DOI:
10.1039/D1NA00235J
(Paper)
Nanoscale Adv., 2021,
3, 3867-3874
Facile room-temperature self-assembly of extended cation-free guanine-quartet network on Mo-doped Au(111) surface†
Received
30th March 2021
, Accepted 5th May 2021
First published on 20th May 2021
Abstract
Guanine-quadruplex, consisting of several stacked guanine-quartets (GQs), has emerged as an important category of novel molecular targets with applications from nanoelectronic devices to anticancer drugs. Incorporation of metal cations into a GQ structure is utilized to form stable G-quadruplexes, while formation of a cation-free GQ network has been challenging. Here we report the room temperature (RT) molecular self-assembly of extended pristine GQ networks on an Au(111) surface. An implanted molybdenum atom within the Au(111) surface is used to nucleate and stabilize the cation-free GQ network. Additionally, decoration of the Au(111) surface with 7-armchair graphene nanoribbons (7-AGNRs) enhances the GQ domain size by suppressing the influence of the disordered phase nucleated from Au step edges. Scanning tunneling microscopy/spectroscopy (STM/STS) and density functional theory (DFT) calculations confirm the formation of GQ networks and unravel the nucleation and growth mechanism. Our work, utilizing a hetero-atom doped substrate, provides a facile approach to enhance the stability and domain size of the GQ self-assembly, which would be applicable for other molecular structures.
Introduction
Self-assembly of organic molecules with sufficient stability is highly desirable to develop intriguingly organized materials or molecular architectures for diverse applications from nanoelectronic circuits to drug design.1–4 The emergence of various RNA/DNA quadruplex structures is a vital discovery in biomolecular science because of their antiproliferative behavior and the gene-regulating features of non-canonical RNA/DNA secondary structures.5–7 Among various RNA/DNA quadruplexes, guanine is the only purine generated in human cells to date,8,9 where stabilizing the G-quadruplex has become a practical rationale for therapeutic approaches to treat cancer with crucial selectivity that is required for clinical development.10,11 In light of molecular electronics, the DNA G-quadruplex is a legitimate candidate for miniaturizing memory storages and electronic devices since it is capable of transporting electrical current and inherits DNA's versatile and programmable structure, while native double-stranded DNA produces no detectable signal.1
In attempts to understand what causes quadruplex self-assembly, several studies have been reported on surface-assembled purines and pyrimidines on Au(111).12–16 Mainly, chain-like networks were observed primarily due to the relatively strong intermolecular hydrogen bonds.17–19 In addition, the impact of impurity atoms on the network patterns has been studied. For instance, the presence of metal ions is not necessary for the fabrication of xanthine-quartets (XQs)18 or the tetrad form of lipophilic phenolic guanosine derivatives,20 but is required to form GQ and hypoxanthine-quartet (HX-Q) networks.19,21,22 Although the cooperative effect, responsible for Watson–Crick base pairing, strengthens the hydrogen bonds in a GQ relative to those in isolated G dimers,23,24 recent reports show that this effect is not sufficient to initiate a stable GQ from its molecular building blocks.18,19,21,22 A pristine form of GQ is electrostatically unfavorable as four oxygen atoms are located towards the central cage; thus, incorporation of a monovalent cation, either K+ or Na+,15,22,25 or post-deposition of transition metal adatoms onto guanine molecules and annealing were required for GQ self-assembly.18,21 The geometry of a stabilized initial seed is an additional crucial factor in the single-step growth of the molecular network at RT, where the nucleation can proceed from either a flat terrace or an edge region. To block surface diffusion towards undesirable nucleation sites, a heterogeneous admixture is employed.26 Stabilizing pure GQ networks without incorporating metal cations can boost opportunities to fabricate more diverse, innovative surface assembled structures with novel applications.
Here we report RT self-assembly of a cation-free GQ network on Au(111) by substituting a minute amount of surface Au atoms by Mo atoms, and it shows robust thermal stability against annealing up to 500 K. The underlying mechanism is demonstrated to be the covalent interaction between G/9H and surface Mo based on our corroborative confirmation with STS measurement and DFT calculations. Furthermore, enlarged domain size of GQ networks was observed in the case of bottom-up synthesis of GNRs prior to G growth. Our work provides a facile approach for enhancing the stability and domain size of GQ networks, which might be applicable to two dimensional self-assembly of other nucleobases that have shown only limited stability and size.
Results and discussion
Guanine self-assembly on pristine Au(111)
Guanine has several molecular tautomers, and the two most stable ones are the canonical G/9H and the non-canonical G/7H forms.22,27,28 These two forms, like many other tautomeric structures of organic compounds, transform into each other through the migration of an H atom and coexist when reaching a certain temperature or coming into contact with specific materials.28–30 Previous studies on 2D assembly show that a GQ network is based on the canonical structure (G/9H) in the presence of alkali metals.22,23 The guanine zigzag structure on Au(111), which is the thermodynamically most stable structure on the surface, has double and triple intermolecular hydrogen bonds and is derived from the G/7H tautomeric form with a mixture of left and right chirality.12,13,22,23 In our experiments, after the deposition of guanine/9H molecules on pristine Au(111) at RT, STM images show a disordered arrangement on the surface (Fig. 1a). Annealing the disordered guanine molecules on Au(111) at 400 K stimulates the molecules to move on the surface more freely and transform into the G/7H tautomeric structure, for which the STM images show a close-packed lattice of G/7H molecules as shown in Fig. 1a. According to our DFT calculations, individual guanine molecules adsorb weakly in a face-down geometry via van der Waals interaction. The vertical distance between the O atom and Au surface layer is 2.6 Å for G/9H and 3.2 Å for the G/7H molecule, with the adsorption energies of −1.38 eV and −1.24 eV, respectively (Fig. 1b). This difference in the adsorption energies of guanine tautomers can be evidenced by the fact that the G/9H has a much stronger molecular dipole moment than any other guanine tautomers, while the G/7H has the smallest molecular dipole moment among them.31,32 Annealing helps adsorbed molecules find each other to form the most stable network connected by hydrogen bonding. The DFT predicted free-standing lattice formation energy is −1.75 eV per molecule for G/7H, stronger than the −1.27 eV per molecule for G/9H (Fig. S1a and b†). The difference (0.48 eV per molecule) is sufficient to stabilize the G/7H lattice over the G/9H lattice, where a single G/9H molecule adsorption was more stable than that of G/7H by 0.14 eV per molecule, which is in good agreement with the observed close-packed lattice after annealing at 400 K (Fig. 1a).
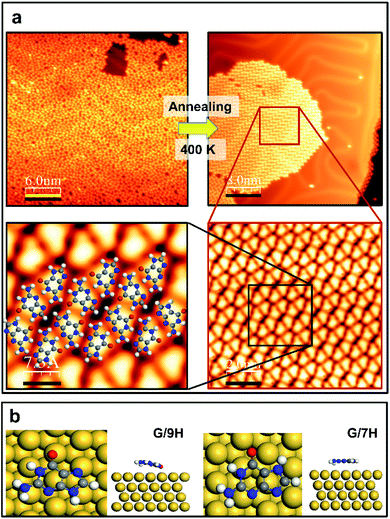 |
| | Fig. 1 Guanine on the pristine Au(111) surface. (a) STM images before and after annealing at 400 K (upper panel). The latter is shown in smaller scales (2.0 nm and 7.3 Å) in the lower panel, depicting that the disordered phase of self-assembled guanine is transformed to the close-packed network on Au(111) with the DFT-optimized structural model superimposed. Scanning conditions (before annealing): tunneling current It = 0.055 nA, sample voltage Vs = +0.9 V; scanning conditions (after annealing): It = 0.15 nA, Vs = +0.9 V. (b) Top and side views of DFT-predicted geometries for the single G/9H (left) and G/7H (right) molecule adsorption on Au(111). Single-molecule adsorption energy (Ea = Emolecule@surface − Esurface − Emolecule) on the surface is calculated to be −1.38 eV (shortest vertical distance h = 2.6 Å) for the G/9H molecule, while −1.24 eV (h = 3.2 Å) for the G/7H molecule. H: white, C: gray, N: blue, O: red, Au: yellow. | |
Guanine self-assembly on Mo-doped Au(111)
Since the chemically stable Au(111) provides a suitable environment for some self-assembled molecular architectures,23,33,34 the possibility of self-assembling of metal-free GQ networks on the Au(111) surface was initially discussed.23 However, later it was found to be dependent on the incorporated metal ions.18,19,21 Furthermore, the growth of a cation-incorporated extensive GQ network was impossible at RT. It needed annealing at a specific temperature.18,19,21,22 Therefore, a mixture of adsorbates (metal ions plus guanine molecules) was found to be the most stable network structure.35,36 When the molecule–surface interaction is weak, like the van der Waals interaction between guanine molecules and the Au(111) surface, G/9H molecules have significant mobility unless the ample kinetic energy is diminished by collisions at RT.33 Consequently, molecules failed to selectively form an ordered G hydrogen bonding network, and so a random molecular structure formed at RT (Fig. 1a). Although the hydrogen bonding in a pristine GQ is substantially four times more stable than the G dimers' hydrogen bond energy,23 the electrostatic repulsions forbid a cation-free GQ network.19,24
For one-step fabrication of a GQ network at RT, we stabilized the initial seed on terrace regions by doping the Au(111) surface with a small amount of Mo atoms (Fig. S2†), where the atomically flat surface and the Au(111) herringbone structure are maintained after depositing Mo clusters and annealing. In order to remove any trace of Mo adatoms that might remain on the Au surface after deposition, the sample was annealed at 700 K for 30 min (Fig. S3†). Our DFT calculations show that the G/9H molecule makes a covalent bond with an embedded Mo atom at the Au surface layer in a face-down geometry with −2.42 eV adsorption energy and a vertical distance of 2.0 Å between O and Mo atoms (Fig. 2c). As we calculated the formation energy of the GQ network in the presence of one Mo–O covalent bond per four G/9H molecules (Fig. 2d), the molecular network becomes more stable with a formation energy of −1.96 eV per molecule on the Mo-doped Au surface, compared to the GQ network on the pristine Au(111) with a formation energy of −1.82 eV per molecule (Table 1). The influence of the Mo–O covalent bond on hydrogen bond strength was investigated by comparing the DFT calculated intraquartet hydrogen bond lengths of adsorbed GQ networks on two surfaces: pristine Au(111) surface and Mo-doped Au(111) surface. The covalent bond between Mo and O6 results in vertical movement of O6 atoms toward the Mo atoms, where the O6–H hydrogen bond length increases from 1.65 Å to 2.0 Å. Thus, the suppressed effective charges (Fig. S9†) and elongated bond length associated with O6–H weaken the hydrogen bond strength. Also, when two or three Mo atoms are embedded in the surface layer, the formation of the GQ network is slightly more enhanced compared to the case of a single Mo (Fig. S4† and Table 1). However, this di-vacancy replacement would hardly take place compared with the mono-vacancy replacement (Fig. S5 and S6†), and the mono-vacancy replacement is entropically favoured at high-temperature annealing (700 K). Overall, there will be more probability of finding the GQ form's initial seeds on the Mo-doped Au(111) surface than on the pristine Au(111) (Fig. S1c and d†). As a result of intraquartet hydrogen bonding and covalent bonding between Mo at the surface and O of G/9H in addition to interquartet hydrogen bonding, a cation-free GQ network was formed at RT, as is evident from Fig. 2a and b, which show the STM image of the cation-free GQ network with the superimposed DFT-optimized structural model.
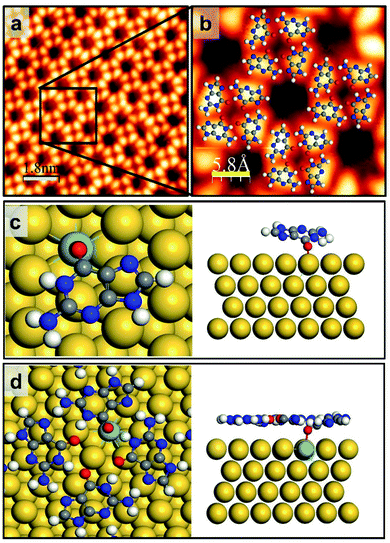 |
| | Fig. 2 G/9H molecule adsorption and GQ network formation on Mo-doped Au(111). (a) Close-up STM image of guanine self-assembly on the Mo-doped Au(111) surface from rare, limited GQ networks surrounded by disordered domains. (b) The GQ network geometry obtained by DFT is superimposed. Scanning conditions: It = 0.125 nA, Vs = +0.8 V. (c and d) Top and side views of DFT-predicted geometry for (c) the single G/9H molecule adsorption (Ea = −2.42 eV and h = 2.0 Å) and (d) the GQ network formation energy Ef = −1.96 eV per molecule on Mo-doped Au(111) (also see Table 1 and Fig. S4†). H: white, C: gray, N: blue, O: red, Mo: azure, Au: yellow. Scale bar: 6.0 nm in panel a, 1.8 nm in panel b, 5.8 Å in panel c. | |
Table 1 GQ network formation energies: free-standing, on pristine Au(111), and on the Mo-doped Au(111) surface with one, two, or three Mo atoms embedded at the surface layer. Free standing lattice formation energy per molecule: Ef = [E2D lattice − 4 × Emolecule]/4; lattice formation energy on the surface per molecule: Ef = [E2D lattice@surface − 4 × Emolecule − Esurface]/4
| GQ network |
Formation energy (eV per molecule) |
| Free-standing |
−1.27 |
| On the pristine Au(111) surface |
−1.82 |
| On the Mo-doped Au(111) surface (single Mo atom at the surface layer) |
−1.96 |
| On the Mo-doped Au(111) surface (two Mo atoms at the surface layer) |
−2.17 |
| On the Mo-doped Au(111) surface (three Mo atoms at the surface layer) |
−2.12 |
Effect of graphene nanoribbon pre-synthesis
It has been known that due to reconstructed patterns and low-coordinated Au atoms, step edges represent preferential nucleation sites for the growth of supramolecular structures.33,34,37,38 Therefore, guanine molecules have no choice other than making hydrogen bonds with a chain array of initial molecules at step edges that are not compatible with the GQ network's geometry. Based on our experiments, the disordered domains initiated from step edges can interfere with the GQ network's growth on the Mo-doped Au(111) surface (Fig. S1d†). This interference can be spatially terminated by placing GNRs since a guanine molecule's interaction with a GNR is weak. The effect of graphene nanoribbon pre-synthesis on guanine self-assembly was initially investigated on the pristine Au(111) surface. The DFT calculation shows that the adsorption energy of a guanine molecule on 7-AGNR adsorbed on Au(111) is −0.94 eV, much weaker than the adsorption energy on Au(111) (−1.38 eV). Therefore, guanine molecules are hardly stacked on GNRs,39,40 as shown in Fig. 3b; hence 7-AGNR acts as a neutral fence of cage area to grow guanine self-assemblies.26 The successful growth of GNRs on the pristine Au(111) surface is confirmed by STM, as shown in Fig. 3a and S8.† The guanine molecules were assembled within a region enclosed by GNRs rather than diffusing towards GNRs (Fig. 3b). It is consistent with the observation that the binding energy of the free-standing GNR–guanine side-to-side configuration is −0.32 eV, while that of a guanine dimer is −0.65 eV. The STM image in Fig. 3c shows that guanine self-assembly is less affected by step edges, which are preferentially covered by GNRs. As the Au surface was free of Mo, we observed the disordered phase of G/9H for RT deposition (Fig. 3b) and a pure G/7H close-packed structure for 400 K annealing (Fig. 3d). Guanine molecules were deposited at RT after the GNR pre-synthesis and Mo-doping to investigate both effects together. Extended GQ islands on the Mo-doped Au(111) surface with 7-AGNR pre-synthesis are shown in Fig. 4a. Decorating the surface with GNRs facilitated the formation of more extensive and homogeneous GQ networks by producing confined areas in which molecular networks nucleated without interacting with the GNR boundaries (Fig. 4a, b and S1f–l†). GQ domains were surrounded inevitably by a minor number of disordered domains (Fig. 4a). Guanine molecules were not influenced by surface step edges after Mo-doping (Fig. 4b), as diffusion passage was blocked by GNRs. Step edges could be easily passivated by the H atoms released from the GNR precursor (DBBA) during the cyclodehydrogenation step of GNR synthesis at 670 K.41,42 Since the Mo concentration of the Au(111) surface layer was minute, it did not affect the on-surface synthesis of 7-AGNRs. Au(111) surface step edges as active adsorption sites are passivated and deactivated once GNRs are synthesized on-surface in any coverage ratio. However, STM images showed that the GQ network could not be grown in confined areas with less than 5.0 × 5.0 nm2 dimensions (Fig. 4c and d).
 |
| | Fig. 3 Effect of GNR pre-synthesis on guanine nucleation and growth on the pristine Au(111) surface at RT. (a) STM image of 7-AGNRs synthesized on Au(111). Scanning conditions: It = 0.03 nA, Vs = +0.75 V. (b) Guanine monolayer islands (disordered phase) nucleated on Au(111) and grown until they reached GNR edges. Self-assembled guanine nucleated on the pristine Au surface between GNRs without binding them. Guanine molecules were not stacked on the GNR surface. Scanning conditions: It = 0.015 nA, Vs = +1.10 V. (c) Step edges are not preferential nucleation zones after GNR pre-synthesis, as shown with yellow dashed lines. Scanning conditions: It = 0.003 nA, Vs = +0.8 V. (d) G/7H close-packed structure after annealing the disordered phase at 400 K. The inset image shows a higher resolution of the zigzag structure of G/7H tautomers. Curing the disordered phase on the pristine Au surface by GNR pre-synthesis is not sufficient to nucleate a GQ network's seed at RT, even though the destructive effect of Au step edges is eliminated. Scanning conditions: It = 0.02 nA, Vs = +0.9 V. | |
 |
| | Fig. 4 Effect of GNR pre-synthesis on GQ network's growth on the Mo-doped Au(111) surface at RT. (a) Extended GQ networks are nucleated and grown between 7-AGNRs and separated from limited disordered domains. Disordered domains terminated by GNRs are shown with yellow dashed lines. Scanning conditions: It = 0.03 nA, Vs = +0.9 V (b) step edges are shown with yellow dashed lines, and they are not preferential nucleation zones for guanine molecules after GNR pre-synthesis. Minute doping plus GNR pre-synthesis effects conjointly led to extended pure GQ networks on the Au(111) surface. Scanning conditions: It = 0.03 nA, Vs = +0.8 V. (c) Dense GNRs on the surface interrupts GQ network's growth. If the confined area has dimensions less than around 5.0 × 5.0 nm2, the GQ networks could not be grown. (d) Higher coverage ratio of GNRs led to isolated GQs with no chance to make a stable network. | |
Annealing GQ networks and disordered domains
Although both Mo-doping and GNR pre-synthesis effects induce the one-step formation of extensive GQ networks at RT, a minor number of disordered domains were still observable (Fig. 4a and S1f–l†). To investigate the origin of the disordered domains, we annealed guanine self-assembly on the Mo-doped Au surface without GNR pre-synthesis (Fig. S1d†). Disordered domains of guanine on Mo-doped Au(111) transformed into a close-packed structure at 400 K, while some disordered phases remained (Fig. 5a). The disordered domains originated from not only the interference of assemblies initiated from step edges but also the locally overdoped Au(111) surface by Mo (Fig. 5a and S7a†). This made adsorbed guanine molecules rather fixed with a lack of conformality to the GQ lattice due to the relatively strong covalent bonds with the substrate. The disordered domains on the Mo-doped Au surface finally transformed into the G/7H structure after annealing at 460 K (Fig. 5b and S7b†). In contrast to the GQ network, the close-packed lattice had the flexibility to adapt with guanine molecules bonded at step edges or Mo atoms (Fig. S7c and d†). DFT calculation also showed that the G/7H molecule makes a covalent bond with an embedded Mo atom in the Au surface layer in a face-down geometry with a vertical distance of 2.1 Å between O and Mo atoms and −2.37 eV adsorption energy (Fig. 5c). For the Mo-doped Au surface with GNR pre-synthesis, annealing at 460 K also resulted in pure GQ networks and a few close-packed islands along with 7-AGNRs (Fig. 5d). At 500 K annealing, GNRs moved towards each other and did not mix/bond with the GQ networks or close-packed G/7H structures. Small GQ networks (less than 10 × 10 nm2 area) were no longer observable at 500 K annealing (Fig. 5e), they either merged or gradually converted to the close-packed lattices. A higher annealing temperature of 520 K resulted in a complete conversion from the extensive metal-free G-quartet network to the zigzag structure with separated and unchanged GNRs (Fig. 5f and S7f†), unlike the case of G-quartet-cations, in which G/9H to G/7H conversion is not allowed as long as a sufficient number of cations are available.22
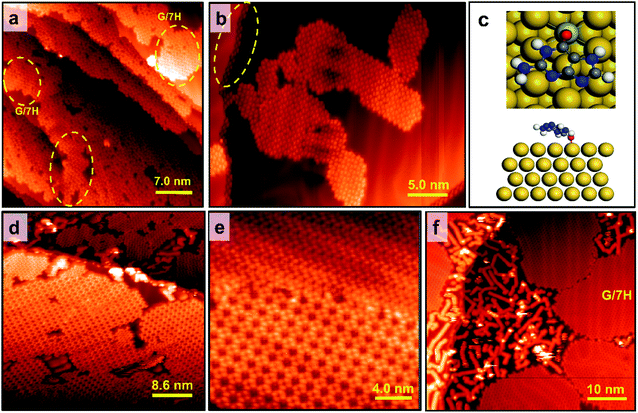 |
| | Fig. 5 Annealing the guanine self-assemblies on the Mo-doped Au surface. (a) At 400 K, a disordered domain was partially transformed into the close-packed structure, while some disordered phases remain due to relatively strong covalent bonds with Mo embedded at the Au surface (on the Mo-doped Au surface without GNR pre-synthesis). GQ networks barely remain at this temperature since their sizes are limited and small. Close-packed islands are highlighted with yellow dashed circles. Scanning conditions: It = 0.019 nA, Vs = +0.9 V. (b) By annealing at 460 K, guanine self-assemblies (disordered phases and small GQ islands) on the Mo-doped Au(111) surface without GNR pre-synthesis were fully transformed into the G/7H structure, which had the flexibility to adapt with guanine molecules bonded at Mo atoms or step edges as indicated with a yellow dashed circle. Scanning conditions: It = 0.02 nA, Vs = +0.9 V. (c) Top and side views of the DFT-predicted geometry for single G/7H molecule adsorption (Ea = −2.37 eV and h = 2.1 Å) on the Mo-doped Au(111) surface. H: white, C: gray, N: blue, O: red, Mo: azure, Au: yellow. (d) At 460 K, disordered domains on Mo-doped Au with GNR pre-synthesis were fully transformed into the G/7H structure, while the pre-formed extensive GQ networks were stable at this temperature. Scanning conditions: It = 0.02 nA, Vs = +1.0 V. (e) At 500 K, the G/7H lattice coexists with the GQ network, while small islands of GQ domains no longer exist. Scanning conditions: It = 0.044 nA, Vs = +0.9 V. (f) At 520 K, large islands of the close-packed structure are formed. GNRs were relocated but stayed segregated from guanine assemblies and gathered around step edges or between large islands of G/7H networks. Scanning conditions: It = 0.035 nA, Vs = +0.9 V. Scale bar: 7.0 nm in panel a, 5.0 nm in panel b, 8.6 nm in panel d, 6.4 nm in panel e, and 10 nm in panel d. | |
Electronic structure
The presence of Mo atoms at the surface layer was demonstrated from the molecules' electronic structure by interplaying STS measurements and DFT calculations. We examined the GQ structure on the Mo-doped Au(111) surface annealed at 400 K, which has mainly shown three structural domains in one sample: disordered phase and ordered ones consisting of G/7H and G/9H molecules. These three types indicated by the colored dots in the inset of Fig. 6a are related to the absence or presence of Mo underneath G. The measured STS data of G/9H molecules in the GQ network, G/7H molecules in the close-packed network, and guanine molecules in disordered domains are shown in Fig. 6a. Although the measured LUMO peaks of the molecule in GQ and close-packed networks are out of the achievable range by STS, the recorded spectrum reveals a new, broad spectroscopic feature in the positive potential for the guanine molecule in the disordered domain. The distinct spectral features are unraveled by DFT density of states calculations. The structural models are shown in Fig. S1e† for the G/9H molecule in a GQ network on Au(111) (black), in Fig. 1b for the G/7H molecule on Au(111) (green), and in Fig. 2d for the G/9H molecule with a covalent bond (in the GQ lattice) with the Mo-doped Au(111) surface (red). The newly emerged broad LUMO peak resulted from the covalent bonding between the guanine molecule and surface (Fig. 6b). The additional coupling of pz orbitals of the O atom of the anchored guanine molecule and the embedded Mo atom at the Au surface (Fig. 6c) lowered the molecule's energy levels (Fig. 6b). Accordingly, LUMO's peak of the chemically bonded molecule with the surface became achievable by STS and identified the anchored guanine molecule on the surface (red curve in Fig. 6a).
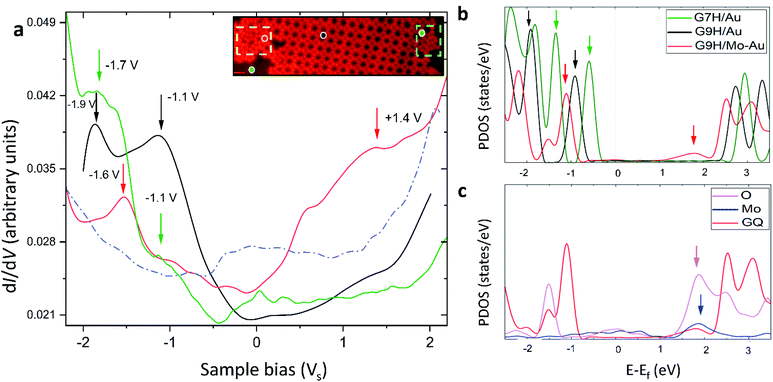 |
| | Fig. 6 Electronic structure of guanine molecules on the Au(111) surface. (a) The black (green) curve shows dI/dV point spectroscopy data collected from the G/9H (G/7H) molecule in a GQ (close-packed) network on Mo-doped Au(111). The red curve is dI/dV point spectroscopy data collected from a G/9H molecule in a disordered domain on the Mo-doped Au surface (inset image shows a GQ network, a zigzag lattice, and anchored guanine molecules in a disordered domain after annealing at 400 K). The dashed blue curve shows a spectrum collected on bare Au(111). In comparison with the spectrum for the bare case, the robust features of molecules are clearly visible with the suppressed peak of the intrinsic Au(111) surface state near −0.4 V. The spectroscopy parameters are Vac = 24 mV and f = 701 Hz; dI/dV point spectra were recorded under open feedback loop conditions. Scanning conditions: It = 0.155 nA, Vs = +1.0 V. All STS data shown here were obtained at T = 77 K. (b) DFT projected DOS (PDOS) for a G/9H molecule in a GQ network (Fig. S1e†) on pristine Au(111) (black), on Mo-doped Au(111) (red) surfaces (Fig. 2d), and a G/7H molecule (green) on the pristine Au(111) surface (Fig. 1b). (c) PDOS of covalently bonded Mo (blue) and O (purple) pz orbitals and PDOS of adsorbed GQ network (red) pz orbitals. The guanine molecule's chemical bond with the surface results in additional pz orbital coupling of Mo and O atoms and yields a newly emerged broad LUMO peak (red arrow in b). This peak is characteristic of the guanine molecule's covalent bonding with the Au surface. Blue and purple arrows in (c) show the origin of the LUMO peak indicated with a red arrow in (b). Green, black, and red arrows in (a) and (b) are peer-to-peer related. | |
Conclusions
In conclusion, we have successfully prepared the cation-free G-quartet network on the Au(111) surface at RT by strengthening first the molecule–surface interaction and controlling the initial seed's stability and geometry. The pre-synthesis of GNRs on the Mo-doped Au surface modified the surface domain spatially and chemically, which prompted effective nucleation and growth of GQ networks. The nucleation and growth mechanisms are unraveled from the interplay of STM imaging, STS measurements, and DFT calculations. GQs and GNRs relocated while stayed segregated from each other at various temperatures. The extensive cation-free GQ network exhibited robust thermal stability against annealing up to 500 K. Interconversion of metal-free networks to a more stable close-packed G/7H network in the presence of GNRs was achieved at 520 K. Our findings could open a new window for applications of graphene nanostructures and bimetallic surfaces in the stabilization of newly emerging surface assembled molecular structures.
Materials and methods
Sample preparation
For all the experiments, a 200 nm thin film of Au(111) epitaxially grown on mica (Phasis, Switzerland) was used as the substrate. Standard Ar+-sputtering/annealing cycles were applied to prepare an atomically clean surface. On-surface 7-AGNR synthesis on the Mo-doped Au(111) surface was performed using 10,10′-dibromo-9,9′-bianthracene (DBBA) precursor monomers (purchased from Sigma Aldrich, purity >98%) deposited via DCT at RT (Fig. S8†).43 The sample subsequently annealed gradually by dehalogenation below 370 K as well as polymerization at 470 K and cyclodehydrogenation at 670 K for 10 min,41,42,44 in the same way as the GNR synthesis on the bare Au(111) surface. The guanine 9H molecules (purchased from Sigma Aldrich, purity >98%) were first degassed thoroughly, and then thermally sublimated onto the clean Au(111) substrate at RT from a tantalum crucible heated at 500 K, resulting in a deposition rate of 0.01 molecules per min (0.01 ML min−1) on bare Au(111) and 0.03 molecules per min (0.03 ML min−1) on the Mo-doped Au(111) surface with/without GNR pre-synthesis. For binary surface preparation, a molybdenum target with purity >99% was repeatedly annealed/Ar+-sputtered in UHV for use as a Mo source. Mo clusters sputtered and deposited on the Au surface heated at 520 K (operating pressure 5.0 × 10−6 mbar) were alloyed with the Au surface during post-annealing first at 830 K/10 min for herringbone reconstruction of the Au(111) surface, and then at 700 K for 30 min to get large clean terraces and to remove any trace of Mo adatoms that might remain on the Au surface (Fig. S3†). The sample was subsequently transferred to the STM chamber within UHV for STM measurements at 77 K.
SPM characterization and measurements
All STM experiments were performed in a UHV chamber (base pressure 1.0 × 10−10 mbar) equipped with a low-temperature STM from Omicron Nanotechnology using electrochemically etched W tips. STS measurements were recorded using a lock-in amplifier with a modulation frequency of 701 Hz and a bias modulation amplitude of Vac = 24 mV. dI/dV point spectra were recorded under open feedback loop conditions. WSxM and Vernissage software from Omicron were used to process STM images.45
Surface characterization and measurements
TEM sample preparation was performed using the FIB-SEM technique. A gallium ion beam was employed to cut a thin slab of bulk Au. HR-TEM/HAADF STEM verified that the Au surface was doped with pure Mo, and EDS elemental mapping analysis showed the Mo concentration in the subsurface layer. The TOF-SIMS technique was also used to analyze the composition of the Mo-doped Au(111) surface (Fig. S2g†).
Density functional theory (DFT) calculations
DFT calculations were performed using the Vienna Ab initio Simulation Package (VASP). Tkatchenko and Scheffler (TS) dispersion correction46 and the Perdew–Burke–Ernzerhof (PBE) exchange functional47 were used to describe the adsorption of organic molecules on the Au(111) surface. In the Au(111) slab supercell, four layers of Au atoms were considered, and the top two layers were optimized. More than 12 Å of vacuum space were considered to avoid the interaction between periodic slabs. The plane-wave energy cut-off was 550 eV.
Author contributions
A. G. initiated/conducted on-surface synthesis, SPM experiments, and electronic/structural characterization analysis. He drafted the manuscript. E. O. performed DFT calculations and analysis for molecular self-assemblies and the surface structure. D. Y. K. prepared supporting calculations for immiscibility. G. L. validated the DFT calculations, and H. S. verified the on-surface synthetic experiments. K. S. K., H. S., and G. L. supervised the project. A. G. and K. S. K. are responsible for the project concept.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Research Foundation (NRF) of Korea (National Honor Scientist Program: 2010-0020414, Basic Science Research Program: 2018R1D1A1B07045983) and KISTI (KSC-2021-CRE-0193, KSC-2020-CRE-0185, KSC-2020-CRE-0049, KSC-2020-CRE-0146). A. G. would like to thank Saeed Pourasad for a fruitful discussion on theoretical simulations.
Notes and references
- G. I. Livshits, A. Stern, D. Rotem, N. Borovok, G. Eidelshtein, A. Migliore, E. Penzo, S. J. Wind, R. Di Felice, S. S. Skourtis, J. C. Cuevas, L. Gurevich, A. B. Kotlyar and D. Porath, Nat. Nanotechnol., 2014, 9, 1040–1046 CrossRef CAS PubMed.
- L. Martínez-Fernández, L. Esposito and R. Improta, Photochem. Photobiol. Sci., 2020, 19, 436–444 CrossRef PubMed.
- A. Ciesielski, M. El Garah, S. Masiero, P. Samorì and P. Samori, Small, 2016, 12, 83–95 CrossRef CAS PubMed.
- L. Stefan and D. Monchaud, Nat. Rev. Chem., 2019, 3, 650–668 CrossRef.
- D. Varshney, J. Spiegel, K. Zyner, D. Tannahill and S. Balasubramanian, Nat. Rev. Mol. Cell Biol., 2020, 21, 459–474 CrossRef CAS PubMed.
- M. L. Bochman, K. Paeschke and V. A. Zakian, Nat. Rev. Genet., 2012, 13, 770–780 CrossRef CAS PubMed.
- V. Brázda, L. Hároníková, J. C. C. Liao and M. Fojta, Int. J. Mol. Sci., 2014, 15, 17493–17517 CrossRef PubMed.
- G. Biffi, D. Tannahill, J. McCafferty and S. Balasubramanian, Nat. Chem., 2013, 5, 182–186 CrossRef CAS PubMed.
- R. Hänsel-Hertsch, M. Di Antonio and S. Balasubramanian, Nat. Rev. Mol. Cell Biol., 2017, 18, 279–284 CrossRef PubMed.
- S. Neidle, Nat. Rev. Chem., 2017, 1, 1–10 CrossRef.
- F. R. Winnerdy, P. Das, B. Heddi and A. T. Phan, J. Am. Chem. Soc., 2019, 141, 18038–18047 CrossRef CAS PubMed.
- W. Xu, J. G. Wang, M. F. Jacobsen, M. Mura, M. Yu, R. E. A. Kelly, Q. Q. Meng, E. Lægsgaard, I. Stensgaard, T. R. Linderoth, J. Kjems, L. N. Kantorovich, K. V. Gothelf and F. Besenbacher, Angew. Chem., Int. Ed., 2010, 49, 9373–9377 CrossRef CAS PubMed.
- W. Xu, R. E. A. Kelly, H. Gersen, E. Laegsgaard, I. Stensgaard, L. N. Kantorovich, F. Besenbacher, E. Lægsgaard, I. Stensgaard, L. N. Kantorovich and F. Besenbacher, Small, 2009, 5, 1952–1956 CrossRef CAS PubMed.
- A. I. Livshits and L. Kantorovich, J. Phys. Chem. C, 2013, 117, 5684–5692 CrossRef CAS.
- C. Zhang, L. Wang, L. Xie, H. Kong, Q. Tan, L. Cai, Q. Sun and W. Xu, ChemPhysChem, 2015, 16, 2099–2105 CrossRef CAS PubMed.
- Y. Ding, L. Xie, C. Zhang and W. Xu, Chem. Commun., 2017, 53, 9846–9849 RSC.
- H. Kong, C. Zhang, Q. Sun, X. Yu, L. Xie, L. Wang, L. Li, S. Hu, H. Ju, Y. He, J. Zhu and W. Xu, ACS Nano, 2018, 12, 9033–9039 CrossRef CAS PubMed.
- C. Chen, H. Sang, P. Ding, Y. Sun, M. Mura, Y. Hu, L. N. Kantorovich, F. Besenbacher and M. Yu, J. Am. Chem. Soc., 2018, 140, 54–57 CrossRef CAS PubMed.
- C. Chen, P. Ding, M. Mura, Y. Chen, Y. Sun, L. N. Kantorovich, H. Gersen, F. Besenbacher and M. Yu, Angew. Chem., Int. Ed. Engl., 2018, 57, 16015–16019 CrossRef CAS PubMed.
- M. El Garah, R. C. Perone, A. S. Bonilla, S. Haar, M. Campitiello, R. Gutierrez, G. Cuniberti, S. Masiero, A. Ciesielski and P. Samorì, Chem. Commun., 2015, 51, 11677–11680 RSC.
- L. Wang, H. Kong, C. Zhang, Q. Sun, L. Cai, Q. Tan, F. Besenbacher and W. Xu, ACS Nano, 2014, 8, 11799–11805 CrossRef CAS PubMed.
- C. Zhang, L. Xie, L. Wang, H. Kong, Q. Tan and W. Xu, J. Am. Chem. Soc., 2015, 137, 11795–11800 CrossRef CAS PubMed.
- R. Otero, M. Schöck, L. M. Molina, E. Lœgsgaard, I. Stensgaard, B. Hammer and F. Besenbacher, Angew. Chem., Int. Ed., 2005, 44, 2270–2275 CrossRef CAS PubMed.
- C. Fonseca Guerra, H. Zijlstra, G. Paragi and F. M. Bickelhaupt, Chem.–Eur. J., 2011, 17, 12612–12622 CrossRef CAS PubMed.
- J. L. Mergny and D. Sen, Chem. Rev., 2019, 119, 6290–6325 CrossRef CAS PubMed.
- S. Haq, B. Wit, H. Sang, A. Floris, Y. Wang, J. Wang, L. Pérez-García, L. Kantorovitch, D. B. Amabilino and R. Raval, Angew. Chem., 2015, 127, 7207–7211 CrossRef.
- J. R. Blas, F. J. Luque and M. Orozco, J. Am. Chem. Soc., 2004, 126, 154–164 CrossRef CAS PubMed.
- H. Kong, Q. Sun, L. Wang, Q. Tan, C. Zhang, K. Sheng and W. Xu, ACS Nano, 2014, 8, 1804–1808 CrossRef CAS PubMed.
- E. D. Raczyńska, W. Kosińska, B. Ośmiałowski and R. Gawinecki, Chem. Rev., 2005, 105, 3561–3612 CrossRef PubMed.
- T. Kumagai, F. Hanke, S. Gawinkowski, J. Sharp, K. Kotsis, J. Waluk, M. Persson and L. Grill, Nat. Chem., 2014, 6, 41–46 CrossRef CAS PubMed.
- J. Franz and F. A. Gianturco, Eur. Phys. J. D, 2014, 68, 279 CrossRef.
- H. Vovusha, R. G. Amorim, R. H. Scheicher and B. Sanyal, RSC Adv., 2018, 8, 6527–6531 RSC.
- J. Camarillo-Cisneros, W. Liu and A. Tkatchenko, Phys. Rev. Lett., 2015, 115, 1–5 CrossRef PubMed.
- C. Tegenkamp, J. Phys.: Condens. Matter, 2009, 21, 013002 CrossRef PubMed.
- W. Xu, H. Kong, C. Zhang, Q. Sun, H. Gersen, L. Dong, Q. Tan, E. Lægsgaard and F. Besenbacher, Angew. Chem., Int. Ed., 2013, 52, 7442–7445 CrossRef CAS PubMed.
- H. M. Lee, A. Kumar, M. Kołaski, D. Y. Kim, E. C. Lee, S. K. Min, M. Park, Y. C. Choi and K. S. Kim, Phys. Chem. Chem. Phys., 2010, 12, 6278–6287 RSC.
- M. E. Cañas-Ventura, W. Xiao, D. Wasserfallen, K. Müllen, H. Brune, J. V. Barth and R. Fasel, Angew. Chem., Int. Ed., 2007, 46, 1814–1818 CrossRef PubMed.
- J. Matos and A. Kara, J. Phys.: Condens. Matter, 2016, 28, 445001 CrossRef PubMed.
- S. K. Min, W. Y. Kim, Y. Cho and K. S. Kim, Nat. Nanotechnol., 2011, 6, 162–165 CrossRef CAS PubMed.
- Y. Cho, S. K. Min, J. Yun, W. Y. Kim, A. Tkatchenko and K. S. Kim, J. Chem. Theory Comput., 2013, 9, 2090–2096 CrossRef CAS PubMed.
- J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Müllen and R. Fasel, Nature, 2010, 466, 470–473 CrossRef CAS PubMed.
- L. Talirz, P. Ruffieux and R. Fasel, Adv. Mater., 2016, 28, 6222–6231 CrossRef CAS PubMed.
- J. D. Teeter, P. S. Costa, P. Zahl, T. H. Vo, M. Shekhirev, W. Xu, X. C. Zeng, A. Enders and A. Sinitskii, Nanoscale, 2017, 9, 18835–18844 RSC.
- P. Ruffieux, J. Cai, N. C. Plumb, L. Patthey, D. Prezzi, A. Ferretti, E. Molinari, X. Feng, K. Müllen, C. A. Pignedoli and R. Fasel, ACS Nano, 2012, 6, 6930–6935 CrossRef CAS PubMed.
- I. Horcas, R. Fernández, J. M. Gómez-Rodríguez, J. Colchero, J. Gómez-Herrero and A. M. Baro, Rev. Sci. Instrum., 2007, 78, 013705 CrossRef CAS PubMed.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 6–9 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00235j |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Elham
Oleiki
a,
Elham
Oleiki





