
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe evolution of nucleosidic analogues: self-assembly of prodrugs into nanoparticles for cancer drug delivery
Milad
Baroud
 a,
Elise
Lepeltier
*a,
Sylvain
Thepot
bcd,
Yolla
El-Makhour
e and
Olivier
Duval
ab
a,
Elise
Lepeltier
*a,
Sylvain
Thepot
bcd,
Yolla
El-Makhour
e and
Olivier
Duval
ab
aMicro et Nanomédecines Translationnelles, MINT, UNIV Angers, UMR INSERM 1066, UMR CNRS 6021, Angers, France. E-mail: elise.lepeltier@univ-angers.fr
bUniversity Hospital of Angers, Hematology, 49933 Angers, France
cUniversité d'Angers, Inserm, CRCINA, 49000 Angers, France
dFédération Hospitalo-Universitaire ‘Grand Ouest Against Leukemia’ (FHU GOAL), France
eEnvironmental Health Research Lab (EHRL), Faculty of Sciences V, Lebanese University, Nabatieh, Lebanon
First published on 22nd February 2021
Abstract
Nucleoside and nucleotide analogs are essential tools in our limited arsenal in the fight against cancer. However, these structures face severe drawbacks such as rapid plasma degradation or hydrophilicity, limiting their clinical application. Here, different aspects of nucleoside and nucleotide analogs have been exposed, while providing their shortcomings. Aiming to improve their fate in the body and combating their drawbacks, two different approaches have been discussed, the prodrug and nanocarrier technologies. Finally, a novel approach called “PUFAylation” based on both the prodrug and nanocarrier technologies has been introduced, promising to be the supreme method to create a novel nucleoside or nucleotide analog based formulation, with enhanced efficacy and highly reduced toxicity.
1. Introduction
Cancer has proven to be a detrimental disease that has had severe impacts on a global scale. It occurs as cells lose their regulatory ability and start to proliferate and multiply in an unrestrained fashion, followed potentially by the invasion of adjacent organs and tissues, eventually spreading throughout the body and impeding the vital functions of the affected areas. This loss in regulatory ability springs from the cumulative defects occurring at the genetic level with various genes being mutated leading to a loss of normal function and the abnormal cells exhibiting characteristics and functions that differ from their past behavior.1This odd growth of cells will lead to the formation of a tumor, an unnatural mass of cells that can be either benign or malignant. Moles and lipomas are a common example of benign tumors; such tumors are restricted to their original location without spreading to adjacent areas, whereas malignant tumors also identified as cancers have the ability to invade surrounding tissues and spread throughout the body (metastasis) utilizing the lymphatic and circulatory systems.
In 2015, the assessment of the World Health Organization (WHO) showed that cancer is the first or second prominent cause of death before the age of 70 years in 91 of 172 countries. By 2018, there were an estimated 18.1 million new cancer cases and 9.6 million cancer deaths worldwide, with the prevailing type of cancer observed being lung cancer.2,3
Concerning the treatment of cancer, the rise of systemic adjuvant therapy, in combination with local surgery and/or radio therapy, caused a plummet in radical surgery as both had similar results.
Of these adjuvant therapies, nucleosidic anti-cancer agents have become a staple chemotherapeutic treatment for many cancers. Anticancer nucleosides can be divided into two categories: analogs of purine and pyrimidine nucleosides and nucleobases. Concerning purine analogs, cladribine and fludarabine are two of the most prominent drugs being used as a treatment for low-grade malignant blood disorders.4 With regard to pyrimidine analogs, cytarabine is a principal drug in the treatment of acute leukemia, gemcitabine is used in the treatment of various solid tumors and hematological cancers and diseases, and finally, fluorouracil and its prodrug capecitabine have shown activity in colorectal and breast cancers. The improvements that traditional analogs have undergone, coupled with the ability of using them in the treatment of a wide range of cancers, have solidified the role of nucleoside analogs as a significant chemotherapeutic agent, not to mention a growth in their popularity that occurred with the understanding of their exact mechanism of action.5–9
However, traditional chemotherapeutics display a high toxicity profile with a small therapeutic window, leading to different severe side effects and a lowered efficacy due to the cancer cell's ability of drug efflux through active pumps called “multidrug resistance”.10,11 Consequently, several novel approaches are stemming to address and overcome these shortcomings, namely the prodrug strategy and nanoparticle drug delivery systems (nano-DDSs).12,13 Prodrugs are pharmacologically inactive molecules that can be metabolized into their active form in vivo. The novelty of prodrugs emerges from their ability to overcome the flaws present in the parent drugs, for instance chemical instability, poor water solubility, severe toxicity, and low permeability.12,14–16 In addition, benefiting from the rapid development of biomaterials and nanotechnology, nano-DDSs have shown distinct advantages in anticancer drug delivery, including improved drug availability, prolonged systemic circulation time, increased tumor accumulation, and spatiotemporally controlled drug release. On top of that, prodrug-based nanoparticles incorporate the advantages gained from both approaches (Fig. 1) allowing for a more stable drug and a further decrease in toxicity as the specificity of the drug increases while offering maximal drug loading capabilities.17
 | ||
| Fig. 1 Strategies applied to improve the delivery efficiency of anticancer drugs: prodrugs, encapsulation and self-assembled prodrug-based nano-DDSs. | ||
2. An overview of nucleosides
Nucleosides and nucleotides are organic molecules that play a key role in several bodily functions, as they are included in the signaling pathways inside cells, metabolism, DNA and RNA synthesis, and enzymatic regulation.Synthetic equivalents of these compounds have been produced; these analogs are capable of mimicking the activity of the original compounds. Thus, they are able to interact with several key enzymes (such as kinases, ribonucleotide reductase, DNA methyl transferases, purine and pyrimidine nucleoside phosphorylases and thymidylate synthase). Additionally, they gain the ability of “hijacking” the conventional metabolic pathways of the original nucleotides owing to the structural similarity they share, leading to the inhibition of cellular division by assimilating into DNA or RNA. This ability of analogs has given rise to numerous medicinal applications including the capacity to inhibit the growth of cancer cells.18,19
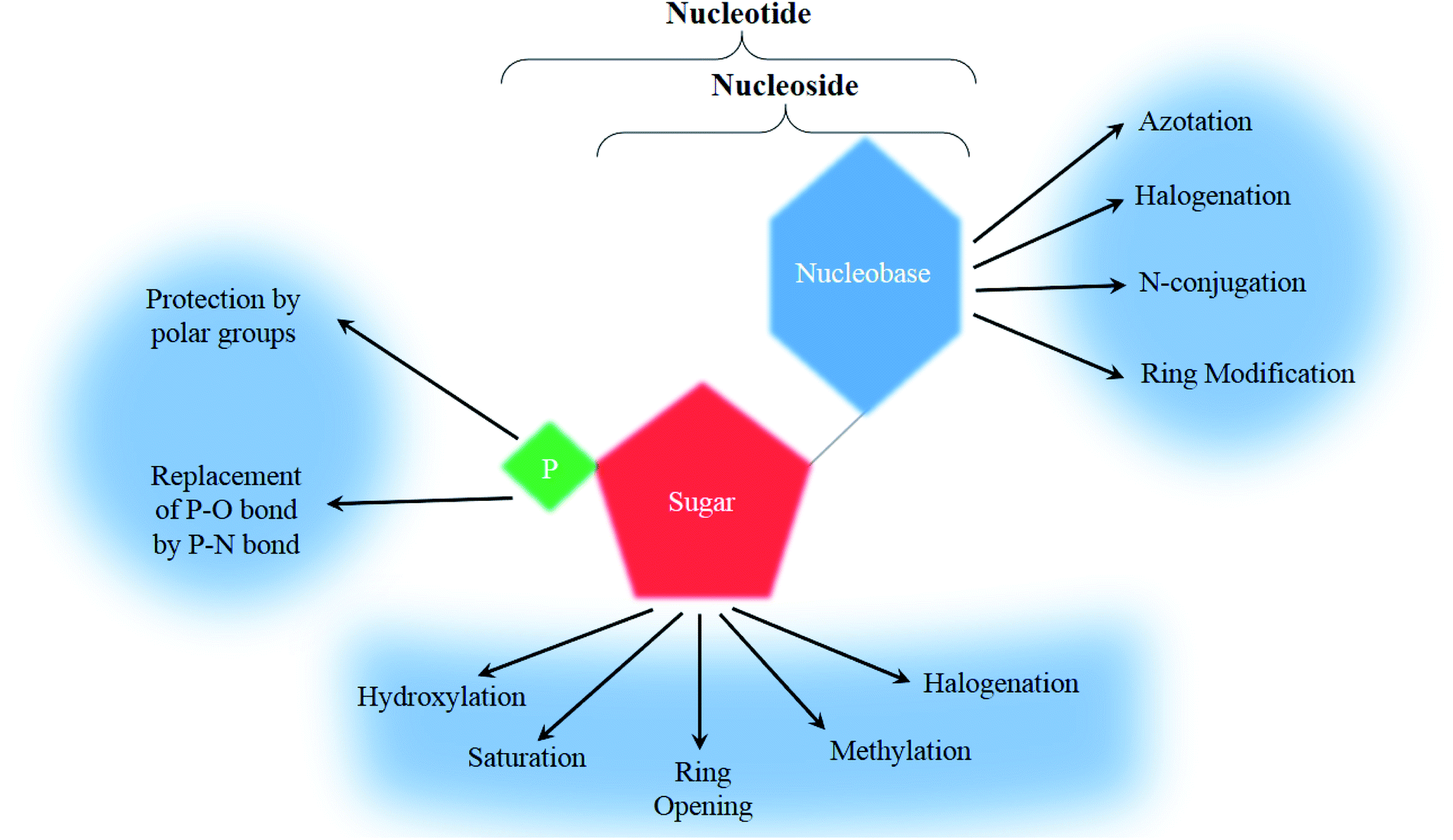
To form a nucleoside, a nucleobase, either a purine (adenine or guanine) or a pyrimidine (cytosine, uracil or thymine), is needed to bind to a pentose sugar ring (a ribose or a deoxyribose). A further modification of the primary alcohol from the sugar ring with a phosphoric acid yields a nucleotide (Fig. 2).20
 | ||
| Fig. 2 General structure and chemical modifications of nucleosides and nucleotides to form analogs. | ||
2.1. Nucleobases
A nucleobase, also called a nitrogen base, is a carbon-based molecule containing a nitrogen atom and reacting with the chemical properties of a Lewis base due to the lone pair of electrons of the mentioned nitrogen atom.Nucleobases can either be a purine or a pyrimidine, characterized by a weak reactivity to electrophilic aromatic substitution and are thus considered weak bases. Owing to their cyclic nature they are non-polar, and function to bond nucleic acids together.21,22
Regarding analogs, the nitrogen bases that are incorporated are rarely the natural ATGC-bases and are frequently modified with 4 major categories being observed ranging between halogenation (with fluorine and chlorine being the most commonly used), azotation, N-conjugation, and ring modification (Fig. 2). Few other modifications that don't fit into the main groups have been observed and will be mentioned separately.
2.2. Sugar ring
The second building block of a nucleoside, the pentose sugar ring can be either a ribose or 2′-deoxyribose (Fig. 3). The major function of the 2′-deoxyribose is becoming the backbone of the DNA molecule, while the ribose becomes the backbone of the RNA molecule. Similar to the nucleobases, the sugar ring is rarely intact and is often modified with the main changes being halogenation, methylation, saturation, ring opening, and hydroxylation/de-hydroxylation (Fig. 2).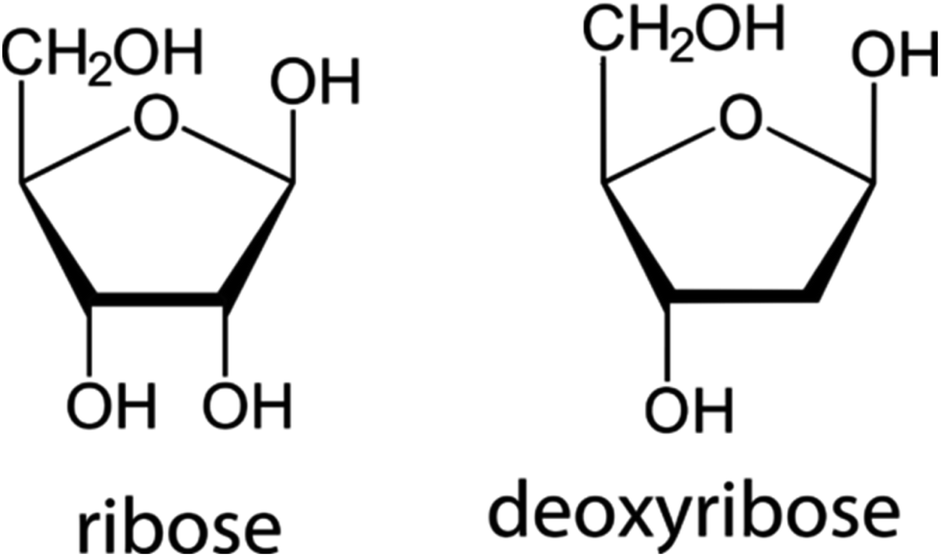 | ||
| Fig. 3 Structure of sugar rings: ribose and deoxyribose. | ||
3. Chemotherapeutic nucleoside analogs
Most of the present natural nucleoside analogs that are being used or are under development are derived from one of the following nucleosides presented in Fig. 4. The joining of a purine (either an adenine or guanine) or a pyrimidine (cytosine, thymine or uracil) base with a pentose ring (β-D-deoxyribofuranose or β-D-ribofuranose for DNA or RNA, respectively) forms the nucleic acid. These nucleic acids can then be modified variously (i.e. azotation of the nucleobase, halogenation of the sugar ring…) as described in Fig. 2 to obtain the nucleosidic analogs utilized in the course of cancer treatment.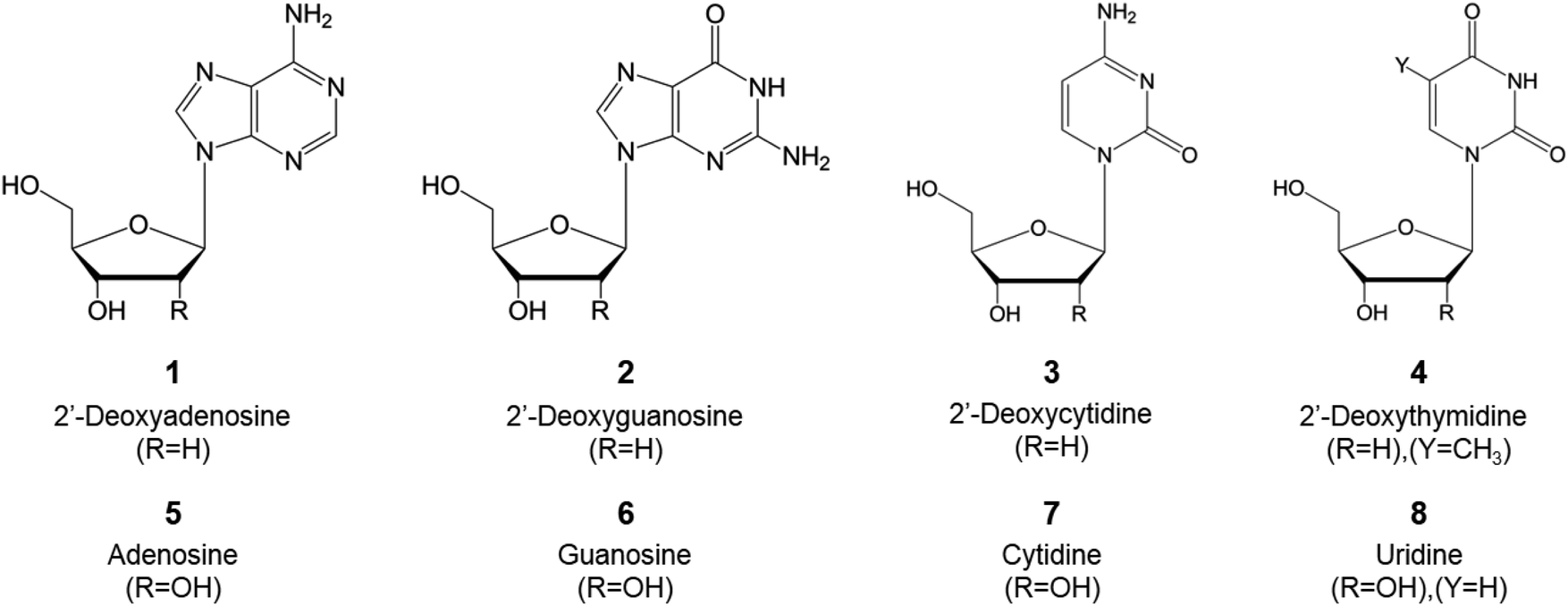 | ||
| Fig. 4 Different types of natural nucleoside that constitute nucleic acids. | ||
3.1. Advancements in therapeutic nucleoside analogs for cancer treatment
Fluorouracil opened the door for advancements in the field of therapeutic nucleoside analogs for cancer treatment as it was the first FDA approved one in 1962, a uracil nucleobase modified by fluorine halogenation; it was widely used in the treatment of different cancers including colon, esophageal, and cervical cancers. Following that, several other nucleoside based drugs emerged: floxuridine, a prime example of these drugs based on fluorouracil and having it as the metabolite, is used for the treatment of colon cancer. The rest of the FDA and EMA approved drugs are presented in Table 1. Despite the limited number of approved nucleoside analogs for the treatment of various cancerous tumors, numerous other ones are being developed and tested in countless clinical trials (Table 2) either separately or in association for their synergistic effects, such as 8-chloroadenosine, a promising new analogue undergoing phase I/II trials on patients with relapse or refractory acute myeloid leukemia.| Name | Structure | Statusa | Chemical modification | Targeted cancer | Mechanism of action | |
|---|---|---|---|---|---|---|
| a Approval status was verified by the European Medicines Agency (http://www.ema.europa.eu) and the US Food and Drug Administration (http://www.fda.gov). | ||||||
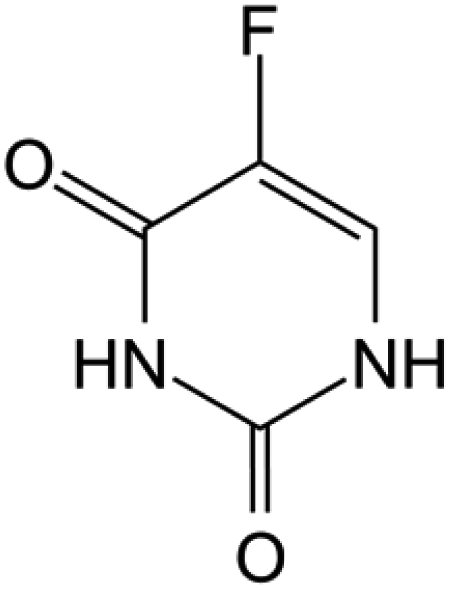
| Fluorouracil |
|
FDA approved (1962) | • Uracil modification by F-halogenation | • Colon, esophageal, stomach, pancreatic, breast, and cervical cancers | • Enzymatic inhibition | 37 |
| EMA approved (NA) | ||||||
| Floxuridine |
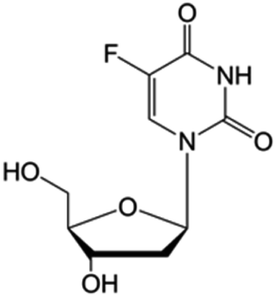
|
FDA approved (1970) | • Cytosine modification by F-halogenation | • Colon cancer | • Enzymatic inhibition | 38 |
| • DNA synthesis inhibition | ||||||
| Cytarabine |
|
FDA approved (1969) | • Replacement of the ribose sugar ring with an arabinose | • Acute myelogenous leukemia | • DNA polymerase inhibition | 5 |
| EMA approved (2001) | • Lymphoblastic leukemia | |||||
| Gemcitabine |
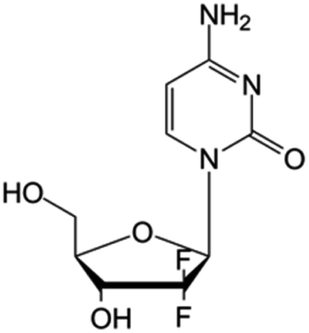
|
FDA approved (1996) | • Sugar ring F-halogenation | • Pancreatic, lung, breast, and bladder cancers | • Enzymatic inhibition | 7 |
| EMA approved (2008) | ||||||
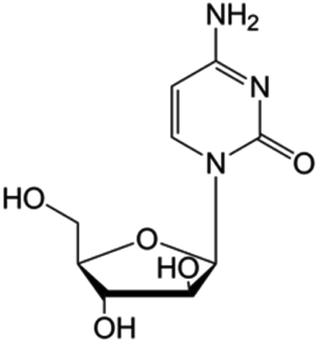
| Azacitidine |
|
FDA approved (2004) | • Cytosine modification by azotation | • Myelodysplastic syndromes (MDS) | • DNA hypomethylation | 39 |
| EMA approved (2008) | • Acute myeloid leukemia (AML) | |||||
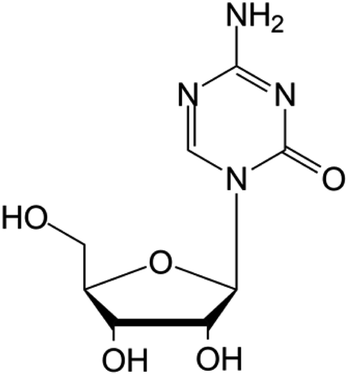
| Decitabine |
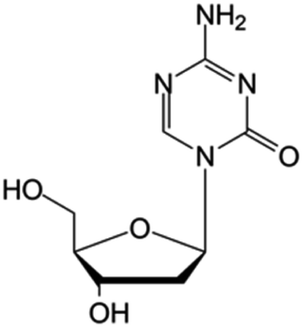
|
FDA approved (2006) | • Cytosine modification by azotation | • Myelodysplastic syndromes (MDS) | • DNA hypomethylation | 6 |
| EMA approved (2006) | • Acute myeloid leukemia (AML) | |||||
| Cladribine |
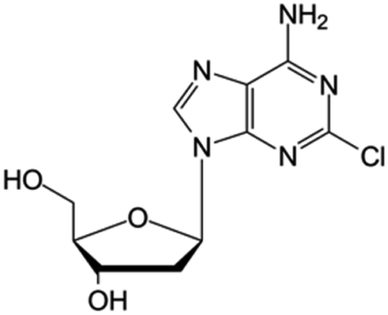
|
FDA approved (1993) | • Adenine modification by Cl-halogenation | • Hairy-cell leukemia | • Enzymatic depletion | 40 |
| EMA approved (2004) | • Non-Hodgkin lymphoma | • ATP accumulation | ||||
| Fludarabine |
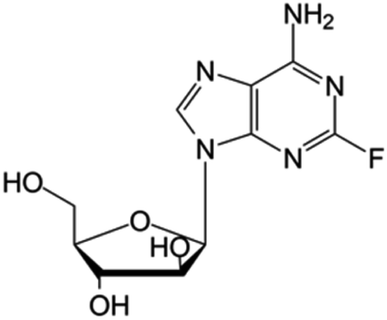
|
FDA approved (1991) | • Adenine modification by F-halogenation | • Chronic lymphocytic leukemia | • Nucleic acid synthesis inhibition | 41 |
| EMA approved (2014) | • Replacement of the ribose sugar ring with an arabinose | • Enzymatic inhibition | ||||
| Nelarabine |
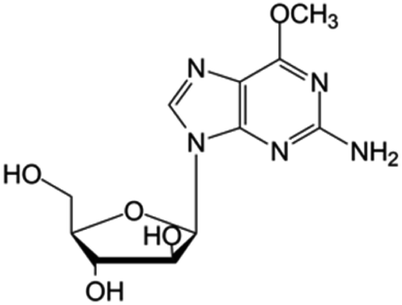
|
FDA approved (2005) | • Guanine by modification of a functional group | • T-cell acute lymphoblastic leukemia and lymphoma | • DNA synthesis inhibition | 42 |
| • Sugar ring is an arabinose instead of ribose | ||||||
| Name | Structure | Statusa | Chemical modification | Targeted cancer | Mechanism of action | |
|---|---|---|---|---|---|---|
| a Trial status was verified by the NIH – U.S. National Library of Medicine clinical trials database (http://www.clinicaltrials.gov). | ||||||
| Tezacitabine |
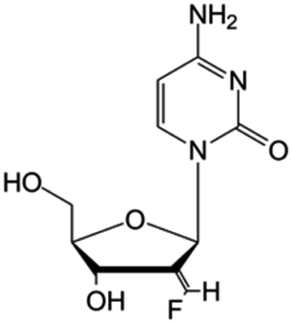
|
Phase I/II trials | • Sugar ring modification by fluoro-methylene-halogenation | • Refractory solid tumors | • Enzymatic inhibition | 43 |
| Troxacitabine |
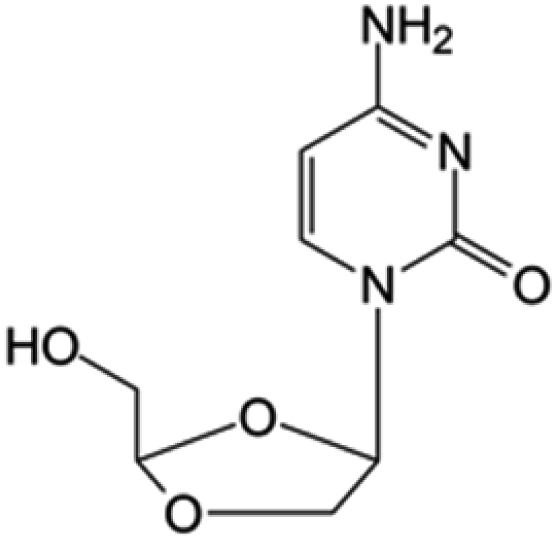
|
Phase I/II trials | • Sugar ring modification by dihydroxylation | • Acute myelogenous leukemia (AML) | • Inhibitor of DNA polymerases | 44 and 45 |
| • L-Enantiomer | ||||||
| rx-3117 |
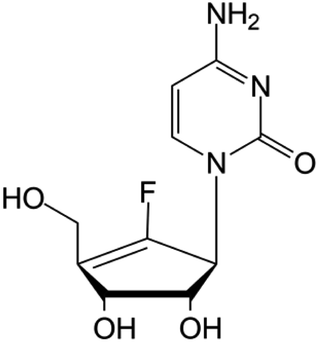
|
Phase I/II trials | • Sugar replacement by a cyclopentene ring and F-halogenation | • Metastatic bladder cancer | • DNA hypomethylation | 46 and 47 |
| • Metastatic pancreatic cancer | ||||||
| 8-Chloroadenosine |
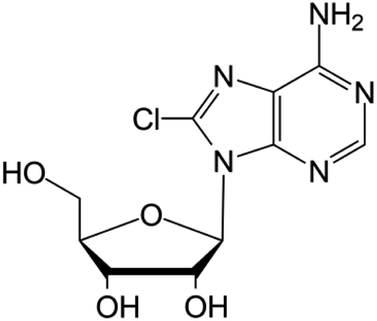
|
Phase I/II trials | • Adenine modification by Cl-halogenation | • Recurrent acute myeloid leukemia | • Enzymatic inhibition | 48 |
3.2. Mechanisms of action
In the course of cancer treatment, the analogs function as antimetabolites, via mimicking the metabolic pathway of the intrinsic nucleotides. Nucleosidic transporters facilitate the entry of selected nucleoside analogs (NAs),15 and they are phosphorylated by several kinases: 5NT – 5′-nucleotidase, nucleoside monophosphate kinase (NMPK), nucleoside diphosphate kinase (NDPK), and finally creatine kinase or 3-phosphoglycerate kinase, leading to the building up of di- and tri-phosphorylated nucleoside analogs in cancer cells. These molecules will then act, after obtaining their active forms as mono-, di- and tri-phosphorylated nucleosides, by inhibiting intracellular enzymes, like polymerases or ribonucleotide reductase (RNR), as well as by being assimilated into newly synthesized DNA and RNA. The assimilation of nucleoside or nucleotide analogs into DNA may induce the termination of chain elongation16 (Fig. 5). An in-depth knowledge of the mechanism of action of currently used compounds is of great value for the development of novel molecules. These data have led to the development of molecules that act independently of membrane transporters or activating kinases and are less susceptible to degradation. A better understanding of the mechanism of action of these compounds will also contribute to the rational development of synergistic combinations of nucleoside or nucleotide analogs with drugs that have different and/or complementary action.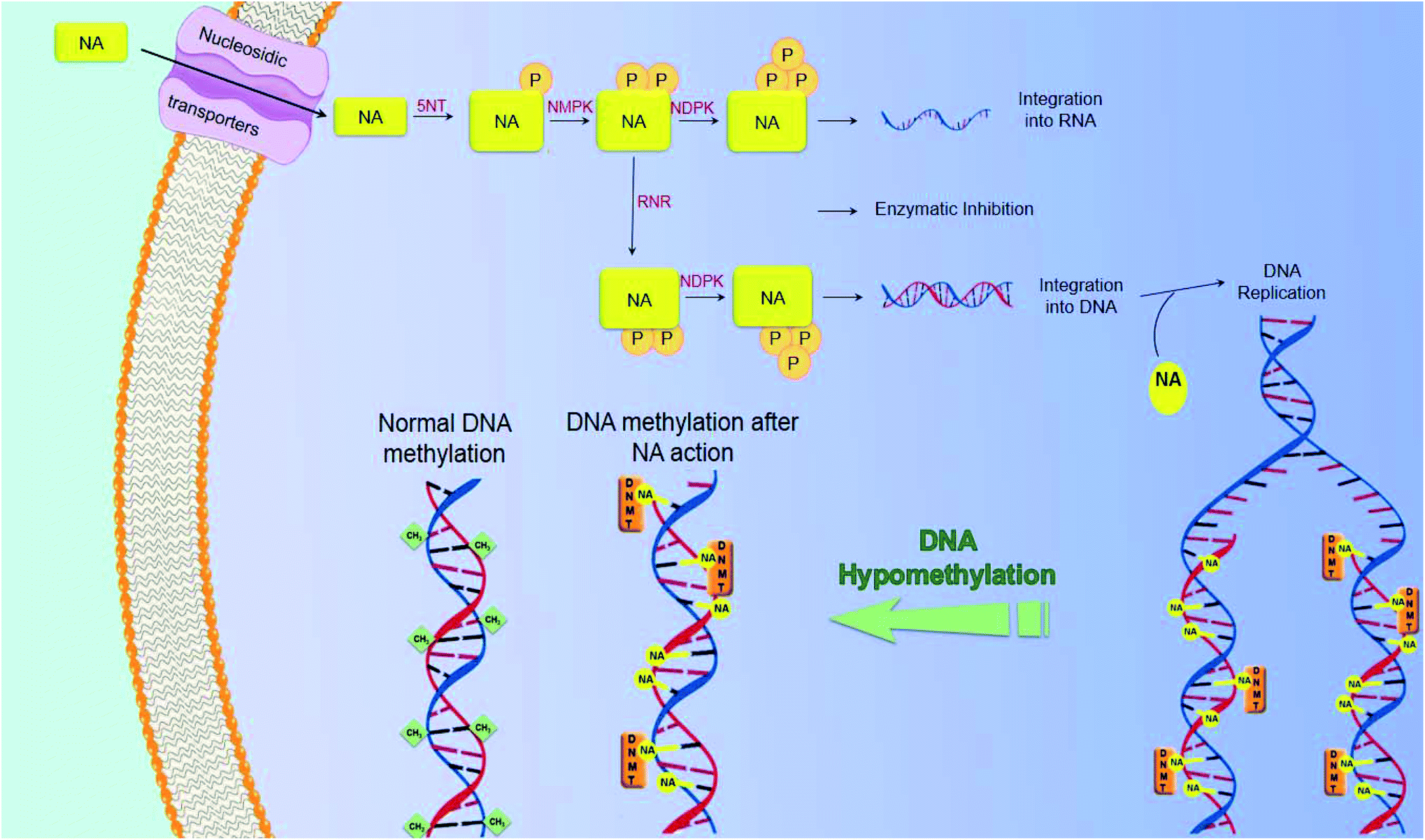 | ||
| Fig. 5 Mechanism of action of nucleosidic analogs. | ||
3.3. Mechanism of resistance
It is vital that a deeper understanding of the mechanism of resistance towards nucleoside and nucleotide analogs is reached.Three broad mechanisms of resistance arise after considering cell line and clinical studies.
The first resistance mechanism emerges from a lack in the intracellular levels of nucleic acid triphosphates owing to either a decreased level of activating enzymes, insufficient cellular uptake of the therapeutic molecule, or enhanced nucleic acid degradation due to higher levels of 5-nucleotidase or deaminases.23,24 Such processes have been deeply observed in the case of cancer resistance to cytarabine.25–27
The second resistance mechanism arises from an insufficient alteration to the DNA strands, mainly as a result of inadequate ribonucleotide reductase inhibition, as observed by Heinemann et al. in the case of gemcitabine resistance.28,29
The third resistance mechanism results from a decreased apoptosis induction. It comes as a result of an increase in the levels of pro-apoptotic molecules such as Bax, coupled with a decrease in the levels of anti-apoptotic molecules like Bcl-2: these level alterations occur when DNA damage is detected leading to a surge in the expression of the p53 gene. Building on this, several studies have uncovered that mutations in the p53 gene in different cancers have given rise to nucleoside analog resistance, namely showing that the fludarabine resistance gained by chronic lymphocytic leukemia cells was due to mutations in the p53 gene.30–33
Though most causes behind cancer resistance to nucleoside and nucleotide analogs seem to fall under one of these chief mechanisms, different and less frequent mechanisms do occur in several odd cases and have been extensively compiled by Tsesmetzis et al.34
Several approaches have risen to bypass these resistance mechanisms including restrictions in nucleoside transport and metabolism, such as masking the negative charge of the molecule thus resulting in neutral and membrane-permeable nucleotides. Indeed, nucleotide and nucleoside analog-based prodrugs such as capecitabine35,36 and other similar molecules have been developed and are undergoing continuous studies and clinical trials.
4. The prodrug approach
The term prodrug was introduced 50 years ago in 1958 when Adrien Albert suggested that the prodrug approach could be used to alter certain drug properties or drug toxicity.49A prodrug is a biologically inert, small molecule that is metabolized in vivo through a spontaneous process (e.g. hydrolytic degradation) or via a bio-catalytic mechanism to release the pharmacologically active molecule near the targeted tissue, consequently allowing a specific release of the active entity at its target site: among different strategies to improve the selectivity of chemotherapy drugs, the targeted prodrug strategy is a particularly favorable approach for highly selective chemotherapy.
The general design of a prodrug is depicted in Fig. 6(a). A chemotherapy prodrug may contain four components: the parent drug or its derivative that exhibits the pharmacological effect; a metabolically labile chemical linker which binds the functional group of the parent drug (hydroxyl, carboxylic, amine, carbonyl, phosphate groups, etc.) to the rest of the prodrug designated as the “promoiety”; a polymer spacer, or an enzymatically cleavable spacer that can release the parent drug in the presence of a tumor-specific enzyme; and an optional targeting moiety for specific delivery to tumor cells.14,15,50 With that in mind, there exist several key points to consider going into the design process:
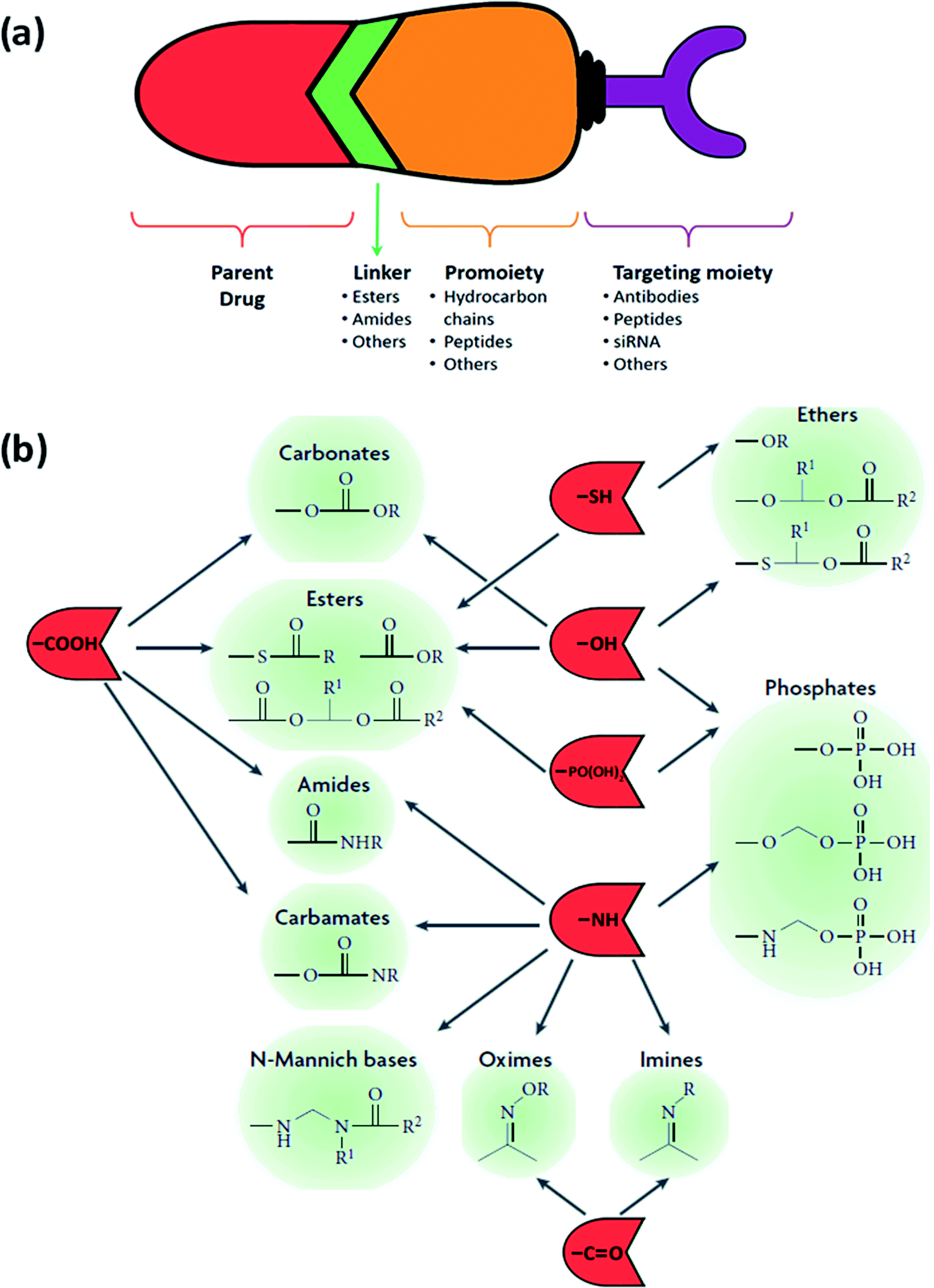 | ||
| Fig. 6 (a) The general design of a prodrug. (b) Functional groups on parent drugs that are used in prodrug design (shown in red) and conjugated to the desired promoieties using compatible linkers (shown in green). Adapted from ref. 53. | ||
• Parent drug: what are the functional groups present in this molecule that will allow a chemical prodrug derivatization?
• Promoiety: choosing a molecule that is safe and is readily excreted from the body. The disease state, dose and the duration of therapy should also be taken into account.
• Parent and prodrug: an expansive understanding of the pharmacokinetics is crucial, especially absorption, distribution, metabolism, and excretion.
• Degradation by-products: a thorough consideration of this category is crucial as these by-products and their interactions can affect chemical and physical stability and lead to the formation of new degradation products.
Given their fundamental characteristic, the inert pre-metabolism form, prodrugs improve the pharmacological properties of their active parent molecule through a simple chemical modification. The objective behind the traditional prodrug approach is to improve solubility in water or diffusion through the lipid membrane, chemical stability, oral or local absorption, brain permeability, and to reduce unacceptable taste, irritation or pain, pre-systemic metabolism, and toxicity.
Prodrugs allow the targeting of specific antigens, peptide transporters, or enzymes that are over-expressed on tumor cells or the microenvironment: it can be achieved by conjugating a tumor-specific ligand or a polymer to the chemotherapy drug via a cleavable linker.51
4.1. Classification based on linkers
The choice of the functional linking group between the promoiety and the parent drug has an imperative impact on the design of the prodrug. To ensure that the active drug will be released in the desired area and under the desired conditions, this functional group should be enzymatically/chemically cleavable or be self-immolative (pH). This will occur either at the surface of the cancerous cells owing to a specific ligand–receptor combination or upon reaching and interacting with the tumor's microenvironment, due to a change in the pH or the presence of an overexpressed enzyme. As the linker should stay intact in the course of its circulation until it arrives at the desired site, its stability is of vital significance. Therefore, a handful of functional groups that present such characteristics are used in the design of prodrugs (Fig. 6(b)), with the two main and heavily utilized families being esters and amides.A variety of links can be established, as they are usually introduced via conjugation using charged groups such as phosphates or phosphate salts, hemi-esters, and sulfates. These functional groups can be easily hydrolyzed by the action of sulfatases, ubiquitous esterase, and phosphatases usually present in the liver, blood and other tissues and organs, permitting the release of the active parent drugs. The ester bond's half-life can vary from several minutes to several hours depending on the accessibility of this bond determined by the environmental conditions and differences in the structure of the prodrug.28,29
In the case of carboxylic acid esters, the precise prediction of pharmacokinetic characteristics in humans is very challenging, due to the variation between humans and other species used in the preclinical studies.54,55
Concerning phosphate ester prodrugs, they are mainly employed in the augmentation of the aqueous solubility of the parent molecule, as the presence of the dianionic phosphate promoiety increases the aqueous solubility, thus improving the oral or parenteral administration. Namely, drugs with a poor water solubility stemming from their hydroxyl and amine functional groups are the target of such an approach. They exhibit an improved chemical stability and a swift metabolism back to the parent drug by phosphatases present in the liver or the intestinal brush border. Furthermore, as phosphate esters are hydrolyzed by alkaline phosphatases at analogous rates in humans and other species used in the preclinical studies, they gain an advantage over carboxylic acid esters.53,56,57
Alternatively, carbamates are derivatives of carboxylic acids and amines, while carbonates are derivatives of carboxylic acids and alcohols, and both have a greater enzymatic stability when compared to other esters, due to the heteroatoms on both sides of the carbonyl group. However, they are more vulnerable to hydrolysis than amides.33,34
4.2. Perks of the prodrug approach
The prodrug approach has proven to be a great aid in overcoming the drawbacks of traditional drugs in the pharmacokinetic and pharmacodynamic domains, for instance poor stability and solubility, insufficient blood–brain barrier permeability, deficient oral absorption and early metabolism.62When it comes to nucleoside analogs, their therapeutic effect relies on successive phosphorylation via different kinases until they reach their triphosphate form. But this process is kinetically led at the first phosphorylation step. Thus, nucleoside analogs regularly either have phosphonate present or are delivered as their monophosphorylated form.64
However, this approach has been widely applied to pronucleosides and pronucleotides in the anti-viral domain (the work of Hostetler et al. is a prime example65 where traditional anti-viral nucleosides underwent esterification with an alkoxyalkyl group), essentially masking them as lysophospholipids, allowing these molecules to be readily taken up in the gastrointestinal tract and improving their circulation time in plasma. The active metabolite similarly has a longer half-life within cells, allowing intermittent dosing. Limited research in the domain of anti-cancer pronucleosides exists, though the work of Perigaud et al. on gemcitabine phosphoester prodrugs offers great insights.66,67
By masking the polar groups of the parent drugs, the lipophilicity of these molecules is enhanced. Hydrophilic groups like carboxyl, phosphate, hydroxyl, or amine groups have been conjugated to more lipophilic alkyl or aryl esters or N-acyl derivatives, which under the action of peptidases or esterases are readily hydrolyzed back to the parent.16
Vig et al. explored this point, where amino acid ester prodrugs of floxuridine were developed to target the oligopeptide transporters that are greatly expressed in the epithelial cells constituting the intestinal brush border membrane and are involved in the transport of ACE inhibitors, di and tri-peptides and an assortment of other drugs across the intestine.69
Zuwala et al. worked on the antiretroviral nucleosidic analogue agent azidothymidine, and they showed that the conjugation of azidothymidine to poly(methacrylic acid) leads to an increased duration of action when compared to the free azidothymidine.72 Though this work is against viral diseases, the advantages gained can be readily translated to impact the cancer field.
Xu et al. utilized the increased presence of the cathepsin B enzyme in the pancreatic cancer tissue to develop a cholesteryl hemisuccinate-1 gemcitabine prodrug that will release the gemcitabine mainly at the cancer site after contact with cathepsin B that will cleave the amide bond.75
4.3. Advancements in pro-nucleosides for cancer treatment
Arising from a need to exploit the mentioned advantages of prodrugs, a great deal of research has been performed on nucleoside analog based prodrugs. Given that nucleotides and nucleosides are hydrophilic in nature, this gives rise to several limitations including restricted diffusion, limited cellular internalization and a rapid enzymatic degradation. Thus nucleoside analog based prodrugs arise from a need to improve the drug's diffusion and to protect it from metabolic degradation via its conjugation to a hydrophobic moiety thus giving an amphiphilic prodrug.76–78 Capecitabine is the only FDA approved pro-nucleosidic drug since 1998 with an amide bond linker joining the parent drug based on fluorouracil to a chloroformate based promoiety: it is used for the treatment of colonic, breast, colorectal, and stomach neoplasms. Other prodrugs are in clinical trials such as guadecitabine (SGI-110) synthesized by the conjugation of decitabine to a deoxyguanosine via a phosphodiester bond,79 being tested in phase III trials on different cancers including acute myeloid leukemia (AML) and myelodysplastic syndrome. A selection of such promising prodrugs is presented in Table 3; these prodrugs use several types of the mentioned linker, and improve upon the parent drug by the addition of various perks.| Name | Structure | Statusa | Parent drug | Linker | Targeted cancer | |
|---|---|---|---|---|---|---|
| a Trial status was verified by the NIH – U.S. National Library of Medicine clinical trials database (http://www.clinicaltrials.gov). Approval status was verified by the European Medicines Agency (http://www.ema.europa.eu) and the US Food and Drug Administration (http://www.fda.gov). | ||||||
| Capecitabine |

|
FDA approved (1998) | • Fluorouracil | • Amide bond | • Colonic neoplasms | 36 |
| EMA approved (2001) | • Breast neoplasms | |||||
| • Colorectal neoplasms | ||||||
| • Stomach neoplasms | ||||||
| Thymectacin |
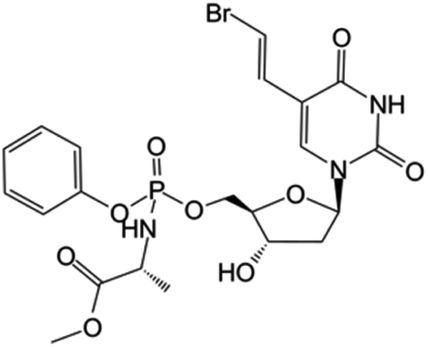
|
Clinical trials | • Brivudine | • Phosphate ester bond | • Cancers overexpressing the thymidylate synthase enzyme | 80 and 81 |
| Phase I/II | ||||||
| LY2334737 |
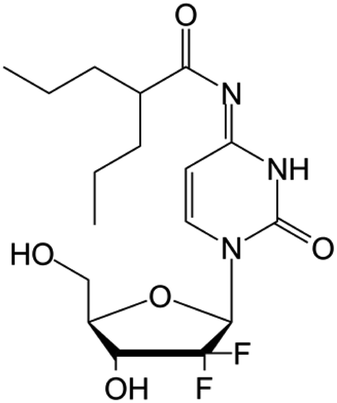
|
Clinical trials | • Gemcitabine | • Amide bond | • Malignant solid tumors | 82 |
| Phase I | • Metastatic tumors | |||||
| CP-4126 |
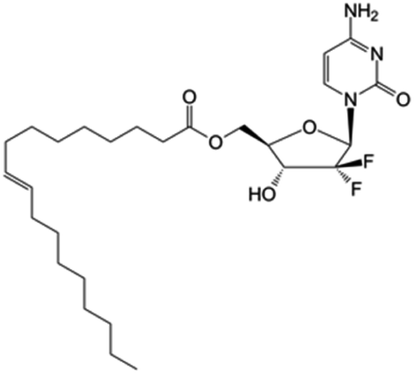
|
Clinical trials | • Gemcitabine | • Carboxyl ester bond | • Advanced adenocarcinoma of pancreas | 83 |
| Phase I/II | ||||||
| Nuc-1031 |
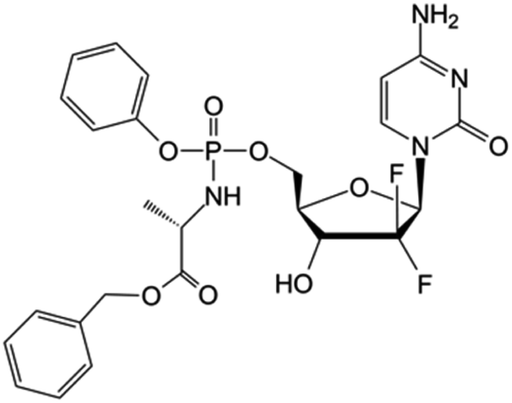
|
Clinical trials | • Gemcitabine | • Phosphate ester bond | • Ovarian cancer | 84 |
| Phase I/II/III | • Biliary tract cancer | |||||
| • Gallbladder cancer | ||||||
| • Cholangiocarcinoma | ||||||
| • Ampullary cancer | ||||||
| Sapacitabine |
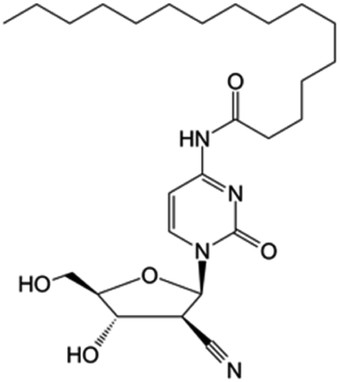
|
Clinical trials | • Cndac (deoxycytidine analog) | • Amide bond | • Myelodysplastic syndromes | 85 |
| Phase I/II/III | • Acute myeloid leukemia | |||||
| Guadecitabine (SGI-110) |
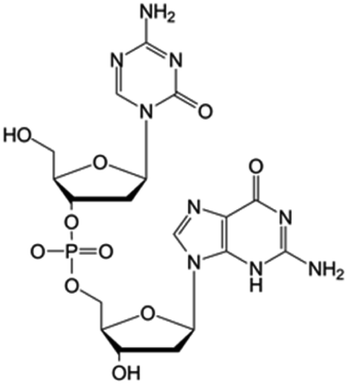
|
Clinical trials | • Decitabine | • Phosphate ester bond | • Small cell lung cancer | 79 and 86 |
| Phase I/II/III | • Ovarian cancer | |||||
| • Hepatocellular carcinoma | ||||||
| • Acute myeloid leukemia | ||||||
| ACV-TP-T |
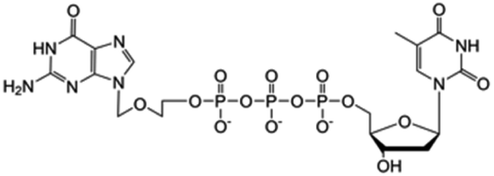
|
Preclinical | • Acylcovir | • Phosphodiester bond | • Pancreatic tumor | 87 |
4.4. The drawbacks of the prodrug approach
Prodrugs in numerous cases are hindered at several levels, as some need specific enzymatic cleavage, chemical activation or a specific transport mechanism in order to achieve sufficient intracellular delivery. Adding to that, the presence of esterases in the serum or tumor microenvironment leads to rapid degradation of the said prodrugs before gaining entry into the cells. Additionally, despite having enhanced levels of phosphorylated nucleoside analogs released from the prodrugs inside the cell, an efficient cytotoxic concentration isn't achieved owing to the intensive catabolic processes present, such as increased 5-nucleotidase activity. Consequently, despite being an enhanced approach in comparison to traditional drugs, the prodrug approach is far from perfect and needs further innovation to reach the therapeutic level needed to elevate the medical health domain.5. The nanoparticle approach
Setting a fixed definition for nanoparticles is tricky, as the size varies between different sources ranging from a few nanometers up to anything less than a micrometer. Not to mention, for some, one dimension at the nanoscale is enough while for others it should cover two or all three dimensions.In 2011 the Commission of the European Union defined a stricter definition than was previously approved: under that definition a nano-object needs only one of its characteristic dimensions to be in the range 1–100 nm to be classed as a nanoparticle, even if its other dimensions are outside that range.88,89
This ever-evolving field of nanomedicine has progressed through four generations thus far, each improving on the previous one, starting with the first generation in the early 1970s that included liposomes, micelles and polymer nanoparticles that were made of biodegradable excipients able to passively circulate with an improved intracellular delivery and accumulate in the liver, but weren't specific to a targeted tissue and were readily eliminated by macrophages (opsonization). The second generation, two decades later utilized the physico-chemical concept of the steric repulsion to enhance upon the first generation of nanocarriers with the introduction of a new molecule, PEG (polyethylene glycol), thus taking advantage of the “Enhanced Permeability and Retention” (EPR) effect, and allowed the targeting of tumors and other inflammatory diseases. Later, the third generation of nanocarriers arrived with the decoration of the nanocarrier surface with different bio-recognition modalities providing them with additional functionalities including evasion of the immune system (stealth) and the extension of the blood circulation duration aiming to accumulate in the targeted tissue, as well as an improved targeting ability. Recently, the fourth generation provided even more advanced functionalities with stimuli-responsive nanomedicine, such as silicon nanocarriers that are able to deploy multiple waves of nanoparticles and to stimulate a signaling or biological cascade.90–93
The small size of nanoparticles is of great benefit in the medical domain, allowing them to diffuse through the endothelium, enter cells or have a premeditated ability to bind to specific cells. Such properties have ushered novel advancements in the field of nanomedicine via the development of novel drug delivering techniques, for instance by providing local heating (hyperthermia),94 blocking vasculature to diseased tissues and tumors,95 or via transportation of drugs.96
5.1. Classification of nanoparticles
Nanoparticles can be classified into any of various types depicted in Fig. 7, depending on their size, shape, and material properties. Other classifications distinguish organic, inorganic and metal–organic excipients used to formulate (i) organic nanoparticles including liposomes, micelles, polymeric nanoparticles or dendrimers, (ii) inorganic ones encompassing quantum dots and calcium phosphate among others, and (iii) metal–organic framework nanoparticles (MOFs).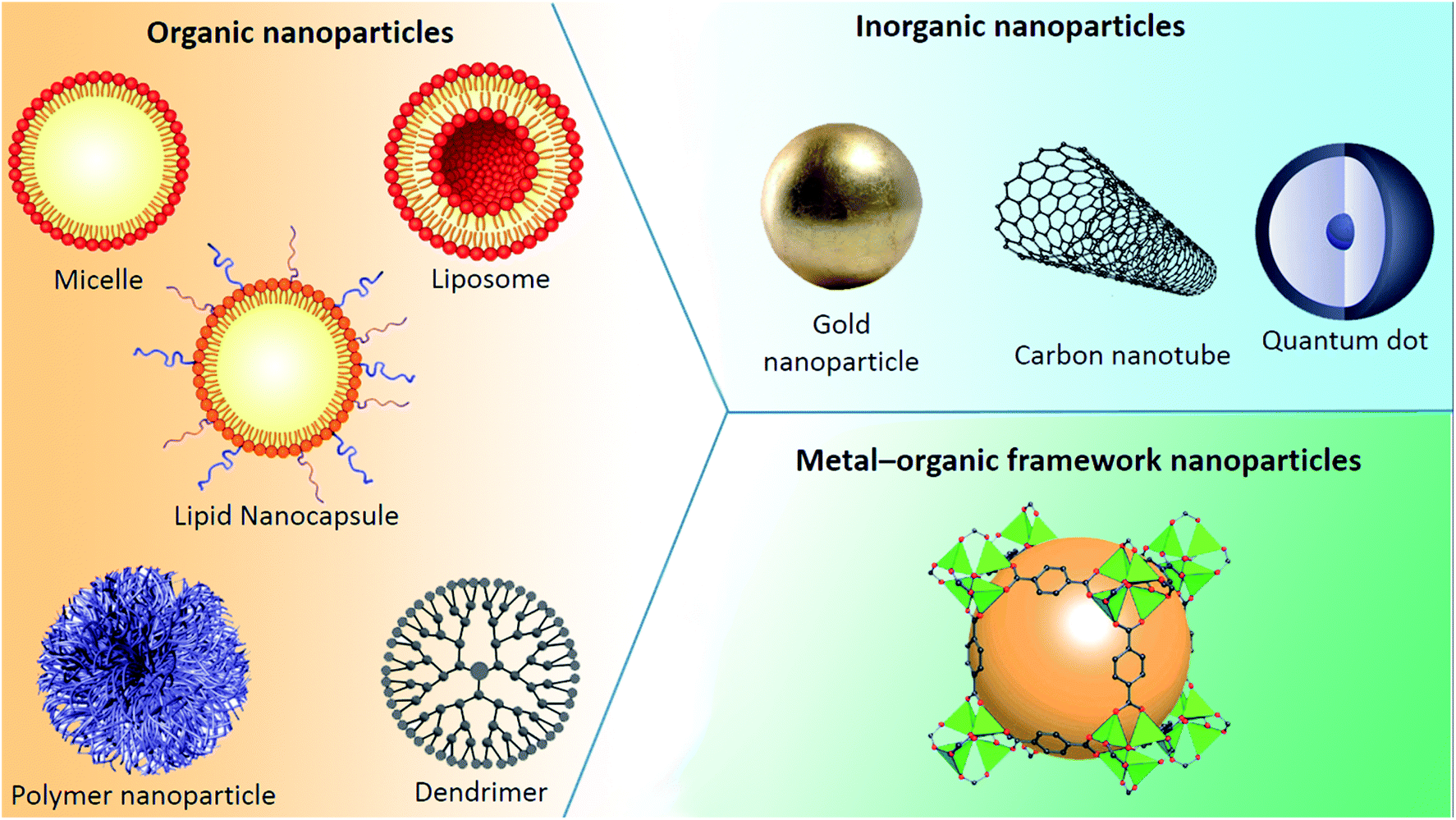 | ||
| Fig. 7 Various types of nanoparticle. | ||
5.1.1.1. Liposomes and lipid nanocapsules. Bangham and his coworkers are credited with the discovery of liposomes during the 1960s:97,98 various studies then followed to characterize them, with alternative methods for the preparation of liposomes being discovered between 1975 and 1985. Subsequently, the study of liposomes spread to different scientific areas: biophysics (study of cell membrane properties), chemistry (energy conversion), colloid science (study of stability and thermodynamics of finite systems), biochemistry (membrane protein functions) and biology (study on cellular trafficking) cumulating in the introduction of the first nanocarrier drug: a doxorubicin HCl liposome injection (commercial name: Doxil® (FDA approved in 1995) or Caelyx® (EMA approved in 1996)).99
Liposomes are made up of lipids with a hydrophilic tail and a hydrophobic head, creating an amphiphilic molecule that aggregates to form spherical liposomes, due to the interactions occurring upon contact with water. Liposomes have an aqueous core enclosed with a lipid bilayer.
Liposomes, via the modification of drug absorption, reduction of metabolism and the prolongation of the drug's half-life, are able to enhance the efficacy of conventional drugs. Upon encapsulation, the drug distribution ceases to be determined by the physico-chemical characteristics of the original drug molecule, and becomes reliant on the characteristics of the carrier. These factors, along with the ability of liposomes to solubilize both hydrophilic and hydrophobic molecules, secure liposomes as a potent drug delivery platform with application in various diseases such as cancer,100 and fungal101 and bacterial infections.102
When it comes to lipid nanocapsules (LNCs), Heurtault and her coworkers103 are credited with the discovery of this nanocarrier. The size of LNCs ranges from 20 to 100 nm. Compared to liposomes which require an organic solvent for formulation, LNC formulation is solvent-free, producing a carrier with enhanced stability (physical stability up to 18 months). LNCs have a membrane consisting of a combination of lecithin (a phospholipid) and a PEGylated surfactant. This membrane encloses an oily core composed of medium-chain triglycerides and the formulation is founded on the phase-inversion temperature. Owing to these elements, LNCs are characterized by a hybrid structure between polymer nanocapsules and liposomes.104–107
The two drugs that have been approved by the FDA based on the encapsulation approach of nucleosidic analogs are DepoCyt™ based on the encapsulation of cytarabine (approved in 1999), and Vyxeos (cpx-351) based on a double encapsulation of cytarabine and daunorubicin (approved in 2017), and are used in the treatment of lymphomatous meningitis and acute myeloid leukemia, respectively. Both of these drugs use liposomes as the nanocarrier with varying encapsulation efficiency.108–112 Several other encapsulated nucleosidic analogs for cancer treatment in different development and testing stages have been presented in Table 4.
| Name | Active molecule | Statusa | Nanocarrier | Targeted cancer | Encapsulation efficiency | |
|---|---|---|---|---|---|---|
| a Trial status was verified by the NIH – U.S. National Library of Medicine clinical trials database (http://www.clinicaltrials.gov). Approval status was verified by the European Medicines Agency (http://www.ema.europa.eu) and the US Food and Drug Administration (http://www.fda.gov). | ||||||
| Vyxeos (cpx-351) | • Cytarabine | • FDA approved (2017) | • Liposome | • Acute myeloid leukemia | • 5% | 108 and 109 |
| • Daunorubicin | • EMA approved (2018) | |||||
| • Clinical trial phase IV | ||||||
| DepoCyt™ | • Cytarabine | • FDA approved (1999) | • Liposome | • Lymphomatous meningitis | • 86.30% | 110–112 |
| • EMA approved (2008) | ||||||
| • Withdrawn | ||||||
| Gemcitabine-loaded liposomes | • Gemcitabine | • Preclinical | • Liposome | • Anaplastic thyroid carcinoma | • 90% | 156 |
| GemC16-loaded 2N-LPs | • Gemcitabine | • Preclinical | • Liposome | • Pancreatic cancer | • 97.3% | 157 |
| • Loading capacity (8.9%) | ||||||
| Decitabine LNCs | • Decitabine | • Preclinical | • Lipid nanocapsules | • Acute myeloid leukemia | • 88% | 158 |
5.1.1.2. Polymeric nanoparticles (PnPs). Polymers are macromolecules consisting of successive repeating units arranged in a chain-like formation that can take various shapes and can be biodegradable or not according the use desired. Polymeric nanoparticles are being used in drug delivery, providing potential protection of the active agent, a sustained release and even an active targeting approach with a surface modification of the polymeric bone. Polyethylene glycol (PEG) may well be the most well known and frequently used synthetic polymer, owing to the properties it imparts on the nanoparticles such as “stealth”, since the densely packed hydrophilic PEG diminishes the interparticle attractive forces and causes steric repulsion to the surrounding biological components, for instance plasma proteins and antibodies: it stops phagocytosis and decreases clearance from the bloodstream or body.113–115
In his study Y. Haggag et al. encapsulated 5-fluorouracil (5FU) within a polymeric nanoparticle. The PnP formed had a high drug loading, a particle size of 185 nm, with a sustained drug release for 7 days. The in vivo results confirmed the enhanced anticancer activity of the 5FU-loaded NP. Histopathological examinations displayed the damage to tumor tissue after NP treatment. Finally, the PnP was found to be less toxic to liver and kidney tissues compared to the free 5-fluorouracil.116
5.1.1.3. Dendrimers. Dendrimers are characterized by a symmetric core surrounded by an inner and outer shell. At a molecular level, the dendritic branching forms globular structures. A typical dendrimer is made of three different parts, a central core made out of a single atom or an atomic group with a minimum of two identical chemical functions, repeating units forming several interior layers, and various peripheral functional groups.117
Diverse dendrimers exist, with unique biological properties, allowing them to be used as drug delivery agents for the treatment of different diseases such as HIV118,119 and cancer, or as contrast agents with theranostic applications for imaging techniques such as Magnetic Resonance Imaging (MRI).120,121
Concerning the use of dendrimers as nucleoside analogue carriers, Szulc A. et al. have accomplished several strides. Their studies demonstrate that maltose and maltotriose modified poly(propylene imine) dendrimers (PPI dendrimers) are a potential carrier for triphosphate forms of nucleoside analogue drugs. These dendrimers have been chosen owing to their biopermeability, non-toxicity, non-immunogenicity and ability to stay in the blood circulation.122–126
Also worth mentioning is the research of M. Gorzkiewicz and coworkers, where dendrimers served as nano-carriers for fludarabine, while simultaneously improving its cellular entry and enabling both the direct and phosphorylation-independent toxicity.127
5.1.2.1. Carbon nanotubes (CNTs). Carbon nanotubes (CNTs) are cylindrical molecules constructed from carbon atoms forming graphene sheets rolled into a cylindrical shape that can have open or capped endings, with diameters of merely 1 nm but a length of several micrometers.128,129 With such a high surface area, conjugation with different therapeutic molecules is possible upon their functionalization with specific groups able to bind the desired molecule or similarly to Qds seek a specific target on a cell or tissue.130,131
Following Zheng et al.'s discovery of the hybridization phenomenon of single-stranded DNA molecules and CNTs (single-walled carbon nanotubes) in 2003,132 much research has been done in this field in both the theranostic and pharmaceutical domains.135,136 Remarkably, Zhang P. et al.'s work on gemcitabine–lentinan–carbon nanotubes revealed that the combination of drug therapy and near-infrared photothermal therapy possesses great synergistic antitumor efficacy.135
5.1.2.2. Quantum dots. Quantum dots (Qds) are nanocrystals made from semiconducting materials ranging in diameter between 2 and 10 nm, lending them a quantum effect owing to their extremely small size. Meaning that electrons inside the dots are confined and can only occupy defined energy levels. Upon the excitation of these electrons by an external energy source, an intense response from these Qds follows.
Building upon these properties, quantum dots can be enclosed within a shell able to mimic the receptors on the cells. The quantum dots will then track down and attach to the specific receptor. Then, relying on the fluorescent nature of quantum dots the site of concern is made evident. Only a limited number of receptors are needed on the surface of the dot, and when compared to the surface area of the dot, it leaves enough space to place other things such as drugs, allowing for instance quantum dots to find and treat cancer cells. This eludes healthy cells and therefore the side effects associated with cancer treatments will decrease.136–139
Gemcitabine-Qds have been approached by several teams and showed promising results including a high drug loading capacity, reduced cytotoxicity towards normal cells and tissues, a good drug release profile, a higher cellular cytotoxicity and more effective inhibition capacity when compared to free gemcitabine.140,141
5.1.2.3. Gold nanoparticles. Gold nanoparticles (GNPs) are particles with a diameter of 1 to 100 nm and have been reported as potential carriers to small drug molecules or large biomolecules, like proteins, DNA, RNA or nucleic acids. The linkage between the nucleic acids and GNPs is usually through non-covalent bonds, which are easier to break off during cellular uptake.142,143
A prominent application of using gold nanoparticles as a carrier for nucleoside analogues is revealed by S. Song and coworkers, where the gold nanoparticles are used as a vehicle to carry a purine analog called fludarabine for the treatment of hematological cancers, showing improved efficacy when compared to free fludarabine.144
5.1.2.4. SiO2 nanoparticles. Silicon dioxide nanoparticles can be formulated to have a wide range of sizes and their surface chemistry easily modified to fit diverse applications, with mesoporous silica nanoparticles playing a great role in drug delivery and nanomedicine. Jeelani et al.'s work offers a comprehensive look into the different types and applications of silica nanoparticles.145
While in the field of cancer treatment, Azali and coworkers were able to formulate a SiO2 nanoparticle to act as a vehicle for the nucleosidic analogue 5-fluoruracil (5-FU), the aim of their study was to compare the release of 5-FU depending on the solvent medium used to load this drug into the nanoparticle. Transmission electron microscopy (TEM) showed that these nanoparticles had a hexagonal structure. Additionally, adsorption data showed that 95.27% of 5-FU were loaded into the nanoparticles. Interestingly, in ethanol medium, the release rate percentage was lower than in deionized water medium, at 9% and 23%, respectively.146
5.1.2.5. Iron oxide nanoparticles. There are several phases of iron oxide crystallites exhibited as hematite (α-Fe2O3), maghemite (γ-Fe2O3), goethite (Fe[O(OH)]) and magnetite (Fe3O4). Both hematite and magnetite are favored for biomedical applications due to their low cost and non-toxicity, including MRI contrast enhancement, detoxification of biological fluids, tissue repair, cell separation, immunoassay, hyperthermia, and drug delivery.147,148
An interesting application in the field of cancer treatment that stands out is the work of Shahabadi et al.: they were able to formulate a cytarabine-coated Fe3O4 magnetic nanoparticle with a size of 23 nm. The in vitro cytotoxic activity was investigated against HL-60, KG-1, and Raji cell lines, showing a twofold efficacy increase when compared to free cytarabine. Furthermore, in vitro DNA binding studies were carried out, showing that DNA aggregated to the nanoparticles via groove binding mode.149
Rodriguez-Ruiz et al. approached the idea of a gemcitabine loaded MOF, using a monophosphorylated form of gemcitabine that was loaded into MIL-100 iron-trimesate nanoMOFs: the anticancer effect of this nanoparticle was tested on a pancreatic PANC-1 cell line. The formulated nanoparticles acted as “nanosponges”, soaking Gem-MP from its aqueous solution, with a maximal drug loading of 30 wt%, reflecting the strong interaction between the drug and MOFs. Furthermore, these MOFs were nine times more effective against the pancreatic cancer cell line when compared to free gemcitabine.155
5.2. Drawbacks of the nanoparticle approach
The approach of using nanoparticles such as micelles or liposomes, as a means of overcoming the shortcoming present at the levels of intracellular transport or drug activation in tumor cells, does tackle a huge hurdle faced by traditional drugs. Nevertheless, the nanoparticle approach still has its limitations, for instance the drug loading capacity of these vectors is low, as the amount of the encapsulated therapeutic molecule is feeble when considering the amount needed to reach a therapeutic effect. Moreover, excipients needed to formulate nanoparticles have a certain degree of toxicity and undesirable side effects, not to mention the large size of this vector when compared to the size of the therapeutic molecule, allowing its recognition by the immune cells as a foreign body and leading to its clearance especially by macrophages. Therefore, an approach that can combine both the prodrug and nanoparticle techniques, gaining both their advantages, can potentially be the break needed to overcome cancer resistance to nucleotide and nucleoside analogs.6. Self-assembly of nucleolipid prodrugs
Molecular self-assembly is a key concept in supramolecular chemistry. The thermodynamic incompatibility between the different blocks causes a spatial organization into ordered morphologies at the nanoscale level with the production of novel structural features, as demonstrated by recent studies.159 This is because assembly of molecules in such systems is directed through noncovalent interactions (e.g., hydrogen bonding, metal coordination, hydrophobic forces, van der Waals forces, π–π stacking, and/or electrostatic interactions) as well as electromagnetic interactions. Common examples include the formation of micelles, vesicles, liquid crystal phases, and Langmuir monolayers by surfactant molecules. Further examples of supramolecular assemblies demonstrate that a variety of different shapes and sizes can be obtained using molecular self-assembly.160The hydrophobic forces result from an entropic phenomenon and trigger the self-assembly of hydrophobic moieties: the produced nanoparticles will thus be created owing to the collective weak interaction present, as nucleotides, nucleosides and their analogs will interact through π–π stacking, hydrogen bonds, electrostatic interaction, and van der Waals forces.161 Consequently, the various key factors of the self-assemblies including stability, shape, size, charge, viscosity, and surface morphology amongst others will affect their cellular internalization and diffusion, thus impacting the pharmacological activity of the new molecules.162
Therefore, building on the previous idea of the formation of a nucleoside/nucleotide based amphiphilic prodrug, an additional step can be performed via pushing the prodrugs with appropriate physico-chemical properties into self-assembly to form nanoparticles168 (Fig. 8), consequently increasing the drug's diffusion ability as well as protecting it from enzymatic degradation and boosting its bioactivity.
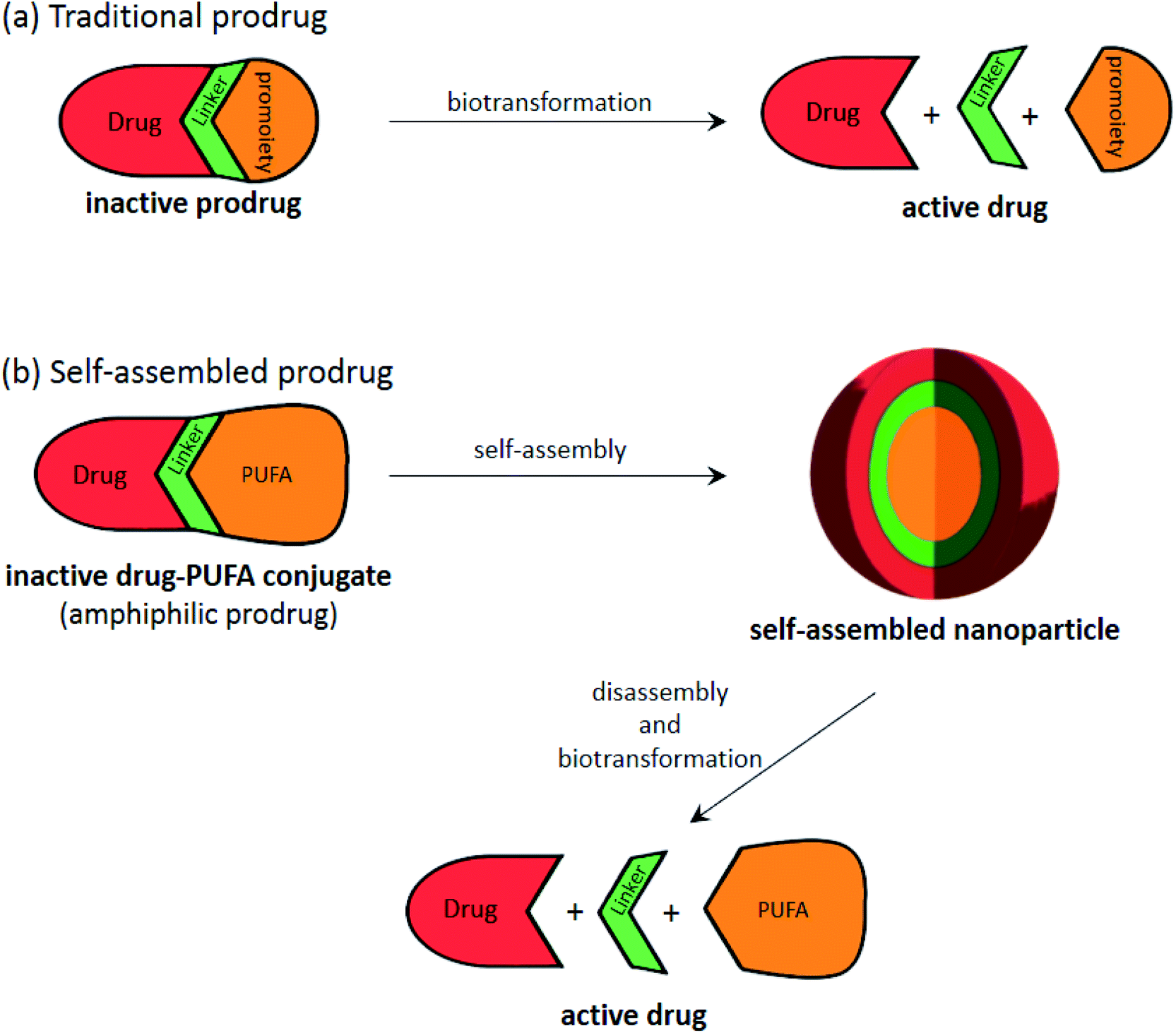 | ||
| Fig. 8 Illustration showing the general process of (a) traditional prodrug biotransformation and (b) self-assembly of prodrugs, that can be applied to “PUFAs”. | ||
Given that nucleobases, nucleosides and nucleotides have a hydrophilic nature, conjugating them to molecules having a hydrophobic nature will produce an amphiphilic molecule that is better suited for self-assembly: examples of that case are the CP-4126, capecitabine and LY2334737 prodrugs mentioned previously in Table 3, where the nucleosides have been conjugated to either a carboxylic or ester group.82 Indeed, the coupling of the nucleoside and nucleotide derivatives to a fatty acid (especially unsaturated ones) seems to be the best method to achieve an amphiphilic prodrug with self-assembling potential.
Several drugs in the preclinical stage of their development are based on this approach; for instance, DOC, a self-assembling pro-nucleosidic drug, was designed by linking clofarabine to oleic acid via an ester bond. The obtained prodrug is able, due to the reasons mentioned above, to self-assemble into spherical nanoparticles and is being tested on breast cancer.167 A selection of similar self-assembling prodrugs is presented in Table 5.
| Name | Prodrug structure | Active drug | Fatty acid | Shape | Targeted cancer | |
|---|---|---|---|---|---|---|
| 5-FCPal |
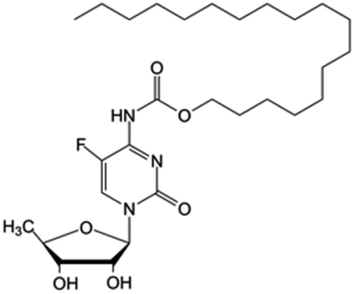
|
• Capecitabine | • Palmitic acid | • Lamellar sheets | • Breast cancer | 163 and 164 |
| LA-Gem |

|
• Gemcitabine | • Linoleic acid | • Spherical | • Pancreatic ductal adenocarcinoma | 165 |
| SQdFdC |
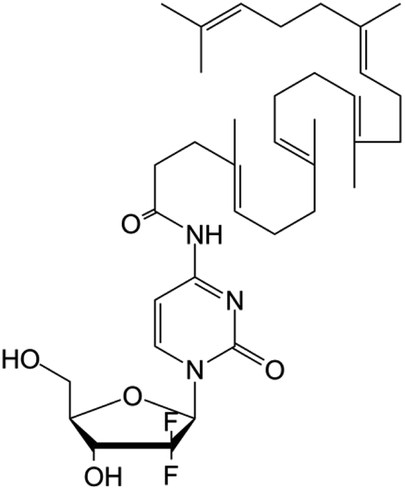
|
• Gemcitabine | • Squalenoyl | • Unilamellar vesicles | • Pancreatic cancer | 166 |
| DOC |
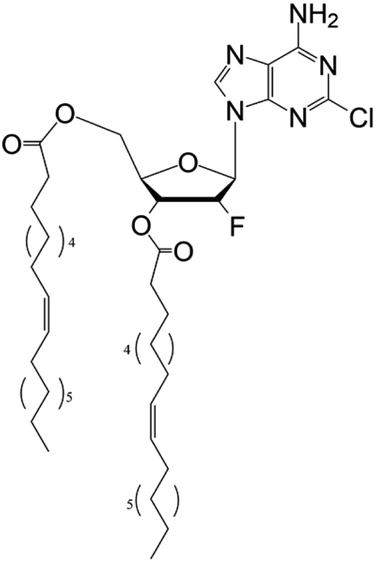
|
• Clofarabine | • Oleic acid | • Spherical | • Breast cancer | 167 |
6.1. The PUFAylation approach
Although the term “PUFAylation” was only very recently introduced by Liming Wu et al., the concept of conjugating a polyunsaturated fatty acid (PUFA) to a nucleoside leading to the spontaneous self-assembly of the created prodrug is one that has been tackled before. Certainly, this innovative strategy was established by Couvreur and colleagues, whereby squalene derivatives (a cholesterol precursor) were conjugated to nucleoside analogs in a process they termed “squalenoylation”. What is more, “PUFAylation”, while permitting this conjugate to self-assemble in water, doesn't require any excipients, an advantage it gains over traditional formulated nanoparticles, thus improving the drug tolerability in animals and demonstrating exceptional effectiveness, with an unbeatable drug loading. Moreover, this method produces prodrugs with an adequate amphiphilicity, allowing for an enhanced in vivo antitumor efficacy compared to the parent drug. When compared to “squalenoylation”, “PUFAylation” seems to have the edge, as the conversion of squalene into squalenylacetic acid is a tiresome multistep reaction, while most PUFAs are commercially accessible. Moreover, most of these “PUFAs” are essential for biological functions and are plentiful in the body meaning that they may reduce the toxicity stemming from the use of adjuvants, and finally the π–π association between PUFAs will increase the stability of the self-assemblies. Based on all that, the PUFA technology is an immensely encouraging one for the manufacturing of prodrugs and their derived self-assembled nanoparticles.162,165,166,169,170Of the several “PUFAylations” presented, the PUFAylation of the gemcitabine prodrug SQdFdC (Table 5) is a definitive example of this concept. The coupling of squalene (a cholesterol precursor) with gemcitabine leads to the spontaneous formation of nanoparticles with a diameter of 120–140 nm in an aqueous medium. The self-assemblies took the shape of an inverse hexagonal supramolecular structure, with an aqueous core that was surrounded by the gemcitabine molecules linked to the squalene moieties. This PUFAylated self-assembly has shown improved efficacy under both in vitro and in vivo conditions when compared to free gemcitabine. In in vitro experiments, SQdFdC exhibited higher cytotoxicity than gemcitabine in both human and murine resistant leukemia cell lines, while in in vivo experiments, SQdFdC revealed notably greater anticancer activity than gemcitabine against both solid subcutaneously grafted tumors and aggressive metastatic leukemia. This increased efficacy of SQdFdC can be attributed to the nanoparticle shape of the self-assembling prodrug, offering protection from deamination and a specific targeting with a prodrug cleaved by cathepsin B, overexpressed in cancer cells.171–175
Mulet et al. also present interesting early research regarding PUFAylation, comparing the molecular structure, phase behavior, and cellular uptake differences between 3′-oleoylthymidine and other non-saturated nucleolipids, showing that the presence of lyotropic liquid crystalline phases was reliant on the number of unsaturated chains. Additionally, the complexity of the ordered systems formed, from the 1D lamellar system presented here to inverse 2D hexagonal or 3D cubic phases, would lead to different bioactive release profiles.176
Wu et al. have similarly shown very promising results for their LA-Gem prodrug “PUFAylated” self-assemblies (Table 5). The self-assemblies showed a high drug loading, a limited metabolism by endogenous enzymes, an improved release and intracellular uptake. Likewise, promising in vivo results were obtained versus a pancreatic ductal adenocarcinoma (PDAC) PDX mouse model.165
Additional insights into PUFAs can be gained from the work of Maksimenko et al.; this research is an advancement on the previous work of Couvreur previously discussed, by taking the idea of “squalenoylation” and moving it to a more generalized “terpenoylation” concept. Several polyisoprenoyl prodrugs of gemcitabine were formulated, with different polyisoprenoyl chain lengths: the obtained nanoparticles were then tested in vitro on murine melanoma cell line B16F10, human pancreatic carcinoma cell line MiaPaCa-2, human lung carcinoma cell line A549 and human breast adenocarcinoma cell line MCF7. It was revealed that the anticancer efficacy of these nanoparticles was dependent on the size of the polyisoprenoyl chains: the polyisoprenoyl prodrug of gemcitabine containing three isoprene units was the most active nanoparticle on all the cancer cell lines tested. The results obtained in vivo (carcinoma xenograft model in mice) were in accordance with the in vitro results.177
6.2. Critical packing parameters dictate the self-assembled nanoparticle shape
As proposed by Israelachvili and Mitchell in the mid 1970s, the architecture of self-assemblies is predicted by the molecular shape of amphiphilic phospholipids with a critical packing parameter (CPP) defined as the equation of CPP = V/(a0lc), where V, lc and a0 are, respectively, the volume and length of the lipophilic chain, and the cross-sectional area of the hydrophilic core of the phospholipid expressed per molecule in the aggregates.178 Altering these different parameters leads to the molecule adopting a different interfacial curvature and therefore a different morphology. The values obtained by the CPP equation will influence the shape of the self-assemblies as shown in Fig. 9: spherical micelles, with high curvature, are formed when CPP ≤ 1/3, cylinders are formed in the range 1/3 < CPP ≤ 1/2, when 1/2 < CPP ≤ 1, vesicles are formed, at CPP ≈ 1 lamellar phases are obtained, and finally for CPP > 1 inverted micelles are obtained.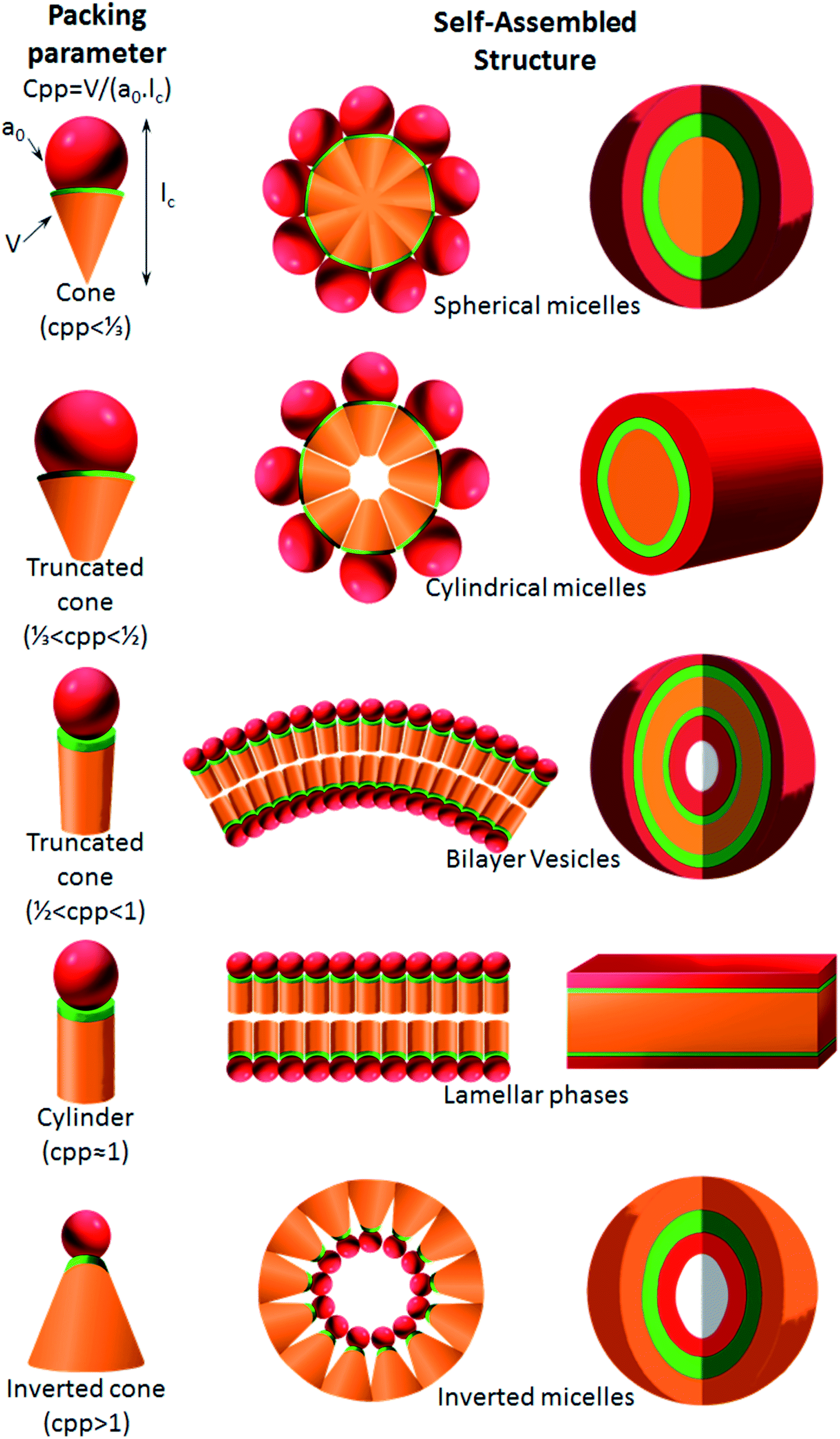 | ||
| Fig. 9 Molecular shape and critical packing parameter (CPP) of amphiphilic molecules, and the self-assembly entities formed of different amphiphiles (v, the lipophilic chain volume; a0, the cross-sectional area of the hydrophilic head group; lc, the length of the lipophilic chain). | ||
7. Conclusion
In conclusion, nucleoside and nucleotide analogs are staple agents for the treatment of various cancers, especially hematologic malignancies, and given the limited arsenal of drugs to combat this disease, it is crucial to enhance the pre-existing drugs. Two technologies were thus approached as a means to solve this issue, the prodrug and nanoparticle strategies, and although both of them did indeed enhance the traditional parent molecule in different aspects, both of them fell short due to different drawbacks. Therefore, a combination of both approaches might be the answer. Certainly, self-assembling PUFAylated molecules are shaping up to be an impressive tool that combines the advantages of both previous approaches while disregarding their drawbacks and it warrants further investigation.Conflicts of interest
The authors declare no conflicts of interest. The authors declare no competing financial interest.Acknowledgements
Financial support by the “La Ligue contre le cancer 49” association is gratefully acknowledged.References
- G. M. Cooper, The Cell: a Molecular Approach, Sinauer Associates, an imprint of Oxford University Press, Oxford, New York, 8th edn, 2019 Search PubMed.
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 394–424 CrossRef.
- J. Ferlay, M. Colombet, I. Soerjomataram, C. Mathers, D. M. Parkin, M. Piñeros, A. Znaor and F. Bray, Int. J. Cancer, 2019, 144, 1941–1953 CrossRef CAS PubMed.
- C. W. Freyer, N. Gupta, M. Wetzler and E. S. Wang, Am. J. Hematol., 2015, 90, 62–72 CrossRef CAS PubMed.
- N. D. Reese and G. J. Schiller, Current Hematologic Malignancy Reports, 2013, 8, 141–148 CrossRef PubMed.
- P.-F. He, J.-D. Zhou, D.-M. Yao, J.-C. Ma, X.-M. Wen, Z.-H. Zhang, X.-Y. Lian, Z.-J. Xu, J. Qian and J. Lin, Oncotarget, 2017, 8, 41498–41507 CrossRef PubMed.
- E. Moysan, G. Bastiat and J.-P. Benoit, Mol. Pharm., 2013, 10, 430–444 CrossRef CAS PubMed.
- Q. Li, H. Yan, W. Liu, H. Zhen, Y. Yang and B. Cao, PLoS One, 2014, 9(8), e104346 CrossRef PubMed.
- M. Bassetto and M. Slusarczyk, Pharm. Pat. Anal., 2018, 7, 277–299 CrossRef CAS PubMed.
- M. Rebucci and C. Michiels, Biochem. Pharmacol., 2013, 85, 1219–1226 CrossRef CAS PubMed.
- S. Mukherjee, The Emperor of All Maladies: a Biography of Cancer, Scribner, New York, 1st edn, 2011 Search PubMed.
- L. Bildstein, C. Dubernet and P. Couvreur, Adv. Drug Delivery Rev., 2011, 63, 3–23 CrossRef CAS PubMed.
- D. T. Bui, J. Nicolas, A. Maksimenko, D. Desmaële and P. Couvreur, Chem. Commun., 2014, 50, 5336–5338 RSC.
- C. E. Muller, Chem. Biodiversity, 2009, 6, 2071–2083 CrossRef PubMed.
- Y. Singh, M. Palombo and P. Sinko, Curr. Med. Chem., 2008, 15, 1802–1826 CrossRef CAS PubMed.
- H. Maag, Drug Discovery Today: Technol., 2012, 9, e121–e130 CrossRef CAS PubMed.
- M.-H. Lin, C.-F. Hung, C.-Y. Hsu, Z.-C. Lin and J. Y. Fang, Future Med. Chem., 2019, 11, 2131–2150 CrossRef CAS PubMed.
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Molecular Biology of the Cell, Garland Science, 4th edn, 2002 Search PubMed.
- G. P. Moss, P. A. S. Smith and D. Tavernier, Pure Appl. Chem., 1995, 67, 1307–1375 Search PubMed.
- R. L. P. Adams, J. T. Knowler and D. P. Leader, The Biochemistry of the Nucleic Acids, Chapman & Hall, London, New York, 11th edn, 1992 Search PubMed.
- F. A. Carey and R. M. Giuliano, Organic Chemistry, McGraw-Hill Education, New York, NY, 10th edn, 2016 Search PubMed.
- D. L. Nelson and M. M. Cox, Lehninger Principles of Biochemistry, W. H. Freeman, New York, 5th edn, 2008 Search PubMed.
- C. Galmarini, J. Mackey and C. Dumontet, Leukemia, 2001, 15, 875–890 CrossRef CAS PubMed.
- C. M. Galmarini, G. Warren, M. T. Senanayake and S. V. Vinogradov, Int. J. Pharm., 2010, 395, 281–289 CrossRef CAS PubMed.
- N. Degwert, E. Latuske, G. Vohwinkel, H. Stamm, M. Klokow, C. Bokemeyer, W. Fiedler and J. Wellbrock, Eur. J. Haematol., 2016, 97, 239–244 CrossRef CAS PubMed.
- D. Nowak, N. L. M. Liem, M. Mossner, M. Klaumünzer, R. A. Papa, V. Nowak, J. C. Jann, T. Akagi, N. Kawamata, R. Okamoto, N. H. Thoennissen, M. Kato, M. Sanada, W.-K. Hofmann, S. Ogawa, G. M. Marshall, R. B. Lock and H. P. Koeffler, Exp. Hematol., 2015, 43, 32–43 CrossRef CAS PubMed.
- M. Levin, M. Stark, B. Berman and Y. G. Assaraf, Cell Death Dis., 2019, 10, 1–14 CrossRef CAS PubMed.
- V. Heinemann, Y.-Z. Xu, S. Chubb, A. Sen, L. W. Hertel, G. B. Grindey and W. Plunkett, Cancer Res., 1992, 52, 533–539 CAS.
- Y. Jia and J. Xie, Genes Dis., 2015, 2, 299–306 CrossRef PubMed.
- M. T. de la Fuente, B. Casanova, E. Cantero, M. Hernández del Cerro, J. Garcia-Marco, A. Silva and A. Garcia-Pardo, Biochem. Biophys. Res. Commun., 2003, 311, 708–712 CrossRef CAS PubMed.
- M. Mraz, K. Cerna, V. Mayerova, K. Musilova, K. Plevova, S. Pavlova, B. Tichy, M. Doubek, Y. Brychtova, J. Malcikova, J. Mayer, M. Trbusek and S. Pospisilova, Blood, 2012, 120, 3883 CrossRef.
- T. Zenz, S. Häbe, T. Denzel, J. Mohr, D. Winkler, A. Bühler, A. Sarno, S. Groner, D. Mertens, R. Busch, M. Hallek, H. Döhner and S. Stilgenbauer, Blood, 2009, 114, 2589–2597 CrossRef CAS PubMed.
- I. Sturm, A. G. Bosanquet, S. Hermann, D. Güner, B. Dörken and P. T. Daniel, Cell Death Differ., 2003, 10, 477–484 CrossRef CAS PubMed.
- N. Tsesmetzis, C. Paulin, S. Rudd and N. Herold, Cancers, 2018, 10, 240 CrossRef PubMed.
- M. Venturini, Eur. J. Cancer, 2002, 38, 3–9 CrossRef PubMed.
- N. S. Siddiqui, A. Godara, M. M. Byrne and M. W. Saif, Expert Opin. Pharmacother., 2019, 20, 399–409 CrossRef PubMed.
- D. B. Longley, D. P. Harkin and P. G. Johnston, Nat. Rev. Cancer, 2003, 3, 330–338 CrossRef CAS PubMed.
- T. L. Whited and D. J. Taylor, Mol. Cell. Oncol., 2018, 5, e1536844 CrossRef PubMed.
- L. J. Scott, Drugs, 2016, 76, 889–900 CrossRef CAS PubMed.
- C. Knies, H. Reuter, K. Hammerbacher, E. Bender, G. A. Bonaterra, R. Kinscherf and H. Rosemeyer, Chem. Biodiversity, 2019, e1800497 CrossRef PubMed.
- J. Lukenbill and M. Kalaycio, Leuk. Res., 2013, 37, 986–994 CrossRef CAS PubMed.
- F. J. Hernandez-Ilizaliturri and M. S. Czuczman, Clin. Med.: Ther., 2009, 1 Search PubMed.
- G. I. Rodriguez, R. E. Jones, E. K. Orenberg, M. L. Stoltz and D. J. Brooks, Clin. Cancer Res., 2002, 8, 2828–2834 CAS.
- R. Swords, E. Apostolidou and F. Giles, Hematology, 2006, 11, 321–329 CrossRef CAS PubMed.
- J. E. Karp, Acute Myelogenous Leukemia, Springer Science & Business Media, 2009 Search PubMed.
- G. J. Peters, D. Sarkisjan, B. E. Hassouni, R. J. Honeywell, J. R. Julsing, B. Balboni, D. J. D. Klerk, S. Zekanovic, K. Smid, E. Giovannetti, Y. B. Lee and D. J. Kim, Cancer Res., 2019, 79, 1267 Search PubMed.
- H. M. Babiker, P. J. Schlegel, L. G. Hicks, A. J. Bullock, N. Burhani, E. Benaim, C. Peterson, C. Heaton and A. J. Ocean, J. Clin. Oncol., 2019, 37, 420 Search PubMed.
- R. Buettner, C.-C. Chen, B. Kumar, L. S. Chen, K. Wennerberg, R. Thompson, T. Pemovska, L. Li, V. Gandhi, G. Marcucci, V. Pullarkat and S. T. Rosen, Blood, 2015, 126, 792 CrossRef.
- A. Albert, Nature, 1958, 182, 421–423 CrossRef CAS PubMed.
- M. A. Lesniewska-Kowiel and I. Muszalska, Eur. J. Med. Chem., 2017, 129, 53–71 CrossRef CAS PubMed.
- R. Mahato, W. Tai and K. Cheng, Adv. Drug Delivery Rev., 2011, 63, 659–670 CrossRef CAS PubMed.
- P. Ettmayer, G. L. Amidon, B. Clement and B. Testa, J. Med. Chem., 2004, 47, 2393–2404 CrossRef CAS PubMed.
- J. Rautio, H. Kumpulainen, T. Heimbach, R. Oliyai, D. Oh, T. Järvinen and J. Savolainen, Nat. Rev. Drug Discovery, 2008, 7, 255–270 CrossRef CAS PubMed.
- B. M. Liederer and R. T. Borchardt, J. Pharm. Sci., 2006, 95, 1177–1195 CrossRef CAS PubMed.
- P. Potter and R. Wadkins, Curr. Med. Chem., 2006, 13, 1045–1054 CrossRef CAS PubMed.
- T. Heimbach, D.-M. Oh, L. Y. Li, N. Rodríguez-Hornedo, G. Garcia and D. Fleisher, Int. J. Pharm., 2003, 261, 81–92 CrossRef CAS PubMed.
- D. Petrović, K. Szeler and S. C. L. Kamerlin, Chem. Commun., 2018, 54, 3077–3089 RSC.
- D. Gupta, S. V. Gupta, K.-D. Lee and G. L. Amidon, Mol. Pharm., 2009, 6, 1604–1611 CrossRef CAS PubMed.
- J. Soul-Lawton, E. Seaber, N. On, R. Wootton, P. Rolan and J. Posner, Antimicrob. Agents Chemother., 1995, 39, 2759–2764 CrossRef CAS PubMed.
- C. Y. Yang, A. H. Dantzig and C. Pidgeon, Pharm. Res., 1999, 16, 1331–1343 CrossRef CAS PubMed.
- S. Mahesh, K.-C. Tang and M. Raj, Mol. J. Synth. Chem. Nat. Prod. Chem., 2018, 23(10), 2615 Search PubMed.
- K. M. Huttunen and J. Rautio, Curr. Top. Med. Chem., 2011, 11, 2265–2287 CrossRef CAS PubMed.
- T. Heimbach, D.-M. Oh, L. Y. Li, M. Forsberg, J. Savolainen, J. Leppänen, Y. Matsunaga, G. Flynn and D. Fleisher, Pharm. Res., 2003, 20, 848–856 CrossRef CAS PubMed.
- A. R. Van Rompay, M. Johansson and A. Karlsson, Pharmacol. Ther., 2000, 87, 189–198 CrossRef CAS PubMed.
- K. Y. Hostetler, Antiviral Res., 2009, 82, A84–A98 CrossRef CAS PubMed.
- E. Moysan, G. Bastiat and J.-P. Benoit, Mol. Pharm., 2013, 10, 430–444 CrossRef CAS PubMed.
- C. Perigaud, S. Peyrottes, C. Dumontet, WO2009053654A2, 2009. https://patents.google.com/patent/WO2009053654A2/en.
- K. Sugano, M. Kansy, P. Artursson, A. Avdeef, S. Bendels, L. Di, G. F. Ecker, B. Faller, H. Fischer, G. Gerebtzoff, H. Lennernaes and F. Senner, Nat. Rev. Drug Discovery, 2010, 9, 597–614 CrossRef CAS PubMed.
- B. S. Vig, P. J. Lorenzi, S. Mittal, C. P. Landowski, H.-C. Shin, H. I. Mosberg, J. M. Hilfinger and G. L. Amidon, Pharm. Res., 2003, 20, 1381–1388 CrossRef CAS PubMed.
- K. S. Liu, O. Y. P. Hu, S. T. Ho, J. I. Tzeng, Y. W. Chen and J. J. Wang, Br. J. Anaesth., 2004, 92, 712–715 CrossRef CAS PubMed.
- K.-S. Liu, J.-I. Tzeng, Y.-W. Chen, K.-L. Huang, C.-H. Kuei and J.-J. Wang, Anesth. Analg., 2006, 102, 1445–1451 CrossRef CAS PubMed.
- K. Zuwala, A. A. A. Smith, A. Postma, C. Guerrero-Sanchez, P. Ruiz-Sanchis, J. Melchjorsen, M. Tolstrup and A. N. Zelikin, Adv. Healthcare Mater., 2015, 4, 46–50 CrossRef CAS PubMed.
- L.-H. Shao, S.-P. Liu, J.-X. Hou, Y.-H. Zhang, C.-W. Peng, Y.-J. Zhong, X. Liu, X.-L. Liu, Y.-P. Hong, R. A. Firestone and Y. Li, Cancer, 2012, 118, 2986–2996 CrossRef CAS PubMed.
- Y.-J. Zhong, L.-H. Shao and Y. Li, Internet J. Oncol., 2013, 42, 373–383 CrossRef CAS PubMed.
- Y. Xu, J. Geng, P. An, Y. Xu, J. Huang, W. Lu, S. Liu and J. Yu, RSC Adv., 2014, 5, 6985–6992 RSC.
- Q. Zhu, Y. Lu, X. He, T. Liu, H. Chen, F. Wang, D. Zheng, H. Dong and J. Ma, Sci. Rep., 2017, 7, 1–10 CrossRef CAS PubMed.
- D. Desai, M. Åkerfelt, N. Prabhakar, M. Toriseva, T. Näreoja, J. Zhang, M. Nees and J. Rosenholm, Pharmaceutics, 2018, 10, 237 CrossRef CAS PubMed.
- R. Zhang, X. Qin, F. Kong, P. Chen and G. Pan, Drug Delivery, 2019, 26, 328–342 CrossRef CAS PubMed.
- G. S. Daher-Reyes, B. M. Merchan and K. W. L. Yee, Expert Opin. Invest. Drugs, 2019, 28, 835–849 CrossRef CAS PubMed.
- S. T. C. Neuteboom, P. L. Karjian, C. R. Boyer, M. Beryt, M. Pegram and G. M. Wahl, Mol. Cancer Ther., 2002, 1, 377–384 CAS.
- U. Pradere, E. C. Garnier-Amblard, S. J. Coats, F. Amblard and R. F. Schinazi, Chem. Rev., 2014, 114, 9154–9218 CrossRef CAS PubMed.
- S. J. Faivre, A. J. Olszanski, K. Weigang-Köhler, H. Riess, R. B. Cohen, X. Wang, S. P. Myrand, E. R. Wickremsinhe, C. L. Horn, H. Ouyang, S. Callies, K. A. Benhadji and E. Raymond, Invest. New Drugs, 2015, 33, 1206–1216 CrossRef CAS PubMed.
- F. E. Stuurman, E. E. Voest, A. Awada, P. O. Witteveen, T. Bergeland, P.-A. Hals, W. Rasch, J. H. M. Schellens and A. Hendlisz, Invest. New Drugs, 2013, 31, 959–966 CrossRef CAS PubMed.
- M. Slusarczyk, M. H. Lopez, J. Balzarini, M. Mason, W. G. Jiang, S. Blagden, E. Thompson, E. Ghazaly and C. McGuigan, J. Med. Chem., 2014, 57, 1531–1542 CrossRef CAS PubMed.
- T. M. Kadia, G. M. Borthakur, E. Jabbour, M. Y. Konopleva, F. Ravandi, C. D. DiNardo, D. Zheleva, D. Blake and J. H. Chiao, Blood, 2019, 134, 3926 CrossRef.
- G. Garcia-Manero, G. Roboz, K. Walsh, H. Kantarjian, E. Ritchie, P. Kropf, C. O'Connell, R. Tibes, S. Lunin, T. Rosenblat, K. Yee, W. Stock, E. Griffiths, J. Mace, N. Podoltsev, J. Berdeja, E. Jabbour, J.-P. J. Issa, Y. Hao, H. N. Keer, M. Azab and M. R. Savona, Lancet Haematology, 2019, 6, e317–e327 CrossRef PubMed.
- S. Polvani, M. Calamante, V. Foresta, E. Ceni, A. Mordini, A. Quattrone, M. D'Amico, C. Luchinat, I. Bertini and A. Galli, Gastroenterology, 2011, 140, 709–720 CrossRef CAS PubMed.
- Definition – Nanomaterials – Environment – European Commission, https://ec.europa.eu/environment/chemicals/nanotech/faq/definition_en.htm.
- Nat. Nanotechnol., 2019, 14, 193 Search PubMed.
- J. O. Martinez, B. S. Brown, N. Quattrocchi, M. Evangelopoulos, M. Ferrari and E. Tasciotti, Chin. Sci. Bull. Kexue Tongbao, 2012, 57, 3961–3971 CrossRef CAS PubMed.
- A. Sanchis, J.-P. Salvador and M.-P. Marco, Colloids Surf., B, 2019, 173, 825–832 CrossRef CAS PubMed.
- Y. Diebold and M. Calonge, Prog. Retinal Eye Res., 2010, 29, 596–609 CrossRef CAS PubMed.
- P. Couvreur, J. Controlled Release, 2019, 311–312, 319–321 CrossRef CAS PubMed.
- J. Chen, J. Liu, Y. Hu, Z. Tian and Y. Zhu, Sci. Technol. Adv. Mater., 2019, 20, 1043–1054 CrossRef CAS PubMed.
- R. S. Darweesh, N. M. Ayoub and S. Nazzal, Int. J. Nanomed., 2019, 14, 7643–7663 CrossRef CAS PubMed.
- L. Ruiz-Gatón, S. Espuelas, J. Huarte, E. Larrañeta, N. Martin-Arbella and J. M. Irache, Int. J. Pharm., 2019, 571, 118699 CrossRef PubMed.
- A. D. Bangham and R. W. Horne, J. Mol. Biol., 1964, 8, 660–668 CrossRef CAS PubMed.
- R. W. Horne, A. D. Bangham and V. P. Whittaker, Nature, 1963, 200, 1340 CrossRef CAS.
- V. Weissig, Methods Mol. Biol., 2017, 1522, 1–15 CrossRef CAS PubMed.
- G. Ricciuti, E. Finolezzi, S. Luciani, E. Ranucci, M. Federico, M. D. Nicola, I. A. L. Zecca and F. Angrilli, Hematol. Oncol., 2018, 36, 44–48 CrossRef CAS PubMed.
- S. Ambati, A. R. Ferarro, S. E. Kang, J. Lin, X. Lin, M. Momany, Z. A. Lewis and R. B. Meagher, mSphere, 2019, 4(1), e00025-19 CrossRef PubMed.
- N. Osborne, R. Catania, S. Stolnik-Trenkic, F. H. Falcone and K. Robinson, Access Microbiol., 2019, 1, 882 Search PubMed.
- B. Heurtault, P. Saulnier, J.-P. Benoit, J.-E. Proust, B. Pech and J. Richard, US Pat., US8057823B2, 2011, https://patents.google.com/patent/US8057823B2/en Search PubMed.
- B. Heurtault, P. Saulnier, B. Pech, J.-E. Proust and J.-P. Benoit, Pharm. Res., 2002, 19, 875–880 CrossRef CAS PubMed.
- B. Heurtault, P. Saulnier, B. Pech, J. E. Proust and J. P. Benoît, Int. J. Pharm., 2002, 242, 167–170 CrossRef CAS.
- B. Heurtault, P. Saulnier, B. Pech, M.-C. Venier-Julienne, J.-E. Proust, R. Phan-Tan-Luu and J.-P. Benoît, Eur. J. Pharm. Sci., 2003, 18, 55–61 CrossRef CAS.
- N. T. Huynh, C. Passirani, P. Saulnier and J. P. Benoit, Int. J. Pharm., 2009, 379, 201–209 CrossRef CAS PubMed.
- G. Howell, C. Oliai and G. Schiller, Anticancer Res., 2018, 38, 6927–6930 CrossRef PubMed.
- J. E. Lancet, G. L. Uy, J. E. Cortes, L. F. Newell, T. L. Lin, E. K. Ritchie, R. K. Stuart, S. A. Strickland, D. Hogge, S. R. Solomon, R. M. Stone, D. L. Bixby, J. E. Kolitz, G. J. Schiller, M. J. Wieduwilt, D. H. Ryan, A. Hoering, K. Banerjee, M. Chiarella, A. C. Louie and B. C. Medeiros, J. Clin. Oncol., 2018, 36, 2684–2692 CrossRef CAS PubMed.
- A. Hamada, T. Kawaguchi and M. Nakano, Clin. Pharmacokinet., 2002, 41, 705–718 CrossRef CAS PubMed.
- B. Salehi, Z. Selamoglu, K. S. Mileski, R. Pezzani, M. Redaelli, W. C. Cho, F. Kobarfard, S. Rajabi, M. Martorell, P. Kumar, N. Martins, T. Subhra Santra and J. Sharifi-Rad, Biomolecules, 2019, 9, 773 CrossRef CAS PubMed.
- M. C. Chamberlain, J. Neuro-Oncol., 2012, 109, 143–148 CrossRef CAS PubMed.
- Z. Amoozgar and Y. Yeo, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2012, 4, 219–233 CAS.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS.
- S. Salmaso and P. Caliceti, J. Drug Delivery, 2013, 374252 Search PubMed.
- Y. A. Haggag, M. A. Osman, S. A. El-Gizawy, A. E. Goda, M. M. Shamloula, A. M. Faheem and P. A. McCarron, Biomed. Pharmacother., 2018, 105, 215–224 CrossRef CAS PubMed.
- D. A. Tomalia, Prog. Polym. Sci., 2005, 30, 294–324 CrossRef CAS.
- E. Vacas-Córdoba, M. Maly, F. J. De la Mata, R. Gómez, M. Pion and M. Á. Muñoz-Fernández, Int. J. Nanomed., 2016, 11, 1281–1294 Search PubMed.
- J. L. Jiménez, M. Pion, F. J. de la Mata, R. Gomez, E. Muñoz, M. Leal and M. A. Muñoz-Fernandez, New J. Chem., 2012, 36, 299–309 RSC.
- E. Wiener, M. W. Brechbiel, H. Brothers, R. L. Magin, O. A. Gansow, D. A. Tomalia and P. C. Lauterbur, Magn. Reson. Med., 1994, 31, 1–8 CrossRef CAS.
- C. Kojima, B. Turkbey, M. Ogawa, M. Bernardo, C. A. S. Regino, L. H. Bryant, P. L. Choyke, K. Kono and H. Kobayashi, Nanomedicine, 2011, 7, 1001–1008 CrossRef CAS.
- A. Szulc, M. Signorelli, A. Schiraldi, D. Appelhans, B. Voit, M. Bryszewska, B. Klajnert-Maculewicz and D. Fessas, Int. J. Pharm., 2015, 495, 940–947 CrossRef CAS PubMed.
- A. Szulc, L. Pulaski, D. Appelhans, B. Voit and B. Klajnert-Maculewicz, Int. J. Pharm., 2016, 513, 572–583 CrossRef CAS.
- B. Klajnert, D. Appelhans, H. Komber, N. Morgner, S. Schwarz, S. Richter, B. Brutschy, M. Ionov, A. K. Tonkikh, M. Bryszewska and B. Voit, Chem.–Eur. J., 2008, 14, 7030–7041 CrossRef CAS PubMed.
- A. Janaszewska, K. Mączyńska, G. Matuszko, D. Appelhans, B. Voit, B. Klajnert and M. Bryszewska, New J. Chem., 2012, 36, 428–437 RSC.
- A. Janaszewska, B. Ziemba, K. Ciepluch, D. Appelhans, B. Voit, B. Klajnert and M. Bryszewska, New J. Chem., 2012, 36, 350–353 RSC.
- M. Gorzkiewicz and B. Klajnert-Maculewicz, Eur. J. Pharm. Biopharm., 2017, 114, 43–56 CrossRef CAS PubMed.
- G. Herlem, F. Picaud, C. Girardet and O. Micheau, in Nanocarriers for Drug Delivery, ed. S. S. Mohapatra, S. Ranjan, N. Dasgupta, R. K. Mishra and S. Thomas, Elsevier, 2019, pp. 469–529 Search PubMed.
- M. Inagaki, F. Kang, M. Toyoda and H. Konno, in Advanced Materials Science and Engineering of Carbon, ed. M. Inagaki, F. Kang, M. Toyoda and H. Konno, Butterworth-Heinemann, Boston, 2014, pp. 15–40 Search PubMed.
- A. Guven, G. J. Villares, S. G. Hilsenbeck, A. Lewis, J. D. Landua, L. E. Dobrolecki, L. J. Wilson and M. T. Lewis, Acta Biomater., 2017, 58, 466–478 CrossRef CAS PubMed.
- A. Kazemi-Beydokhti, S. Zeinali Heris and M. R. Jaafari, Chem. Eng. Res. Des., 2016, 112, 56–63 CrossRef CAS.
- M. Zheng, A. Jagota, E. D. Semke, B. A. Diner, R. S. Mclean, S. R. Lustig, R. E. Richardson and N. G. Tassi, Nat. Mater., 2003, 2, 338–342 CrossRef CAS PubMed.
- H. Wang and A. Ceulemans, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 195419 CrossRef.
- K. Umemura, Nanomaterials, 2015, 5, 321–350 CrossRef CAS PubMed.
- P. Zhang, W. Yi, J. Hou, S. Yoo, W. Jin and Q. Yang, Int. J. Nanomed., 2018, 13, 3069–3080 CrossRef CAS PubMed.
- Y. Ruan, W. Yu, F. Cheng, X. Zhang, T. Rao, Y. Xia and S. Larré, IET Nanobiotechnol., 2011, 5, 47–51 CrossRef CAS PubMed.
- H. Zhang, D. Yee and C. Wang, Nanomedicine, 2008, 3, 83–91 CrossRef CAS PubMed.
- A. E. O'Connor, W. M. Gallagher and A. T. Byrne, Photochem. Photobiol., 2009, 85, 1053–1074 CrossRef PubMed.
- M. Fang, M. Chen, L. Liu and Y. Li, J. Biomed. Nanotechnol., 2017, 13, 1–16 CrossRef CAS PubMed.
- S. Samimi, M. S. Ardestani and F. A. Dorkoosh, J. Drug Delivery Sci. Technol., 2021, 61, 102287 CrossRef CAS.
- U. Yunus, M. A. Zulfiqar, M. Ajmal, M. H. Bhatti, G.-S. Chaudhry, T. S. T. Muhammad and Y. Y. Sung, Biomed. Mater., 2020, 15, 065004 CrossRef CAS PubMed.
- P. Ghosh, G. Han, M. De, C. K. Kim and V. M. Rotello, Adv. Drug Delivery Rev., 2008, 60, 1307–1315 CrossRef CAS PubMed.
- K. Pal, F. Al-suraih, R. Gonzalez-Rodriguez, S. K. Dutta, E. Wang, H. S. Kwak, T. R. Caulfield, J. L. Coffer and S. Bhattacharya, Nanoscale, 2017, 9, 15622–15634 RSC.
- S. Song, Y. Hao, X. Yang, P. Patra and J. Chen, J. Nanosci. Nanotechnol., 2016, 16, 2582–2586 CrossRef CAS PubMed.
- P. G. Jeelani, P. Mulay, R. Venkat and C. Ramalingam, Silicon, 2020, 12, 1337–1354 CrossRef CAS.
- N. Syazaliyana Azali, N. Hidayatul Nazirah Kamarudin, A. Rasyidah Abdul Rahim, N. Syifa'a Jamal Nasir, S. Najiha Timmiati and N. Farhana Jaafar, Mater. Today: Proc., 2019, 19, 1722–1729 CAS.
- P. Sangaiya and R. Jayaprakash, J. Supercond. Novel Magn., 2018, 31, 3397–3413 CrossRef CAS.
- L. Shen, B. Li and Y. Qiao, Materials, 2018, 11, 324 CrossRef PubMed.
- N. Shahabadi, M. Falsafi and K. Mansouri, Colloids Surf., B, 2016, 141, 213–222 CrossRef CAS PubMed.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213–1214 RSC.
- H. Kumagai, C. J. Kepert and M. Kurmoo, Inorg. Chem., 2002, 41, 3410–3422 CrossRef CAS PubMed.
- O. M. Yaghi, H. Li and T. L. Groy, J. Am. Chem. Soc., 1996, 118, 9096–9101 CrossRef CAS.
- D. Bradshaw, J. B. Claridge, E. J. Cussen, T. J. Prior and M. J. Rosseinsky, Acc. Chem. Res., 2005, 38, 273–282 CrossRef CAS PubMed.
- V. Rodriguez-Ruiz, A. Maksimenko, R. Anand, S. Monti, V. Agostoni, P. Couvreur, M. Lampropoulou, K. Yannakopoulou and R. Gref, J. Drug Targeting, 2015, 23, 759–767 CrossRef CAS PubMed.
- M. Celano, M. G. Calvagno, S. Bulotta, D. Paolino, F. Arturi, D. Rotiroti, S. Filetti, M. Fresta and D. Russo, BMC Cancer, 2004, 4, 63 CrossRef PubMed.
- P. Li, S. Luo, L. Xiao, B. Tian, L. Wang, Z. Zhang and Y. Zeng, Acta Pharmacol. Sin., 2019, 40, 1448–1456 CrossRef CAS PubMed.
- T. Briot, E. Roger, N. Lautram, A. Verger, A. Clavreul and F. Lagarce, Int. J. Nanomed., 2017, 12, 8427–8442 CrossRef CAS PubMed.
- J. Katsaras and T. Gutberlet, Lipid Bilayers: Structure and Interactions, Springer-Verlag, Berlin Heidelberg, 2001 Search PubMed.
- K. Ariga, J. P. Hill, M. V. Lee, A. Vinu, R. Charvet and S. Acharya, Sci. Technol. Adv. Mater., 2008, 9, 014109 CrossRef PubMed.
- S. Sivakova and S. J. Rowan, Chem. Soc. Rev., 2005, 34, 9–21 RSC.
- E. Lepeltier, C. Bourgaux, V. Rosilio, J. H. Poupaert, F. Meneau, F. Zouhiri, S. Lepêtre-Mouelhi, D. Desmaële and P. Couvreur, Langmuir, 2013, 29, 14795–14803 CrossRef CAS PubMed.
- X. Gong, M. J. Moghaddam, S. M. Sagnella, C. E. Conn, S. J. Danon, L. J. Waddington and C. J. Drummond, Colloids Surf., B, 2011, 85, 349–359 CrossRef CAS PubMed.
- S. M. Sagnella, X. Gong, M. J. Moghaddam, C. E. Conn, K. Kimpton, L. J. Waddington, I. Krodkiewska and C. J. Drummond, Nanoscale, 2011, 3, 919–924 RSC.
- L. Wu, F. Zhang, X. Chen, J. Wan, Y. Wang, T. Li and H. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 3327–3340 CrossRef CAS PubMed.
- A. Maksimenko, J. Caron, J. Mougin, D. Desmaële and P. Couvreur, Int. J. Pharm., 2015, 482, 38–46 CrossRef CAS PubMed.
- D. Wang, B. Liu, Y. Ma, C. Wu, Q. Mou, R. Wang, D. Yan, C. Zhang and X. Zhu, J. Am. Chem. Soc., 2017, 139(40), 14021–14024 CrossRef CAS PubMed.
- D. Lombardo, M. A. Kiselev, S. Magazù and P. Calandra, Adv. Condens. Matter Phys., 2015, 2015, 1–22 CrossRef.
- B. Xie, J. Wan, X. Chen, W. Han and H. Wang, Mol. Cancer Ther., 2020, 19, 822–834 CrossRef CAS PubMed.
- H. Wang, Z. Lu, L. Wang, T. Guo, J. Wu, J. Wan, L. Zhou, H. Li, Z. Li, D. Jiang, P. Song, H. Xie, L. Zhou, X. Xu and S. Zheng, Cancer Res., 2017, 77, 6963–6974 CrossRef CAS PubMed.
- L. Bildstein, C. Dubernet, V. Marsaud, H. Chacun, V. Nicolas, C. Gueutin, A. Sarasin, H. Bénech, S. Lepêtre-Mouelhi, D. Desmaële and P. Couvreur, J. Controlled Release, 2010, 147, 163–170 CrossRef CAS PubMed.
- B. Pili, C. Bourgaux, H. Amenitsch, G. Keller, S. Lepêtre-Mouelhi, D. Desmaële, P. Couvreur and M. Ollivon, Biochim. Biophys. Acta, Biomembr., 2010, 1798, 1522–1532 CrossRef CAS PubMed.
- V. Allain, C. Bourgaux and P. Couvreur, Nucleic Acids Res., 2012, 40, 1891–1903 CrossRef CAS PubMed.
- L. Bildstein, V. Marsaud, H. Chacun, S. Lepêtre-Mouelhi, D. Desmaële, P. Couvreur and C. Dubernet, Soft Matter, 2010, 6, 5570–5580 RSC.
- L. H. Reddy, C. Dubernet, S. L. Mouelhi, P. E. Marque, D. Desmaele and P. Couvreur, J. Controlled Release, 2007, 124, 20–27 CrossRef CAS PubMed.
- X. Mulet, T. Kaasgaard, C. E. Conn, L. J. Waddington, D. F. Kennedy, A. Weerawardena and C. J. Drummond, Langmuir, 2010, 26, 18415–18423 CrossRef CAS PubMed.
- A. Maksimenko, J. Mougin, S. Mura, E. Sliwinski, E. Lepeltier, C. Bourgaux, S. Lepêtre, F. Zouhiri, D. Desmaële and P. Couvreur, Cancer Lett., 2013, 334, 346–353 CrossRef CAS PubMed.
- J. N. Israelachvili and D. J. Mitchell, Biochim. Biophys. Acta, Biomembr., 1975, 389, 13–19 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |