
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA comparative study of tough hydrogen bonding dissipating hydrogels made with different network structures†
Badri Narayanan
Narasimhan
 ab,
Gerrit Sjoerd
Deijs
ab,
Sesha
Manuguri‡
ab,
Matthew Sheng Hao
Ting
ab,
M. A. K.
Williams
bc and
Jenny
Malmström
*ab
ab,
Gerrit Sjoerd
Deijs
ab,
Sesha
Manuguri‡
ab,
Matthew Sheng Hao
Ting
ab,
M. A. K.
Williams
bc and
Jenny
Malmström
*ab
aDepartment of Chemical and Materials Engineering, University of Auckland, Auckland, New Zealand. E-mail: j.malmstrom@auckland.ac.nz
bMacDiarmid Institute for Advanced Materials and Nanotechnology, 6140 Wellington, New Zealand
cSchool of Fundamental Sciences, Massey University, PN461, Private Bag 11222, Palmerston North 4442, New Zealand
First published on 29th March 2021
Abstract
Hydrogels are excellent soft materials to interface with biological systems. Precise control and tunability of dissipative properties of gels are particularly interesting in tissue engineering applications. In this work, we produced hydrogels with tunable dissipative properties by photopolymerizing a second polymer within a preformed cross-linked hydrogel network of poly(acrylamide). We explored second networks made with different structures and capacity to hydrogen bond with the first network, namely linear poly(acrylic acid) and branched poly(tannic acid). Gels incorporating a second network made with poly(tannic acid) exhibited excellent stiffness (0.35 ± 0.035 MPa) and toughness (1.64 ± 0.26 MJ m−3) compared to the poly(acrylic acid) counterparts. We also demonstrate a strategy to fabricate hydrogels where the dissipation (loss modulus) can be tuned independently from the elasticity (storage modulus) suitable for cell culture applications. We anticipate that this modular design approach for producing hydrogels will have applications in tailored substrates for cell culture studies and in load bearing tissue engineering applications.
Introduction
Hydrogels made of cross-linked polymeric networks have found applications as, for example, drug release systems,1,2 soft actuators3,4 and scaffolds in tissue engineering studies.5,6 The mechanical properties of hydrogels play a vital role in determining the usability of hydrogels in various applications. For instance, the tunability of the viscoelastic properties of hydrogels is important for cell culture studies6–8 and tissue engineering.9,10 In the case of orthopedic load-bearing applications, hydrogels with high stiffness and toughness are desired with stable mechanical properties over a considerable time.11,12 However, stiffness and toughness are generally inversely related in elastic materials.13 Stiffness refers to the ability of a material to resist deformation under load while toughness correlates with the energy dissipated before fracture.14 Therefore, toughness requires a material to be both strong and ductile. For gels, a high cross-linking ratio leads to high stiffness, but low toughness due to the lack of energy dissipation mechanisms. Consequently, this limits the working strain range of such gels.15 Recent attempts to improve the toughness of hydrogels include producing double network gels,16,17 sliding ring hydrogels,18,19 nanocomposite gels20,21 and hydrogels based on other non-covalent interactions.22,23While many of the elegant approaches adopted either involve chemical functionalization or complex fabrication procedures,24,25 a relatively easier route to produce tough hydrogels is by using the double network (DN) scheme. DN gels consist of two cross-linked polymer networks, where one network is designed to dissipate energy through sacrificial bonds.14 The strategies adopted to achieve dissipation in double network gels fall into two groups. The first group involves introducing a second network with fewer cross-links relative to the first network. In such DN gels, the relatively less cross-linked network dissipates energy and hinders crack propagation of the more cross-linked network. As a result, such gels have been observed to be stiff, yet ductile.15 In the second strategy, non-covalent/supramolecular associations between networks enable the tuning of dissipative properties, leading to stiff and tough hydrogels.26–28 Both strategies have been adopted in various applications such as scaffolds for tissue engineering, drug delivery, and mechanical sensing applications.29 Although significant progress has been made using double network schemes, the spatial modulation of mechanical properties and using the dissipative properties of double network gels to study mechanobiology have not been explored.
In the field of mechanobiology, researchers study how physical forces and mechanical properties of cells and tissues affect cell development and differentiation, and more broadly, physiology and disease. This research field has been primarily motivated by the fabrication of hydrogels with tunable stiffness.5,30 However, in vivo, cells reside in an extracellular matrix with a far more complex mechanical behavior. Recent mechanobiology literature therefore underscores the need for the development of hydrogel substrates with control over both the elastic and viscous components to further the knowledge of the field.31,32
The present study explored the effects of hydrogen bonding in polymeric semi-interpenetrating network gels, to produce materials where both storage and loss moduli can be precisely tuned. Specifically, we have explored the effect of photopolymerizing monomers, with different structures and hydrogen bonding capacity, inside a cross-linked hydrogel network (for schematic, see Fig. 1). This type of semi-interpenetrating network gel enables the tunability of dissipation in the gel. To aid the mechanistic understanding of these gels, we have chosen three different monomers to be polymerized inside a cross-linked poly(acrylamide) (PAAm) hydrogel network, acrylic acid, acrylamide, and tannic acid. Poly(acrylic acid) (PAA) was chosen due to its charged state at physiological pH, and its known ability to form intermolecular complexes with PAAm.33 The carboxylic acid groups of PAA serve as hydrogen bond donors, while the amide groups of PAAm serve as hydrogen bond acceptors.34,35 Tannic acid, a polyphenol capable of UV-polymerization, with multiple hydrogen bonding donor sites, was chosen as the second type of hydrogen bonding network. After polymerization, tannic acid is expected to form short, branched polymers (as compared to the long linear poly(acrylic acid)).36 Polymerization of acrylamide as a second network was chosen as a control, where little or no intermolecular hydrogen bonding interaction between the two acrylamide networks is expected, due to the weak hydrogen bond donor characteristics of PAAm.33
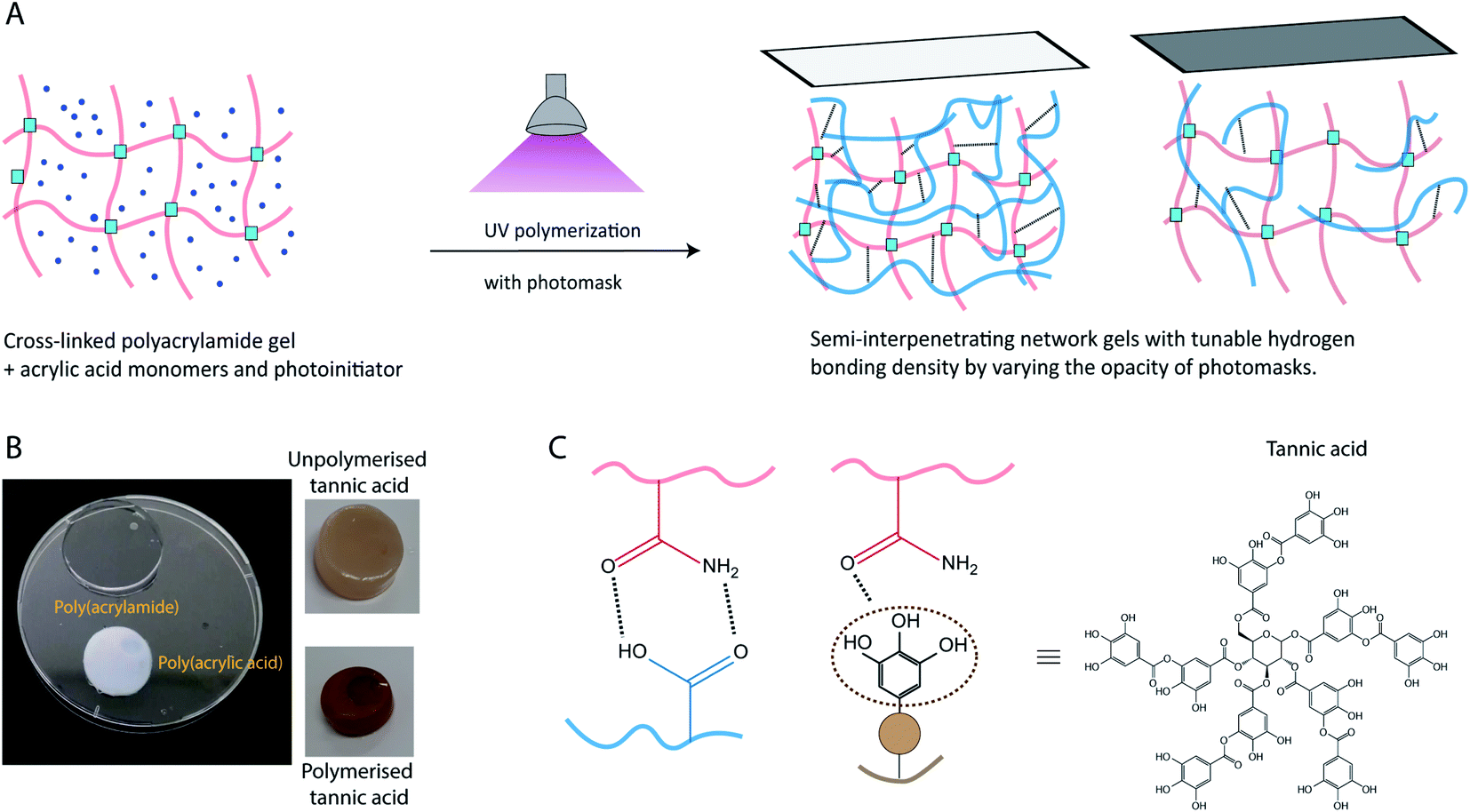 | ||
| Fig. 1 Scheme for production of semi-interpenetrating network hydrogels with poly(acrylic acid) or poly(tannic acid). (a) The dissipation can be tuned by polymerizing under a mask of varied transparency. (b) Photos of hydrogels made with different second networks. (c) Graphical representation of hydrogen bonding interactions between polymer networks. | ||
The gels in this study were characterized using rheology, compression testing, and tensile measurements. The gels with hydrogen bonding second networks were found to exhibit higher strength, toughness, and modulus. Furthermore, the use of photo-polymerization of the second network allows for patterning of the second network, which can be used to create gradients or patterns of mechanical properties. This, combined with the temporally stable mechanical properties of the produced gels, is of particular importance for future cell culture studies.
Experimental
Materials
Acrylamide (40%), bis-acrylamide, acrylic acid (100%), tannic acid (Chinese gallnut source), tetramethylethylenediamine (TEMED), ammonium persulfate (APS), 2-ketoglutaric acid, phosphate buffer saline (PBS, pH 7.4), and dichlorodimethylsilane (DCDMS) were purchased from Sigma Aldrich, New Zealand and were used as received. All solutions were prepared using type 1/ultrapure water (18.2 MΩ cm, from a Milli-Q Direct 8 water purification system). Water and buffer solutions used in gel preparation were deoxygenated by bubbling with N2 for at least 30 min.Functionalization of substrates and molds
Hydrogels were made using either laser-cut stainless rings (20 mm diameter, 1 mm thick) or acrylic molds (dog-bone shaped or circular molds). Silicon wafer, stainless steel molds, and glass coverslips were first subjected to UV-ozone for 30 minutes and hydrophobized by adding a few drops of DCDMS and gently wiping the surfaces with Kimwipes. The hydrophobic functionalization lasts for two or three castings of the hydrogels. After that, the above procedure is repeated again.Stock solutions
A bis-acrylamide stock solution was prepared by dissolving 20 mg of bis-acrylamide in 1 mL of deoxygenated water followed by filtering through a 0.22 μm PTFE syringe filter. TEMED (20%, v/v) and APS (10%, w/v) solutions were prepared separately in deoxygenated water and solubilized by vortexing.Preparation of hydrogels
The first PAAm network was synthesized by mixing acrylamide and bis-acrylamide in deoxygenated type 1 water or PBS buffer. After mixing, the TEMED solution was added, followed by the APS solution with mixing via vortexing for 1 minute between each step. Following mixing, the solution was poured into molds consisting of a stainless-steel ring on top of glass or silicon wafer substrates. The solution was left undisturbed to polymerize for at least 45 minutes. In a typical reaction, 5 μL of TEMED stock solution and 10 μL of APS (1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of TEMED/APS) were added to 1 mL of acrylamide and bis-acrylamide solution. Specific concentrations for the low and high cross-linking of the first network are presented in Table 1. Preliminary experiments revealed that PAAm-PAA semi-interpenetrating networks made from first network cross-linking ratios in Table 1 yielded a similar storage modulus. Hence, the cross-linking ratios in Table 1 were chosen for the experiments.
:1 molar ratio of TEMED/APS) were added to 1 mL of acrylamide and bis-acrylamide solution. Specific concentrations for the low and high cross-linking of the first network are presented in Table 1. Preliminary experiments revealed that PAAm-PAA semi-interpenetrating networks made from first network cross-linking ratios in Table 1 yielded a similar storage modulus. Hence, the cross-linking ratios in Table 1 were chosen for the experiments.
| Annotation | [Acrylamide] (M) | [Bis-acrylamide] (mM) | [APS] (mM) | [TEMED] (mM) |
|---|---|---|---|---|
| Low-crosslinking (LC) | 1.688 | 1.945 | 4.38 | 6.67 |
| High cross-linking (HC) | 1.125 | 16.215 | 4.38 | 6.67 |
To polymerize the second network inside the PAAm gels (low or high cross-linking), the gels were immersed in a deoxygenated solution (type 1 water or PBS) of monomer (acrylic acid, tannic acid, or acrylamide) and photoinitiator for 24 hours, followed by polymerization by exposure to UV light. A photomask defined by 10% and 20% opacities (Adobe illustrator, USA) was printed on a transparency film using a laser printer (MPC3003, Ricoh, USA). For the acrylic acid polymerized gels, a DCDMS treated quartz glass is placed above the gel over which the printed photomask is placed. For tannic acid polymerized gels, the photomask is placed directly over the top of the gel and tannic acid solution was placed around the gel to avoid drying of the gels. Complete conditions for the second networks can be found in Table 2. In Table 2, PAA, UTA and PTA correspond to poly(acrylic acid), unpolymerized tannic acid and polymerized tannic acid respectively. Following photopolymerization, the hydrogels were then taken out of the molds carefully and swelled as reported for each technique.
| Annotation | Monomer | [Monomer] (M) | Photoinitiator | [Photoinitiator] (mM) | Light source | Light exposure time |
|---|---|---|---|---|---|---|
| PAAmLC-PAA-1 | Acrylic acid | 0.844 | 2-Ketoglutaric acid | 34.2 | 365 nm, 10 mW cm−2, ABM mask aligner | 200 seconds for gels made in PBS, 150 seconds for gels made in H2O |
| PAAmLC-PAA-2 | 1.688 | |||||
| PAAmLC-PAA-3 | 3.376 | |||||
| PAAmHC-PAA-1 | Acrylic acid | 0.56 | 2-Ketoglutaric acid | 34.2 | 350 seconds in both PBS and water | |
| PAAmHC-PAA-2 | 1.12 | |||||
| PAAmHC-PAA-3 | 2.24 | |||||
| PAAmLC-UTA-1 | Tannic acid | 0.044 | n/a | n/a | 254 nm, Boekel Scientific UV cross-linker | n/a for UTA, 3 h for PTA |
| PAAmLC-PTA-1 | ||||||
| PAAmLC-UTA-2 | 0.088 | |||||
| PAAmLC-PTA-2 | ||||||
| PAAmLC-UTA-3 | 0.0176 | |||||
| PAAmLC-PTA-3 | ||||||
| PAAmLC-UTA-1 | Tannic acid | 0.044 | n/a | n/a | n/a for UTA, 3 h for PTA | |
| PAAmLC-PTA-1 | ||||||
| PAAmLC-UTA-2 | 0.088 | |||||
| PAAmLC-PTA-2 | ||||||
| PAAmLC-PAAm | Acrylamide | 1.688 | 2-Ketoglutaric acid | 34.2 | 365 nm, 10 mW cm−2, ABM mask aligner | 150 seconds |
Rheological testing
Rheological measurements were performed on the hydrogels using an AR-G2, stress-controlled rheometer (TA Instruments, New Castle, England) equipped with a 20 mm parallel plate smooth geometry. The hydrogels were loaded on the Peltier plate, and a normal force of 0.2–0.3 N was applied to ensure contact between the sample and geometry before measurements. Strain sweep measurements were carried out from 0.1 to 100% of strain at 1 Hz of frequency to determine the linear viscoelastic limit of the hydrogels. Frequency sweep measurements were performed from 0.05 Hz to 10 Hz at 1% strain. Three individual gels were characterized separately for each condition and the mean and standard deviations were calculated and reported.Tensile and compression tests
Tensile and compression measurements were performed on the hydrogels using an Instron 5543 tensile testing machine (Instron, Norwood, USA). The hydrogels used for tensile and compression testing were all produced from water solutions and were equilibrated in water for 24 hours before characterization. For the tensile tests, the gels were cast in a dog bone-shaped acrylic mold (35 mm × 6 mm) with a thickness of 2–3 mm, according to the procedure described in Takahashi et al.37 The dimensions of the hydrogels were measured using a digital Vernier scale before the experiments. The hydrogels were stretched until break at a speed of 50 mm min−1. The presented results are the mean and standard deviation of three gels of each composition.Cylindrical hydrogels were made by casting the gels in acrylic molds of 5 mm thickness and 10 mm diameter for the compression tests. The thickness of the gels was measured using a digital Vernier scale before measurements. Compression testing was performed at a constant speed of 1 mm min−1 using either a 50 N or a 1 kN load cell, until fracture. To assess stress-relaxation, the hydrogels were compressed to an equivalent of 1.5% of the gel thickness with a deformation rate of 1 mm min−1. The compressive strain was held constant at this position (for up to 60 min depending on the gel response) while the corresponding stress was monitored. Compressive modulus and stress-relaxation data were both generated on three independent gels for each condition and presented as mean and standard deviations.
Ultraviolet-visible (UV-Vis) spectroscopy
Gels for UV-Vis were prepared in rectangular acrylic molds of 5 mm thickness and 12 mm in length. The prepared gels were equilibrated in water or PBS for 24 hours before characterization. UV-Vis spectra were recorded using a Shimadzu UV-3600 plus spectrophotometer (Shimadzu, Japan) equipped with a thermoelectric temperature controller. The hydrogel to be measured was placed in a quartz cuvette with either water or PBS, depending on which solution the gel was prepared from. The temperature was ramped from 25 °C to 50 °C at a ramp rate of 0.5 °C per minute while recording the absorbance at 550 nm. Two gels were tested for each condition to assess the reproducibility of the experiments. For the tannic acid solutions, spectra were recorded from 200 nm to 800 nm. For the calculation of leached tannic acid, three individual gels per condition were used and the leachate concentration was reported from the mean and standard deviation of the three gels.Fourier transform infrared spectroscopy (FTIR)
FTIR spectra of the gels were obtained using a built-in diamond attenuated total reflectance accessory (ATR) (Thermo Scientific Nicolet iS50, USA). Prior to measurements, the gels were flash-frozen in liquid nitrogen and then dried in a commercial freeze drier (Virtis, Benchtop). FTIR spectra were analyzed in the wavenumber range 400 to 4000 cm−1. All spectra were obtained at room temperature and the baseline was recorded in air.Swelling studies
The dry weight (Wd) of the hydrogels was measured by thoroughly drying out the prepared hydrogels in an oven at 50 °C for two days. The wet weight (Ws) of the hydrogels was measured by re-swelling the gels in water for two days. The swelling degree was calculated by: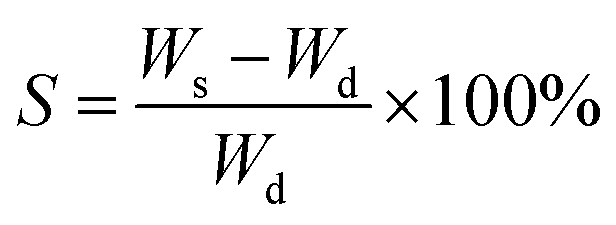
The water content in the gels was calculated by:
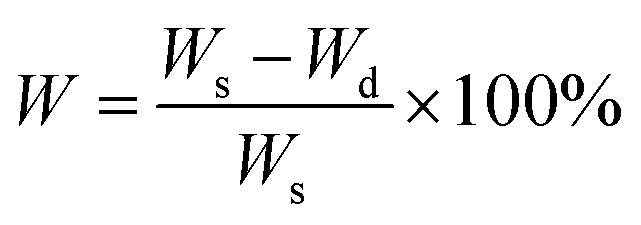
The data are reported as mean and standard deviations from three separate gels from each condition.
Results and discussion
Characterization of hydrogels
To investigate the inclusion of a second network within the PAAm gels, the fabricated gels were thoroughly investigated; chemically using ATR FTIR and physically using a suite of mechanical characterization methods.Radical polymerization of acrylamide and bis-acrylamide monomers results in a covalently cross-linked PAAm gel network (Fig. S1 reaction scheme†). The produced single network PAAm gels were swollen for 24 hours in solutions of the monomers for subsequent polymerization and the production of the second networks. For the concentrations used in this work, the pore size of the PAAm gels is expected to be in the range of 100 s of nanometers.38 Since the hydrodynamic size of monomers of acrylic acid (72 Da) and tannic acid (less than 2 nm39) is small compared to the pore size of the gels, we assume complete diffusion of monomers into the PAAm gels.
While the PAAm gels (single and semi-interpenetrating network) are transparent and colorless, the hydrogels made with second networks of poly(acrylic acid) and poly(tannic acid) are both opaque(Fig. 1C), assumingly due to the associative interactions between the two networks.35 The slight brown color exhibited by PAAm-poly(tannic acid) (PAAm-PTA) gel is due to the concentration of tannic acid used in this work. The visible color change of the gel is a strong indication of the successful incorporation of the second network into the gels. To further confirm the presence of the second network in the gels, FTIR spectra were recorded of PAAmLC, PAAmLC-PAA-2, PAAmLC-UTA-2 and PAAmLC-PTA-2 gels (as per Tables 1 and 2) as well as of pure PAA polymer and pure tannic acid. For the pure PAAm FTIR spectra, the peaks at 1603 cm−1 and 1645 cm−1 were assigned to the N–H bending and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of the amide group.35 In the PAA spectrum, the peaks at 1234 cm−1 and 1695 cm−1 were attributed to the C–O stretch and CO stretch of the carboxylic acid group.35 The FTIR spectrum of PAAmLC-PAA-2 showed signatures of both the PAA and PAAm, which confirmed the incorporation of PAA inside the PAAm gel (Fig. 2A).
O stretching of the amide group.35 In the PAA spectrum, the peaks at 1234 cm−1 and 1695 cm−1 were attributed to the C–O stretch and CO stretch of the carboxylic acid group.35 The FTIR spectrum of PAAmLC-PAA-2 showed signatures of both the PAA and PAAm, which confirmed the incorporation of PAA inside the PAAm gel (Fig. 2A).
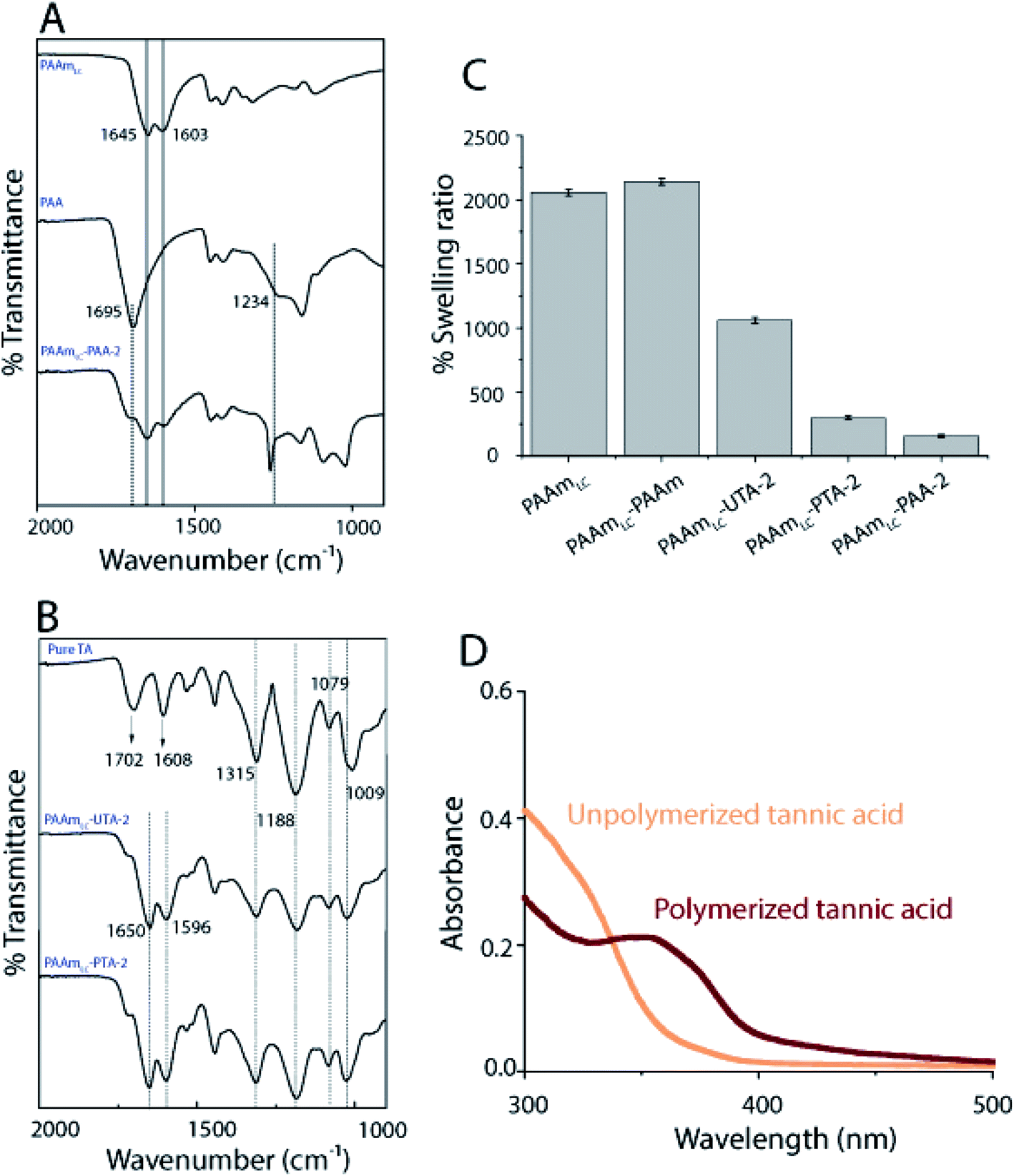 | ||
| Fig. 2 Characterization of hydrogels. (A) FTIR of hydrogels with second networks made of PAA (B) FTIR of hydrogels made with UTA or PTA. The concentrations correspond to PAAmLC-PAA-2, PAAmLC-UTA-2 and PAAmLC-PTA-2 respectively. (C) Plots of the swelling ratio of gels. (D) UV-Vis spectra of UTA and PTA at 0.01 mg mL−1 made in PBS. | ||
For the PAAmLC-UTA-2 and PAAmLC-PTA-2 gels, the FTIR spectra displayed peaks attributed to the presence of aromatic rings in the tannic acid,40 indicating the presence of tannic acid in both gels (refer Table S1 for detailed assignment of peaks†).
The inclusion of the second network may increase or decrease the swelling ratio and water content depending on the interactions between the first and second networks. In this case, the swelling studies revealed that PAA and PTA incorporated gels swelled significantly less than pure PAAm gels. It was further observed that the gels with polymerized tannic acid swelled less than the gels where the tannic acid was not polymerized (Fig. 2C). The fact that the tannic acid is polymerized allows the hydrogen bonding to couple the two networks over longer length scales (that of the polymerized tannic acid vs. monomeric tannic acid in the unpolymerized case). The same trend was observed for the corresponding PAAmHC gels as well (Fig. S5B†). In the case of PAAm incorporated gels, the swelling ratio was found to be slightly higher than that of PAAmLC gels and significantly higher when compared to those of PAA and PTA incorporated gels, which can be attributed to the absence of interactions between the first and the second networks.
To further affirm the polymerization of tannic acid, UV-Vis was performed on the solutions of unpolymerized and polymerized tannic acid (Fig. 2D). It has been suggested that a UV-Vis peak observed at 350 nm is correlated with the quinone formation which is an indication of polymerization of phenolic compounds.36 The appearance of a distinct 350 nm peak for the PTA when compared to the UTA clearly confirms the presence of polymeric species (Fig. 2D).
Finally, we performed dynamic light scattering (DLS) on the solution mixtures of PAAm and PAA, unpolymerized tannic acid (UTA) or PTA to analyze the size of the hydrogen bonded aggregates (Fig. S1B†). DLS of PAAm-PAA showed a wide size distribution of aggregates, which may be attributed to the random radical polymerization of the monomers. However, the DLS of PAAm-UTA and PAAm-PTA exhibited more narrow size distributions and a lower polydispersity. Specifically, the size of the aggregates in the PAAm-PTA mixture (∼330 nm) was found to be larger than those in the PAAm-UTA mixture (∼210 nm). The narrower size distribution for PAA-PTA compared to the PAA counterpart may be attributed to that the polymerization of tannic acid results in shorter polymers.
Tunable mechanical properties by tuning the density of hydrogen bonds
To evaluate the functional outcomes of the inclusion of hydrogen bonding second networks in the gels, the mechanical properties were characterized. The gels were prepared with different concentrations of acrylic acid or tannic acid, as per descriptions in Table 2, and the tannic acid was either polymerized or not. The control gels with the second network of PAAm were found to be very slippery (challenging to clamp), presumably due to the high water content and tensile testing of these gels was not successful for the concentrations used in this work. Similar issues have been reported for the case of physical entanglement hydrogels (PAAm nanogels incorporated inside PAAm gels).41 Example tensile testing stress–strain curves can be seen in Fig. 3, along with strength, toughness and Young's modulus values extracted from the curves. The PAAmLC parent gel displays a largely linear curve due to the elastic nature of the covalently cross-linked gel. This is in agreement with the expectations for a mostly elastic material. With the inclusion of a hydrogen bonding second network/component, most gels exhibit non-linear stress–strain relationships as expected from dissipating materials. However, for gels with unpolymerized tannic acid (gels PAAmLC-UTA-1 and PAAmLC-UTA-2 in Fig. 3A) the curves appear fairly linear. The other clear difference in the curves is the point at which the gels break. When compared to the parent gel, the semi-interpenetrating network gels exhibited higher tensile strength and strain at break (Fig. 3A and B), although this effect was small for the gels with unpolymerized tannic acid. Strength, toughness and stiffness were derived from the stress–strain curves and are presented as the mean with error bars representing the standard deviation (n = 3) in Fig. 3. The strength is defined as the stress at break, the toughness as the area under the stress–strain curve until break and the stiffness is calculated from the gradient of the curve in the most linear region of the curve.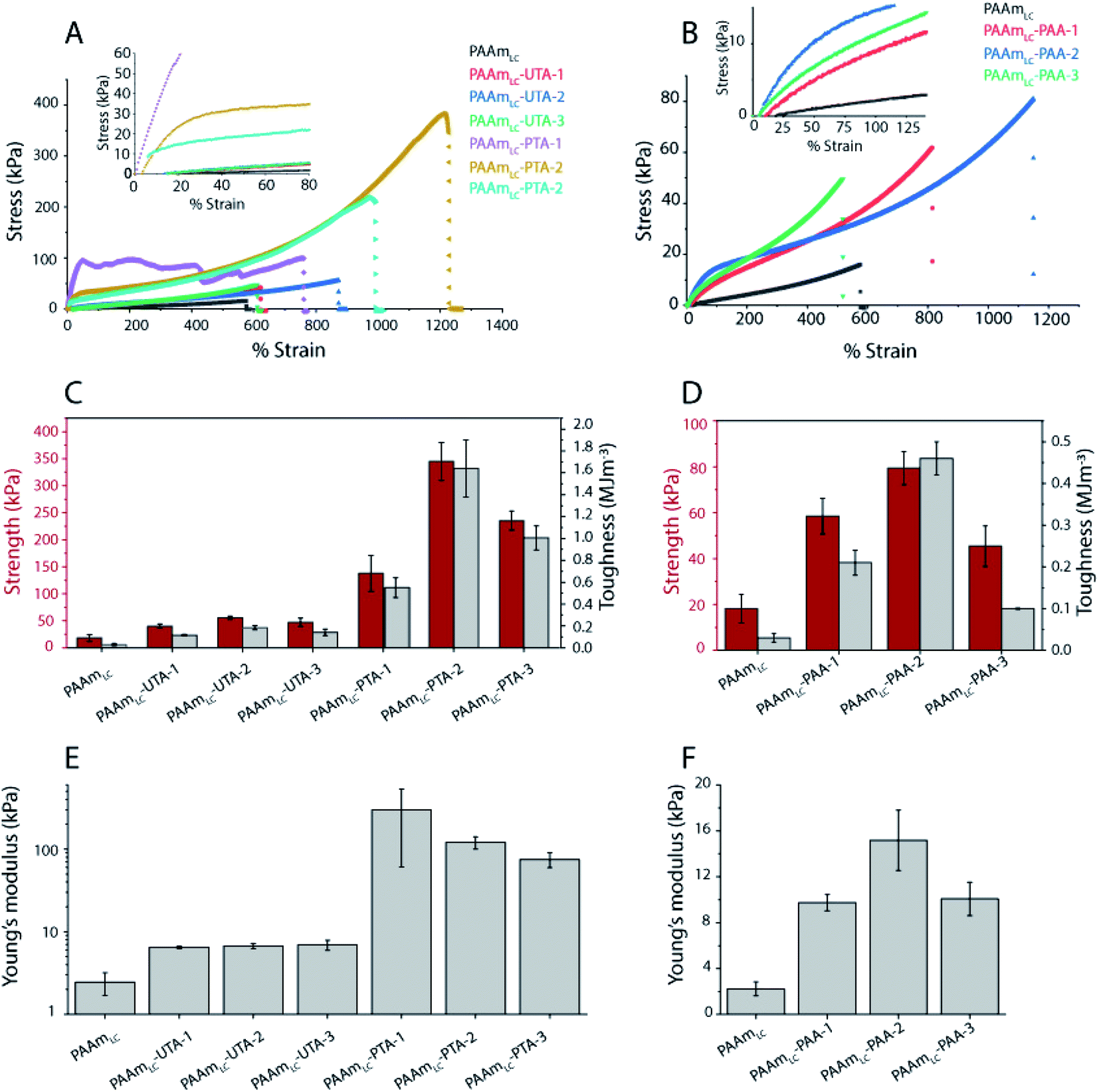 | ||
| Fig. 3 Tensile properties of hydrogels. (A and B) Tensile stress–strain curves of hydrogels incorporated with UTA/PTA and PAA of different compositions. (C) Plots of tensile strength and toughness of UTA/PTA gels and (D) tensile strength and toughness of PAA incorporated gels. The toughness was measured by calculating the area under the stress–strain curve. (E) Young's modulus of UTA/PTA gels, where Young's modulus was obtained from the slope of the linear region between 5 and 15% of strain. (F) Young's modulus of PAA incorporated gels. Young's modulus was obtained from the slope of the linear region between 20 and 100% of strain. | ||
From Fig. 3 it can be observed that PAAmLC-UTA gels exhibited only a modest increase in strength, stiffness, and toughness compared to PAAmLC. This change in material properties is likely due to hydrogen bonding interactions between the parent network and tannic acid, a branched molecule with multiple hydroxyl groups capable of hydrogen bonding with the amides in PAAm.42 This observation is in agreement with a recent study that showed such enhancement of stiffness and toughness for PAAm gels immersed in tannic acid of different concentrations.43 A much more significant increase in strength, stiffness, and toughness was observed for the PAAmLC-PTA gels. This may be due to the fact that the polymerization of tannic acid is able to interact with the first network across longer length scales (that of the polymerized tannic acid). It is clear from the tensile curves that the yield strength is particularly high for PAAmLC-PTA-1. However, due to large variation in that data, this does not correlate with a significantly higher stiffness (Young's modulus) of this sample compared to PAAmLC-PTA-2 or 3. For these higher concentrations of PTA, the gels exhibited a clear yield point after which they enter a non-linear and stiffening regime, resulting in stronger and tougher gels. It is important to note that the stiffness in this case was derived from an area of low strain in the elastic region. For clarity, the individual tensile stress–strain curve for PAAmLC-PTA-2 has been annotated with the individual sections as defined in this work (Fig. S6A†). When the concentration of PTA is increased even more in the gel (PAAmLC-PTA-3), the gel exhibited lower tensile strength, modulus, and toughness when compared to PAAmLC-PTA-2 (Fig. 3A and E). The lowering of tensile properties with an increase in concentration may be correlated with the phase separation of the polymers at higher loading, the change in the network structure or a suboptimal ratio between hydrogen bond donors and acceptors. A recent study made using poly(acrylic acid) hydrogel with incorporated poly(vinyl alcohol) showed a similar decrease in tensile properties of gels above a certain concentration threshold, which was attributed to a change in the network structure of the gels.44
Of the tannic acid incorporated gels discussed thus far, PAAmLC-PTA-2 was the strongest and toughest with a tensile strength of 0.35 ± 0.035 MPa, a toughness of 1.64 ± 0.26 MJ m−3 and an average tensile strain at break of 1200%. We also characterized the fracture energy of hydrogels using the tear test for the PAAmLC-PTA-2 hydrogels using the procedure described in Sun et al.45 The fracture energy using the tear test was calculated to be 2372 ± 244 J m−2 (details in the ESI, Fig. S11†). The fracture energy obtained was comparable to polydopamine incorporated PAAm gels reported in the literature.46
The literature values for a similar concentration of the first network (10–15%) made with gelatin methacrylate and unpolymerized tannic acid as the second network show a tensile strength of between 0.1 and 0.2 MPa and tensile strain% in the range of 200–250%.47 Like PAAm in our case, gelatin has functional groups that can form hydrogen bonds with tannic acid. In the gelatin case, the hydrogen bonding may be more complex due to the available functional groups in protein materials. In that case, a much higher concentration of tannic acid was also used 100% (w/v) compared to 15% (w/v) in our case. Thus, while the studies have differences in the concentrations used, it appears that the polymerization of tannic acid in our case resulted in the production of stiff and tough hydrogels by simple one-step polymerization.
A similar trend, but at a different magnitude, can be observed for the PAAmLC-PAA gels. Fig. 3B, D and F clearly show an increase in strength, toughness and stiffness for PAAmLC-PAA gels compared to PAAmLC. Also, here, a concentration dependence is observed, with the highest tensile properties observed for PAAmLC-PAA-2. Specifically, the tensile strength, Young's modulus, and toughness for PAAmLC-PAA-2 were found to be 78.4 ± 7.1 kPa, 15.17 ± 2.6 kPa, and 0.46 ± 0.04 MJ m−3 respectively. The enhanced tensile properties may be attributed to the ratio of hydrogen bond donors to acceptors being optimal for the PAAmLC-PAA-2 composition which has also been reported in other studies.26,48 The PAAmLC-PTA and PAAmLC-PAA gels also showed enhancement in mechanical properties in compression testing measurements (Fig. 4). The PAAmLC-UTA-1 and PAAmLC-PTA-1 gels did not exhibit any significant change in differences in compressive properties compared to PAAmLC. PAAmLC-UTA-2 and PAAmLC-PTA-2 both displayed increased compressive strength, toughness and stiffness. The highest values were obtained for PAAmLC-PTA-2 with a compressive strength of 25.7 ± 0.6 MPa along with a toughness of 1.63 ± 0.2 MJ m−3 and compressive modulus of 0.07 ± 0.018 MPa. These values are comparable to those of stiff and tough composite hydrogels made with gelatin,49,50 although in the gelatin case, the second network was ionically cross-linked. It is remarkable to observe that such high tensile and compressive properties can still be achieved for PTA hydrogels purely based on hydrogen bonding interactions, without crosslinking the second network.
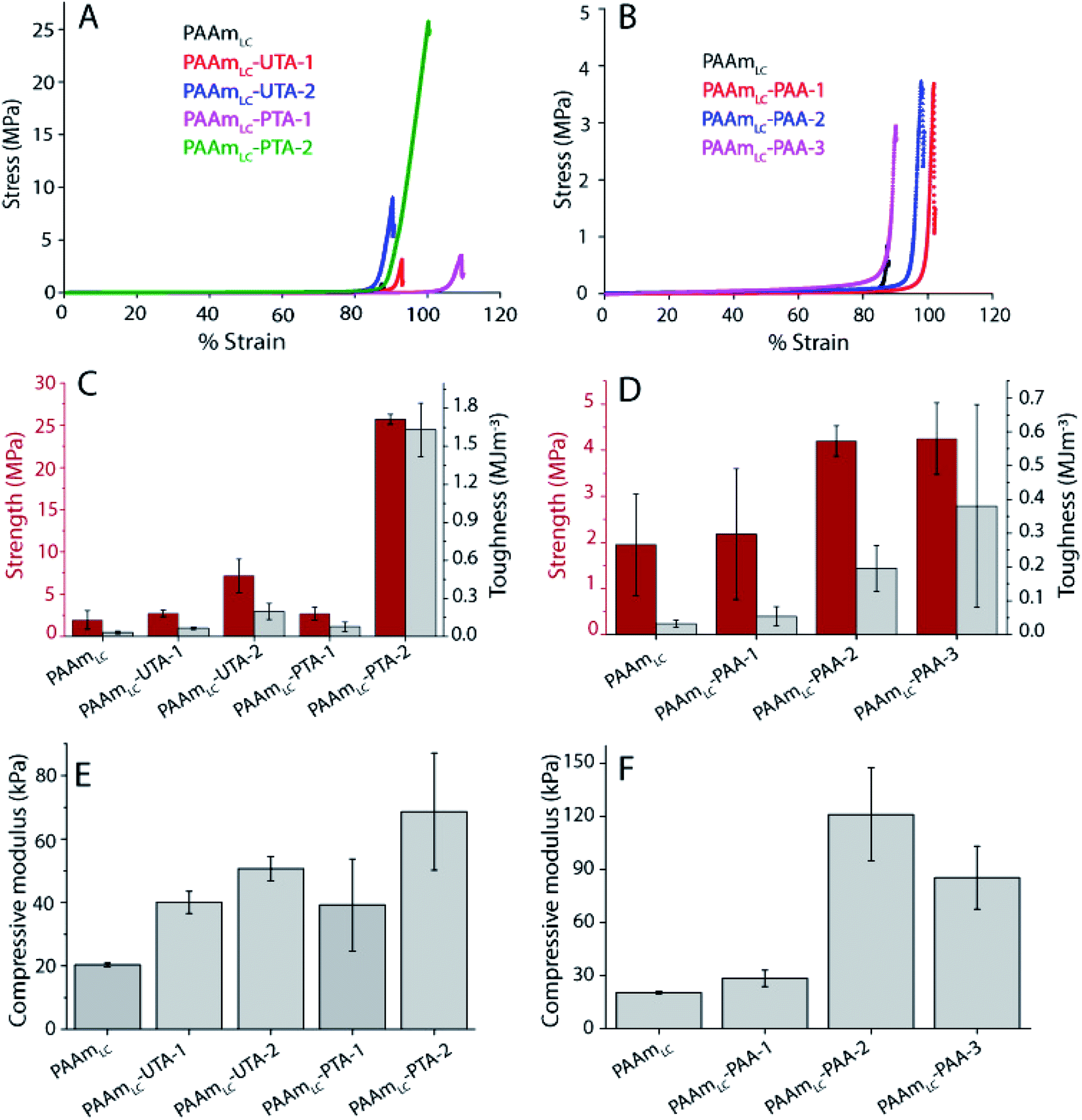 | ||
| Fig. 4 Compressive properties of hydrogels. (A and B) Compressive stress–strain curves of UTA, PTA and PAA incorporated hydrogels with different compositions. (C and D) Plots of compressive strength and toughness of tannic acid incorporated and PAA incorporated gels. (E and F) Comparison of compressive moduli of gels made with tannic acid and PAA incorporated gels. The compressive modulus was obtained from the slope of the stress–strain curve between 20% and 40% of compressive strain. | ||
While the standard deviation for the strength and toughness of the PAAmLC-PAA gels makes conclusions challenging, a similar trend to those observed in tensile testing can be observed. The compressive modulus shows the same increase for the intermediate concentration. Compression testing was also possible for the PAAmLC-PAAm control, and the compressive properties of those gels were similar to those of PAAmLC hydrogels (1.95 ± 1.1 MPa strength, 0.032 ± 0.01 MJ m−3 toughness, 20.3 ± 0.6 kPa modulus for PAAmLC gels and 1.63 ± 0.29 MPa strength, 0.017 ± 0.005 MJ m−3 toughness, 20.16 ± 4.6 kPa modulus for PAAmLC-PAAm gels). To investigate the influence of the crosslinking of the first network on the mechanical properties of the semi-interpenetrating network gels, a first network with a higher cross-linking ratio (PAAmHC, cross-linking ratio 69:1) was prepared. It is to be noted that a lower concentration of acrylamide monomers was used for the highly cross-linked gels compared to the loosely cross-linked gels (1.12 M vs. 1.69 M). This lower concentration was used since gels with the same high cross-linking ratio and the same monomer concentration as the PAAmLC were found to be too brittle to handle and test, in particular as semi-interpenetrating network gels. As expected from a more cross-linked gel, PAAmHC was found to be stiffer than PAAmLC, with higher Young's and compressive moduli of 19.6 ± 5 kPa and 52.9 ± 4.7 kPa respectively (Fig. S6 and S7†), compared to 2.23 ± 0.6 kPa and 20.3 ± 0.6 kPa for PAAmLC (Fig. 3 and 4).
A significant increase in tensile strength, toughness, and modulus was observed for PAAmHC-PTA, compared to both PAAmHC and PAAmHC-UTA (Fig. 5A and S6A†). However, no significant change in compressive properties was seen compared to PAAmHC (Fig. 5C and S6C†). In the case of PAA incorporated gels, a significant enhancement of both tensile and compressive properties was observed for PAAmHC-PAA-2 and 3 (Fig. 5B, D, S6B and D†).
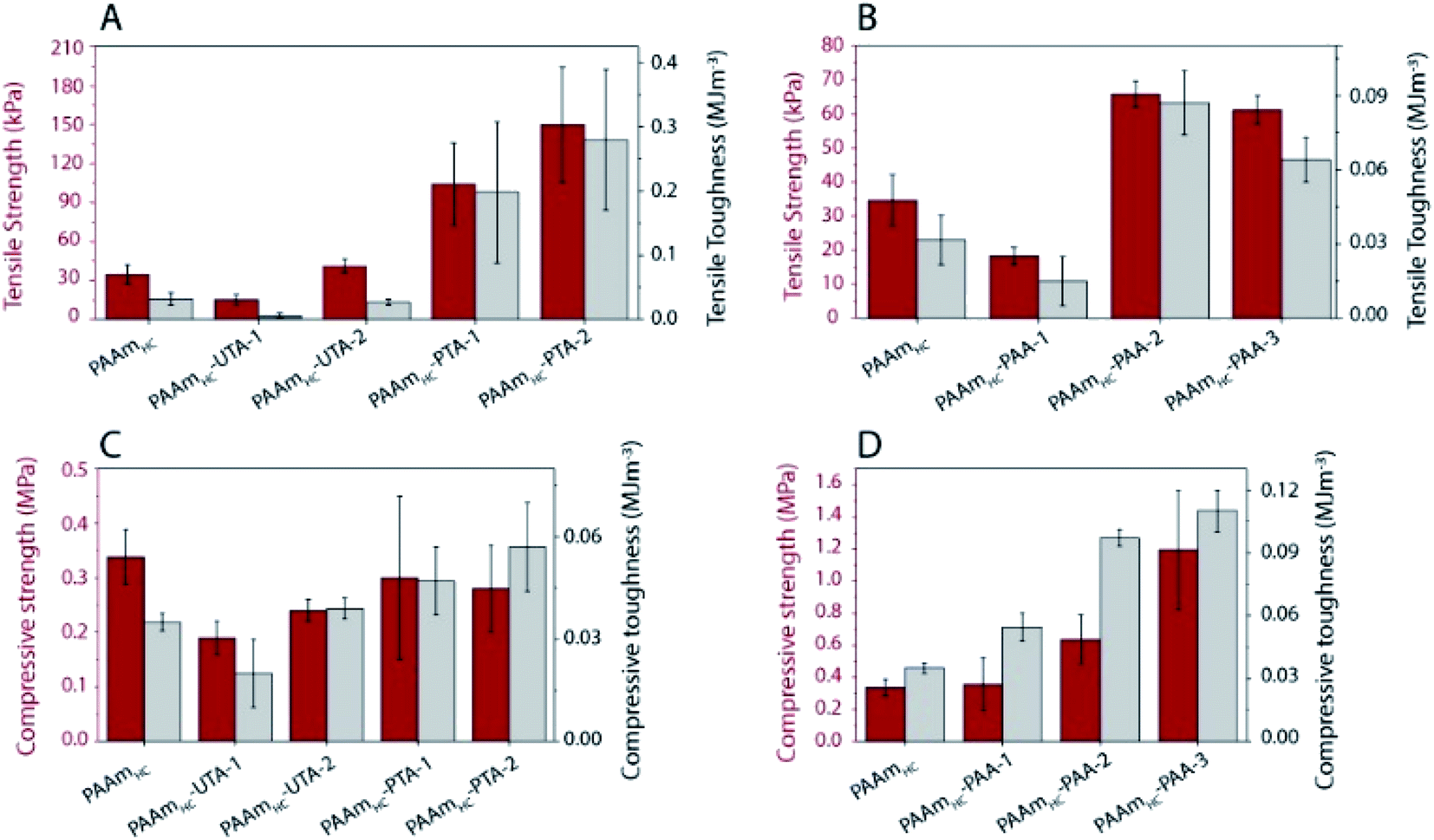 | ||
| Fig. 5 Mechanical properties of highly cross-linked first network gels. (A)Tensile strength and toughness of tannic acid polymerized gels (B) and acrylic acid polymerized gels. (C) Compressive strength and toughness of tannic acid polymerized gels. (D) Compressive strength and toughness of acrylic acid polymerized gels. | ||
It has previously been demonstrated that both monomer and cross-linking concentrations of the first network significantly affect the tensile properties of double network gels.51 A direct comparison of properties between both crosslinking ratios in our case is not possible as we used different concentrations of monomers for the two different crosslinking ratios. However, the results clarify that irrespective of the cross-linking ratio of the first network, the PAA and PTA incorporated gels exhibit enhanced mechanical properties.
Time-dependent mechanical properties
Hydrogels with tunable time-dependent properties are particularly interesting for mechanotransduction studies.52,53 Many studies use the plastic deformation exhibited by ionic cross-linking to tune the stress relaxation properties.53,54 Since the gels in this study exhibit hydrogen bonding interactions, a relaxation behavior may be expected as a result of breaking of hydrogen bonds in response to the applied stress. Stress relaxation tests were performed using compression testing fixtures to determine the relaxation times of the semi-interpenetrating network gels with both cross-linking ratios of the first network. The relaxation time is defined as the time taken by the gels to relax to half of the peak stress.The single network PAAmLC gels did not exhibit stress relaxation (Fig. S8A†), in line with elastic materials. In fact, of the loosely cross-linked gels, only PAAmLC-PAA-2 and 3 exhibited significant stress relaxation (Fig. S8C†). For the highly cross-linked first network, none of the gels displayed significant stress relaxation (Fig. S8B†).
The average relaxation times of PAAmLC-PAA-2 and PAAmLC-PAA-3 gels were found to be around 1000 seconds after one day of swelling (Fig. S8C†). The relaxation behavior observed in these gels is attributed to the breaking of hydrogen bonds between PAAm and PAA in response to applied stress. Long polymer chains with ionic cross-links exhibit substantial stress relaxation due to the breaking of ionic cross-links with applied stress.55 The fact that polymerized tannic acid gels (PAAmLC-PTA-1 and PAAmLC-PTA-2) did not exhibit this type of relaxation behavior may be an additional indication that polymerization of tannic acid did not yield long chain polymer networks.
Gels with tunable viscous properties by adjusting the opacity of photomasks
As a proof of concept, we tested the possibility of producing hydrogels with tunable dissipation. Gels with such a tunable loss modulus will be of interest for cell mechanotransduction studies. To this end, the photopolymerization of the second network of the gels was tuned by adding a mask of varying opacity. Frequency sweep oscillatory shear rheology was used to characterize the gels.In order to produce gels with stable mechanical properties under cell culture conditions, all gels were prepared under physiological conditions by replacing water with PBS. Fig. S9A† demonstrates the change in mechanical properties due to the osmotic swelling for gels prepared using water. Frequency sweep rheology of tannic acid polymerized gels made with two different opacities of transparent masks (10% and 20%) exhibited similar storage moduli but different loss moduli in the low frequency regime (Fig. 6A). Since tannic acid is expected to polymerize into short polymer species that could diffuse out of the gel, these hydrogels were characterized after equilibration for 24 hours in PBS. A frequency of 0.16 Hz was chosen to compare the gels' mechanical properties, as it has been suggested that cells probe the mechanical properties of substrates close to 0.16 Hz.32 Compared to the loosely cross-linked single network PAAm gels (PAAmLC), the semi-interpenetrating network gels with polymerized tannic acid exhibited a small increase in storage modulus and a noticeable increase in loss modulus (Fig. 6B). The controls in this case, were the PAAmLC gels with unpolymerized tannic acid (PAAmLC-UTA in Fig. 6B), which showed only a slight increase in loss modulus (G′′ average −105 Pa, compared to polymerized tannic acid gels with G′′ average – 706 Pa and 370 Pa for 10% and 20% masks respectively). However, for the highly cross-linked first network (PAAmHC gels), a significant difference in loss modulus was not observed using the photomask approach (data not shown).
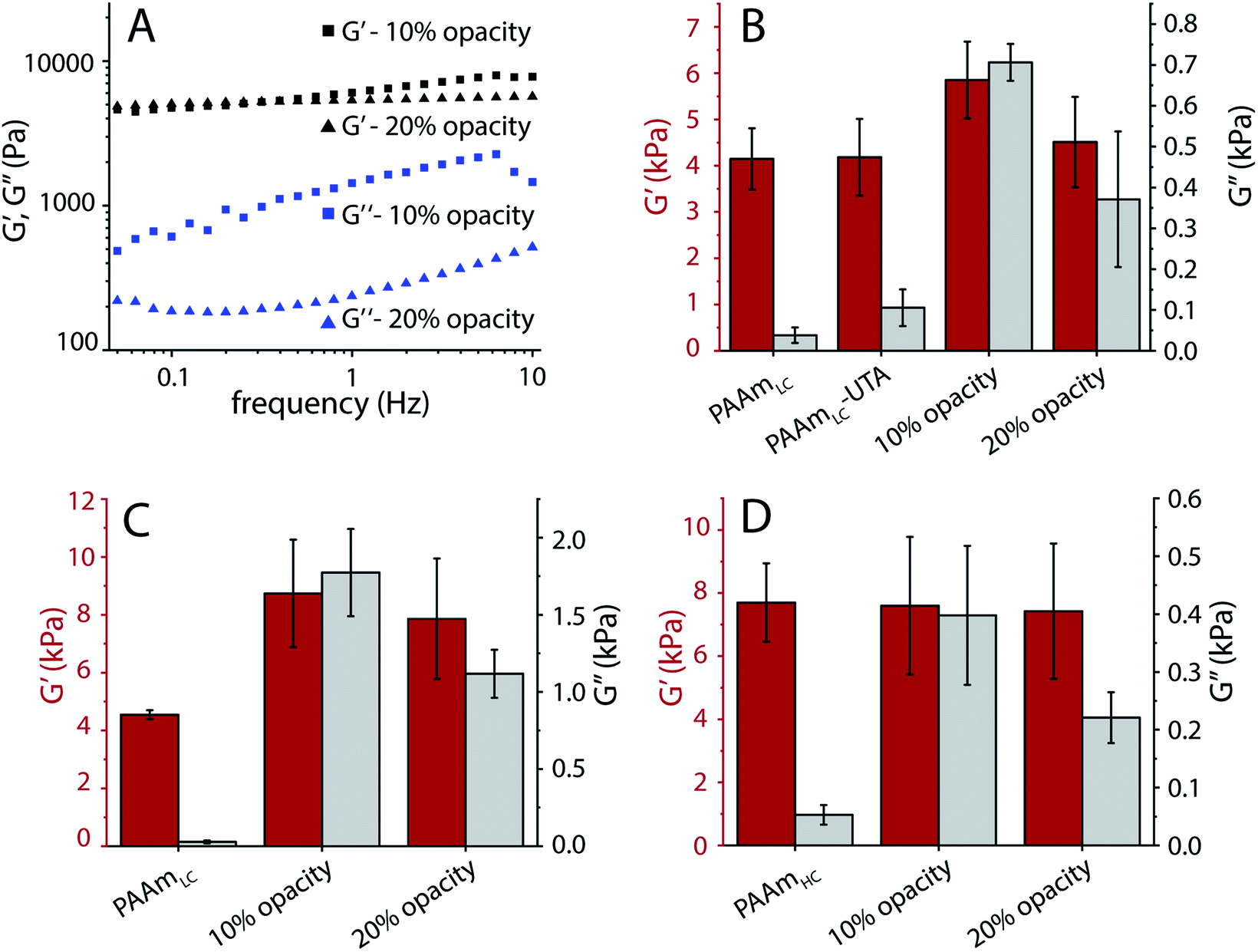 | ||
| Fig. 6 Rheological properties of hydrogels with tunable viscous properties. (A) Frequency sweep of tannic acid polymerized gels under photomasks of varying opacity. (B) Storage and loss moduli of tannic acid polymerized gels (PAAmLC-PTA-2) and controls after 24 hours of swelling in PBS. (C) Storage and loss moduli of acrylic acid polymerized gels (PAAmLC-PAA-2) with different photomasks after 4 hours of swelling in PBS. (D) Storage and loss moduli of acrylic acid polymerized (high cross-linking) gels (PAAmHC-PAA-2) with different photomasks after 4 hours of swelling in PBS. | ||
Gels with poly(acrylic acid) as the second network also displayed marked differences in loss modulus between hydrogels produced with 10% and 20% opacity photomasks, both in the case of low and high cross-linking of the first PAAm network (Fig. 6C and D). While the PAAmHC gels displayed a constant storage modulus after polymerization of the second network, in the case of PAAmLC gels, a noticeable change of the storage modulus was observed. This may be attributed to the higher swelling of the gel with the lower cross-linking, leading to a larger uptake of acrylic acid. Such a noticeable change in storage modulus was not observed for PAA incorporated PAAmHC gels when compared with PAAmHC gels. While the obtained loss modulus for the PAAmHC-PAA gels was low compared to the PAAmLC-PAA gels (G′′ average – 398 Pa and 221 Pa for 10% and 20% mask, Fig. 7D), it is evident that gels with a similar storage modulus and different loss moduli can be fabricated, irrespective of the first network crosslinking ratio.
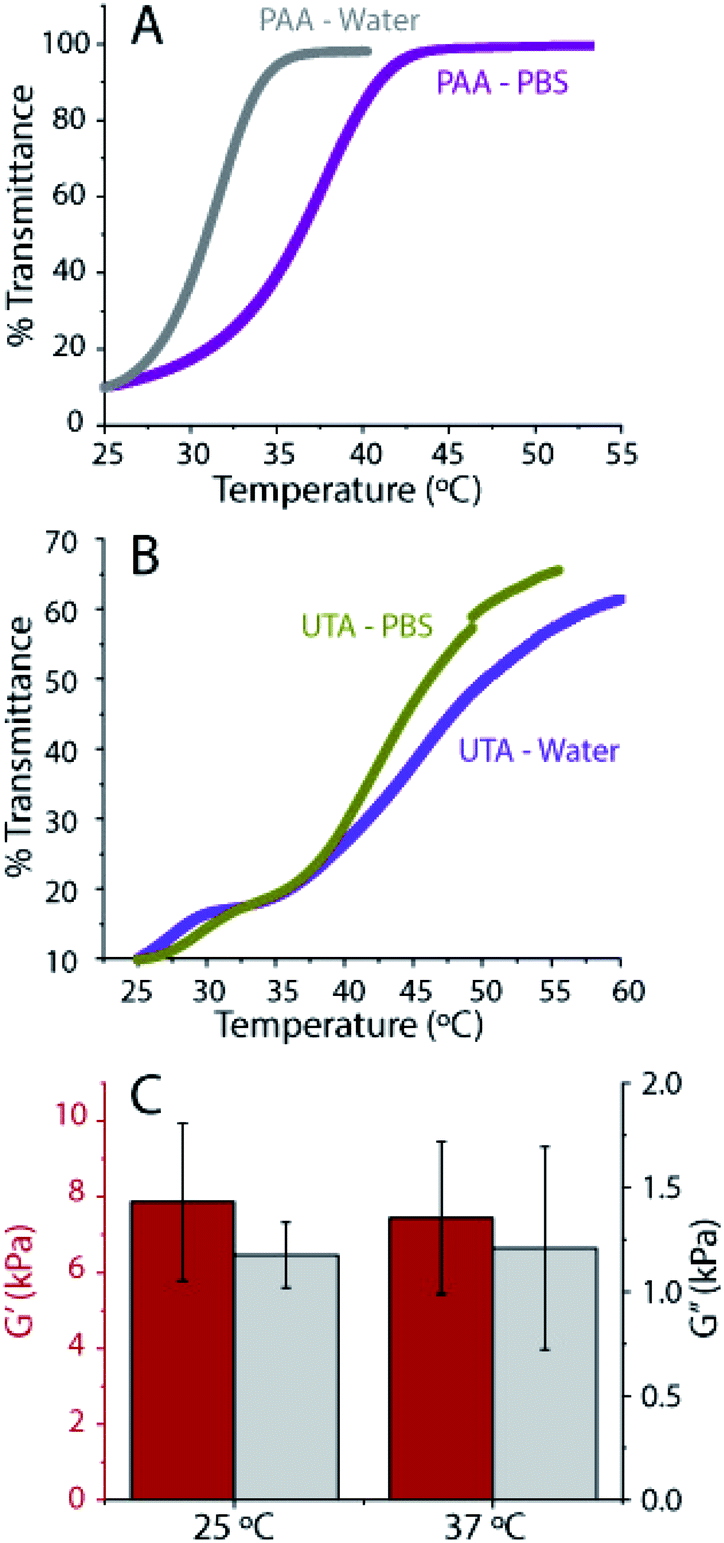 | ||
| Fig. 7 Stability of hydrogels. (A) Temperature controlled UV-Vis of PAA incorporated gels made with water and PBS measured. (B) Temperature controlled UV-Vis of tannic acid incorporated gels measured. The temperature was ramped at 0.5 °C per minute and the absorbance at 550 nm was measured in both cases. (C) Comparison of storage and loss moduli at 0.16 Hz for 4 hours swollen gels at 25 °C and 24 hours swollen gels at 37 °C, measured at 37 °C. The gels are made by photopolymerizing acrylic acid under a 20% mask (PAA incorporated gels). | ||
UV-Vis of the PBS solution used for swelling the gels was performed to evaluate the amount of tannic acid that gets leached out of the PAAmLC-UTA and PAAmLC-PTA gels (Fig. S10A† shows the calibration curve of tannic acid in PBS). Both unpolymerized and polymerized tannic acid gels leached out similar amounts of tannic acid after one day and the leachate did not show any traces of polymeric species in the case of polymerized tannic acid gels (Fig. S10B†). Therefore, we assume that all polymerized tannic acid species remain in the gel.
Temperature-controlled UV-Vis spectroscopy was performed to determine the transition temperature of the gels. Hydrogen bonds or electrostatic interactions between polymers result in a transition temperature, similar to the so-called upper critical solution temperature (UCST), above which, the components in a mixture are miscible in all proportions. Here, we attribute this temperature transition to the breaking of associative interactions at high temperatures.56 We define our upper phase transition temperature, as the temperature during the heating ramp at which the transmission reaches 50%. From the UV-Vis, we found the upper phase transition temperature to be close to 37 °C for the PAAmLC-PAA gels made in PBS (compared to 31 °C for the gels made in water) (Fig. 7A). Previous studies have shown that the UCST for hydrogels made by random copolymerization of PAAm and PAA was affected by hydrogen bonding and that it was proportional to the amount of poly(acrylic acid) and cross-linking.57 The presence of ions is well known to affect polymer transition temperatures.56 The tannic acid containing PAAmLC-UTA gels exhibit a much broader transition with a transition temperature of around 49 °C for water made gels and 45 °C for PBS made gels (Fig. 7B). The polymerized tannic acid PAAmLC hydrogels exhibit an even more complex behavior (Fig. S10C†), emphasizing the complexity of the processes involved when the associative interactions break at high temperature which warrants further investigation.
As the UCST of PAAmLC-PAA gels was close to that of cell culture conditions, the mechanical properties of the gels after swelling in PBS at 37 °C for 24 hours were tested (Fig. 7C). The frequency sweep rheology revealed that the mechanical properties of the gels did not change significantly when compared to the mechanical properties of gels obtained at room temperature after 4 hours of swelling in PBS. Hence, it can be concluded that both PAA incorporated and tannic acid hydrogels hold promise as cell culture substrates.
Polymer gels consisting of two cross-linked polymer networks have been shown to exhibit excellent toughness and stiffness.43,49,58,59 However, the influence of using uncross-linked polymers with different structures as second networks has not previously been studied in depth. We systematically studied poly(acrylamide) gels with the incorporation of three different second networks: linear polymers that can hydrogen bond with the first network (PAA, molecular weight 3.43 × 106 g mol−1 obtained by using a viscometer with k and a values obtained from the literature,60 see Fig. S4†), linear polymers that do not form hydrogen bonds with the first network (PAAm), and short branched polymers that can hydrogen bond with the first network (PTA). We found that hydrogen bonding gels exhibited higher stiffness and toughness, in agreement with expectations. An optimum concentration of the hydrogen bonding second network was also observed (PAAmLC-PAA-2, PAAmLC-PTA-2) where the highest values of stiffness and toughness were achieved. When compared to the single network PAAmLC gels, the PAAmLC-PTA-2 hydrogels exhibited a 19 times tensile strength enhancement, and a 55 times increase in toughness. Similarly, when compared to the unpolymerized tannic acid incorporated gels (PAAmLC-UTA-2), PTA gels showed a 6.3 times increase in tensile strength and a 9 times increase in toughness. When compared to PAAmLC-PAA-2 gels, a 4.3 times increase in strength and a 3.6 times enhancement in toughness were observed. Finally, Young's modulus was calculated to be 0.12 ± 0.02 MPa which is about 20 times higher than that of UTA gels and 50 times higher than that of the parent gels. In addition to that, the water content of PAAmLC-PTA-2 gels (average of 75%, Fig. S5A†) was found to be slightly higher than those of previously reported hydrogels with similar compression properties.49 From the tensile properties of polymerized tannic acid gels, it is clear that the gels exhibit high stiffness and toughness. The yielding behavior and subsequent energy dissipation observed in these gels are attributed to the breaking of weak hydrogen bonded aggregates between PAAm and polymerized tannic acid. Double networks composed of poly(methacrylic acid) and poly(dimethacrylamide) have previously been found to exhibit high stiffness and toughness, which was attributed to the varied sizes of the hydrogen bonded aggregates.26 Indeed, the DLS revealed that polymerized tannic acid produced larger aggregates with poly(acrylamide) when compared to the poly(acrylic acid) (Fig. S1B†) which might in part explain the increased stiffness and toughness of PAAmLC-PTA-2 gels when compared to PAAmLC-PAA-2 gels. The incorporation of polymerized tannic acid into PAAm gels results in increased hydrogen bonding density.
Polymerized tannic acid incorporated inside PAAm gels is similar to self-polymerized dopamine incorporated inside PAAm gels.46 However, for polydopamine incorporated gels, though an enhancement of toughness was observed, the stiffness was found to decrease when compared with the control gels without polydopamine. In contrast, in our case, we observed that incorporation of polymeric tannic acid enhances both stiffness and toughness. A recent study by Cui et al.61 showed that oligomeric lignin, hydrogen bonding elastomers, showed a similar enhancement of stiffness and toughness. The authors argue that the enhancement is ascribed to the rigidity and network forming ability of the oligomeric lignin species. We believe that polymerized tannic acid incorporated inside PAAm gels enhances the stiffness and toughness through a similar mechanism, although further structural characterization, such as small angle X-ray and neutron scattering, is needed to confirm this hypothesis.
Furthermore, we also speculate that the structure of the polymerized polyphenol results in more entanglement within the first network. However, the exact mechanism of how the polymerized polyphenol's structure contributes to the mechanical properties is still not clear at this point. Our results further suggest that for the rational design of hydrogels for specific applications, the structure of second networks should also be considered in addition to the cross-linker concentration and the density of hydrogen bonds.
Conclusions
Recently there has been a growing interest in making substrates with a tunable loss modulus for mechanotransduction studies.10,62 The most common method for achieving this is by producing gels with fewer cross-links. However, such gels can only achieve tuning of loss modulus from 50–600 Pa with tan delta (G′′/G′) from 0.01 to 0.1.31,63 Moreover, such systems also suffer from swelling induced changes in mechanical properties. With our system, we have achieved tuning of loss modulus from 800 Pa to 2000 Pa for a similar storage modulus (tan delta of 0.25) by simply changing the opacity of the photomasks during polymerization. This wide range of loss moduli would help in properly discerning the cell response to the substrate loss modulus more clearly than the previously reported studies. In the future, such systems can be used to study cell responses to loss modulus gradients or dissipation gradients. We also found that by adjusting the density of the hydrogen bonds, we can achieve tuning of the stress relaxation times which was also shown to critically contribute to cellular mechanotransduction.6,52 In terms of comparison against natural tissues, our hydrogel systems exhibit mechanical properties similar to that of soft tissues and the relaxation times are comparable to that of skin.64In summary, we have demonstrated a simple, yet versatile, strategy to produce hydrogels with tunable dissipation. By careful selection of the second network, this study addresses the critical gap in the literature in adopting semi-interpenetrating network gels for cell culture studies, which are mainly hindered by swelling. While the current study focused on understanding the effect of an uncross-linked second network with different structures using a variety of mechanical characterization techniques, the simplicity of this approach enables easy adaptability of such gels to various cellular mechanotransduction studies. Furthermore, we found that the polymerization of polyphenols, which results in short polymers, yields a remarkable increase in stiffness and toughness. Finally, to the best of our knowledge, we report for the first time that it is possible to achieve tunable dissipation properties using UV polymerization of polyphenols.
Author contributions
B. N. and J. M. designed and planned the research. B. N., S. D. and M. T. performed the experiments. B. N., S. M., M. W. and J. M. analysed and interpreted data. B. N and J. M wrote the manuscript with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support by the Marsden Fund Council and the Rutherford Discovery Fellowship, from Government funding, managed by Royal Society Te Apārangi. J. M. acknowledges MacDiarmid institute for Advanced Materials and Nanotechnology for their support of this research. The authors thank Dr Sophia Rodrigues and Dr Josh Workman for the useful discussions.Notes and references
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 1–17 Search PubMed.
- B. Xue, M. Qin, T. Wang, J. Wu, D. Luo, Q. Jiang, Y. Li, Y. Cao and W. Wang, Adv. Funct. Mater., 2016, 26, 9053–9062 CrossRef CAS.
- Y. S. Kim, M. Liu, Y. Ishida, Y. Ebina, M. Osada, T. Sasaki, T. Hikima, M. Takata and T. Aida, Nat. Mater., 2015, 14, 1002–1007 CrossRef CAS PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- O. Chaudhuri, Biomater. Sci., 2017, 5, 1480–1490 RSC.
- A. R. Cameron, J. E. Frith, G. A. Gomez, A. S. Yap and J. J. Cooper-White, Biomaterials, 2014, 35, 1857–1868 CrossRef CAS PubMed.
- T. E. Brown, B. J. Carberry, B. T. Worrell, O. Y. Dudaryeva, M. K. McBride, C. N. Bowman and K. S. Anseth, Biomaterials, 2018, 178, 496–503 CrossRef CAS PubMed.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef CAS PubMed.
- E. Hui, K. I. Gimeno, G. Guan and S. R. Caliari, Biomacromolecules, 2019, 20, 4126–4134 CrossRef CAS PubMed.
- W. Wang, Y. Zhang and W. Liu, Prog. Polym. Sci., 2017, 71, 1–25 CrossRef.
- E. M. Boazak, V. K. Greene and D. T. Auguste, PLoS One, 2019, 14, 1–11 CrossRef PubMed.
- A. Lake and G. J. Thomas, Proc. R. Soc. London, Ser. A, 1967, 300, 108–119 Search PubMed.
- C. Creton, Macromolecules, 2017, 50, 8297–8316 CrossRef CAS.
- J. P. Gong, Soft Matter, 2010, 6, 2583–2590 RSC.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. Bin Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485–487 CrossRef CAS.
- K. Ito, Curr. Opin. Solid State Mater. Sci., 2010, 14, 28–34 CrossRef CAS.
- J. Wang, Z. Chen, X. Li, M. Liu, Y. Zhu and L. Jiang, ACS Appl. Mater. Interfaces, 2019, 11, 41659–41667 CrossRef CAS PubMed.
- M. K. Shin, G. M. Spinks, S. R. Shin, S. I. Kim and S. J. Kim, Adv. Mater., 2009, 21, 1712–1715 CrossRef CAS.
- I. Jeon, J. Cui, W. R. K. Illeperuma, J. Aizenberg and J. J. Vlassak, Adv. Mater., 2016, 28, 4678–4683 CrossRef CAS PubMed.
- S. Y. Zheng, H. Ding, J. Qian, J. Yin, Z. L. Wu, Y. Song and Q. Zheng, Macromolecules, 2016, 49, 9637–9646 CrossRef CAS.
- L. Han, X. Lu, K. Liu, K. Wang, L. Fang, L. T. Weng, H. Zhang, Y. Tang, F. Ren, C. Zhao, G. Sun, R. Liang and Z. Li, ACS Nano, 2017, 11, 2561–2574 CrossRef CAS PubMed.
- D. Gan, T. Xu, W. Xing, M. Wang, J. Fang, K. Wang, X. Ge, C. W. Chan, F. Ren, H. Tan and X. Lu, J. Mater. Chem. B, 2019, 7, 1716–1725 RSC.
- X. Hu, M. Vatankhah-varnoosfaderani, J. Zhou, Q. Li and S. S. Sheiko, Adv. Mater., 2015, 27, 6899–6905 CrossRef CAS PubMed.
- Q. Chen, X. Yan, L. Zhu, H. Chen, B. Jiang, D. Wei, L. Huang, J. Yang, B. Liu and J. Zheng, Chem. Mater., 2016, 28, 5710–5720 CrossRef CAS.
- C. B. Rodell, N. N. Dusaj, C. B. Highley and J. A. Burdick, Adv. Mater., 2016, 28, 8419–8424 CrossRef CAS PubMed.
- Z. Gu, K. Huang, Y. Luo, L. Zhang, T. Kuang, Z. Chen and G. Liao, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2018, 10, 1–15 Search PubMed.
- D. E. Discher, P. Janmey and Y. Wang, Science, 2005, 310, 1139–1144 CrossRef CAS PubMed.
- A. R. Cameron, J. E. Frith and J. J. Cooper-White, Biomaterials, 2011, 32, 5979–5993 CrossRef CAS PubMed.
- E. E. Charrier, K. Pogoda, R. G. Wells and P. A. Janmey, Nat. Commun., 2018, 9, 1–13 CrossRef CAS PubMed.
- C. Zhao, Z. Ma and X. X. Zhu, Prog. Polym. Sci., 2019, 90, 269–291 CrossRef CAS.
- F. Iimain, T. Tanaka and E. Kokufuta, Nature, 1991, 349, 400–401 CrossRef.
- Y. Xu, O. Ghag, M. Reimann, P. Sitterle, P. Chatterjee, E. Nofen, H. Yu, H. Jiang and L. L. Dai, Soft Matter, 2017, 14, 151–160 RSC.
- F. Behboodi-Sadabad, H. Zhang, V. Trouillet, A. Welle, N. Plumeré and P. A. Levkin, Adv. Funct. Mater., 2017, 27, 1–11 CrossRef.
- R. Takahashi, T. Ikai, T. Kurokawa, D. R. King and J. P. Gong, J. Mater. Chem. B, 2019, 7, 6347–6354 RSC.
- N. C. Stellwagen, Electrophoresis, 1998, 19, 1542–1547 CrossRef CAS PubMed.
- M. Oćwieja, Z. Adamczyk and M. Morga, J. Colloid Interface Sci., 2015, 438, 249–258 CrossRef PubMed.
- A. Ricci, K. J. Olejar, G. P. Parpinello, P. A. Kilmartin and A. Versari, Appl. Spectrosc. Rev., 2015, 50, 407–442 CrossRef CAS.
- F. Puza, Y. Zheng, L. Han, L. Xue and J. Cui, Polym. Chem., 2020, 11, 2339–2345 RSC.
- D. Zhang, Z. Xu, H. Li, C. Fan, C. Cui, T. Wu, M. Xiao, Y. Yang, J. Yang and W. Liu, Biomater. Sci., 2020, 8, 1455–1463 RSC.
- H. Fan, J. Wang and Z. Jin, Macromolecules, 2018, 51, 1696–1705 CrossRef CAS.
- H. Xu, F. K. Shi, X. Y. Liu, M. Zhong and X. M. Xie, Soft Matter, 2020, 16, 4407–4413 RSC.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. Bin Ihsan, T. Akasaki, K. Sato, A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 1–6 CrossRef PubMed.
- L. Han, L. Yan, K. Wang, L. Fang, H. Zhang, Y. Tang, Y. Ding, L. T. Weng, J. Xu, J. Weng, Y. Liu, F. Ren and X. Lu, NPG Asia Mater., 2017, 9, e372 CrossRef CAS.
- B. Liu, Y. Wang, Y. Miao, X. Zhang, Z. Fan, G. Singh, X. Zhang, K. Xu, B. Li, Z. Hu and M. Xing, Biomaterials, 2018, 171, 83–96 CrossRef CAS PubMed.
- Y. You, J. Yang, Q. Zheng, N. Wu, Z. Lv and Z. Jiang, Sci. Rep., 2020, 10, 1–8 CrossRef PubMed.
- L. Xu, C. Wang, Y. Cui, A. Li, Y. Qiao and D. Qiu, Sci. Adv., 2019, 5, eaau3442 CrossRef PubMed.
- Q. He, Y. Huang and S. Wang, Adv. Funct. Mater., 2018, 28, 1–10 Search PubMed.
- H. Xin, S. Z. Saricilar, H. R. Brown, P. G. Whitten and G. M. Spinks, Macromolecules, 2013, 46, 6613–6620 CrossRef CAS.
- O. Chaudhuri, L. Gu, M. Darnell, D. Klumpers, S. A. Bencherif, J. C. Weaver, N. Huebsch and D. J. Mooney, Nat. Commun., 2015, 6, 1–7 Search PubMed.
- O. Chaudhuri, L. Gu, D. Klumpers, M. Darnell, S. A. Bencherif, J. C. Weaver, N. Huebsch, H. P. Lee, E. Lippens, G. N. Duda and D. J. Mooney, Nat. Mater., 2016, 15, 326–334 CrossRef CAS PubMed.
- O. Chaudhuri, L. Gu, M. Darnell, D. Klumpers, S. A. Bencherif, J. C. Weaver, N. Huebsch and D. J. Mooney, Nat. Commun., 2015, 6, 6365 CrossRef CAS PubMed.
- X. Zhao, N. Huebsch, D. J. Mooney and Z. Suo, J. Appl. Phys., 2010, 107, 063509 CrossRef PubMed.
- J. Niskanen and H. Tenhu, Polym. Chem., 2017, 8, 220–232 RSC.
- C. Echeverria, D. López and C. Mijangos, Macromolecules, 2009, 42, 9118–9123 CrossRef CAS.
- X. N. Zhang, Y. J. Wang, S. Sun, L. Hou, P. Wu, Z. L. Wu and Q. Zheng, Macromolecules, 2018, 51, 8136–8146 CrossRef CAS.
- Y. J. Wang, X. N. Zhang, Y. Song, Y. Zhao, L. Chen, F. Su, L. Li, Z. L. Wu and Q. Zheng, Chem. Mater., 2019, 31, 1430–1440 CrossRef CAS.
- J. Brandrup, E. H. Immergut and E. A. Grulke, Polymer Handbook, ed. A. Akihiro and D. R. Bloch, John Wiley & Sons, Inc, 2005 Search PubMed.
- M. Cui, N. A. Nguyen, P. V. Bonnesen, D. Uhrig, J. K. Keum and A. K. Naskar, ACS Macro Lett., 2018, 7, 1328–1332 CrossRef CAS.
- M. Cantini, H. Donnelly, M. J. Dalby and M. Salmeron-Sanchez, Adv. Healthcare Mater., 2020, 9, 1901259 CrossRef CAS PubMed.
- K. Mandal, Z. Gong, A. Rylander, V. B. Shenoy and P. A. Janmey, Biomater. Sci., 2020, 8, 1316–1328 RSC.
- O. Chaudhuri, J. Cooper-White, P. A. Janmey, D. J. Mooney and V. B. Shenoy, Nature, 2020, 584, 535–546 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00103e |
| ‡ Present address: Department of Neuroscience and Biomedical Engineering, Aalto University, Espoo, 0076, Finland. |
| This journal is © The Royal Society of Chemistry 2021 |