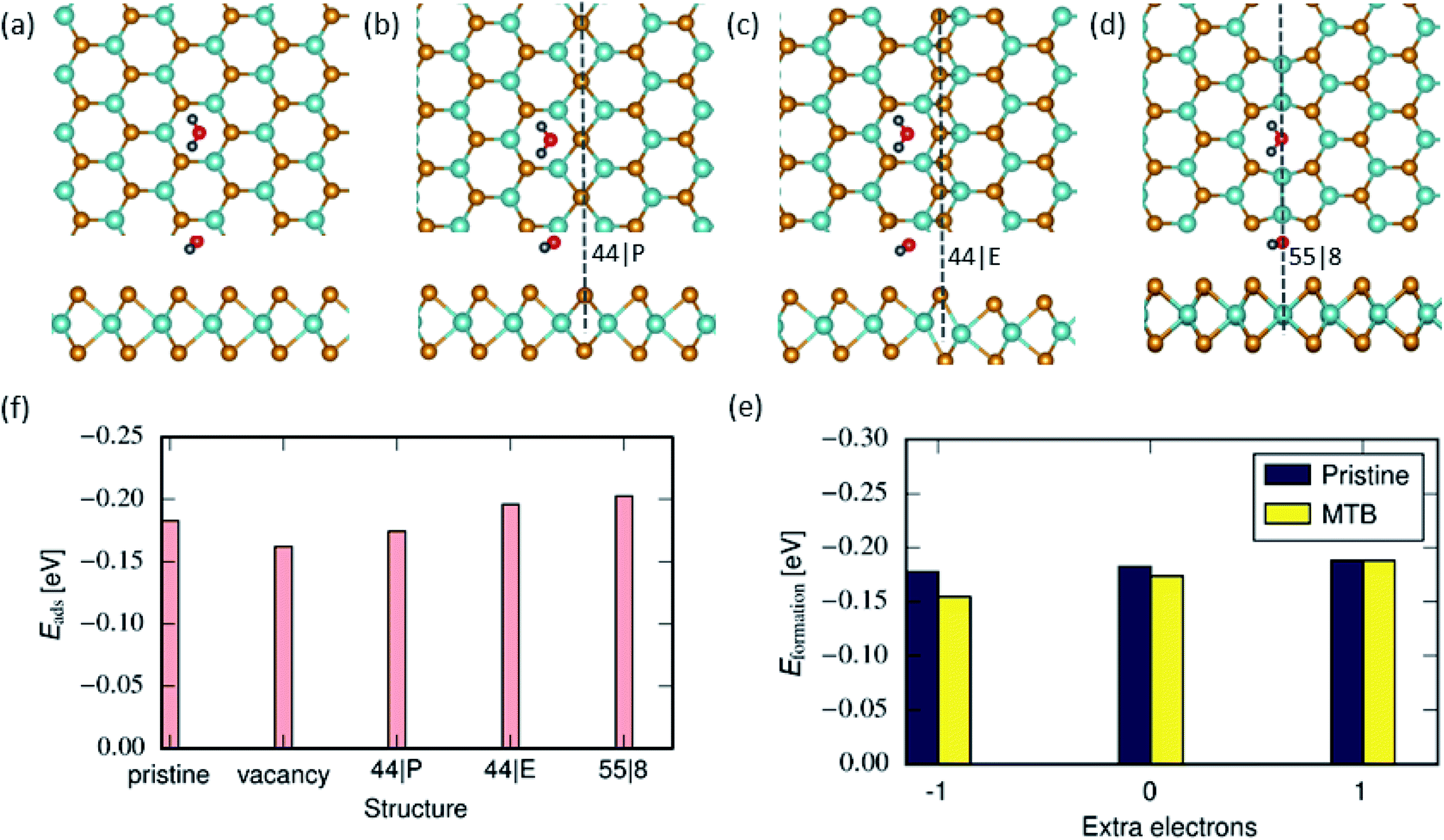
We first determined the atomic configurations of water adsorption on pristine MoSe2. To identify the active sites for water adsorption, various initial molecule orientations on the top of the basal plane are investigated, see Fig. 1 and S1 in the ESI.† In the most stable configuration, the H2O molecule is located ≈2.6 Å (the distance between the lower atom of the molecule and the uppermost Se atoms) above the center of the hexagon with the H atom pointing to the Se atoms of the monolayer. This is due to the electrostatic interaction between positively charged H atoms in water (+0.65 e) between and negatively charged Se plane, in agreement with the previous reports.55
The interaction of water with the basal plan of pristine MoSe2 is mainly physisorption with an adsorption energy of ≈−0.18 eV at the DFT-D2 level. Calculations using a more accurate DFT-TS method provided an adsorption energy of −0.15 eV. These values are consistent with the earlier theoretical reports56 of water adsorption on MoS2, indicating that perfect TMD monolayers have a hydrophobic character due to the chemically inert surface with saturated chalcogen ions. Similarly, in the case of a single Se vacancy, the small size of the defect does not permit the molecule to interact with positively charged Mo atoms causing a weak adsorption energy of ≈−0.16 eV.
Contrary to edges or vacancies, the MTBs are devoid of undercoordinated atoms at the surface of the TMD monolayer. As a result, the adsorption energies for water molecules on top of MTBs differ only slightly (≈0.05 eV) in comparison to the pristine monolayer, as evident from Fig. 1. Fig. 1(b)–(d) illustrate the energetically favorable adsorption configurations of water molecule on various MTBs. It is found that the adsorption energy on the 55| 8 is the lowest followed by the 44| E and 44| P MTB. Unlike the adsorption configuration on the 44| E and 44| P MTBs, the plane of the molecule is almost parallel to the MoSe2 basal plane and located on top of the 55| 8 MTB. The effect can be attributed to the presence of Mo–Mo bonds in the structure which are positively charged. Since MTBs have metallic states at the Fermi level22,39–41 and additional charges can be localized in these structures, we also investigated the charging effects on the adsorption energy in 44| P MTB. It is found that extra electrons or holes (with a linear density of 0.05 e nm−1) have minor effects on the adsorption energy on the 44| P MTB as evident from Fig. 1(e). The possible reason is that the Mo atomic layer is sandwiched between two Se atomic layers, and the extra charge mainly accumulated at Mo atoms is screened by Se atoms. It should be mentioned that in the case of additional positive charges, water molecules prefer adsorption in the pristine areas over that on 44| P MTBs.
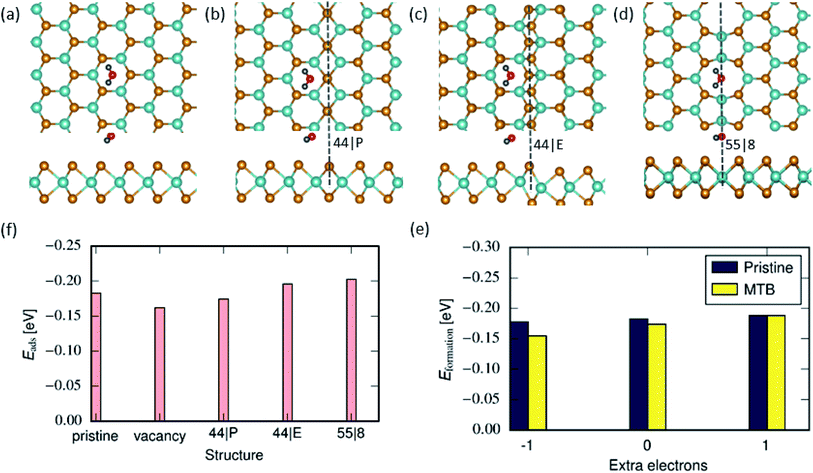 |
| | Fig. 1 Water adsorption on MoSe2 monolayer: Atomic structures of 2D MoSe2 with adsorbed water monomer on pristine (a), 44| P (b), 44| E (c) and 55| 8 (d) MTBs. (f) Adsorption energy of water monomers on top of MoSe2 in the pristine area, vacancy and next to the MTB. (e) Adsorption energy of H2O molecule on 44| P MTB when additional charge is present. | |
3.2 Adsorption of hydroxyl groups
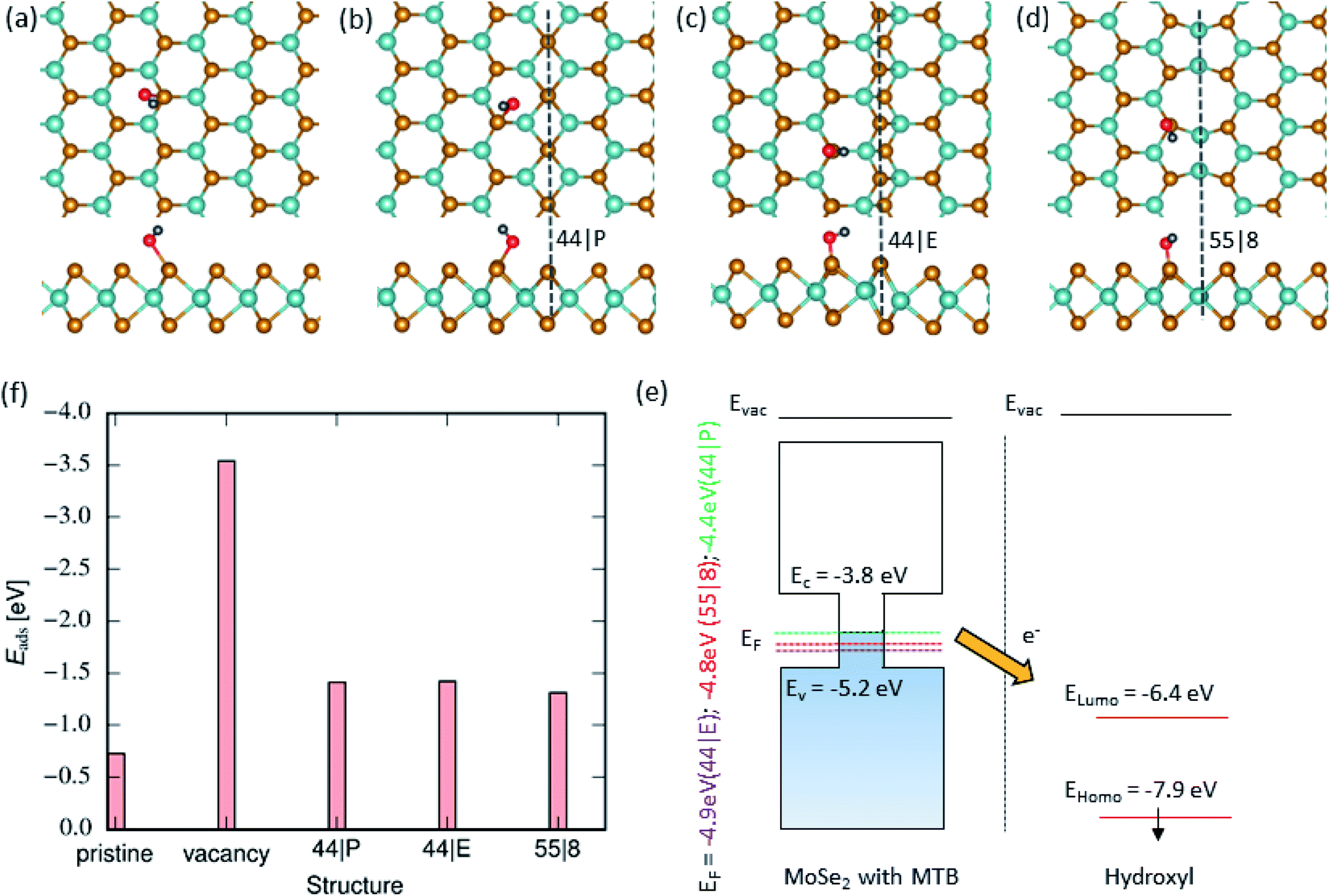
Fig. 2(a–d) show the lowest-energy configurations for hydroxyl groups adsorbed on the pristine basal plane, vacancy and various MTBs. The high chemical activity of OH as compared to that of H2O originates from the difference in electronic structures of the molecules. The gap between the highest occupied (HOMO) and the lowest unoccupied molecular orbitals (LUMO) of hydroxyl is 1.53 eV which is significantly smaller than that of a water molecule (6.20 eV). Unlike water, HOMO and LUMO of hydroxyl are not center-symmetrical and are localized more on the oxygen than on the hydrogen atom. Accordingly, the interaction of hydroxyl with the basal plane of the monolayer is stronger than for a water molecule. We found that the position of OH is energetically most preferable on top of the nearest-neighbor Se atoms next to the MTB. The adsorbed hydroxyl is oriented in such a way that the O atom is facing the Se atoms. Fig. 2(f) presents adsorption energies on different MTBs, which are very close to each other, with a difference of only ≈0.1 eV. The difference between adsorption energy at MTB to the pristine part is about 0.7 eV, as also illustrated in Fig. 2(f). This gives rise to a preferential adsorption of OH groups on MTBs. Moreover, the two spin-polarized LUMO states of the OH molecule are below the Fermi level, indicating a charge transfer from the MoSe2 to the adsorbent, as seen in Fig. 2(f). In order to rationalize the strong molecule–MTB interaction, we also analyzed the electronic structure of the system after the adsorption of the molecule. Bader analysis was used to assess the charge transfer between molecules and the substrate. The hydroxyl group creates a strong bond by transferring charge from the MTB to the hydroxyl group. Since MTBs are metallic, the charge transfer is larger as compared to the pristine system. In the case of a single vacancy, the anchoring atom i.e. oxygen, occupy the vacancy forming a strong bond with Mo atoms. The adsorption energy at vacancy sites is lower than that on MTBs by ≈2 eV.
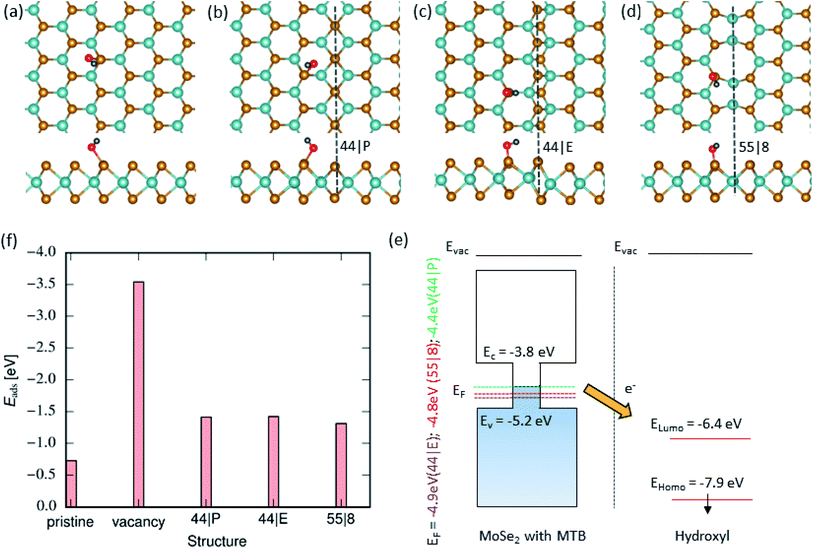 |
| | Fig. 2 Hydroxyl adsorption on MoSe2 monolayer. (a)–(d) Atomic structures of 2D MoSe2 with adsorbed hydroxyl radicals (top and side views). From left-to-right, the panels represent the lowest energy configurations of adsorbed hydroxyl groups in the pristine area and on the 44| P, 44| E and 55| 8 MTBs, respectively. (f) Adsorption energy of hydroxyl on top of MoSe2 in the pristine area, vacancy and next to the MTBs. (e) Schematically representation of the electronic structure of MoSe2 with various MTBs and isolated OH group. | |
3.3 Adsorption of oxygen
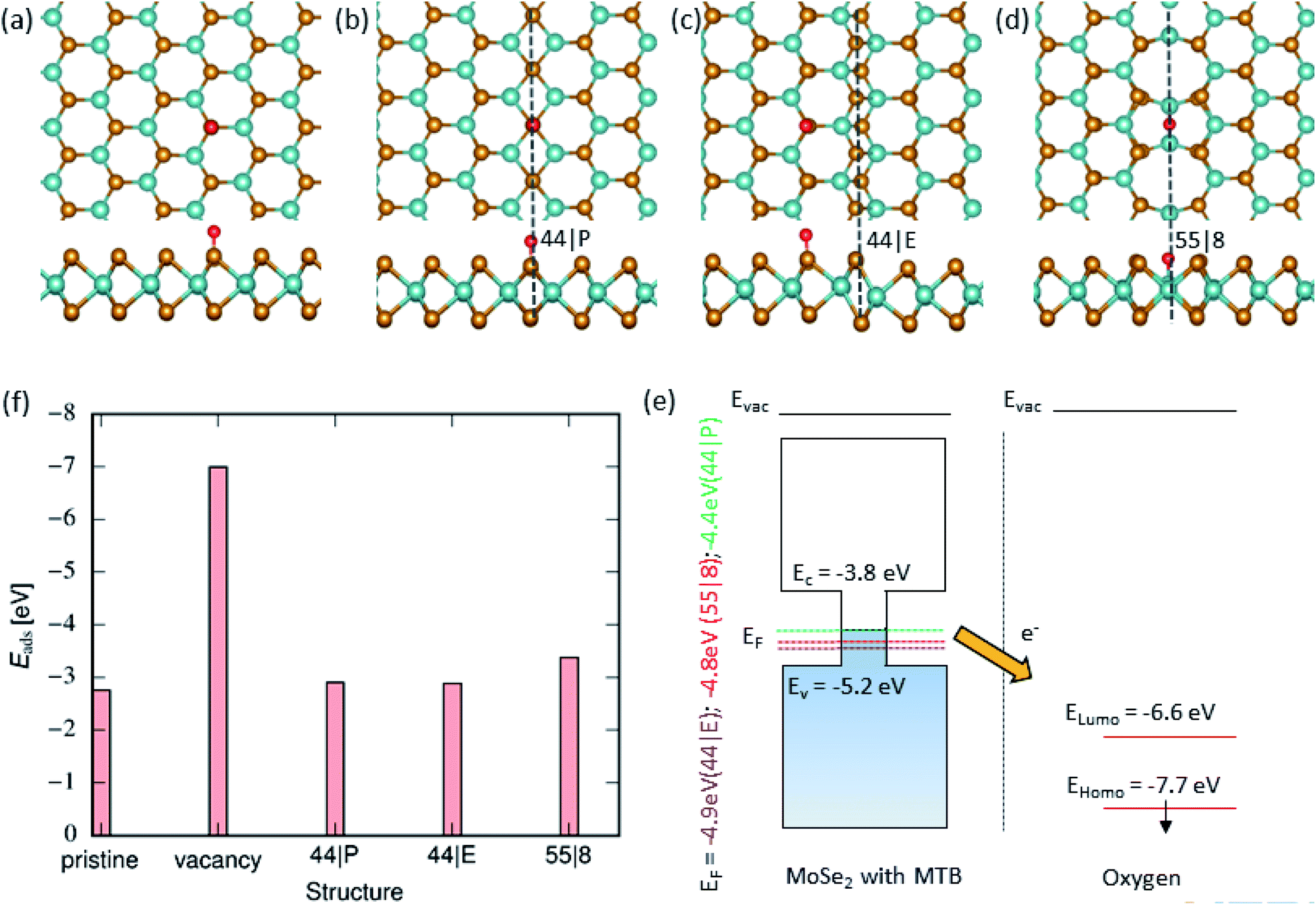
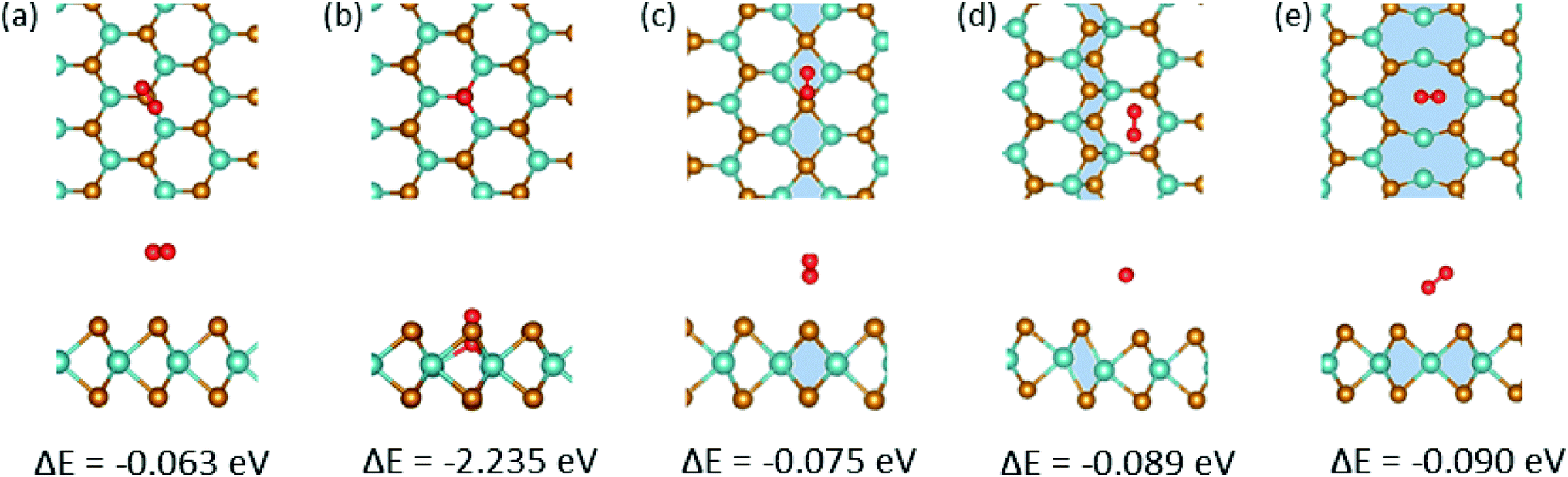
Next, we explored the adsorption of an O atom on the MoSe2 monolayer. It is known that point defects in TMDs, such as S-vacancies, can be active sites for dissociation of O2 molecules.57 Our calculations indicate that one needs 3.04 eV per atom for an isolated O2 molecule to dissociate. Thus, in agreement with the results of earlier studies,58 it is energetically unfavorable for the molecule to dissociate on pristine MoSe2 monolayer, as evident from Fig. 3(f), but the dissociation is exothermic on a vacancy, assuming that one O atoms takes the substitutional position and forms strong covalent bonds with the three adjacent Mo atoms, and the other is adsorbed on the surface. After dissociation, the other O atoms can diffuse to the surrounding area on the sample surface. Therefore, it is imperative to explore the interaction of an adsorbed O atom with MTBs in the MoSe2 monolayer. We found that the oxygen atom prefers to sit on top of the chalcogen atom as shown in Fig. 3(a–c). The only exception is the 55| 8 MTB, where oxygen atom is adsorbed in the middle of the Mo–Mo stripe, Fig. 3(d). The adsorption energy of the oxygen atom is naturally lower than that of the water molecule as the atom is chemisorbed on the surface. As compared to hydroxyl, the lower adsorption energies indicate stronger interaction of oxygen with MoSe2. The strong interaction with the 55| 8 MTB can also be deduced from the charge transfer picture given in Fig. 3. In addition, the adsorption of the O2 molecule on the 55| 8 MTB was studied. The adsorption energy of −0.090 eV was found. From Fig. S3,† by comparing the adsorption energies of the O2 molecule and two O atoms, it is clear that the adsorbed molecule may spontaneously dissociate to form two adsorbed O atoms and releasing energy of 0.86 eV.
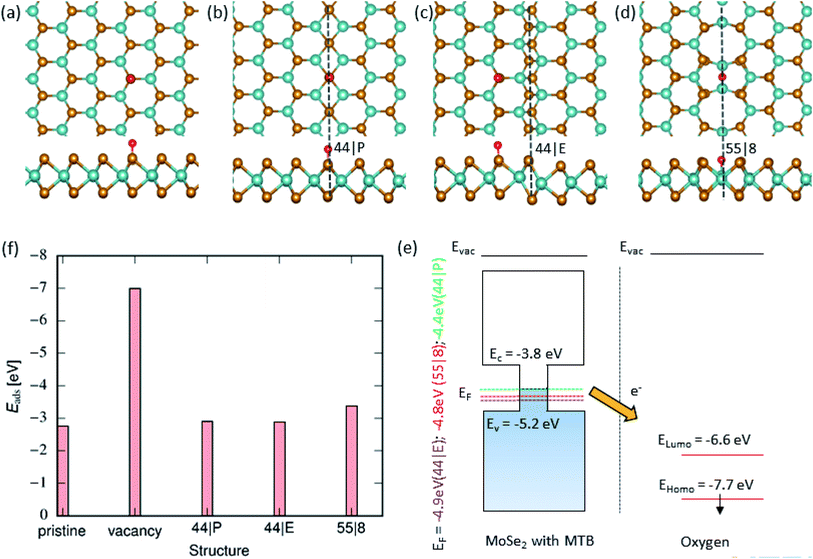 |
| | Fig. 3 Oxygen adsorption on MoSe2 monolayer. (a)–(d) Atomic structures of 2D MoSe2 with adsorbed oxygen (top and side views). From left to right, the panels shown the lowest energy configurations of adsorbed oxygen in the pristine area, on the 44| P, 44| E and 55| 8 MTBs, respectively. (f) Adsorption energy of oxygen on top of MoSe2 in the pristine area, vacancy and next to the MTBs. (f) Schematically illustration of the electronic structure of MoSe2 with MTB and isolated O atom. | |
Fig. 3(f) presents a comparison of adsorption energies at different MTBs. Among all the MTBs, the 55| 8 MTB shows the lowest adsorption energy, −3.4 eV, which corresponds to the strongest interaction between O and surface. Unlike OH adsorption, the difference in O adsorption energy between pristine and on the 44| P or 44| E MTBs is small ≈0.1 eV. The adsorption energy on the 55| 8 MTB is ≈0.6 eV lower than for the pristine sheet, suggesting preferential adsorption of O at the MTB. Besides, the adsorption of O is strongly influenced by presence of Se vacancies. The adsorption energy at vacancy sites is larger in comparison to MTB by ≈4 eV.
3.4 Adsorption of hydrogen
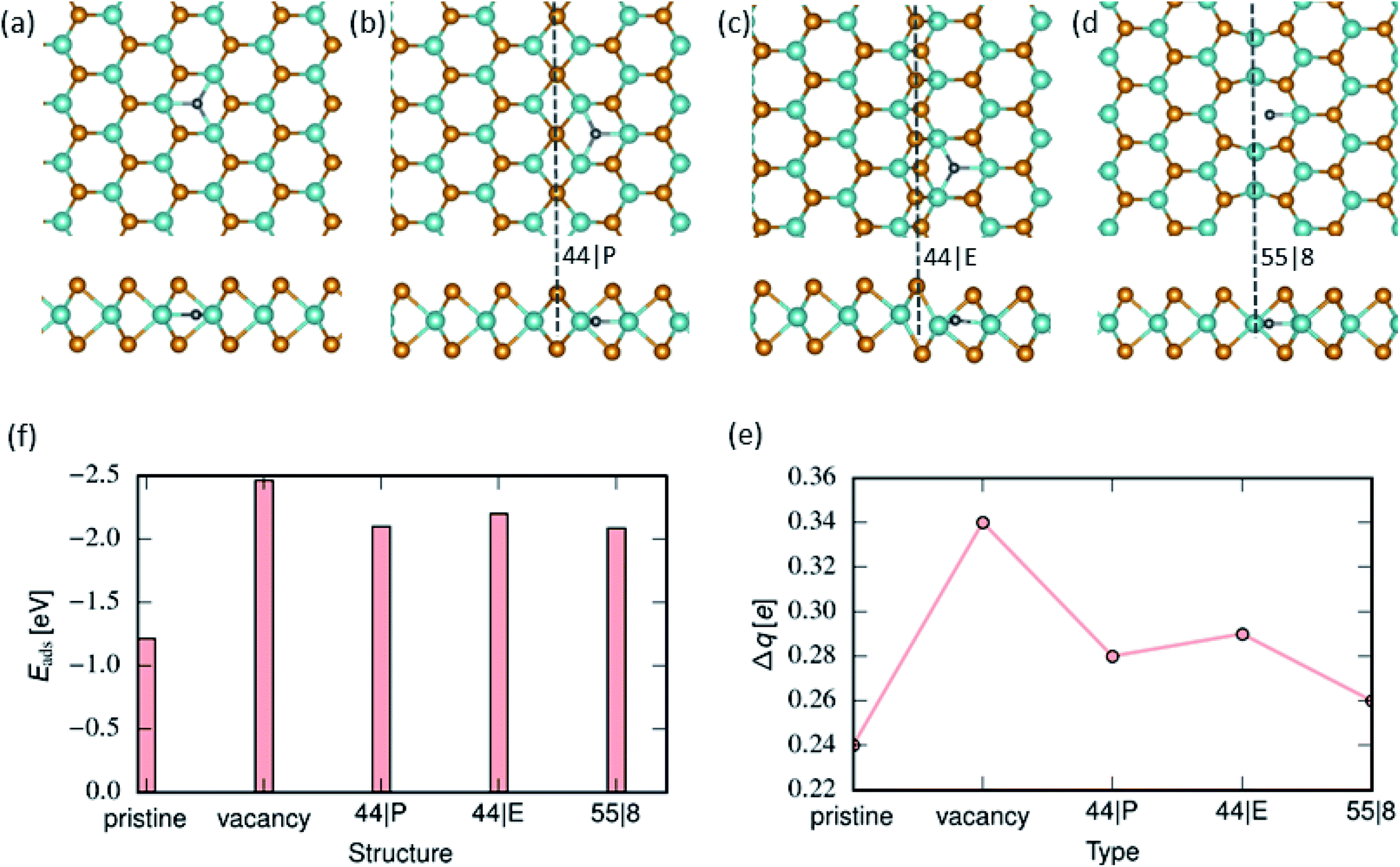
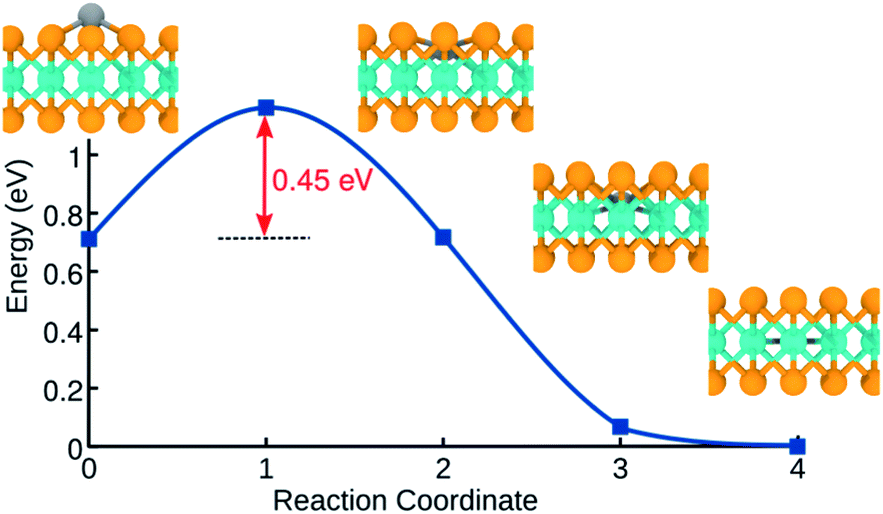
We further examined the energetics of adsorbed H atoms on the pristine MoSe2 monolayer. The calculations indicate that H atoms in the interstitial configuration are energetically more favorable by 0.72 eV than adatom configurations. Using NEB method, we calculated the energy barrier along the path between adatom configuration in the hollow site and the corresponding interstitial position, as shown in Fig. 5. The data indicates that the transition between two configurations requires overcoming an energy barrier of ca. 0.45 eV. As compared to MoS2, the larger primitive cell of MoSe2 provide more free space for H in the interstitial position. Similar behavior has been reported for H12 and transition metal atoms adsorbed on MoTe2.59 The lowest-energy position of H atoms in the Mo-atom plane indicates that H permeability of MoSe2 should be quite different from that of MoS2.
The strongest binding of an H atom to the 44| P and 44| E MTBs occurs at the center of the hexagon next to the MTB. For the 55| 8 MTB, however, H prefers to be embedded into the 8-fold ring of the MTB. Nevertheless, the adsorption energies of H atoms were found to be very similar for all types of MTBs. The selective adsorption of hydrogen can be attributed to the states available at the Fermi level. Similar to edge defects previously studied,33,60 there are many sharp peaks near the Fermi level, indicating the existence of many free electrons that can readily redistribute under any chemical perturbation leading to selective adsorption. The calculated adsorption energies of H, Fig. 4(f) indicate that Se vacancy is the most favourable adsorption site, followed by MTBs. We further analyzed the charge transfer from substrate MoSe2 to H atom. The results are presented in 4(e). H adsorption on Se vacancy site results in the largest charge transfer due to a strong interaction between the H atom and three adjacent Mo atoms. Moreover, the binding of hydrogen is stronger than that of the hydroxyl group, but weaker than the oxygen atom.
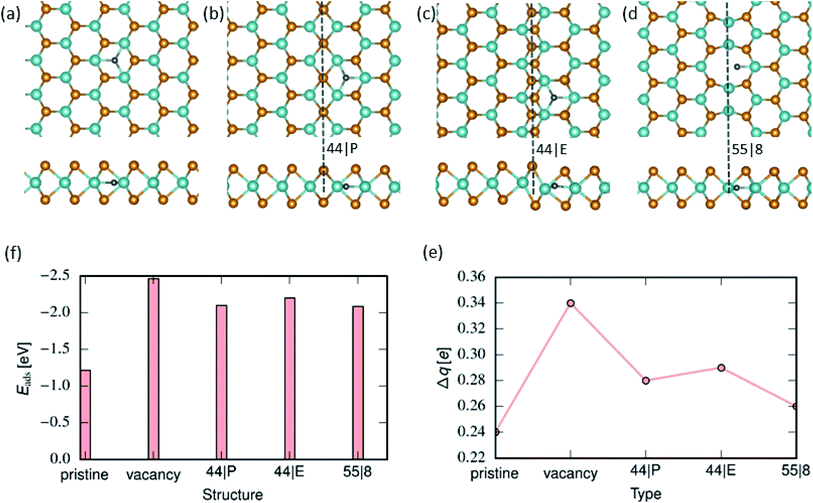 |
| | Fig. 4 Hydrogen adsorption on MoSe2 monolayer. (a)–(d) Atomic structures of 2D MoSe2 with an adsorbed hydrogen atom (top and side views). From left to right, the panels represent lowest energy configurations of adsorbed hydrogen atoms on pristine, 44| P, 44| E and 55| 8 MTBs respectively. (f) Adsorption energy of hydrogen atoms in the pristine area, on vacancy and next to MTBs. (e) Charge transferred from MoSe2 to H group, as revealed by the Bader analysis. | |
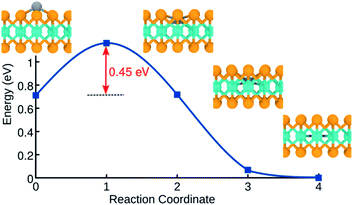 |
| | Fig. 5 Calculated energy profile of diffusion process for an adsorbed H atom on monolayer MoSe2. | |
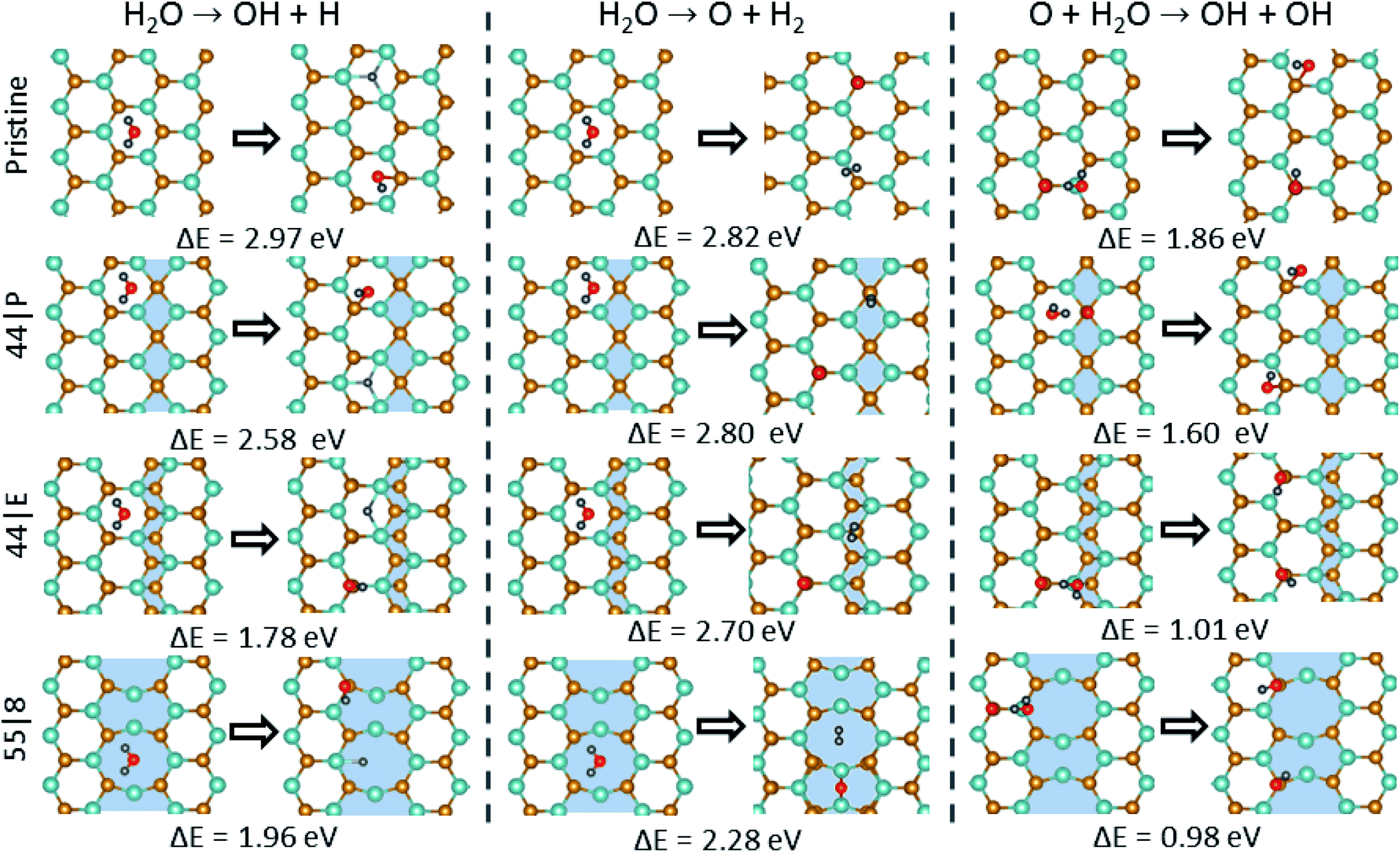
3.5 Water dissociation
Water dissociation is a key issue not only in the green production of hydrogen from water, but also because it plays an essential role in numerous catalytic reactions. The typical procedure for the hydrogen production from water on catalytic surfaces includes the splitting of water into adsorbed H and OH species (H2O + 2* ↔ H* + OH*). The adsorbed hydrogen atoms can join to form a molecule. Alternatively, the hydrogen atoms in the water molecule can be directly attached to form a hydrogen molecule thereby the separated O atom is adsorbed on the surface (H2O + * ↔ O* + H2). We evaluated the feasibility of these mechanisms that lead to the dissociation of a H2O molecule at MTBs. Assuming that O2 molecules adsorbed on vacancy sites can dissociate and the atoms diffuse on the outer Se plane, the important question is how these O atoms affect the dissociation of water on MoSe2. Here we considered two possible scenarios, as shown in Fig. 6: the dissociation of water on (1) the pristine surface, and (2) the oxidized surface. First, the energy barrier for H2O dissociation to OH radical and H on the pristine surface was evaluated. Our results show that an energy barrier of 2.97 eV is required to be overcome for the dissociation of water on the perfect basal plane, indicating that the direct dissociation is highly unlikely on a pristine MoSe2 monolayer. In the case of MTBs, the dissociation barrier is reduced, where for instance the lowest barrier of 1.96 eV was found for the 55| 8 MTB. Similarly, the energy barrier for water dissociation to O atom and hydrogen molecule is lower for MTBs than the pristine sheet. We further investigated the effect of oxidation on the dissociation of the H2O molecule. The presence of oxygen decreases the dissociation energy barrier by 1 eV as compared to the basal plane of the monolayer. In the case of MTBs, the energy barrier is further lowered by about 1 eV reaching to 0.98 eV for the 55| 8 MTB. Our findings suggest that the 55| 8 MTBs could be suitable defects for the dissociation of H2O where a strong chemical reaction with OH is accomplished by the interchange of electrons with the adjacent Mo atoms resulting in the separation of H and O atoms.
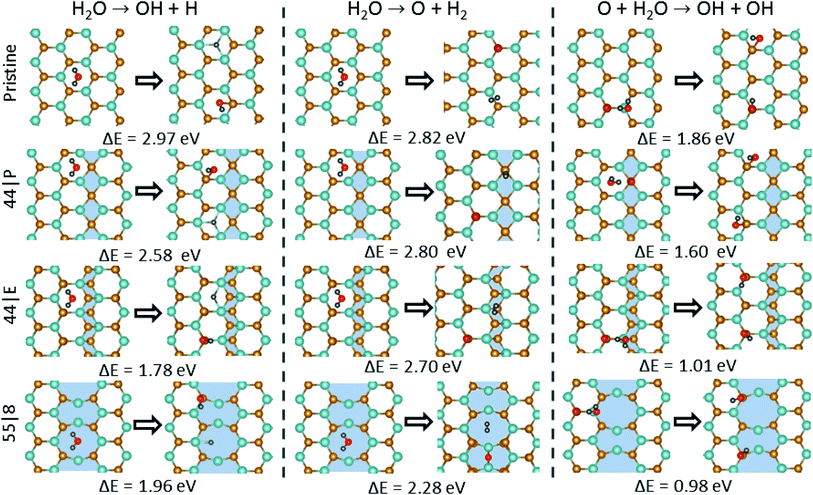 |
| | Fig. 6 Schematic view of water association and dissociation reactions and the energetics for the pristine system and that with 44| P, 44| E and 55| 8 MTBs. | |
3.6 Oxygen reduction reaction
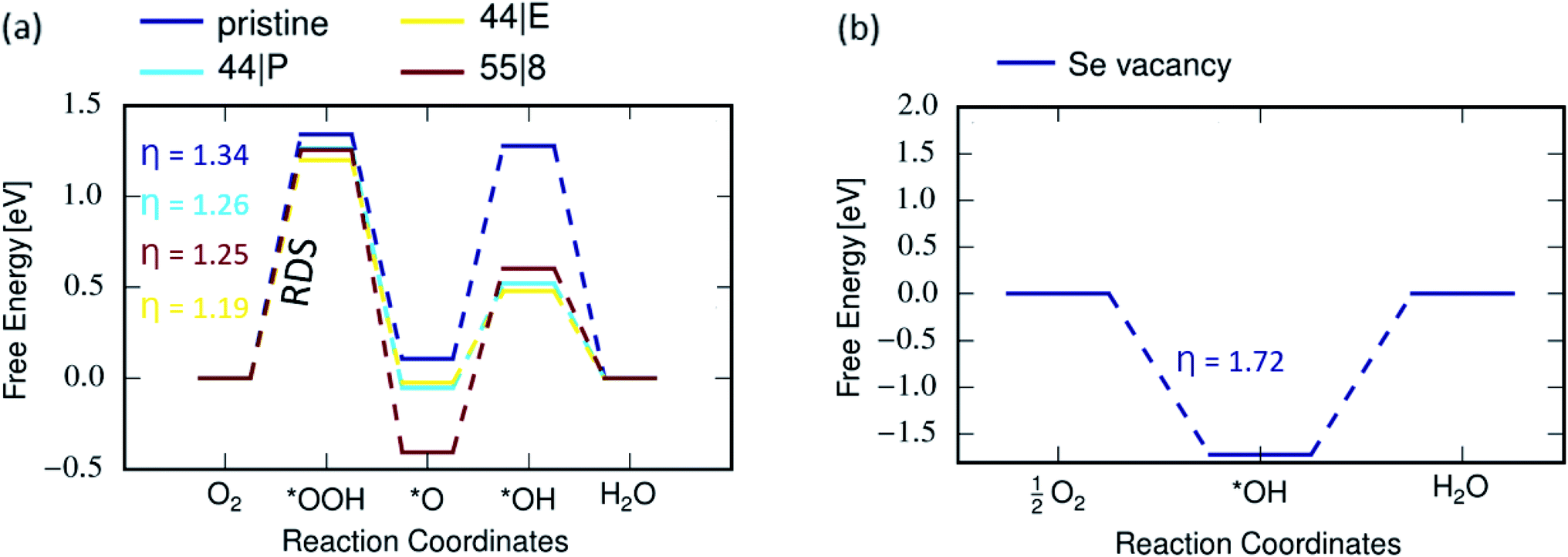
The first step for ORR process involves the adsorption of oxygen molecules on the electrode. The corresponding structures and energies of the MoSe2 sheets representing the electrodes are shown in Fig. 7. From calculation it is seen that oxygen molecules prefers to adsorb on vacancies in MoSe2. The free-energy diagram for the associative ORR mechanism is shown in Fig. 8. The ORR performance for the pristine system and with MTBs is calculated at equilibrium potential of 1.23 V as mentioned in the method section. The overpotential is the minimum additional energy barrier required to be overcome for the ORR process,53 meaning that the higher the overpotential, the more difficult the reaction becomes. With the 4e-process described in Fig. 8, the rate-determining step (RDS) is the protonation of the adsorbed oxygen to *OOH which requires the energy of 1.34 eV in the pristine case.This can be explained due to the weak vdW binding of O2 molecule and the strong *OOH binding to the catalyst.51,53 Hydrogen peroxide can be formed as an intermediate of ORR only by the 2e− associative mechanism in eqn (4) instead of *O + H2O. Hydrogen peroxide has been detected during ORR, but it remains unclear whether it is a key intermediate of the dominant ORR mechanism or a side-product.61 The strong hydroperoxyl adsorption on MoSe2 as shown in Fig. S2† favours O–O bond dissociation in the adsorbed *OOH. Thus all catalysts with strong interaction between the reaction site and O species could easily dissociate the O2 molecule leading to H2O formation, while a too-weak one leads to H2O2 reaction path.53 Here, in this case due to the above mentioned property, a 4e− oxygen reduction to water is preferred. Also, the kinetics for the H2O2 and H2O pathways is shown in Fig. S4.† Although the determining step is the same as in the ORR process on MTBs, the overpotentials are reduced, following the order 44| P > 55| 8 > 44| E. Therefore, the ORR process on 44| E MTB is found to be more efficient, with the overpotential of 1.19 eV, in comparison to the pristine system and other investigated MTBs. Our results show that ORR in presence of Se vacancy follows a 2e− process because of the absence of stable OOH adsorbates and strong adsorption of O2 at the vacancy site (Fig. 8(b)).
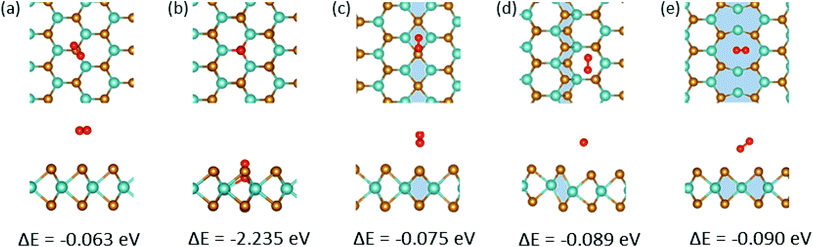 |
| | Fig. 7 Adsorption of O2 molecules on MoSe2 at various sites. (a) Pristine (b) Se vacancy (c) 44| P, (d) 44| E, (e) 55| 8 MTBs. | |
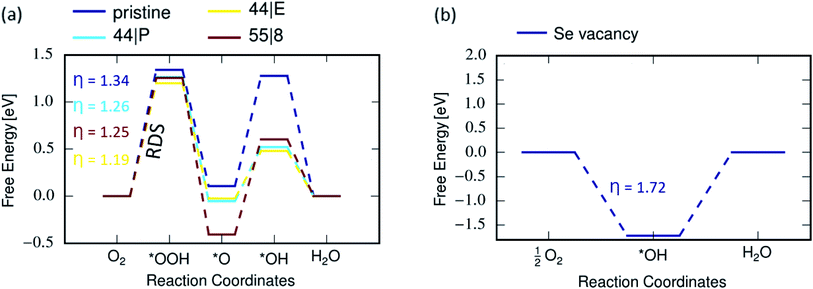 |
| | Fig. 8 Gibbs free energy diagrams of ORR along the (a) 4e− pathway on pristine and defective MoSe2; (b) 2e− pathway for Se vacancy, where η denotes the overpotential. | |
The overpotential for pristine MoSe2 is 1.34 V, significantly higher than 0.45 V of Pt electrocatalyst,53 0.47 V of defective graphene,62 and 0.67 V of N-doped graphene.63 The features of the Gibbs free energy diagram for MoSe2 are similar to those reported for the defective WTe2.38 In the case of MTBs, the ORR overpotential is found to be ≈1.2 V indicating better ORR characteristics than at Se vacancy.
4 Conclusions
In conclusion, we investigated the adsorption of H2O, OH, O, H and O2 species on monolayer MoSe2. The adsorption of H2O is the weakest, with an adsorption energy ranging from −0.18 eV to −0.20 eV. On the other hand, adsorption of O is the strongest, with the adsorption energy being in the range of −2.8 eV to −3.4 eV. It is found that for all the considered adsorbates except for water, the most preferred site of adsorption are Se vacancies. We demonstrated that the adsorbed OH and O species can be used as anchor points for selective adsorption of water at MTBs (55| 8 MTB for O adsorption). We further investigated the water dissociation and association on different MTBs. From our calculation, it is evident that energy required for dissociation of water molecules is reduced by ≈1.0 eV as compared to the pristine area when it happens on a 55| 8 MTB. With regard to the behavior of O2, calculations show that oxygen prefers to adsorb as dissociated atoms on the 55| 8 MTB rather than as a molecule. Moreover, ORR process on pristine MoSe2 and MTBs has lower overpotential than on a Se vacancy, thus indicating better ORR efficiency than at vacancies. Although, based on the results of previous studies,30,31 the ORR reactivity at edges is expected to be higher than at MTBs, potentially much higher concentration of MTBs, which can be produced during or after the growth by TM atom deposition26 or electron beam irradiation,23–25 in comparison to edges, suggests that MTBs can have a strong influence on the ORR efficiency of MoSe2 sheets. Overall, our findings provide important insights into the adsorption of water and its derivatives on MoSe2 monolayers with MTBs which should be accounted for when assessing the catalytic performance of this material in electrochemical energy applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge funding from the German Research Foundation (DFG), projects KR 4866/2-1 and KR 4866/6-1. M. B. further acknowledges support from the National Science Foundation under awards NSF-CHE 1801199 and 2140038. The computational support from the Technical University of Dresden computing cluster (TAURUS), from High Performance Computing Center (HLRS) in Stuttgart, Germany, and CSC Finland, is gratefully appreciated.
Notes and references
- K. F. Mak, C. Lee, J. Hone, J. Shan and T. F. Heinz, Phys. Rev. Lett., 2010, 105, 136805–136809 CrossRef PubMed.
- A. Kuc, N. Zibouche and T. Heine, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 245213–245217 CrossRef.
- D. J. Late, Y.-K. Huang, B. Liu, J. Acharya, S. N. Shirodkar, J. Luo, A. Yan, D. Charles, U. V. Waghmare, V. P. Dravid and C. N. R. Rao, ACS Nano, 2013, 7, 4879–4891 CrossRef CAS PubMed.
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699–712 CrossRef CAS PubMed.
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed.
- Z. Li, Y. Xiao, Y. Gong, Z. Wang, Y. Kang, S. Zu, P. M. Ajayan, P. Nordlander and Z. Fang, ACS Nano, 2015, 9, 10158–10164 CrossRef CAS PubMed.
- Z. Yin, H. Li, H. Li, L. Jiang, Y. Shi, Y. Sun, G. Lu, Q. Zhang, X. Chen and H. Zhang, ACS Nano, 2011, 6, 74–80 CrossRef PubMed.
- A. Avsar, C.-y. Cheon, M. Pizzochero, M. Tripathi, A. Ciarrocchi, O. V. Yazyev and A. Kis, Nat. Commun., 2020, 11, 4806 CrossRef CAS PubMed.
- P. M. Coelho, H.-P. Komsa, K. Lasek, V. Kalappattil, J. Karthikeyan, M. Phan, A. V. Krasheninnikov and M. Batzill, Adv. Electron. Mater., 2019, 5, 1900044 CrossRef.
- Y. Kang, Y. Gong, Z. Hu, Z. Li, Z. Qiu, X. Zhu, P. M. Ajayan and Z. Fang, Nanoscale, 2015, 7, 4482–4488 RSC.
- S. Helveg, J. V. Lauritsen, E. Lægsgaard, I. Stensgaard, J. K. Nørskov, B. S. Clausen, H. Topsøe and F. Besenbacher, Phys. Rev. Lett., 2000, 84, 951–954 CrossRef CAS PubMed.
- T. Kosmala, H. C. Diaz, H.-P. Komsa, Y. Ma, A. V. Krasheninnikov, M. Batzill and S. Agnoli, Adv. Energy Mater., 2018, 8, 1800031 CrossRef.
-
A. Kolobov and J. Tominaga, Two-Dimensional Transition Metal Dichalcogenides, Springer, 2016 Search PubMed.
- V. Sorkin, H. Pan, H. Shi, S. Y. Quek and Y. W. Zhang, Crit. Rev. Solid State Mater. Sci., 2014, 39, 319–367 CrossRef CAS.
- Z. Lin, B. R. Carvalho, E. Kahn, R. Lv, R. Rao, H. Terrones, M. A. Pimenta and M. Terrones, 2D Materials, 2016, 3, 022002 CrossRef.
- A. V. Krasheninnikov, Nanoscale Horiz., 2020, 5, 1447–1452 RSC.
- A. M. van der Zande, P. Y. Huang, D. A. Chenet, T. C. Berkelbach, Y. You, G.-H. Lee, T. F. Heinz, D. R. Reichman, D. A. Muller and J. C. Hone, Nat. Mater., 2013, 12, 554–561 CrossRef CAS PubMed.
- W. Zhou, X. Zou, S. Najmaei, Z. Liu, Y. Shi, J. Kong, J. Lou, P. M. Ajayan, B. I. Yakobson and J.-C. Idrobo, Nano Lett., 2013, 13, 2615–2622 CrossRef CAS PubMed.
- O. Yazyev and Y. P. Chen, Nat. Nanotechnol., 2014, 9, 755–767 CrossRef CAS PubMed.
- K. Lasek, J. Li, S. Kolekar, P. M. Coelho, L. Guo, M. Zhang, Z. Wang and M. Batzill, Surf. Sci. Rep., 2021, 100523 CrossRef CAS.
- M. Chubarov, T. H. Choudhury, D. R. Hickey, S. Bachu, T. Zhang, A. Sebastian, A. Bansal, H. Zhu, N. Trainor, S. Das, M. Terrones, N. Alem and J. M. Redwing, ACS Nano, 2021, 15, 2532–2541 CrossRef CAS PubMed.
- H.-P. Komsa and A. V. Krasheninnikov, Adv. Electron. Mater., 2017, 3, 1600468 CrossRef.
- O. Lehtinen, H.-P. Komsa, A. Pulkin, M. B. Whitwick, M.-W. Chen, T. Lehnert, M. J. Mohn, O. V. Yazyev, A. Kis, U. Kaiser and A. V. Krasheninnikov, ACS Nano, 2015, 9, 3274–3283 CrossRef CAS PubMed.
- J. Lin, S. T. Pantelides and W. Zhou, ACS Nano, 2015, 9, 5189–5197 CrossRef CAS PubMed.
- Y.-C. Lin, T. Björkman, H.-P. Komsa, P.-Y. Teng, C.-H. Yeh, F.-S. Huang, K.-H. Lin, J. Jadczak, Y.-S. Huang, P.-W. Chiu, A. V. Krasheninnikov and K. Suenaga, Nat. Commun., 2015, 6, 6736 CrossRef CAS PubMed.
- P. M. Coelho, H.-P. Komsa, H. Coy Diaz, Y. Ma, A. V. Krasheninnikov and M. Batzill, ACS Nano, 2018, 12, 3975–3984 CrossRef CAS PubMed.
- U. Paulus, A. Wokaun, G. Scherer, T. Schmidt, V. Stamenkovic, N. Markovic and P. Ross, Electrochim. Acta, 2002, 47, 3787–3798 CrossRef CAS.
- H. R. Colón-Mercado and B. N. Popov, J. Power Sources, 2006, 155, 253–263 CrossRef.
- X. Tong, X. Zhan, D. Rawach, Z. Chen, G. Zhang and S. Sun, Prog. Nat. Sci.: Mater. Int., 2020, 30, 787–795 CrossRef CAS.
- Y. Hao, L.-C. Xu, J. Pu, L. Wang and L.-F. Huang, Electrochim. Acta, 2020, 338, 135865 CrossRef CAS.
- A. Y. S. Eng, A. Ambrosi, Z. Sofer, P. Šimek and M. Pumera, ACS Nano, 2014, 8, 12185–12198 CrossRef CAS PubMed.
- W. Shi, K. Fan and Z. Wang, Phys. Chem. Chem. Phys., 2018, 20, 29423–29429 RSC.
- C. Tsai, K. Chan, F. Abild-Pedersen and J. K. Nørskov, Phys. Chem. Chem. Phys., 2014, 16, 13156–13164 RSC.
- B. Mohanty, M. Ghorbani-Asl, S. Kretschmer, A. Ghosh, P. Guha, S. K. Panda, B. Jena, A. V. Krasheninnikov and B. K. Jena, ACS Catal., 2018, 8, 1683–1689 CrossRef CAS.
- H. Huang, X. Feng, C. Du and W. Song, Chem. Commun., 2015, 51, 7903–7906 RSC.
- H. Huang, X. Feng, C. Du, S. Wu and W. Song, J. Mater. Chem. A, 2015, 3, 16050–16056 RSC.
- Z. Wang, J. Zhao, Q. Cai and F. Li, J. Mater. Chem. A, 2017, 5, 9842–9851 RSC.
- O. M. Na, N. T. X. Huynh, P. T. Thi, V. Chihaia and D. N. Son, RSC Adv., 2020, 10, 8460–8469 RSC.
- S. Barja, S. Wickenburg, Z.-F. Liu, Y. Zhang, H. Ryu, M. M. Ugeda, Z. Hussain, Z.-X. Shen, S.-K. Mo, E. Wong, M. B. Salmeron, F. Wang, M. F. Crommie, D. F. Ogletree, J. B. Neaton and A. Weber-Bargioni, Nat. Phys., 2016, 12, 751–756 Search PubMed.
- W. Jolie, C. Murray, P. S. Weiß, J. Hall, F. Portner, N. Atodiresei, A. V. Krasheninnikov, C. Busse, H.-P. Komsa, A. Rosch and T. Michely, Phys. Rev. X, 2019, 9, 011055 Search PubMed.
- Y. Ma, H. C. Diaz, J. Avila, C. Chen, V. Kalappattil, R. Das, M.-H. Phan, T. Čadež, J. M. P. Carmelo, M. C. Asensio and M. Batzill, Nat. Commun., 2017, 8, 14231 Search PubMed.
- G. Ye, Y. Gong, J. Lin, B. Li, Y. He, S. T. Pantelides, W. Zhou, R. Vajtai and P. M. Ajayan, Nano Lett., 2016, 16, 1097–1103 CrossRef CAS PubMed.
- W. Zhan, F. Jia, Y. Yuan, C. Liu, K. Sun, B. Yang and S. Song, J. Hazard. Mater., 2020, 384, 121382 CrossRef CAS PubMed.
- J. Petö, T. Ollár, P. Vancsó, Z. I. Popov, G. Z. Magda, G. Dobrik, C. Hwang, P. B. Sorokin and L. Tapasztó, Nat. Chem., 2018, 10, 1246–1251 CrossRef PubMed.
- J. Li, T. Joseph, M. Ghorbani-Asl, S. Kolekar, A. V. Krasheninnikov and M. Batzill, Nanoscale, 2021, 13, 1038–1047 RSC.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comp. Mat. Sci., 1996, 6, 15 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
- Y. Hao, P.-L. Gong, L.-C. Xu, J. Pu, L. Wang and L.-F. Huang, ACS Appl. Mater. Interfaces, 2019, 11, 46327–46336 CrossRef CAS PubMed.
- S. Tian, C. Deng, Y. Tang and Q. Tang, J. Phys. Chem. C, 2020, 124, 24899–24907 CrossRef CAS.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS PubMed.
-
M. Chase, NIST-JANAF Thermochemical Tables, American Institute of Physics, vol. 1, 1998, 4th edn Search PubMed.
- C. Ataca and S. Ciraci, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 195410 CrossRef.
- G. Levita, P. Restuccia and M. Righi, Carbon, 2016, 107, 878–884 CrossRef CAS.
- B. Akdim, R. Pachter and S. Mou, Nanotechnology, 2016, 27, 185701 CrossRef PubMed.
- R. C. Longo, R. Addou, S. KC, J.-Y. Noh, C. M. Smyth, D. Barrera, C. Zhang, J. W. P. Hsu, R. M. Wallace and K. Cho, 2D Materials, 2017, 4, 025050 CrossRef.
- J. Karthikeyan, H.-P. Komsa, M. Batzill and A. V. Krasheninnikov, Nano Lett., 2019, 19, 4581–4587 CrossRef CAS PubMed.
- H. Shu, D. Zhou, F. Li, D. Cao and X. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 42688–42698 CrossRef CAS PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- Y. Jia, L. Zhang, A. Du, G. Gao, J. Chen, X. Yan, C. L. Brown and X. Yao, Adv. Mater., 2016, 28, 9532–9538 CrossRef CAS PubMed.
- S. Sinthika, U. V. Waghmare and R. Thapa, Small, 2018, 14, 1703609 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00429h |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
M.
Ghorbani-Asl
a,
M.
Ghorbani-Asl