
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Inhaled antibiotic-loaded polymeric nanoparticles for the management of lower respiratory tract infections
Mohammad Zaidur
Rahman Sabuj
 ab and
Nazrul
Islam
*abc
ab and
Nazrul
Islam
*abc
aPharmacy Discipline, School of Clinical Sciences, Queensland University of Technology (QUT), Brisbane, QLD, Australia. E-mail: nazrul.islam@qut.edu.au
bInstitute of Health and Biomedical Innovation (IHBI), Queensland University of Technology (QUT), Brisbane, QLD, Australia
cCentre for Immunology and Infection Control (CIIC), Queensland University of Technology (QUT), Brisbane, QLD, Australia
First published on 17th May 2021
Abstract
Lower respiratory tract infections (LRTIs) are one of the leading causes of deaths in the world. Currently available treatment for this disease is with high doses of antibiotics which need to be administered frequently. Instead, pulmonary delivery of drugs has been considered as one of the most efficient routes of drug delivery to the targeted areas as it provides rapid onset of action, direct deposition of drugs into the lungs, and better therapeutic effects at low doses and is self-administrable by the patients. Thus, there is a need for scientists to design more convenient pulmonary drug delivery systems towards the innovation of a novel treatment system for LRTIs. Drug-encapsulating polymer nanoparticles have been investigated for lung delivery which could significantly reduce the limitations of the currently available treatment system for LRTIs. However, the selection of an appropriate polymer carrier for the drugs is a critical issue for the successful formulations of inhalable nanoparticles. In this review, the current understanding of LRTIs, management systems for this disease and their limitations, pulmonary drug delivery systems and the challenges of drug delivery through the pulmonary route are discussed. Drug-encapsulating polymer nanoparticles for lung delivery, antibiotics used in pulmonary delivery and drug encapsulation techniques have also been reviewed. A strong emphasis is placed on the impact of drug delivery into the infected lungs.
 Mohammad Zaidur Rahman Sabuj | Mohammad Zaidur Rahman Sabuj obtained his Master of Research degree from the Biological Sciences Department, Macquarie University, Australia. In 2019, he moved to Queensland University of Technology (QUT), Brisbane, where he is currently working as a PhD researcher in the Pharmacy Discipline. His research is focused on the development and characterization of drug-encapsulating polymer nanoparticles. His research interests are in the in vitro evaluation of the drug release from polymeric nanoparticles, determining the aerosolization properties of pulmonary drugs, and in vitro evaluation of cytotoxicity and antimicrobial efficacy using drug-encapsulating polymeric nanoparticles. |
 Nazrul Islam | Nazrul Islam completed his PhD at the Monash Institute of Pharmaceutical Sciences, Monash University, Australia. Currently, he is a senior lecturer in the Faculty of Health, Queensland University of Technology (QUT), Brisbane, Australia. His research is focused on nanoparticulate pulmonary drug delivery and understanding the mechanism of micronized drug dispersion from powder agglomerates to increase the delivery efficiency from dry powder inhaler (DPI) formulations. |
1. Introduction
Lower respiratory tract infections (LRTIs) are one of the major lung diseases caused by the pathogenic bacteria Pseudomonas aeruginosa and Streptococcus pneumonia, which are associated with cystic fibrosis (CF), chronic obstructive pulmonary disease (COPD) and bronchiectasis. Some other microorganisms are also considered as the causative agents of LRTIs including respiratory syncytial virus, fungus, and mycoplasmas. However, some environmental substances (tobacco smoke, dust, chemicals, vapours, allergens, and air pollution) can also cause inflammation and damage lung cells, which produce excessive mucus in the small air sacs and lead to an infection.1 The Global Burden of Diseases, Injuries and Risk Factors Study (GBD) defined LRTIs more precisely as pneumonia or bronchitis, which is one of the leading causes of morbidity and mortality worldwide.2S. pneumonia, the major causative agent of pneumonia, was listed as the leading cause of LRTIs, which contributed to more deaths than all other diseases.3 In addition, LRTIs were ranked as the sixth leading cause of death for all ages and the major cause of death among children younger than 5 years and contributed to the total recorded death of 2.38 million.2 Antibiotics are the first line treatments for LRTIs. In severe cases, patients need to be hospitalized where they are treated with oral or intravenous antibiotics.1 Currently available treatment systems require long term administration of drugs at high doses. Thus, the dose-dependent toxicity of drugs makes the currently available treatment system less efficient. Therefore, the establishment of a proper management system for LRTIs is of pivotal importance for researchers.Pulmonary drug delivery technology is based on the delivery of inhalable formulations (micronized dry powders, solutions, or suspensions) which are aerosolized by suitable devices and deposited into the deep lungs. This route of drug delivery is efficient in delivering drugs directly into the deep lungs. Therefore, pulmonary drug delivery has several advantages over traditional systemic drug delivery methods, including rapid onset of drug action at a very low dose, reduced dose related adverse effects and improved patient compliance.
Pulmonary drug delivery for LRTIs using nanoparticles is one of the emerging strategies to fight against the antibiotic resistant microorganisms, especially P. aeruginosa.4 Nanoparticle based drug delivery includes several carriers in nano-size, such as polymeric micelles, drug polymer conjugates, liposomes, and dendrimers.5 The drug loaded polymer nanoparticles can provide several benefits for drug delivery systems including protecting the drug from degradation under unfavourable conditions, increasing drug solubility and absorption through epithelium by providing easy diffusion, preferential distribution of drugs within the target cells and improved therapeutic effects. In addition, polymer-based drug nanoparticle surfaces are modifiable, which improves the drug release pattern in a more controlled fashion; thus the desired therapeutic effects can be achieved for a long time.6–8
This review provides an overview on LRTIs, available treatments against this disease and their drawbacks. Then, a brief description on the pulmonary drug delivery system in light of the suitability to introduce it against LRTI treatment and the challenges of pulmonary drug delivery in the infected lungs are discussed. Polymer based inhalation delivery, excipients, and drug–polymer conjugation to form nanoparticles for pulmonary delivery are also described. Finally, common preparation techniques used to synthesize drug-encapsulating polymer nanoparticles for lung delivery are described.
2. An overview of the respiratory tract infections
Respiratory tract infection (RTI) is an infection within the lungs which affects both upper and lower respiratory tracts.9 More precisely it is regarded as any respiratory illness which refers to a variety of infections in the nose, sinuses, throat, airways and lungs. Most of the illness related to RTIs do not need medication and get better gradually.10 However, in some severe cases antibiotics are the only drugs used for the treatment. Rapid diagnosis is important to identify the causative agents and provide timely therapeutic intervention.11 Both viruses and bacteria can cause RTIs and in most cases they spread through direct contact, airborne particles and droplets from an infected person.12 Viral RTIs constitute a major public health issue because of their extensive incidence, ease of transmission and significant rate of morbidity and mortality.13 Children are two or three times more susceptible than adults with acute viral RTIs.14 Common upper respiratory tract infections (URTIs) include cold, sinusitis, tonsillitis and laryngitis, while LRTIs include influenza, pneumonia, bronchitis and bronchiolitis.15 Pneumonia, the common LRTI is caused by virus or bacteria and less commonly by fungi.16 The alveoli in the lungs fill with secretions and fluid, decreasing the ability for oxygen to be transported across the tissue to adequately oxygenate vital organs.17 Symptoms include shortness of breath and hypoxia and patients often need oxygen therapy and ventilation support. As such, pneumonia is considered the most severe case of LRTIs.183. Details on lower respiratory tract infections (LRTIs)
LRTI (Fig. 1) is a fatal lung infection that lasts for up to several weeks and appropriate diagnosis and treatments are required, otherwise the serious infection causes the patient's death. It usually shows symptoms such as coughing and sputum production, palpitation, wheeze, chest pain and shortness of breath. Symptoms of LRTIs usually depend on the type of infection and its severity. It is often associated with other lung disorders such as COPD and CF.19 Both Gram-positive and Gram-negative bacteria are the causative agents of LRTIs, whereas viral infections are also found in COPD.20 Microorganisms that are responsible for typical bacterial infection include Staphylococcus aureus, Klebsiella pneumonia, and Haemophilus influenza; whereas, atypical bacterial infection is caused by Chlamydophila pneumonia, Mycoplasma pneumonia, Legionella pneumonia and Chlamydia psittaci.21 Accurate detection of the causative agents can be done using molecular diagnosis of the infected respiratory tract.22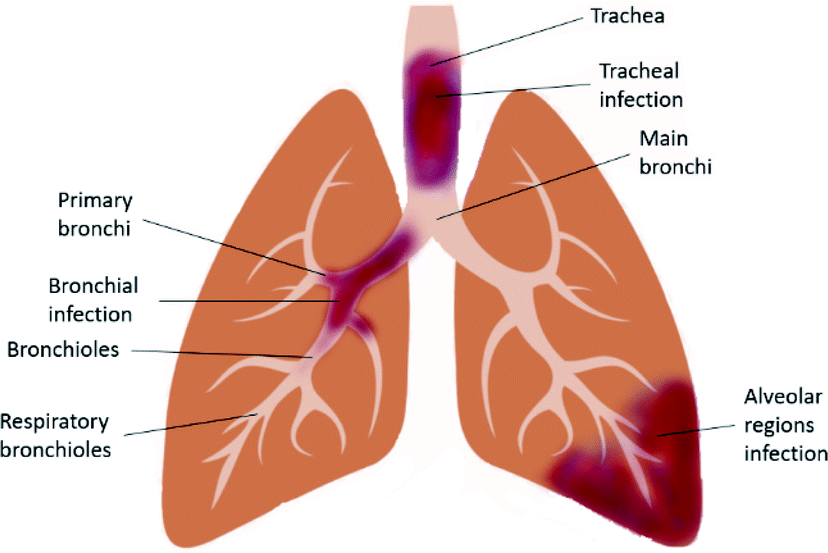 | ||
| Fig. 1 Schematic diagram of the infected lower respiratory tract. | ||
4. Currently available treatments for LRTIs and their limitations
Antibiotics are the mainstay treatment for bacterial lung infection and are commonly selected considering the severity of the infections; in addition, the patient's age and other complications are considered while prescribing.23 Currently, there are two types of antibiotic treatment available for LRTIs; the first one is antibiotics for community acquired pneumonia (CAP) and another one is for hospital acquired pneumonia (HAP). CAP is treated with several antibiotics including fluoroquinolone, nemonoxacin, zabofloxacin, delafloxacin, tetracycline, macrolide, cephalosporin, pleuromutilin, and streptogramins.24 However, most recent investigation shows that amoxicillin and clarithromycin are the most commonly prescribed antibiotics from these drug groups for acute LRTIs.25 In contrast, HAP treatment is limited to some specific groups of antibiotics including dihydrofolate-reductase inhibitors, lipoglycopeptides and oxazolidinone.24 Most of these antibiotics target the bacterial DNA gyrase enzyme and show activity by inhibiting DNA replication,26 thus reducing mutant selection and toxic side effects.27 Both oral and parenteral therapies are available for LRTIs with dosage requirement for 8–12 hours for 7–10 days with variable doses from 400–875 mg according to the severity of the patient's symptoms.28 However, evidence shows that the antibiotics administered orally do not always help patients to recover baseline lung function, owing to the inaccurate drug concentration to the target site leading to the bacterial resistance against the antibiotics.29 In addition, these currently available oral antibiotic therapies are administered with high doses and frequent dosing.30 Increasing doses of drugs cannot overcome the problem; rather, it contributes to microbial resistance.31 Researchers prioritize their attention on the selection of the most effective and the least toxic antibiotics,32 but there is still a need to find better delivery methods to minimize dose related adverse effects. Health-care professionals specialised in antimicrobial stewardship still seek help to determine appropriate antibiotic choice, dose and minimizing resistance.33LRTIs have huge impact on the global economy as a large amount of cost is associated in the management of LRTIs.34 Treatment for LRTIs causes enormous economic burden including direct and indirect financial losses; the estimated financial cost for antibiotic treatment associated with this disease is more than US$109 million each year globally.35 Because of the limitations of the currently available treatment systems, it is necessary to develop a better management system for the successful treatment of LRTIs at a very low dose of drugs with reduced costs. Pulmonary drug delivery could be utilized to substitute the currently available treatment system for LRTIs as it provides rapid onset of action, better therapeutic effects at a very low dose owing to the large surface area for drug absorption and is user friendly.36 In addition, inhaled antibiotics are administered in low doses and do not show the potential drug-related toxicity risks.37 Very limited studies have been done on pulmonary drug delivery for the effective treatment of LRTIs. Therefore, extensive studies are warranted to extend the application of pulmonary drug delivery for the management of LRTIs.
5. Pulmonary drug delivery system
Pulmonary delivery of drugs is a well-established drug delivery technology for the management of asthma, COPD and CF. It has been reported that pulmonary drug delivery is effective in treating the respiratory tracts.38 It has attracted significant attention in the last three decades for both the local and systemic therapeutic outcomes by offering lungs an optimistic pathway of non-invasive drug delivery. Thus, inhaled medication has become an interesting alternative route of drug administration for managing diseases related to respiratory tracts i.e. asthma, COPD, cystic fibrosis and lung infections such as pneumonia.39–43 The pulmonary route of drug delivery using biotherapeutics and macromolecules (e.g. growth hormones, protein, vaccine, and gene therapy)44–47 has shown promising effects both in local and systemic illness. Currently, only one inhaled dry powder inhaler product (tobramycin, Tobi Podhaler) is available for the treatment of P. aeruginosa infection associated with CF. It has shown greater efficiency compared to that of other routes by achieving the bioavailability and efficacy at a very low dose. However, successful delivery of drugs through the pulmonary route is governed by three factors, drug formulations, suitable drug delivery devices and the patient's inspiratory force,48 which are detailed in the following sub-sections.5.1. Drug formulations
Drug formulations are considered as the most important factor for the successful delivery of drugs into the infected airways. Inhaled formulations of the micronized drugs (<5 μm) include dissolved/suspended drug particles in a suitable solvent (suspensions or emulsions) or powder formulation mixed with a large carrier (lactose) with improved flow properties for efficient aerosolization. Micronized or nanosized dry powders offer maximum potentials to deliver the formulated drug particles into the lower parts of the lungs as they can avoid the defense mechanisms of the lungs, being very negligible in size. The distribution of the inhaled drug particles in the lung depends on the characteristics of the inhaled particles, such as drug particle diameter, mass, shape, density and hygroscopicity,49 the physiology of the respiratory tract, and breathing patterns of the patients.50 Increased flow properties of the formulated drugs ensure that a desirable amount of drugs could be deposited into the targeted sites.51 To increase the flow properties, the formulation of drugs can be engineered by several methods including mixing with dispersibility enhancers, preparing particles by spray drying, freeze-drying and supercritical fluids to get controlled properties including size and shape. In a study, Islam et al.52 investigated the effect of fine particles of lactose on the aerosolization of salmeterol xinafoate from the dry powder inhaler formulation, and fine lactose played a key role in increasing the drug dispersion; however, the size of the large carriers had a limited impact on the drug aerosolization.53 Islam et al.54 continued their studies to determine the drug carrier force by using atomic force microscopy (AFM), which showed a significant impact of the carrier particle surface roughness on the dispersion of the adhered drug. However, these studies were extensively based on lactose carriers and could not provide a clear understanding of the effects of other carriers. Excipients could be used as additives with the formulated drug to get better flow properties.55 Only a limited number of excipients are available to increase the flow properties of the formulated drug particles, especially for pulmonary routes.56 The formulated drug particle size should be less than 5 μm, which is highly cohesive and cause poor flow properties which results in poor dispersion and dose variation; therefore, the desired amount of drug could not reach into the deep lungs.57 So far, various excipients have been used to improve the efficiency of pulmonary drug delivery including polymers, surfactants, sugars, amino acids, lipids and absorption enhancers.56 Evidence shows that carriers or excipients with active pharmaceutical ingredients used in pulmonary formulations could provide better therapeutic effects than the conventional drug delivery systems.58 Lactose is the most preferred excipient for pulmonary drug formulations which has been extensively used to increase the flow properties of poorly dispersible drug particles.59 However, more needs to be done to increase the flowability of the formulated drugs and their excipients.5.2. Drug delivery devices
Three principal devices are available for pulmonary drug delivery: nebulizers, metered dose inhalers and dry powder inhalers.5.3. Patient's inspiratory force
Another important factor for successful drug deposition into the infected airways is the patient's inspiratory force. Upon inhalation from the delivery devices, the formulated drugs are introduced, and the inspiration force aerosolizes the powder bed by shear and turbulence60 and particles enter into the patient's airway. After inhalation, the particle size of the inhaled drug determines the deposition of drug in different regions of the lungs. Generally, particles need to be less than 2 μm for their successful deposition into the deep lungs.78 A slow breathing pattern is incapable of carrying the drugs into the infected areas, especially into the deep lungs; instead, they are deposited into the small airways, bronchioles and alveolar regions with suitable size and mass.79 Drug insufflation, carrier-based DPI formulation drugs are detached from the surface of the large carrier particles and deposited into the deep lungs while the large carriers impact the oropharynx and are cleared.806. Challenges of drug delivery by the pulmonary route in the infected lungs
The challenges of delivering drugs within the targeted area include minimizing its degradability, increasing its bioavailability, and furthermore reducing its cellular toxicity. In a study, Douafer et al.63 described various devices and techniques which have been used to overcome the challenges and achieve proper therapeutic effects of the administered drugs. However, lung physiology needs to be considered as a controlling factor to utilize any devices or techniques for lung drug delivery. Lung delivery of drugs also faces challenges including multiple filtrations through the respiratory tract, an innate immunological response and rapid exhalation from the lungs.81 However, three mechanisms such as impaction, sedimentation and diffusion have been demonstrated to overcome these challenges.82 Thus, the challenges for successful drug delivery into the infected lungs could be discussed in two sections: drug deposition in the infected airways and bacterial colonies/biofilms.6.1. Drug deposition in the infected airways
Inhaled particles <5 μm in size through the pulmonary route normally reach the deep lungs in healthy patients. However, the lung's natural defense mechanism also creates a barrier for the inhaled particles as foreign particles. There is a thick mucus protective layer in the upper airways (from the windpipe to the tertiary bronchi) that trap and clear foreign particles by either coughing or swallowing.83,84 The clearance of particles from this region is also governed by the number of cilia and the ciliary beat frequency, as well as the quality and quantity of mucus.85 Bissonnette et al.86 reported that the alveolar region serves as barriers for the transportation of molecules in the deeper areas of the lungs, e.g. a barrier lining of a variety of proteins and lipids, the compact junction present in the epithelial cell, alveolar macrophages, etc. Additionally, lung infection causes mucosal swelling of airway which results in narrowing of the cross-sectional diameter of the airways than that of a healthy lung. Excessive mucus production and deposition in infected lungs cause more turbulence of the inhaled air.87 Consequently, inhalable particles (<5 μm) deposit on the central airway mucosa instead of reaching the deep lungs. Thus, infected airways create more barriers for the inhaled drugs to reach the targeted site. Besides, they can cause local detrimental effects including bronchospasm and coughing due to deposition in the upper respiratory tracts, which may uplift patient compliance.88 Nanotechnology and particle engineering techniques were found to be efficient in overcoming this barrier of infected lungs for pulmonary drug deposition,89–91 and drug nanoparticles with a diameter of 1–200 nm can avoid entrapment in the upper airway and it can reach the alveolar sacs for absorption and rapid onset of action.92 Nanotechnology was also found advantageous for the objective of improving drug solubility, dissolution profiles and pharmacokinetic profiles and reducing the premature mucociliary clearance of hydrophobic drugs.936.2. Bacterial colonies/biofilms
Bacterial colonies inside the infected lungs are immersed in a dense immobilized mucus layer which acts as the solid barrier to the antibiotic exposure.94 Polynomic contents of the mucus (e.g. mucin, actin, and DNA) physically bind to antibiotic molecules by both electrostatic and hydrophobic interactions95 and hence prevent antibiotic molecules from reaching the bacterial colonies. Besides, the obstructed circulation of the antibiotic particles due to the presence of the mucus barrier makes them highly vulnerable to the lung phagocytic clearance, causing their short retention time in the lung.96,97 However, DPI nanoparticles (<200 nm) are found effective in mucus penetration due to their smaller size.92 Even they can avoid unfavourable mucociliary clearance and phagocytic clearance98 by remaining in the lung lining fluid until dissolution99 or translocation by the epithelial cells.100 It is desirable that nanotechnology could overcome bacterial colony forming challenges easily using micron sized DPI formulations, as the aim of antibiotic DPIs for lung infection treatment is to exert a bactericidal effect and better management of the disease.7. Polymer–drug conjugated nanoparticles for pulmonary delivery
Drug formulations for pulmonary delivery can be developed as a controlled release (CR) profile which ensures the release of the formulated drugs gradually and predictably over extended periods, thus maintaining a constant plasma concentration. The CR provides the formulated drugs greater effectiveness to treat persistent conditions as the medication is given consistently. It adds benefit over immediate drug release by reducing side effects while improving patient compliance as doses are in a simplified schedule. CR profiles are dependent on the formulation of drugs with specified particle size, shape, and surface properties. Polymers have been used extensively as carriers to investigate their CR patterns. Both synthetic and natural polymers showed encouraging CR patterns including, poly(lactic-co-glycolic acid) (PLGA), poly(ε-caprolactone) (PCL), polyvinyl alcohol (PVA), poly(lactic acid) (PLA), poly(ether-anhydride), chitosan, sodium hyaluronate and albumin.101–103 Particle engineering of biodegradable polymers as carriers has shown protection to the encapsulated drugs and stable drug delivery from the formulations and efficient transportation to the targeted sites. However, micron size drug particles with suitable carriers still face challenges to achieve CR, thus requiring more suitable nano-sized drug formulations. Drug-encapsulating polymer nanoparticles could be an alternative tool to overcome this limitation with the desired CR profile. Drug loaded polymer micro- or nano-particles are the most recent advanced technology of drug formulations towards pulmonary drug delivery. Thus, the selection of a proper drug delivery technique is a critical issue to avoid the defense system of the lungs which have a strict clearance mechanism and multiple barriers against foreign insertion.104,105 Nanotechnology has a promising scope to utilize it in pulmonary drug delivery to avoid first pass metabolism and rapid absorption because of its small size, and physical and chemical characteristics including large surface area.106,107 Nanoparticles in the form of DPI formulations (Fig. 2) are considered as the most feasible delivery method of drugs embedded within a carrier rather than the direct pulmonary administration of nanoparticles.108 Nanoparticles showed agglomerates and compromised deposition behaviour of particles because of the high surface energy, while appropriate excipients in DPI formulations showed promising deposition and deagglomeration behaviour.109 Drug loaded polymeric nanoparticles can carry the drug into the infected lungs either by loading the drug molecules on the surface of the polymers or encapsulating the drug molecules within its matrix.110 Interestingly, after deposition into the infected airways or into the deep lungs where a humid environment exists, the polymer matrix dissolves and releases the nanoparticulate drug molecules.111 The polymer encapsulated siRNA targeting lung cancer showed less toxic effects within the cells because of the preferential accumulation of the drug in the target cells and also found reduced toxic effects of celecoxib-loaded PLGA nanoparticles on lung cancers.112,113 Thus, the engineered polymeric nanoparticles are capable of carrying the encapsulated drugs into the target areas and make them suitable to provide better therapeutic effects at a very low dose and minimize dose associated toxic effects. Polymeric nanoparticles are designed for targeted delivery,114 and sustained delivery58,115,116 of drugs in the deep lungs109 by DPIs. Wu et al.117 demonstrated that DPI formulations of polymeric cyclosporine drug nanoparticles (1–100 nm) are effective in producing highly aerosolized particles. The surface charge of modified polymer drug nanoparticles was found to improve antibiotic delivery in the infected lungs.118 However, this study could not provide the cytotoxic effects of the synthetic lung surfactant-mimic phospholipid, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine and 1,2-dipalmitoyl-sn-glycero-3-(phosphor-rac-1-glycerol) sodium salt. Salvati et al.119 studied the surface properties of the polymeric drug nanoparticle (cationic charged/neutral) and found it to adhere to the mucus layer of the lungs and exert sustained drug release. In another study, Huang et al.120 found that inhaled nanoparticles were effective in the alteration of drug interactions with target cells, owing to the preferential accumulation. Thus, polymer–drug conjugated nanoparticles are suitable to be utilized in pulmonary delivery with precise delivery for both local and systemic effects.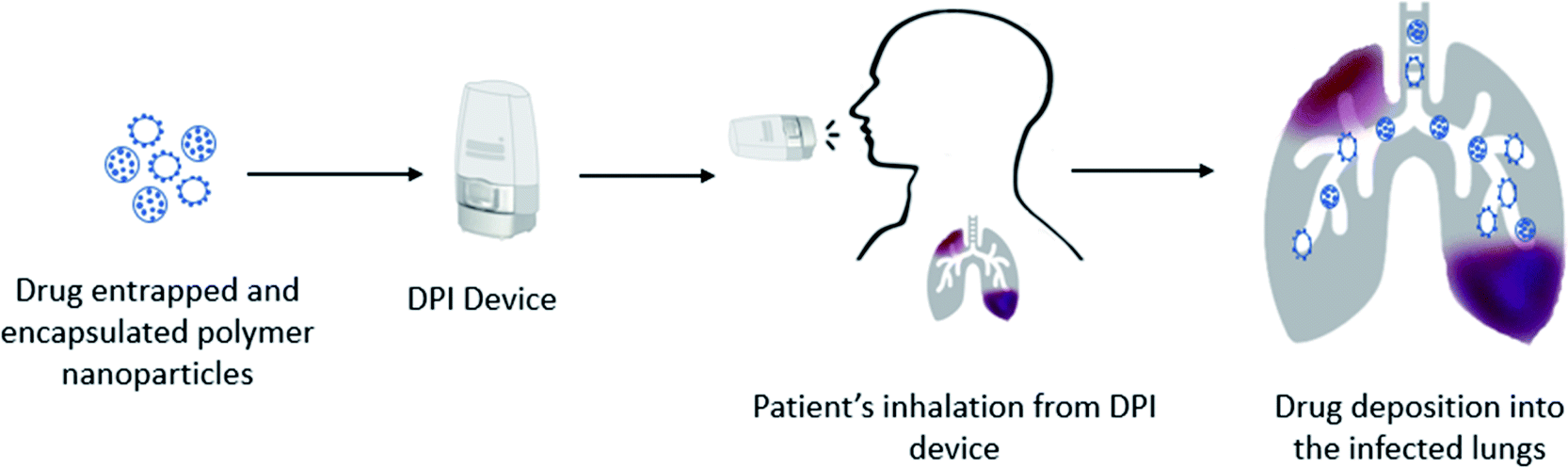 | ||
| Fig. 2 Schematic diagram of pulmonary drug delivery from DPI formulations. The formulation consists of drug entrapped and encapsulated in polymer nanoparticles. Nanoparticles are aerosolized using a DPI device and patient's inspiratory force and deposited into the infected lung. | ||
8. Polymer based pulmonary delivery
Polymers are considered as attractive carriers of drugs to the lung. They offer easy encapsulation strategies for drugs within them in various forms including nanoparticles, microparticles and nano-embedded microparticles.111,121 In addition, polymers could slow the systemic absorption of conjugated delivery, thus increasing the drug sustainability within the lung. This is helpful for the treatment of lung related illness, but the drug needs to be deposited within the lung for a longer period rather than instant absorption.122 Both natural and synthetic polymers have been extensively studied for pulmonary drug delivery as carriers or excipients for DPIs,123 facilitating the aerodynamic properties, inhibiting particle aggregation, and thus increasing particle dispersion and deposition.124 However, synthetic polymers showed more effective drug release profiles in a sustained way over the natural polymers.122 The choice of suitable polymer carriers for drug encapsulation depends on their biodegradability, encapsulation efficacy, drug release pattern and easy formulation properties.1258.1. Natural polymers for pulmonary delivery
Among natural polymers, chitosan and alginate have extensively been studied for their biocompatibility, biodegradability, and low-toxicity behaviour for lung drug delivery. Rohani et al.152 demonstrated the enhanced fine particle fraction (FPF) (46–81%) of the spray-dried chitosan encapsulated insulin powder formulation. They suggested that excipients including mannitol, sodium alginate and sodium citrate helped increase the flow properties by changing the surface properties of the formulated powder. Al-Qadi et al.126,127 showed the improved aerosolization properties of the protein-loaded chitosan nanoparticles over other formulations. The improved absorption of the nanoparticles occurred due to the interaction between the cationic chitosan with the target cells. For example, protein encapsulated chitosan showed improved systemic absorption with prolonged drug release upon lung delivery.128 Thus, chitosan has been positioned as a promising carrier for pulmonary delivery of various drugs. Antibiotic-encapsulating chitosan microparticles and nanoparticles showed promising effects to improve drug deposition, dispersibility, tissue uptake, and modified surface characteristics for better aerosolization.91 In addition, anticancer, anti-asthma antifungal and antihypertensive drugs were also investigated for pulmonary delivery. Promising characteristics of chitosan to improve drug delivery of pulmonary drugs encouraged its use in other delivery routes, as most recently modified chitosan in nanobioconjugate photosensitive nanocarriers was studied to determine its compatibility with other biological carriers including azobenzene molecules, thus showing improved therapeutic effects in cardiac delivery.129 However, safety issues related to chitosan and its derivatives in lung delivery makes it a less preferable choice in recent studies.91Another natural polymer which has been extensively investigated for pulmonary drug delivery is alginate due to its low cost, ease of preparation, mucoadhesiveness, biocompatibility and nontoxic characteristics.130,131 However, alginate loaded micro- or nano-particles also limits its use due to its rapid drug release characteristics.132 Although various studies have been done to improve its drug release pattern, still a lot needs to be done to determine the alginate improved drug release pattern in combination with other polymer carriers.130Table 1 shows some of the natural polymers which have been used as carriers in pulmonary drug delivery.
| Polymer | Encapsulated molecules | Main findings | Ref. |
|---|---|---|---|
| a Abbreviation: PLGA – poly(lactic-co-glycolic acid). | |||
| Chitosan | Isoniazid | Prolonged drug release and improved aerosolization | 136 |
| Crosslinked chitosan | Levofloxacin | Improved aerosolization | 137 |
| Chitosan | Ciprofloxacin | Improved aerosolization and therapeutic effects | 138 |
| Chitosan | Isoniazid and rifampicin | Improved bioavailability and cellular uptake | 139 |
| Chitosomes (chitosan–xanthan gum) | Liposomes | Improved aerosolization | 140 |
| Chitosan | Ethambutol dihydrochloride | Improved bioavailability and cellular uptake | 141 |
| Chitosan | Dapsone | Prolonged drug release and improved aerosolization | 142 |
| Chitosan | Ofloxacin | Improved aerosolization and cellular uptake | 143 |
| Chitosan | Moxifloxacin | Improved cellular uptake | 144 |
| Chitosan | Rifampicin and rifabutin | Prolonged drug release; improved aerosolization and bioavailability | 145 |
| Chitosan/fucoidan | Gentamicin | Prolonged drug release and improved bioavailability | 120 |
| Chitosan | Vancomycin | Improved bioavailability and cellular uptake | 146 |
| Alginate | Paclitaxel | Prolonged drug release and improved bioavailability | 147 |
| Alginate/chitosan | Tobramycin | Improved bioavailability and cellular uptake | 148 |
| Alginate | Isoniazid, rifampicin, and pyrazinamide | Prolonged drug release | 149 |
| Alginate/PLGA | Amikacin, ciprofloxacin and polymyxin | Improved aerosolization | 150 |
| Alginate/chitosan | PR8 influenza virus | Improved bioavailability | 151 |
| Chitosan | Insulin | Improved dispersibility | 152 |
| Alginate | Poloxamer | Prolonged drug release | 153 |
8.2. Synthetic polymers for pulmonary delivery
Synthetic polymers showed promising effects over natural polymers for being easily synthesizable and cost effective. Various synthetic polymers have shown promising effects, being biocompatible and versatile including polyanhydrides, poly(lactic acid) (PLA) and poly(lactic-co-glycolic-acid) (PLGA).133Table 2 shows some of the synthetic polymers which have been used as carriers in pulmonary drug delivery. Synthetic polymer nanoparticles showed enormously positive characteristics over microparticles; thus, research nowadays is extensively focused on nanoparticle based pulmonary delivery.134 Recent findings from aerosol delivery of polymer-conjugated drug particles encouraged Zhang et al.135 to study it in neurodegenerative diseases using baicalein loaded poly(ethylene glycol)-block-poly(D, L-lactide) (PEG–PLA) conjugated particles, and the findings were promising against oxidative stress and inflammation.| Polymer | Encapsulated molecules | Main findings | Ref. |
|---|---|---|---|
| a Abbreviation: PLA – polylactic acid; PLGA – poly(lactic-co-glycolic acid); PVA – poly(vinyl alcohol); PEG – poly(ethylene glycol); PCL – polycaprolactone. | |||
| PLA/PLGA | Hepatitis B vaccine | Prolonged drug release and improved bioavailability | 154 |
| PLGA | Rifampicin, isoniazid and pyrazinamide | Improved bioavailability | 155 |
| PLGA | Rifampicin | Prolonged drug release | 156 |
| PLGA | Pirfenidone | Prolonged drug release and improved bioavailability | 157 |
| PLGA | Voriconazole | Prolonged drug release and improved aerosolization | 158 |
| PLGA/chitosan | Calcitonin (peptide) | Prolonged drug release | 159 |
| PLGA/chitosan | Exendin-4 | Prolonged drug release | 160 |
| PLGA/polyethyleneimine | DNA vaccine | Increased therapeutic effects | 161 |
| PLGA/polyethyleneimine | siRNA | Increased therapeutic effects | 112 |
| PLGA/PEG | Velcade | Increased therapeutic effects | 162 |
| PEG | Plasmid DNA | Improved cellular uptake | 163 |
| PLGA/Fe3O4 | Quercetin | Increased therapeutic effects | 164 |
| PVA/PLGA | siRNA | Improved bioavailability | 165 |
| PVA/PLGA | Salbutamol | Prolonged drug release and improved therapeutic effects | 166 |
9. Antibiotics used in DPI formulations for pulmonary delivery
Antibiotics are considered as the first line treatment for lung related diseases, especially for LRTIs. As discussed earlier, currently available antibiotic therapies are in high doses and need frequent administration, thus increasing cellular toxicity and antimicrobial resistance. To overcome the limitations of the currently available antibiotic formulations, research has been focused on the nanoparticulate DPI formulations of these antibiotics. Table 3 shows some of the antibiotics which have been sufficiently formulated into aerosolized nanoparticles for pulmonary delivery. Both natural and synthetic polymers were studied extensively to improve the therapeutic effects of the formulated antibiotic-loaded nanoparticles. Besides, in many cases, excipients were used to increase the powder flowability of the prepared formulations while formulating aerosol particles for lung delivery.167,168 However, still their effectiveness needs to be tested in a standard lab and human trials to determine their complete safety profile.| Antibiotics/drugs | Carrier | Formulation technique | Excipients | Findings | Ref. |
|---|---|---|---|---|---|
| a Abbreviation: DPPC – 1,2-dipalmitoyl-sn-glycero-3-phosphocholine; PLGA – poly(lactic-co-glycolic acid); PVA – poly(vinyl alcohol); TPP – tripolyphosphate; HPMC – hydroxypropyl methylcellulose; PCL – polycaprolactone; PEG – poly(ethylene glycol). | |||||
| Vancomycin and clarithromycin | DPPC | Spray drying | — | Improved aerosolization | 178 |
| Tobramycin and azithromycin | Organic solution | Spray drying | — | Improved aerosolization | 89 |
| Rifampicin | PLGA | Solvent evaporation/ spray drying | PVA and L-leucine | Prolonged drug release | 156 |
| Moxifloxacin and ofloxacin | DPPC | Spray drying | — | Improved aerosolization | 179 |
| Isoniazid | Chitosan/TPP | Spray drying | Lactose, mannitol, maltodextrin, and leucine, glycerine | Improved aerosolization | 136 |
| Isoniazid and rifampicin | HPMC | Precipitation | Mannitol, leucine, and Tween 80 | Improved aerosolization and prolonged drug release | 180 |
| Tobramycin | PLGA/Chitosan | Emulsion/solvent evaporation | — | Improved aerosolization | 181 |
| Aspirin and salbutamol | Polyacrylate | Spray drying | Tween 20 | Prolonged drug release | 182 |
| Levofloxacin | PCL/PVA | Emulsion/solvent evaporation | D-Mannitol and L-leucine | Improved aerosolization | 183 |
| Ciprofloxacin and levofloxacin | PLGA/PCL | Emulsion/solvent evaporation | — | Improved drug penetration | 92 |
| Levofloxacin | PLGA/Lecithin (lipid) | Spray drying/Spray freeze drying | D-Mannitol, L-leucine, and PVA | Improved aerosolization | 184 |
| Rifampicin | PLGA/PVA | Emulsion/solvent evaporation | Lactose | Improved aerosolization | 185 |
| Clarithromycin | PLGA/PVA | Emulsion/solvent evaporation | Mannitol, L-leucine, and lactose | Improved aerosolization | 186 |
| Ciprofloxacin | Polyacrylate | Spray drying | L-Leucine and lactose | Improved aerosolization and prolonged drug release | 187 |
| Levofloxacin | PCL | Spray drying | Pluronic F-68, D-mannitol, lactose, and L-leucine | Improved aerosolization | 188 |
| Levofloxacin | PCL | Emulsion/solvent evaporation | L-Leucine, PVA, α-lactose monohydrates, and D-mannitol | Improved aerosolization | 189 |
| Rifampicin | Chitosan | Ionotropic gelation | — | Prolonged drug release and improved cellular uptake | 190 |
| Isoniazid, rifampicin, and pyrazinamide | Alginate | Cation-induced gelification | Chitosan | Prolonged drug release | 191 |
| Rifampicin, isoniazid, and pyrazinamide | PLGA | Emulsion/solvent evaporation | — | Improved bioavailability | 155 |
| Pirfenidole | PLGA | Emulsion/solvent evaporation | PVA | Improved bioavailability | 157 |
| Salbutamol | Poly(vinyl sulfonate-co-vinyl alcohol)-g-PLGA | Modified solvent evaporation | — | Prolonged drug release | 166 |
| Ibuprofen | PEG–PLGA | Emulsion/solvent evaporation | — | Improved mucus penetration | 192 |
| Ethionamide | PLGA | Emulsion/solvent evaporation | Lactose | Prolonged drug release | 193 |
| Tobramycin | PEG–PLGA | Emulsion/solvent evaporation | — | Improved bioavailability | 194 |
| Tobramycin | PLGA | Spray drying | — | Improved cellular uptake | 195 |
| Rifampicin | PLGA | Spray drying | — | Prolonged drug release and improved cellular uptake | 196 |
| Rifampicin | PLGA | Emulsion/solvent evaporation | L(+)-Arginine and L-leucine | Improved aerosolization | 197 |
| Ciprofloxacin | PLGA | Nanoprecipitation | Pluronic F-68 | Improved aerosolization | 198 |
| Ciprofloxacin | PLGA | Nanoprecipitation | Pluronic F-68 | Improved aerosolization | 199 |
10. Preparation techniques for drug-encapsulating polymer nanoparticles
Drug conjugated polymer nanoparticles for pulmonary delivery have some special features that should be considered during formulation and design. Several methods have been developed to control the particle size distribution, increase stability, and improve the CR profile and targeted delivery with enhanced bioavailability. So far, spray drying has been the most popular technique for preparing inhalable powders. Freeze-drying or lyophilization has also been investigated as a method to produce a solid dry powder that could be administered through lung delivery or after rehydration in the appropriate buffer. The selection of methods depends on the characteristics of the polymers, solubility of the drugs and stability of the formulated powder nanoparticles. The following methods have mostly been used for polymer-based drug nanoparticle formulations for lung delivery.10.1. Double emulsion/solvent evaporation method
Solvent evaporation is appropriate for encapsulating both hydrophilic and lipophilic drugs with high efficiency.169 This method is performed in a water-in-oil (w/o) emulsion of the polymer and is being prepared using an appropriate surfactant. The stable emulsion of the polymer is cross-linked by an appropriate cross-linking agent such as glutaraldehyde to harden the droplets. The polymer nanoparticles are acquired by evaporation of the oil phase. The size of the prepared nanoparticles depend on the extent of the cross-linking agent, speed of stirring and aqueous droplet size; thus, formulated nanoparticles have been shown to demonstrate enhanced mucoadhesiveness or cellular absorption upon drug delivery.170 However, drawbacks of this method include a tedious procedure, harsh cross-linking agents and difficulty to wash away the cross-linking agent.17110.2. Spray-drying method
Spray-drying is a process where mechanical high energy input is avoided. Therefore, this technique is appropriate for thermolabile materials and macromolecules such as peptides and proteins. Polymers with/without drugs are primarily dissolved or dispersed in an organic solvent (e.g. acetic acid/dichloromethane), and then a suitable cross-linking agent is added to this formulation. An outflux of hot air atomises the solution into small droplets of free-flowing nanoparticulate powders. Particle size depends upon the size of the nozzle, spray flow rate, atomisation pressure and inlet air temperature, and extent of cross-linking. The hot air can degrade the heat-liable substance.17210.3. Antisolvent precipitation/salting-out method
The anti-solvent method is widely used in pharmaceuticals to produce very fine particles with specific particle surface morphology and physical state. Acetone is commonly chosen as the water-miscible organic solvent in this method as acetone is pharmaceutically acceptable in terms of toxicity.173 Poly(vinyl alcohol) is added in the organic solvent (acetone), which contains the polymer in it and highly concentrated salt solution. High salt concentration in the aqueous phase prevents the mixing of acetone with pure water despite being miscible. After emulsification, the addition of water in a sufficient quantity causes acetone to diffuse into the aqueous phase, resulting in the formation of nanoparticles.17410.4. Ionotropic gelation (polyelectrolyte complexation: TPP method)
This method encapsulates a drug by the interaction of an ionic polymer with oppositely charged ion to initiate cross linking.175 To prepare nanoparticles, a polymer (e.g. chitosan) is dissolved in an aqueous solution (e.g. cation of chitosan); then a poly anionic tripolyphosphate (TPP) is added dropwise to the solution under constant stirring. As a result of complexation between oppositely charged species, the polymer undergoes ionic gelation and precipitates to form spherical particles. The resulting polymer particle suspension needs to be centrifuged and dried subsequently. This method is environment friendly as water can be used as the solvent. Prepared nanoparticles have been demonstrated to enhance mucoadhesiveness or cellular absorption upon pulmonary delivery.176 The self-assembly method of drug-encapsulating polymer nanoparticles is a recent addition in the field of nanotechnology. Hydrogen bonding may be used to form nanoparticles between the neutral polymers and tannic acid. This method was used to prepare doxorubicin-encapsulating poly(2-oxazoline) nanoparticles for cancer treatment.177 This technique could be used for drug-encapsulating polymer nanoparticles for pulmonary delivery as well.11. Conclusion and future directions
This article provides a comprehensive critical review of the current status of LRTIs, the limitation of the current management system and the development of better delivery technology. LRTIs are still considered a life-threatening disease, and the currently available treatment system has dose-related adverse effects. Besides, the direct and indirect cost associated with the management system of the LRTIs has a huge economic impact on the world economy. Thus, the establishment of a cost-effective management system for LRTIs is on the priority list of the scientists. Inhaled antibiotics have drawn the attention of researchers as an efficient management system to overcome the economic burden. Research in developing drug-encapsulating polymer nanoparticles for pulmonary delivery is progressing and has achieved considerable success so far. Nanoparticulate drugs can diffuse through the mucus layer of the infected lungs, release drugs in the cells and produce better therapeutic action at a low dose. However, some commonly used polymers such as chitosan are raising concerns on their biodegradability characteristics. Therefore, in vivo optimization is required to determine the safety of the polymer nanoparticles for lung cells. Given the potential for improved treatment by delivering drugs into the deep lungs, further research is essential to develop efficient inhaled antibiotic formulations for the proper management of LRTIs in the future.Author contributions
Mohammad Zaidur Rahman Sabuj: conceptualization, design, original draft preparation, editing and revision of manuscript. Nazrul Islam: supervision, conceptualization, review & editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Mohammad Zaidur Rahman Sabuj was supported by a postgraduate research scholarship from the Queensland University of Technology.References
- A. Biggers and J. Fletcher, Lower respiratory tract infections: what to know, 2019 Search PubMed.
- M. Naghavi, A. A. Abajobir, C. Abbafati, K. M. Abbas, F. Abd-Allah, S. F. Abera, V. Aboyans, O. Adetokunboh, A. Afshin and A. Agrawal, Lancet, 2017, 390, 1151–1210 Search PubMed.
- G. B. D. L. R. I. Collaborators, Lancet Infect. Dis., 2018, 18, 1191–1210 Search PubMed.
- F. Andrade, D. Rafael, M. Videira, D. Ferreira, A. Sosnik and B. Sarmento, Adv. Drug Delivery Rev., 2013, 65, 1816–1827 Search PubMed.
- X. Yu, I. Trase, M. Ren, K. Duval, X. Guo and Z. Chen, J. Nanomater., 2016, 2016 Search PubMed.
- M. C. Roco, Curr. Opin. Biotechnol., 2003, 14, 337–346 Search PubMed.
- S. S. Suri, H. Fenniri and B. Singh, J. Occup. Med. Toxicol., 2007, 2, 16 Search PubMed.
- V. Torchilin, in Multifunctional Pharmaceutical Nanocarriers, Springer, 2008, pp. 1–32 Search PubMed.
- P. Little, Prescribing of antibiotics for self-limiting respiratory tract infections in adults and children in primary care, Centre for Clinical Practice at NICE, NHS, 2008 Search PubMed.
- M. Ruopp, K. Chiswell, J. T. Thaden, K. Merchant and E. L. Tsalik, Ann. Am. Thorac. Soc., 2015, 12, 1852–1863 Search PubMed.
- D. Low, Clin. Microbiol. Infect., 2008, 14, 298–306 Search PubMed.
- P. Cariani, The respiratory tract and its infections, Harvard Medical School, United States, 2017 Search PubMed.
- J. Richter, C. Panayiotou, C. Tryfonos, D. Koptides, M. Koliou, N. Kalogirou, E. Georgiou and C. Christodoulou, PLoS One, 2016, 11, e0147041 Search PubMed.
- J. S. Tregoning and J. Schwarze, Clin. Microbiol. Rev., 2010, 23, 74–98 Search PubMed.
- M. C. Gulliford, M. V. Moore, P. Little, A. D. Hay, R. Fox, A. T. Prevost, D. Juszczyk, J. Charlton and M. Ashworth, BMJ, 2016, 354, i3410 Search PubMed.
- D. Behera, Text Book of Pulmonary Medicine, 2010, p. 1834 Search PubMed.
- Centres for Disease Control and Prevention, Complications and Treatments of Sickle Cell Disease, 2019 Search PubMed.
- O. Ruuskanen, E. Lahti, L. C. Jennings and D. R. Murdoch, Lancet, 2011, 377, 1264–1275 Search PubMed.
- A. Mahashur, Lung India, 2018, 35, 143 Search PubMed.
- B. Kocsis and D. Szabo, Expert Opin. Pharmacother., 2017, 18, 1345–1355 Search PubMed.
- R. S. Bedi, Lung India, 2006, 23, 130–131 Search PubMed.
- A. Camporese, Le Infezioni in Medicina, 2012, 20, 237–244 Search PubMed.
- K. Christiansen, Aust. Prescr., 1996, 19, 48–51 Search PubMed.
- Y. Liu, Y. Zhang, W. Zhao, X. Liu, F. Hu and B. Dong, Front. Pharmacol., 2019, 10 Search PubMed.
- R. Denholm, E. T. van der Werf and A. D. Hay, Respir. Res., 2020, 21, 4 Search PubMed.
- M. H. Kollef and K. D. Betthauser, Curr. Opin. Infect. Dis., 2019, 32, 169–175 Search PubMed.
- M. A. Pfaller, R. E. Mendes, M. Castanheira, R. K. Flamm, R. N. Jones and H. S. Sader, Pediatr. Infect. Dis. J., 2017, 36, 486–491 Search PubMed.
- J. Yuan, B. Mo, Z. Ma, Y. Lv, S.-L. Cheng, Y. Yang, Z. Tong, R. Wu, S. Sun and Z. Cao, J. Microbiol., Immunol. Infect., 2019, 52, 35–44 Search PubMed.
- S. Stanojevic, A. McDonald, V. Waters, S. MacDonald, E. Horton, E. Tullis and F. Ratjen, Thorax, 2017, 72, 327–332 Search PubMed.
- C. Feldman and G. Richards, F1000Research, 2018, 7, F1000 Faculty Rev-1121 Search PubMed.
- E. M. Kraus, S. Pelzl, J. Szecsenyi and G. Laux, PLoS One, 2017, 12, e0174584 Search PubMed.
- N. J. Rowbotham, S. Smith, P. A. Leighton, O. C. Rayner, K. Gathercole, Z. C. Elliott, E. F. Nash, T. Daniels, A. J. Duff and S. Collins, Thorax, 2018, 73, 388–390 Search PubMed.
- W. Bullington, A. Smyth, S. Elborn, P. Drevinek, S. Hempstead and M. Muhlebach, Open Forum Infectious Diseases, 2019, 6, S382 Search PubMed.
- C. Trucchi, C. Paganino, A. Orsi, D. Amicizia, V. Tisa, M. F. Piazza, D. Gallo, S. Simonetti, B. Buonopane and G. Icardi, BMC Health Serv. Res., 2019, 19, 585 Search PubMed.
- W. H. Organization, Pneumonia, WHO, 2019 Search PubMed.
- M. E. Aulton and K. M. Taylor, Aulton's Pharmaceutics E-Book: The Design and Manufacture of Medicines, Elsevier Health Sciences, 2017 Search PubMed.
- S. Smith, N. J. Rowbotham and E. Charbek, Cochrane Database Syst. Rev., 2018, 10, 3–57 Search PubMed.
- P. Mehta, J. Drug Delivery, 2016, 2016, 8290963 Search PubMed.
- H. Heijerman, E. Westerman, S. Conway and D. Touw, J. Cystic Fibrosis, 2009, 8, 295–315 Search PubMed.
- M. H. Kollef, C. W. Hamilton and A. B. Montgomery, Curr. Opin. Infect. Dis., 2013, 26, 538–544 Search PubMed.
- J. P. J. Mogayzel, E. T. Naureckas, K. A. Robinson, G. Mueller, D. Hadjiliadis, J. B. Hoag, L. Lubsch, L. Hazle, K. Sabadosa, B. Marshall and t. P. C. P. G. Committee, Am. J. Respir. Crit. Care Med., 2013, 187, 680–689 Search PubMed.
- B. S. Quon, C. H. Goss and B. W. Ramsey, Ann. Am. Thorac. Soc., 2014, 11, 425–434 Search PubMed.
- A. Shakshuki and R. U. Agu, Pulmonary Therapy, 2017, 3, 267–281 Search PubMed.
- T. Gessler, W. Seeger and T. Schmehi, J. Aerosol Med. Pulm. Drug Delivery, 2008, 21, 1–12 Search PubMed.
- V. P. Guntur and R. Dhand, Respiratory Care, 2007, 52, 911–922 Search PubMed.
- L. D. Mastrandrea and T. Quattrin, Adv. Drug Delivery Rev., 2006, 58, 1061–1075 Search PubMed.
- J. S. Patton, J. G. Bukar and M. A. Eldon, Clin. Pharmacokinet., 2004, 43, 781–801 Search PubMed.
- N. Islam and M. J. Cleary, Med. Eng. Phys., 2012, 34, 409–427 Search PubMed.
- N. R. Labiris and M. B. Dolovich, Br. J. Clin. Pharmacol., 2003, 56, 588–599 Search PubMed.
- J. Löndahl, W. Möller, J. H. Pagels, W. G. Kreyling, E. Swietlicki and O. Schmid, J. Aerosol Med. Pulm. Drug Delivery, 2014, 27, 229–254 Search PubMed.
- N. R. Labiris and M. B. Dolovich, Br. J. Clin. Pharmacol., 2003, 56, 600–612 Search PubMed.
- N. Islam, P. Stewart, I. Larson and P. Hartley, Pharm. Res., 2004, 21, 492–499 Search PubMed.
- N. Islam, P. Stewart, I. Larson and P. Hartley, J. Pharm. Sci., 2004, 93, 1030–1038 Search PubMed.
- N. Islam, P. Stewart, I. Larson and P. Hartley, J. Pharm. Sci., 2005, 94, 1500–1511 Search PubMed.
- A. Haywood and B. D. Glass, Aust. Prescr., 2011, 34, 112–114 Search PubMed.
- G. Pilcer and K. Amighi, Int. J. Pharm., 2010, 392, 1–19 Search PubMed.
- H. Schiavone, S. Palakodaty, A. Clark, P. York and S. T. Tzannis, Int. J. Pharm., 2004, 281, 55–66 Search PubMed.
- H. Wang, J. Holgate, S. Bartlett and N. Islam, Eur. J. Pharm. Biopharm., 2020, 154, 175–185 Search PubMed.
- Y. Rahimpour and H. Hamishehkar, Adv. Pharm. Bull., 2012, 2, 183–187 Search PubMed.
- M. J. Telko and A. J. Hickey, Respiratory Care, 2005, 50, 1209–1227 Search PubMed.
- M. Ali, in Handbook of Non-Invasive Drug Delivery Systems, ed. V. S. Kulkarni, William Andrew Publishing, Boston, 2010, pp. 209–246, DOI:10.1016/B978-0-8155-2025-2.10009-5.
- M. A. Johnson, S. P. Newman, R. Bloom, N. Talaee and S. W. Clarke, Chest, 1989, 96, 6–10 Search PubMed.
- H. Douafer, V. Andrieu and J. M. Brunel, J. Controlled Release, 2020, 325, 276–292 Search PubMed.
- J. N. Pritchard, R. H. Hatley, J. Denyer and D. v. Hollen, Ther. Delivery, 2018, 9, 121–136 Search PubMed.
- C. O'Callaghan and P. W. Barry, Thorax, 1997, 52(suppl. 2), S31–S44 Search PubMed.
- A. Mullen, Devices for Inhaled Medications (Asthma Inhalers, COPD Inhalers), National Jewish Health, 1400 Jackson Street, Denver, Colorado, 2018 Search PubMed.
- S. P. Newman, A. R. Clark, N. Talaee and S. W. Clarke, Thorax, 1989, 44, 706 Search PubMed.
- C. Leach, Respiratory Drug Delivery, 1996, pp. 133–144 Search PubMed.
- A. B. Watts, J. T. McConville and R. O. Williams III, Drug Dev. Ind. Pharm., 2008, 34, 913–922 Search PubMed.
- N. Islam and S. Rahman, Drug Discoveries Ther., 2012, 6, 123–132 Search PubMed.
- N. Islam and E. Gladki, Int. J. Pharm., 2008, 360, 1–11 Search PubMed.
- M. C. Manning, K. Patel and R. T. Borchardt, Pharm. Res., 1989, 6, 903–918 Search PubMed.
- H. D. Smyth and A. J. Hickey, Controlled pulmonary drug delivery, Springer, 2011 Search PubMed.
- S. P. Newman and W. W. Busse, Respiratory Medicine, 2002, 96, 293–304 Search PubMed.
- A. M. Healy, M. I. Amaro, K. J. Paluch and L. Tajber, Adv. Drug Delivery Rev., 2014, 75, 32–52 Search PubMed.
- K. Iida, Y. Hayakawa, H. Okamoto, K. Danjo and H. Luenbergerb, Chem. Pharm. Bull., 2003, 51, 1455–1457 Search PubMed.
- X. M. Zeng, G. P. Martin, C. Marriott and J. Pritchard, Int. J. Pharm., 2000, 200, 93–106 Search PubMed.
- J. Brain, P. Valberg and S. Sneddon, Aerosols in Medicine: Principles, Diagnosis Therapy, 1985, pp. 123–147 Search PubMed.
- W. Yang, J. I. Peters and R. O. Williams III, Int. J. Pharm., 2008, 356, 239–247 Search PubMed.
- U. H. Cegla, Respiratory Medicine, 2004, 98, S22–S28 Search PubMed.
- P. Muralidharan, M. Malapit, E. Mallory, D. Hayes Jr and H. M. Mansour, Nanomedicine, 2015, 11, 1189–1199 Search PubMed.
- A. J. Hickey, J. Controlled Release, 2014, 190, 182–188 Search PubMed.
- S. Favre-Bonté, T. Köhler and C. Van Delden, J. Antimicrob. Chemother., 2003, 52, 598–604 Search PubMed.
- J. M. Zuckerman, Infectious Disease Clinics of North America, 2004, 18, 621–649 Search PubMed.
- G. Amsden, J. Antimicrob. Chemother., 2005, 55, 10–21 Search PubMed.
- E. Y. Bissonnette, J.-F. Lauzon-Joset, J. S. Debley and S. F. Ziegler, Front. Immunol., 2020, 11, 583042 Search PubMed.
- K. W. Tsang and D. Bilton, Respirology, 2009, 14, 637–650 Search PubMed.
- S. Claus, C. Weiler, J. Schiewe and W. Friess, Eur. J. Pharm. Biopharm., 2014, 86, 1–6 Search PubMed.
- X. Li, F. G. Vogt, D. Hayes Jr and H. M. Mansour, Eur. J. Pharm. Sci., 2014, 52, 191–205 Search PubMed.
- M. M. Bailey and C. J. Berkland, Med. Res. Rev., 2009, 29, 196–212 Search PubMed.
- N. Islam and V. Ferro, Nanoscale, 2016, 8, 14341–14358 Search PubMed.
- W. S. Cheow, M. W. Chang and K. Hadinoto, J. Biomed. Nanotechnol., 2010, 6, 391–403 Search PubMed.
- J. Sung, B. Pulliam and D. Edwards, Trends Biotechnol., 2008, 25, 563–570 Search PubMed.
- A. A. Pragman, J. P. Berger and B. J. Williams, Clin. Pulm. Med., 2016, 23, 57–66 Search PubMed.
- B. Hunt, A. Weber, A. Berger, B. Ramsey and A. Smith, Antimicrob. Agents Chemother., 1995, 39, 34–39 Search PubMed.
- P. G. Bhat, D. R. Flanagan and M. D. Donovan, J. Pharm. Sci., 1996, 85, 624–630 Search PubMed.
- J. Levy, J. Pediatr., 1986, 108, 841–846 Search PubMed.
- R. Grenha, V. M. Levdikov, M. J. Fogg, E. V. Blagova, J. A. Brannigan, A. J. Wilkinson and K. S. Wilson, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2005, 61, 459–462 Search PubMed.
- N. Tsapis, D. Bennett, B. Jackson, D. A. Weitz and D. Edwards, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12001–12005 Search PubMed.
- G. Oberdörster, E. Oberdörster and J. Oberdörster, Environ. Health Perspect., 2005, 113, 823–839 Search PubMed.
- J. M. Chan, L. Zhang, K. P. Yuet, G. Liao, J.-W. Rhee, R. Langer and O. C. Farokhzad, Biomaterials, 2009, 30, 1627–1634 Search PubMed.
- H. K. Makadia and S. J. Siegel, Polymers, 2011, 3, 1377–1397 Search PubMed.
- H. Peniche and C. Peniche, Polym. Int., 2011, 60, 883–889 Search PubMed.
- C. Loira-Pastoriza, J. Todoroff and R. Vanbever, Adv. Drug Delivery Rev., 2014, 75, 81–91 Search PubMed.
- J. S. Patton and P. R. Byron, Nat. Rev. Drug Discovery, 2007, 6, 67–74 Search PubMed.
- L. M. Ensign, C. Schneider, J. S. Suk, R. Cone and J. Hanes, Adv. Mater., 2012, 24, 3887–3894 Search PubMed.
- H. H. Sigurdsson, J. Kirch and C.-M. Lehr, Int. J. Pharm., 2013, 453, 56–64 Search PubMed.
- F. Shiehzadeh and M. Tafaghodi, Curr. Pharm. Des., 2016, 22, 2549–2560 Search PubMed.
- M. K. Al-Hallak, M. K. Sarfraz, S. Azarmi, W. H. Roa, W. H. Finlay and R. Löbenberg, Ther. Delivery, 2011, 2, 1313–1324 Search PubMed.
- K. Hadinoto, P. Phanapavudhikul, Z. Kewu and R. B. Tan, Int. J. Pharm., 2007, 333, 187–198 Search PubMed.
- J. C. Sung, B. L. Pulliam and D. A. Edwards, Trends Biotechnol., 2007, 25, 563–570 Search PubMed.
- J. Das, S. Das, A. Paul, A. Samadder, S. S. Bhattacharyya and A. R. Khuda-Bukhsh, Toxicol. Lett., 2014, 225, 454–466 Search PubMed.
- J. Emami, A. Pourmashhadi, H. Sadeghi, J. Varshosaz and H. Hamishehkar, Pharm. Dev. Technol., 2015, 20, 791–800 Search PubMed.
- P. M. Young, R. O. Salama, B. Zhu, G. Phillips, J. Crapper, H.-K. Chan and D. Traini, Drug Dev. Ind. Pharm., 2015, 41, 859–865 Search PubMed.
- C. Kumaresan and K. Sathishkumar, Indian J. Pharm. Sci., 2016, 78, 136 Search PubMed.
- M. D. A. Muhsin, G. George, K. Beagley, V. Ferro, H. Wang and N. Islam, Mol. Pharm., 2016, 13, 1455–1466 Search PubMed.
- X. Wu, W. Zhang, D. Hayes Jr and H. M. Mansour, Int. J. Nanomed., 2013, 8, 1269–1283 Search PubMed.
- N. Sanders, C. Rudolph, K. Braeckmans, S. C. De Smedt and J. Demeester, Adv. Drug Delivery Rev., 2009, 61, 115–127 Search PubMed.
- A. Salvati, C. Åberg, T. dos Santos, J. Varela, P. Pinto, I. Lynch and K. A. Dawson, Nanomedicine, 2011, 7, 818–826 Search PubMed.
- Y.-C. Huang, R.-Y. Li, J.-Y. Chen and J.-K. Chen, Carbohydr. Polym., 2016, 138, 114–122 Search PubMed.
- M. Paranjpe and C. C. Müller-Goymann, Int. J. Mol. Sci., 2014, 15, 5852–5873 Search PubMed.
- E. Rytting, J. Nguyen, X. Wang and T. Kissel, Expert Opin. Drug Delivery, 2008, 5, 629–639 Search PubMed.
- F. Emami, S. J. M. Yazdi and D. H. Na, J. Pharm. Invest., 2019, 1–16 Search PubMed.
- K. Kadota, A. Senda, H. Tagishi, J. O. Ayorinde and Y. Tozuka, Int. J. Pharm., 2017, 517, 8–18 Search PubMed.
- Z. Liang, R. Ni, J. Zhou and S. Mao, Drug Discovery Today, 2015, 20, 380–389 Search PubMed.
- S. S. R. Rohani, K. Abnous and M. Tafaghodi, Int. J. Pharm., 2014, 465, 464–478 Search PubMed.
- S. Al-Qadi, A. Grenha, D. Carrión-Recio, B. Seijo and C. Remuñán-López, J. Controlled Release, 2012, 157, 383–390 Search PubMed.
- M. Murata, K. Nakano, K. Tahara, Y. Tozuka and H. Takeuchi, Eur. J. Pharm. Biopharm., 2012, 80, 340–346 Search PubMed.
- P. Mena-Giraldo, S. Pérez-Buitrago, M. Londoño-Berrío, I. C. Ortiz-Trujillo, L. M. Hoyos-Palacio and J. Orozco, Sci. Rep., 2020, 10, 1–12 Search PubMed.
- D. M. Hariyadi and N. Islam, Adv. Pharmacol. Sci., 2020, 2020, 8886095 Search PubMed.
- C. d. C. Spadari, L. B. Lopes and K. Ishida, Front. Microbiol., 2017, 8, 97 Search PubMed.
- F. Abasalizadeh, S. V. Moghaddam, E. Alizadeh, E. Akbari, E. Kashani, S. M. B. Fazljou, M. Torbati and A. Akbarzadeh, J. Biol. Eng., 2020, 14, 8 Search PubMed.
- H. M. Abdelaziz, M. Gaber, M. M. Abd-Elwakil, M. T. Mabrouk, M. M. Elgohary, N. M. Kamel, D. M. Kabary, M. S. Freag, M. W. Samaha and S. M. Mortada, J. Controlled Release, 2018, 269, 374–392 Search PubMed.
- I. d'Angelo, C. Conte, A. Miro, F. Quaglia and F. Ungaro, Curr. Top. Med. Chem., 2015, 15, 386–400 Search PubMed.
- L. Zhang, S. Yang, L. Huang and P. C.-L. Ho, Int. J. Pharm., 2020, 591, 119981 Search PubMed.
- P. S. Pourshahab, K. Gilani, E. Moazeni, H. Eslahi, M. R. Fazeli and H. Jamalifar, J. Microencapsulation, 2011, 28, 605–613 Search PubMed.
- M. C. Gaspar, J. J. Sousa, A. A. Pais, O. Cardoso, D. Murtinho, M. E. S. Serra, F. Tewes and J.-C. Olivier, Eur. J. Pharm. Biopharm., 2015, 96, 65–75 Search PubMed.
- R. Osman, P. L. Kan, G. Awad, N. Mortada, E.-S. Abd-Elhameed and O. Alpar, Int. J. Pharm., 2013, 449, 44–58 Search PubMed.
- T. Garg, G. Rath and A. K. Goyal, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 997–1001 Search PubMed.
- M. L. Manca, M. Manconi, D. Valenti, F. Lai, G. Loy, P. Matricardi and A. M. Fadda, J. Pharm. Sci., 2012, 101, 566–575 Search PubMed.
- M. I. Ahmad, T. Nakpheng and T. Srichana, Inhalation Toxicol., 2014, 26, 908–917 Search PubMed.
- M. Ortiz, D. S. Jornada, A. R. Pohlmann and S. S. Guterres, AAPS PharmSciTech, 2015, 16, 1033–1040 Search PubMed.
- J.-H. Park, H.-E. Jin, D.-D. Kim, S.-J. Chung, W.-S. Shim and C.-K. Shim, Int. J. Pharm., 2013, 441, 562–569 Search PubMed.
- C. A. Ventura, S. Tommasini, E. Crupi, I. Giannone, V. Cardile, T. Musumeci and G. Puglisi, Eur. J. Pharm. Biopharm., 2008, 68, 235–244 Search PubMed.
- R. V. Pai, R. R. Jain, A. S. Bannalikar and M. D. Menon, J. Aerosol Med. Pulm. Drug Delivery, 2016, 29, 179–195 Search PubMed.
- M. de Jesús Valle, J. G. González, F. G. López and A. S. Navarro, J. Antibiot., 2013, 66, 447–451 Search PubMed.
- S. Alipour, H. Montaseri and M. Tafaghodi, Colloids Surf., B, 2010, 81, 521–529 Search PubMed.
- M. Hill, M. Twigg, E. A. Sheridan, J. G. Hardy, J. S. Elborn, C. C. Taggart, C. J. Scott and M. E. Migaud, Pharmaceutics, 2019, 11, 379 Search PubMed.
- Z. Ahmad and G. Khuller, Expert Opin. Drug Delivery, 2008, 5, 1323–1334 Search PubMed.
- Q. Zhou, S. S. Y. Leung, P. Tang, T. Parumasivam, Z. H. Loh and H.-K. Chan, Adv. Drug Delivery Rev., 2015, 85, 83–99 Search PubMed.
- J. Mosafer, A.-H. Sabbaghi, A. Badiee, S. Dehghan and M. Tafaghodi, Asian J. Pharm. Sci., 2019, 14, 216–221 Search PubMed.
- S. S. R. Rohani, K. Abnous and M. Tafaghodi, Int. J. Pharm., 2014, 465, 464–478 Search PubMed.
- K. Moebus, J. Siepmann and R. Bodmeier, Eur. J. Pharm. Sci., 2012, 45, 358–366 Search PubMed.
- C. Thomas, A. Rawat, L. Hope-Weeks and F. Ahsan, Mol. Pharm., 2011, 8, 405–415 Search PubMed.
- R. Pandey, A. Sharma, A. Zahoor, S. Sharma, G. Khuller and B. Prasad, J. Antimicrob. Chemother., 2003, 52, 981–986 Search PubMed.
- J. C. Sung, D. J. Padilla, L. Garcia-Contreras, J. L. VerBerkmoes, D. Durbin, C. A. Peloquin, K. J. Elbert, A. J. Hickey and D. A. Edwards, Pharm. Res., 2009, 26, 1847–1855 Search PubMed.
- R. Trivedi, E. F. Redente, A. Thakur, D. W. Riches and U. B. Kompella, Nanotechnology, 2012, 23, 505101 Search PubMed.
- B. Sinha, B. Mukherjee and G. Pattnaik, Nanomedicine, 2013, 9, 94–104 Search PubMed.
- H. Yamamoto, Y. Kuno, S. Sugimoto, H. Takeuchi and Y. Kawashima, J. Controlled Release, 2005, 102, 373–381 Search PubMed.
- C. Lee, J. S. Choi, I. Kim, K. T. Oh, E. S. Lee, E.-S. Park, K. C. Lee and Y. S. Youn, Int. J. Nanomed., 2013, 8, 2975 Search PubMed.
- M. Bivas-Benita, M. Y. Lin, S. M. Bal, K. E. van Meijgaarden, K. L. Franken, A. H. Friggen, H. E. Junginger, G. Borchard, M. R. Klein and T. H. Ottenhoff, Vaccine, 2009, 27, 4010–4017 Search PubMed.
- N. Vij, T. Min, R. Marasigan, C. N. Belcher, S. Mazur, H. Ding, K.-T. Yong and I. Roy, J. Nanobiotechnol., 2010, 8, 1–18 Search PubMed.
- J. S. Suk, A. J. Kim, K. Trehan, C. S. Schneider, L. Cebotaru, O. M. Woodward, N. J. Boylan, M. P. Boyle, S. K. Lai and W. B. Guggino, J. Controlled Release, 2014, 178, 8–17 Search PubMed.
- N. K. Verma, K. Crosbie-Staunton, A. Satti, S. Gallagher, K. B. Ryan, T. Doody, C. McAtamney, R. MacLoughlin, P. Galvin and C. S. Burke, J. Nanobiotechnol., 2013, 11, 1–12 Search PubMed.
- J. Nguyen, T. W. Steele, O. Merkel, R. Reul and T. Kissel, J. Controlled Release, 2008, 132, 243–251 Search PubMed.
- E. Rytting, M. Bur, R. Cartier, T. Bouyssou, X. Wang, M. Krüger, C.-M. Lehr and T. Kissel, J. Controlled Release, 2010, 141, 101–107 Search PubMed.
- T. Peng, S. Lin, B. Niu, X. Wang, Y. Huang, X. Zhang, G. Li, X. Pan and C. Wu, Acta Pharm. Sin. B, 2016, 6, 308–318 Search PubMed.
- Q. T. Zhou and D. A. Morton, Adv. Drug Delivery Rev., 2012, 64, 275–284 Search PubMed.
- C. Li, W. Wang, X. Wang, H. Jiang, J. Zhu and S. Lin, Eur. Polym. J., 2015, 68, 409–418 Search PubMed.
- Y. Wang, P. Li, T. Truong-Dinh Tran, J. Zhang and L. Kong, Nanomaterials, 2016, 6, 26 Search PubMed.
- D. Pooja, L. Tunki, H. Kulhari, B. B. Reddy and R. Sistla, Data in Brief, 2016, 6, 15–19 Search PubMed.
- M. E. Ali and A. Lamprecht, Eur. J. Pharm. Biopharm., 2014, 87, 510–517 Search PubMed.
- D. Zasadowski, J. Yang, H. Edlund and M. Norgren, Carbohydr. Polym., 2014, 113, 411–419 Search PubMed.
- S. Galindo-Rodriguez, E. Allemann, H. Fessi and E. Doelker, Pharm. Res., 2004, 21, 1428–1439 Search PubMed.
- A. Trapani, S. Di Gioia, N. Ditaranto, N. Cioffi, F. M. Goycoolea, A. Carbone, M. Garcia-Fuentes, M. Conese and M. J. Alonso, Int. J. Pharm., 2013, 447, 115–123 Search PubMed.
- T. V. Nguyen, T. T. H. Nguyen, S.-L. Wang, T. P. K. Vo and A. D. Nguyen, Res. Chem. Intermed., 2017, 43, 3527–3537 Search PubMed.
- L. Liu, L. Yin, H. Bian and N. Zhang, Chin. Chem. Lett., 2020, 31, 501–504 Search PubMed.
- C.-W. Park, X. Li, F. G. Vogt, D. Hayes Jr, J. B. Zwischenberger, E.-S. Park and H. M. Mansour, Int. J. Pharm., 2013, 455, 374–392 Search PubMed.
- J. Duan, F. G. Vogt, X. Li, D. Hayes Jr and H. M. Mansour, Int. J. Nanomed., 2013, 8, 3489 Search PubMed.
- R. Kaur, T. Garg, U. Das Gupta, P. Gupta, G. Rath and A. K. Goyal, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 182–187 Search PubMed.
- F. Ungaro, I. d'Angelo, C. Coletta, R. d. E. di Villa Bianca, R. Sorrentino, B. Perfetto, M. A. Tufano, A. Miro, M. I. La Rotonda and F. Quaglia, J. Controlled Release, 2012, 157, 149–159 Search PubMed.
- K. Hadinoto, K. Zhu and R. B. Tan, Int. J. Pharm., 2007, 341, 195–206 Search PubMed.
- K. Kho and K. Hadinoto, Powder Technol., 2011, 214, 169–176 Search PubMed.
- Y. Wang, K. Kho, W. S. Cheow and K. Hadinoto, Int. J. Pharm., 2012, 424, 98–106 Search PubMed.
- K. Tomoda, T. Ohkoshi, Y. Kawai, M. Nishiwaki, T. Nakajima and K. Makino, Colloids Surf., B, 2008, 61, 138–144 Search PubMed.
- P. H. Moghaddam, V. Ramezani, E. Esfandi, A. Vatanara, M. Nabi-Meibodi, M. Darabi, K. Gilani and A. Najafabadi, Powder Technol., 2013, 239, 478–483 Search PubMed.
- L. Ely, W. Roa, W. H. Finlay and R. Löbenberg, Eur. J. Pharm. Biopharm., 2007, 65, 346–353 Search PubMed.
- K. Kho, W. S. Cheow, R. H. Lie and K. Hadinoto, Powder Technol., 2010, 203, 432–439 Search PubMed.
- W. S. Cheow, M. L. L. Ng, K. Kho and K. Hadinoto, Int. J. Pharm., 2011, 404, 289–300 Search PubMed.
- M. A. Moretton, D. A. Chiappetta, F. Andrade, J. Das Neves, D. Ferreira, B. Sarmento and A. Sosnik, J. Biomed. Nanotechnol., 2013, 9, 1076–1087 Search PubMed.
- A. Zahoor, S. Sharma and G. Khuller, Int. J. Antimicrob. Agents, 2005, 26, 298–303 Search PubMed.
- E. F. Craparo, B. Porsio, C. Sardo, G. Giammona and G. Cavallaro, Biomacromolecules, 2016, 17, 767–777 Search PubMed.
- S. K. Debnath, S. Saisivam and A. Omri, J. Pharm. Biomed. Anal., 2017, 145, 854–859 Search PubMed.
- J. Ernst, M. Klinger-Strobel, K. Arnold, J. Thamm, A. Hartung, M. W. Pletz, O. Makarewicz and D. Fischer, Eur. J. Pharm. Biopharm., 2018, 131, 120–129 Search PubMed.
- D. E. Geller, M. W. Konstan, J. Smith, S. B. Noonberg and C. Conrad, Pediatric Pulmonology, 2007, 42, 307–313 Search PubMed.
- K. Ohashi, T. Kabasawa, T. Ozeki and H. Okada, J. Controlled Release, 2009, 135, 19–24 Search PubMed.
- I. Takeuchi, Y. Tetsuka, T. Nii, M. Shinogase and K. Makino, Colloids Surf., A, 2017, 529, 387–393 Search PubMed.
- N. G. Türeli, A. E. Türeli and M. Schneider, Int. J. Pharm., 2016, 515, 343–351 Search PubMed.
- N. G. Türeli, A. Torge, J. Juntke, B. C. Schwarz, N. Schneider-Daum, A. E. Türeli, C.-M. Lehr and M. Schneider, Eur. J. Pharm. Biopharm., 2017, 117, 363–371 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |