
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design of tandem catalysts using a core–shell structure approach†
Esteban
Gioria
 ab,
Liseth
Duarte-Correa
c,
Najmeh
Bashiri
ae,
Walid
Hetaba
cd,
Reinhard
Schomaecker
e and
Arne
Thomas
*a
ab,
Liseth
Duarte-Correa
c,
Najmeh
Bashiri
ae,
Walid
Hetaba
cd,
Reinhard
Schomaecker
e and
Arne
Thomas
*a
aTechnische Universität Berlin, Fakultät II, Institut für Chemie: Funktionsmaterialen, Sekretariat BA2, Hardenbergstraße 40, 10623 Berlin, Germany. E-mail: e.gioria@tu-berlin.de; arne.thomas@tu-berlin.de
bInstitute of Research on Catalysis and Petrochemistry, INCAPE, UNL-CONICET, Santiago del Estero 2829, 3000 Santa Fe, Argentina. E-mail: egioria@fiq.unl.edu.ar
cFritz Haber Institute of the Max Planck Society, Department of Inorganic Chemistry, Faradayweg 4-6, 14195 Berlin, Germany
dMax Planck Institute for Chemical Energy Conversion, Department of Heterogeneous Reactions, Stiftstraße 34-36, 45470 Mülheim an der Ruhr, Germany
eTechnische Universität Berlin, Fakultät II, Institut für Chemie, Sekretariat TC 8 Straße des 17. Juni 124, 10623 Berlin, Germany
First published on 25th May 2021
Abstract
A facile and rational approach to synthesize bimetallic heterogeneous tandem catalysts is presented. Using core–shell structures, it is possible to create spatially controlled ensembles of different nanoparticles and investigate coupled chemocatalytic reactions. The CO2 hydrogenation to methane and light olefins was tested, achieving a tandem process successfully.
Supported metal catalysts are widely employed for diverse chemical reactions. Different techniques are used for their preparation, such as incipient wetness impregnation and subsequent reduction of a metallic precursor on pre-formed supports, but also co-precipitation, sol–gel processes, and many others.1–3 However, the conventional techniques for the preparation of supported metal catalysts have several limitations, especially regarding the control of particle size and particle distribution on the support, which makes it difficult to relate the catalytic activity and selectivity to a well-defined active site.4,5 In this respect, the deposition of pre-formed monodisperse metal nanoparticles has some advantages,6,7 especially regarding the control on their particle size.8–11
Many important chemical processes involve more than one reaction and therefore two or more different catalytic centers are required to obtain the desired product. In this context, tandem catalysts have gained a lot of attention in recent years.12–16 These multifunctional catalysts are able to carry out two or more consecutive chemical transformations that cannot be achieved using a single catalyst. Such one-pot tandem reactions are of major interest in sustainable chemistry, as they can reduce the number of complicated and energy-consuming separation and purification steps and the amount of side- or waste products. Coupling of several chemical reactions in one single system furthermore enables a more efficient heat and mass transfer control, thus in principle can be carried out with lower costs.17,18
However, performing consecutive reactions in ‘one pot’ greatly reduces the degrees of freedom for the catalysts and the catalysed reactions. To achieve acceptable catalytic turnovers, both catalysts must be present during the entire reaction sequence without interference by any reactant, intermediate, or product. Furthermore, unfavourable interactions between the two catalysts caused by mutual corrosion, inhibition, or competition for the substrates must be avoided. Finally, both catalysts must show comparable or adjusted activity under the same reaction conditions, i.e., temperature, pressure, solvent, or gas composition. Due to these delicate prerequisites, the number of successful examples for ‘one-pot’ multiple catalytic reactions is still limited. So far, most of the reported ‘one-pot’ reactions involve molecular catalysts, while the above-mentioned obstacles might be easier alleviated using solid systems. Therefore the development of heterogeneous tandem materials is of great interest.19,20
As mentioned before, homogeneity in particle size is an important parameter to control catalytic reactions. However, in a solid tandem catalyst, it can be assumed that also the distance between the different active sites is a key factor. This is not considered in conventional multicomponent catalysts, in which different metallic nanoparticles are immobilized on specific support without further spatial control. Indeed, there are only a few works that report techniques to control the particle distance at the nanometric scale.21–24 Beaumont et al. demonstrated the hydrogen spillover and surface diffusion phenomena onto silica using the kinetics of CO2 methanation on size selected platinum and cobalt nanoparticles.25 The pre-formed nanoparticles were loaded on silica by co-impregnation and compared to mechanical mixtures of Co/SiO2 and Pt/SiO2. They found that by increasing the spatial separation between cobalt and platinum entities, the apparent activation energy dropped drastically. This is certainly a facile synthesis strategy; however, the nanoparticle distance cannot be finely controlled since they are randomly distributed at the mesoscale on the co-impregnated catalyst and at the microscale in the mechanical mixture.
Reducing the structural complexity of multicomponent catalysts might allow better control of the synergistic effects between two chemo-catalytically active sites and thus gain a deeper understanding of reaction mechanisms. In this work, we report a simple, robust and scalable approach using core-NP–shell-NP structures to tune the distance between platinum nanoparticles (PtNP) and cobalt nanoparticles (CoNP). Both actives centers are spaced by a silica mesoporous shell and form an orthogonal tandem catalyst.26 The shell allows the access of the reactants to the first active center located on the core, and the diffusion of the intermediary molecules to the second one at the outer shell. This methodology can be applied to the design of several multi-metallic materials, suitable for the study of diverse chemical reactions.
As one important reaction, the hydrogenation of carbon dioxide employing green hydrogen is a potential path for the sustainable production of methane and further added-value products like light olefins and alcohols.27,28 Therefore, in this work we report the design, synthesis and catalytic performance of a tandem catalyst for the CO2 hydrogenation to form methane and light olefins.
For many core–shell type catalysts reported in the literature, monometallic nanoparticles as core are surrounded with a metal oxide or silica shell.29–33 Thus, core–shell structures with two or more types of active metals are still limited. Recently, Xie et al. reported a tandem catalyst based on a 35 nm ceria core with platinum nanoparticles, covered by a 25 nm silica shell and cobalt nanoparticles on top.34 However, the entire core–shell structures have still diameters of just a few nanometres and therefore the issues regarding handling, separation and upscaling remain.35
To circumvent these problems, here we decided not to use the first metal nanoparticle catalyst as the core, but uniform, monodisperse silica particles of significantly larger size (∼200 nm) prepared by a Stöber process. Using these silica particles as both support and core of the core–shell catalyst allowed that they can easily be separated after the synthesis and that the core–shell catalysts can be prepared on large scales. Thus, the dense silica core was decorated with platinum nanoparticles, followed by the synthesis of a uniform mesoporous silica shell. Then, pre-formed cobalt nanoparticles were deposited on top. The mesoporous silica shell not only acts as a physical spacer between both active centers but also as a protective layer against platinum nanoparticle agglomeration, while its mesoporosity ensures sufficient diffusion of the molecules between both sites.36 Finally, the catalysts were tested for the carbon dioxide reduction to methane or light hydrocarbons.
Fig. 1 indicates the different steps for the synthesis of the core–shell catalyst. Silica spheres of 200 nm were obtained following a modified Stöber method, from tetraethyl orthosilicate (TEOS) as silica precursor. The nanospheres were characterized by scanning electron microscopy (SEM) (Fig. 2a and S1†) and nitrogen adsorption, indicating a low surface area of 20 m2 g−1 (Fig. S2†). The relatively large particle size was chosen to gain a reproducible synthesis, monodisperse particle size distribution, and most importantly to facilitate isolation and purification of the material.
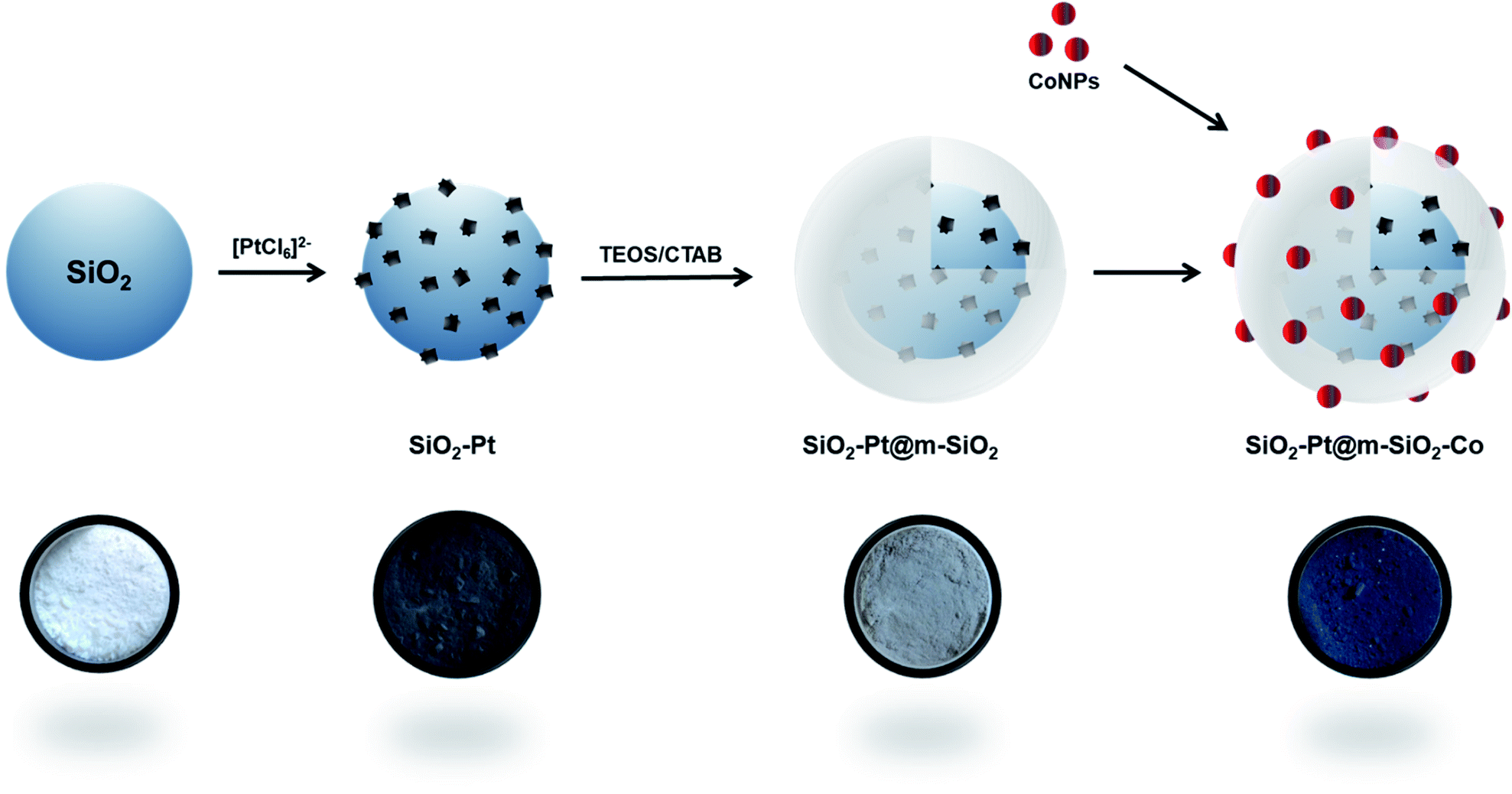 | ||
| Fig. 1 Steps applied for the synthesis of the core–shell catalyst and their respective digital photographs. | ||
 | ||
| Fig. 2 SEM and TEM micrographs of the different structures: (a) SiO2, (b) SiO2–NH2, (c and d) SiO2–Pt, (e and f) SiO2–Pt@m-SiO2, (g and h) SiO2–Pt@m-SiO2–Co. | ||
In the following step, the surface of the silica nanospheres was grafted with amino groups employing (3-aminopropyl)triethoxysilane (APTES) and ethanol as solvent. The particle size distribution was not modified (Fig. 2b and S3†) and the grafting effectiveness was corroborated by X-ray photoelectron spectroscopy (XPS) (Fig. S4†).
The functionalization of the surface allows the in situ formation of platinum nanoparticles employing the strong electrostatic adsorption method, as it can be seen in the schematic representation in Fig. S5.† Under acidic conditions, the platinum complex [PtCl6]2− is strongly attracted to the positively charged amino groups. As a next step, the metallic complex was in situ reduced at room temperature employing ethanol as solvent and NaBH4 as a reducing agent. In these conditions, small and monodispersed PtNP of 3.9 ± 1.1 nm were formed. The platinum nanoparticles deposition was corroborated by transmission electron microscopy (TEM) (Fig. 2c and d) and X-ray diffraction (XRD) (Fig. S6†).
For the platinum nanoparticle deposition, both conventional magnetic stirring and ultrasound techniques were compared. Also, water and ethanol were employed as solvents. However, ethanol and conventional magnetic stirring were selected due to the simplicity and quality of the final SiO2–Pt spheres. In contrast to the use of ethanol as solvent, water tends to form larger and agglomerated Pt nanoparticles (Fig. S7†), showing that the nanoparticles' growth rate strongly depends on the polarity and dielectric constant of the solvent molecule.37 Notably, the use of a 200 nm silica support allows easy recovery of the catalyst using mild centrifugation conditions (Fig. S8†).
The SiO2–Pt structure was further encapsulated with a mesoporous silica shell obtained by a sol–gel process. The nanospheres were dispersed in a mixture containing ethanol, deionized water, and cetyltrimethylammonium bromide (CTAB) as a soft-templating agent for the mesoporous channels. After calcination treatment for 2 h at 350 °C in air, a uniform silica shell with a mean thickness of ca. 50 nm was obtained. The calcination temperature was selected considering the thermogravimetric analysis of the organic template in air (Fig. S9a and b†). Furthermore, the platinum nanoparticles did not show any changes after the silica shell is formed, confirmed by TEM and scanning-TEM energy dispersive X-ray spectroscopy (EDS) measurements (Fig. 2e, f and S10†). Also, there is no evidence of migration or leaching of the PtNP into the channels of the mesoporous structure. Nitrogen adsorption studies reveal that the surface area increases from 20 m2 g−1 to 503 m2 g−1 with a monomodal pore size distribution of 2.3 nm, indicating the mesoporous nature of the shell structure (Fig. S11†).
Finally, cobalt nanoparticles were pre-formed in order to obtain monodisperse active centers. In the Fischer–Tropsch reaction, it is reported that the particle size plays an important role in the catalytic performance. In general terms, CO conversion decreases for cobalt nanoparticles below 10 nm.38–40 Therefore, 15 nm CoNP were adopted as an appropriate value. Regarding the CoNP synthesis, it is well-known that it is not possible to obtain stable colloids of monodispersed cobalt nanoparticles using water as the solvent, as oxidation and agglomeration is promoted. Therefore, CoNP were formed in a hydrophobic medium employing a thermal decomposition route.41 The metallic precursor, dicobalt octacarbonyl Co2(CO)8 was dissolved in o-dichlorobenzene and quickly injected in a hot solution of oleic acid in o-dichlorobenzene. Using this approach, the metallic complex is thermally decomposed to form metallic CoNP. Oleic acid acts as a capping agent, controlling the growth kinetics of the metallic nanoparticles. After recovering and dispersion in hexane, monodisperse cobalt nanoparticles of 15.5 ± 3.3 nm were obtained (Fig. S12†).
In a final step, both SiO2–Pt@m-SiO2 and the CoNP were suspended in hexane, mixed and dried at room temperature. A final calcination step was applied to remove all the organic compounds. Nitrogen adsorption studies of the final structure reveal a surface area of 167 m2 g−1, with a monomodal pore size of 2.4 nm (Fig. S13†).
The SiO2–Pt@m-SiO2–Co catalyst have a metal loading of 0.23% Pt and 12% Co, determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES). Besides, the cobalt nanoparticles were successfully dispersed on the surface of the shell, corroborated by TEM and SEM (Fig. 2g, h and S14†).
Following this protocol, it is possible to obtain a specific arrangement of metal nanoparticles spaced by a mesoporous silica shell at a nanometric scale, as it is verified by STEM-EDS studies (Fig. 3).
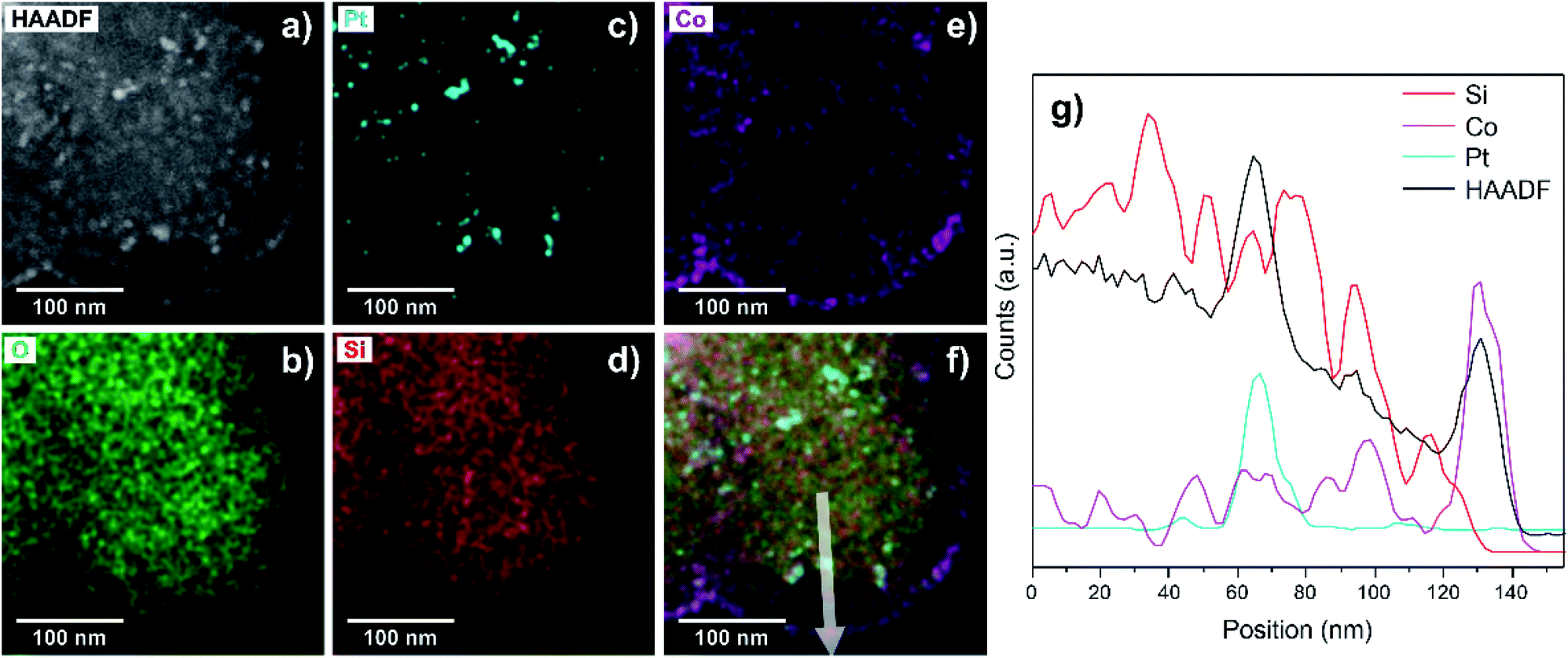 | ||
| Fig. 3 STEM image and EDS analysis of the core-Pt–shell-Co structures. (a) High-Angle Annular Dark-Field (HAADF)-STEM image and corresponding elemental maps of (b) O, (c) Pt, (d) Si, (e) Co, (f) mixed coloured mapping of HAADF, Si, O, Pt and Co, and (g) line-scan profile of Si, Co, Pt and HAADF signals. | ||
Regarding the catalytic activity, the reduction of CO2 employing hydrogen was carried out in a fixed bed reactor at 350 °C with a N2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO2:H2 molar ratio of 1:1:3. The space velocity was fixed in 15000 mL g−1 h−1 and a total pressure of 6 bar.
:CO2:H2 molar ratio of 1:1:3. The space velocity was fixed in 15000 mL g−1 h−1 and a total pressure of 6 bar.
It is frequently reported that when carbon dioxide hydrogenation takes place on platinum supported nanoparticles, carbon monoxide is produced as the main product following the reverse water–gas shift reaction.25,42–45 However, when carbon dioxide is hydrogenated over supported cobalt nanoparticles, methane is forming as the main product following the Sabatier reaction.46–50
The conversion and selectivity products of our tandem catalyst can be seen in Fig. 4. Not only methane is formed but also light olefins (C2–C4) as products. Carbon dioxide reaches a conversion of 19% with 60% selectivity to carbon monoxide and 40% to hydrocarbons. The formed hydrocarbons are distributed in 86% of methane and 14% of C2–C4 olefins.
 | ||
| Fig. 4 Catalytic performance of SiO2–Pt@m-SiO2–Co catalyst in the CO2 hydrogenation. P = 6 bar, T = 350 °C, CO2:H2 ratio of 1:3, GHSV = 15000 mL g−1 h−1. (a) CO2 conversion and product selectivity; (b) hydrocarbon distribution. | ||
It is assumed that in a first step, carbon dioxide reacts with hydrogen on platinum nanoparticles at the core through the reverse water–gas shift reaction, forming carbon monoxide as the main product. In a second step, CO diffuses through the mesoporous shell structure until it contacts the cobalt nanoparticles on the outer shell. The carbon monoxide should be here converted to methane and light olefins (C2–C4) through the Fischer–Tropsch process.51–53 Therefore, the CO2 conversion and product distribution indicate that a tandem process involving both active sites was achieved.
Changes in the core–shell catalyst after 20 h of time on stream were studied by STEM-EDS (Fig. S15†). There is no clear evidence of platinum nanoparticles agglomeration, probably due to the mesoporous silica shell that acts as a protective layer against sintering.33,54,55 However, the high temperature and prolonged TOS leads to a partial migration and coalescence of the outer cobalt nanoparticles.
The catalytic performance in terms of selectivity agrees with the observed structural changes. Within the first 12 h of TOS, selectivity of CO increases and selectivity of hydrocarbons decreases (Fig. 4a). Regarding hydrocarbons, selectivity of CH4 increases and selectivity of C2–C4 decreases (Fig. 4b). The selectivity to C2–C4 olefins is related to the CO conversion through the Fischer–Tropsch reaction on the outer cobalt sites. Due to their coalescence and agglomeration, the metal active surface area and thus olefin yield decrease until a constant value is reached. A similar trend was observed on supported, size-controlled Co and Fe-based catalysts, where selectivity loss of olefins was related to an increase in the particle size as well.56–58 Selectivity of CO through the reverse water–gas shift reaction increases until it reaches a constant value, promoted by stable platinum sites protected by the mesoporous silica shell.
Xie et al. reported an analogous reaction pathway under similar reaction conditions corroborating the consecutive CO2 to CO formation followed by methane and C2–C4.34 However, our synthesis can yield 550 mg of catalyst per batch and is notably nine-time superior in terms of production of value-added olefins, which is of great importance because of the high price of nobel metals (139 mol C2–C4 per mol Pt per h in this work, vs. 15 mol C2–C4 per mol Pt per h for the reported CeO2–Pt@m-SiO2–Co).
Dispersion and size of the metal in Pt/SiO2 are crucial factors for the reverse water–gas shift reaction, determining the availability of surface metal atoms i.e. the number of active sites at the Pt–silica interface.42,59 Thus, the promising performance of our core–shell catalyst can be related to an efficient dispersion of the platinum nanoparticles on the silica surface. CO chemisorption measurements were performed yielding platinum surface areas of 1.38 m2 g−1 and 1.09 m2 g−1 for SiO2–Pt and SiO2–Pt@m-SiO2, respectively. Hence, the accessibility to platinum sites was slightly affected during the preparation of the shell structure.
Furthermore, the spatial arrangement of the active centers can play an important role. More studies are being carried out to elucidate the influence of the distance between metallic nanoparticles in the performance of coupled catalytic reactions at a nanometric scale.
Conclusions
A facile and scalable route for the preparation of multifunctional catalysts is developed, allowing to control the distance between two different metal nanoparticles in one material. This is exemplified by the preparation of Pt and CoNP spatially separated by a mesoporous silica layer. The catalyst has been tested on the environmental and commercial relevant CO2 hydrogenation reaction, indicating that a successful tandem process is achieved. This protocol can be easily extended to other metallic nanoparticle combinations as active centers for diverse chemocatalytic reactions. Furthermore, our study represents a useful approach to the precise tuning of the distance between different active sites. This is of great importance to gain a deeper understanding of coupled catalytic reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. G. thanks to the Deutsch-Argentinisches Hochschulzentrum (CUAA-DAHZ) and the German Academic Exchange Service (DAAD). L. D.-C. and W. H. thank Prof. Dr R. Schlögl for his support. Funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2008 – 390540038 – UniSysCat is acknowledged.References
- J. A. Schwarz, C. Contescu and A. Contescu, Chem. Rev., 1995, 95, 477–510 CrossRef CAS.
- F. Pinna, Catal. Today, 1998, 41, 129–137 CrossRef CAS.
- M. Rafti, A. Brunsen, M. C. Fuertes, O. Azzaroni and G. J. A. A. Soler-Illia, ACS Appl. Mater. Interfaces, 2013, 5, 8833–8840 CrossRef CAS PubMed.
- B. A. T. Mehrabadi, S. Eskandari, U. Khan, R. D. White and J. R. Regalbuto, in Advances in Catalysis, Elsevier Inc., 1st edn, 2017, vol. 61, pp. 1–35 Search PubMed.
- S. Eskandari, G. Tate, N. R. Leaphart and J. R. Regalbuto, ACS Catal., 2018, 8, 10383–10391 CrossRef CAS.
- E. Gioria, F. Marchesini, A. Soldati, A. Giorello, J. Hueso and L. Gutierrez, Appl. Sci., 2019, 9, 4075 CrossRef CAS.
- N. Miguel-Sancho, G. Martinez, V. Sebastian, A. Malumbres, I. Florea, R. Arenal, M. C. Ortega-Liebana, J. L. Hueso and J. Santamaria, ACS Appl. Mater. Interfaces, 2017, 9, 41529–41536 CrossRef CAS PubMed.
- M. Rahaman, A. Dutta and P. Broekmann, ChemSusChem, 2017, 10, 1733–1741 CrossRef CAS PubMed.
- J. Liu, Y. Zheng and S. Hou, RSC Adv., 2017, 7, 37823–37829 RSC.
- L. Bai, S. Zhang, Q. Chen and C. Gao, ACS Appl. Mater. Interfaces, 2017, 9, 9710–9717 CrossRef CAS PubMed.
- H. Mistry, R. Reske, P. Strasser and B. Roldan Cuenya, Catal. Today, 2017, 288, 30–36 CrossRef CAS.
- Z. Ma and M. D. Porosoff, ACS Catal., 2019, 9, 2639–2656 CrossRef CAS.
- W. Leng, R. Ge, B. Dong, C. Wang and Y. Gao, RSC Adv., 2016, 6, 37403–37406 RSC.
- F. Z. Jin, C. Q. Chen, Q. Zhao, J. L. Kan, Y. Zhou and G. J. Chen, Catal. Commun., 2018, 111, 84–89 CrossRef CAS.
- Y. Hu, J. Zhang, H. Huo, Z. Wang, X. Xu, Y. Yang, K. Lin and R. Fan, Catal. Sci. Technol., 2020, 10, 315–322 RSC.
- M. Li, C. Pischetola, F. Cárdenas-Lizana and M. A. Keane, Appl. Catal., A, 2020, 590, 117368 CrossRef CAS.
- S. Sithambaram, R. Kumar, Y. C. Son and S. L. Suib, J. Catal., 2008, 253, 269–277 CrossRef CAS.
- M. Yang, H. Qi, F. Liu, Y. Ren, X. Pan, L. Zhang, X. Liu, H. Wang, J. Pang, M. Zheng, A. Wang and T. Zhang, Joule, 2019, 3, 1937–1948 CrossRef CAS.
- M. J. Climent, A. Corma, S. Iborra and M. J. Sabater, ACS Catal., 2014, 4, 870–891 CrossRef CAS.
- S.-C. Ke, T.-T. Luo, G.-G. Chang, K.-X. Huang, J.-X. Li, X.-C. Ma, J. Wu, J. Chen and X.-Y. Yang, Inorg. Chem., 2020, 59, 1736–1745 CrossRef CAS PubMed.
- L. Guerrini, F. McKenzie, A. W. Wark, K. Faulds and D. Graham, Chem. Sci., 2012, 3, 2262–2269 RSC.
- H. Mistry, F. Behafarid, R. Reske, A. S. Varela, P. Strasser and B. Roldan Cuenya, ACS Catal., 2016, 6, 1075–1080 CrossRef CAS.
- P. Olk, J. Renger, M. T. Wenzel and L. M. Eng, Nano Lett., 2008, 8, 1174–1178 CrossRef CAS PubMed.
- M.-F. Tsai, C. Hsu, C.-S. Yeh, Y.-J. Hsiao, C.-H. Su and L.-F. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 1508–1519 CrossRef CAS PubMed.
- S. K. Beaumont, S. Alayoglu, C. Specht, N. Kruse and G. A. Somorjai, Nano Lett., 2014, 14, 4792–4796 CrossRef CAS PubMed.
- T. L. Lohr and T. J. Marks, Nat. Chem., 2015, 7, 477–482 CrossRef CAS PubMed.
- W. Li, H. Wang, X. Jiang, J. Zhu, Z. Liu, X. Guo and C. Song, RSC Adv., 2018, 8, 7651–7669 RSC.
- M. Bowker, ChemCatChem, 2019, 11, 4238–4246 CrossRef CAS PubMed.
- Y. Xu, J. Ma, Y. Xu, L. Xu, L. Xu, H. Li and H. Li, RSC Adv., 2013, 3, 851–858 RSC.
- K. Xu, J.-X. Wang, X.-L. Kang and J.-F. Chen, Mater. Lett., 2009, 63, 31–33 CrossRef CAS.
- C. C. Crane, F. Wang, J. Li, J. Tao, Y. Zhu and J. Chen, J. Phys. Chem. C, 2017, 121, 5684–5692 CrossRef CAS.
- H. Sakai, T. Kanda, H. Shibata, T. Ohkubo and M. Abe, J. Am. Chem. Soc., 2006, 128, 4944–4945 CrossRef CAS PubMed.
- S. H. Joo, J. Y. Park, C.-K. Tsung, Y. Yamada, P. Yang and G. A. Somorjai, Nat. Mater., 2009, 8, 126–131 CrossRef CAS PubMed.
- C. Xie, C. Chen, Y. Yu, J. Su, Y. Li, G. A. Somorjai and P. Yang, Nano Lett., 2017, 17, 3798–3802 CrossRef CAS PubMed.
- A. Flegler, S. Wintzheimer, M. Schneider, C. Gellermann and K. Mandel, in Handbook of Nanomaterials for Industrial Applications, Elsevier, 2018, pp. 137–150 Search PubMed.
- S. H. Joo, J. Y. Park, C. K. Tsung, Y. Yamada, P. Yang and G. A. Somorjai, Nat. Mater., 2009, 8, 126–131 CrossRef CAS PubMed.
- J. Liu, C. Liang, X. Zhu, Y. Lin, H. Zhang and S. Wu, Sci. Rep., 2016, 6, 1–10 CrossRef PubMed.
- T. Herranz, X. Deng, A. Cabot, J. Guo and M. Salmeron, J. Phys. Chem. B, 2009, 113, 10721–10727 CrossRef CAS PubMed.
- G. Melaet, A. E. Lindeman and G. A. Somorjai, Top. Catal., 2014, 57, 500–507 CrossRef CAS.
- J. Yang, V. Frøseth, D. Chen and A. Holmen, Surf. Sci., 2016, 648, 67–73 CrossRef CAS.
- V. Iablokov, S. K. Beaumont, S. Alayoglu, V. V. Pushkarev, C. Specht, J. Gao, A. P. Alivisatos, N. Kruse and G. A. Somorjai, Nano Lett., 2012, 12, 3091–3096 CrossRef CAS PubMed.
- S. Kattel, B. Yan, J. G. Chen and P. Liu, J. Catal., 2016, 343, 115–126 CrossRef CAS.
- G. Pekridis, K. Kalimeri, N. Kaklidis, E. Vakouftsi, E. F. Iliopoulou, C. Athanasiou and G. E. Marnellos, Catal. Today, 2007, 127, 337–346 CrossRef CAS.
- Y. Wang, Y. Zhai, D. Pierre and M. Flytzani-Stephanopoulos, Appl. Catal., B, 2012, 127, 342–350 CrossRef CAS.
- H. Su, Y. Ye, K.-J. Lee, J. Zeng, B. S. Mun and E. J. Crumlin, J. Catal., 2020, 391, 123–131 CrossRef CAS.
- G. Garbarino, T. Cavattoni, P. Riani and G. Busca, Catal. Today, 2020, 345, 213–219 CrossRef CAS.
- S. K. Beaumont, S. Alayoglu, C. Specht, W. D. Michalak, V. V. Pushkarev, J. Guo, N. Kruse and G. A. Somorjai, J. Am. Chem. Soc., 2014, 136, 9898–9901 CrossRef CAS PubMed.
- W. Li, X. Nie, X. Jiang, A. Zhang, F. Ding, M. Liu, Z. Liu, X. Guo and C. Song, Appl. Catal., B, 2018, 220, 397–408 CrossRef CAS.
- P. Riani, G. Garbarino, T. Cavattoni, F. Canepa and G. Busca, Int. J. Hydrogen Energy, 2019, 44, 27319–27328 CrossRef CAS.
- C. Vogt, M. Monai, G. J. Kramer and B. M. Weckhuysen, Nat. Catal., 2019, 2, 188–197 CrossRef CAS.
- E. Iglesia, Appl. Catal., A, 1997, 161, 59–78 CrossRef CAS.
- J. S. Jung, S. W. Kim and D. J. Moon, Catal. Today, 2012, 185, 168–174 CrossRef CAS.
- W. Chen, I. A. W. Filot, R. Pestman and E. J. M. Hensen, ACS Catal., 2017, 7, 8061–8071 CrossRef CAS PubMed.
- H. Wang, M. Liu, Y. Ma, K. Gong, W. Liu, R. Ran, D. Weng, X. Wu and S. Liu, ACS Catal., 2018, 8, 2796–2804 CrossRef CAS.
- H. Zhao, D. Wang, C. Gao, H. Liu, L. Han and Y. Yin, J. Mater. Chem. A, 2016, 4, 1366–1372 RSC.
- J. Xie, H. M. Torres Galvis, A. C. J. Koeken, A. Kirilin, A. I. Dugulan, M. Ruitenbeek and K. P. de Jong, ACS Catal., 2016, 6, 4017–4024 CrossRef CAS PubMed.
- D. Moodley, M. Claeys, E. van Steen, P. van Helden, D. Kistamurthy, K.-J. Weststrate, H. Niemantsverdriet, A. Saib, W. Erasmus and J. van de Loosdrecht, Catal. Today, 2020, 342, 59–70 CrossRef CAS.
- M. Wolf, H. Kotzé, N. Fischer and M. Claeys, Faraday Discuss., 2017, 197, 243–268 RSC.
- M. Zhu, Q. Ge and X. Zhu, Trans. Tianjin Univ., 2020, 26, 172–187 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00310k |
| This journal is © The Royal Society of Chemistry 2021 |