
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding intracellular nanoparticle trafficking fates through spatiotemporally resolved magnetic nanoparticle recovery
Emily
Sheridan†
 a,
Silvia
Vercellino†
ab,
Lorenzo
Cursi
a,
Laurent
Adumeau
a,
James A.
Behan
*a and
Kenneth A.
Dawson
*a
a,
Silvia
Vercellino†
ab,
Lorenzo
Cursi
a,
Laurent
Adumeau
a,
James A.
Behan
*a and
Kenneth A.
Dawson
*a
aCentre for BioNano Interactions, School of Chemistry, University College Dublin, Belfield, Dublin 4, Ireland. E-mail: james.behan@cbni.ucd.ie; Kenneth.a.dawson@cbni.ucd.ie
bUCD Conway Institute of Biomolecular and Biomedical Research, School of Biomolecular and Biomedical Science, University College Dublin, Belfield, Dublin 4, Ireland
First published on 3rd March 2021
Abstract
The field of nanomedicine has the potential to be a game-changer in global health, with possible applications in prevention, diagnostics, and therapeutics. However, despite extensive research focus and funding, the forecasted explosion of novel nanomedicines is yet to materialize. We believe that clinical translation is ultimately hampered by a lack of understanding of how nanoparticles really interact with biological systems. When placed in a biological environment, nanoparticles adsorb a biomolecular layer that defines their biological identity. The challenge for bionanoscience is therefore to understand the evolution of the interactions of the nanoparticle–biomolecules complex as the nanoparticle is trafficked through the intracellular environment. However, to progress on this route, scientists face major challenges associated with isolation of specific intracellular compartments for analysis, complicated by the diversity of trafficking events happening simultaneously and the lack of synchronization between individual events. In this perspective article, we reflect on how magnetic nanoparticles can help to tackle some of these challenges as part of an overall workflow and act as a useful platform to investigate the bionano interactions within the cell that contribute to this nanoscale decision making. We discuss both established and emerging techniques for the magnetic extraction of nanoparticles and how they can potentially be used as tools to study the intracellular journey of nanomaterials inside the cell, and their potential to probe nanoscale decision-making events. We outline the inherent limitations of these techniques when investigating particular bio-nano interactions along with proposed strategies to improve both specificity and resolution. We conclude by describing how the integration of magnetic nanoparticle recovery with sophisticated analysis at the single-particle level could be applied to resolve key questions for this field in the future.
 Emily Sheridan | Emily Sheridan obtained her B.A. (Mod.) in Biochemistry from Trinity College Dublin (TCD) in 2017, her research project focusing on novel G-quadruplex binding molecules as anti-cancer therapeutics. Before joining CBNI in 2018, she held research positions in protein crystallography in the Trinity Biomedical Sciences Institute, TCD and molecular cell biology in the Smurfit Institute of Genetics, TCD. She is now completing her PhD at the Centre for BioNano Interactions (CBNI), UCD with a research focus on the nanoscale molecular recognition that governs the intracellular trafficking of nanoparticles. |
 Silvia Vercellino | Silvia Vercellino obtained her BSc and MSc in Pharmaceutical Chemistry from Universitá di Torino in 2017. Her project was focused on the synthesis of core–shell magnetic nanoparticles for drug delivery and thermo therapy. She investigated the interaction on nanoparticles and cells at the Centre for BioNano Interactions (CBNI), UCD funded by Erasmus+ program. She is now a PhD student at the Conway Institute of Biomolecular and Biomedical Research (UCD). Her research aims to isolate and characterize re-engineered nanoparticles and to unravel the biological interaction of complex nanostructures and living systems. |
 Lorenzo Cursi | Lorenzo Cursi obtained his BSc in Chemistry and Materials Chemistry and his MSc in Chemistry from the University of Bologna in 2016. His research projects were focused on the development of gravitational field-flow fractionation and the isolation and characterization of magnetosomes through the use of asymmetrical flow field-flow fractionation coupled with multi-angle light scattering (AF4-MALS). He joined the Centre for BioNano Interactions in September 2017 as a PhD student at UCD funded by Celtic Advanced Life Science Innovation Network (CALIN). His research focuses on the development of tools and methods for understanding bionano interactions with a focus on the magnetic toolset. |
 Laurent Adumeau | Laurent Adumeau is currently a postdoctoral researcher at the Centre for BioNano Interactions (CBNI), UCD. After completing a MSc in Biochemistry, he obtained his PhD at the University of Bordeaux, with research focusing on the synthesis of inorganic nanoparticles and their controlled surface biofunctionalization for biological applications. Laurent joined Prof. Kenneth Dawson's group at CBNI in 2017. His interdisciplinary research focuses on the investigation of the mechanisms involved in the engineered nanoparticle recognition by cells. |
 James A. Behan | James A. Behan joined the Centre for BioNano Interactions (CBNI), UCD in April 2019 as a Postdoctoral Fellow. Previously he obtained a B.A. (First Class Hons) in Chemistry from Trinity College Dublin (TCD) in 2014 with a Foundation Scholarship. He went on to complete his PhD in Physical Chemistry in TCD in 2018, where his work focused on the preparation of non-crystalline carbon systems with controlled structure and surface chemistry and their application in electrocatalysis and biosensing. In 2020 James won a postdoctoral fellowship from the Irish Research Council for research into the tailoring of nanoparticle surface organization for targeted applications in biology at CBNI. |
 Kenneth A. Dawson | Professor Kenneth A. Dawson is both the founding and current Director of the Centre for BioNano Interactions (CBNI) based at University College Dublin (UCD), Ireland. He is also Chair of Physical Chemistry at UCD. The scientific focus of the CBNI is to understand the interaction of nanoparticles with living systems (www.ucd.ie/cbni). The Centre seeks to discover those unique architectural and other nanoscale features that control these biological interactions and determine biological outcomes. Professor Dawson is the discoverer of the ‘biomolecular corona’ effect, for which he was awarded the US National Academy Cozzarelli Prize. |
1. Introduction
Nanoparticles (NPs) are promising tools for many biomedical applications including diagnostics, drug delivery and as treatment agents.1–4 An enormous effort has been made to take advantage of the unique features of NPs to enhance drug efficiency by designing and synthesizing a number of delivery systems,3 with the objective of delivering therapeutics exclusively to the target tissue.5 However, despite these endeavors and the considerable resources invested, the current state-of-the-art NP-based therapies show a low delivery efficiency,6 and only a few specific NP-based formulations are currently FDA approved and available on the market.7From the moment of administration, nanomedicine delivery efficiency involves a multitude of complex factors pertaining to in vivo transport and biodistribution. One of the most important factors contributing to low delivery efficiency arises from the interaction of NPs with complex biological milieu: it is now widely accepted that the suspension of NPs in biological fluids results in the formation of an adsorbed layer of biomolecules known as the biomolecular corona.8 The composition of this corona and the spatial orientation of the biomolecules that comprise it are both critical parameters in the cell-NP recognition process,9 as particular protein motifs exposed on the surface are recognized by cell membrane receptors.10 Alterations in the organization of the corona – for example, due to pre-incubation of NPs with different serum concentrations11 or with specific individual proteins12 – is known to influence cell uptake.13 Hence, irrespective of the biological identity imparted to a NP-based therapy through the attachment of targeting ligands or biomolecules, consequent NP internalization depends on the nature of the biomolecular corona formed in situ. Moreover, once formed, the composition of the biomolecular corona changes dynamically through constant desorption and adsorption depending on the biomolecular environment encountered as the NP is trafficked through the cell,8,14,15 which has been shown to result in divergent intracellular sorting outcomes depending on the presence of organelle-specific proteins introduced into the NP-corona.16 Additionally, since protein exchange kinetics can last hours, the NP corona also retains the memory of the environment to which the NPs have been exposed. This memory is what we will refer to as the ‘biomolecular barcode’ in this article.
The overwhelming outcome of corona formation for nanomedicine is a lack of both targeting specificity and signaling clarity for the targeted cell. In the majority of cases, this leads to the segregation of NPs after cell uptake within certain cytoplasmic compartments, particularly within the endolysosomal pathway.17–19 Interestingly, a minor population of NPs do not follow this traditional intracellular route18 but are instead diverted down alternative trafficking pathways. The initiator of these alternative pathways remains unknown, and the rarity and transient nature of these events make their study challenging, as they are buried in the complexity of the bulk analysis of the cell mechanisms, which can only reveal the dominant routes. Moreover, the in situ study of intracellular events is complicated by the difficulty in accessing intracellular compartments.
Magnetic nanoparticles (MNPs) compose a subclass of NPs that is the subject of intense research, in particular within the field of nanomedicine.20,21 They have properties which are amenable to a variety of applications, such as magnetic resonance imaging (MRI),22,23 targeted hyper-thermic treatment of tumours,20,24–28 site-specific-drug delivery and in-cell manipulation through the application of external magnetic fields. However, here we will not focus on applications of MNPs in nanomedicine, but we will instead reflect on how MNPs, once integrated into an overall workflow, can offer a potential platform to elucidate intracellular trafficking events, which is crucial for nanomedicine applications. Indeed, the magnetic nature of MNPs provides the potential to selectively recover the NPs from their intracellular environment29 to allow a more detailed analysis than is possible with non-magnetic internalized NPs. In principle, by exploiting magnetic extraction at different times in the intracellular trafficking process, it is possible to recover and translate the biomolecular barcode into an understanding of the trafficking lineage of the nanoparticle through multiple intracellular vesicles, unravelling alterations in the corona which redirect the NPs to rarer trafficking pathways.
We believe that applying selective MNP-based extraction techniques will provide the capacity to investigate rare NP trafficking events and understand the mechanisms responsible for divergent NP sorting. This knowledge in turn will enable the development of targeted nanomedicines capable of avoiding the lysosomal pathway. In this article we provide an overview of the application of MNPs in the study of intracellular bionano interactions; we introduce the fundamentals underlying MNP-based extraction as well as emerging technologies that exploit magnetic properties for this purpose. We then describe the challenges inherent to the task of achieving spatiotemporal resolution in the extraction, including issues in the recovery of pure subpopulations and sample contamination, both of which may bias downstream analyses. Finally, we emphasize the need for single particle analysis in order to deal with the complex heterogeneity of the events happening in the cell. Some low-throughput techniques such as electron microscopy combined with immunostaining already allow for single nanoparticle characterization. However, we argue that a move towards a high throughput and high dimensional analysis workflow at the level of individual extracted NPs will afford the best chance for the field to unravel the precise intracellular bionano interactions governing intracellular trafficking decisions.
2. Magnetic isolation of nanoparticles from intracellular compartments
Magnetic separation using commercially available micro- or nanoparticles (the mechanism of which is depicted in Fig. 1a) is one of the most common techniques used in targeted biological extractions30,31 and has also shown promise in the isolation of cellular organelles.29 It offers clear advantages versus alternative techniques (e.g. centrifuge-based techniques), such as the ability to distinguish between organelle sub-populations that share similar buoyancy,32 no requirement for expensive equipment such as ultracentrifuges, and time efficiency, especially when working with large sample volumes.33 Magnetic separation can provide samples enriched in specific organelles, such as mitochondria34 and lysosomes,29 with good yields and purity, often superior to those provided by centrifuge-based techniques. However, the lack of a standardized procedure for magnetic separation itself and differences in sample processing (e.g. lysis procedures) can lead to contradictory results.34,35 Furthermore, due to the small size of the species involved, the magnetic separation of sub-cellular species requires the use of high magnetic field gradients that can become a source of contamination and mechanical damage, such as vesicle rupture or object compaction (Fig. 1b).32,35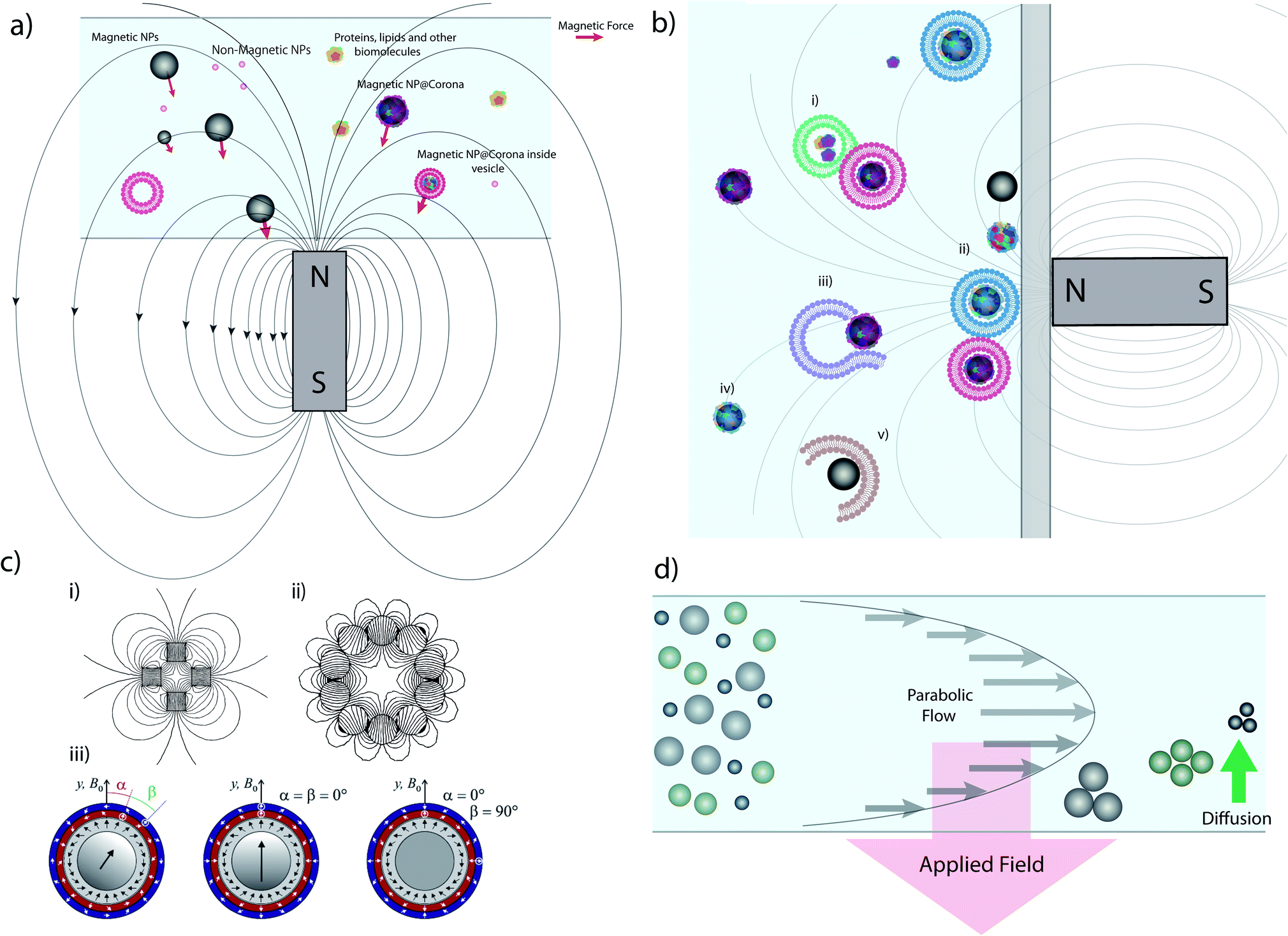 | ||
| Fig. 1 Magnetic extraction system and limitations. (a) Illustration of the principle of magnetic extraction using a simple bar magnet. MNPs and MNPs with biomolecular corona or internalized within vesicles are selectively attracted toward the magnet by a force (the red vector size is a representation of the magnetic force) in relation to the magnetic field gradient and particles size, as discussed in the main text. (b) Common issues affecting magnetic separation: (i) vesicles/biomolecules not related to the MNP are extracted due to adsorption; (ii) diverse populations with different vesicles/coronas are all extracted simultaneously in bulk, reducing resolution and implying that downstream analysis is invariably an average, and prolonged exposure to large forces at the wall may damage vesicles; (iii) ruptured or damaged vesicles during lysis may not be extracted; (iv) particles may not be captured in the timescale of the experiment depending on their distance from the magnet and their size; and (v) MNPs may pick up or adsorb materials during the extraction process which do not reflect their intracellular trafficking pathway and so the results obtained are misleading. (c) Different magnet geometries showing (i) a quadrupole, (ii) a circular Halbach array generating an hexapole and (iii) combination of different Halbach arrays forming concentric quadrupoles. (d) Schematic representation of FFF separation in normal mode. Parts c (i) and c (ii) of this figure were simulated using Finite Element Method Magnetics36 based on ref. 37 and 38. Part c (iii) reprinted from O. Baun and P. Blümler, Permanent magnet system to guide superparamagnetic particles, Journal of Magnetism and Magnetic Materials, 439, 294–304,39 Copyright (2017), with permission from Elsevier. | ||
Other techniques have been developed for the isolation of organelles, such as fluorescence-assisted organelle sorting (FAOS)40–42 and free-flow electrophoresis,43,44 but these lead to high levels of cross contamination.45 By contrast, magnetic separation techniques, such as immunoprecipitation with magnetic beads, allow for the rapid and targeted isolation of organelles with high purity45 while remaining fully compatible with downstream analysis.
In short, despite some limitations to which we have just alluded and will discuss in more detail in the following section, magnetic separation currently provides a significant route for the isolation and identification of ultra-rare species in biology, due to its capacity for processing larger volumes of samples and because of its targeting versatility: any species can be targeted for magnetic extraction as long as it can be labelled, internally or externally, with MNPs and other constructs.
2.1. Principles of magnetic extraction
Every material possesses magnetic properties, so the term “magnetic nanoparticles” is not in itself a precise definition. Depending mainly on their electronic configuration, a dia- or paramagnetic material will respond weakly to a magnetic field, whereas a ferro- or ferrimagnetic material will respond strongly to it. However, it is commonly understood that “magnetic material”, and by extension, “magnetic nanoparticle” refers to the second category, and that is what we refer to in this article. The migration of suspended MNPs and other nanomaterials in a fluid in the presence of a magnetic field is known as magnetophoresis. Here we will restrict our discussion to superparamagnetic materials under typical conditions for the extraction of biological samples such as vesicles, viz. MNPs possessing a single magnetic domain, above the blocking temperature. For discussions of paramagnets and other weakly-magnetic materials, the reader is directed to other excellent review articles and monographs on the subject.46,47Under the ascribed conditions, the mobility of a MNP induced by a magnetic field depends on the net magnetic force, Fm, exerted on the particle which can be approximated as:48,49
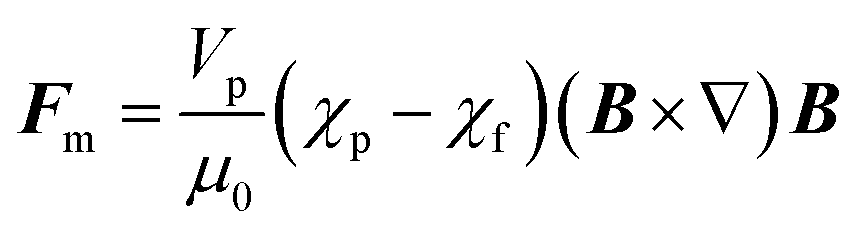 | (1) |
The motion of a particle undergoing magnetophoresis within a fluid is opposed by drag forces. Supposing the magnetic particle is spherical, and the fluid flow is laminar, the viscous drag force Fd experienced is given by the Stokes' law:54
| Fd = 3πηdhvs | (2) |
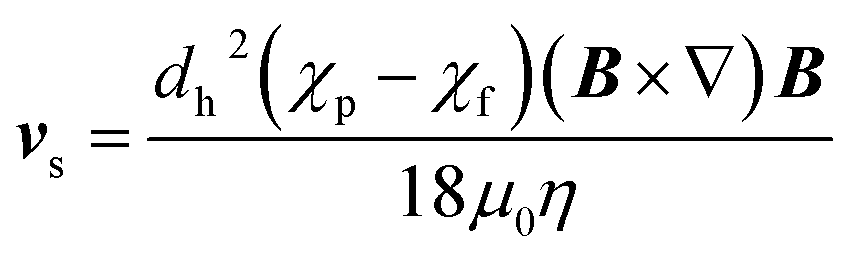 | (3) |
Rapid and effective magnetic separation is of particular importance in vesicle extraction because these structures have limited lifetimes in non-physiological conditions and are susceptible to damage from both mechanical and thermal stress. Examining eqn (3), it is clear that the recovery time is inversely proportional to vs, and hence to the magnetic field gradient. It is possible to increase the gradient of the magnetic field by assembling several permanent magnets in precise geometries such as quadrupoles,37,56 Halbach arrays38,57 and their combinations39 (Fig. 1c).
Another alternative for increasing the magnetic field exerted by permanent magnets, is by employing columns loaded with beads or wires with a high magnetic permeability that can generate local high magnetic field gradients within the column – so-called ‘magnetic chromatography’.47,58,59 An example of this approach is found in many commercial products such as MACS separators.60,61 Different magnet arrangements have been successfully exploited for the separation of magnetically tagged cells,37,62 viruses,63 DNA64,65 and subcellular fractions from cell lysate such as mitochondria,66 exosomes67 and endocytic organelles.29,68
As a result, some magnetic species may spend significantly more time accumulated on the wall of the test tube or the surface of a magnetic bead under the influence of a high magnetic gradient, risking damage to biological structures and potential distortion of results due to the aggregation of distinct subpopulations into clusters. Immunoprobe extractions with MNPs may alleviate this issue somewhat, since they typically have specific biological targets which may not be present in sufficiently large concentrations to form significant aggregates. However, even in this more restricted case, magnetic probes will target multiple subpopulations that share common targets. Due to these limitations, magnetic extraction as currently employed is an effective technique for separating magnetic materials from a diamagnetic milieu but lacks discriminatory power between magnetic subpopulations. Such subpopulations may possess extraordinarily divergent biological identities despite apparent similarities in magnetophoretic behaviour, and as such the refinement of the magnetic separation based on characteristics intrinsic to a given population, such as particle size, density, or the presence of specific patterns of receptors, is highly desirable. Despite the limitations just described, magnetic separation techniques are still in many ways superior to centrifugation-based techniques in terms of their selectivity and level of contamination. Moreover, as described in the following section, there are novel techniques for magnetic separation in flow which may help to mitigate these issues.
2.2 New techniques exploiting magnetism in extraction
The degree of fractionation can be understood in terms of the retention or accumulation of MNPs, as described by:70
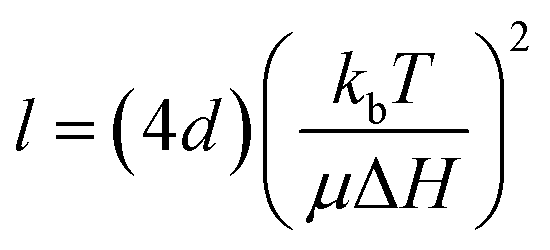 | (4) |
However, subpopulations with profound differences in their biological identity will nevertheless exist due to variations in intracellular trafficking history, even among fractionated MNP samples with comparable size and magnetic moment isolated from biological milieu. Identification and isolation of these subpopulations is within the purview of FFF and related techniques.
For non-magnetic NPs, asymmetric FFF (aFFF) and other flow-based techniques have been successfully employed in the gentle, size-based fractionation even of delicate biological samples, such as extracellular vesicles and exomeres.79 In addition, sedimentation FFF (sFFF), which employs centrifugal forces, introduces density as a variable in NP separation as well as size, allowing for particles with similar size but different densities to be separated. Here we mention these techniques in the context of ‘hyphenation’ with mFFF, where two or more of the techniques are run in series. This may be considered as the ‘flow’ analogue of the combination of different batch techniques, such as centrifugation followed by magnetic extraction, to achieve a higher degree of sample purity. In principle, the hyphenated approach enables a more refined multi-variable subpopulation fractionation on the basis of subtle differences in susceptibility, density, size, and shape. Indeed, hyphenation of asymmetric FFF with single-particle inductively-coupled mass spectrometry (sp-ICPMS) enables subpopulations of metal NPs to be quantified on a particle-by-particle basis. We envision that refined MNP separation by FFF and related techniques may be coupled with downstream analysis techniques, including multiplex epitope mapping (discussed below), allowing quantitative subpopulations identified on the basis of physicochemical parameters to be associated with a particular biological identity.
3. Challenges associated with the unravelling of bionano interactions with magnetic nanoparticles
3.1 Maintaining the biomolecular integrity of isolated samples
It is important to remember that the purpose of the magnetic separation of the particles from the cell environment is not to collect a material, but to isolate particles associated with individual trafficking events whilst preserving the biomolecular traces associated with their intracellular environment. Hence, disturbances during extraction and isolation steps must be minimized or, when unavoidable, taken into consideration in the analysis of results. Apart from potential mechanical damage incurred during the extraction, most of this interference comes from the inevitable environmental changes happening during the cell lysis or during the extraction itself. Cell lysis induces the release of massive amounts of biomolecules, organelles and cell debris that were previously contained in separate compartments, all of which have the capacity to adsorb non-specifically onto MNPs or MNP-vesicles of interest. Such interference can be minimized through a two-stage approach: initially, by applying an appropriate cell-lysis technique prior to magnetic extraction, and then following this by careful washing and handling of the sample post-extraction.Cell lysis procedures for magnetic extraction experiments must reach a balance between incomplete lysis and excessive lysis. Incomplete or partial lysis will leave some intact, whole cells which will be potentially magnetically extracted with the sample of interest. While harsh lysis conditions can induce excessive damage to the lipid membranes of intracellular vesicles, resulting in damage to samples and absorption from surrounding cellular debris. Ideal cell lysis should break only the plasma membrane, releasing the cellular contents untouched and allowing for extraction of intact compartments containing MNPs free from contaminating biomolecules. Time is also an important factor as rapid lysis methods limit damage caused by protease and phosphatase enzymes and changes to the native state of vesicles. Although an old technique, homogenization of osmotically swollen cells remains one of the preferred methods of cell disruption for downstream vesicle analysis as it is gentle, can lyse large volumes rapidly, and requires often standard buffer components and equipment. This technique also presents the strong advantage of avoiding the use of detergents usually used in alternative lysis methods in order to compensate for the need for large mechanical forces to break cells. The use of detergent during the lysis would damage the integrity of vesicle membranes and would also limit downstream analysis.
Following magnetic extraction of nanoparticle-containing compartments, careful washing of samples must be carried out to remove contaminating biomolecules. However, even when care is taken, any approach is liable to result in the loss of weakly interacting partners. Both cell lysis and/or extraction and washing induce a consequent dilution and equilibrium displacement on objects of interest and their interacting partners which can lead to their dissociation. A compromise between sample purity and loss of information must be accepted. Limiting sample damage and losses throughout the steps has heightened importance once dealing with single object analysis. Indeed, while bulk analysis of sample populations can withstand losses and partial damage without jeopardizing the results, with single event analysis even small losses and damage to biomolecules from samples will critically alter the result of the populations based on the result of omics analysis.
Newer microfluidic and lysis-on-a-chip style approaches are being investigated for cell lysis, but the methods employed are often better suited for nucleic acid or protein extraction as they are low throughput, expensive, and challenging to fabricate, and can damage vesicular membranes in the lysis process. Any application would require extensive optimization for subcellular compartment analysis to be reliably achieved.84 There are potential applications for these emerging techniques downstream of vesicle extraction, for the rupturing of the vesicle membrane for MNP release and subsequent intracellular corona studies. If incorporated with additional functionalities that could analyse or sort single NPs and vesicles at small volumes, they could become crucial aspects in single NP corona analysis.
3.2 Challenges associated with the complex population heterogeneity of intracellular nanoparticles
As we have described in the introduction to this article, the endolysosomal pathway is the dominant trafficking route for MNPs in cells85 but small subpopulations have been observed to be sorted to other cellular compartments such as the Golgi,86 and endoplasmic reticulum (ER),87 or to undergo exocytosis from the cell.88 The heterogeneity in behavior between individual NPs begins at the plasma membrane, where even single suspensions of NPs are internalized by different uptake processes and at varying rates (Fig. 2a, top panel). This initial disparity within the population is the result of the differences by which the cell ‘sees’ individual coronas based not only on their biomolecular composition, but also on how the NP is orientated for interaction with cell surface receptors.11,89 The causes of these phenomena remain largely unknown and unravelling them is a significant challenge for the field, with tremendous potential for exciting breakthroughs if it is overcome. Due to the low frequency of these divergent trafficking events, the tools used to gain a comprehensive insight into intracellular NP interactions must be sensitive enough to both detect and quantify even single events. Such events may first be observed through light microscopy techniques with fluorescently labelled NPs, with further confirmation and exploration accomplished with complementary downstream techniques.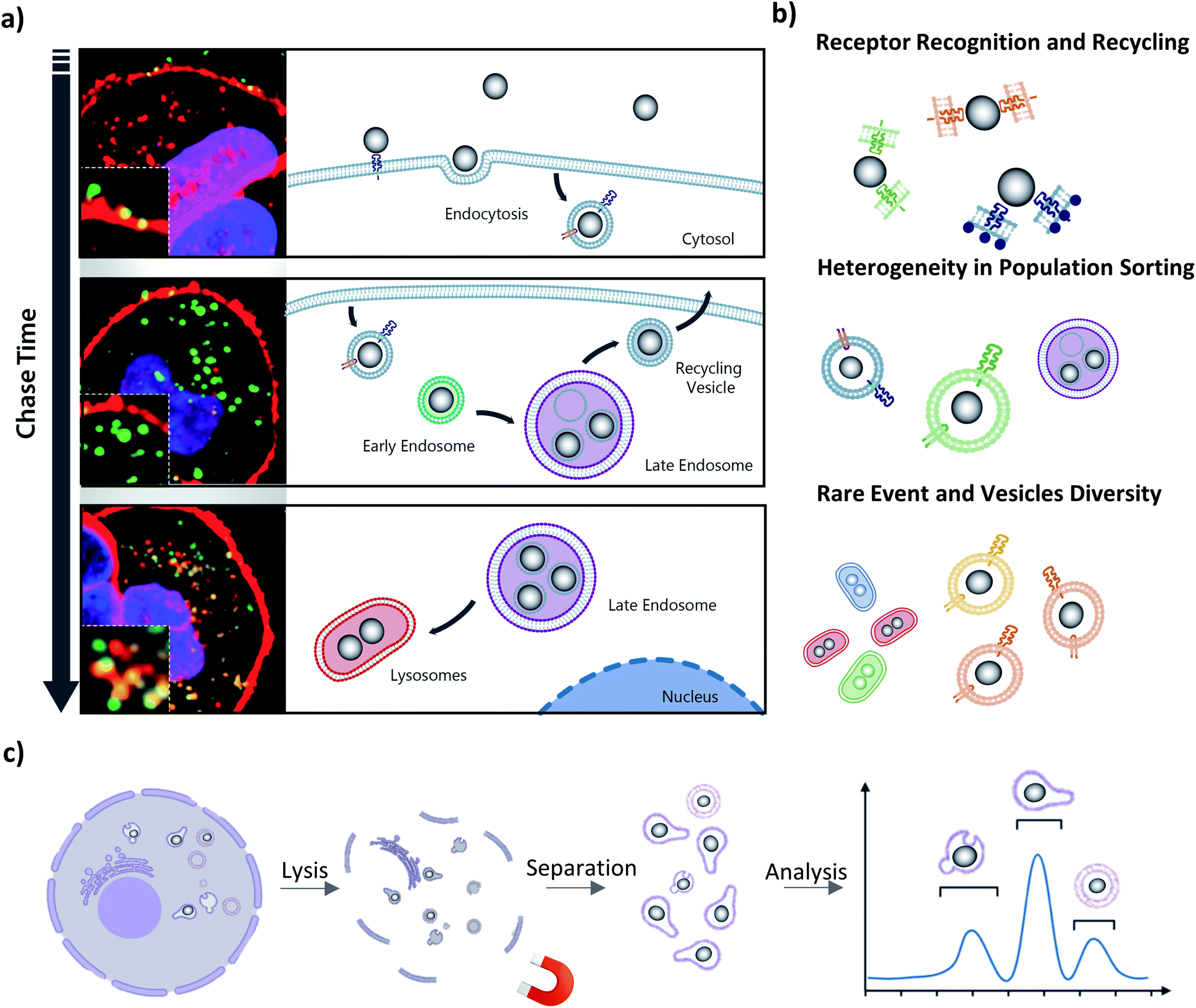 | ||
| Fig. 2 Spatiotemporal heterogeneity and MNP microenvironment. (a) Time resolved recovery of heterogenous populations can reveal MNP trafficking information. (b) Multi-level complexity of NPs interactions with cells. (c) Cell-free approach towards single-particle analysis. | ||
Challenges arise not only in capturing rare, transient events in nanoparticle trafficking but also in our capacity to understand the role of endosomal trafficking vesicle subpopulations in individual trafficking decisions. Imaging approaches are limited by the parameters that can be detected and analyzed at one time, which allows for only few markers available for identifying individual vesicle compartments. It has long been established that endosomal trafficking vesicles are more dynamic than a few surface proteins can reveal, and their “kiss and run” interactions, which result in surface protein and cargo sharing, further complicates their identification.90,91 Moreover, even in unstimulated cells, intracellular vesicles are highly heterogenous within the same vesicle subtype,92 and these subtypes have subtle but significant differences in their functioning.93 This introduces significant complexity into the analysis and emphasizes the need for extraction and downstream characterisation of single NPS.
The important but often subtle differences within a single class of vesicles (Fig. 2b) can include their size, architecture, motility, spatial localization, functional activity, enzymatic activity and pH. There is an ongoing revolution in our understanding of intracellular vesicles and their range of associated trafficking networks, with slight alterations in functioning already being identified as hallmarks of disease states, highlighting the need to reinvestigate old pathways with a new eye for detail.94–99 For instance, lysosomes, once considered solely as the degradative arm of the cell, are now known to be far more dynamic and broader-functioning compartments, and play a key role in metabolic sensing for the cell.100,101 These advances in our understanding are built largely on advanced imaging-based investigations but have been difficult to dissect further due to our inability to isolate and chemically resolve subpopulations in a cell-free environment.102,103 As discussed previously, isolation of vesicle populations is currently achieved through density-based fractionation or with immunopurification using a limited range of membrane protein markers to define a population, and relying on analysis of these bulk populations results in averaged measurements and a consequent loss of intricacies within samples.
The introduction of time-resolved magnetic recovery does allow, however, for enriching of subpopulations of vesicles, especially those associated with rare intracellular events, and allows for time-resolved investigation of consecutive trafficking “decision-making” steps to help unravel the mechanistic response to biomolecular corona composition. The temporal distribution can be investigated with imaging-based techniques to track over time the intracellular trajectory of single NPs, as a means to identify the critical steps in the trafficking process for extraction times of interest. The need for a multi-dimensional and multi-parametric analysis to investigate the activity and interaction of subcellular structures and NPs is clearly emerging in this field.
4. Integrating technology towards single magnetic nanoparticle analysis
From the previous discussion, it is evident that even if the use of MNPs allows for NP recovery from cells (and thus in principle allows for the analysis of NP-related intracellular events), bulk analysis techniques are inherently incapable of identifying effects associated with small sub-populations of NPs and cannot resolve rare events. Indeed, by averaging the overall diversity of events, bulk analysis can only reveal the dominant trafficking and decision-making mechanisms, whilst precious information associated with rare events remains hidden. We believe that to extend further the knowledge that MNPs can bring to in-cell bionano interactions, their use must be supported by a development of single particle analysis techniques (Fig. 2c). Indeed, single MNP analysis is the only way to reliably relate biomolecular identity with ultimate cell fate decisions.Throughout intracellular trafficking the NP corona will become altered through the addition and loss of biomolecules from the endocytic trafficking vesicles it interacts with8,16 and will lead to the previously described “biomolecular barcode”. Decoding this barcode requires an understanding of its time-resolved composition of multiple biomolecules. The evolving corona can become a host for multiple bioactive molecules including lipids, and post-translationally modified proteins (for instance differential glycosylation of the corona) resulting in differences in cellular uptake and responses.104,105 With the large impact of individual biomolecules on the interaction between NPs and cells it is necessary to have an in-depth analysis of the multi-layered identity of individual coronas to fully understand the range of interactions of NPs in cells.
4.1 Subcellular and single nanoparticle omics
Omics technologies are increasingly striving for single event analysis, with their limits pushed by the requirement for high-throughput, accurate analysis of low abundance biomolecules. While single cell genomics and transcriptomics are now changing the face of science today, single cell proteomics remains elusive, even if some promising techniques are emerging.106,107 While we wait for single event analysis to come to fruition, several clever approaches in mass spectrometry (MS) have allowed us to get closer than ever to high resolution subcellular event profiling, with novel methods of analysis of isotope labelled samples such as LOPIT and SCOPE-MS,108–111 providing new insights into the complex organelle proteome.The powerful methodology of these approaches could be applied to temporally resolved fractions of magnetically-extracted vesicles to compare and sample protein distribution throughout the endocytic trafficking pathway of NPs. This would allow an overall view of the biomolecular components of the pathway and, if compared to nanoparticle-depleted fractions, there is the possibility of identifying possible subtypes of vesicles involved in NP trafficking and creating reference “maps” of NP journeys through the cell, a powerful resource in bionano interaction studies.
Performing single-corona proteomics is far more complex than complete single vesicle analysis. Integration of the numerous approaches of advanced downstream analysis with magnetic extraction, under optimal conditions as we have outlined, could aid in identifying single NP biomolecular microenvironments within a complex mixture. Incorporation of analysis of lipidomics, proteomics and post-translational modification states of corona proteins and vesicle carriers will help form the basis of ultramodern NP interactomics.
4.2 Quantitative evaluation of the biological identity of recovered magnetic nanoparticles through surface mapping
One of the most significant problems in nanoscale event analysis via conventional flow cytometers is their fluidic system, which is engineered to hydrodynamically focus cells into single file for their individual analysis and detection within the flow cell, the detection chamber. Flow cytometers, which are mainly in use for immune cell phenotyping and analysis, are typically built with three radius settings based on different standard cell sizes. This means the radius is much greater than the width of organelles or nanoscale vesicles, making it possible for multiple particles to be detected simultaneously as one event. Therefore, flow cytometry experiments require extensive preparation and optimization to prevent coincident events. For this reason, dedicated nano cytometers are being commercialized and fabricated to overcome limitations of current technologies, incorporating flow-based systems for high-throughput analysis of single nanoscale particles.123 The ultimate achievement of nanoscale cytometry would be the incorporation of nanoscale flow-assisted cell sorting (FACS), or nanoFACS, which could sort identified rare populations for further analysis. Current FACS technology is being employed for use in sorting exosome populations, but its accuracy in sorting is still below the threshold to allow reliable sorting of small particles.124
Mass cytometry, often known by its commercial name, cytometry by time of flight (CyTOF), is driving major advances in single-cell profiling, and is beginning to be utilized for single-organelle analysis. It is more powerful than conventional fluorescence cytometry, which is usually limited by dye spectral overlap and detector configuration, as it is based on analysing cell samples with a panel of isotope labelled antibodies, the wide range of potential isotope labels allows for a broader range of simultaneous measurements per sample, from a maximum of approximately 12 labels for fluorescence cytometry up to 40+ with CyTOF.42,125 Although the hurdle of antibody absorption remains present for experimental design, it provides interesting potential for high-dimensional epitope mapping.
Through identifying compartment specific biomolecules from the surface of extracted coronas, there is the potential to relate back to trafficking steps made by single NPs. This form of “lineage mapping” could be achieved by integrating knowledge acquired in single nanoparticle analysis to read the corona as a biomolecular barcode. This high-dimensional data could then be processed through dimensionality reduction analyses to create full lineage maps of a nanoparticle population with the potential to create interactome maps for multiple preparations of nanoparticle corona under investigation.126 Understanding the full range of interactions of introduced nanomedicines is currently the main roadblock towards clinical translation and therefore it is vital to map the full bionano interactome of engineered nanomaterials to cross this clinical barrier.
5. Conclusion
The lack of in-depth knowledge of the in situ biological interactions of nanostructures has been a major factor hampering both the advancement of the field and nanoscale clinical translation. Through their unique properties, magnetic nanoparticles provide an anchor for researchers for the selective and temporally-resolved recovery of delicate intracellular bio-nanostructures, providing insights into intracellular decision-making events and trafficking pathways. We have outlined the state-of-the-art in magnetic extractions, along with both its key advantages and shortcomings, and have shown how magnetic recovery may be integrated into a workflow combining low-throughput super resolution microscopy with high-throughput omics analysis and nanoparticle surface mapping. This approach in principle enables the quantitative association of the molecular barcode of trafficked nanoparticles with key biological decision-making events (Fig. 3). This knowledge will have profound implications for the design of novel targeted nanomedicines. While the technological, instrumental, and methodological developments are progressing, there is now an interesting opportunity (and need) for a much deeper and stronger connection between those developments, and the key bionanoscience questions: in this regard, we have emphasized the need for analysis to be refined to the level of single cellular sub-compartments, narrow populations of intracellular nanoparticles, and ultimately single-particle analysis, if the field is to truly advance. The combination of teams of scientists that are able to bring the relevant concepts, skills, and infrastructure together to develop our understanding of mechanisms could now significantly affect the pace of overall development of nanoscale biomedical research and translation. This represents a significant opportunity for those in relevant communities.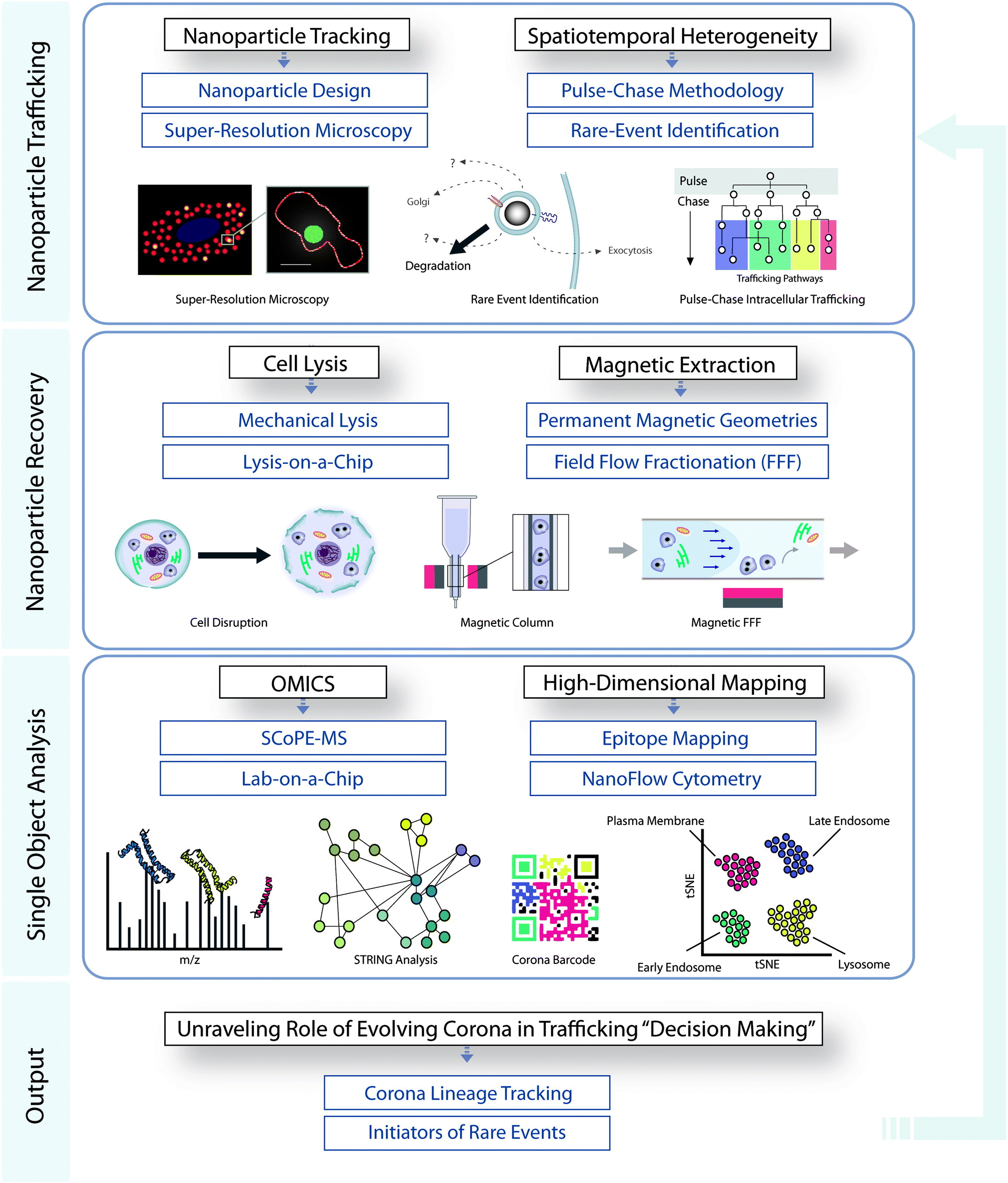 | ||
| Fig. 3 Integrated workflow for studying the role of evolving corona in intracellular decision making. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
KAD, LA, ES and SV acknowledge that this publication has emanated from research supported in part by grants from Science Foundation Ireland [17/NSFC/4898 (KAD, LA and ES), 17/ERCD/4962 (KAD) and 15/SIRG/3423 (SV)]. LC acknowledges this work was part-funded by the Celtic Advanced Life Science Innovation Network (CALIN), an EU INTERREG project part-funded by Welsh government through the Ireland-Wales Programme. SV also acknowledges support by the PhD Research Scholarship from the UCD School of Biomolecular and Biomedical Science. JAB acknowledges the support of the Irish Research Council under Grant Number GOIPD/2020/434. Fig. 2c and the TOC were created with http://BioRender.com.References
- G. Chen, I. Roy, C. Yang and P. N. Prasad, Chem. Rev., 2016, 116, 2826–2885 CrossRef CAS PubMed.
- J. Yoo, C. Park, G. Yi, D. Lee and H. Koo, Cancers, 2019, 11, 640 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941 CrossRef CAS PubMed.
- R. Tietze, J. Zaloga, H. Unterweger, S. Lyer, R. P. Friedrich, C. Janko, M. Pöttler, S. Dürr and C. Alexiou, Biochem. Biophys. Res. Commun., 2015, 468, 463–470 CrossRef CAS PubMed.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. Chan, Nat. Rev. Mater., 2016, 1, 16014 CrossRef CAS.
- D. Bobo, K. J. Robinson, J. Islam, K. J. Thurecht and S. R. Corrie, Pharm. Res., 2016, 33, 2373 CrossRef CAS PubMed.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- P. M. Kelly, C. Åberg, E. Polo, A. O'Connell, J. Cookman, J. Fallon, Ž. Krpetić and K. A. Dawson, Nat. Nanotechnol., 2015, 10, 472 CrossRef CAS PubMed.
- S. Lara, F. Alnasser, E. Polo, D. Garry, M. C. Lo Giudice, D. R. Hristov, L. Rocks, A. Salvati, Y. Yan and K. A. Dawson, ACS Nano, 2017, 11, 1884–1893 CrossRef CAS PubMed.
- V. Francia, K. Yang, S. Deville, C. Reker-Smit, I. Nelissen and A. Salvati, ACS Nano, 2019, 13, 11107–11121 CrossRef CAS PubMed.
- S. Ritz, S. Schöttler, N. Kotman, G. Baier, A. Musyanovych, J. Kuharev, K. Landfester, H. Schild, O. Jahn, S. Tenzer and V. Mailänder, Biomacromolecules, 2015, 16, 1311–1321 CrossRef CAS PubMed.
- Y. Zhang, J. L. Y. Wu, J. Lazarovits and W. C. W. Chan, J. Am. Chem. Soc., 2020, 142, 8827–8836 CrossRef PubMed.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggård, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- H. Mohammad-Beigi, Y. Hayashi, C. M. Zeuthen, H. Eskandari, C. Scavenius, K. Juul-Madsen, T. Vorup-Jensen, J. J. Enghild and D. S. Sutherland, Nat. Commun., 2020, 11, 4535 CrossRef CAS PubMed.
- M. Qin, J. Zhang, M. Li, D. Yang, D. Liu, S. Song, J. Fu, H. Zhang, W. Dai, X. Wang, Y. Wang, B. He and Q. Zhang, Theranostics, 2020, 10, 1213–1229 CrossRef CAS PubMed.
- S. Barua and S. Mitragotri, Nano Today, 2014, 9, 223–243 CrossRef CAS PubMed.
- P. Sandin, L. W. Fitzpatrick, J. C. Simpson and K. A. Dawson, ACS Nano, 2012, 6, 1513–1521 CrossRef CAS PubMed.
- P. Foroozandeh and A. A. Aziz, Nanoscale Res. Lett., 2018, 13, 339 CrossRef PubMed.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS PubMed.
- Q. A. Pankhurst, N. T. K. Thanh, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2009, 42, 224001 CrossRef.
- J. Reguera, D. Jiménez de Aberasturi, M. Henriksen-Lacey, J. Langer, A. Espinosa, B. Szczupak, C. Wilhelm and L. M. Liz-Marzán, Nanoscale, 2017, 9, 9467–9480 RSC.
- C. Sun, J. S. H. Lee and M. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1252–1265 CrossRef CAS PubMed.
- A. Espinosa, J. Reguera, A. Curcio, Á. Muñoz-Noval, C. Kuttner, A. Van de Walle, L. M. Liz-Marzán and C. Wilhelm, Small, 2020, 16, 1904960 CrossRef CAS PubMed.
- F. Gazeau, M. Lévy and C. Wilhelm, Nanomedicine, 2008, 3, 831–844 CrossRef CAS PubMed.
- R. Di Corato, A. Espinosa, L. Lartigue, M. Tharaud, S. Chat, T. Pellegrino, C. Ménager, F. Gazeau and C. Wilhelm, Biomaterials, 2014, 35, 6400–6411 CrossRef CAS PubMed.
- Z. Nemati, J. Alonso, I. Rodrigo, R. Das, E. Garaio, J. A. Garcia, I. Orue, M.-H. Phan and H. Srikanth, J. Phys. Chem. C, 2018, 122, 2367 CrossRef CAS.
- F. M. Kievit and M. Zhang, Acc. Chem. Res., 2011, 44, 853–862 CrossRef CAS PubMed.
- F. Bertoli, G.-L. Davies, M. P. Monopoli, M. Moloney, Y. K. Gun'ko, A. Salvati and K. A. Dawson, Small, 2014, 10, 3307–3315 CrossRef CAS PubMed.
- I. Šafařík and M. Šafaříková, J. Chromatogr. B: Biomed. Sci. Appl., 1999, 722, 33–53 CrossRef.
- A. A. Neurauter, M. Bonyhadi, E. Lien, L. Nøkleby, E. Ruud, S. Camacho and T. Aarvak, in Cell separation, Springer, 2007, pp. 41–73 Search PubMed.
- J. Fritsch, V. Tchikov, L. Hennig, R. Lucius and S. Schutze, Traffic, 2019, 20, 246–258 CrossRef CAS PubMed.
- W. W. Chen, E. Freinkman and D. M. Sabatini, Nat. Protoc., 2017, 12, 2215–2231 CrossRef.
- H. T. Hornig-Do, G. Gunther, M. Bust, P. Lehnartz, A. Bosio and R. J. Wiesner, Anal. Biochem., 2009, 389, 1–5 CrossRef CAS PubMed.
- L. Kappler, J. Li, H. U. Haring, C. Weigert, R. Lehmann, G. Xu and M. Hoene, Sci. Rep., 2016, 6, 21107 CrossRef CAS PubMed.
- D. C. Meeker, Finite Element Method Magnetics, 2018, http://www.femm.info/wiki/HomePage, (accessed December 2020) Search PubMed.
- M. Zborowski and J. J. Chalmers, Anal. Chem., 2011, 83, 8050–8056 CrossRef CAS PubMed.
- P. Joshi, P. S. Williams, L. R. Moore, T. Caralla, C. Boehm, G. Muschler and M. Zborowski, Anal. Chem., 2015, 87, 9908–9915 CrossRef CAS PubMed.
- O. Baun and P. Blümler, J. Magn. Magn. Mater., 2017, 439, 294–304 CrossRef CAS.
- D. J. Gauthier, J. A. Sobota, F. Ferraro, R. E. Mains and C. Lazure, Proteomics, 2008, 8, 3848–3861 CrossRef CAS PubMed.
- M. Degtyarev, M. Reichelt and K. Lin, PLoS One, 2014, 9, e87707 CrossRef PubMed.
- H. M. G. Brown and E. A. Arriaga, Anal. Chem., 2018, 90, 13315–13321 CrossRef CAS PubMed.
- R. Wildgruber, G. Weber, P. Wise, D. Grimm and J. Bauer, Proteomics, 2014, 14, 629–636 CrossRef CAS PubMed.
- M. Islinger, R. Wildgruber and A. Volkl, Electrophoresis, 2018, 39, 2288–2299 CrossRef CAS PubMed.
- A. K. Tharkeshwar, K. Gevaert and W. Annaert, Proteomics, 2018, 18, e1700113 CrossRef PubMed.
- J. G. Webster, M. Zborowski and J. J. Chalmers, in Wiley Encyclopedia of Electrical and Electronics Engineering, 2015, pp. 1–23, DOI:10.1002/047134608x.W8236.
- L. Borlido, A. M. Azevedo, A. C. Roque and M. R. Aires-Barros, Biotechnol. Adv., 2013, 31, 1374–1385 CrossRef CAS PubMed.
- X. Xuan, Micromachines, 2019, 10, 744 CrossRef PubMed.
- M. Hejazian, W. Li and N. T. Nguyen, Lab Chip, 2015, 15, 959–970 RSC.
- L. M. Bronstein, X. Huang, J. Retrum, A. Schmucker, M. Pink, B. D. Stein and B. Dragnea, Chem. Mater., 2007, 19, 3624–3632 CrossRef CAS.
- J. T. Jang, H. Nah, J. H. Lee, S. H. Moon, M. G. Kim and J. Cheon, Angew. Chem., Int. Ed., 2009, 48, 1234–1238 CrossRef CAS PubMed.
- S. Kralj and D. Makovec, ACS Nano, 2015, 9, 9700–9707 CrossRef CAS PubMed.
- J. Park, K. An, Y. Hwang, J.-G. Park, H.-J. Noh, J.-Y. Kim, J.-H. Park, N.-M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891–895 CrossRef CAS PubMed.
- J. Lunacek, M. Lesnak, P. Jandacka, R. Dvorsky, J. Repkova, J. Seidlerova and N. Vitkovska, Sep. Sci. Technol., 2015, 50, 2606–2615 CAS.
- J. S. Park, S. H. Song and H. I. Jung, Lab Chip, 2009, 9, 939–948 RSC.
- H. Felice, G. Ambrosio, M. Anerella, R. Bossert, S. Caspi, D. W. Cheng, D. R. Dietderich, P. Ferracin, A. K. Ghosh, R. Hafalia, C. R. Hannaford, V. Kashikhin, J. Schmalze, S. Prestemon, G. L. Sabbi, P. Wanderer and A. V. Zlobin, IEEE Trans. Appl. Supercond., 2009, 19, 1235–1239 CAS.
- J. E. Hilton and S. M. McMurry, J. Magn. Magn. Mater., 2012, 324, 2051–2056 CrossRef CAS.
- A. Ditsch, S. Lindenmann, P. E. Laibinis, D. I. C. Wang and T. A. Hatton, Ind. Eng. Chem. Res., 2005, 44, 6824–6836 CrossRef CAS.
- G. D. Moeser, K. A. Roach, W. H. Green, T. Alan Hatton and P. E. Laibinis, AIChE J., 2004, 50, 2835–2848 CrossRef CAS.
- C. V. Mura, M. I. Becker, A. Orellana and D. Wolff, J. Immunol. Methods, 2002, 260, 263–271 CrossRef CAS.
- M. Lewin, N. Carlesso, C.-H. Tung, X.-W. Tang, D. Cory, D. T. Scadden and R. Weissleder, Nat. Biotechnol., 2000, 18, 410–414 CrossRef CAS PubMed.
- R. Hanamsagar, T. Reizis, M. Chamberlain, R. Marcus, F. O. Nestle, E. de Rinaldis and V. Savova, Sci. Rep., 2020, 10, 2219 CrossRef CAS PubMed.
- C. Wang, C. Wang, X. Wang, K. Wang, Y. Zhu, Z. Rong, W. Wang, R. Xiao and S. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 19495–19505 CrossRef CAS PubMed.
- J. H. Min, M. K. Woo, H. Y. Yoon, J. W. Jang, J. H. Wu, C. S. Lim and Y. K. Kim, Anal. Biochem., 2014, 447, 114–118 CrossRef CAS PubMed.
- S. Ghahari, S. Ghahari and G. A. Nematzadeh, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2018, 1102–1103, 125–134 CrossRef CAS PubMed.
- B. Banik and S. Dhar, Curr. Protoc. Cell Biol., 2017, 76, 25.24.21–25.24.20 Search PubMed.
- S. Zong, L. Wang, C. Chen, J. Lu, D. Zhu, Y. Zhang, Z. Wang and Y. Cui, Anal. Methods, 2016, 8, 5001–5008 RSC.
- A. K. Tharkeshwar, J. Trekker, W. Vermeire, J. Pauwels, R. Sannerud, D. A. Priestman, D. Te Vruchte, K. Vints, P. Baatsen, J. P. Decuypere, H. Lu, S. Martin, P. Vangheluwe, J. V. Swinnen, L. Lagae, F. Impens, F. M. Platt, K. Gevaert and W. Annaert, Sci. Rep., 2017, 7, 41408 CrossRef CAS PubMed.
- S. P. Yeap, S. S. Leong, A. L. Ahmad, B. S. Ooi and J. Lim, J. Phys. Chem. C, 2014, 118, 24042–24054 CrossRef CAS.
- T. M. Vickrey and J. A. Garcia-ramirez, Sep. Sci. Technol., 1980, 15, 1297–1304 CrossRef CAS.
- H. Kato, in Characterization of Nanoparticles, ed. V.-D. Hodoroaba, W. E. S. Unger and A. G. Shard, Elsevier, 2020, pp. 249–264, DOI:10.1016/b978-0-12-814182-3.00016-x.
- C. Contado, Anal. Bioanal. Chem., 2017, 409, 2501–2518 CrossRef CAS PubMed.
- F. Del Giudice, H. Madadi, M. M. Villone, G. D'Avino, A. M. Cusano, R. Vecchione, M. Ventre, P. L. Maffettone and P. A. Netti, Lab Chip, 2015, 15, 1912–1922 RSC.
- G. D. Chen, C. J. Alberts, W. Rodriguez and M. Toner, Anal. Chem., 2010, 82, 723–728 CrossRef CAS PubMed.
- N. Pamme, Lab Chip, 2006, 6, 24–38 RSC.
- T. Salafi, K. K. Zeming and Y. Zhang, Lab Chip, 2017, 17, 11–33 RSC.
- T. C. Schunk, J. Gorse and M. F. Burke, Sep. Sci. Technol., 1984, 19, 653–666 CrossRef CAS.
- A. H. Latham, R. S. Freitas, P. Schiffer and M. E. Williams, Anal. Chem., 2005, 77, 5055–5062 CrossRef CAS PubMed.
- H. Zhang, D. Freitas, H. S. Kim, K. Fabijanic, Z. Li, H. Chen, M. T. Mark, H. Molina, A. B. Martin, L. Bojmar, J. Fang, S. Rampersaud, A. Hoshino, I. Matei, C. M. Kenific, M. Nakajima, A. P. Mutvei, P. Sansone, W. Buehring, H. Wang, J. P. Jimenez, L. Cohen-Gould, N. Paknejad, M. Brendel, K. Manova-Todorova, A. Magalhães, J. A. Ferreira, H. Osório, A. M. Silva, A. Massey, J. R. Cubillos-Ruiz, G. Galletti, P. Giannakakou, A. M. Cuervo, J. Blenis, R. Schwartz, M. S. Brady, H. Peinado, J. Bromberg, H. Matsui, C. A. Reis and D. Lyden, Nat. Cell Biol., 2018, 20, 332–343 CrossRef CAS PubMed.
- Q. Feng, S. S. Lee and B. Kornmann, Micromachines, 2019, 10, 538 CrossRef PubMed.
- J. Lim, C. Lanni, E. R. Evarts, F. Lanni, R. D. Tilton and S. A. Majetich, ACS Nano, 2011, 5, 217–226 CrossRef CAS PubMed.
- F. Etoc, D. Lisse, Y. Bellaiche, J. Piehler, M. Coppey and M. Dahan, Nat. Nanotechnol., 2013, 8, 193–198 CrossRef CAS.
- X. Wang, C. Ho, Y. Tsatskis, J. Law, Z. Zhang, M. Zhu, C. Dai, F. Wang, M. Tan, S. Hopyan, H. McNeill and Y. Sun, Sci. Robot., 2019, 4, eaav6180 CrossRef.
- M. Shehadul Islam, A. Aryasomayajula and P. R. Selvaganapathy, Micromachines, 2017, 8, 83 CrossRef.
- S. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Chem. Soc. Rev., 2017, 46, 4218–4244 RSC.
- A. Paillard, F. Hindré, C. Vignes-Colombeix, J.-P. Benoit and E. Garcion, Biomaterials, 2010, 31, 7542–7554 CrossRef CAS PubMed.
- Y. Cui, W. Shan, R. Zhou, M. Liu, L. Wu, Q. Guo, Y. Zheng, J. Wu and Y. Huang, Nanoscale, 2018, 10, 1494–1507 RSC.
- T. Yong, X. Zhang, N. Bie, H. Zhang, X. Zhang, F. Li, A. Hakeem, J. Hu, L. Gan and H. A. Santos, Nat. Commun., 2019, 10, 1–16 CrossRef CAS PubMed.
- C. C. Fleischer and C. K. Payne, J. Phys. Chem. B, 2014, 118, 14017–14026 CrossRef CAS PubMed.
- A. Gupta, F. Rivera-Molina, Z. Xi, D. Toomre and A. Schepartz, Nat. Chem. Biol., 2020, 16, 408–414 CrossRef CAS PubMed.
- J. Klumperman and G. Raposo, Cold Spring Harbor Perspect. Biol., 2014, 6, a016857 CrossRef PubMed.
- H. Wu, P. Carvalho and G. K. Voeltz, Science, 2018, 361, eaan5835 CrossRef PubMed.
- N. Naslavsky and S. Caplan, J. Cell Sci., 2018, 131, jcs216499 CrossRef PubMed.
- R. Willett, J. A. Martina, J. P. Zewe, R. Wills, G. R. V. Hammond and R. Puertollano, Nat. Commun., 2017, 8, 1580 CrossRef PubMed.
- J. C. Stinchcombe, E. Majorovits, G. Bossi, S. Fuller and G. M. Griffiths, Nature, 2006, 443, 462–465 CrossRef CAS PubMed.
- J. S. Bonifacino and J. Neefjes, Curr. Opin. Cell Biol., 2017, 47, 1–8 CrossRef CAS PubMed.
- J. Neefjes, M. M. L. Jongsma and I. Berlin, Trends Cell Biol., 2017, 27, 580–594 CrossRef CAS PubMed.
- D. Dong, X. Huang, L. Li, H. Mao, Y. Mo, G. Zhang, Z. Zhang, J. Shen, W. Liu, Z. Wu, G. Liu, Y. Liu, H. Yang, Q. Gong, K. Shi and L. Chen, Light: Sci. Appl., 2020, 9, 11 CrossRef CAS PubMed.
- R. Sannerud, C. Esselens, P. Ejsmont, R. Mattera, L. Rochin, A. K. Tharkeshwar, G. De Baets, V. De Wever, R. Habets, V. Baert, W. Vermeire, C. Michiels, A. J. Groot, R. Wouters, K. Dillen, K. Vints, P. Baatsen, S. Munck, R. Derua, E. Waelkens, G. S. Basi, M. Mercken, M. Vooijs, M. Bollen, J. Schymkowitz, F. Rousseau, J. S. Bonifacino, G. Van Niel, B. De Strooper and W. Annaert, Cell, 2016, 166, 193–208 CrossRef CAS PubMed.
- R. E. Lawrence and R. Zoncu, Nat. Cell Biol., 2019, 21, 133–142 CrossRef CAS PubMed.
- K. Dan, A. T. Veetil, K. Chakraborty and Y. Krishnan, Nat. Nanotechnol., 2019, 14, 252–259 CrossRef CAS PubMed.
- K. Leung, K. Chakraborty, A. Saminathan and Y. Krishnan, Nat. Nanotechnol., 2019, 14, 176–183 CrossRef CAS PubMed.
- A. M. Valm, S. Cohen, W. R. Legant, J. Melunis, U. Hershberg, E. Wait, A. R. Cohen, M. W. Davidson, E. Betzig and J. Lippincott-Schwartz, Nature, 2017, 546, 162–167 CrossRef CAS PubMed.
- S. Wan, P. M. Kelly, E. Mahon, H. Stockmann, P. M. Rudd, F. Caruso, K. A. Dawson, Y. Yan and M. P. Monopoli, ACS Nano, 2015, 9, 2157–2166 CrossRef CAS PubMed.
- F. Bertoli, D. Garry, M. P. Monopoli, A. Salvati and K. A. Dawson, ACS Nano, 2016, 10, 10471–10479 CrossRef CAS.
- N. Slavov, Science, 2020, 367, 512 CrossRef CAS PubMed.
- B. Budnik, E. Levy, G. Harmange and N. Slavov, Genome Biol., 2018, 19, 161 CrossRef PubMed.
- E. Lundberg and G. H. H. Borner, Nat. Rev. Mol. Cell Biol., 2019, 20, 285–302 CrossRef CAS PubMed.
- A. Geladaki, N. Kočevar Britovšek, L. M. Breckels, T. S. Smith, O. L. Vennard, C. M. Mulvey, O. M. Crook, L. Gatto and K. S. Lilley, Nat. Commun., 2019, 10, 331 CrossRef PubMed.
- L. J. Foster, C. L. de Hoog, Y. Zhang, Y. Zhang, X. Xie, V. K. Mootha and M. Mann, Cell, 2006, 125, 187–199 CrossRef CAS PubMed.
- T. P. Dunkley, R. Watson, J. L. Griffin, P. Dupree and K. S. Lilley, Mol. Cell. Proteomics, 2004, 3, 1128–1134 CrossRef CAS PubMed.
- L. M. Herda, D. R. Hristov, M. C. Lo Giudice, E. Polo and K. A. Dawson, J. Am. Chem. Soc., 2017, 139, 111–114 CrossRef CAS PubMed.
- N. Arraud, R. Linares, S. Tan, C. Gounou, J.-M. Pasquet, S. Mornet and A. R. Brisson, J. Thromb. Haemostasis, 2014, 12, 614–627 CrossRef CAS PubMed.
- S. P. Perfetto, P. K. Chattopadhyay and M. Roederer, Nat. Rev. Immunol., 2004, 4, 648–655 CrossRef CAS PubMed.
- M. C. Lo Giudice, L. M. Herda, E. Polo and K. A. Dawson, Nat. Commun., 2016, 7, 13475 CrossRef CAS PubMed.
- P. Holliger and P. J. Hudson, Nat. Biotechnol., 2005, 23, 1126–1136 CrossRef CAS PubMed.
- S. Muyldermans, Annu. Rev. Biochem., 2013, 82, 775–797 CrossRef CAS PubMed.
- G. P. Smith, Science, 1985, 228, 1315–1317 CrossRef CAS PubMed.
- R. H. Hoess, Chem. Rev., 2001, 101, 3205–3218 CrossRef CAS PubMed.
- S. L. Deutscher, Chem. Rev., 2010, 110, 3196–3211 CrossRef CAS PubMed.
- R. Stoltenburg, C. Reinemann and B. Strehlitz, Biomol. Eng., 2007, 24, 381–403 CrossRef CAS PubMed.
- S. Tombelli, M. Minunni and M. Mascini, Biosens. Bioelectron., 2005, 20, 2424–2434 CrossRef CAS PubMed.
- H. Lian, S. He, C. Chen and X. Yan, Annu. Rev. Anal. Chem., 2019, 12, 389–409 CrossRef CAS PubMed.
- A. Morales-Kastresana, T. A. Musich, J. A. Welsh, W. Telford, T. Demberg, J. C. S. Wood, M. Bigos, C. D. Ross, A. Kachynski, A. Dean, E. J. Felton, J. Van Dyke, J. Tigges, V. Toxavidis, D. R. Parks, W. R. Overton, A. H. Kesarwala, G. J. Freeman, A. Rosner, S. P. Perfetto, L. Pasquet, M. Terabe, K. McKinnon, V. Kapoor, J. B. Trepel, A. Puri, H. Kobayashi, B. Yung, X. Chen, P. Guion, P. Choyke, S. J. Knox, I. Ghiran, M. Robert-Guroff, J. A. Berzofsky and J. C. Jones, J. Extracell. Vesicles, 2019, 8, 1597603 CrossRef CAS PubMed.
- R. Gadalla, B. Noamani, B. L. MacLeod, R. J. Dickson, M. Guo, W. Xu, S. Lukhele, H. J. Elsaesser, A. R. A. Razak, N. Hirano, T. L. McGaha, B. Wang, M. Butler, C. J. Guidos, P. S. Ohashi, L. L. Siu and D. G. Brooks, Front. Radiat. Oncol., 2019, 9 DOI:10.3389/fonc.2019.00415..
- S. Palit, C. Heuser, G. P. de Almeida, F. J. Theis and C. E. Zielinski, Front. Immunol., 2019, 10 DOI:10.3389/fimmu.2019.01515.
Footnote |
| † Emily Sheridan and Silvia Vercellino contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2021 |