DOI:
10.1039/D0NA00992J
(Paper)
Nanoscale Adv., 2021,
3, 1464-1472
Stable lead-halide perovskite quantum dots as efficient visible light photocatalysts for organic transformations†
Received
26th November 2020
, Accepted 16th January 2021
First published on 18th January 2021
Abstract
Lead halide perovskite (LHP) based colloidal quantum dots (CQDs) have tremendous potential for photocatalysis due to their exceptional optical properties. However, their applicability in catalysis is restricted due to poor chemical stability and low recyclability. We report halide-passivated, monodisperse CsPbBr3CQDs as a stable and efficient visible-light photocatalyst for organic transformations. We demonstrate oxidative aromatization of a wide range of heterocyclic substrates including examples which are poor hydrogen transfer (HAT) reagents. Two to five-fold higher rate kinetics were observed for reactions catalyzed by CsPbBr3CQDs in comparison with bulk-type CsPbBr3 (PNCs) or conventionally synthesized CsPbBr3CQDs and other metal organic dyes (rhodamine 6G and [Ru(bpy)3]2+). Furthermore, these CQDs exhibit improved air-tolerance and photostability and in turn show a higher turnover number (TON) of 200, compared to conventionally prepared CQDs (TON = 166) and state-of-the-art bulk-type perovskite-based catalyst (TON = 177). Our study paves the way for the practical applicability of energy-level tunable, size-controlled LHP CQDs as efficient photocatalysts in organic synthesis.
Introduction
Lead halide perovskites (LHPs) have exceptional light absorbing/emitting properties.1–5 In particular, the ability of LHPs to facilitate efficient charge separation and transfer upon photon absorption6 has been extensively explored in the domain of photovoltaics7–11 with power conversion efficiencies approaching the theoretical limit.12,13 These unique photophysical properties of LHPs are also very appealing for activating organic substrates in photoredox catalysis where light absorption, charge separation and transfer are equally important.14–19 LHP based colloidal quantum dots (CQDs) with well-defined size and shape, prepared for example by the hot-injection method,20 may have additional advantages compared to larger sized and polydisperse LHP nanocrystals (PNCs) in photocatalysis: first, naturally they have a larger surface area and the energy levels can be tuned as a function of size9,21,22 to control electronic transfer processes with the substrates. Second, these CQDs are highly dispersible in many organic solvents forming a homogeneous colloidal solution, yet with heterogeneous catalytic attributes such as recyclability and separability. And third, due to their monodisperse, well-defined size, shape and surface morphology, surface facet-controlled catalysis23–25 with high specificity and affinity for various substrates could be explored. Unfortunately, conventionally prepared size- and shape-controlled LHP CQDs have poor air and moisture stability,14,18 limiting their practical application in the photocatalysis. Furthermore, these nanocrystals are typically prepared in small scale compared to their bulk or polydisperse counterparts.14,18 In fact, so far efficient photocatalysis of organic transformations were all achieved using highly polydisperse LHP PNCs (ca. 2–100 nm) with an average size too large to be in quantum confinement regime.14,18 Conventionally prepared colloidal LHP CQDs, in turn, exhibited poor catalytic efficiency due to their poor tolerance of moisture, oxygen, substrates and solvents compared to these larger sized PNCs. In the literature, various strategies to improve air and moisture stability of LHPs can be found, specially of CsPbI3 and CsPbBr3 based perovskites. These strategies encompass the use of additives such as halide salt,26 phosphinic acid,27 ammonium halide,28 2,2′-iminodibenzoic acid,29 sulphides and metal ions30 and polymers31 or imply the application of specific post-synthetic purification steps.9,32 While these approaches have led to improved performances of LHP based optoelectronic devices (e.g., solar cells, LEDs), they could not yet be exploited in the field of photocatalysis.
Herein, we report the use of halide passivated air-stable, CsPbBr3 perovskite CQDs for the synthesis of a broad range of azaheterocycles via oxidative aromatization. Photocatalytic aromatization reactions involving nitrogen heterocycles such as 1,2-dihydropyridines (1,2-DHP), 1,4-dihydropyridines (1,4-DHP), dihydrobenzothiazoles and pyrazolines are important, however, kinetically slow reactions, requiring a strong oxidant and/or efficient catalyst. Although, air is a benign source of molecular oxygen which can act as an oxidant in the singlet state,33–35 conventionally prepared perovskite-based CQDs and bulk halide perovskites are highly sensitive to O2 and moisture, limiting their use in photocatalysis.14,18,36,37 We demonstrate the practical applicability of monodispersed, air-stable CsPbBr3CQDs prepared using the individual Cs, Pb and Br precursors (“three precursors method”) in the oxidative aromatization of azaheterocycles under visible light in open air. The nitrogen containing heterocycles (substrates) and heteroaromatics (products) under study in this report form a highly important class of organic compounds with ubiquitous presence in bioactive natural products or pharmaceutical drugs38–42 and are at the same time the prototypes for the study of biochemical redox reactions.43–45 Previous studies, which mostly focused on the aromatization of 1,2-dihydropyridine (1,2-DHP), 1,4-DHP and 1,3,5-triaryl-pyrazolines as substrates, used Ru-, Pd-, and Pt-based molecular catalysts with some degree of success,45–48 however, at the cost of the use of expensive metal complexes exhibiting oxygen intolerance, absence of recyclability, and fixed redox potentials.
Results and discussion
Synthesis and characterization of air-stable photocatalyst
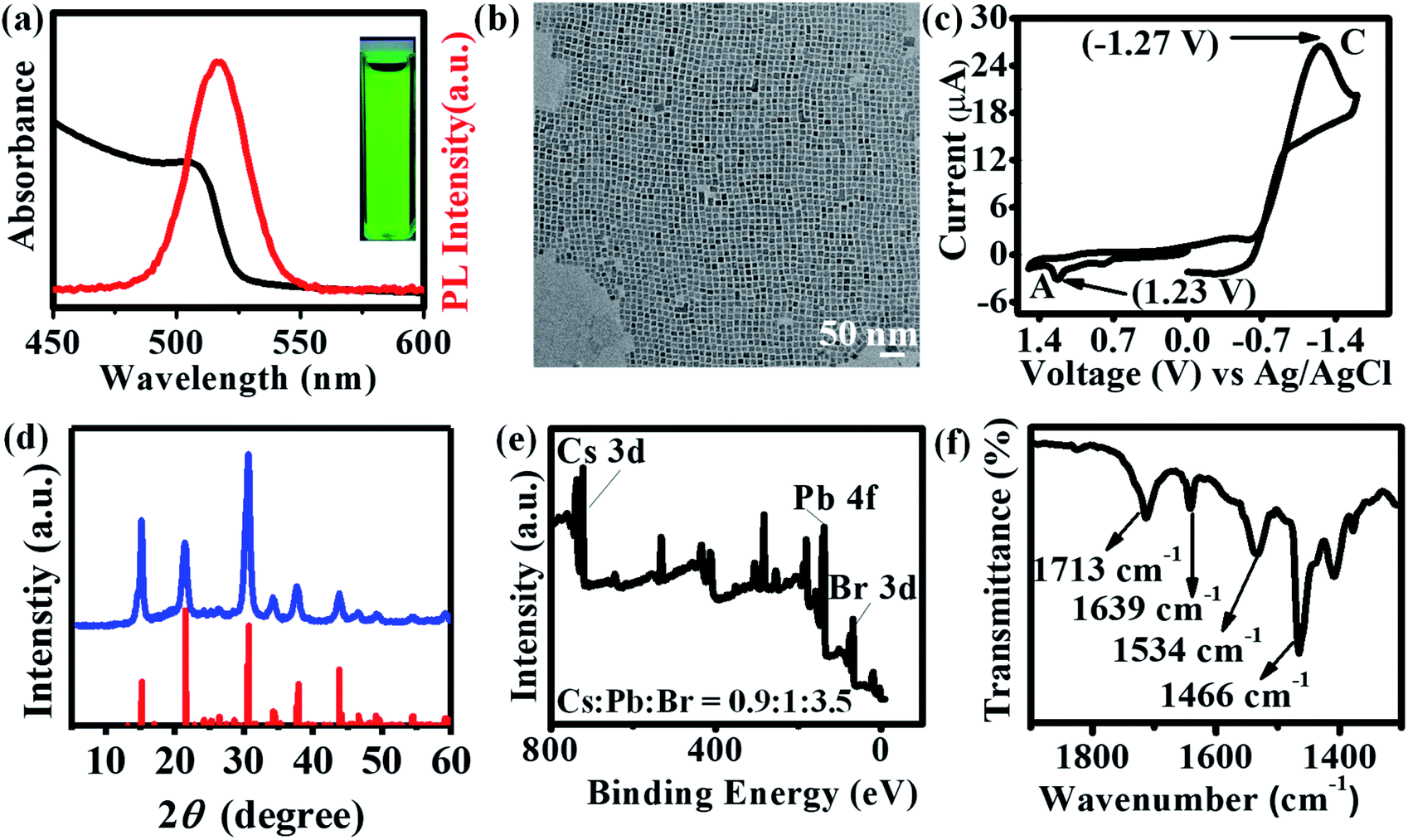
Air-stable, halide-passivated CsPbBr3 NCs (QD1) were prepared under optimized and scaled-up conditions via the “three precursors method” using bromine/oleylamine solution as bromide source based on our previous report (cf. ESI†).22 As opposed to the conventional “two precursors method”20 where three elements (viz., Cs, Pb and Br) are obtained from two precursors (viz., Cs-oleate and PbBr2), the “three precursors method” allows for independent tuning of all three elements and achieving the desired halide-rich synthetic conditions.22,49,50QD1 was prepared under halide-excess condition (Cs![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pb:Br ∼ 1:1:6) at high temperature (200 °C), based on the detailed reaction conditions-stability correlation study from our previous report.22 The colloidal dispersion of QD1 in hexane exhibited a UV-Vis absorption peak at 505 nm and a PL emission peak at 517 nm with an optical band gap of 2.46 eV (Fig. 1a). QD1 exhibits long-term stability in ambient air conserving fluorescence quantum yield (Fig. S1 and S2†). TEM studies show that the nanocrystals have an edge length ∼9.4 ± 0.4 nm (Fig. 1b), which is in the moderate to weak quantum confinement regime.51 Increasing the size of NCs to ∼15.7 ± 0.7 nm (QD2, Fig. S3a and b†) led to a slight red-shift of the emission maximum (521 nm) and decreasing the size to ∼7.2 ± 0.5 nm (QD3, Fig. S3c and d†) resulted in a blue shift (510 nm) attributed to the quantum confinement effect.51–54 In photocatalysis, the knowledge of the relative energy level positions of the catalyst with respect to the substrate is crucial for evaluating and maximizing the driving force of the reaction.18 Cyclic voltammetry (CV) was used to determine the valence band maximum (VBM) and conduction band minimum (CBM) of the QDs.55 The cyclic voltammogram of QD1 (Fig. 1c) shows the anodic peak (A) at 1.23 V and cathodic peak (C) at −1.27 V for one complete cycle. The peak potential difference between these two peaks (A–C) results in an electrochemical bandgap of 2.50 eV, which is consistent with the obtained optical bandgap of QD1.55 The exceptional air-stability of these CQDs (Fig. 1d) is attributed to halide/amine passivated surfaces as confirmed by X-ray photoelectron spectroscopy (XPS) and Fourier transform infrared spectroscopy (FTIR)22 (Fig. 1e and f). The Pb 4f core level spectra of QD1 show peaks at 138.39 and 143.25 eV assigned to the Pb 4f7/2 and Pb 4f5/2 levels, respectively. These values are slightly higher than for conventional CsPbBr3 NCs (QD4) which show peaks at 138.34 and 143.20 eV (Fig. S4c†). This increase in binding energy value (shift by 0.05 eV) is consistent with literature reports for halide-ion rich CsPbBr3 NCs.22,26 The analysis of the Cs 3d, Pb 4f, and Br 3d peaks further confirmed that QD1 displays a higher bromide ion rich surface (Cs:Pb:Br ∼ 0.9:1:3.5, Fig. 1e) in comparison to the QD4 (Cs:Pb:Br ∼ 1.2:1:2.7, Fig. S4b†). Bromine is a volatile liquid and to ensure sufficient bromine precursor, we used excess of bromine (∼6 times) compared to Pb or Cs precursor. Fig. 1f shows a characteristic FTIR band at 1639 cm−1 confirming the presence of protonated amine groups (–NH3+). This observation, taken together with XPS results, confirm the presence of oleylammonium halide on the NC surface which is crucial for stability.56 In addition, the peaks at 1713 cm−1 and 1534 cm−1 are attributed to the C
:Pb:Br ∼ 1:1:6) at high temperature (200 °C), based on the detailed reaction conditions-stability correlation study from our previous report.22 The colloidal dispersion of QD1 in hexane exhibited a UV-Vis absorption peak at 505 nm and a PL emission peak at 517 nm with an optical band gap of 2.46 eV (Fig. 1a). QD1 exhibits long-term stability in ambient air conserving fluorescence quantum yield (Fig. S1 and S2†). TEM studies show that the nanocrystals have an edge length ∼9.4 ± 0.4 nm (Fig. 1b), which is in the moderate to weak quantum confinement regime.51 Increasing the size of NCs to ∼15.7 ± 0.7 nm (QD2, Fig. S3a and b†) led to a slight red-shift of the emission maximum (521 nm) and decreasing the size to ∼7.2 ± 0.5 nm (QD3, Fig. S3c and d†) resulted in a blue shift (510 nm) attributed to the quantum confinement effect.51–54 In photocatalysis, the knowledge of the relative energy level positions of the catalyst with respect to the substrate is crucial for evaluating and maximizing the driving force of the reaction.18 Cyclic voltammetry (CV) was used to determine the valence band maximum (VBM) and conduction band minimum (CBM) of the QDs.55 The cyclic voltammogram of QD1 (Fig. 1c) shows the anodic peak (A) at 1.23 V and cathodic peak (C) at −1.27 V for one complete cycle. The peak potential difference between these two peaks (A–C) results in an electrochemical bandgap of 2.50 eV, which is consistent with the obtained optical bandgap of QD1.55 The exceptional air-stability of these CQDs (Fig. 1d) is attributed to halide/amine passivated surfaces as confirmed by X-ray photoelectron spectroscopy (XPS) and Fourier transform infrared spectroscopy (FTIR)22 (Fig. 1e and f). The Pb 4f core level spectra of QD1 show peaks at 138.39 and 143.25 eV assigned to the Pb 4f7/2 and Pb 4f5/2 levels, respectively. These values are slightly higher than for conventional CsPbBr3 NCs (QD4) which show peaks at 138.34 and 143.20 eV (Fig. S4c†). This increase in binding energy value (shift by 0.05 eV) is consistent with literature reports for halide-ion rich CsPbBr3 NCs.22,26 The analysis of the Cs 3d, Pb 4f, and Br 3d peaks further confirmed that QD1 displays a higher bromide ion rich surface (Cs:Pb:Br ∼ 0.9:1:3.5, Fig. 1e) in comparison to the QD4 (Cs:Pb:Br ∼ 1.2:1:2.7, Fig. S4b†). Bromine is a volatile liquid and to ensure sufficient bromine precursor, we used excess of bromine (∼6 times) compared to Pb or Cs precursor. Fig. 1f shows a characteristic FTIR band at 1639 cm−1 confirming the presence of protonated amine groups (–NH3+). This observation, taken together with XPS results, confirm the presence of oleylammonium halide on the NC surface which is crucial for stability.56 In addition, the peaks at 1713 cm−1 and 1534 cm−1 are attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching (free acid) and oleate anion (surface-bound) vibrations respectively.57 The strong peak at 1466 cm−1 is due to bending vibration of C–H in CH2 group which is integral part of the ligands. Importantly, QD1 showed improved photostability under blue LED compared to PNCs and conventionally prepared CsPbBr3CQDs (QD4) (Fig. S5a–d†).
O stretching (free acid) and oleate anion (surface-bound) vibrations respectively.57 The strong peak at 1466 cm−1 is due to bending vibration of C–H in CH2 group which is integral part of the ligands. Importantly, QD1 showed improved photostability under blue LED compared to PNCs and conventionally prepared CsPbBr3CQDs (QD4) (Fig. S5a–d†).
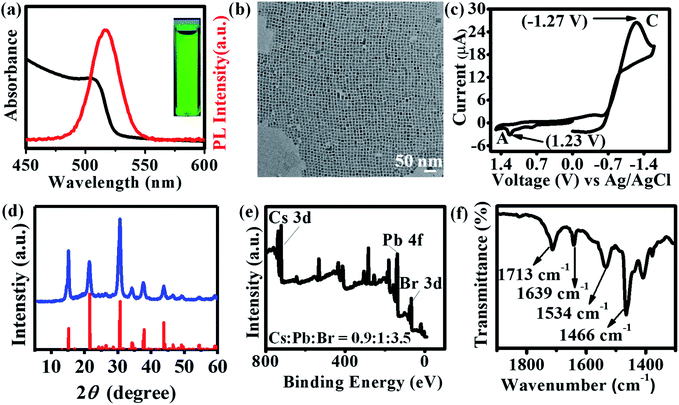 |
| | Fig. 1 Characterization of the CsPbBr3 perovskite QDs (QD1): (a) UV-Vis absorption (black) and photoluminescence emission (red) spectra of the colloidal dispersion (excitation wavelength: 400 nm), inset: colloidal solution under UV lamp; (b) TEM image (cube edge length = ∼9.4 ± 0.4 nm); (c) cyclic voltammogram of the nanocrystals dispersed in acetonitrile/toluene (1:4) solution using tetrabutylammonium perchlorate (TBAP) as supporting electrolyte with a scan rate of 50 mV s−1; (d) powder X-ray diffractogram of the QDs indexed to orthorhombic phase (JCPDS no. 00-054-0752) indicated as red bar; (e) XPS survey spectrum; (f) FTIR spectrum. | |
Screening and optimization of the photoredox reaction
To assess the photocatalytic utility of QD1 in aromatization reactions under air, we selected Hantzsch ester i.e., diethyl 2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (1a) as a model substrate due to its synthetic accessibility. 1a is also a popular NAD(P)H-type model for redox processes based on the dihydropyridine skeleton43–45 and is well known in pharmacology as calcium channel blockers for curing cardiovascular diseases.58,59 Furthermore, CV studies (Fig. S6†) show that the redox potential of 1a is above the VBM of QD1 and therefore, it acts as a potential hole accepting substrate for QD1.
To initiate the photocatalytic studies, 1a was dissolved in dichloromethane (DCM) and illuminated with a blue LED lamp in the presence of a catalytic amount (1 mg) of QD1. NMR studies confirmed the formation of the aromatized product 2a with 85% yield (Scheme 1). The reaction was carried out using off-the-shelf solvent, in air at room temperature without the use of an additional external oxidant. Although Hantzsch ester (1a) is a known hydrogen atom transfer (HAT) reagent, yet it is very slowly oxidized without the catalyst demonstrating the necessity to identify suitable catalysts.45–47 In fact, the aromatization of 1a performed in the absence of catalyst furnished 2a in poor yield (20% yield, entry 3, Table S1†) implying the essential role of QD1 to activate the organic substrate 1a. We screened the reaction of 1a under different synthetic conditions to optimize the catalytic efficiency of QD1. The results of the optimization studies are detailed in Table S1.† In the absence of light, only a trace amount of 2a was detected confirming that the presence of light is necessary to favor the progress of reaction (entry 2, Table S1†). Different solvents of varying polarity and reactivity such as toluene, CHCl3, 1,4-dioxan, THF, 1,2-dichloroethane (DCE) and ethyl acetate were also tested and the successful formation of 2a from 1a was observed in each case with high to excellent yields (entries 5–9, Table S1†). Although, the dispersion of QD1 showed improved photostability in hexane as compared to DCE (Fig. S5†), the photocatalytic reaction was not performed in hexane solvent owing to its non-polar attributes. The precursors (organic substrates) were not soluble in hexane solvent. For the substrate 1a, the best result in terms of reaction yield was observed in 1,2-dichloroethane (DCE) solvent affording the desired product 2a in 95% yield. These parameters were marked as the optimum photocatalytic reaction conditions for further studies (entry 4, Table S1†).
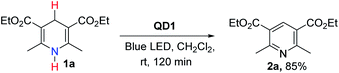 |
| | Scheme 1 Initial result of oxidative aromatization of 1a in the presence of QD1 acting as the photocatalyst. | |
Substrate scope: extension to the less reactive HAT reagents
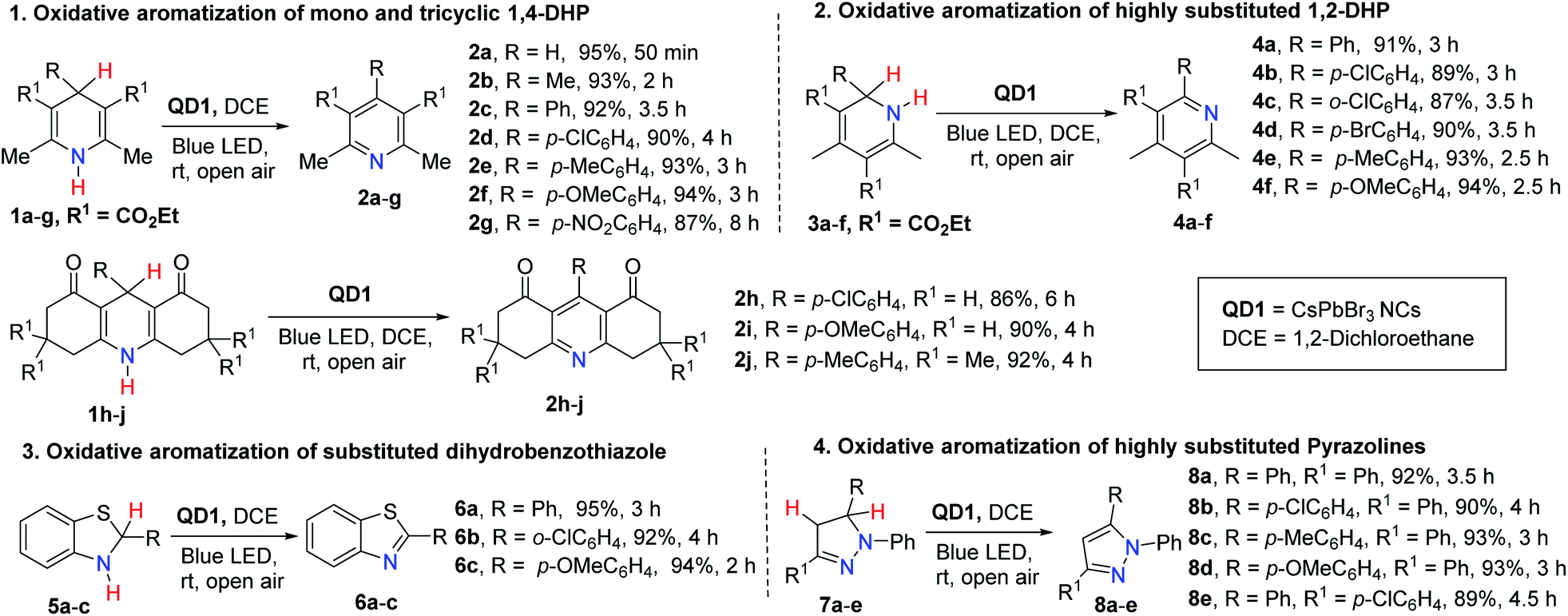
The present photocatalytic protocol is highly efficient and compatible for aromatizing a broad range of heterocyclic substrates. In the following, we demonstrate the successful aromatization of a library of substrates consisting of substituted 1,4-DHP (1a–j), 1,2-DHP (3a–f), 2-aryl-2,3-dihydrobenzothiazole (5a–c) and 1,3,5-triaryl-pyrazoline (7a–e), using QD1 under optimized reaction conditions (solvent: DCE, catalyst: 1 mg QD1, room temperature). Most significantly, the overwhelming majority of these substrates are poor HAT reagents compared to 1a without the catalyst. In fact, only traces of product were detected even when reaction was prolonged to more than 5 h. With the catalyst, the reaction showed excellent yields at much shorter reaction times (86–95%, Fig. 2). Highly pure products were directly isolated from the reaction mixture via centrifugation, without the need of column chromatographic separation techniques. We observed that the reaction rates were different for different substituents, underlining the strong influence of the electronic nature of the substituents (R) on the progress (rate) of the reaction. To understand the quantitative relation between structure and reactivity, detailed kinetic studies were performed on 1,4-DHP based substrates.
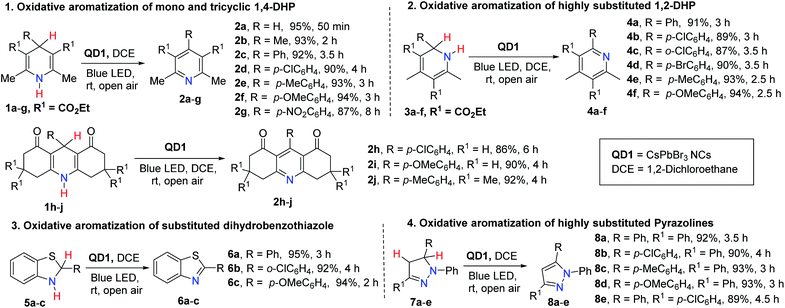 |
| | Fig. 2 Aromatization of a broad variety of substrates comprising 1,4-DHP, 1,2-DHP, dihydrobenzothiazole and pyrazoline derivatives catalyzed by QD1 photocatalyst under blue light irradiation affording a library of corresponding aromatized azaheterocycles. The maximum absorption wavelength and the illumination intensity of the blue LED light (40 W) are 461 nm and 0.363 mW cm−2 at a distance of 100 cm respectively. | |
Structure-reactivity correlation study
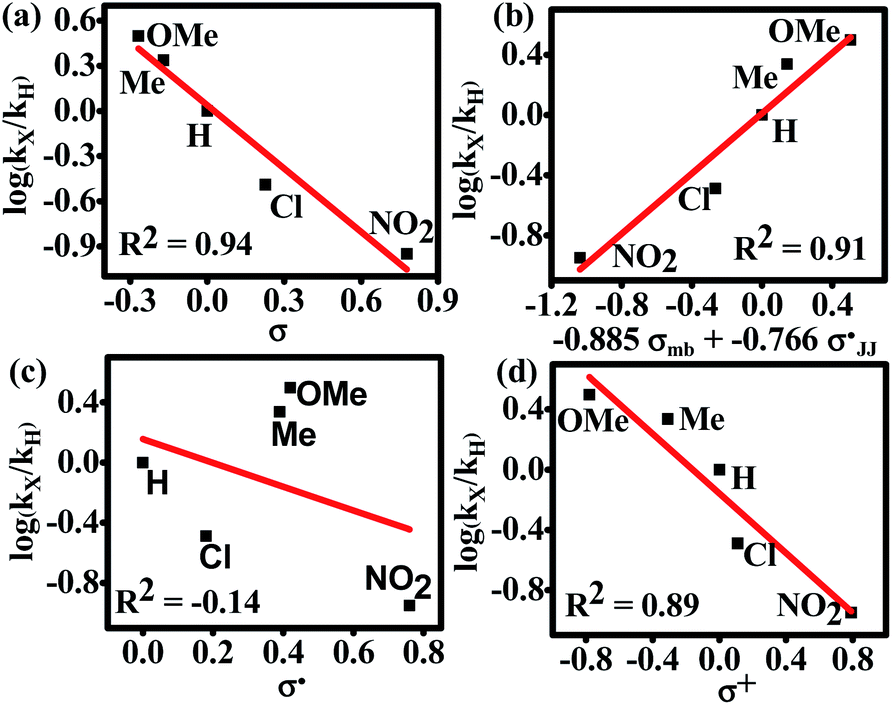
We observed that the rate of oxidative aromatization was faster in the presence of electron donating groups (EDGs) and the reverse effect was noted in the presence of electron withdrawing groups (EWGs) as the substituent. Evidently, kinetic studies reveal that the reaction rate constants of para-substituted 1,4-aryl-DHP (1c–1g) (R = p-X-C6H4, where X = OMe, Me, H, Cl and NO2) decreases four fold as the para-substituent becomes progressively more electron withdrawing (Fig. S7 and Tables S2, S3†). Applying the Hammett equation for this transformation, a linear relationship between logarithms of first order rate constant and Hammett substituent constant (σ)60 was observed, corroborating the strong electronic effect (Fig. 3a). To obtain deeper mechanistic insights from these kinetic data, the more advanced Jiang's dual-parameter61 that considers both polar and spin delocalization effects was used (Fig. 3b). Again, a linear correlation was obtained using a Jiang's dual-parameter, underlining the possible involvement of both ionic and/or radical intermediates in the reaction. In Jiang's dual-parameter formalism, positive ln(kX/kH) experimental values suggest that the reaction is accelerated by EWGs and vice versa. The negative value of the polar reaction constant (ρmb = −0.885) indicates a modest positive charge developed in the transition state, while the negative spin delocalization constant 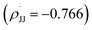 suggests that para-substitution has no effect on the delocalization of the radical formed during the reaction. The ratio of the constants
suggests that para-substitution has no effect on the delocalization of the radical formed during the reaction. The ratio of the constants 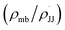 which is slightly more than unity (1.15) strongly indicates that the polar delocalization effect is more dominant compared to the spin delocalization effect. This observation is further corroborated by the fact that a non-linear correlation (Fig. 3c) was obtained when only spin delocalization parameters were considered, whereas a linear correlation was obtained when only polar substitution was considered (Fig. 3d). In other words, these data suggest that the reaction possibly proceeds via radical cation or radical anion intermediates where the electronic effect on reactivity is predominant due to polar effects.
which is slightly more than unity (1.15) strongly indicates that the polar delocalization effect is more dominant compared to the spin delocalization effect. This observation is further corroborated by the fact that a non-linear correlation (Fig. 3c) was obtained when only spin delocalization parameters were considered, whereas a linear correlation was obtained when only polar substitution was considered (Fig. 3d). In other words, these data suggest that the reaction possibly proceeds via radical cation or radical anion intermediates where the electronic effect on reactivity is predominant due to polar effects.
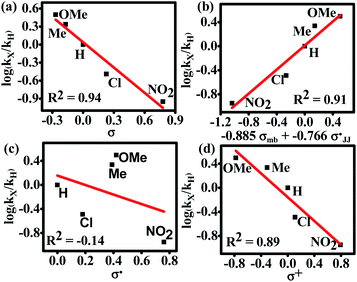 |
| | Fig. 3 Hammett single and dual correlations of the aromatization of para-substituted 1,4-aryl-DHP such as 1c (H), 1d (Cl), 1e (Me), 1f (OMe), and 1g (NO2). (a) Simple Hammett plot corroborating the influence of the inductive effect of the substituents on reactivity; (b) construction of the Hammett plot employing Jiang's dual-parameter; (c) non-linear correlation of the Hammett plot of log(KX/KH) versus σ˙ indicating no spin delocalization; (d) Hammett plot of log(KX/KH) versus σ+ signifying the dominance of polar effects over spin delocalization effects. | |
Driving force and mechanism
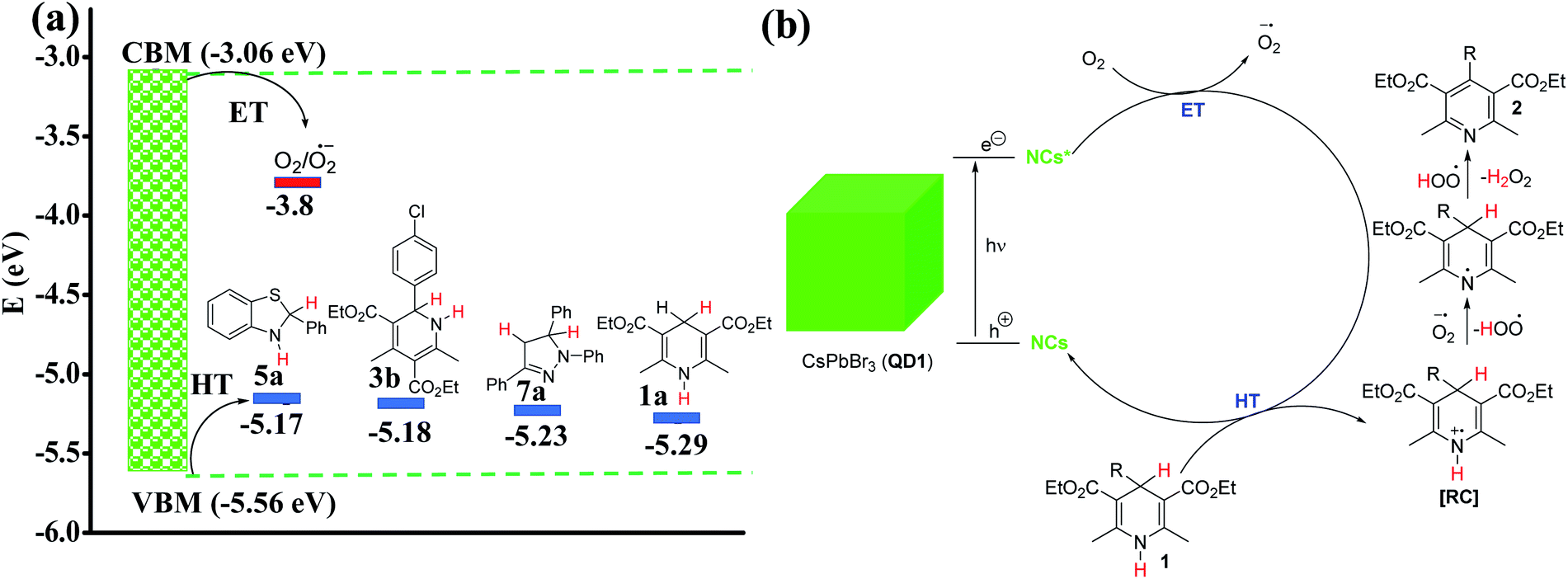
CV studies on four different model azaheterocycle substrates suggested that most of these heterocycles are potential hole acceptors with their redox potential value well above the VBM of the QD1 (Fig. S6, S11b, S12 and S15†). The driving force for electron and hole transfer is provided in Fig. 4a based on electrochemical data. The hole accepting tendency of the substrate is further confirmed by Stern–Volmer studies64 (Fig. S21†). Since the reaction takes place in the presence of air and based on the redox potential values it is safe to conclude that oxygen accepts the electron from the excited QD1 photocatalyst to form a superoxide anion radical [O2˙−] intermediate,33–35 while the substrate (1) accepts the hole to form a radical cation intermediate (RC). This leads to the successful charge separation and activation of the substrate. The in situ generated superoxide anion radical is responsible for the subsequent aromatization of RC to deliver the corresponding aromatized product (2) along with the production of H2O2 (Fig. 4b). A similar mechanism operates for the aromatization of the other substrates such as 3, 5 and 7 (Fig. S22a and b†). The proposed mechanism is also consistent with the previous reports.35,48 Additionally, QD1 catalysed photo-aromatization of 1c (a control experiment) was performed under nitrogen atmosphere, using solvents purged with the nitrogen gas. As expected, the formation of the product 2c was suppressed considerably under strictly air-free conditions. However, the same reaction under open air condition afforded the aromatized product 2c in 92% yield in 3.5 h (Fig. S23†) indicating the key role of O2 in aromatizing the organic substrate. Direct evidence of the radical intermediate was achieved using 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO), which is a widely used radical scavenger. In the presence of TEMPO, the photo-aromatization of 1a was significantly suppressed yielding only 30% of product within 2 h compared to 95% in 50 min in the absence of the radical quencher. The proposed mechanism is further supported by the detection of H2O2 in the reaction mixture (Fig. S24†). It is well-known that 3,3′,5,5′-tetramethylbenzidine (TMB) gives a blue colored diimine complex with a maximum absorbance at 652 nm in the presence of H2O2.65,66 To confirm the formation of H2O2 in our reaction, we mixed TMB with the extract of the reaction solution after 1 h of the aromatization reaction of 1a in acetate buffer (pH = 4) and monitored the UV-Vis absorbance during 12 h. The detection and evolution of the characteristic peak at 652 nm confirms the formation of H2O2. We also performed control experiments with laboratory grade 30% H2O2 and without H2O2 under similar conditions to rule out any ambiguity.
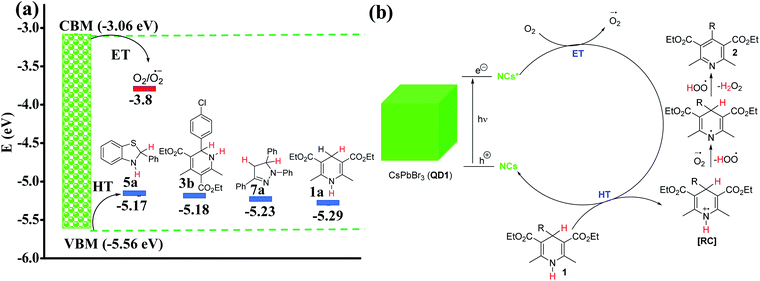 |
| | Fig. 4 (a) Redox potential (eV) of different substrates compared to the valence band maximum (VBM) and conduction band minimum (CBM) of QD1. The data obtained from cyclic voltammetry is marked with blue bars and the literature value for the O2/O2˙− redox pair by a red bar. The measured potentials (V) from CV were converted to HOMO/LUMO energy levels (eV) with respect to a Fc/Fc+ reference (−4.8 eV to vacuum);62,63 (b) plausible mechanism for the formation of aromatized products (2) in the presence of QD1 as photocatalyst under blue light irradiation. (RC = radical cation, ET = electron transfer and HT = hole transfer). | |
Comparison of the catalytic activity of QD1 with other photocatalysts
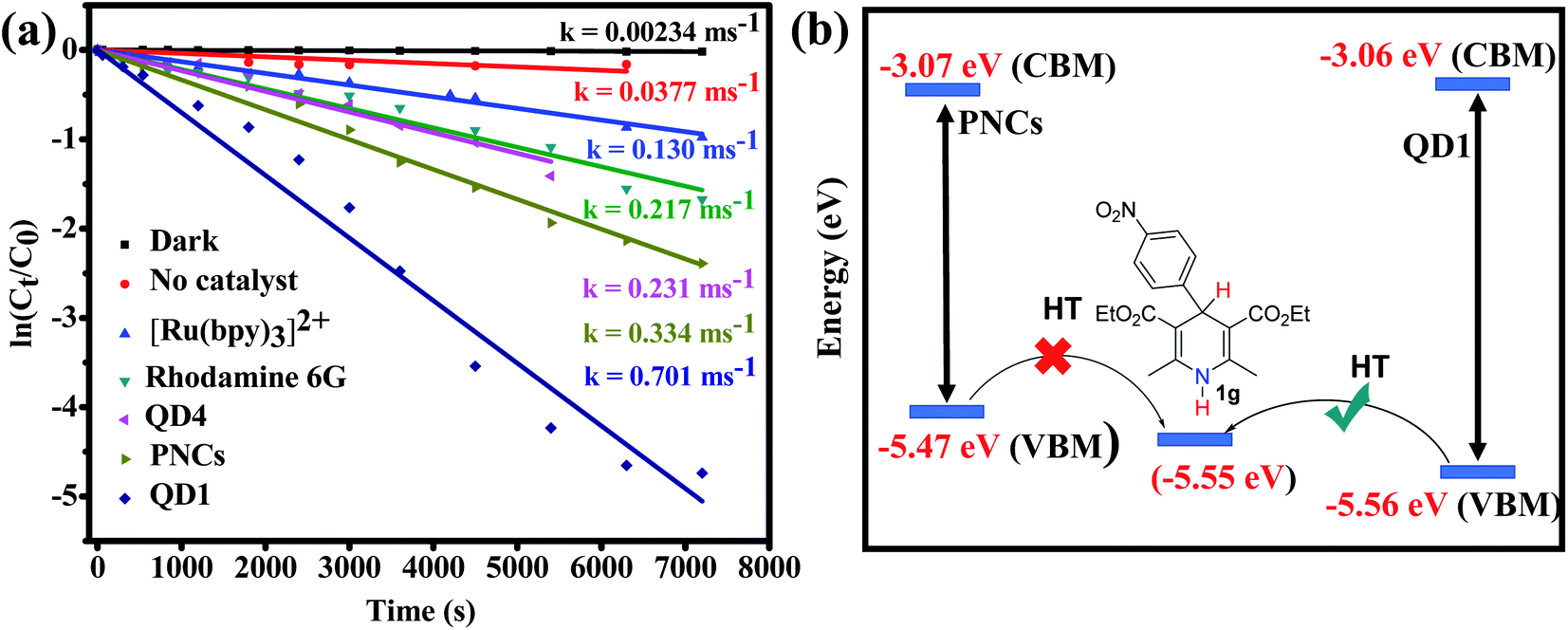
For comparison, oxidative aromatization of 1b was carried out in the presence of various commonly used state-of-the-art catalysts such as the ruthenium complex ([Ru(bpy)3][PF6]2),67 polydisperse CsPbBr3 NCs (PNCs)14,18 and rhodamine 6G (Rh-6G).68,69 The photocatalysts were either purchased or synthesized using standard literature protocols and thoroughly characterized using UV-Vis spectroscopy, PL, NMR or TEM wherever it was applicable (Fig. S25–S28†). The kinetic data were obtained by monitoring the progress of the oxidative aromatization of 1b using UV-Vis absorption spectroscopy (Fig. S29†). The gradual decrease of the distinct absorption peak at 339 nm as a function of time signifies of the substrate and progress of the reaction. Fig. 5a compares the reaction rate of QD1 with those of other photocatalysts. The pseudo first order rate constants (k) were calculated by plotting ln(Ct/C0) as a function of the reaction time. Polydisperse CsPbBr3 NCs (PNCs) are air-stable with higher catalytic efficiency, compared to conventionally prepared LHP QDs, transition metal complexes and other state-of-the-art quantum dots.14,18 In their detailed work, Yan and co-workers have demonstrated the use of PNCs for a wide range of organic transformations including C–C, C–N and C–O bond formations.14,18 In our case, as expected, compared to the conventionally prepared QD4 and the Ru-based molecular complex [Ru(bpy)3][PF6]2, the (2–5 times) higher rate of reaction was noted when PNCs were applied as the catalyst under similar conditions. The lower reaction rate in the presence of QD4 is attributed to its poor stability. The XRD studies revealed that QD4 underwent a complete degradation within 10 days under ambient condition to a mixture of tetragonal CsPb2Br5 and orthorhombic CsPbBr3 phases (Fig. S26†). Interestingly, halide-passivated QD1 shows an even higher (two-fold) rate compared to PNCs. We underline that these results were highly reproducible and consistent using different batches of catalyst samples prepared under the same conditions. The observed rate increase is attributed to ultrafast interfacial electron and hole transfer,6 the increase of the surface area and the more homogeneous nature of QD1 as compared to PNCs. The difference in efficiency was even more dramatic in substrates with EWG (1g). To give an example, the oxidative aromatization of 1g with R = p-NO2–C6H4 at 4 position as EWG using PNCs was unsuccessful even after 8 h. On the other hand, the same reaction resulted in 85% yield with QD1 under the same conditions. This stark difference can be attributed to the more favorable band alignment of QD1 with respect to the redox potential of substrate 1g, favoring the hole transfer (Fig. 5b). Due to quantum confinement effects, QD1 has a slightly higher band gap compared to PNCs as demonstrated in Fig. 5b. Furthermore, QD1 was 5 times more efficient as compared to the Ru-based transition metal complexes (Fig. 5a). [Ru(bpy)3][PF6]2 is widely used as photocatalyst in various organic transformations and is also a known photocatalyst for the oxidative aromatization reaction. Therefore, the direct comparison of QD1 with [Ru(bpy)3][PF6]2 gives us insight into the catalytic potential of QD1 for organic transformations in general. We attribute the better catalytic efficiency of QD1 over these molecular catalysts to its strong O2-tolerance. The efficiency of Ru(bpy)32+ is known to diminish in O2-rich conditions due to the strong competition between the quenching of the catalyst by O2 and the catalytic reaction, thereby demanding N2-sparging.70 We also employed Rh-6G, a popular air-tolerant organic dye,68,69 for comparison. Compared to Rh-6G we found 3 times higher reaction rate with QD1, indicating that these LHP based QDs have a high potential as alternatives to existing molecular catalysts for use in various organic transformations.
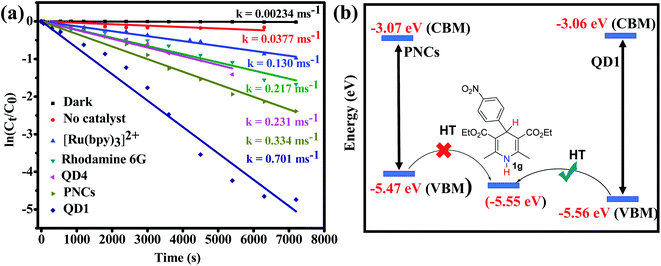 |
| | Fig. 5 (a) Plots of ln(Ct/C0) versus reaction time of the aromatization of 1b comparing the catalytic activity of QD1 with other photocatalysts; (b) more advantageous band alignment of QD1 with respect to bulk-type PNCs favouring hole transfer to substrates containing strong EWGs (1g). CBM and VBM for QD1 obtained from cyclic voltammetry and for PNCs from the literature.18 | |
Consistent with the kinetic data above, QD1 demonstrated a higher turnover number (TON) compared to PNCs and conventionally synthesized QD4. 0.1 mmol of 1a and QD1 (1 mg) in 3 mL of DCE were subjected to blue light irradiation (procedure detailed in Fig. S30 and S31†) for 4 consecutive cycles and the yield of the aromatized product (2a) was determined for each cycle using 1H NMR analysis of the crude reaction mixture. The reaction yielded 96%, 93%, 86% and 70% for 1st, 2nd, 3rd and 4th cycle, respectively, indicating that the catalytic activity of QD1 remained robust at least for 4 cycles. However, QD1 on long exposure to the polar solvent (DCE) and light under optimal reaction condition undergoes degradation due to the desorption of the passivating agents (bromide, oleic acid and oleylamine) from the surface of QD1 minimizing its stability. The presence of capping ligands on the surface of the QDs plays a decisive role in imparting the long-term stability of QDs.71 The TON of QD1 (200) was higher than that of the polydisperse NCs (PNCs, TON = 177) and of conventionally synthesized CsPbBr3 NCs (QD4, TON = 166) for the aromatization of 1a under optimized reaction conditions (Table 1). The TON was calculated based on molecular weight of CsPbBr3 (579.8 g mol−1, for details see ESI note†).18 Finally, we studied the comparative efficiency of the three different sized CQDs (QD1, QD2 and QD3) (Fig. S32†) using 1b. As expected, compared to QD1, larger sized CQDs (QD2) showed a slower reaction rate, which is ascribed to the decrease of the surface area in the latter and/or to the less favorable energy level alignment due to reduced quantum confinement.18 With smaller CQDs (QD3) the observed decrease of the catalytic efficiency appears at a first glance surprising. It is likely due to the decrease of the air-stability with decreasing size.18 We noticed that smaller CQDs (QD3) have poor photostability compared to larger ones (QD1), possibly due to the lower temperature synthetic conditions employed. In fact, previous studies have demonstrated that pronounced stability of LHP CQDs was only observed at high temperature synthetic conditions.22
Table 1 Comparison of the TON of QD1 with polydisperse CsPbBr3 NCs (PNCs) and with conventionally prepared CsPbBr3CQDs (QD4) for the aromatization of 1a
| Photo-catalyst |
Yield% (1st cycle) |
Yield% (2nd cycle) |
Yield% (3rd cycle) |
Yield% (4th cycle) |
TON |
|
QD1
|
96 |
93 |
86 |
70 |
200 |
|
PNCs
|
91 |
79 |
74 |
62 |
177 |
|
QD4
|
93 |
72 |
69 |
53 |
166 |
Conclusions
Despite the recent surge of development in synthetic protocols, mainly aimed at improving performances in optoelectronic devices, to date the wide application of LHP CQDs as photocatalysts in organic synthesis is not successful, mainly due to their poor air- and photostability. Here we demonstrated that stable LHP CQDs prepared under optimum temperature and halide-rich conditions are very promising photocatalysts for organic transformation. We tested the applicability of these CQDs by carrying out oxidative aromatization of a variety of azaheterocycles in air using off-the-shelf substrates and solvent. They outperform many state-of-the-art molecular and nanomaterials-based catalysts in terms of reaction rate, yield, reusability and turnover number. The variation of the substituents of the substrates showed that the rate of oxidative aromatization was faster in the presence of electron donating groups. Further analysis of the kinetic data indicated the presence of radical cation or radical anion intermediates and the predominance of electronic effects governing the reactivity. Our study also unraveled the underlying mechanisms confirming the role of the azaheterocycle substrates as hole acceptors and that of ambient oxygen as electron acceptor from photoexcited LHP CQDs culminating in the aromatization of the substrate and concomitant liberation of H2O2. Both high photo- and chemical stability as well as favorable energy level alignment of the QDs with respect to the organic substrate are required to achieve efficient photocatalysis. Concluding, this study provides a significant step towards the practical applicability of energy-level tunable, size-controlled LHP CQDs as efficient photocatalysts in organic synthesis. We expect that these stable CQDs are equally effective for C–C, C–O, C–N, C–S bond formation, C–H functionalization, thiol coupling and various other important organic reactions.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
SP and ST acknowledge SERB-DST, Government of India for research funding (EEQ/2016/000751 and EMR/2016/002505). SB would like to thank Department of Science and Technology, Government of India (DST/INSPIRE/03/2016/001207) [IF160689] for financial support under DST-INSPIRE Scheme. DB, BG and STh acknowledges Sikkim University for fellowship. AP, SR and DS acknowledges SERB-DST (EEQ/2016/000685) and DST-Inspire (DST/INSPIRE/04/2015/002674), for financial assistance. PR acknowledges funding from the French Research Agency ANR (grants SuperSansPlomb: ANR-15-CE05-0023-01, PERSIL: ANR-16-CE05-0019-02), from LABEX Lanef and H2020 (GreQue, Marie Sklodowska-Curie grant agreement no. 754303). WLL thank the CEA DRF Impulsion program for financial support. The TEM work used the platforms of the Grenoble Instruct-ERIC Centre (ISBG; UMS 3518 CNRS-CEA-UGA-EMBL) with support from FRISBI (ANR-10-INSB-05-02) and GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB). The IBS electron microscope facility is supported by the Auvergne-Rhône-Alpes Region, the fonds FEDER, the Fondation Recherche Médicale (FRM), C GIS-IBISA. Authors thank Karan Chhetri for his initial help with photocatalysis experiments.
References
- H. Huang, M. I. Bodnarchuk, S. V. Kershaw, M. V. Kovalenko and A. L. Rogach, ACS Energy Lett., 2017, 2, 2071–2083 CrossRef CAS.
- Y. Wang, X. Li, J. Song, L. Xiao, H. Zeng and H. Sun, Adv. Mater., 2015, 27, 7101–7108 CrossRef CAS.
- J. S. Manser, J. A. Christians and P. V. Kamat, Chem. Rev., 2016, 116, 12956–13008 CrossRef CAS.
- Q. A. Akkerman, G. Rainò, M. V. Kovalenko and L. Manna, Nat. Mater., 2018, 17, 394–405 CrossRef CAS.
- X. Zhang, H. Lin, H. Huang, C. Reckmeier, Y. Zhang, W. C. H. Choy and A. L. Rogach, Nano Lett., 2016, 16, 1415–1420 CrossRef CAS.
- K. Wu, G. Liang, Q. Shang, Y. Ren, D. Kong and T. Lian, J. Am. Chem. Soc., 2015, 137, 12792–12795 CrossRef CAS.
- Y. Wu, X. Yang, W. Chen, Y. Yue, M. Cai, F. Xie, E. Bi, A. Islam and L. Han, Nat. Energy, 2016, 1, 16148 CrossRef CAS.
- J. B. Hoffman, G. Zaiats, I. Wappes and P. V. Kamat, Chem. Mater., 2017, 29, 9767–9774 CrossRef CAS.
- A. Swarnkar, A. R. Marshall, E. M. Sanehira, B. D. Chernomordik, D. T. Moore, J. A. Christians, T. Chakrabarti and J. M. Luther, Science, 2016, 354, 92–95 CrossRef CAS.
- T. C. Sum and N. Mathews, Energy Environ. Sci., 2014, 7, 2518–2534 RSC.
- J. Burschka, N. Pellet, S. J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Grätzel, Nature, 2013, 499, 316–319 CrossRef CAS.
- Q. Tai, K. C. Tang and F. Yan, Energy Environ. Sci., 2019, 12, 2375–2405 RSC.
- Y. Cheng, F. So and S. W. Tsang, Mater. Horiz., 2019, 6, 1611–1624 RSC.
- X. Zhu, Y. Lin, Y. Sun, M. C. Beard and Y. Yan, J. Am. Chem. Soc., 2019, 141, 733–738 CrossRef CAS.
- K. Chen, X. Deng, G. Dodekatos and H. Tüysüz, J. Am. Chem. Soc., 2017, 139, 12267–12273 CrossRef CAS.
- H. Lu, X. Zhu, C. Miller, J. San Martin, X. Chen, E. M. Miller, Y. Yan and M. C. Beard, J. Chem. Phys., 2019, 151, 204305 CrossRef.
- Y. F. Xu, M. Z. Yang, B. X. Chen, X. D. Wang, H. Y. Chen, D. Bin Kuang and C. Y. Su, J. Am. Chem. Soc., 2017, 139, 5660–5663 CrossRef CAS.
- X. Zhu, Y. Lin, J. San Martin, Y. Sun, D. Zhu and Y. Yan, Nat. Commun., 2019, 10, 2843 CrossRef.
- W.-B. Wu, Y.-C. Wong, Z.-K. Tan and J. Wu, Catal. Sci. Technol., 2018, 8, 4257–4263 RSC.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R.
X. Yang, A. Walsh and M. V Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS.
- A. Dutta, S. K. Dutta, S. Das Adhikari and N. Pradhan, ACS Energy Lett., 2018, 3, 329–334 CrossRef CAS.
- S. Thapa, K. Bhardwaj, S. Basel, S. Pradhan, C. J. Eling, A. M. Adawi, J.-S. G. Bouillard, G. J. Stasiuk, P. Reiss, A. Pariyar and S. Tamang, Nanoscale Adv., 2019, 1, 3388–3391 RSC.
- K. Zhou and Y. Li, Angew. Chem., Int. Ed., 2012, 51, 602–613 CrossRef CAS.
- R. Long, K. Mao, X. Ye, W. Yan, Y. Huang, J. Wang, Y. Fu, X. Wang, X. Wu, Y. Xie and Y. Xiong, J. Am. Chem. Soc., 2013, 135, 3200–3207 CrossRef CAS.
- Z. Quan, Y. Wang and J. Fang, Acc. Chem. Res., 2013, 46, 191–202 CrossRef CAS.
- J. Y. Woo, Y. Kim, J. Bae, T. G. Kim, J. W. Kim, D. C. Lee and S. Jeong, Chem. Mater., 2017, 29, 7088–7092 CrossRef CAS.
- C. Wang, A. S. R. Chesman and J. J. Jasieniak, Chem. Commun., 2017, 53, 232–235 RSC.
- A. Dutta, S. K. Dutta, S. Das Adhikari and N. Pradhan, Angew. Chem., Int. Ed., 2018, 57, 9083–9087 CrossRef CAS.
- J. Pan, Y. Shang, J. Yin, M. De Bastiani, W. Peng, I. Dursun, L. Sinatra, A. M. El-zohry, M. N. Hedhili, A. Emwas, O. F. Mohammed, Z. Ning and O. M. Bakr, J. Am. Chem. Soc., 2018, 140, 562–565 CrossRef CAS.
- H. Sun, Z. Li, L. Kong, B. Wang, C. Zhang, Q. Yuan, S. Huang, Y. Liu and L. Li, Chem. Commun., 2018, 54, 9345–9348 RSC.
- B. Li, Y. Zhang, L. Fu, T. Yu, S. Zhou, L. Zhang and L. Yin, Nat. Commun., 2018, 9, 1076 CrossRef.
- A. Dutta and N. Pradhan, ACS Energy Lett., 2019, 4, 709–719 CrossRef CAS.
- M. Hayyan, M. A. Hashim and I. M. Alnashef, Chem. Rev., 2016, 116, 3029–3085 CrossRef CAS.
- A. S. Jalilov, C. Zhang, E. L. G. Samuel, W. K. A. Sikkema, G. Wu, V. Berka, T. A. Kent, A. L. Tsai and J. M. Tour, ACS Appl. Mater. Interfaces, 2016, 8, 15086–15092 CrossRef CAS.
- K. Gu, Y. Wang, J. Shen, J. Zhu, Y. Zhu and C. Li, ChemSusChem, 2020, 13, 682–687 CrossRef CAS.
- F. Lang, O. Shargaieva, V. V. Brus, H. C. Neitzert, J. Rappich and N. H. Nickel, Adv. Mater., 2018, 30, 1702905 CrossRef.
- C. C. Boyd, R. Cheacharoen, T. Leijtens and M. D. McGehee, Chem. Rev., 2019, 119, 3418–3451 CrossRef CAS.
- S. Aiello, G. Wells, E. L. Stone, H. Kadri, R. Bazzi, D. R. Bell, M. F. G. Stevens, C. S. Matthews, T. D. Bradshaw and A. D. Westwell, J. Med. Chem., 2008, 51, 5135–5139 CrossRef CAS.
- C. Allais, J. M. Grassot, J. Rodriguez and T. Constantieux, Chem. Rev., 2014, 114, 10829–10868 CrossRef CAS.
- Y. Yan, J. Yang, Z. Yu, M. Yu, Y. T. Ma, L. Wang, C. Su, J. Luo, G. P. Horsman and S. X. Huang, Nat. Commun., 2016, 7, 13083 CrossRef CAS.
- L. Da Costa, E. Scheers, A. Coluccia, A. Casulli, M. Roche, C. Di Giorgio, J. Neyts, T. Terme, R. Cirilli, G. La Regina, R. Silvestri, C. Mirabelli and P. Vanelle, J. Med. Chem., 2018, 61, 8402–8416 CrossRef CAS.
- T. Wu, C. Jiang, L. Wang, S. L. Morris-Natschke, H. Miao, L. Gu, J. Xu, K. H. Lee and Q. Gu, J. Nat. Prod., 2015, 78, 1593–1599 CrossRef CAS.
- F. P. Guengerich, M. V. Martin, P. H. Beaune, P. Kremers, T. Wolff and D. J. Waxman, J. Biol. Chem., 1986, 261, 5051–5060 CrossRef CAS.
- D. M. Stout and A. I. Meyers, Chem. Rev., 1982, 82, 223–243 CrossRef CAS.
- X. Q. Zhu, B. J. Zhao and J. P. Cheng, J. Org. Chem., 2000, 65, 8158–8163 CrossRef CAS.
- N. Nakamichi, Y. Kawashita and M. Hayashi, Org. Lett., 2002, 4, 3955–3957 CrossRef CAS.
- D. Zhang, L. Z. Wu, L. Zhou, X. Han, Q. Z. Yang, L. P. Zhang and C. H. Tung, J. Am. Chem. Soc., 2004, 126, 3440–3441 CrossRef CAS.
- R. Ananthakrishnan and S. Gazi, Catal. Sci. Technol., 2012, 2, 1463–1471 RSC.
- M. Imran, V. Caligiuri, M. Wang, L. Goldoni, M. Prato, R. Krahne, L. De Trizio and L. Manna, J. Am. Chem. Soc., 2018, 140, 2656–2664 CrossRef CAS.
- Q. A. Akkerman, L. Mart, L. Goldoni, M. Imran, D. Baranov, H. J. Bolink, F. Palazon and L. Manna, Chem. Mater., 2018, 30, 6915–6921 CrossRef CAS.
- J. Butkus, P. Vashishtha, K. Chen, J. K. Gallaher, S. K. K. Prasad, D. Z. Metin, G. Laufersky, N. Gaston, J. E. Halpert and J. M. Hodgkiss, Chem. Mater., 2017, 29, 3644–3652 CrossRef CAS.
- B. Zorman, M. V. Ramakrishna and R. A. Friesner, J. Phys. Chem., 1995, 99, 7649–7653 CrossRef CAS.
- X. Luo, R. Lai, Y. Li, Y. Han, G. Liang, X. Liu, T. Ding, J. Wang and K. Wu, J. Am. Chem. Soc., 2019, 141, 4186–4190 CrossRef CAS.
- Y. Dong, T. Qiao, D. Kim, D. Parobek, D. Rossi and D. H. Son, Nano Lett., 2018, 18, 3716–3722 CrossRef CAS.
- V. K. Ravi, G. B. Markad and A. Nag, ACS Energy Lett., 2016, 1, 665–671 CrossRef CAS.
- D. Yoo, J. Y. Woo, Y. Kim, S. W. Kim, S. H. Wei, S. Jeong and Y. H. Kim, J. Phys. Chem. Lett., 2020, 11, 652–658 CrossRef CAS.
- J. Liu, K. Song, Y. Shin, X. Liu, J. Chen, K. X. Yao, J. Pan, C. Yang, J. Yin, L. J. Xu, H. Yang, A. M. El-Zohry, B. Xin, S. Mitra, M. N. Hedhili, I. S. Roqan, O. F. Mohammed, Y. Han and O. M. Bakr, Chem. Mater., 2019, 31, 6642–6649 CrossRef CAS.
- D. Vo, W. C. Matowe, M. Ramesh, N. Iqbal, M. W. Wolowyk, S. E. Howlett and E. E. Knaus, J. Med. Chem., 1995, 38, 2851–2859 CrossRef CAS.
- R. H. Bocker and F. P. Guengerich, J. Med. Chem., 1986, 29, 1596–1603 CrossRef CAS.
- D. H. McDaniel and H. C. Brown, J. Org. Chem., 1958, 23, 420–427 CrossRef CAS.
- X. K. Jiang, Acc. Chem. Res., 1997, 30, 283–289 CrossRef CAS.
- C. Fujisue, T. Kadoya, T. Higashino, R. Sato, T. Kawamoto and T. Mori, RSC Adv., 2016, 6, 53345–53350 RSC.
- M. L. Tang, A. D. Reichardt, P. Wei and Z. Bao, J. Am. Chem. Soc., 2009, 131, 5264–5273 CrossRef CAS.
- F. C. L. Luiz, L. S. Garcia, L. S. G. Filho, L. R. Teixeira and S. R. W. Louro, J. Fluoresc., 2011, 21, 1933–1940 CrossRef CAS.
- L. Gao, J. Wu and D. Gao, ACS Nano, 2011, 5, 6736–6742 CrossRef CAS.
- D. Josephy, T. Eling and R. Mason, J. Biol. Chem., 1982, 257, 3669–3675 CrossRef.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS.
- I. Ghosh and B. König, Angew. Chem., Int. Ed., 2016, 55, 7676–7679 CrossRef CAS.
- M. Beija, C. A. M. Afonso and J. M. G. Martinho, Chem. Soc. Rev., 2009, 38, 2410–2433 RSC.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
- R. Grisorio, M. E. Di Clemente, E. Fanizza, I. Allegretta, D. Altamura, M. Striccoli, R. Terzano, C. Giannini, M. Irimia-Vladu and G. P. Suranna, Nanoscale, 2019, 11, 986–999 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization of photocatalyst, copies of NMR, CV spectra of substrates, kinetic data and turnover number (TON) calculation. See DOI: 10.1039/d0na00992j |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Lakshi
Saikia
c,
Lakshi
Saikia
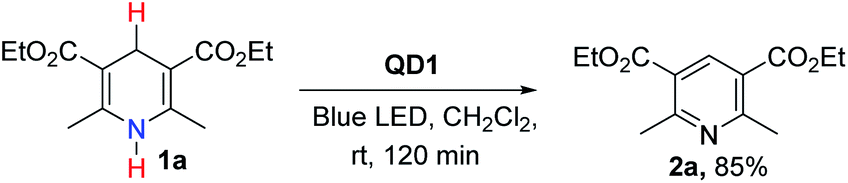
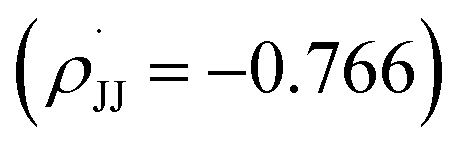 suggests that para-substitution has no effect on the delocalization of the radical formed during the reaction. The ratio of the constants
suggests that para-substitution has no effect on the delocalization of the radical formed during the reaction. The ratio of the constants 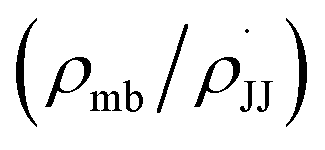 which is slightly more than unity (1.15) strongly indicates that the polar delocalization effect is more dominant compared to the spin delocalization effect. This observation is further corroborated by the fact that a non-linear correlation (Fig. 3c) was obtained when only spin delocalization parameters were considered, whereas a linear correlation was obtained when only polar substitution was considered (Fig. 3d). In other words, these data suggest that the reaction possibly proceeds via radical cation or radical anion intermediates where the electronic effect on reactivity is predominant due to polar effects.
which is slightly more than unity (1.15) strongly indicates that the polar delocalization effect is more dominant compared to the spin delocalization effect. This observation is further corroborated by the fact that a non-linear correlation (Fig. 3c) was obtained when only spin delocalization parameters were considered, whereas a linear correlation was obtained when only polar substitution was considered (Fig. 3d). In other words, these data suggest that the reaction possibly proceeds via radical cation or radical anion intermediates where the electronic effect on reactivity is predominant due to polar effects.