
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVersatile precursor-dependent copper sulfide nanoparticles as a multifunctional catalyst for the photocatalytic removal of water pollutants and the synthesis of aromatic aldehydes and NH-triazoles†
Soniya
Agarwal
,
Parmita
Phukan
,
Diganta
Sarma
 * and
Kalyanjyoti
Deori
*
* and
Kalyanjyoti
Deori
*
Department of Chemistry, Dibrugarh University, Dibrugarh-786004, Assam, India. E-mail: dsarma22@gmail.com; kalchemdu@gmail.com; kalchemdu@dibru.ac.in
First published on 18th May 2021
Abstract
A series of copper sulfide (CS) nanoparticles (NPs) were synthesized just by varying the amount of the sulfur precursor and have been explored for the first time as a three-way heterogeneous catalyst in the photocatalytic oxidation of a number of aromatic alcohols, photocatalytic degradation and the reduction of water pollutants, and the facile synthesis of pharmaceutically important moiety 4-aryl-NH-1,2,3-triazoles. The green and novel protocol was successfully developed for the synthesis of covellite (CuS, Cu2+) and the covellite-villamaninite (CuS–CuS2) (copper in Cu2+, Cu1+) phases of copper sulfide, employing EDTA both as the chelating and capping agent via a simple precipitation method at room temperature using water as the solvent. A blue shift in the absorption spectra and band gap in the range of 2.02–2.07 eV prompted the investigation of the as-synthesized CS nanoparticles as the photocatalyst under visible light irradiation. In the absence of any oxidizing or reducing agent, covellite CuS nanoparticles showed the highest photocatalytic efficiency for the degradation of methylene blue (MB) and the reduction of carcinogenic and mutagenic Cr(VI) to non-toxic Cr(III). Interestingly, the mixed phase of CS (CuS–CuS2), where Cu is present in both +1 and +2 oxidation states, was found to be the most efficient catalyst compared to CuS toward the visible light-mediated selective oxidation of various benzyl alcohols to their corresponding aldehydes. However, in the synthesis of substituted NH-1,2,3-triazoles, single-phase CS nanoparticles (i.e., CuS) provided the best catalytic result. This significant outcome certainly opens up the scope for realizing the present demand of low-cost multifunctional semiconductor nano-materials, which will have a huge impact on the economy and environment when they show more than two potential applications.
Introduction
21st century demands the concept of sustainability and versatility in scientific research. Cultivating one single eco-friendly catalyst or photocatalyst, in better words, a multifunctional catalyst, which can find application in varied areas such as industries, environmental remediation, and medicine, will be highly beneficial to mankind. Many issues of environmental and economic concerns can be tackled through this model. Such sustainable models are not very common but can be proved to be highly beneficial in the development of competent technologies. One of the important fundamental transformations in synthetic chemistry is the selective oxidation of organic compounds. To manufacture such large-scale industrially significant oxidized products, harsh reaction conditions are employed (high temperature and pressure), resulting in unnecessary energy consumption and a toxic environment. In order to tackle this situation, a greener route using visible light as the cleanest energy source has to come in practice. Benzaldehyde, the second most important chemical after vanillin in the cosmetic and perfumery industries and also an important intermediate in the synthesis of other chemicals, has been traditionally synthesized employing harsh oxidizing agents such as KMnO4, MnO2, CrO3 and catalysts comprising noble metals along with few methods involving temperature as the driving force.1–3 Therefore, developing an economic and greener route for benzaldehyde synthesis is important in industrial research.Besides the selective photocatalytic oxidation of benzyl alcohol to benzaldehyde, environmental remediation by the degradation of mutagenic dyes and Cr(VI) reduction has attracted the attention of the scientific community. Increasing water crisis is a distressing situation and demands immediate attention. Water scarcity is increasing each passing day but still there is no solution to the problem. One of the major contributors to this problem is water contamination due to unused dye and carcinogenic Cr(VI) released from various industries using them for catalysis, pigmentation, dyeing, leather tanning, etc.4,5 These organic dyes are toxic, mutagenic, carcinogenic, and do not break down effortlessly in the nature. Thus, due to the stubborn nature of these dyes, they persist in the environment for a very long duration, causing harmful effects to both human health and the ecosystem, thus making water unfit for further use. An emerging sustainable remedy to this challenge is semiconductor photocatalysis. In addition to being simple and cost-effective, it uses green and renewable energy source for waste water treatment. Traditional inorganic semiconductor materials such as TiO2, metal oxides, metal sulphides, and metal organic frameworks are used for the purpose.6,7 Among these, transition metal chalcogenides are promising semiconductors because of their unique physicochemical and optical properties, tuneable band gap, and stability to arrest UV-visible radiation.
Copper sulfide (CS) is one brilliant example of this category. It is a cheap and non-toxic p-type semiconductor with a band gap of 2.2 eV, which can be tailored by varying different synthetic parameters such as the concentration of the copper precursor and the nature of the solvent.8,9 Copper sulfide occurs in many phases from copper-rich to copper-poor, i.e., from chalcocite (Cu2S) to villamaninite (CuS2), with other phases such as djurleite (Cu1.96S), digenite (Cu1.8S), anilite (Cu1.75S), geerite (Cu1.6S), spionkopite (Cu1.39S), yarrowite (Cu1.12S), and covellite (CuS) in between.10,11 These phases can be synthesized with varying morphology and band gap. So far, flower,12 branch structure,13 hexagonal plates,14,15 sphere,16 dandelion nanotubes,17 snowflake-like pattern, and hollow sphere18 type morphologies have been reported by several groups. One of the striking features of CuS is the ability to arrest solar energy in the visible region; therefore, it is of marked importance to synthesize CuS with enhanced optical properties. A number of methods such as sonochemical,19 solid-state synthesis,20 hydrothermal,21 solvothermal,22 chemical vapor deposition (CVD),23 template and surfactant-assisted24 have been reported to synthesize micro- and nanostructured CuS. Moreover, the synthesis of the villamaninite (CuS2) phase of CS nanoparticles by controlling the precursors' stoichiometry is a challenging task. The cubic pyrite phase of CuS2 (villamaninite) has monovalent copper with the electronic structure of Cu+(S2)−, where, one S–S bonded moiety is connected with copper in a single unit, as shown in Fig. 1.25,26 A sulfur-rich phase of CS (CuS2) can restrict the exposure of the metal (Cu) for better chemical and physical stability. As we dove deep into the subject matter, we hardly found any pure or bare CuS2 synthesized to date.27,28 Unveiling the structural features and the catalytic activity of CuS2 nanoparticles and their applications as a model system in a wide range of catalytic applications for many commercially important reactions thus remain important goals of modern scientific research.
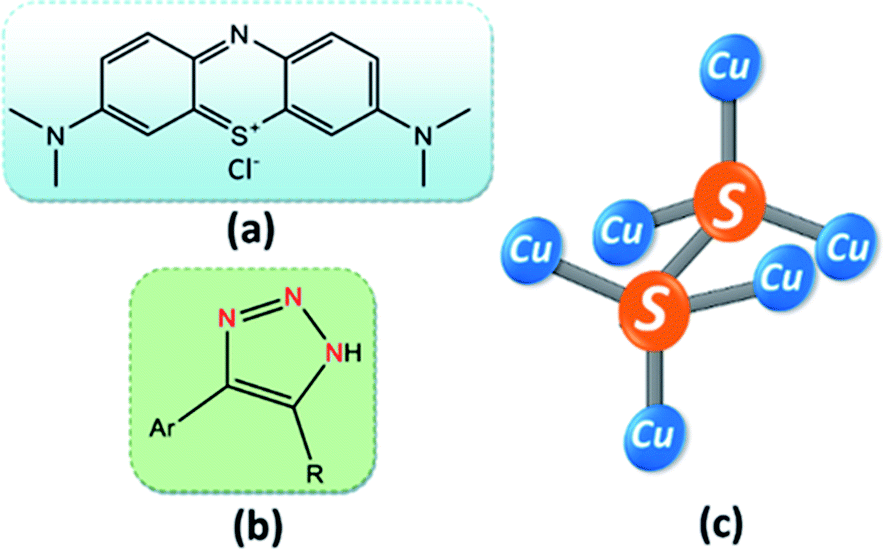 | ||
| Fig. 1 Molecular structure of (a) methylene blue, (b) 4-aryl-NH-1,2,3-triazole, and (c) CuS2. | ||
Despite having an interesting band gap at the nano range, there are only a few reports available on the photocatalytic activity of CuS in organic reactions.14 Also, more economical and eco-friendly synthetic protocols for synthesizing pure phases of CuS and CuS2 nanoparticles are still required. CuS has broad applications in solar cells, biosensors,29 photocatalysis,30 lithium-ion batteries,31 and drug delivery.32 Though it has applications in several fields, it is very much unexplored in the field of catalysis. Very few catalytic transformations such as H2 evolution,33 electrochemical CO2 reduction,34 and NH3 production35 have utilized CS as a catalyst.
Apart from a few reports available employing CS as a catalyst in organic synthesis, there is no single report on CS as a three-way heterogeneous catalyst and the catalytic activity of CuS2 nanoparticles is still not known. Hence, in addition to the photocatalytic synthesis of benzaldehyde, we wanted to explore CS's ability to catalyze other important reactions for developing medicinally important organic products. One such pharmaceutically important moiety, NH-triazole, serves as integral component of many biologically essential compounds such as anti-bacterial, anti-allergic, anti-cancer, and anti-malarial agents.36–39 Due to their attractive physical and chemical properties such as hydrogen bonding ability, strong dipole moment, and chemical stability, NH-triazoles are of utmost importance in the current areas of research and development.40,41
In this work, we have reported a cost-effective, effortless, and environmentally benign protocol for the synthesis of different phases of copper sulfide NPs by varying the sulfur precursor concentration. These synthesized NPs turned out to be optically active under visible light irradiation and were found to be very efficient as a three-way heterogeneous catalyst. The photocatalytic activity of single phase CuS was significant in the degradation of methylene blue (MB) (Fig. 1a) and the reduction of carcinogenic Cr(VI) to harmless Cr(III), while the mixed-phase CS (CuS–CuS2) (see Fig. 1c for the chemical structures of CuS2) was most efficient toward the selective oxidation of various aromatic alcohols to their corresponding aldehydes. In addition to photocatalytic processes, a smooth and effective catalytic system was developed to explore the efficiency of the as-synthesized CS NPs in the synthesis of NH-triazoles (Fig. 1b), where CuS NP is the more preferred catalyst out of the two phases.
Experimental section
Chemicals
Cupric chloride dihydrate (CuCl2·2H2O, 99%, Sigma Aldrich), sodium sulphide (Na2S, 70%, Merck), polyethylene glycol-400 (PEG-400, Merck), nitromethane (CH3NO2, 98%, TCI), nitroethane (C2H5NO2, 98%, Spectrochem), sodium azide (NaN3, 99%, Spectrochem). EDTA (disodium salt dihydrate, 99.5%), NH4OH solution, methylene blue (MB), and potassium dichromate (K2Cr2O7, 99.9%) were purchased from Rankem, benzyl alcohol (99%) and all the related substrates were purchased from TCI. All aldehydes were purchased from Spectrochem with 98% purity. All chemicals were used as received without further purification. The products were isolated by column chromatography (60–200 mesh) over silica gel. Thin-layer chromatography was performed using silica gel 60F254 plates and visualized under UV light.Catalyst preparation
CS NPs were prepared employing a very simple and hassle-free precipitation technique. 1 mmol each of CuCl2·2H2O and EDTA were dissolved in 10 mL of deionized water and stirred until a clear blue solution was obtained. The pH of the solution was adjusted to 7 by adding one to two drops of NH4OH. A solution of 2 mmol Na2S in 10 mL deionized water was added to the blue solution and a black precipitate was obtained. It was further stirred for 15 min, and then centrifuged and washed with water three times. A part of the obtained sample was dispersed in acetone for further characterization and the remaining was dried at 60 °C for 12 h. This sample was named as CS2. In a similar fashion, CS NPs with sulfur precursor concentration 5 mmol, 10 mmol, and 20 mmol were synthesized and respectively named as CS5, CS10, and CS20.Instrumentation
Selective photocatalytic oxidation of various benzyl alcohols to corresponding benzaldehydes
To test the feasibility of the synthesized CS NPs toward benzyl alcohol oxidation, a series of reactions for a variety of benzyl alcohols (4-methoxybenzyl alcohol, 4-methylbenzyl alcohol, 4-chlorobenzyl alcohol, benzyl alcohol, 4-fluorobenzyl alcohol, etc.) was conducted. In a model reaction, 1 mmol of benzyl alcohol was taken in a round bottom flask and to it, 5 mL acetonitrile (ACN) as the solvent, TBHP as the oxidizing agent, and 20 mg of the obtained CS NPs as the photocatalyst were added. The mixture was stirred at room temperature under continuous irradiation of visible light. The progress of the reaction was monitored by TLC under UV light. The product was confirmed by 1H and 13C NMR spectra. The conversion and selectivity were calculated for the performed reactions according to the following equations.| Conversion (%) = [(So − Salcohol)/So] × 100 |
| Selectivity (%) = [Saldehyde/(So − Salcohol)] × 100 |
Photocatalytic degradation of MB and Cr(VI) reduction
The photocatalytic activity of the as-synthesized CS2 and CS20 NPs was checked for cationic dye MB and K2Cr2O7. At room temperature, 50 mg of the as-synthesized catalyst was added to a 50 mL aqueous solution of MB (1.25 × 10−5 M) and K2Cr2O7(15 × 10−5 M) under vigorous stirring and visible light irradiation. From few initial trials, the time of visible light irradiation was optimized to 30 min and 60 min for MB and K2Cr2O7, respectively. After certain time intervals, 1 mL of the solution was taken out and centrifuged to remove the photocatalyst from the dye solution; then, it was subjected to optical absorption measurements and the UV-visible spectrum was recorded.For all photocatalytic measurements, a commercial LED lamp (7 W, 630 lumens) with wavelength distribution 400–700 nm (white light) was used as the light source. The distance between the light source and the reaction chamber was 20 cm. A commercial solar meter was used to exactly measure the optical irradiance at the sample position, which was found to be 0.06 mW cm−2. Room temperature (30 °C) was maintained throughout the reaction by fixing a cold-water circulating tank with the reaction chamber.
Catalytic measurement for triazole synthesis
The mixture of benzaldehyde (1 mmol), NaN3 (3 mmol), nitroalkane (2 mmol), and CS2 (10 mg) was stirred in 2 mL PEG-400 solvent at 100 °C under aerobic conditions. The reaction progress was monitored by TLC. Upon completion of the reaction, the reaction mixture was allowed to cool to room temperature and then extracted with ethyl acetate (3 × 10 mL). The organic layer was then dried over anhydrous Na2SO4. The organic solvent was evaporated under reduced pressure. The obtained product was purified by column chromatography over silica gel using ethyl acetate/hexane mixture. The products were characterized by NMR spectroscopy.Results and discussion
A single step simple precipitation procedure has been developed to synthesize CS NPs in two different pure states (+1 and +2) for studying the role of different oxidation states on the selectivity and efficiency of catalytic reactions. For this purpose, we synthesized four CS samples by altering only the amount of the sulfur precursor (Na2S). It was found that when the sulfur precursor's concentration was 2 mmol for the sample CS2, only the pure phase of CuS having copper in +2 oxidation state was obtained, which can be clearly confirmed from the XRD pattern shown in Fig. 2a. The sharp and distinct peaks of the obtained pattern revealed the high crystalline nature of the samples. Peaks at 27.6, 29.17, 31.8, 32.7, and 47.8 are well-indexed to (100), (102), (103), (006), and (110) crystal planes, respectively, and are in complete agreement with the bulk XRD pattern of the hexagonal covellite CuS (JCPDS card no. 24-0060, space group: P6(3)/mmc, a = b = 3.79; c = 16.34). No additional peaks in the obtained XRD pattern confirmed the purity of the sample. The peak for the (110) plane has the highest intensity, revealing its exposure in the crystal structure. EDTA might be responsible for the capping of other low intensity planes. This capping of planes by EDTA in the crystal plays a key role in the formation of the obtained morphology. On increasing the sulfur precursor concentration from the CS2 (2 mmol) to the CS5 (5 mmol) sample, we have observed two very low intensity peaks at 2θ values of 23.97° and 25.67° (black rhombohedral mark in Fig. 2a), respectively, in addition to all other peaks measured for CuS. The appearance of these extra peaks in the CS5 sample may be attributed to the formation of a small amount of crystalline intermediate between CuS and CuS2. Moreover, the crystallite size calculated from this obtained XRD pattern using Scherrer's formula was found to be 9.8 nm for CS2 and 10.2 nm for CS5. The peak broadening of the as-synthesized CuS revealed its nanocrystalline nature (Fig. 2a).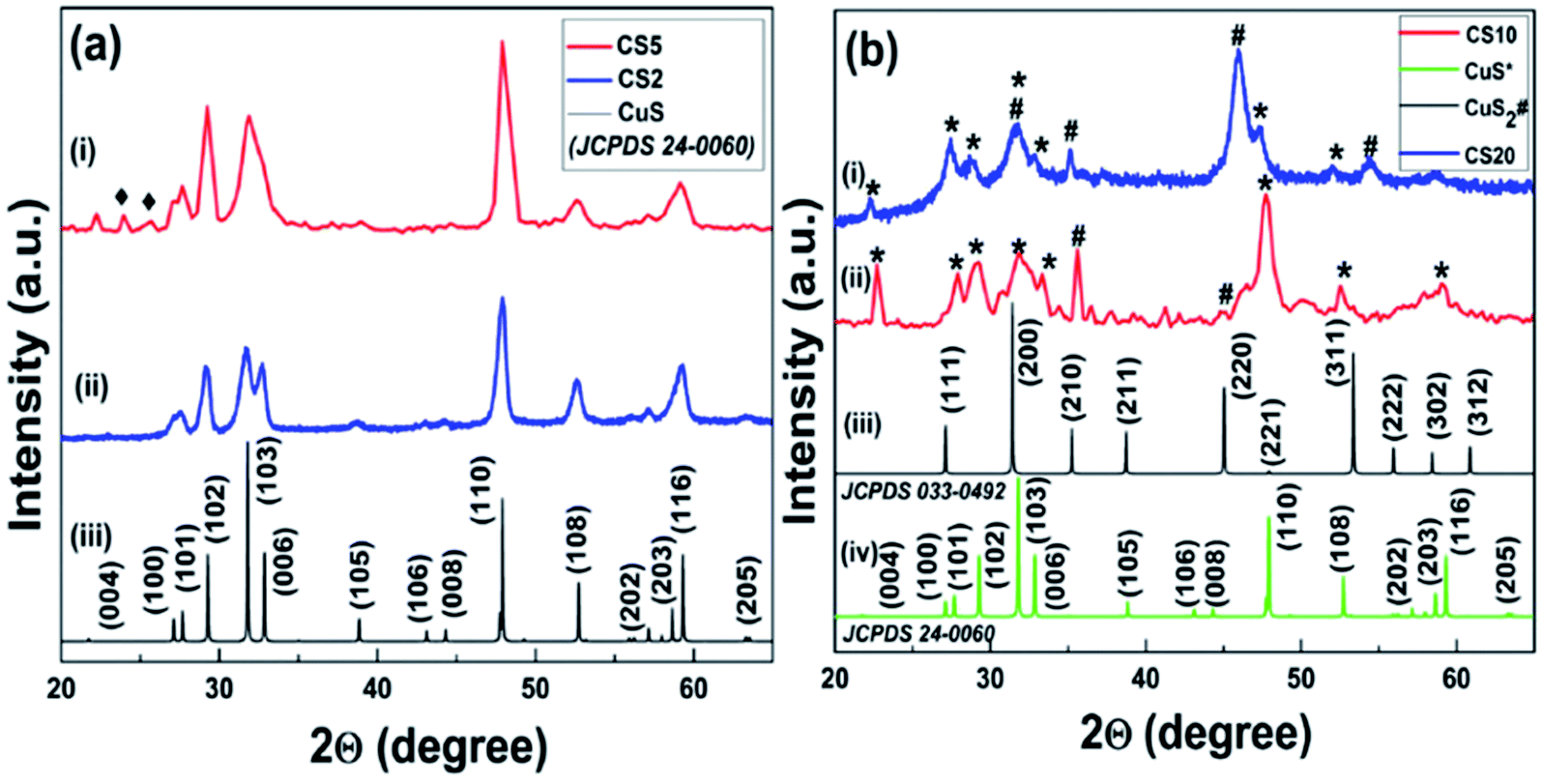 | ||
| Fig. 2 (a) Powder XRD pattern of the as-synthesized sample (i) CS2, (ii) CS5, and (iii) bulk CuS. (b) CS20 (i) and CS10 (ii) samples with bulk patterns of CuS2 (iii) and CuS (iv). | ||
In a similar way, when the sulfur precursor concentration was further increased to 10 mmol in the case of the CS10 sample, a new phase of CuS2 was obtained along with initial CuS. This significant outcome mainly developed our interest to study the sulfur concentration dependence structural properties. The appearance of some unassigned low intensity peaks in the CS 10 sample in between 36–43° (Fig. 2b) cannot be ignored completely. These peaks were matched with the (105), (106), and (006) planes of simulated bulk CuS with a slight shift in the x-axis position. The formation of the intermediate phases in a small amount is also one of the many possibilities. Any roughness seen in the experimental XRD patterns might be due to the instrumental/background noise or negligible amount of sulphur containing crystalline impurities. Interestingly, these extra peaks were found only in the case of the intermediate samples, i.e., CS 5 and CS 10 samples. Therefore, we believe that these additional peaks are mainly for intermediate or other unidentified non-stoichiometric phases of copper sulfide, which could not be recognized properly.
It is noteworthy to mention here that when the concentration of the sulfur precursor was again increased to 20 mmol for the sample CS20, the development of a new phase of CS nanoparticles was seen. When examined, the XRD patterns of CS20 show the formation of cubic pyrite CuS2 along with covellite CuS. No impurity peaks were found other than CuS and CuS2. Peaks at 27.3, 31.6, 35.2, and 45.9 correspond to the (111), (200), (210), and (220) crystal planes, respectively, and confirm the presence of cubic pyrite CuS2 (JCPDS card no. 033-0492, space group: Pa3, a = b = c = 5.69). The peaks marked * and # are distinct peaks for CuS and CuS2, respectively, confirming the formation of mixed phase CS NPs. For the sample CS10, the peak with the highest intensity is that for CuS but for CS20, it is for CuS2 (Fig. 2b). This difference can be attributed to the fact that the transformation of CuS to the CuS2 phase is possible upon increasing the sulfur concentration in the samples. The copper-deficient phase (CuS2) is more dominant in CS20 than in CS10, as evident from the XRD pattern and it also shows the contribution of excess sulfur in forming a new phase, i.e., CuS2 along with pre-existing CuS. Hence, samples CS10 and CS20 resulted in mixed phase CuS–CuS2 NPs. This incongruity in the peak intensity for the two samples also confirms the dependence of sulfur concentration on the mechanism of crystal growth. The respective calculated crystallite size for samples CS10 and CS20 was 10.2 nm and 6.9 nm.
To further confirm the valence states and the composition of the CS samples, XPS was performed. Fig. 3a shows the wide survey scan XPS spectrum of the CS2 and CS20, samples confirming the presence of the Cu 2p and S 2p peaks along with the signal for the O 1s and C 1s states. The obtained high resolution XPS spectra were calibrated with reference to the C(1s) line at 284.6 eV (Fig. 3b). First, for the CS2 sample, two distinct peaks at 931.9 eV and 951.9 eV correspond to Cu 2p3/2 and Cu 2p1/2, respectively, in accordance to the literature data (see Fig. 3c). Reports suggest that the difference between the peaks for Cu 2p3/2 and Cu 2p1/2 must be 20 eV, which is also true in our case.30 These data confirm that Cu in the synthesized sample (CS2) is in the +2 state. Also, the survey scan of the S 2p region shown in Fig. 3d exhibits two peaks with binding energy values of 161.7 eV and 162.7 eV, confirming the presence of S in the −2 state.42 The evidence of CuS formation was also furnished by the FT-IR data (Fig. S1†). The characteristic peak for Cu–S is observed at 619 cm−1.43 The other peaks in the spectra at 1415 cm−1 (O–H bending), 1640 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), and 3426 cm−1 (O–H stretching) are due to EDTA adsorbed on CuS during the synthesis. This demonstrates the function of EDTA as a capping agent as well.
O), and 3426 cm−1 (O–H stretching) are due to EDTA adsorbed on CuS during the synthesis. This demonstrates the function of EDTA as a capping agent as well.
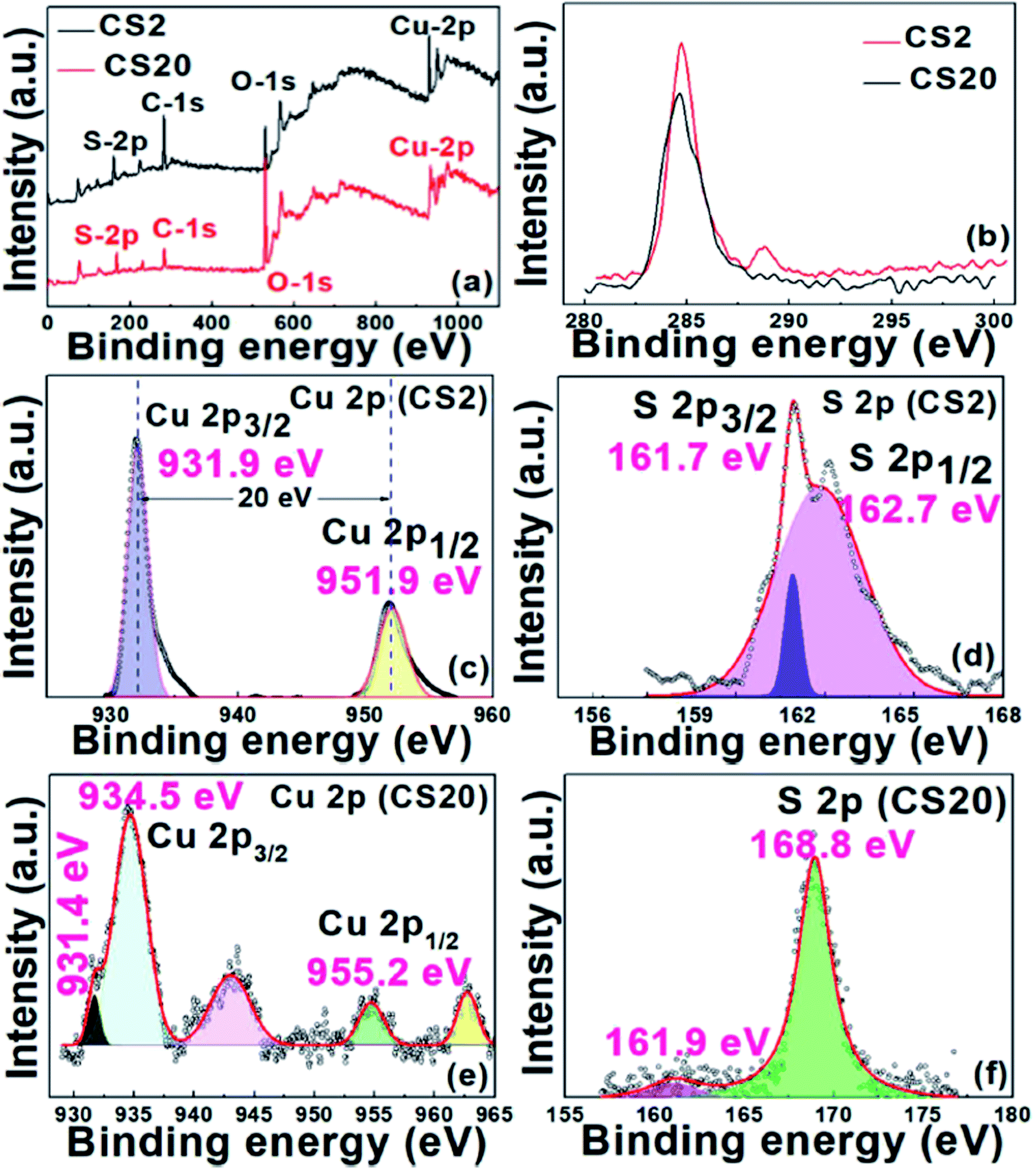 | ||
| Fig. 3 (a) Wide survey scan XPS spectra of the CS2 and CS20 samples. High resolution XPS spectra of (b) C 1s of the CS2 and CS20 samples, (c) Cu 2p and (d) S 2p of the CS2 sample. (e) Cu 2p and (f) S 2p spectra of the CS20 sample. | ||
Further, the XPS scan of the CS20 sample revealed binding energy peaks at 931.4 and 953.9 eV, as shown in Fig. 3e and f, which corresponds to Cu+ for Cu 2p3/2 and Cu 2p1/2, respectively. Other set of peaks at 934.5 and 955.2 eV relate to Cu2+ for Cu 2p3/2 and Cu 2p1/2, respectively. Further, the presence of two satellite peaks at 942.7 and 962.5 eV can also be attributed to the presence of Cu2+ and Cu+.11,30 The probable reason for the low intensity Cu+ peak at 931.49 eV is the surface oxidation of the sample during the analysis. This surface oxidation phenomenon is also observed in case of S, where a peak at 168.8 eV is due to sulfate.44
The HR-TEM images give an insight into the morphology and particle size of the as-synthesized CS NPs. As can be observed from the HR-TEM image of CS2 depicted in Fig. 4a, flat bean-shaped particles are formed having diameter in the range of 20–30 nm. After nucleation, crystal growth takes place from crystallites of size 9.8 nm to give particle size ranging between 20 and 30 nm. The lattice fringes spaced 0.32 nm correspond to the (100) plane of hexagonal CuS (Fig. 4b). 2D fast Fourier transform (FFT) (inset Fig. 4b) pattern was generated from a selected area of the HRTEM image and the d-spacing was calculated according to a recent report, which also confirms the presence of only the (100) exposed plane.45 The SAED pattern further demonstrates a spotty hexagonal pattern due to the crystalline nature in the hexagonal phase of the sample (Fig. 4c). Also, the HR-TEM image of the as-synthesized sample CS20 revealed the formation of an irregular sheet-like structure with particle size in the range of 25–35 nm and an interplanar d-spacing of 0.33 nm corresponding to the (111) plane of the CuS2 cubic pyrite phase (Fig. 4d and e). From the FFT pattern shown in the inset of Fig. 4e, the exact interplanar distance for the CS20 sample was calculated and found to be 0.328 nm, which is close to 0.33 nm. Spotty rings in the SAED pattern (Fig. 4f) correspond to planes, as observed in the XRD pattern. Compared to the CS2 sample, the change in the spotty SAED pattern of the CS20 sample revealed that a part of the sample is transformed into the cubic pyrite phase at the expense of the hexagonal covellite phase. The near-transparent nature of the images suggests the very low thickness of the sheet-type NPs. The aggregation of the NPs is due to the contribution of strong intermolecular forces such as π–π interaction and van der Waals attraction.
 | ||
| Fig. 4 (a) Low-resolution TEM image of CS2, (b) phase-contrast HRTEM image of CS2 (inset: corresponding 2D-FFT), (c) SAED pattern of CS2, (d) TEM image of CS20, (e) phase-contrast HRTEM image of CS20 (inset: corresponding 2D-FFT), and (f) SAED pattern of CS20. | ||
From the above discussion and with the evidence of our experimental data, we proposed a probable mechanism for the formation of CS NPs. CuCl2·2H2O and EDTA were ionized in water to get a Cu–EDTA complex due to chelation and further addition of NH4OH facilitates the formation of a complex by ionizing EDTA. The role of NH4OH is to assist the construction of the Cu–EDTA complex. Next, the inclusion of sulfur precursor, Na2S, releases S2− ions. These S2− ions combine with Cu2+ ions due to favorable interaction and precipitate as CuS. The morphology of the synthesized samples depends on the rate of release of Cu2+ and S2− ions. A slow-release rate of any of these ions results in irregular morphology. But for the CS10 and CS20 samples, where excess of sulfur precursor was added, mixed phase CuS–CuS2 nanosheets were obtained. The mechanism for the formation of the CuS phase was the same as that discussed above. CuS2 in the sample was responsible due to the excess of the sulfur precursor. An (S–S)− linkage was formed similar to the pyrite-type structure between the sulfide ions and electron exchange reduced Cu2+ to Cu+. Hence, this new Cu+(S2)− phase, together with CuS, gave irregular nanosheets (Fig. 5).
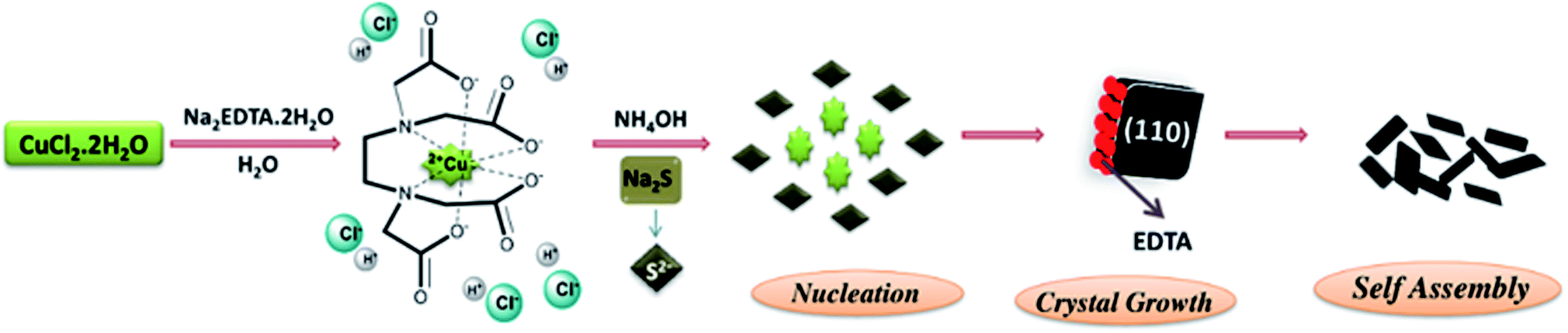 | ||
| Fig. 5 Schematic diagram showing the formation mechanism of CS NPs. | ||
Optical study of the as-synthesized samples
For UV-visible spectrophotometry, the as-synthesized fresh sample was dispersed in acetone and ultrasonicated for 2 min. The absorption spectrum was obtained using a UV-visible spectrophotometer. The dispersed CS20 sample shows two absorption peaks, one in the visible region (near 470 nm) and the other in the UV or near-visible region (near 326 nm), as shown in Fig. 6b. Further, there is also an absorption in the near IR region, which is a characteristic of covellite CuS.43 The appearance of two absorption peaks can be attributed to (i) charge transfer between the (S2)− moiety and Cu+ (at 326 nm), and (ii) S2− to Cu2+ charge transfer (at 470 nm) processes. There is a slight blue shift in the absorption due to the quantum confinement effect. The corresponding direct band gap was calculated to be 2.07 eV (inset Fig. 6b) from Tauc's equation, which is in accordance with its photocatalytic activity in the presence of visible light and also within the reported values. On the contrary, only one broad absorption spectrum was visible for the as-synthesized sample CS2, i.e., pure phase CuS (Fig. 6a). The corresponding band gap using Tauc's equation was found to be 2.11 eV.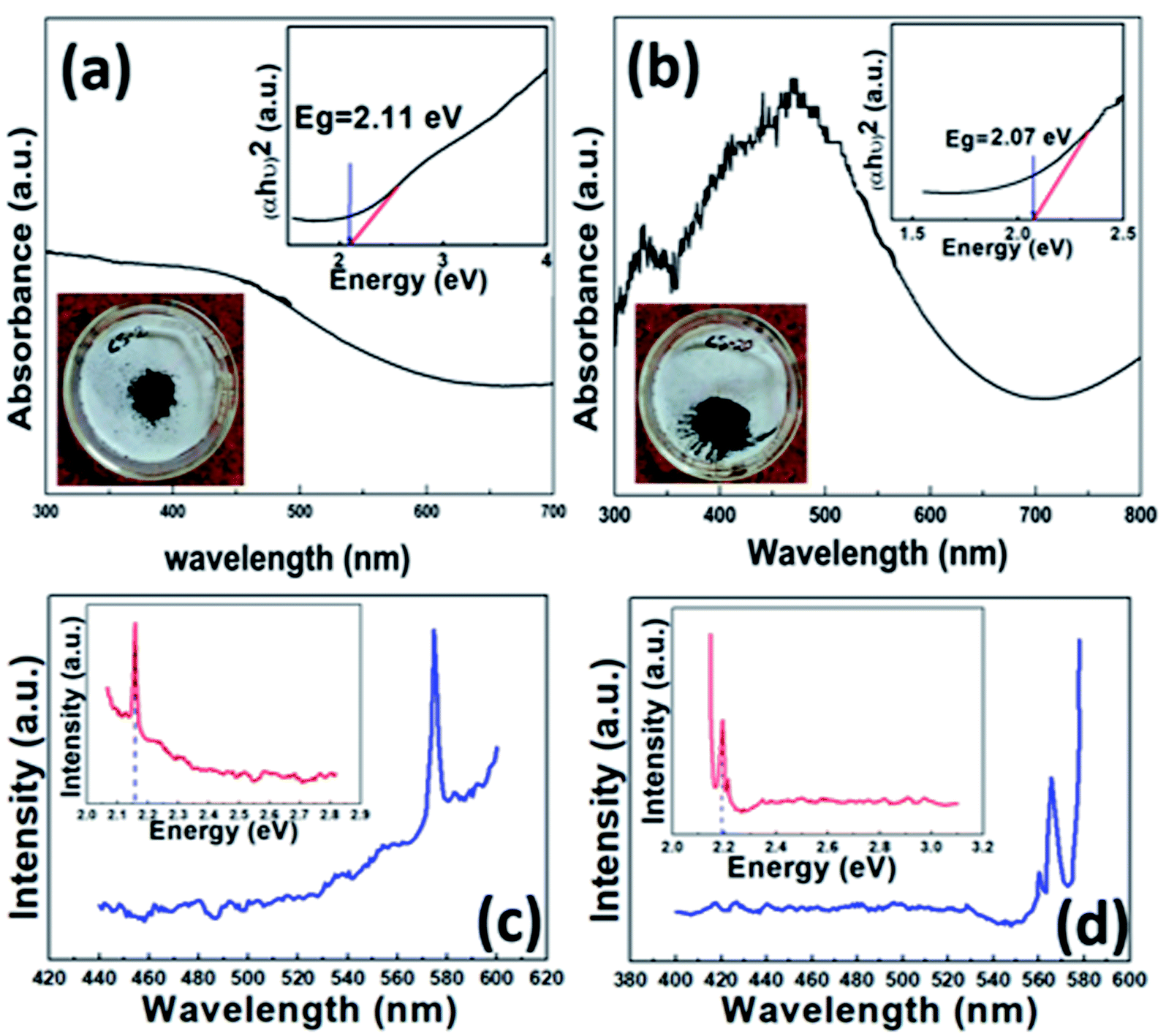 | ||
| Fig. 6 UV-visible absorption spectra (inset top right: corresponding band gap of the as-synthesized sample, inset bottom left: synthesized powdered sample): (a) CS2 and (b) CS20. PL spectra of (c) CS2 and (d) CS20 (inset: band gap calculated from the corresponding PL spectra). | ||
To further confirm the real optical band gap of both the samples, room temperature photoluminescence (RT-PL) analysis was performed on the finely ground powdered sample. For the CS2 sample, the emission peak at 574 nm was observed for 350 nm exitonic wavelength (Fig. 6c) and for the CS20 sample, at 300 nm excitation wavelength, emission of 565 nm was observed. The band gaps were calculated by following the equation given below.
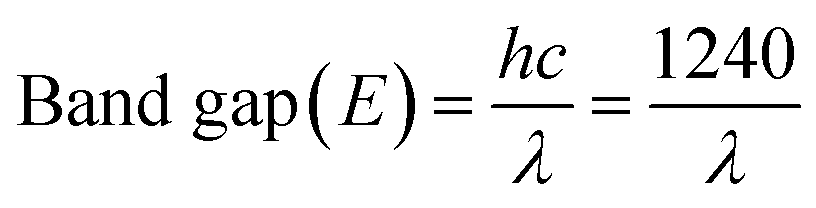 | (1) |
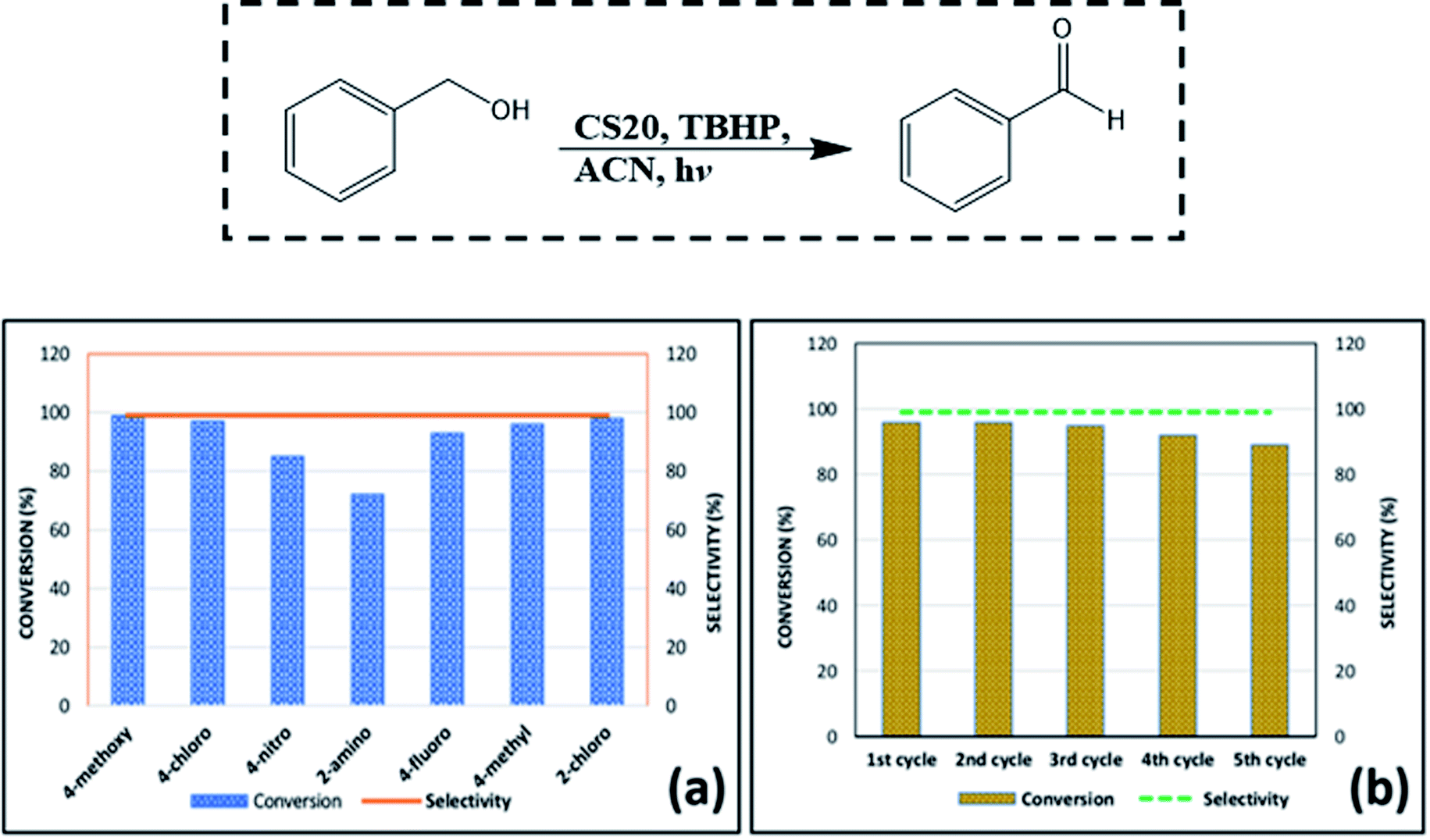 | ||
| Fig. 7 (a) Conversion and selectivity of other substituted benzyl alcohols in accordance with the optimized conditions. (b) Recyclability test for CS20, taking benzyl alcohol as the model substrate. | ||
ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) C0/C = kt C0/C = kt | (2) |
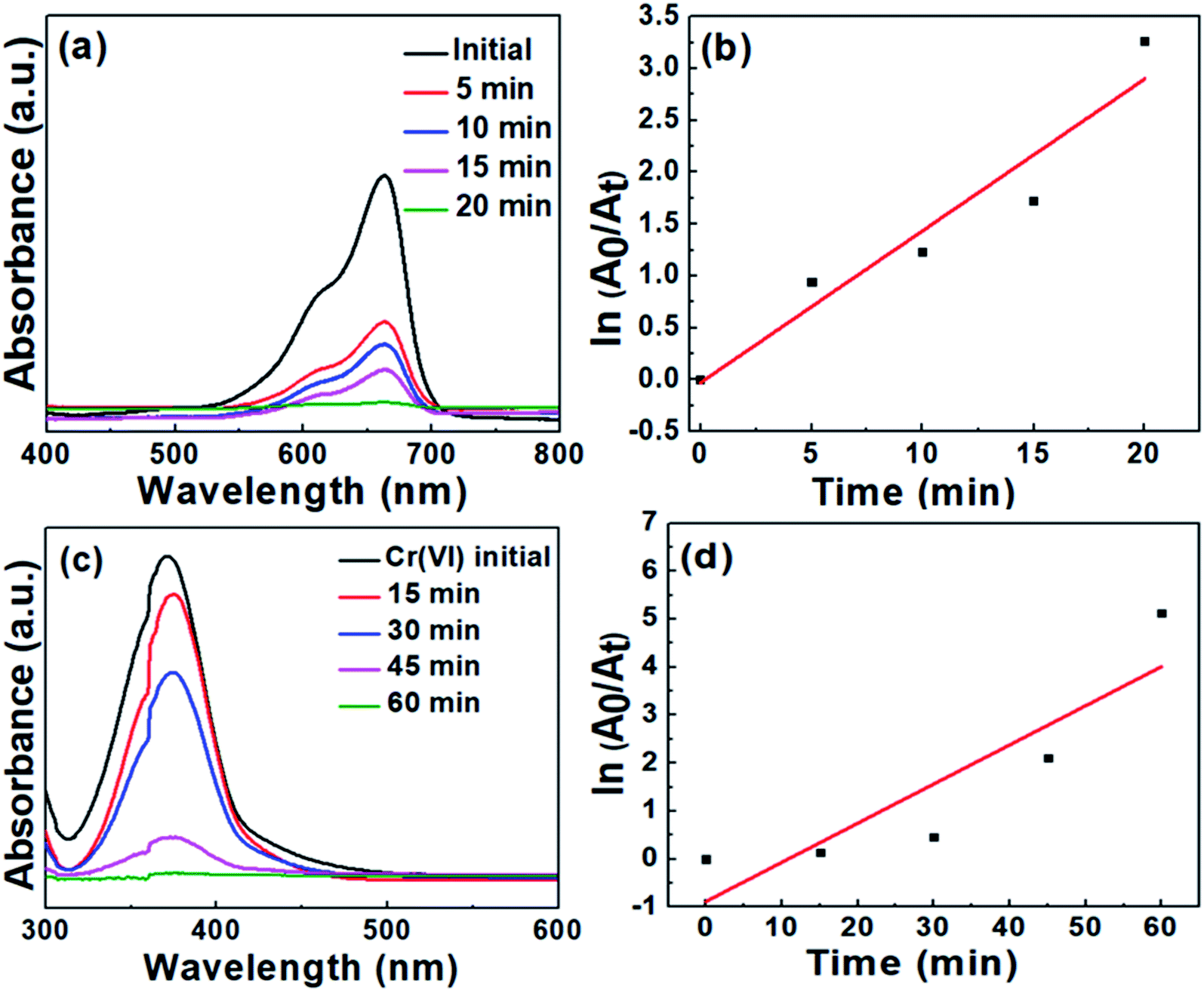 | ||
| Fig. 8 (a) Time-dependent optical absorption spectra for the degradation of MB, (b) kinetic curve for MB degradation, (c) time-dependent optical absorption spectra for Cr(VI) reduction, (b) kinetic curve for Cr(VI) reduction. | ||
In a similar manner, the degradation of MB was conducted and monitored employing CS20 as the catalyst. Contrary to the degradation with CS2, it took a longer time duration to initiate the degradation of MB. These results led us to the conclusion that the semiconductor photocatalyst CS2 is more efficient toward cationic dye degradation rather than CS20.
It was observed that the curves of both MB degradation and Cr(VI) reduction were not well-matched with the pseudo first-order kinetic model (see Fig. 8b and d). The probable reasons behind this deviation are as follows: (i) the formation of various intermediate states during the dye degradation reaction affects the reaction kinetics. At the initial stage of reaction, there is no involvement of any intermediate states but when the reaction progresses, a number of intermediate states are formed, which are then adsorbed on the catalyst surface and thereby reduce the photodegradation rate. (ii) A large amount of dye can be adsorbed on the surface of the catalyst at high concentration during the initial moment of the reaction. It prevents the adsorbed dye molecules to be available for the reaction with free radicals, electrons, and holes generated from the photocatalyst. As a result, a slower degradation rate is observed at the initial moment, which changes its behavior at the later stage of the reaction. A similar kinetic behavior is very much common in the case of MB or methyl orange degradation reactions, as per earlier reports.48,49
To obtain the real photocatalytic activity of our as-synthesized CS samples and as a control reaction, the MB degradation and Cr(VI) reduction experiments were also carried out by adding the photocatalysts to the respective solution and continuously stirring for 1 hour under dark condition. The representative data shown in Fig. S2† confirms the negligible amount of methylene blue degradation and Cr(VI) reduction even after 1 hour of stirring, which excludes any strong influence of surface adsorption. From the above observations, it can be concluded that (i) in the absence of the catalyst, MB degradation and Cr(VI) reduction do not occur, and (ii) the reaction does not occur at dark conditions even in the presence of the catalyst.
The above observations suggest that the as-synthesized nanoparticles are most probable candidates as photocatalysts. However, the quality of the photocatalyst can be mainly measured from the charge transfer efficiency between the valence band and the conduction band of the material, as shown in the representative Fig. S3.† Hence, our curiosity highly motivated us to see the real-time efficiency of our as-synthesized photocatalysts experimentally through some more controlled reactions for MB degradation and Cr(VI) reduction using a higher intensity light source. To our surprise, when we carried out all the photocatalytic reactions again using a commercial 250 watt high-pressure mercury vapor visible lamp (HPMVL) with wavelength distribution of 400–700 nm as the light source, excellent results were obtained. As shown in Fig. S4a and b,† within 5 and 10 min, the complete degradation of MB and the reduction of Cr(VI) were respectively observed. This experimental result definitely indicates the high charge transfer efficiency within the band structure of our as-synthesized photocatalysts.
After carrying out all the photocatalytic reactions, it is very important to understand the stability and long-term durability of the as-synthesized CS2 and CS20 nanoparticles in their respective photocatalytic environment. From a series of controlled photocatalytic experiments, it was found that the as-synthesized CS2 and CS20 nanoparticles are very much stable under the reaction conditions for long durations of time. For the durability test, the MB degradation and Cr(VI) reduction experiments were again carried out in the presence of CS2 nanoparticles under the same photocatalytic conditions. Even after the completion of both the reactions, the reaction solutions were further kept in the stirring mode for another two hours with continuous irradiation of visible light. The catalyst was then separated from the solution mixture and reused in a new set of MB degradation and Cr(VI) reduction reactions. The reused CS2 nanoparticles showed almost similar efficiency toward both the reactions. After the durability test, the CS2 catalyst was recovered, dried, and analyzed for the structural and morphological study. In a similar way, the CS20 catalyst was also recovered after the recyclability test (after the 5th photocatalytic cycle) for the benzyl alcohol oxidation reaction. As shown in Fig. S5a and b,† the retention of the original structure in both the recovered CS2 and CS20 catalysts could be clearly ascertained from the powder XRD patterns. When the morphology of both the recovered catalysts, as can be seen in Fig. S5c and d,† was compared with Fig. 4a and d, a similar retention behavior was observed.
A model reaction of 4-bromo-benzaldehyde, nitromethane, and sodium azide was chosen to screen the reaction parameters and the results are summarized in Table S4.† Thus, after screening the suitable reaction parameters, 10 mg of the CS2 catalyst in the green solvent PEG-400 was chosen as the best reaction condition throughout this work. Once we established the best reaction parameters for NH-triazole synthesis, we then explored the efficiency of the catalyst in a wide range of electronically diverse substrates, the results of which are summarized in Fig. 9a–j. Interestingly, it was found that the substrates showed satisfactory outcomes, resulting in good to excellent yield of the requisite NH-triazole products. Aldehydes bearing electron withdrawing groups such as cyanides and halogens afforded the desired products in excellent yields within a short period of time. Moreover, salicylaldehyde and 2,4-dichlorobenzaldehyde also afforded the product in satisfactory yields (Fig. 9, entries e and g). In addition, the efficiency of the catalyst was also investigated in heterocyclic moieties such as thiophen-2-aldehyde and furan-2-aldehyde; it was found that the heterocyclic moieties also delivered the NH-triazole products quite smoothly in satisfactory yields (Fig. 9, entries h and i). We also carried out the reaction using another nitro compound, i.e., nitroethane, with benzaldehyde under standard reaction conditions and found that nitroethane was also quite suitable for this transformation (Fig. 9, entry j).
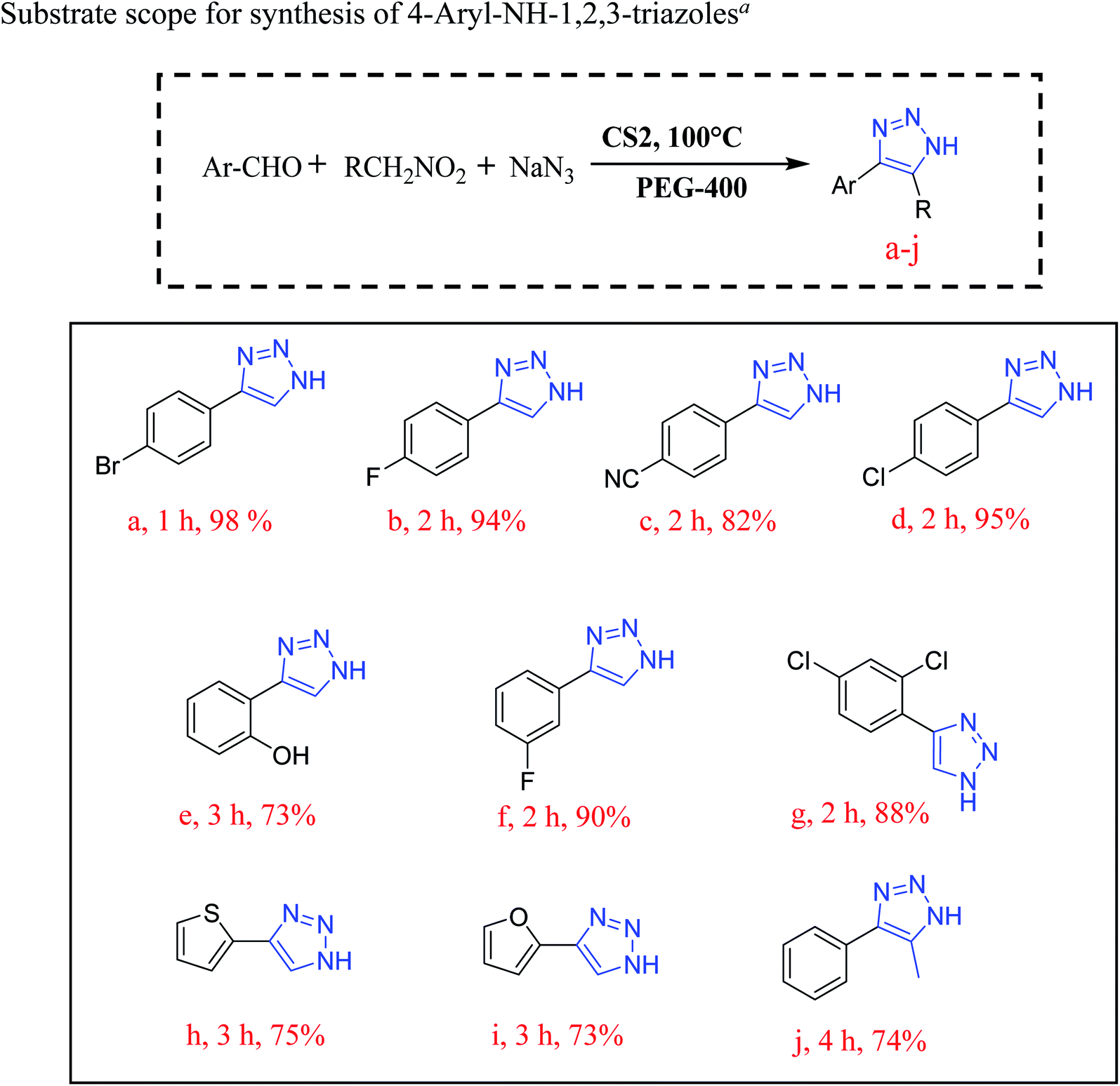 | ||
| Fig. 9 Effect of the substituent on the synthesis of NH-triazoles. aReaction conditions: substituted benzaldehyde (1 mmol), nitromethane (2 mmol), NaN3 (3 mmol), CS2 (10 mg), solvent (3 mL), in air. | ||
Conclusion
In conclusion, we were successful in establishing a green and economic protocol for the synthesis of eco-friendly CS NPs involving an effortless precipitation process. This semiconductor photocatalyst proved to be a consistent multifunctional catalyst and is also suitable for large scale production in a completely green environment. Four samples of copper sulfide NPs were synthesized just by varying the amount of the sulfur precursor and their effect on the phase and composition of the samples was studied. The composition, phase, purity, and size of the NPs were confirmed by various characterization techniques. Further, the pure phase of CuS nanoparticles was found to be a potential candidate for visible light-mediated photocatalytic degradation of rigid organic dye MB and the reduction of carcinogenic Cr(VI) to harmless Cr(III) in the absence of any reducing agent. The same CuS nanomaterial was also proven to be advantageous in the synthesis of the pharmaceutically important 1,2,3-triazole moiety. On the other hand, mixed phase CS NPs (CuS–CuS2) were found to be most efficient catalyst in the selective oxidation of a number of aromatic alcohols to their corresponding aldehydes. This approach of photocatalysis was proved to be efficient without the involvement of any noble metal. Moreover, the role of different oxidizing states of Cu in the catalysis of reactions offering selectivity has been demonstrated.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
S. A. is thankful to SERB, India for research fellowship. K. D. thanks SERB-DST, India (Grant EEQ/2018/000326) and UGC, India (Grant No. F.30-467/2019-BSR) for financial support. D. S. is thankful to the DST, New Delhi, India, for a research grant (no. EMR/2016/002345). We thank Dibrugarh University for providing all the infrastructural facility, STIC-Cochin for PXRD, HRTEM facility, IIT Roorkee for XPS analysis and CSIC Dibrugarh University for NMR measurements. The authors are grateful to the Department of Science and Technology for financial support under DST-FIST programme and UGC, New Delhi for Special Assistance Programme (UGC-SAP) to the Department of Chemistry, Dibrugarh University.References
- P. Wu, Y. Cao, L. Zhao, Y. Wang, Z. He, W. Xing, P. Bai, S. Mintova and Z. Yan, J. Catal., 2019, 375, 32–43 CrossRef CAS.
- M. Marelli, A. Jouve, A. Villa, R. Psaro, A. Balerna, L. Prati and C. Evangelisti, J. Phys. Chem. C, 2019, 123, 2864–2871 CrossRef CAS.
- S. Iraqui, S. S. Kashyap and M. H. Rashid, Nanoscale Adv., 2020, 2, 5790–5802 RSC.
- A. Ajmal, I. Majeed, R. N. Malik, H. Idriss and M. A. Nadeem, RSC Adv., 2014, 4, 37003–37026 RSC.
- S. Sakthivel, B. Neppolian, M. V. Shankar, B. Arabindoo, M. Palanichamy and V. Murugesan, Sol. Energy Mater. Sol. Cells, 2003, 77, 65–82 CrossRef CAS.
- S. Balanand, K. B. Babitha, M. Jeen Maria, A. A. P. Mohamed and S. Ananthakumar, ACS Sustainable Chem. Eng., 2018, 6, 143–154 CrossRef CAS.
- A. T. Kuvarega, R. W. Krause and B. B. Mamba, J. Phys. Chem. C, 2011, 115, 22110–22120 CrossRef CAS.
- S. Khanchandani, S. Kumar and A. K. Ganguli, ACS Sustainable Chem. Eng., 2016, 4, 1487–1499 CrossRef CAS.
- Y. B. Chen, L. Chen and L. M. Wu, Cryst. Growth Des., 2008, 8, 2736–2740 CrossRef CAS.
- Z. Shalabayev, M. Baláž, N. Daneu, E. Dutková, Z. Bujňáková, M. Kaňuchová, Z. Danková, L. Balážová, F. Urakaev, L. Tkáčiková and M. Burkitbayev, ACS Sustainable Chem. Eng., 2019, 7, 12897–12909 CrossRef CAS.
- M. Wang, F. Xie, W. Li, M. Chen and Y. Zhao, J. Mater. Chem. A, 2013, 1, 8616–8621 RSC.
- Z. K. Yang, Y. Teng and J. Xia, J. Mater. Chem. A, 2014, 2, 20004–20009 RSC.
- X. Zhang, F. Yang, S. Cui, W. Wei, W. Chen and L. Mi, Sci. Rep., 2016, 6, 1–11 CrossRef CAS PubMed.
- M. Basu, A. K. Sinha, M. Pradhan, S. Sarkar, Y. Negishi and T. Pal, Environ. Sci. Technol., 2010, 44, 6313–6318 CrossRef CAS PubMed.
- Z. Hosseinpour, S. Hosseinpour, M. Maaza and A. Scarpellini, RSC Adv., 2016, 6, 42581–42588 RSC.
- X. Meng, G. Tian, Y. Chen, R. Zhai, J. Zhou, Y. Shi, X. Cao, W. Zhou and H. Fu, CrystEngComm, 2013, 15, 5144–5149 RSC.
- J. Kundu and D. Pradhan, ACS Appl. Mater. Interfaces, 2014, 6, 1823–1834 CrossRef CAS PubMed.
- H. Zhu, J. Wang and D. Wu, Inorg. Chem., 2009, 48, 7099–7104 CrossRef CAS PubMed.
- H. Wang, J. R. Zhang, X. N. Zhao, S. Xu and J. J. Zhu, Mater. Lett., 2002, 55, 253–258 CrossRef CAS.
- W. Wang and L. Ao, Mater. Chem. Phys., 2008, 109, 77–81 CrossRef CAS.
- X. Yan, E. Michael, S. Komarneni, J. R. Brownson and Z. F. Yan, Ceram. Int., 2013, 39, 4757–4763 CrossRef CAS.
- W. He, H. Jia, X. Li, Y. Lei, J. Li, H. Zhao, L. Mi, L. Zhang and Z. Zheng, Nanoscale, 2012, 4, 3501–3506 RSC.
- M. Xu, H. Wu, P. Da, D. Zhao and G. Zheng, Nanoscale, 2012, 4, 1794–1799 RSC.
- J. Gao, Q. Li, H. Zhao, L. Li, C. Liu, Q. Gong and L. Qi, Chem. Mater., 2008, 20, 6263–6269 CrossRef CAS.
- H. Ueda, M. Nohara, K. Kitazawa, H. Takagi, A. Fujimori, T. Mizokawa and T. Yagi, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 155104 CrossRef.
- J. C. W. Folmer, F. Jellinek and G. H. M. Calis, J. Solid State Chem., 1988, 72, 137–144 CrossRef CAS.
- S. Khanal, G. Casillas, N. Bhattarai, J. J. Velázquez-Salazar, U. Santiago, A. Ponce, S. Mejia-Rosales and M. José-Yacamán, Langmuir, 2013, 29, 9231–9239 CrossRef CAS PubMed.
- C. Xing, Y. Zhang, Z. Wu, D. Jiang and M. Chen, Dalton Trans., 2014, 43, 2772–2780 RSC.
- J. Liu and D. Xue, J. Mater. Chem., 2011, 21, 223–228 RSC.
- S. Adhikari, D. Sarkar and G. Madras, ACS Omega, 2017, 2, 4009–4021 CrossRef CAS PubMed.
- Y. Chen, C. Davoisne, J. M. Tarascon and C. Guéry, J. Mater. Chem., 2012, 22, 5295–5299 RSC.
- S. Ramadan, L. Guo, Y. Li, B. Yan and W. Lu, Small, 2012, 8, 3143–3150 CrossRef CAS PubMed.
- K. S. Bhat and H. S. Nagaraja, J. Sci.: Adv. Mater. Devices, 2020, 5, 361–367 Search PubMed.
- Y. Chen, K. Chen, J. Fu, A. Yamaguchi, H. Li, H. Pan, J. Hu, M. Miyauchi and M. Liu, Nano Mater. Sci., 2020, 2, 235–247 CrossRef.
- H. S. Kim, J. Choi, J. Kong, H. Kim, S. J. Yoo and H. S. Park, ACS Catal., 2020, 11, 435–445 CrossRef.
- H. M. Savanur, K. N. Naik, S. M. Ganapathi, K. M. Kim and R. G. Kalkhambkar, ChemistrySelect, 2018, 3, 5296–5303 CrossRef CAS.
- T. Gregorić, M. Sedić, P. Grbčić, A. T. Paravić, S. K. Pavelić, M. Cetina, R. Vianello and S. Raić-Malić, Eur. J. Med. Chem., 2017, 125, 1247–1267 CrossRef PubMed.
- N. Upmanyu, S. Kumar, M. D. Kharya, K. Shah and P. Mishra, Acta Pol. Pharm., 2011, 68, 213–221 CAS.
- S. V. Akolkar, A. A. Nagargoje, V. S. Krishna, D. Sriram, J. N. Sangshetti, M. Damale and B. B. Shingate, RSC Adv., 2019, 9, 22080–22091 RSC.
- P. Phukan, S. Agarwal, K. Deori and D. Sarma, Catal. Lett., 2020, 150, 2208–2219 CrossRef CAS.
- A. Garg, D. Sarma and A. A. Ali, Curr. Res. Green Sus. Chem., 2020, 3, 100013 Search PubMed.
- L. An, L. Huang, P. Zhou, J. Yin, H. Liu and P. Xi, Adv. Funct. Mater., 2015, 25, 6814–6822 CrossRef CAS.
- M. Saranya, R. Ramachandran, E. J. J. Samuel, S. K. Jeong and A. N. Grace, Powder Technol., 2015, 279, 209–220 CrossRef CAS.
- S. Wang, J. Wang, X. Ji, J. Meng, Y. Sui, F. Wei, J. Qi, Q. Meng, Y. Ren, Y. He and D. Zhuang, J. Mater. Sci.: Mater. Electron., 2020, 31, 10489–10498 CrossRef CAS.
- J. Li, W. Yang, A. Wu, X. Zhang, T. Xu and B. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 8583–8591 CrossRef CAS PubMed.
- Y. Hui, S. Zhang and W. Wang, Adv. Synth. Catal., 2019, 361, 2215–2235 CrossRef CAS.
- P. Wu, L. Song, Y. Wang, X. Liu, Z. He, P. Bai and Z. Yan, Appl. Surf. Sci., 2021, 537, 148059 CrossRef CAS.
- R. S. Dariani, A. Esmaeili, A. Mortezaali and S. Dehghanpour, Optik, 2016, 127, 7143–7154 CrossRef CAS.
- P. Kush, K. Deori, A. Kumar and S. Deka, J. Mater. Chem. A, 2015, 3, 8098–8106 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00239b |
| This journal is © The Royal Society of Chemistry 2021 |