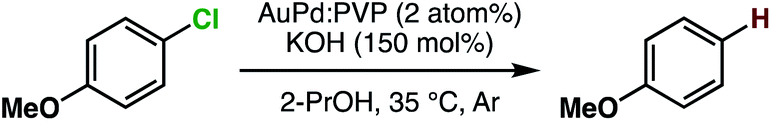DOI:
10.1039/D0NA00951B
(Paper)
Nanoscale Adv., 2021,
3, 1496-1501
Volcano-type correlation between particle size and catalytic activity on hydrodechlorination catalyzed by AuPd nanoalloy†
Received
13th November 2020
, Accepted 18th January 2021
First published on 18th January 2021
Abstract
Although changing the size of metal nanoparticles (NPs) is a reasonable way to tune and/or enhance their catalytic activity, size-selective preparation of NPs possessing random-alloy morphology has been challenging because of the differences in the ionization potential of each metal ion. This study demonstrates a time-controlled aggregation–stabilization method for the size-selective preparation of random alloy NPs composed of Au and Pd, which are stabilized by poly(N-vinyl-2-pyrrolidone) (PVP). By adjusting the mixing time in the presence of a small amount of PVP, aggregation was induced to produce AuPd:PVP with sizes ranging between 1.2 and 8.2 nm at approximately 1 nm intervals. Transmission electron microscopy (TEM), powder X-ray diffraction (PXRD), and extended X-ray absorption fine structure (EXAFS) analyses indicated the formation of various sizes of AuPd nanoalloys, and size-dependent catalytic activity was observed when hydrodechlorination of 4-chloroanisole was performed using 2-propanol as a reducing agent. AuPd:PVP with a size of 3.1 nm exhibited the highest catalytic activity. A comparison of the absorption edges of X-ray absorption near edge structure (XANES) spectra suggested that the electronic state of the Au and Pd species correlated with their catalytic activity, presumably affecting the rate-determining step.
Introduction
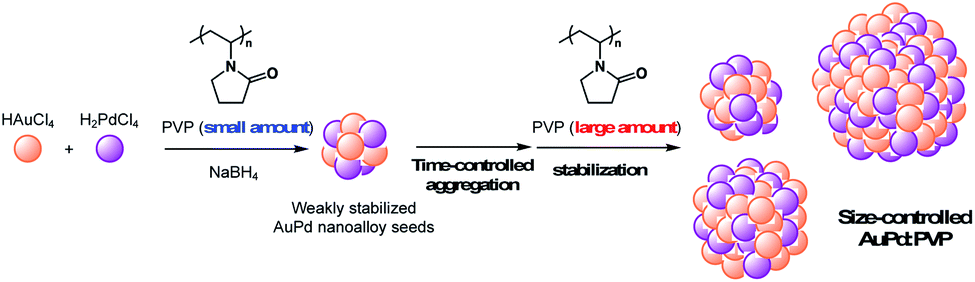
Over the past decade, bimetallic alloy NPs have attracted considerable attention in a wide range of research fields related to catalysis because of their unique catalytic activities.1 Not only the reinforcing effect on the catalytic performance through the interaction of each metal species, the most fascinating aspect of alloy effects is their novel catalytic activity, which monometallic NPs do not exhibit.2 Our early findings of “alloy-specific” reactions for fine chemical synthesis, such as low-temperature Ullmann coupling reaction2a and hydrodechlorination2e of aryl chlorides using quasi-homogeneous AuPd bimetallic nanoalloy catalysts, demonstrated their uniqueness. Shishido's group also reported hydrosilylation of 1,4-unsaturated ketone using AuPd nanoalloy catalysts stabilized on a solid support.2g Since the perturbation of each metal species leads to a drastic change in the electronic structure and surface geometry of NPs (the so-called, ligand effect and ensemble effects),3 one of the facile ways to tune the catalytic activity would be changing the ratio of metal species.2b,g Additionally, the interfacial interaction between metal NPs and stabilizing agents, such as solid supports and polymer matrices, is another way to control the activity,4 however, few options are available and a novel method to control the catalytic activity of alloy NPs is required. Since the catalytic activity of metal NPs is strongly affected by their cluster sizes,5 the development of a method for size-controlled preparation of metal NPs is a promising way to control their catalytic activity. So far, we have established methods for size-selective preparation of AuNPs using poly(N-vinyl-2-pyrrolidone) (PVP) as a stabilizing matrix.6 The thus-prepared Au:PVP catalysts were applied for aerobic oxidation of benzyl alcohol to reveal that the catalytic activity is influenced by the particle size, and also the polymer chain length of PVP (e.g. morphology of PVP).7 Unlike single metal NPs, however, the conventional seed-growth method using small seed NPs and weak reducing agents6b is difficult to apply in the case of a bimetallic alloy system due to large differences in the ionization potential of each metal ion source. Hence, size-selective fabrication of metal nanoalloys is one of the challenges in the field of nanometal science, and a new strategy is required to realize it. Here, we describe a time-controlled strategy for the size-selective preparation of AuPd nanoalloys protected by PVP (Scheme 1), and discuss their size-dependent catalytic effect on hydrodechlorination.
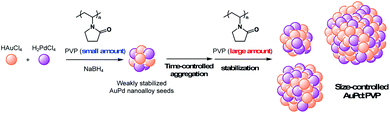 |
| | Scheme 1 Concept of time-controlled aggregation for size-selective preparation of alloy nanoparticles. | |
Results and discussion
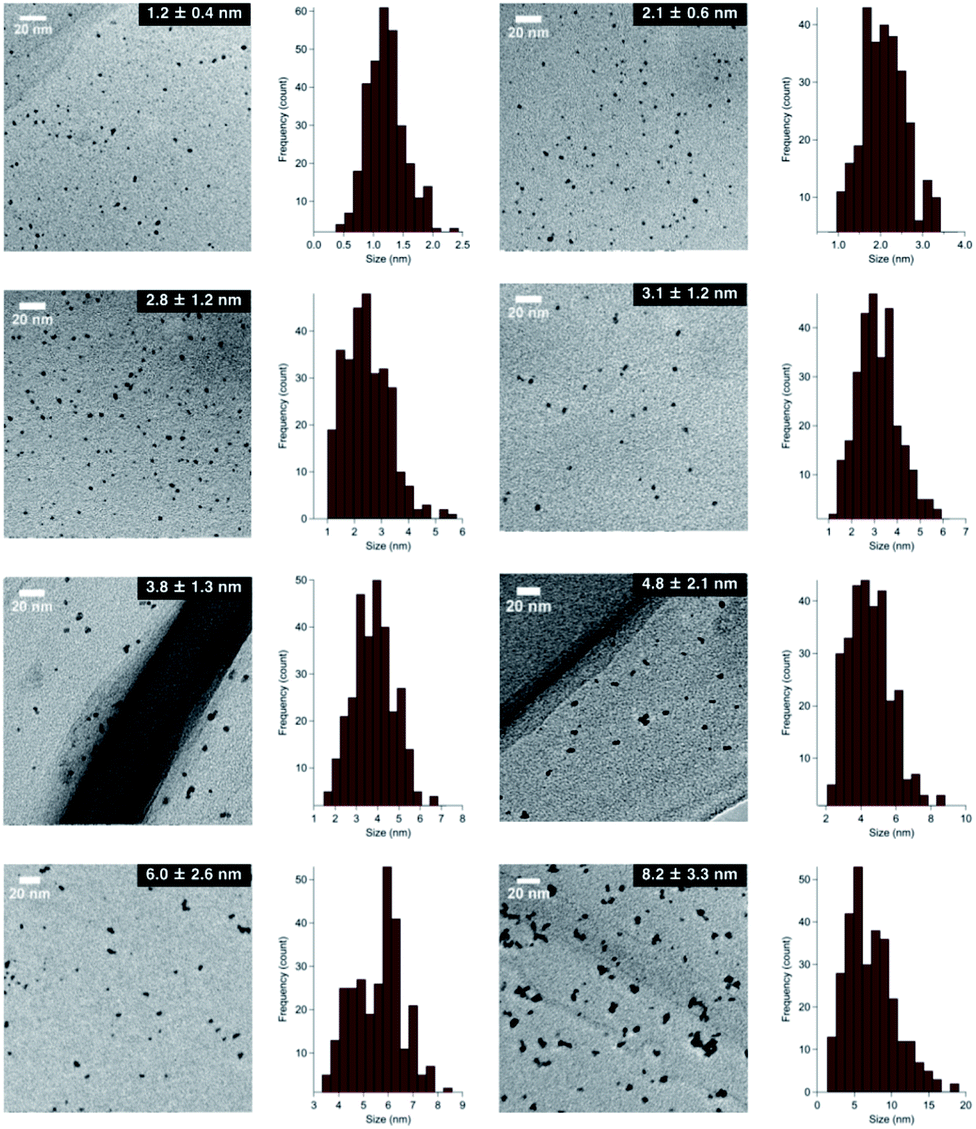
Although several studies have reported the effects of alloy NP sizes, they have arguably not evaluated the actual size effects in the true sense due to the presence of other factors, such as extreme differences in particle sizes, different preparation methods, and differences in morphology, which are thought to significantly affect the catalytic activity.8 To eliminate such uncertain factors to the greatest possible extent, we decided to use NaBH4 as the sole reducing agent for the size-selective preparation of AuPd:PVP. We began by reinvestigating the conventional liquid-phase reduction. In our previous study on the Ullmann coupling reaction, AuPd:PVP(K-30) was prepared using the conventional liquid-phase reduction method with NaBH4 in the presence of equimolar amounts of HAuCl4 and H2PdCl4 at 15 °C.2a Although NPs with an average cluster size of 2.9 ± 0.9 nm were obtained after centrifugal ultrafiltration, scanning TEM-energy dispersive X-ray spectroscope (STEM-EDX) analysis of the thus-prepared nanoalloy revealed a non-uniform composition of Au and Pd atoms due to the difference in their ionization potentials (Au/Au3+ = +1.50 V; Pd/Pd2+ = +0.987 V vs. SHE) (Fig. S1†). To increase the homogeneity of the Au/Pd composition, the temperature of the NaBH4 reduction was optimized to find that a nanoalloy with a similar cluster size (2.8 ± 1.2 nm) was generated at 27 °C reproducibly (Fig. 1 and S3†). The details of the local structure of AuPd:PVP were determined by STEM-EDX and TEM measurements. Judging from the EDX mapping on STEM images, the homogeneity of the nanoalloy's composition was improved compared with that prepared at a lower temperature (Fig. S2†). High-magnification TEM images revealed that the prepared nanoalloy showed a lattice fringe pattern characteristic of a cubooctahedron-shaped crystal, with distances of 2.28–2.34 Å, that is, values between those of single Au (2.35 Å, fcc) and Pd NPs (2.25 Å, fcc), also suggesting the formation of a well-mixed nanoalloy (Fig. S4†).9,10
 |
| | Fig. 1 TEM images of AuPd:PVP nanoalloys. | |
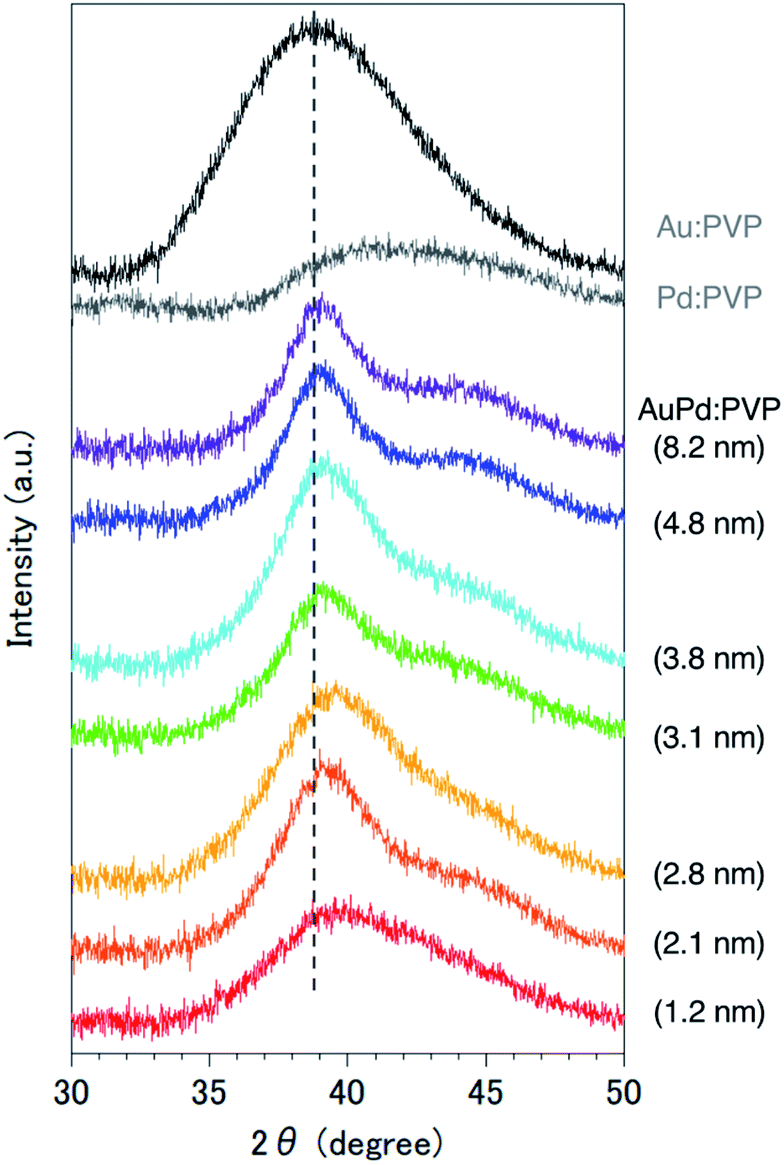
Since it was difficult to obtain the selected area electron diffraction (SAED) image from the nanoalloy due to the weak diffraction intensity of small NPs, next we conducted a survey on the global homogeneity of the nanoalloy using X-ray spectroscopy techniques. Powder X-ray diffraction (PXRD) spectra of AuPd:PVP along with Au:PVP(K-30, 1.3 nm) and Pd:PVP(K-30, 1.5 nm) are shown in Fig. 2. Broadened peaks with the value of 2θ = 38–40° were observed, which is the value between those of the Au(111) and Pd(111) facets with fcc crystal structures. Additionally, peaks that can be assigned to the (200) facet were also detected at the shoulder of the (111) facet (2θ = 44°) in the case of AuPd:PVP, also indicating the fcc crystal structure of the derived nanoalloys.2a,2g,11 To obtain more information about the alloy structure, X-ray absorption spectroscopy (XAS) measurements at Au L3- and Pd K-edges were carried out, and the coordination number (CN) and atomic distance of each metal species were analyzed through curve-fitting in r-space derived from Fourier transformation of k3-weighted EXAFS oscillation. In both elements, the shape of the spectra was apparently different from those of the bulk metals, suggesting the formation of an alloy structure (Fig. S7 and S8†). Clearer evidence for the alloy structure was obtained from the curve-fitting of the EXAFS spectra, showing the existence of Au–Pd bonding (Table 1). The bond length and CN of Au–Pd, derived from both absorption edges, showed acceptable agreement with each other (Au L3 edge: 2.78 Å, CN: 3.0; Pd K-edge: 2.77 Å, CN: 4.4), demonstrating the validity of the fitting results. By collating with the CN of monometallic Au NPs with a diameter of 2.8 nm,7 the sum of the mean CN around the Au atom (8.6) is considered a reasonable value. On the other hand, a relatively small value was observed for the Pd atom (6.5), which indicates that the population of the existing Pd species is to some extent biased toward the surface of the nanoalloy, reflecting the thermodynamic stability of Au–Au bonding and the intrinsic difference in ionization potential. The bond length of Au–Au was estimated to be 2.79 Å, which is shorter than that of bulk Au (2.85 Å). An extension of the bond length of Pd–Pd (2.78 Å; 2.74 Å for Pd foil) was also observed. These observations are consistent with the experimental results of TEM and PXRD described above. Moreover, almost the same curve-fitting results were derived even with other methods of fabricating AuPd:PVP (Table 1), which are described in the next paragraph. Considering all the results, we conclude that AuPd:PVP prepared by liquid-phase reduction using NaBH4 exhibits a random alloy configuration, as revealed by spectroscopic analyses of local and global structures.
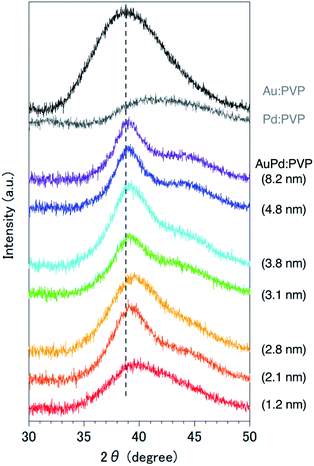 |
| | Fig. 2 PXRD spectra of Au:PVP(K-30, 1.3 nm), Pd:PVP(K-30, 1.5 nm), and AuPd:PVP NPs. | |
Table 1 Parameters obtained from curve-fitting of Fourier transformed k3-weighted EXAFS spectra in r-spacea
| Particle size (nm) |
Shell |
CNb |
R
(Å) |
ΔE0d (eV) |
σ
2
(10−3 Å2) |
χ
v
2
|
R-Factorg (%) |
|
Au L3: k-range = 3–16 Å−1, r-range = 1.5–3.3 Å, S02 = 0.83 (Au foil); Pd K: k-range = 3–14 Å−1, r-range: 1.7–3.2 Å, S02 = 0.75 (Pd foil).
Coordination number.
Bond length.
Absorption edge energy shift.
Debye–Waller factor.
Reduced χ-squared value.
Goodness-of-fit index.
|
| 2.1 |
Au–Au |
5.6 ± 0.51 |
2.77 ± 0.007 |
0.969 ± 1.07 |
9.48 ± 0.500 |
18.2 |
0.70 |
| Au–Pd |
2.7 ± 0.39 |
2.75 ± 0.007 |
6.17 ± 1.05 |
8.24 ± 0.772 |
11.9 |
0.27 |
| Pd–Pd |
1.6 ± 0.35 |
2.77 ± 0.009 |
−4.17 ± 1.49 |
6.69 ± 1.20 |
|
|
| Pd–Au |
4.4 ± 0.51 |
2.77 ± 0.008 |
−4.32 ± 1.33 |
8.00 ± 0.696 |
|
|
| 2.8 |
Au–Au |
5.6 ± 0.61 |
2.79 ± 0.008 |
3.14 ± 1.27 |
10.1 ± 0.631 |
13.7 |
0.85 |
| Au–Pd |
3.0 ± 0.45 |
2.78 ± 0.007 |
6.12 ± 1.12 |
8.56 ± 0.822 |
6.3 |
0.30 |
| Pd–Pd |
2.1 ± 0.43 |
2.78 ± 0.010 |
−2.75 ± 1.43 |
7.98 ± 1.28 |
|
|
| Pd–Au |
4.4 ± 0.58 |
2.77 ± 0.008 |
−2.58 ± 1.41 |
8.22 ± 0.779 |
|
|
| 3.1 |
Au–Au |
5.7 ± 0.54 |
2.78 ± 0.007 |
2.53 ± 1.11 |
9.27 ± 0.503 |
15.1 |
0.76 |
| Au–Pd |
3.0 ± 0.42 |
2.78 ± 0.006 |
6.40 ± 1.02 |
7.97 ± 0.746 |
12.0 |
0.73 |
| Pd–Pd |
2.0 ± 0.64 |
2.77 ± 0.014 |
−3.25 ± 2.20 |
7.22 ± 1.81 |
|
|
| Pd–Au |
4.7 ± 0.90 |
2.77 ± 0.012 |
−2.64 ± 2.12 |
8.04 ± 1.09 |
|
|
| 3.8 |
Au–Au |
5.5 ± 0.46 |
2.79 ± 0.006 |
2.78 ± 0.959 |
8.67 ± 0.402 |
18.3 |
1.05 |
| Au–Pd |
3.2 ± 0.38 |
2.79 ± 0.005 |
6.87 ± 0.859 |
7.71 ± 0.640 |
8.4 |
0.38 |
| Pd–Pd |
2.0 ± 0.47 |
2.77 ± 0.010 |
−2.59 ± 1.65 |
6.88 ± 1.35 |
|
|
| Pd–Au |
4.5 ± 0.63 |
2.77 ± 0.008 |
−4.37 ± 1.58 |
7.29 ± 0.762 |
|
|
| Au foil |
Au–Au |
12 (fix) |
2.85 ± 0.002 |
2.77 ± 0.519 |
8.23 ± 0.202 |
23.6 |
0.37 |
| Pd foil |
Pd–Pd |
12 (fix) |
2.74 ± 0.003 |
−0.943 ± 0.911 |
5.52 ± 0.392 |
543 |
0.43 |
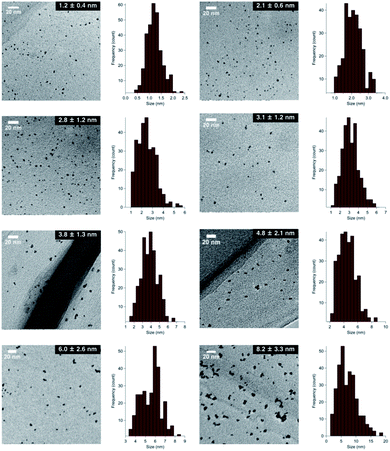
In the case of the conventional liquid-phase reduction method, the NPs generated in situ were immediately stabilized in the presence of an excess amount of PVP (metal ion/PVP = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100) before nucleus growth to produce smaller NPs.6a To fabricate a larger nanoalloy without using other reducing agents, we expected that an aggregation-induced size growth of nanoalloys would be promoted with a very small amount of stabilizing agent, generating a less stable intermediate intensively, and that size control could also be achieved by changing the time of the aggregation phase. We began our investigation with these considerations in mind. A solution of NaBH4 (10 equiv.) was added to a solution of HAuCl4, H2PdCl4, and PVP (3 equiv., as a monomer unit) in H2O, at 27 °C to generate the nanoalloy. After 300 s aging to promote the aggregation, an excess amount of PVP (97 equiv.) was added to stabilize the nanoalloy and cease the aggregation. After the standard purification process, TEM measurements were carried out to ascertain the generation of AuPd:PVP with a particle size of 3.8 ± 1.3 nm (n = 3). Next, we investigated the different aging times of the aggregation (<1, 20, 60, and 180 s) in order to control the particle size (Table S1†). Although no significant size change occurred until 20 and 60 s, AuPd:PVP with a cluster size of 3.3 ± 1.2 nm was obtained after aging for 180 s. In the case of <1 s aging, a smaller nanoalloy was formed, probably due to the lower initial concentration of PVP, increasing the diffusion rate of NaBH4. These results clearly showed that the particle size of the nanoalloy is controllable using the aggregation–stabilization technique. As the mechanism of nucleation is the same as that of the conventional liquid-phase reduction method, the composition of the nanoalloy must be determined at the stage of nucleation and it did not change after aggregation. The PXRD spectra and EXAFS analysis of these nanoalloys exhibited almost the same results as in the case of the conventional method, indicating their global uniformity. TEM measurement of the AuPd:PVP (3.1 ± 1.2 nm) was also performed to reveal the local structure. Although some NPs showed the same pattern of lattice fringe as in the case of conventional reduction, multiply-twinned particles with dodecahedral or regular icosahedral crystal shapes were also observed.12 A reason for the contamination of crystals with different morphologies may be related to the preparation method of utilizing the aggregation of NP seeds. In light of the successful experimental results illustrating the concept of aggregation–stabilization, we anticipated that the size-swelling might be achieved by increasing the number of contacts of the NPs, generating much larger nanoalloys. To verify this assumption, a microfluidic reaction condition was applied in order to perform vigorous mixing before the aging phase, facilitating size growth (Table S2†). A COMET X-01 micromixing unit (Techno Applications Co., Ltd.), which is composed of a T-shaped channel controlling unit connected to a double-lane PTFE tube and micromixer,6c was chosen for this purpose. Maintaining a constant flow rate, the aging time was controlled by changing the channel length to 6.62, 50.8, 183, and 316 cm, which corresponded to 20, 60, 180, and 300 s, producing AuPd:PVP with cluster sizes of 4.8 ± 2.1, 6.0 ± 2.6, 7.1 ± 3.3, and 8.2 ± 3.3 nm, respectively. TEM measurements revealed the shape of the nanoalloy to be a polycrystal consisting of cubooctahedron-shaped crystals, which also reflected the preparation method. The crystallite size was also estimated with the Scherrer equation using the peak assigned to the diffraction of the AuPd(111) facet after deconvolution (Table S3†). Though the crystallite size was consistent with the particle size obtained from TEM measurements in the range of 1.2 to 3.8 nm, the values of crystallite size were small compared to that of particle size in the case of larger nanoparticles (>4.8 nm). This result indicated that AuPd:PVP prepared by the time-controlled aggregation–stabilization techniques consists of the aggregation of small nanoparticles, reflecting the concept of the method.
:100) before nucleus growth to produce smaller NPs.6a To fabricate a larger nanoalloy without using other reducing agents, we expected that an aggregation-induced size growth of nanoalloys would be promoted with a very small amount of stabilizing agent, generating a less stable intermediate intensively, and that size control could also be achieved by changing the time of the aggregation phase. We began our investigation with these considerations in mind. A solution of NaBH4 (10 equiv.) was added to a solution of HAuCl4, H2PdCl4, and PVP (3 equiv., as a monomer unit) in H2O, at 27 °C to generate the nanoalloy. After 300 s aging to promote the aggregation, an excess amount of PVP (97 equiv.) was added to stabilize the nanoalloy and cease the aggregation. After the standard purification process, TEM measurements were carried out to ascertain the generation of AuPd:PVP with a particle size of 3.8 ± 1.3 nm (n = 3). Next, we investigated the different aging times of the aggregation (<1, 20, 60, and 180 s) in order to control the particle size (Table S1†). Although no significant size change occurred until 20 and 60 s, AuPd:PVP with a cluster size of 3.3 ± 1.2 nm was obtained after aging for 180 s. In the case of <1 s aging, a smaller nanoalloy was formed, probably due to the lower initial concentration of PVP, increasing the diffusion rate of NaBH4. These results clearly showed that the particle size of the nanoalloy is controllable using the aggregation–stabilization technique. As the mechanism of nucleation is the same as that of the conventional liquid-phase reduction method, the composition of the nanoalloy must be determined at the stage of nucleation and it did not change after aggregation. The PXRD spectra and EXAFS analysis of these nanoalloys exhibited almost the same results as in the case of the conventional method, indicating their global uniformity. TEM measurement of the AuPd:PVP (3.1 ± 1.2 nm) was also performed to reveal the local structure. Although some NPs showed the same pattern of lattice fringe as in the case of conventional reduction, multiply-twinned particles with dodecahedral or regular icosahedral crystal shapes were also observed.12 A reason for the contamination of crystals with different morphologies may be related to the preparation method of utilizing the aggregation of NP seeds. In light of the successful experimental results illustrating the concept of aggregation–stabilization, we anticipated that the size-swelling might be achieved by increasing the number of contacts of the NPs, generating much larger nanoalloys. To verify this assumption, a microfluidic reaction condition was applied in order to perform vigorous mixing before the aging phase, facilitating size growth (Table S2†). A COMET X-01 micromixing unit (Techno Applications Co., Ltd.), which is composed of a T-shaped channel controlling unit connected to a double-lane PTFE tube and micromixer,6c was chosen for this purpose. Maintaining a constant flow rate, the aging time was controlled by changing the channel length to 6.62, 50.8, 183, and 316 cm, which corresponded to 20, 60, 180, and 300 s, producing AuPd:PVP with cluster sizes of 4.8 ± 2.1, 6.0 ± 2.6, 7.1 ± 3.3, and 8.2 ± 3.3 nm, respectively. TEM measurements revealed the shape of the nanoalloy to be a polycrystal consisting of cubooctahedron-shaped crystals, which also reflected the preparation method. The crystallite size was also estimated with the Scherrer equation using the peak assigned to the diffraction of the AuPd(111) facet after deconvolution (Table S3†). Though the crystallite size was consistent with the particle size obtained from TEM measurements in the range of 1.2 to 3.8 nm, the values of crystallite size were small compared to that of particle size in the case of larger nanoparticles (>4.8 nm). This result indicated that AuPd:PVP prepared by the time-controlled aggregation–stabilization techniques consists of the aggregation of small nanoparticles, reflecting the concept of the method.
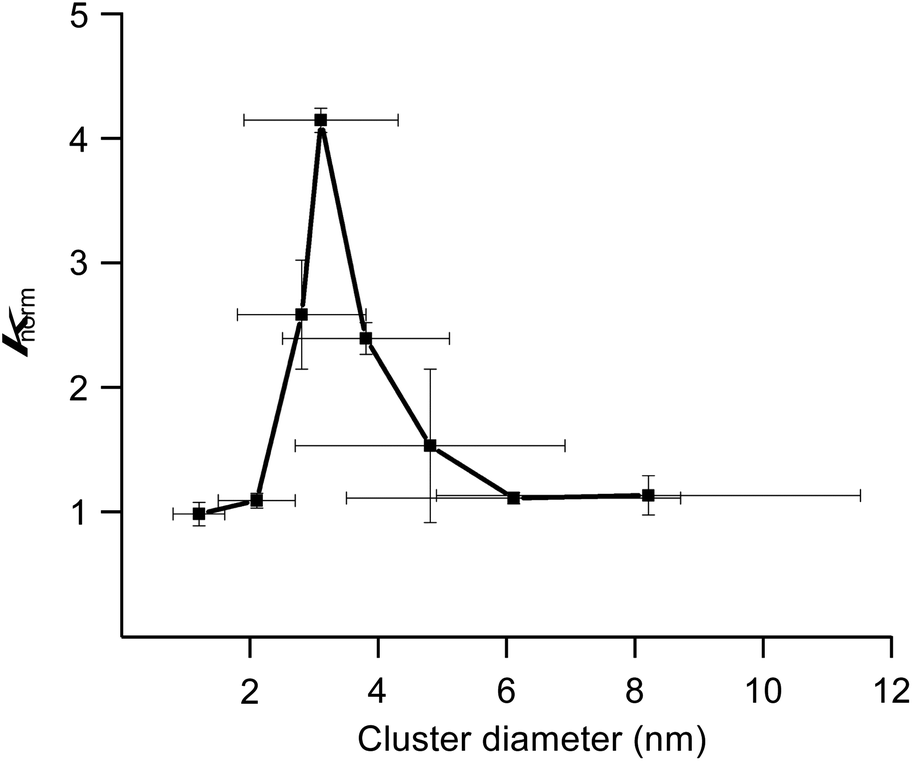
We previously reported hydrodechlorination of aryl chlorides at ambient temperature in the presence of AuPd:PVP nanoalloys using 2-PrOH as a solvent and reducing agent.2e With the different methods of size-selective preparation of AuPd:PVP in this study, a survey on the size dependency of the catalytic effect on hydrodechlorination was conducted using 4-chloroanisole as a substrate. Nakajima et al. previously reported the preparation of ultra-small AuPd:PVP (ca. 0.9 nm) using an ultrafine multichannel microfluidic mixer originally developed by them.13 For the sequential survey on the size-dependence of the catalytic activity and electronic structure, their method was applied with minor modifications to fit our preparation conditions, successfully giving AuPd:PVP(K-30) (1.2 ± 0.4 nm). The reaction was performed at 35 °C in all cases to afford anisole as the sole product, and the rate constant (k) and reaction order (n) of each nanoalloy were derived from the curve-fitting of the time course plot of 1/[A]n−1 using an n-order kinetic equation with using the n value as a variable parameter (Table 2, Fig. S9–S17†). The optimum value was determined as the one when the minimum coefficient of determination (R2) was obtained. As a result, the first order kinetic treatment seemed to be appropriate for small particle sizes, while there seems to be a deviation towards second order kinetics for large particle sizes. This result suggested perhaps the changing of the reaction mechanism (Fig. S18†). In other words, one aryl group was adsorbed on the surface of the metal NPs after oxidative addition in the case of small particle sizes. On the other hand, oxidative addition would successively occur at multiple sites in the case of large particle sizes, affording the multiply adsorbed species because of the increasing of the available active sites on a particle. In addition, the oxidative addition species would be involved in the rate determining step of this reaction. For the sake of comparison, the k values were normalized by the surface areas of the corresponding nanoalloys by assuming the spherical shapes with the diameters, and expressed as values divided by that of the smallest nanoalloy (1.2 nm). As a result, differences in catalytic activity and reaction profile were observed in relation to the particle size. The normalized rate constant (knorm) revealed its maximum value when AuPd:PVP of 3.1 nm was used, and exhibited the so-called “volcano” type of activity profile (Fig. 3).5 Although it has been well recognized that smaller NPs tend to exert better catalytic activity due to the greater number of catalytically reactive sites (i.e. corner and/or edge geometry), size-specific characteristics were observed in this case, probably indicating a significant effect of the electronic structure.
Table 2 Size-dependent catalytic activity in hydrodechlorination of 4-chloroanisole
|
|
| Entry |
Cluster size (nm) |
Colloid sizea (nm) |
k
(h−1) |
k
norm
|
n
|
|
Induced grating (IG) method (0.3 mmol L−1 for PVP).
Derived from the curve-fitting using the equation of 1/[A]n−1 = (n − 1)kt + 1/[A]0n−1 ([A]: concentration of 4-chloroanisole, t: time (h)).
k
norm = (km × dm)/(k1 × d1), k1 = 1.2, d1 = 1.78.
|
| 1 |
1.2 ± 0.4 |
131 ± 30 |
1.78 |
1 |
1.1 |
| 2 |
2.1 ± 0.6 |
111 ± 16 |
1.11 |
1.1 |
1.1 |
| 3 |
2.8 ± 1.2 |
94 ± 24 |
1.85 |
2.4 |
1.2 |
| 4 |
3.1 ± 1.2 |
58 ± 18 |
2.77 |
4.0 |
1.2 |
| 5 |
3.8 ± 1.3 |
68 ± 11 |
1.34 |
2.4 |
1.7 |
| 6 |
4.8 ± 2.1 |
72 ± 12 |
0.907 |
2.0 |
1.6 |
| 7 |
6.0 ± 2.6 |
62 ± 11 |
0.388 |
1.1 |
1.8 |
| 8 |
8.2 ± 3.3 |
103 ± 17 |
0.233 |
0.89 |
2.4 |
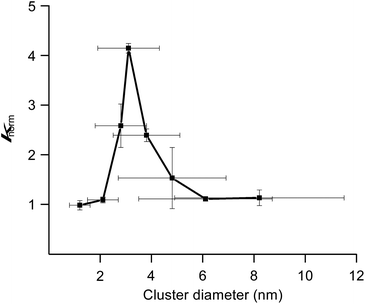 |
| | Fig. 3 Correlation between particle size and normalized rate constant. | |
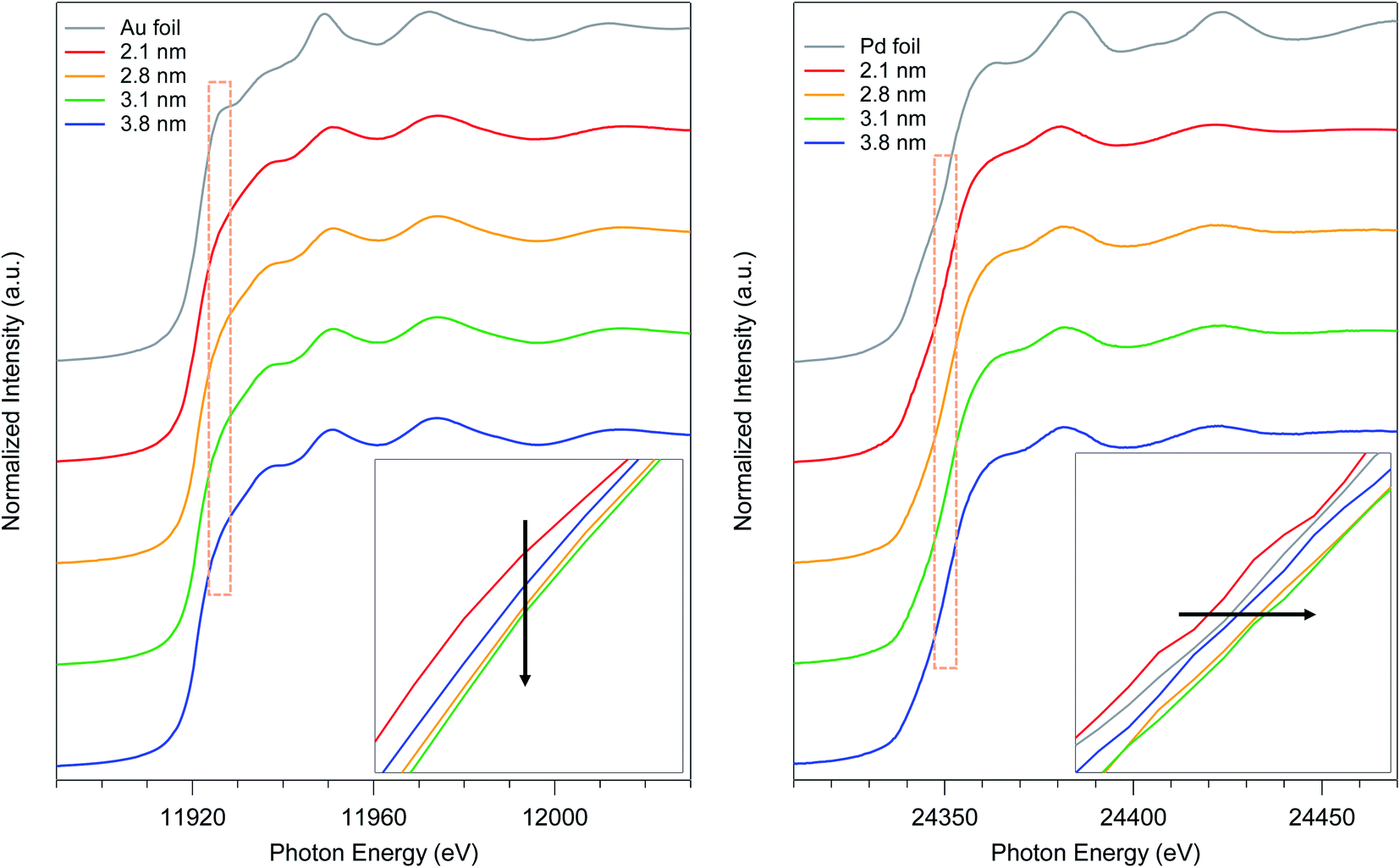
To elucidate the relationship between the particle size and the electronic state of the prepared AuPd:PVPs with several particle sizes, precise analysis of XANES spectra was carried out. The shapes of XANES spectra of AuPd:PVPs quite resemble each other at both absorption edges, which indicated that these Au NPs possess similar density of states.14–16 The Au L3-edge XANES spectra showed a less intense white line signal, suggesting the formation of electron-rich Au species, which was attributed to the electron donation from Pd and PVP.7a The lowest intensity was observed in the case of AuPd:PVP with a diameter of 3.1 nm, which exhibited the highest catalytic activity (Fig. 4, left). Pd K-edge XANES analysis also revealed that AuPd:PVP with a diameter of 3.1 nm showed the highest absorption edge energy at half-height, indicating the formation of electron-deficient Pd species, attributed to the electron-withdrawing effect of the electron-negative Au atoms (Fig. 4, right). Regarding the electronic state of these two metals, the results of XANES spectra at both edges correlated with each other, indicating not only that the electron-rich Au and electron-deficient Pd species might affect the catalytic activity but also that the electronic state of the nanoalloy is perturbed only by changing the cluster size. The colloid sizes in an aqueous solution were also estimated using the induced grating (IG) method to clarify the interfacial morphology between the surrounding PVP and the core metal (Table 2).7b The smallest colloid size (58 ± 18 nm) was observed in the case of the 3.1 nm cluster size, indicating a strong interaction between the metal surface and the PVP matrix. In our research into the “matrix effect of PVP”, we found that the electronic state of the metal surface correlates with the colloid size, probably due to proximity-enhanced electron-donation from carbonyl groups of PVP to the core metal.7 Although in this case, it is difficult to clarify the role of PVP because of the additional electronic influence of the adjacent Au species, we assumed that electron-donation from PVP might facilitate the oxidative addition step. The origin of the size-dependent catalytic activity remains unclear due to multiple factors, such as the quantum size effect, the composition of the metal surface (so-called ensemble effect), and the interfacial morphology, in addition to the issues described above. Therefore, more precise experimental and theoretical investigations are required.
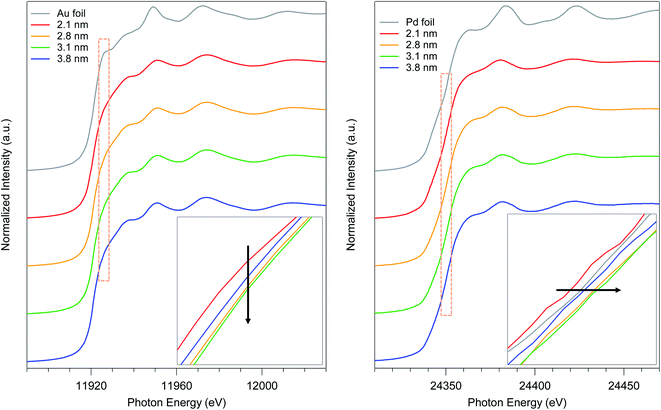 |
| | Fig. 4 Au L3- and Pd K-edge XANES spectra of AuPd:PVP (2.1, 2.8, 3.1, and 3.8 nm) and their magnifications at the absorption edge. | |
Conclusions
We achieved the size-controlled preparation of AuPd nanoalloys protected by PVP with a precision of approximately 1 nm intervals based on the time-controlled aggregation–stabilization technique. Thanks to the use of the same reducing agent, different sizes of AuPd:PVP with almost the same morphology were furnished, allowing us to thoroughly investigate the size-dependent catalytic activity. AuPd:PVP with a size of 3.1 nm showed the highest catalytic effect on hydrodechlorination of 4-chloroanisole. The employed methods are quite practical because of the easy preparation protocol, and enabled us to observe new phenomena. Furthermore, in combination with the trans-deposition method developed by our group,17 it may be possible to prepare a size-specific AuPd nanoalloy deposited on a solid support with retaining its cluster size, thus broadening its range of applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by JSPS-KAKENHI (JP19K22187) and JST (ACT-C) Project (JPMJCR12YI). STEM-EDX measurements at Osaka University were supported by “Nanotechnology Platform Program” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. XAS measurements were performed at SPring-8 BL01B1 beamline with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2015A1134, 2016B1148).
Notes and references
-
(a) N. Toshima and T. Yonezawa, New J. Chem., 1998, 22, 1179–1201 RSC;
(b) R. Ferrando, J. Jellineck and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS;
(c) J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef.
-
(a) R. N. Dhital, C. Kamonsatikul, E. Somsook, K. Bobuatong, M. Ehara, S. Karanjit and H. Sakurai, J. Am. Chem. Soc., 2012, 134, 20250–20253 CrossRef CAS;
(b) A. Murugadoss, K. Okumura and H. Sakurai, J. Phys. Chem. C, 2012, 116, 26776–26783 CrossRef CAS;
(c) R. N. Dhital, C. Kamonsatikul, E. Somsook and H. Sakurai, Catal. Sci. Technol., 2013, 3, 3030–3035 RSC;
(d) R. N. Dhital, K. Bobuatong, M. Ehara and H. Sakurai, Chem.–Asian J., 2015, 10, 2669–2676 CrossRef CAS;
(e) S. Karanjit, A. Jinasan, E. Somsook, R. N. Dhital, K. Motomiya, Y. Sato, K. Tohji and H. Sakurai, Chem. Commun., 2015, 51, 12724–12727 RSC;
(f) N. Agarwal, S. J. Freakley, R. U. McVicker, S. M. Althahban, N. Dimitratos, Q. He, D. J. Morgan, R. L. Jenkins, D. J. Willock, S. H. Taylor, C. J. Kiely and G. J. Hutchings, Science, 2017, 358, 223–227 CrossRef CAS;
(g) H. Miura, K. Endo, R. Ogawa and T. Shishido, ACS Catal., 2017, 7, 1543–1553 CrossRef CAS;
(h) H. Miura, S. Sasaki, R. Ogawa and T. Shishido, Eur. J. Org. Chem., 2018, 1858–1862 CrossRef CAS;
(i) H. Miura, Y. Tanaka, K. Nakahara, Y. Hachiya, K. Endo and T. Shishido, Angew. Chem., Int. Ed., 2018, 57, 6136–6140 CrossRef CAS;
(j) C. Williams, J. H. Carter, N. F. Dummer, Y. K. Chow, D. J. Morgan, S. Yacob, P. Serna, D. J. Willock, R. J. Meyer, S. H. Taylor and G. J. Hutchings, ACS Catal., 2018, 8, 2567–2576 CrossRef CAS;
(k) K. Nakajima, M. Tominaga, M. Waseda, H. Miura and T. Shishido, ACS Sustainable Chem. Eng., 2019, 7, 6522–6530 CrossRef CAS;
(l) T. Sadhukhan, A. Junkaew, P. Zhao, H. Miura, T. Shishido and M. Ehara, Organometallics, 2020, 39, 528–537 CrossRef CAS;
(m) H. Miura, Y. Masaki, Y. Fukuta and T. Shishido, Adv. Synth. Catal., 2020, 362, 2642–2650 CrossRef CAS.
- S. Cao, F. Tao, Y. Tang, Y. Li and J. Yu, Chem. Soc. Rev., 2016, 45, 4747–4765 RSC.
-
(a) T. R. Walsh and M. R. Knecht, Chem. Rev., 2017, 117, 12641–12704 CrossRef CAS;
(b) A. Heuer-Jungemann, N. Feliu, I. Bakaimi, M. Hamaly, A. Alkilany, I. Chakraborty, A. Masood, M. F. Casula, A. Kostopoulou, E. Oh, K. Susumu, M. H. Stewart, I. L. Medintz, E. Stratakis, W. J. Parak and A. G. Kanaras, Chem. Rev., 2019, 119, 4819–4880 CrossRef CAS;
(c) M. Sankar, Q. He, R. V. Engel, M. A. Sainna, A. J. Logsdail, A. Roldan, D. J. Willock, N. Agarwal, C. J. Kiely and G. J. Hutchings, Chem. Rev., 2020, 120, 3890–3938 CrossRef CAS;
(d) L. Jia, T. Zhou, J. Xu, F. Li, Z. Xu, B. Zhang, S. Guo, X. Shen and W. Zhang, Nanomaterials, 2017, 7, 333 CrossRef.
-
(a) M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647–1650 CrossRef CAS;
(b) Y. Liu, H. Tsunoyama, T. Akita, S. Xie and T. Tsukuda, ACS Catal., 2011, 1, 2–6 CrossRef CAS.
-
(a) H. Tsunoyama, H. Sakurai, Y. Negishi and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 9374–9375 CrossRef CAS;
(b) H. Tsunoyama, H. Sakurai and T. Tsukuda, Chem. Phys. Lett., 2006, 429, 528–532 CrossRef CAS;
(c) S. Haesuwannakij, W. Karuehanon, V. L. Mishra, H. Kitahara, H. Sakurai, S. Kanaoka and S. Aoshima, Monatsh. Chem., 2014, 145, 23–28 CrossRef CAS.
-
(a) H. Tsunoyama, N. Ichikuni, H. Sakurai and T. Tsukuda, J. Am. Chem. Soc., 2009, 131, 7086–7093 CrossRef CAS;
(b) S. Haesuwannakij, T. Kimura, Y. Furutani, K. Okumura, K. Kokubo, T. Sakata, H. Yasuda, Y. Yakiyama and H. Sakurai, Sci. Rep., 2017, 7, 9579 CrossRef.
- J. Cheng, X. Gu, X. Sheng, P. Liu and H. Su, J. Mater. Chem. A, 2016, 4, 1887–1894 RSC.
- S.-C. Y. Tsen, P. A. Crozier and J. Liu, Ultramicroscopy, 2003, 98, 63–72 CrossRef CAS.
- D. Zhang, C. Jin, H. Tian, Y. Xiong, H. Zhang, P. Qiao, J. Fan, Z. Zhang, Z. Y. Li and J. Li, Nanoscale, 2017, 9, 6327–6333 RSC.
- A. N. Geraldes, D. Furtunato da Silva, E. S. Pino, J. C. Martins da Silva, R. F. Brambilla de Souza, P. Hammer, E. V. Spinacé, A. O. Neto, M. Linardi and M. Coelho dos Santos, Electrochim. Acta, 2013, 111, 455–465 CrossRef CAS.
- N. Jian and R. E. Palmer, J. Phys. Chem. C, 2015, 119, 11114–11119 CrossRef CAS.
- N. Hayashi, Y. Sakai, H. Tsunoyama and A. Nakajima, Langmuir, 2014, 30, 10539–10547 CrossRef CAS.
- S. Marx and A. Baiker, J. Phys. Chem. C, 2009, 113, 6191–6201 CrossRef CAS.
- F. Liu, D. Wechsler and P. Zhang, Chem. Phys. Lett., 2008, 461, 254–259 CrossRef CAS.
-
(a) P. Dash, T. Bond, C. Fowler, W. Hou, N. Coombs and R. W. J. Scott, J. Phys. Chem. C, 2009, 113, 12719–12730 CrossRef CAS;
(b) T. Balcha, J. R. Strobl, C. Fowler, P. Dash and R. W. J. Scott, ACS Catal., 2011, 1, 425–436 CrossRef CAS.
- S. Haesuwannakij, T. Poonsawat, M. Noikham, E. Somsook, Y. Yakiyama, R. N. Dhital and H. Sakurai, J. Nanosci. Nanotechnol., 2017, 17, 4649–4657 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, magnified TEM and STEM-EDX images, EXAFS spectra, and experimental data for kinetics (PDF). See DOI: 10.1039/d0na00951b |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Sachi
Mouri
a,
Setsiri
Haesuwannakij
a,
Kazu
Okumura
a,
Sachi
Mouri
a,
Setsiri
Haesuwannakij
a,
Kazu
Okumura