
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the synthesis of bi-magnetic manganese ferrite-based core–shell nanoparticles†‡
Marco
Sanna Angotzi
 ab,
Valentina
Mameli
ab,
Claudio
Cara
ab,
Davide
Peddis
bcd,
Huolin L.
Xin
e,
Claudio
Sangregorio
bfg,
Maria Laura
Mercuri
a and
Carla
Cannas
*ab
ab,
Valentina
Mameli
ab,
Claudio
Cara
ab,
Davide
Peddis
bcd,
Huolin L.
Xin
e,
Claudio
Sangregorio
bfg,
Maria Laura
Mercuri
a and
Carla
Cannas
*ab
aDepartment of Chemical and Geological Sciences, University of Cagliari, S.S. 554 bivio per Sestu, 09042 Monserrato (CA), Italy. E-mail: ccannas@unica.it
bConsorzio Interuniversitario Nazionale per la Scienza e Tecnologia dei Materiali (INSTM), Via Giuseppe Giusti 9, 50121 Firenze (FI), Italy
cDipartimento di Chimica e Chimica Industriale, Università di Genova, Via Dodecaneso, 31, 16131 Genova, Italy
dIstituto di Struttura della Materia, Consiglio Nazionale delle Ricerche, Via Salaria Km 29.300, 00015 Monterotondo Scalo (RM), Italy
eDepartment of Physics and Astronomy, University of California, Irvine, CA 92617, USA
fIstituto di Chimica dei Composti OrganoMetallici-Consiglio Nazionale delle Ricerche (ICCOM-CNR), Via Madonna del Piano 10, 50019 Sesto Fiorentino (FI), Italy
gDepartment of Chemistry “U. Schiff”, University of Florence, Via della Lastruccia 3-13, 50019, Sesto Fiorentino (FI), Italy
First published on 21st January 2021
Abstract
Multifunctional nano-heterostructures (NHSs) with controlled morphology are cardinal in many applications, but the understanding of the nanoscale colloidal chemistry is yet to be fulfilled. The stability of the involved crystalline phases in different solvents at mid- and high-temperatures and reaction kinetics considerably affect the nucleation and growth of the materials and their final architecture. The formation mechanism of manganese ferrite-based core–shell NHSs is herein investigated. The effects of the core size (8, 10, and 11 nm), the shell nature (cobalt ferrite and spinel iron oxide) and the polarity of the solvent (toluene and octanol) on the dissolution phenomena of manganese ferrite are also studied. Noteworthily, the combined use of bulk (powder X-ray diffraction, 57Fe Mössbauer spectroscopy, and DC magnetometry) and nanoscale techniques (HRTEM and STEM-EDX) provides new insights into the manganese ferrite dissolution phenomena, the colloidal stability in an organic environment, and the critical size below which dissolution is complete. Moreover, the dissolved manganese and iron ions react further, leading to an inverted core–shell in the mother liquor solution, paving the way to novel synthetic pathways in nanocrystal design. The MnFe2O4@CoFe2O4 core–shell heterostructures were also employed as heat mediators, exploiting the magnetic coupling between a hard (CoFe2O4) and a soft phase (MnFe2O4).
Introduction
Nanoheterostructures (NHSs), because of the possibility of combining several properties (e.g., magnetic1–10 and optical11–16) within the same material, are of ever-growing interest in many fields, ranging from nanomedicine8,17–20 to catalysis21–25 and electronic devices.26–30 The synthetic methodology and the understanding of the mechanism behind the formation of NHSs play a key role in tailoring their final architecture, which can be core–shell,10,17,18 dimer,15,31–36 flower-like,12,37–43etc. Commonly, the seed-mediated growth approach involves the heterogeneous nucleation of a phase onto pre-existing crystals (seeds), which therefore guides the process. The crystalline structure of the two components (or more in multifunctional NHSs) is the principal constraint for the NHS architecture. Indeed, when iso-structural phases are employed, the coherent epitaxial growth of the second component leads to the formation of well-defined core–shell heterostructures, with no or small amount of defects and heterojunctions.44–46 In contrast, when materials having different crystalline phases react, the growth is generally preferred along a particular direction, generating Janus-,47–49 dumbbell-,11,13,22 or flower-like11,33,43,50 NHSs. Nevertheless, other parameters are related to the synthetic operating conditions or materials themselves, and they can affect the nucleation and growth steps. The latter case includes crystal peculiarities or physical properties of the starting materials, like solubility, nucleation rate, and affinity with surfactants. It has been widely demonstrated how the crystal defects (facets, misfit dislocations, and stacking faults) affect the growth of second phases, and therefore, the architecture of the final system, since the nucleation takes place on these active sites.7,43,51–55 Moreover, other synthetic parameters such as temperature, polarity of solvents, surfactants, solution supersaturation degree, and reactant injection rate have been found to be crucial for the growth control.56–58 Indeed, nanosized materials are dynamic entities in solution, especially under solvothermal conditions, and can dissolve and re-crystallize by the Ostwald ripening process.56,59 This phenomenon is known to be size-dependent, but it also depends on the chemical nature of the ions involved, on the precursors and surface ligands used, and the nucleation rate of materials. While there are several theoretical and experimental studies about the stability of nanoparticles and precursors in solution, few of them are devoted to heterostructure formation, when the nucleation and growth of second phases occur.60,61In a previous paper,10 spinel ferrite-based core–shell NHSs were efficiently synthesized by an oleate-based solvothermal approach, starting from either cobalt ferrite of different sizes or manganese ferrite as seeds. In the case of manganese ferrite seeds, a slight dissolution was observed, suggesting a peculiar heterostructure formation mechanism. A systematic study on manganese ferrite stability when it is adopted as a seed material for the growth of a spinel ferrite shell is herein presented as a function of core size, chemical nature of the shell, and solvent polarity. Bulk characterization techniques (XRD, ICP, etc.) and transmission electron microscopy probed down to the nanoscale level (HRTEM and STEM-EDX) have been used to get insights into heterostructure formation and spinel ferrite stability. The core–shell systems in which magnetically hard (CoFe2O4) and soft (MnFe2O4) ferrimagnetic materials are coupled are also investigated as heat mediators under the applied alternating magnetic field.
Experimental
Chemicals
Oleic acid (>99.99%), 1-pentanol (99.89%), hexane (84.67%) and toluene (99.26%) were purchased from Lach-Ner; 1-octanol (>99.99%) and Mn(NO3)2·4H2O (>97.0%) were from Sigma-Aldrich; absolute ethanol and Co(NO3)2·6H2O (99.0%) were from Penta; NaOH (>98.0%) was from Fluka; Fe(NO3)3·9H2O (98.0%) was from Lachema; FeCl2·4H2O (99%) was from Merck; and poly-ethylene glycol-trimethoxysilane (PEG-TMS) was from ABCR GmbH & Co.Methods
Manganese ferrite NPs (MnA, MnB, and MnC, of increasing size) and core–shells were prepared as described in previous work,62–64 starting from metal oleates. In detail, a certain amount of metal oleate was put in a Teflon liner together with organic solvents and water (Table 1). The liner was enclosed in a stainless-steel autoclave (Berghof DAB-2) and put in a preheated oven at 220 °C for 10 hours. After cooling down, the particles were separated by a magnet, and the supernatant mother liquor discarded (except for MnA@Fe1). The particles were washed twice with 10 mL of hexane and 10 mL of ethanol, then collected in 5 mL of hexane (Fig. 1S‡). For the sample MnA@Fe1, the mother liquor has been collected and washed using the same procedure as the other samples and labeled as MnA@Fe1S.| Sample | Seeds (mg) | n-oleatea (mmol) | 1-Pentanol (mL) | Octanol (mL) | Toluene (mL) | Distilled water (mL) | Temperature | Reaction time |
|---|---|---|---|---|---|---|---|---|
| a Referred to CoII–FeIII oleates for MnA@Co or FeII oleate for all the other samples. | ||||||||
| MnA | — | 6 | 10 | 10 | — | 5 | 220 | 10 |
| MnB | — | 1.5 | 10 | — | 10 | 5 | 220 | 10 |
| MnC | — | 3 | 10 | — | 10 | 5 | 220 | 10 |
| Co | — | 6 | 10 | — | 10 | 5 | 220 | 10 |
| MnA@Co | 50 | 2 | 10 | — | 10 | 5 | 220 | 10 |
| MnA@Fe1 | 50 | 2 | 10 | — | 10 | 5 | 220 | 10 |
| MnA@Fe2 | 50 | 2 | 10 | 10 | — | 5 | 220 | 10 |
| MnB@Fe | 25 | 1 | 10 | — | 10 | 5 | 220 | 10 |
| MnC@Fe | 25 | 1 | 10 | — | 10 | 5 | 220 | 10 |
Hydrophobic nanoparticles were converted to hydrophilic nanoparticles by ligand exchange with PEG-TMS.65 6 mg of inorganic particles was dispersed in 30 mL of hexane containing 0.01% (v/v) acetic acid. Then, 0.5% (v/v) of PEG-TMS was added to the mixture and kept under stirring in a rotating shaker for 72 hours, during which the particles were sedimented. The black-brown precipitate was separated using a magnet and washed three times with hexane to remove all silanes in excess. The product was re-dispersed in distilled water. The concentration of colloidal dispersions was 3.4 mg mL−1 for all the samples.
Characterization
The samples were characterized by X-ray diffraction (XRD) using a PANalytical X'Pert PRO with Cu Kα radiation (1.5418 Å), a secondary monochromator, and a PIXcel position-sensitive detector. The peak position and instrumental width were calibrated using powder LaB6 from NIST. Refinement of structural parameters66 was performed by the Rietveld method using MAUD software,67 adopting the recommended fitting procedures.68TEM images were obtained using a JEOL 200CX operating at 160 kV. The particle size distribution was obtained by measuring over 1000 particles with the aid of software Pebbles, setting a spherical shape for the elaboration.69 The volume-weighted particle diameter was calculated as
| DTEM_V = DTEMe(3σ2) | (1) |
HRTEM images were acquired using a JEOL JEM 2010 UHR equipped with a 794 slow-scan CCD camera.
EDX measurements were performed on an FEI Talos F200X in the STEM mode using a field-emission gun operating at 200 kV equipped with a four-quadrant 0.9-sr energy-dispersive X-ray spectrometer. The core size and shell thickness were calculated by examining about 20 particles using software ImageJ.
Fourier-Transform Infrared (FT-IR) spectra were recorded in the 400 to 4000 cm−1 range using a Bruker Equinox 55 spectrophotometer. Samples were measured in a KBr pellet. Spectra were processed using OPUS software.
Thermogravimetric Analysis (TGA) curves were obtained using a PerkinElmer STA 6000, in the 25–850 °C range, with a heating rate of 10 °C min−1 under 40 mL min−1 O2 flow.
Room temperature 57Fe Mössbauer spectroscopy was performed on a Wissel spectrometer using the transmission arrangement and a proportional detector LND-45431. An α-Fe foil was used as a standard, and the fitting procedure was done using the NORMOS program to determine the isomer shift, quadrupole splitting, hyperfine field, and full width at half maximum of the signals.
Inductively coupled plasma-optical emission spectroscopy (ICP-OES) was performed on an Agilent 5110 spectrometer. The samples (around 5 mg) were digested by adding 4 mL of concentrated nitric acid (65%) and stirring for 3 hours at 60 °C. The solution was then filtered with 0.20 μm sieves and diluted for analysis. The calibration curve was done with iron, cobalt, and manganese NIST standards in the range 0.1–2 ppm, employing argon (420.067 nm) as the internal standard and using the following wavelengths for the analysis with the coefficient of determination given in brackets: for cobalt, 228.615 nm (R2 = 0.9999), 230.786 nm (R2 = 0.9998), and 237.863 nm (R2 = 0.9999); for iron, 234.350 nm (R2 = 0.9891), 238.204 (R2 = 0.9893), and 259.940 (R2 = 0.9888); for manganese, 257.610 nm (R2 = 0.9996), 259.372 nm (R2 = 0.9995), and 260.568 nm (R2 = 0.9998). For the calculation of the stoichiometry of the shell in the sample MnA@Co, it was assumed that the stoichiometry of the core did not change.
The magnetic properties were studied using a Quantum Design SQUID magnetometer (Hmax = ±55 kOe). Zero field cooled–field cooled (ZFC–FC) protocols were used to record the magnetization versus temperature curves within 5–300 K under different values of magnetic field. The ZFC curve was obtained by heating the sample from 5 to 300 K under a magnetic field after a cooling process under a zero magnetic field. The FC curve was obtained by cooling the sample from 5 to 300 K under a nonzero magnetic field.
Calorimetric estimation of specific absorption rate (SAR) was carried out using a non-adiabatic experimental set-up built at the LAboratorio di Magnetismo Molecolare (LA.M.M.) using a power supply CELESs MP6/400 (FIVES CELES), a water-cooled heating station connected to the power supply, and an induction coil. Heating curves were recorded under a magnetic field of 17 kA m−1 at 183 kHz for 300 s on water colloidal dispersions of the magnetic NPs. The hydrophobic NPs were made hydrophilic by the ligand exchange procedure with PEG-TMS. The concentration of the colloidal dispersion was 3.4 mg mL−1 for all samples. The temperature of the sample was monitored with an optical fiber probe (OPTOCON-FOTEMP) dipped into the solution. The sample holder was surrounded by polystyrene and placed in a glass Dewar, equipped with an ethylene glycol thermostat, to ensure proper thermal insulation. The SAR, i.e., the thermal power per mass unit, values were estimated by a linear curve fitting in the first 20 s of the heating curves (initial slope method).
Results and discussion
With the aim of studying the formation mechanism of heterostructures having MnFe2O4 as the core and CoFe2O4 or spinel iron oxide as the shell, three samples of MnFe2O4 (MnA, MnB, and MnC) with different particle sizes (8.2 nm, 9.7 nm, and 11.4 nm, respectively) and five related nanoheterostructures (MnA@Co, MnA@Fe1, MnA@Fe2, MnB@Fe, and MnC@Fe) were studied and compared (Table 2), in view of the recent evidence of dissolution phenomena in manganese ferrite nanoparticles.10| Sample | Formula | D TEM_V (nm) | σ TEM (%) | D COREEDX (nm) | D XRD (nm) | ε (10−3) | a (Å) | Fe/Mn | Feshell/Fecore |
|---|---|---|---|---|---|---|---|---|---|
| MnA | Mn0.91Fe2.06O4 | 8.2 | 13 | — | 9.0 (1) | 4.6 (1) | 8.49 (2) | 2.3 | — |
| MnA@Co | Mn0.91Fe2.06O4@Co0.98Fe2.02O4 | 11.6 | 9 | 5.2 (7) | 12.5 (1) | 4.2 (1) | 8.441 (1) | 6.3 | 1.7 |
| MnA@Fe1 | — | 11.8 | 9 | — | 14.3 (1) | 3.8 (1) | 8.382 (1) | 24.4 | 9.6 |
| MnA@Fe1S | — | 6.8 | 12 | — | 7.9 (1) | 6.5 (2) | 8.384 (3) | 12.6 | 4.4 |
| MnA@Fe2 | — | 7.5 | 29 | — | 9.6 (1) | 5.2 (2) | 8.393 (2) | 21.1 | 8.1 |
| MnB | Mn0.95Fe2.03O4 | 9.7 | 18 | — | 10.6 (1) | 4.0 (1) | 8.493 (1) | 2.1 | — |
| MnB@Fe | — | 13.7 | 10 | 6.5 (9) | 13.8 (1) | 4.4 (1) | 8.400 (1) | 11.8 | 4.4 |
| MnC | Mn0.87Fe2.09O4 | 11.4 | 13 | — | 13.5 (1) | 3.8 (1) | 8.489 (1) | 2.4 | — |
| MnC@Fe | — | 13.5 | 13 | 9.2 (9) | 14.5 (1) | 5.1 (1) | 8.435 (1) | 7.1 | 1.9 |
| Co | Co0.95Fe2.03O4 | 11.7 | 12 | — | 14.1 (1) | 3.6 (1) | 8.395 (1) | — | — |
The XRD patterns of MnA, MnB, and MnC displayed in Fig. 1 reveal the typical reflexes of the spinel ferrite phase and the calculated lattice parameter (8.492 ± 0.001 Å) fits well with the manganese ferrite value reported in the literature (8.4990 Å, PDF Card 010-0319). For all samples, microstrain values are in the order of 10−3, in agreement with previous results on spinel ferrite-based heterostructures of similar size and prepared by the oleate-based solvothermal method.43 The particles exhibit a spherical shape and a homogeneous distribution of Fe and Mn (TEM and STEM-EDX chemical mapping in Fig. 1). Similar values of crystallite (DXRD) and particle size (DTEM_V) account for highly crystalline particles, as revealed in the HRTEM images (Fig. 2S‡), which also show the inter-lattice distances typical for spinel ferrites. Moreover, a monolayer of oleate molecules (Fig. 3S and 4S‡) surrounds the particles.10 ICP-OES analysis of samples (Table 2) shows that sub-stoichiometric manganese ferrite (Mn0.91±0.04Fe2.03±0.03O4) is present, similarly to the results already obtained elsewhere.10,63
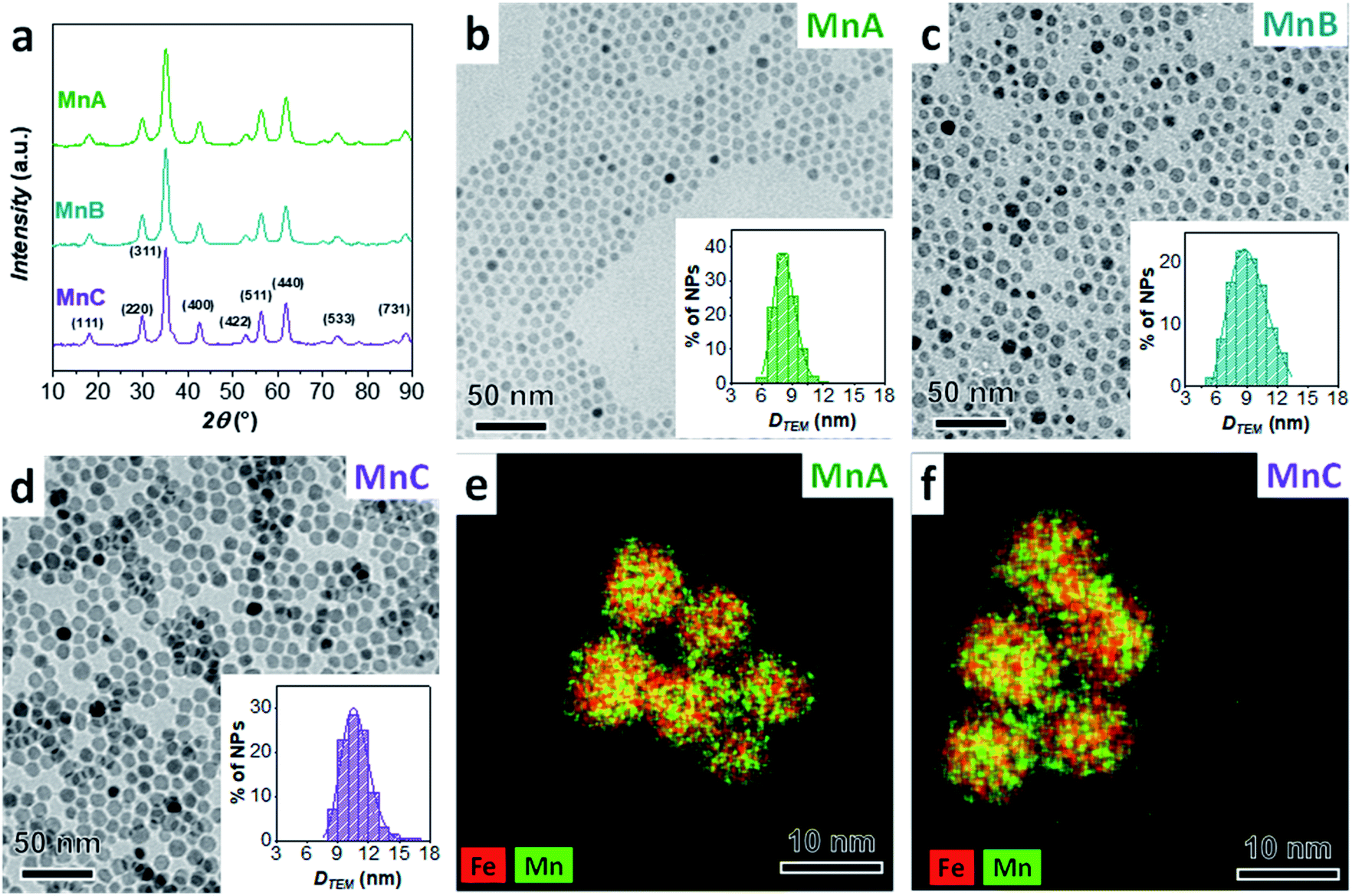 | ||
Fig. 1 XRD pattern (a), TEM image with particle size distribution (b–d), and STEM-EDX chemical mapping (e, f) of manganese ferrite samples (MnA, MnB, and MnC). Number of particles analyzed: 2441 for MnA, 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 146 for MnB, and 1297 for MnC. 146 for MnB, and 1297 for MnC. | ||
MnA-based core–shell nanoparticles
Starting from the MnA sample as seeds, two core–shell samples were prepared with the well-established solvothermal seed-mediated growth method,10 resulting in the samples MnA@Co and MnA@Fe1, with a cobalt ferrite or spinel iron oxide shell, respectively. The XRD patterns and TEM images of both samples (Fig. 2) reveal obtaining spinel ferrite nanocrystals having similar microstrain values, larger crystallite and particle sizes than MnA, lower size dispersity, and smaller cell parameters due to the shrinking of the crystal lattice caused by the presence of cobalt or iron ions (Table 2).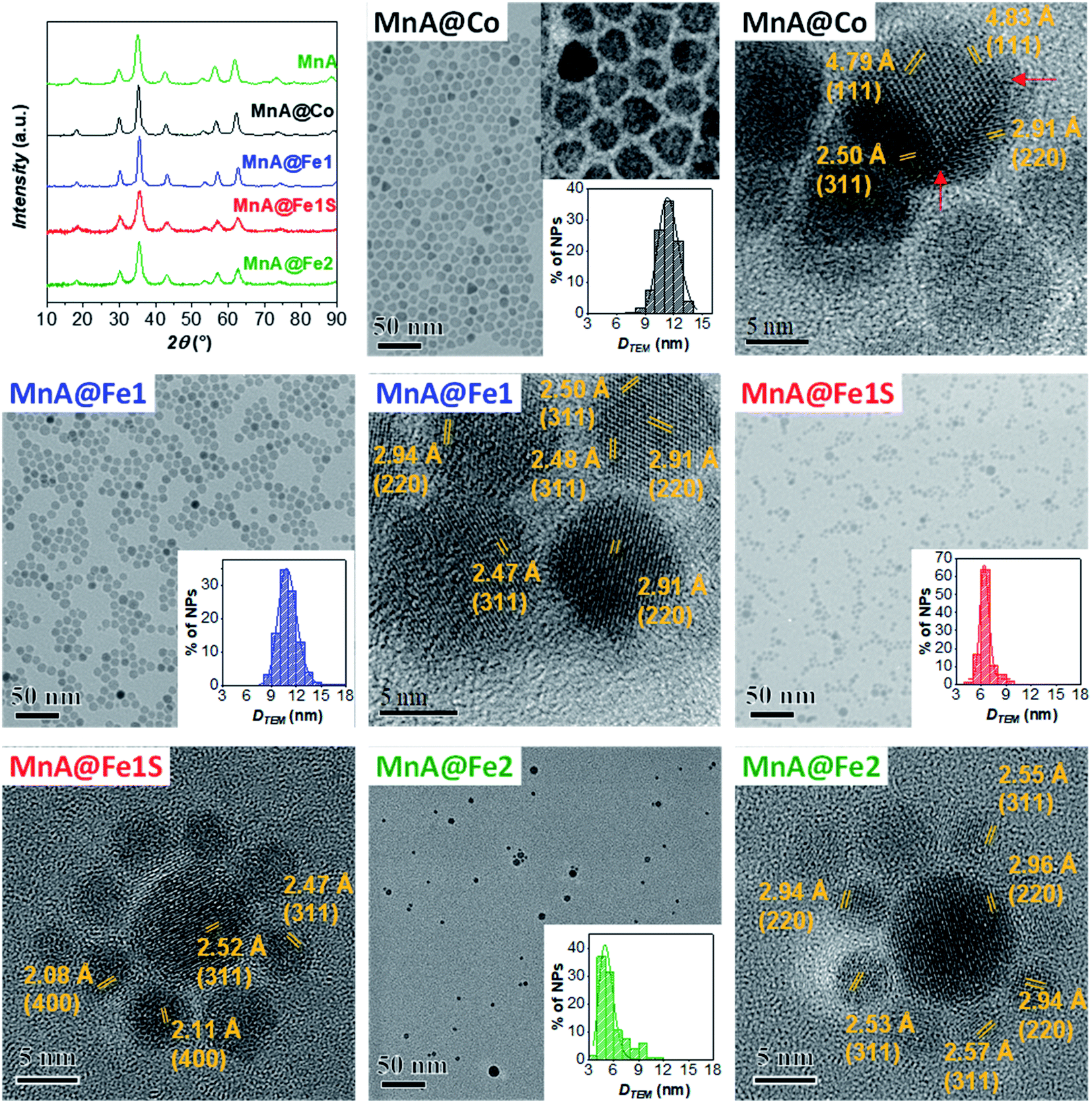 | ||
| Fig. 2 XRD patterns, TEM images with particle size distribution, and HRTEM images with interlattice distances and Miller's indices of the samples MnA@Co, MnA@Fe1, MnA@Fe1S, and MnA@Fe2. Number of particles analyzed: 2133 for MnA@Co, 2392 for MnA@Fe1, 1039 for MnA@Fe1S, and 200 for MnA@Fe2. | ||
Interestingly, as it can be seen by the TEM and HRTEM images reported in Fig. 2 and 3, the sample MnA@Co features different morphologies besides spheroidal, in contrast to MnA@Fe1. To better understand these features and study the heterostructure architecture, STEM-EDX chemical mapping was performed on both samples (Fig. 4). For MnA@Fe1, spherical Mn-free iron oxide nanoparticles were formed, suggesting that manganese was completely dissolved. This dissolution effect is visible, to a less extent, also for MnA@Co (Fig. 4), showing a smaller core size than the original MnA (5.2 nm (DCOREEDX) vs. 8.2 nm (DTEM_V); 74% volume reduction) and asymmetric covering of the shell, which indicates an inhomogeneous growth of cobalt ferrite. These results are also confirmed by ICP-OES analysis of samples (Table 2, vide supra). Indeed, while a minimal amount of manganese was revealed in MnA@Fe1 (Fe/Mn = 24.4 and Feshell/Fecore = 9.6), MnA@Co features a stoichiometric cobalt ferrite shell (Mn0.91Fe2.06O4@Co0.98Fe2.02O4). The latter result indicates that the observed dissolution phenomenon involves the whole manganese ferrite phase and did not affect the stoichiometry of cobalt ferrite.
 | ||
| Fig. 3 HRTEM images with inter-lattice distances and Miller's indices for the sample MnA@Co. A red line highlights the defects and edges of the nanocrystals. | ||
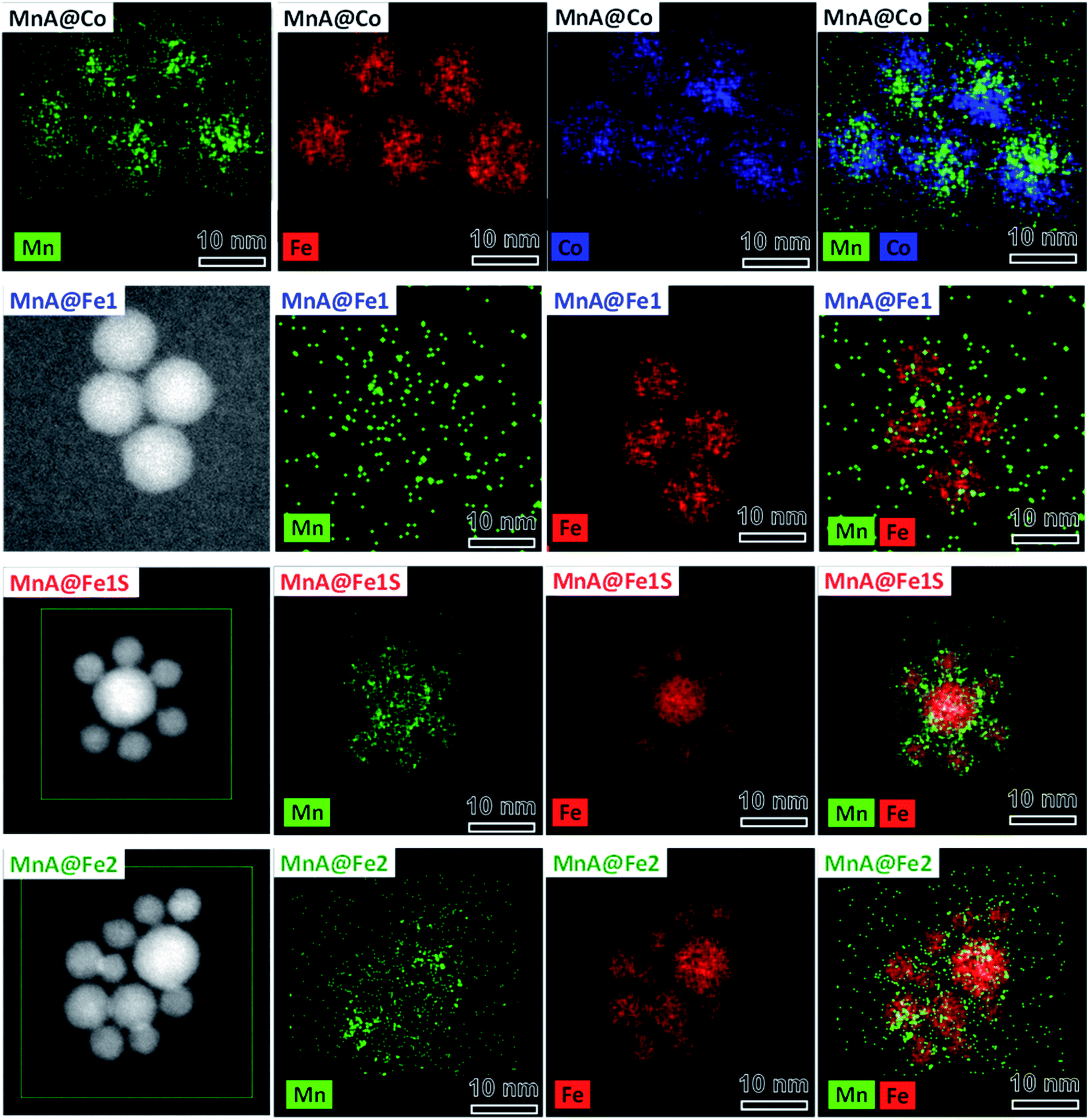 | ||
| Fig. 4 STEM-EDX chemical mapping of the samples MnA@Co, MnA@Fe1, MnA@Fe1S, and MnA@Fe2. Cobalt is represented in blue, manganese in green, and iron in red. The images have been modified and centered in order to have the same scale bar. | ||
Some authors have reported the observation of dissolution phenomena on colloidal dispersions of nanoparticles subjected to thermal treatment.10,70–75 However, in the case of core–shell nanoparticles, the dependence of the dissolution phenomena on the size and chemical nature of the shell has not been treated so far. Here, the systematic study revealed that manganese dissolution is more evident in shells formed by iron oxide rather than cobalt ferrite. Explaining this difference is rather complex since several phenomena take place simultaneously. The following aspects can play a role: (i) solubility of nanostructured metal oxides (core and shell) at a specific temperature, (ii) stabilities of the metal–oleate complexes, (iii) nucleation and growth kinetics of the shell, and (iv) strength of interaction between the NP surface and oleate molecules.
Different authors take into account the hard/soft acid/base theory,76–78 where carboxylates and MnII/FeIII are classified as hard bases/acids, while CoII and FeII are borderline acids. In this context, MnII/FeIII–oleate complexes should be more stable than CoII/FeII–oleate ones. Nevertheless, this interpretation is contradicted when the hydration enthalpies of aqua ions79 or the solubility products of hydroxides are considered. Indeed, H2O and OH− are hard bases (Table 1S‡), and we should expect stronger bonds with MnII and FeIII, considering the hard–soft properties of metal cations. Actually, for both aqua ions and hydroxides, the stability follows the order FeIII > CoII > FeII > MnII. The softer behavior of MnII can be explained in terms of a larger ionic radius (Table 1S‡), which results in higher polarizability. The lower stability of MnII aqua ions and hydroxides should reflect in the lower stability of manganese ferrite if compared to both cobalt ferrite and iron oxide. This trend could explain the observed dissolution phenomenon for manganese ferrite cores. Moreover, the dependence of dissolution phenomena on the chemical nature of the shell can be justified by the higher stability of the CoII species than the FeII ones, which can be reflected in the nucleation kinetics. Indeed, the presence of manganese ferrite in MnA@Co and its absence in MnA@Fe1 can be caused by a faster nucleation rate of cobalt ferrite than iron oxide, which can grow around manganese ferrite before it is completely dissolved. This hypothesis can be confirmed by considering the nucleation and growth mechanism of single-phase NPs. It has already been observed63 that, under the same synthesis conditions, cobalt ferrite gives rise to larger particles than iron oxide. The fast growth of cobalt ferrite when manganese ferrite dissolves progressively can give rise to the formation of asymmetric core shells in MnA@Co, and in some cases, heterojunctions, instead of core–shell architectures (Fig. 3).
The structural and magnetic properties of MnA, MnA@Fe1, and MnA@Co were studied through 57Fe Mössbauer spectroscopy, at room temperature (RT), and DC magnetometry (Fig. 5). At RT, the sample MnA shows a singlet, accounting for nanoparticles in the superparamagnetic state, having an isomer shift of 0.32 mm s−1, typical of FeIII. In contrast, the sample MnA@Co reveals the presence of two overlapped sextets, caused by spinel ferrite nanoparticles in the blocked state, due both to the larger size of particles and the presence of CoII, that cause an increase of the magnetic spin flipping energy barrier (ΔEa = KV, where K is the anisotropy constant and V the particle volume). The two sextets are derived from FeIII in the octahedral and tetrahedral sites of the spinel ferrite structures, as indicated in Table 2S.‡ In contrast, MnA@Fe1, even though larger than MnA, displays a sharp singlet, due to NPs in the superparamagnetic state, with an isomer shift of 0.41 mm s−1, which indicates the presence of FeIII, even though the higher value could be an indication of the presence of a small fraction of FeII. Therefore, both MnA and MnA@Fe1 display superparamagnetic behavior at 300 K, in contrast to MnA@Co, which reveals ferromagnetic behavior due both to the high anisotropy of CoFe2O4 and the rigid coupling between the two ferrimagnetic phases. This has been confirmed by both the hysteresis loop and DCD curves recorded at 5 K, clearly indicating high anisotropy for the MnA@Co sample (Fig. 5).9,17,80 Field-dependent magnetization curves obtained at 10 K (Fig. 5) show no hysteresis for the MnA and MnA@Fe1 samples, typical of magnetically soft ferrites, and a large hysteresis (Hc = 1.14 T) for MnA@Co, due to the presence of the hard cobalt ferrite. The saturation magnetization values found for the samples reach the maximum for the manganese ferrite sample (Ms = 101 emu g−1), as expected for the values found in the literature,81 and the minimum for MnA@Fe1 (Ms = 87 emu g−1), in agreement with the observed scenario of the absence of manganese ferrite in the latter sample and the predominance of FeIII ions. The ZFC curves (Fig. 5) exhibit, for all samples, a single maximum with the absence of a shoulder, indicating a single-particle population. Moreover, there is no separation of ZFC and FC curves at temperatures higher than Tmax, due to the small difference in blocking temperatures of the particles, as also evidenced by the small size dispersity of samples (9–13%). The blocking temperature (Tb) increases in the order of MnA < MnA@Fe1 < MnA@Co, in line with the particle size and effective anisotropy constant. The flatness of the FC curve, especially for MnA@Fe1, indicates high interparticle interactions. It is worth noting that the single maximum in the ZFC curve and the one-stage hysteresis loop of the field-dependent magnetization curve of MnA@Co are important evidence of a single magnetic response caused by the rigid coupling between cobalt and manganese ferrite.
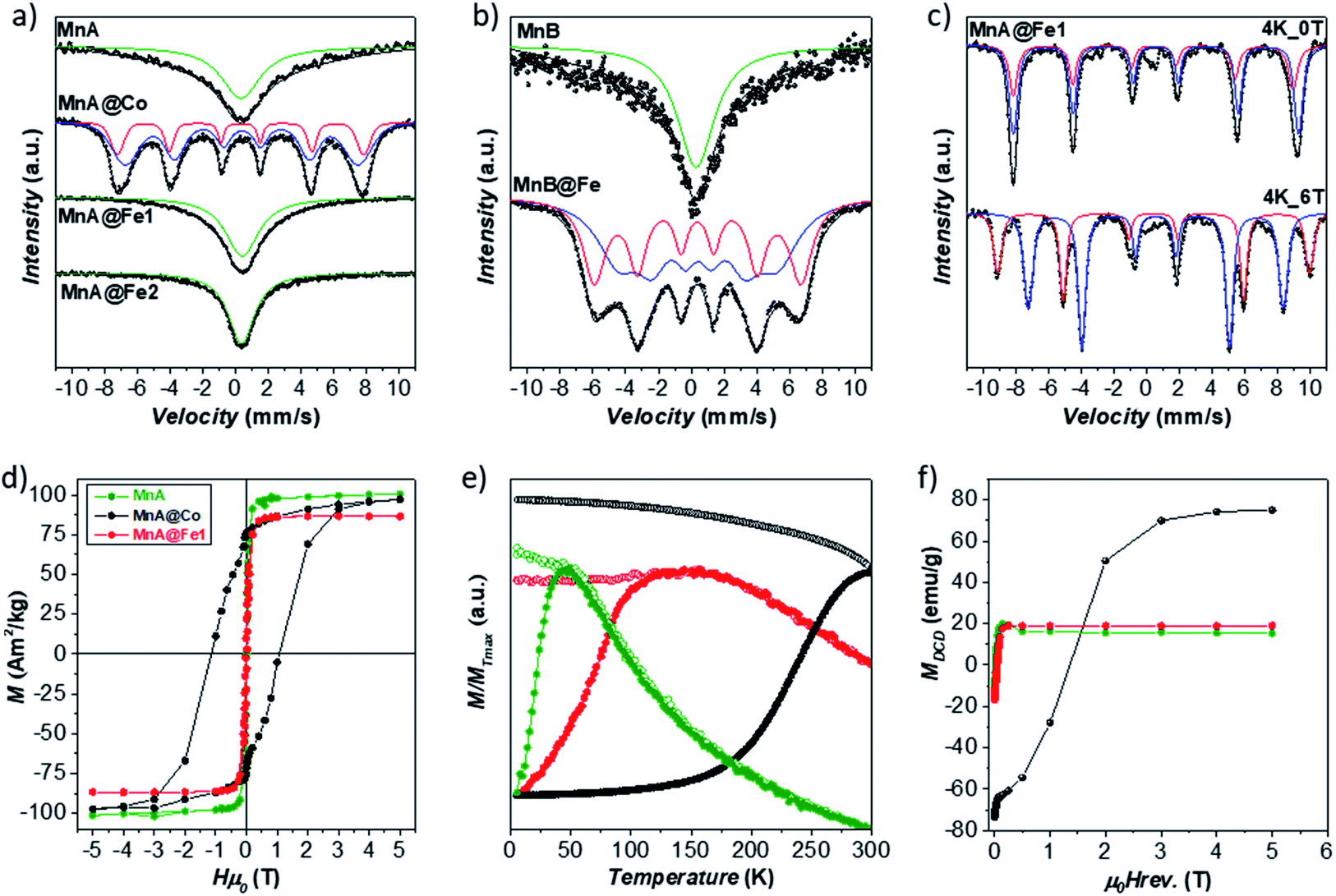 | ||
| Fig. 5 RT 57Fe Mössbauer spectra of samples (a, b). LT 57Fe Mössbauer spectra of samples in the presence and in the absence of an external magnetic field of the sample MnA@Fe1 (c). Field-dependent magnetization curves recorded at 5 K (d), ZFC (full circles) and FC (empty circles) curves recorded at 2.5 mT (e), and DCD curves (f) of the samples MnA, MnA@Co, and MnA@Fe1. | ||
The samples MnA and MnA@Fe1 were analyzed by 57Fe Mössbauer spectroscopy at low temperatures (LT) also to get insights into the site occupancy (Fig. 5, 6S and Table 3S‡).17,82–85 MnA exhibits an iron site occupancy of 22% and 78% for the tetrahedral and octahedral sites, respectively, which are reflected in the inversion degree value of 0.46, close to the values found in the literature for nanosized manganese ferrite.63 For MnA@Fe1, the site occupancy reached 37% for tetrahedral (Td) and 63% for octahedral (Oh) sites, due to the absence of manganese ferrite and the presence of only iron oxide, as observed from STEM-EDX chemical mapping. The ratio Fe(Oh)/Fe(Td), equal to 1.70, is very close to the theoretical γ-Fe2O3 ratio (1.67),81 indicating that, even though the presence of magnetite cannot be excluded, the sample is manly composed of maghemite.
Since the sample MnA@Fe1 did not evidence the presence of manganese ferrite, neither from STEM-EDX chemical mapping nor from 57Fe Mössbauer spectra, the particles present in the mother liquor of synthesis were analyzed. In fact, after the solvothermal treatment, the system is composed of large particles that were separated by a magnet (MnA@Fe1) and a mother liquor containing clear water and dark organic solvents, where smaller particles are present (MnA@Fe1S). This fraction, commonly discharged, has been collected and washed using the same procedure as for the other samples. The XRD pattern and TEM images of MnA@Fe1S are reported in Fig. 2, while the parameters are presented in Table 2. The sample features a spinel structure and a spheroidal particle shape, but with lower crystallite and particle sizes than both MnA and MnA@Fe1, and a higher microstrain value, probably due to the different formation mechanism. Interestingly, through STEM-EDX analysis (Fig. 4), it is possible to observe the formation of core–shell nanoparticles having iron oxide as the core and a thin layer (∼1 nm) of manganese ferrite as the shell. Therefore, the manganese ferrite that dissolved during the solvothermal synthesis of the core–shell grew around the just-formed iron oxide nanoparticles.
Another core–shell sample, MnA@Fe2, was prepared under the same synthesis conditions as for MnA@Fe1, but changing toluene with octanol, to study the effect of the use of a more polar solvent mixture (octanol and pentanol) on dissolution phenomena, and therefore, on core–shell formation. The as-obtained nanoparticles feature a spinel structure, a spheroidal shape, and a similar lattice parameter as MnA@Fe1 (Fig. 2 and Table 2) but a smaller particle size and a broader size distribution (29%), indicating that octanol is not a suitable solvent for producing high-quality core–shell nanoparticles. The STEM-EDX chemical mapping (Fig. 4) also exhibits, in this case, almost complete dissolution of manganese ferrite, as also evidenced by the ICP-OES data that show a similar content of manganese for MnA@Fe2 if compared with MnA@Fe1 (Fe:Mn = 21.1 and Feshell:Fecore = 8.1).
MnB- and MnC-based core–shell nanoparticles
Since the sample MnA (DTEM_V = 8.2 nm) evidenced dissolution phenomena when it undergoes a second solvothermal treatment, two larger samples (MnB, DTEM_V = 9.7 nm, and MnC, DTEM_V = 11.4 nm) were tested as seed precursors for the preparation of core–shell nanoparticles. The characterization of MnB@Fe and MnC@Fe is reported in Table 2 and Fig. 6.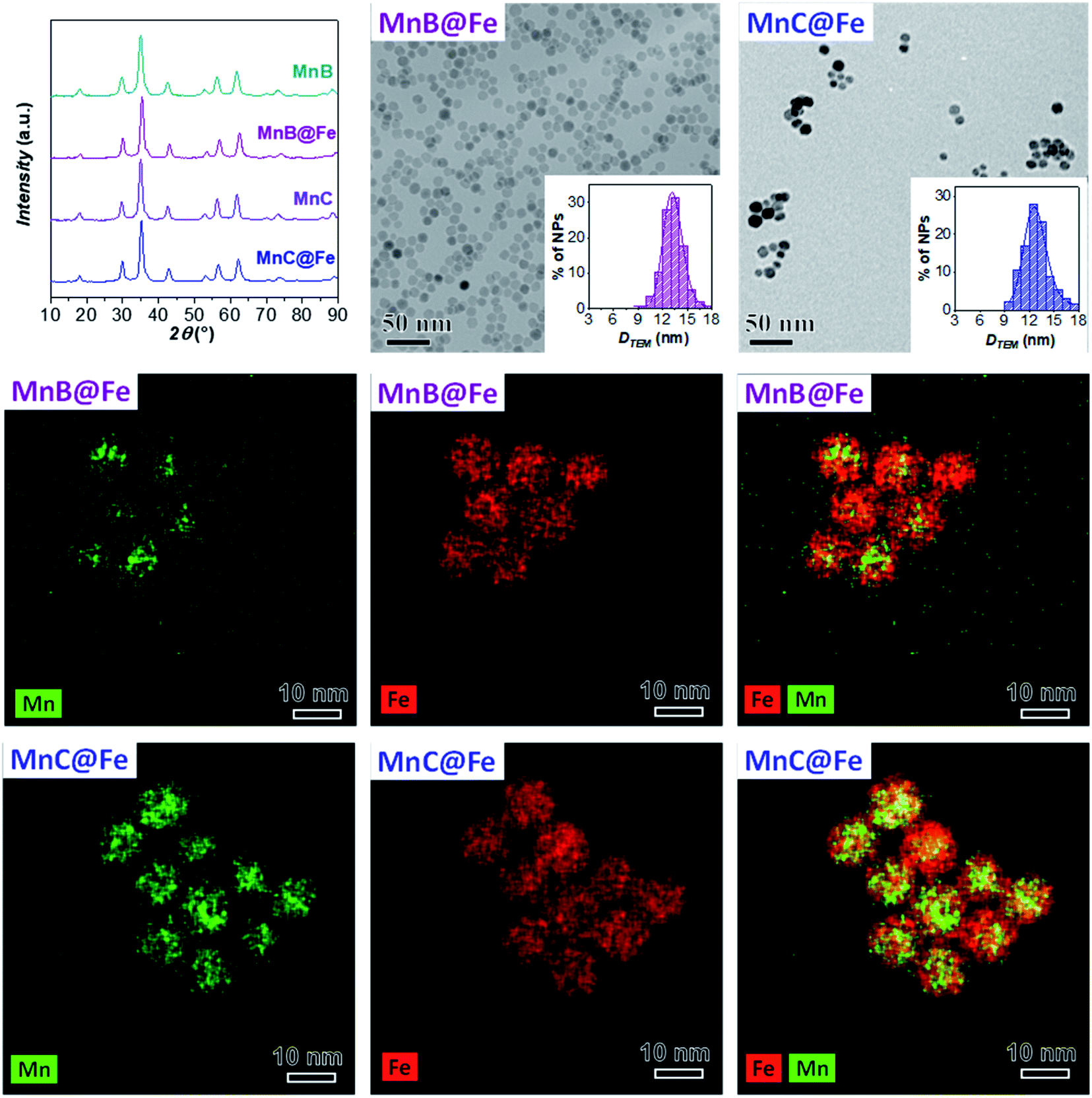 | ||
| Fig. 6 XRD patterns, TEM images with size distributions, and STEM-EDX chemical mapping of MnB@Fe and MnC@Fe. Number of particles analyzed: 2553 for MnB@Fe and 258 for MnC@Fe. The images have been modified and centered in order to have the same scale bar. | ||
Similar to MnA@Fe1, MnB@Fe and MnC@Fe also feature larger crystallite and particle sizes and a lower size dispersity than the respective cores, suggesting a homogeneous growth of particles after the second solvothermal treatment. The STEM-EDX chemical mapping image, shown in Fig. 6, indicates the architecture of the core–shell nanoparticles in which, as evidenced above, the dissolution of manganese took place, but to a less extent in comparison with MnA@Fe1 and MnA@Fe2. Indeed, the core size of MnB@Fe decreased from 9.7 nm (DTEM_V) up to 6.5 nm (DCOREEDX; 70% volume reduction, Table 2) with a dispersity of 22% (similar to that of MnB), even though the final core–shell size dispersity is about 10% (σMnB = 18%). Regarding MnC@Fe, the core size decreased from 11.4 nm to 9.2 nm (DCOREEDX), with 47% volume reduction and 14% dispersity (σMnC = 13%). These results indicate that (i) the dissolution of manganese ferrite is size-dependent, and a linear trend has been found between the volume reduction due to dissolution and the DTEM_V of the cores (Fig. 7S‡); (ii) under the synthetic conditions employed, below a critical size (∼8 nm) the manganese dissolution is complete and a theoretical critical size of about 14 nm should be reached to hamper dissolution phenomena; (iii) a size-regularization process took place and led to core–shell nanoparticles with narrow size dispersity (10% and 14%), although the core size and shell thickness dispersity are relatively high. The size dependence on the manganese dissolution is also confirmed also by ICP-OES data, revealing an increase in the manganese content as the particle size increases (Fe/Mn = 24.4, 11.8, and 7.1; Feshell/Fecore = 9.6, 4.4, and 1.9 for MnA@Fe1, MnB@Fe, and MnC@Fe, respectively).
Magnetic heat dissipation
The sample MnA@Co, which combines a hard and a soft magnetic material, shows rigid exchange coupling and, given the recent studies of core–shell NPs for magnetic heat dissipation,17 was tested as a heat mediator. Since MnA alone did not heat up, it is interesting to compare the heat dissipated by MnA@Co with cobalt ferrite (Co, Fig. 8S‡) and manganese ferrite (MnC) of similar size (DTEM_V in the range 11.4–11.7 nm, Table 2), in terms of SAR, or intrinsic loss power (ILP), which is independent of the field and the amplitude of the applied magnetic field.86It has lately been demonstrated17,18,87 that the exchange coupling between a hard and a soft magnetic material enhances the heating performance with respect to the corresponding core, but the behavior of a core–shell nanoparticle having a soft core and a hard shell if compared to a hard nanoparticle of the same size led to different results in the literature. Indeed, Zhang et al.88 found better performances for a cobalt ferrite sample of 15 nm than those of MnFe2O4@CoFe2O4 of about 14 nm (core 7 nm) due to the lower anisotropy constant of the first nanoparticles. In contrast, Angelakeris and co-workers,89 comparing CoFe2O4 of 15 nm and a core–shell of 16 nm having a 10 nm MnFe2O4 core and a CoFe2O4 shell, obtained higher heating release for the latter sample, despite the lower saturation magnetization. Again, discording results were discovered when studying the effect of the amount of hard-CoFe2O4 shell in spinel iron oxide@cobalt ferrite systems. In one case, the group of Solopan90 revealed an initial increase and then a decrease when the thickness of the cobalt ferrite shell was increased from 0.1 nm to 1 nm and 2.5 nm. In the second case, a constant gain in the heat loss was found with decreasing cobalt ferrite shell thickness, by Yelenich et al.91 Generally, these studies are conducted in water, but sometimes hydrophobic particles in organic dispersions are analyzed.
In this work, magnetic heat dissipation performances were studied in the aqueous colloidal dispersion, after the ligand exchange reaction of oleate molecules with the PEG-TMS one, to render the nanoparticle surface hydrophilic.65Fig. 7 reports the heating curves obtained for the MnA@Co, Co, and MnC dispersions (Cox = 3.4 mg mL−1) under an alternating magnetic field of 183 kHz and 17 kA m−1 (Table 3). The resultant SAR value of the core–shell nanoparticles (43 W g−1, ILP = 0.81 nH m2 kgox−1) was higher, almost double, than that of the cobalt ferrite sample (24 W g−1, ILP = 0.45 nH m2 kgox−1), while MnC did not heat up at all. These results further confirm the efficiency of magnetic exchange coupling to boost the heat dissipation performances.
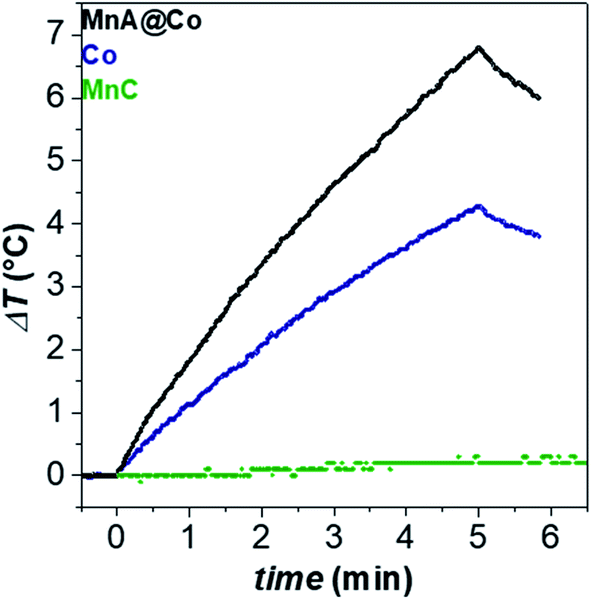 | ||
| Fig. 7 Heating curves of aqueous colloidal dispersions of samples (Cox = 3.4 mg mL−1) at 30 °C. The magnetic field was 183 kHz and 17 kA m−1. | ||
| Sample | D TEM_V (nm) | σ (%) | D DLS (nm) | D (μ2 s−1) | τ B (s) | SAR (W gox−1) | ILP (nH m2 kgox−1) |
|---|---|---|---|---|---|---|---|
| MnA@Co | 11.6 | 9 | 23(6) | 7.7 | 1.1 × 10−4 | 43(5) | 0.81(9) |
| Co | 11.7 | 12 | 33(8) | 3.5 | 1.2 × 10−3 | 24(1) | 0.45(2) |
| MnC | 11.4 | 13 | 42(10) | 2.6 | 3.0 × 10−3 | 0 | 0 |
Conclusions
In this work, the synthesis of manganese ferrite-based heterostructures through a solvothermal approach and their characterization by nanoscale chemical mapping, 57Fe Mössbauer spectroscopy, and DC magnetometry are reported. Manganese dissolution is investigated as a function of the size of manganese ferrite seeds, chemical nature of the shell (cobalt ferrite and iron oxide), and polarity of the solvent. The manganese ferrite nanoparticles have size in the 8–11 nm DTEM_V range, a spherical shape, and oleate molecules as capping agents, and when subjected to solvothermal treatment to obtain the corresponding heterostructures, they underwent dissolution phenomena to a different extent. When a shell of iron oxide was selected, the smallest particles (8.2 nm) dissolved completely, leading to almost pure maghemite, while the others kept a manganese ferrite core but with significant volume reduction (70 and 47%) dependent on the starting core size (9.7 nm and 11.4 nm). The dissolved manganese was observed in the supernatant mother liquor of synthesis as a thin shell of new-formed spinel iron oxide nanoparticles. The dissolution phenomena are also dependent on the chemical nature of the shell, which are less evident when cobalt ferrite was employed instead of iron oxide. This behavior was ascribed to the higher stability and faster nucleation and growth of cobalt ferrite with respect to iron oxide. The use of a more polar solvent mixture in the synthesis confirmed, once again, the almost complete dissolution of manganese ferrite. Interestingly, MnA@Co, where a hard and a soft magnetic material are strongly coupled in the same heterostructure, paves the way to develop heat mediators based on core–shell NPs, showing performances with increased efficiency by tuning the magnetic exchange coupling.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are much obliged to Prof. Daniel Niznanský from the Charles University of Prague for his invaluable scientific contribution on 57Fe Mössabuer spectroscopy and X-ray diffraction. CESA project RAS-Piano Sulcis, PON AIM (PON Ricerca e Innovazione 2014–2020 – Azione I.2 – DD n. 407 del 27 febbraio 2018 “Attraction and International Mobility”, Cult-GeoChim project AIM1890410-3), and Fluorsid S.p.A. are gratefully acknowledged for financing the fellowships of M. Sanna Angotzi, V. Mameli, and C. Cara, respectively. University of Cagliari (UniCA) and Fondazione di Sardegna (FdS) are acknowledged for the financial support – Project: CUP F72F20000240007(2019): “Surface-tailored Materials for Sustainable Environmental Applications”. This research used resources of the Center for Functional Nanomaterials, which is a U.S. DOE Office of Science Facility, at Brookhaven National Laboratory under Contract No. DE-SC0012704.References
- O. Masala and R. Seshadri, J. Am. Chem. Soc., 2005, 127, 9354–9355 CrossRef CAS.
- H. T. Hai, H. T. Yang, H. Kura, D. Hasegawa, Y. Ogata, M. Takahashi and T. Ogawa, J. Colloid Interface Sci., 2010, 346, 37–42 CrossRef CAS.
- L. Wang, X. Wang, J. Luo, B. N. Wanjala, C. Wang, N. A. Chernova, M. H. Engelhard, Y. Liu, I. T. Bae and C. J. Zhong, J. Am. Chem. Soc., 2010, 132, 17686–17689 CrossRef CAS.
- B. P. Pichon, O. Gerber, C. Lefevre, I. Florea, S. Fleutot, W. Baaziz, M. Pauly, M. Ohlmann, C. Ulhaq, O. Ersen, V. Pierron-Bohnes, P. Panissod, M. Drillon and S. Begin-Colin, Chem. Mater., 2011, 23, 2886–2900 CrossRef CAS.
- N. F. Troitiño, B. Rivas-Murias, B. Rodríguez-González and V. Salgueiriño, Chem. Mater., 2014, 26, 5566–5575 CrossRef.
- W. Baaziz, B. P. Pichon, Y. Liu, J.-M. Greneche, C. Ulhaq-Bouillet, E. Terrier, N. Bergeard, V. Halte, C. Boeglin, F. Choueikani, M. Toumi, T. Mhiri and S. Begin-Colin, Chem. Mater., 2014, 26, 5063–5073 CrossRef CAS.
- E. Wetterskog, C. Tai, J. Grins, L. Bergström and G. Salazar-Alvarez, ACS Nano, 2013, 7, 7132–7144 CrossRef CAS.
- S. Noh, W. Na, J. Jang, J.-H. Lee, E. J. Lee, S. H. Moon, Y. Lim, J.-S. Shin and J. Cheon, Nano Lett., 2012, 12, 3716–3721 CrossRef CAS.
- A. López-Ortega, M. Estrader, G. Salazar-Alvarez, S. Estradé, I. V. Golosovsky, R. K. Dumas, D. J. Keavney, M. Vasilakaki, K. N. Trohidou, J. Sort, F. Peiró, S. Suriñach, M. D. Baró and J. Nogués, Nanoscale, 2012, 4, 5138–5147 RSC.
- M. Sanna Angotzi, A. Musinu, V. Mameli, A. Ardu, C. Cara, D. Niznansky, H. L. Xin and C. Cannas, ACS Nano, 2017, 11, 7889–7900 CrossRef CAS.
- H. Yu, M. Chen, P. M. Rice, S. X. Wang, R. L. White and S. Sun, Nano Lett., 2005, 5, 379–382 CrossRef CAS.
- Z. Xu, Y. Hou and S. Sun, J. Am. Chem. Soc., 2007, 129, 8698–8699 CrossRef CAS.
- C. Xu, B. Wang and S. Sun, J. Am. Chem. Soc., 2009, 131, 4216–4217 CrossRef CAS.
- X. F. Zhang, L. Clime, H. Q. Ly, M. Trudeau and T. Veres, J. Phys. Chem. C, 2010, 114, 18313–18317 CrossRef CAS.
- F. Lin, W. Chen, Y.-H. Liao, R. Doong and Y. Li, Nano Res., 2011, 4, 1223–1232 CrossRef CAS.
- Y. Zhang, Y. Zhao, Y. Yang, J. Shen, H. Yang, Z. Zhou and S. Yang, Sens. Actuators, B, 2015, 220, 622–626 CrossRef CAS.
- M. Sanna Angotzi, V. Mameli, C. Cara, A. Musinu, C. Sangregorio, D. Niznansky, H. L. Xin, J. Vejpravova and C. Cannas, Nanoscale Adv., 2020, 2, 3191–3201 RSC.
- J.-H. Lee, J.-T. Jang, J.-S. Choi, S. H. Moon, S.-H. Noh, J.-W. Kim, J.-G. Kim, I.-S. Kim, K. I. Park and J. Cheon, Nat. Nanotechnol., 2011, 6, 418–422 CrossRef CAS.
- F. Fabris, E. Lima, E. De Biasi, H. E. Troiani, M. Vásquez Mansilla, T. E. Torres, R. Fernández Pacheco, M. R. Ibarra, G. F. Goya, R. D. Zysler and E. L. Winkler, Nanoscale, 2019, 11, 3164–3172 RSC.
- J. Robles, R. Das, M. Glassell, M. H. Phan and H. Srikanth, AIP Adv., 2018, 8, 2–8 Search PubMed.
- A. Amarjargal, L. D. Tijing, I. T. Im and C. S. Kim, Chem. Eng. J., 2013, 226, 243–254 CrossRef CAS.
- C. Wang, H. Yin, S. Dai and S. Sun, Chem. Mater., 2010, 22, 3277–3282 CrossRef CAS.
- M. Sahu, M. Shaikh, A. Rai and K. V. S. Ranganath, J. Inorg. Organomet. Polym. Mater., 2020, 30, 1002–1007 CrossRef CAS.
- W. Zang, G. Li, L. Wang and X. Zhang, Catal. Sci. Technol., 2015, 5, 2532–2553 RSC.
- A. Silvestri, S. Mondini, M. Marelli, V. Pifferi, L. Falciola, A. Ponti, A. M. Ferretti and L. Polito, Langmuir, 2016, 32, 7117–7126 CrossRef CAS.
- Virginia Commonwealth University, US Pat.2015 0001437A1, 2015 Search PubMed.
- V. G. Harris, US Pat., 2012O16867OA1, 2012 Search PubMed.
- Industry-University Cooperation Foundation, Hanyang University Erica Campus and LG Electronics INC., US Pat., 20140286817A1, 2014 Search PubMed.
- Fujifilm Corporation, and Tohoku University, US Pat., 20110027588A1, 2011 Search PubMed.
- S. Liu, US Pat., 2006OOO5898A1, 2006 Search PubMed.
- O. Moscoso-Lodoño, D. Muraca, P. Tancredi, C. Cosio-Castañeda, K. R. Pirota and L. M. Socolovsky, J. Phys. Chem. C, 2014, 118, 13168–13176 CrossRef.
- M. E. F. Brollo, R. López-Ruiz, D. Muraca, S. J. A. Figueroa, K. R. Pirota and M. Knobel, Sci. Rep., 2015, 4, 6839 CrossRef.
- P. Tancredi, O. Moscoso Londoño, P. C. Rivas Rojas, U. Wolff, L. M. Socolovsky, M. Knobel and D. Muraca, J. Phys. D: Appl. Phys., 2018, 51(29), 295303 CrossRef.
- C.-H. Lai, T.-F. Wu and M.-D. Lan, IEEE Trans. Magn., 2005, 41, 3397–3399 CAS.
- L. Pan, J. Tang and Y. Chen, Sci. China: Chem., 2013, 56, 362–369 CrossRef CAS.
- S. Peng, C. Lei, Y. Ren, R. E. Cook and Y. Sun, Angew. Chem., Int. Ed., 2011, 50, 3158–3163 CrossRef CAS.
- Y. Mao, P. Yi, Z. Deng and J. Ge, CrystEngComm, 2013, 15, 3575 RSC.
- M. Kaloti and A. Kumar, J. Phys. Chem. C, 2016, 120, 17627–17644 CrossRef CAS.
- H. Gu, Z. Yang, J. Gao, C. K. Chang and B. Xu, J. Am. Chem. Soc., 2005, 127, 34–35 CrossRef CAS.
- L. Pan, Y. Chen and F. Wang, Mater. Chem. Phys., 2012, 134, 177–182 CrossRef CAS.
- J. Huang, Y. Sun, S. Huang, K. Yu, Q. Zhao, F. Peng, H. Yu, H. Wang and J. Yang, J. Mater. Chem., 2011, 21, 17930 RSC.
- Y. Shan, Y. Yang, Y. Cao and Z. Huang, RSC Adv., 2015, 5, 102610–102618 RSC.
- M. Sanna Angotzi, V. Mameli, C. Cara, V. Grillo, S. Enzo, A. Musinu and C. Cannas, Sci. Rep., 2020, 10, 17015 CrossRef CAS.
- L. Carbone and P. D. Cozzoli, Nano Today, 2010, 5, 449–493 CrossRef CAS.
- R. Scarfiello, C. Nobile and P. D. Cozzoli, Front. Mater., 2016, 3, 1–29 Search PubMed.
- P. Mélinon, S. Begin-Colin, J. L. Duvail, F. Gauffre, N. H. Boime, G. Ledoux, J. Plain, P. Reiss, F. Silly and B. Warot-Fonrose, Phys. Rep., 2014, 543, 163–197 CrossRef.
- H. Zhang, Z. Yang, Y. Ju, X. Chu, Y. Ding, X. Huang, K. Zhu, T. Tang, X. Su and Y. Hou, Adv. Sci., 2018, 5, 1–9 Search PubMed.
- D. Jishkariani, Y. Wu, D. Wang, Y. Liu, A. Van Blaaderen and C. B. Murray, ACS Nano, 2017, 11, 7958–7966 CrossRef CAS.
- C. Jin, Y. Qu, M. Wang, J. Han, Y. Hu and R. Guo, Langmuir, 2016, 32, 4595–4601 CrossRef CAS.
- X. Huang, S. Li, Y. Huang, S. Wu, X. Zhou, S. Li, C. L. Gan, F. Boey, C. A. Mirkin and H. Zhang, Nat. Commun., 2011, 2, 292–296 CrossRef.
- H. Shi, B. Dong and W. Wang, Nanoscale, 2012, 4, 6389–6392 RSC.
- E. Fantechi, C. Innocenti, G. Bertoni, C. Sangregorio and F. Pineider, Nano Res., 2020, 13, 785–794 CrossRef CAS.
- B. Muzzi, M. Albino, C. Innocenti, M. Petrecca, B. Cortigiani, C. d. J. Fernández, G. Bertoni, R. Fernandez-Pacheco, A. Ibarra, C. Marquina, M. R. Ibarra and C. Sangregorio, Nanoscale, 2020, 14076–14086 RSC.
- F. Qian, S. Gradečak, Y. Li, C. Y. Wen and C. M. Lieber, Nano Lett., 2005, 5, 2287–2291 CrossRef CAS.
- C. Zhu, M. Yu, J. Zhou, Y. He, Q. Zeng, Y. Deng, S. Guo, M. Xu, J. Shi, W. Zhou, L. Sun, L. Wang, Z. Hu, Z. Zhang, W. Guo and Z. Liu, Nat. Commun., 2020, 11, 1–10 Search PubMed.
- S. G. Kwon and T. Hyeon, Small, 2011, 7, 2685–2702 CrossRef CAS.
- C. Cara, A. Musinu, V. Mameli, A. Ardu, D. Niznansky, J. Bursik, M. A. Scorciapino, G. Manzo and C. Cannas, Cryst. Growth Des., 2015, 15, 2364–2372 CrossRef CAS.
- G. Muscas, G. Singh, W. R. Glomm, R. Mathieu, P. A. Kumar, G. Concas, E. Agostinelli and D. Peddis, Chem. Mater., 2015, 27, 1982–1990 CrossRef CAS.
- R. M. Cornell and U. Schwertmann, The Iron Oxides, Wiley, 2003, vol. 39 Search PubMed.
- J. A. Gomes, G. M. Azevedo, J. Depeyrot, J. Mestnik-Filho, F. L. O. Paula, F. A. Tourinho and R. Perzynski, J. Phys. Chem. C, 2012, 116, 24281–24291 CrossRef CAS.
- J. Yang, E. H. Sargent, S. O. Kelley and J. Y. Ying, Nat. Mater., 2009, 8, 683–689 CrossRef CAS.
- M. Sanna Angotzi, V. Mameli, A. Musinu and D. Nizňnanský, J. Nanosci. Nanotechnol., 2019, 19, 5008–5013 CrossRef.
- M. Sanna Angotzi, V. Mameli, C. Cara, A. Ardu, D. Nizňnanský and A. Musinu, J. Nanosci. Nanotechnol., 2019, 19, 4954–4963 CrossRef.
- V. Mameli, M. Sanna Angotzi, C. Cara and C. Cannas, J. Nanosci. Nanotechnol., 2019, 19, 4857–4887 CrossRef.
- R. De Palma, S. Peeters, M. J. Van Bael, H. Van Den Rul, K. Bonroy, W. Laureyn, J. Mullens, G. Borghs and G. Maes, Chem. Mater., 2007, 19, 1821–1831 CrossRef CAS.
- T. A. S. Ferreira, J. C. Waerenborgh, M. H. R. M. Mendonça, M. R. Nunes and F. M. Costa, Solid State Sci., 2003, 5, 383–392 CrossRef CAS.
- L. Lutterotti and P. Scardi, J. Appl. Crystallogr., 1990, 23, 246–252 CrossRef CAS.
- R. A. Young and D. B. Wiles, J. Appl. Crystallogr., 1982, 15, 430–438 CrossRef CAS.
- S. Mondini, A. M. Ferretti, A. Puglisi and A. Ponti, Nanoscale, 2012, 4, 5356 RSC.
- J. A. Gomes, G. M. Azevedo, J. Depeyrot, J. Mestnik-Filho, F. L. O. Paula, F. A. Tourinho and R. Perzynski, J. Phys. Chem. C, 2012, 116, 24281–24291 CrossRef CAS.
- J. A. Gomes, M. H. Sousa, F. A. Tourinho, R. Aquino, G. J. Da Silva, J. Depeyrot, E. Dubois and R. Perzynski, J. Phys. Chem. C, 2008, 112, 6220–6227 CrossRef.
- Z. X. Tang, C. M. Sorensen, K. J. Klabunde and G. C. Hadjipanayis, J. Colloid Interface Sci., 1991, 146, 38–52 CrossRef CAS.
- G. Gnanaprakash, J. Philip and B. Raj, Mater. Lett., 2007, 61, 4545–4548 CrossRef CAS.
- A. L. Swindle, A. S. E. Madden, I. M. Cozzarelli and M. Benamara, Environ. Sci. Technol., 2014, 48, 11413–11420 CrossRef CAS.
- E. A. Grulke, M. J. Beck, R. A. Yokel, J. M. Unrine, U. M. Graham and M. L. Hancock, Environ. Sci.: Nano, 2019, 6, 1478–1492 RSC.
- H. Chang, B. H. Kim, H. Y. Jeong, J. H. Moon, M. Park, K. Shin, S. I. Chae, J. Lee, T. Kang, B. K. Choi, J. Yang, M. S. Bootharaju, H. Song, S. H. An, K. M. Park, J. Y. Oh, H. Lee, M. S. Kim, J. Park and T. Hyeon, J. Am. Chem. Soc., 2019, 141, 7037–7045 CrossRef CAS.
- A. Ullrich, M. M. Rahman, P. Longo and S. Horn, Sci. Rep., 2019, 9, 1–9 CrossRef.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- H. F. Halliwell and S. C. Nyburg, Trans. Faraday Soc., 1963, 59, 1126–1140 RSC.
- A. López-Ortega, M. Estrader, G. Salazar-Alvarez, A. G. Roca and J. Nogués, Phys. Rep., 2015, 553, 1–32 CrossRef.
- J. M. D. Coey, Magnetism and magnetic materials, Cambridge University Press, Cambridge, 2009 Search PubMed.
- C. Cara, V. Mameli, E. Rombi, N. Pinna, M. Sanna Angotzi, D. Nižňanský, A. Musinu and C. Cannas, Microporous Mesoporous Mater., 2020, 298, 110062 CrossRef CAS.
- V. Mameli, A. Musinu, D. Niznansky, D. Peddis, G. Ennas, A. Ardu, C. Lugliè and C. Cannas, J. Phys. Chem. C, 2016, 120, 27635–27645 CrossRef CAS.
- M. Fantauzzi, F. Secci, M. Sanna Angotzi, C. Passiu, C. Cannas and A. Rossi, RSC Adv., 2019, 9, 19171–19179 RSC.
- N. Monni, V. Mameli, S. A. Sahadevan, S. Gai, C. Cannas and M. L. Mercuri, J. Nanosci. Nanotechnol., 2019, 19, 5043–5047 CrossRef CAS.
- M. Kallumadil, M. Tada, T. Nakagawa, M. Abe, P. Southern and Q. A. Pankhurst, J. Magn. Magn. Mater., 2009, 321, 1509–1513 CrossRef CAS.
- S. Liébana-Viñas, K. Simeonidis, U. Wiedwald, Z.-A. Li, Z. Ma, E. Myrovali, A. Makridis, D. Sakellari, G. Vourlias, M. Spasova, M. Farle and M. Angelakeris, RSC Adv., 2016, 6, 72918–72925 RSC.
- Q. Zhang, I. Castellanos-Rubio, R. Munshi, I. Orue, B. Pelaz, K. I. Gries, W. J. Parak, P. Del Pino and A. Pralle, Chem. Mater., 2015, 27, 7380–7387 CrossRef CAS.
- M. Angelakeris, Z.-A. A. Li, M. Hilgendorff, K. Simeonidis, D. Sakellari, M. Filippousi, H. Tian, G. Van Tendeloo, M. Spasova, M. Acet and M. Farle, J. Magn. Magn. Mater., 2015, 381, 179–187 CrossRef CAS.
- S. O. Solopan, N. Nedelko, S. Lewińska, A. Ślawska-Waniewska, V. O. Zamorskyi, A. I. Tovstolytkin and A. G. Belous, J. Alloys Compd., 2019, 788, 1203–1210 CrossRef CAS.
- O. V. Yelenich, S. O. Solopan, J. M. Greneche and A. G. Belous, Solid State Sci., 2015, 46, 19–26 CrossRef CAS.
Footnotes |
| † The present work is dedicated to Prof. Anna Musinu, on the occasion of her retirement, for her precious and unique contribution to research and teaching activities of the Solid State Chemistry and Nanomaterials Group. |
| ‡ Electronic supplementary information (ESI) available: Scheme of synthesis. Additional TEM and STEM-EDX figures. FTIR spectra. TGA curves. Rietveld refinements of XRD patterns. Magnetic properties. Magnetic fluid hyperthermia. See DOI: 10.1039/d0na00967a |
| This journal is © The Royal Society of Chemistry 2021 |