
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFabrication of polyethylene terephthalate (PET) nanoparticles with fluorescent tracers for studies in mammalian cells†
Leah M.
Johnson
 *a,
Jeffrey B.
Mecham
a,
Sai Archana
Krovi
a,
Maria M.
Moreno Caffaro
a,
Shyam
Aravamudhan
b,
Alexander L.
Kovach
a,
Timothy R.
Fennell
a and
Ninell P.
Mortensen
a
*a,
Jeffrey B.
Mecham
a,
Sai Archana
Krovi
a,
Maria M.
Moreno Caffaro
a,
Shyam
Aravamudhan
b,
Alexander L.
Kovach
a,
Timothy R.
Fennell
a and
Ninell P.
Mortensen
a
aRTI International 3040 East Cornwallis Drive, Research Triangle Park, NC 27709, USA. E-mail: leahjohnson@rti.org
bJoint School of Nanoscience and Nanoengineering, North Carolina A&T State University, 2907 East Gate City Blvd, Greensboro, NC 27401, USA
First published on 15th December 2020
Abstract
Fluorescent nanoparticles (NPs) comprising polyethylene terephthalate (PET) with a hydrodynamic diameter of 158 ± 2 nm were synthesized in a bottom-up approach. Concentration-dependent uptake and cytotoxicity of PET NPs in macrophages are shown. The fabrication of well-characterized NPs, derived from high-commodity polymers, will support future studies to assess effects on biological systems.
Introduction
Plastics are globally ubiquitous and have supported advancements in society by improving the performance of food and beverage packaging, building materials, healthcare and personal care products, and transportation vehicles.1 Despite these benefits, there exists an emerging concern involving the fate of plastic debris in the environment.2–7 An estimated 4900 million metric tons (Mt) of discarded plastics currently reside in landfills or the environment, with the most abundant non-fiber resins including polystyrene (PS), polyethylene (PE), polyvinyl chloride (PVC), polyurethane (PU), polypropylene (PP), and polyethylene terephthalate (PET).8 In particular, the PET thermoplastic is a semicrystalline, lightweight, and water resistant material9 that critically serves global markets for packaging, films, and textiles with the worldwide production of the PET resin reaching over 30 million tons in recent years.10 While recycling tactics exist for PET,11,12 many PET products are intended for swift disposal and consequently accumulate in the environment, where the material is susceptible to degradation through photochemical, thermal, and mechanical mechanisms.13–18 Degradation processes could generate randomized fragments of PET contaminants with wide size distributions and morphologies.Numerous reports highlight the presence of fragmented plastics, termed ‘microplastics’, in drinking water19–21 and beverages,22,23 food,24–27 and the natural environment.28 Environmental microplastics can originate from two key sources: intentionally formulated products (e.g., microbead scrubs, toothpaste, cleaners) and plastic debris that physically or chemically degrades after environmental exposures (e.g., UV irradiation, heat, mechanical stress, humidity).29–32 Because decomposition processes can produce fragments of diverse size, morphology, and composition, efforts are underway to standardize definitions and harmonize scientific communication.33 This classification of microplastics could ultimately encompass many physicochemical properties,33,34 but thus far plastic fragments are typically categorized by size. One highly recognized definition by the National Oceanic and Atmospheric Administration (NOAA) refers to microplastic as fragments with diameters <5 mm.35 Because the size of particles is interrelated to physicochemical properties, a refinement to size classifications will support the full understanding of the fate and behavior of these plastic fragments. For example, recent reports indicate the potential of nano-scale plastics in the environment.36 Nanomaterials can penetrate biological membranes and be taken up by cells and accumulate within tissues, as evident from the wealth of nanotoxicity studies with various metal NPs.37–41 Moreover, the high surface area of nanomaterials can serve as carriers of various environmental constituents due to the absorption to the surface of the materials. The ultimate consequence of plastic fragments, together with chemicals absorbed to these plastic vectors,42 on biological systems remains unknown.
Despite the growing concern over plastic NPs as widespread environmental contaminants, very few studies have investigated the biological effects of nanomaterials that comprise high-volume commodity plastics (e.g., PET, PP, PVC, PU). To date, the majority of reports have focused on the biological responses to commercially available fluorescently-labeled PS NPs.43,44 One key hurdle that hinders further testing of other commodity-based nanoplastics involves the limited availability of highly characterized and biological traceable nanomaterials. In the case of PET, for example, very few reports currently exist that describe the fabrication of PET NPs suited for biological studies. The fabrication of PET NPs has occurred via laser ablation45 and catalytic emulsion polymerization,46 however, these approaches do not incorporate tracers into the NPs to monitor spatial distribution in cells or small organisms. One encouraging study from Rodríguez-Hernández et al. recently reported the synthesis of fluorescently labeled PET NPs for studies in macrophages, but their results also showed aggregation of the NPs.47
Herein, we report the synthesis of disperse PET NPs with a tight size distribution using a facile, bottom-up fabrication approach. We further show the incorporation of fluorescent tracers into the NPs that enables visualization and characterization of these PET NPs within mammalian cells.
Materials and methods
Fabrication of PET NPs
A solution of PET was prepared by mixing 0.58 g PET fiber (IZO Home Goods) with 35 mL hexafluoroisopropanol (HFIP) (Sigma-Aldrich, St. Louis, MO, USA) in a 40 mL scintillation vial equipped with a magnetic stir bar that was coated with polytetrafluoroethylene (PTFE). PET solution (10 mL) was added dropwise at 1 mL min−1 using a syringe pump (Model # NE-300, New Era Pump Systems, Inc., Farmingdale, NY, USA) with a Poulten & Graf GmbH Fortuna® Optima® 10 mL glass syringe into ultrapure deionized water (75 mL, 18.2 MΩ cm resistivity) at room temperature, resulting in precipitation of PET NPs. The entire contents of the precipitation vessel were transferred to a 250 mL round-bottomed flask and rotary evaporated under vacuum at 55 °C to remove residual HFIP. Upon reduction of the volume in the round-bottomed flask (∼30 mL), ultrapure deionized water (∼75 mL) was added, and the flask was subjected to rotary evaporation for a second time. The concentrated suspension of particles was pipetted into a 20 mL scintillation vial. Particles containing rhodamine B (Sigma-Aldrich, St. Louis, MO, USA) were formulated using a similar approach as specified above. The tracer solution in HFIP (0.05 mg mL−1) was prepared from a stock solution of 1 mg mL−1. An aliquot of the 0.05 mg mL−1 tracer solution (1 mL) was then added to the PET solution prior to precipitation into ultrapure deionized water.Purification of PET NPs
To remove residual HFIP, the suspension of particles was centrifuged and resuspended. Each wash step consisted of centrifuging the suspension at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 rpm for 5 minutes at room temperature, removing the supernatant, and resuspending in an equal volume of 0.5 mg mL−1 Bovine Serum Albumin (BSA) (pH 8.2) to maintain the concentration of the particles in suspension. The particles were resuspended by a 30 second vortex step followed by discrete sonication in a cup horn sonicator (Ultrasonic Liquid Processor S-400, Misonic Inc., Farmingdale, NY) delivering a total of 840 J mL−1. For the first wash step, the initial particle suspension was spiked with BSA to a final concentration of 0.5 mg mL−1 before the first centrifugation step. The particles were washed three times and resuspended in a final solution of 0.5 mg mL−1 BSA (pH 8.2).
100 rpm for 5 minutes at room temperature, removing the supernatant, and resuspending in an equal volume of 0.5 mg mL−1 Bovine Serum Albumin (BSA) (pH 8.2) to maintain the concentration of the particles in suspension. The particles were resuspended by a 30 second vortex step followed by discrete sonication in a cup horn sonicator (Ultrasonic Liquid Processor S-400, Misonic Inc., Farmingdale, NY) delivering a total of 840 J mL−1. For the first wash step, the initial particle suspension was spiked with BSA to a final concentration of 0.5 mg mL−1 before the first centrifugation step. The particles were washed three times and resuspended in a final solution of 0.5 mg mL−1 BSA (pH 8.2).
Characterization of PET NPs
000× to 300000×.
:10 and 1:100 in BSA, and BSA was used as the blank. A slit width of 2 nm was used with a data interval of 0.5 nm.
Studies in mammalian cells
:200 DAPI (Life Technologies, Grand Island, NY) for 15 min at room temperature. Cells were washed three times in PBS before bright field and fluorescence imaging with a 40× objective. Imaging was conducted using an Olympus IX71 inverted microscope with a CCD Microscopy Camera (INFINITY3-3URF, 3.0 Megapixel, CoolLED). Image processing was performed using ImageJ (NIH).
Results and discussion
Fabrication and characterization of PET NPs
The PET NPs were fabricated with a precipitation method, wherein a solution of PET in HFIP was slowly added to ultrapure water resulting in the formation of NPs. Multiple washing steps were used to remove residual HFIP solvent from the NP formulations, resulting in an undetectable fluorine signal via19F-NMR. While washing the PET NPs with ultrapure water, the particles aggregated and therefore a solution of BSA protein at 0.5 mg mL−1 was used instead to maintain particle dispersion during washing steps. Here, utilization of BSA was compatible with subsequent studies in cell culture, as discussed in the following section. However, use of species-specific proteins or alternative surfactants as stabilizing agents of these NPs may be required to align with other biological systems under investigation in future work. To enable detection of the PET NPs within cells, the particles were labeled with rhodamine B (PET-RB) by incorporation of the tracer into the NPs during fabrication.The spherical morphology of the PET-RB NPs was evident via SEM (Fig. 1A) and TEM (Fig. 1B) and no morphological differences were apparent for the PET NPs without tracer (ESI 2†). After washing and resuspending the particles in a BSA solution, the hydrodynamic diameters were 170 ± 3 nm for PET-NPs and 158 ± 2 nm for PET-RB NPs (Fig. 1C and ESI 2†). The washing steps with the BSA solution slightly increased the hydrodynamic diameters, as compared to those of the unwashed samples, but the average size distributions remained below 200 nm with polydispersity indices at 0.2 and 0.1 for PET NP and PET-RB NP, respectively. The average diameters of NPs were also calculated from the SEM images at 95 ± 14 nm for PET NPs and 88 ± 14 nm for PET-RB NPs. The differences between the hydrodynamic diameters and the diameters calculated from SEM images (i.e., dried samples) are expected and could result, in part, from the existence of a BSA corona within in the particle suspensions48 or from the indirect measurements associated with DLS that relies on fluctuations in the particle scattering intensity in solution.49 The zeta potential for NPs suspended within the BSA solution was −37 mV for PET NPs and −38 mV for PET-RB NPs, which aligns with the high stability and dispersion of the particles. For example, the PET-RB NPs measured 162 ± 2 nm (polydispersity index of 0.2) after two months of storage at 4 °C.
 | ||
| Fig. 1 (A) SEM, (B) TEM, and (C) the DLS curve for PET-RB NPs. The DLS profile is an average of three DLS runs. | ||
To explore the composition of the NPs, FT-IR analysis was performed (Fig. 2). The FT-IR profiles of NPs showed characteristic absorption bands of the PET bulk polymer (ESI 3†),50–52 including at 1715 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), 1578 cm−1 (stretching of CC in ring), and 1505 cm−1 (in-plane bending of C–H in ring; stretching of CC in ring), and 1240 cm−1 (CO in-plane bending, C–C stretching, C(O)–O stretching).52 As shown in Fig. 2, the prominent IR absorption bands were similar between PET and PET-RB NPs. Interestingly, bands associated with rhodamine B, such as near 1590 cm−1 (COO– stretching)53 were not clearly present for the PET-RB NPs, despite the verification of the fluorescent tracer via fluorescence microscopy. The lack of prominent rhodamine B absorbance bands in the FT-IR could be due to the low concentration of the tracer, which was undetectable in the IR spectrum. Additional testing using Raman spectroscopy (ESI 4†), Pyro-GC/MS (ESI 5†), and XPS (ESI 6†) also confirmed various moieties within the PET NPs. These in-depth analyses of the PET NPs support steps towards more standardized NPs for studies in biological systems, as discussed in the next section.
O stretching), 1578 cm−1 (stretching of CC in ring), and 1505 cm−1 (in-plane bending of C–H in ring; stretching of CC in ring), and 1240 cm−1 (CO in-plane bending, C–C stretching, C(O)–O stretching).52 As shown in Fig. 2, the prominent IR absorption bands were similar between PET and PET-RB NPs. Interestingly, bands associated with rhodamine B, such as near 1590 cm−1 (COO– stretching)53 were not clearly present for the PET-RB NPs, despite the verification of the fluorescent tracer via fluorescence microscopy. The lack of prominent rhodamine B absorbance bands in the FT-IR could be due to the low concentration of the tracer, which was undetectable in the IR spectrum. Additional testing using Raman spectroscopy (ESI 4†), Pyro-GC/MS (ESI 5†), and XPS (ESI 6†) also confirmed various moieties within the PET NPs. These in-depth analyses of the PET NPs support steps towards more standardized NPs for studies in biological systems, as discussed in the next section.
 | ||
| Fig. 2 FT-IR spectra of PET NPs without tracer (top) and with rhodamine B (bottom). | ||
Evaluation of PET NP and PET-RB NP in mammalian cells
Prior to evaluation in mammalian cells, it is critical to ensure the absence of contaminants. To this end, a kinetic turbidity LAL assay was used to ascertain potential endotoxin contamination in the NPs. Although levels of endotoxins were detectable, the values were below the recommended FDA limit of 0.5 EU per mL for medical devices54,55 showing 0.1 EU per mL and 0.064 EU per mL for PET-NPs and PET-RB NPs, respectively. The cytotoxicity and uptake of PET NPs were evaluated in murine alveolar macrophages, RAW264.7 in a dose–response manner. Cytotoxicity was evaluated by determining cell membrane integrity (LDH release) and metabolic activity (MTS) (Fig. 3). A significant increase in LDH release was observed at 0.0625 mg mL−1 for PET NP (P-value = 0.0016) and at 0.0010 mg mL−1 for PET-RB NP (P-value = 0.0034). We speculate that the PET-RB NP interfered with the cell membrane even at the low concentrations. At a concentration of 0.125 mg mL−1 for both PET NPs (160 ± 27.5% of control for PET-NP and 178 ± 18.3% of control for PET-RB NPs) the LDH release continued to scale with increasing concentration, so that LDH release for 0.5 mg mL−1 was 506 ± 85% of control for PET NP and 447 ± 46.1% of control for PET-RB NPs. On the other hand, a slight increase in the MTS assay was observed for the lowest concentration of PET NPs. This might be due to an interference between PET NPs and MTS but requires further investigation. Only at the highest tested concentration of PET NPs of 0.5 mg mL−1 did the MTS assay show a significant decrease in mitochondrial activity (82.9 ± 8.77% of control for PET-NP and 71.3 ± 29.4% of control for PET-RB NPs). Together these findings suggest that the cell membrane integrity was impacted at a lower NP concentration before mitochondrial activity was altered.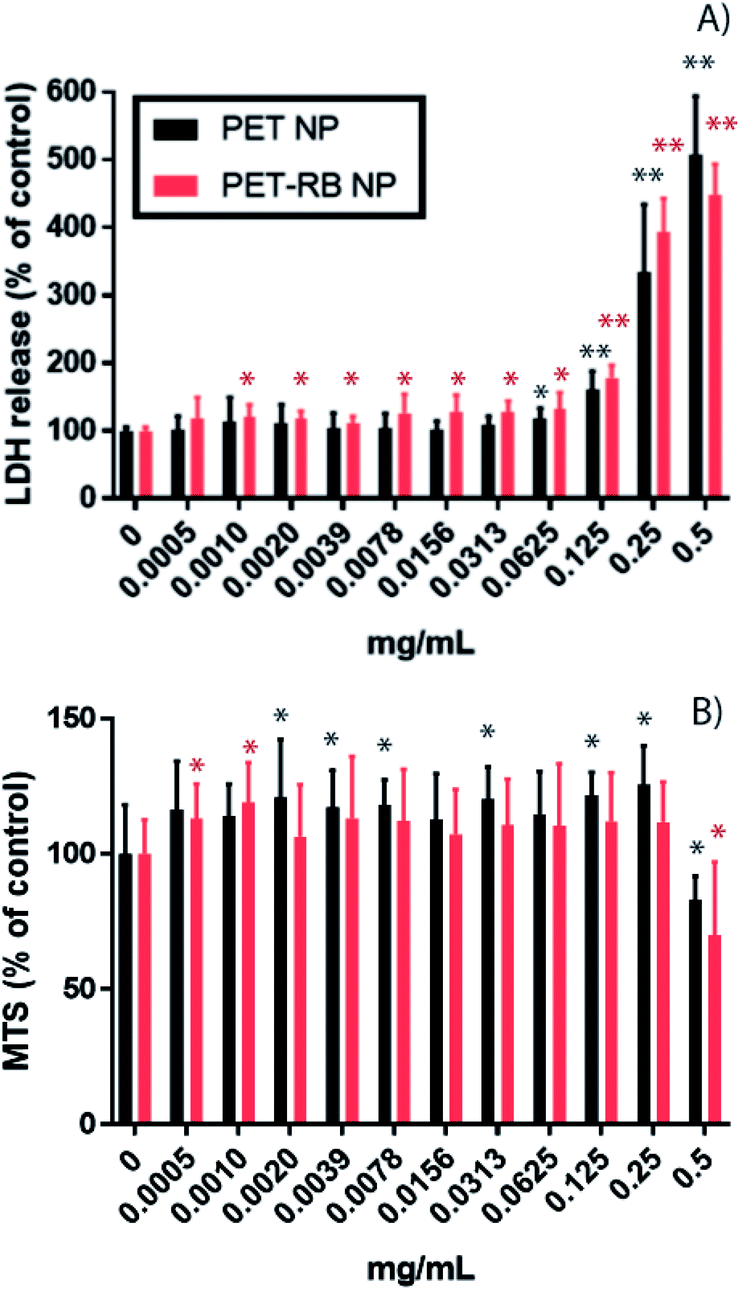 | ||
| Fig. 3 Cytotoxicity of PET-NP (black bars) and PET-RB NPs (red bars) tested by (A) membrane integrity (LDH release) and (B) metabolic activity (MTS assay). The graphs show mean ± standard deviation. One asterisk indicates P-values <0.05 and two asterisks indicate P-value <0.001. | ||
The cellular uptake of PET-RB NPs and resulting morphological changes in RAW264.7 cells were evident from bright field and fluorescence microscopy. Following exposure to a low concentration of 0.005 mg mL−1 PET-RB NPs, particles were visible in the cell cytoplasm (Fig. 4D), however at concentrations of 0.05 and 0.5 mg mL−1 PET-RB NPs large clusters of NPs were observed intracellularly both in bright field and fluorescence microscopy (Fig. 4, individual fluorescence channels are shown in ESI 7† and overlay of bright field and fluorescence images in ESI 8†). The fluorescence intensity of the larger NP aggregates caused oversaturation at the exposure time needed to visualize individual PET-RB NPs particles, making the aggregates look larger in the fluorescence microscopy images compared to the bright field images. Since the PET-RB NPs showed a low level of autofluorescence in the green wavelength it was not possible to determine if PET-RB NPs were associated with lysosomes. At 0.05 mg mL−1 PET-RB NPs, particles were observed inside phagocytic bodies, but while several cells had formed a tight phagosome around the NPs, vacuoles with large empty spaces surrounding the NPs were observed at the higher concentration. At the highest concentration, phagosomes enlarged and caused elongated, crescent-shaped nuclei in the periphery of the cells. Morphological changes such as blebs were observed at 0.005 mg mL−1, indicating cell membranes delaminate from cortical cytoskeletal structures.56 These blebs became more numerous at 0.05 mg mL−1, but not at 0.5 mg mL−1. At 0.5 mg mL−1, condensation and increased fluorescence intensity of the nuclei were observed supporting the cytotoxicity data, indicating that a number of cells were not viable at this concentration.
 | ||
| Fig. 4 Bright field (A, C, E and G) and fluorescence (B, D, F and H) microscopy of RAW 264.7 cells exposed to control (A + B), 0.005 mg mL−1 (C + D), 0.05 mg mL−1 (E + F), and 0.5 mg mL−1 PET-RB NPs (G + H). Cell nuclei are blue, PET-RB NPs are red, and cell cytoplasm is green (the images from individual fluorescence channels are shown in ESI 7†). | ||
Conclusions
The environmental presence of fragmented plastics, derived from high-commodity polymers, is an emerging concern with unknown consequences for human health. As a crucial high-commodity polymer and contributor of plastic waste, PET has infiltrated drinking water, food, and beverages in the form of small-scale debris (i.e., microplastics), as shown in various reports. Although current reports have focused on micron-scale PET and other plastics, a potential exists for environmental contamination of nanoscale PET, as well.This manuscript reports the bottom-up synthesis of PET NPs with hydrodynamic diameters below 200 nm. To support studies in cell models, we incorporated the rhodamine B fluorescent tracer into the PET NPs and measured uptake within RAW264.7 macrophages. Our results showed uptake of PET-RB NPs in the macrophages in a dose–response manner. Our findings suggest that a lower concentration of PET NPs (0.0010 mg mL−1) was required to impact the integrity of the cell membrane of macrophages, as compared to concentrations of PET NPs required to alter mitochondrial activity (0.5 mg mL−1). Clear morphological changes occurred at higher concentrations of PET-RB NPs (0.5 mg mL−1), showing enlarged phagosomes that caused elongation of nuclei and likely cell death. Although more studies are required to further elucidate the effects of the NPs in cell models and other biological systems, this study shows that mammalian macrophage cells are affected by PET nanoscale plastics. The generation of well characterized PET NPs will support future studies to assess downstream biological responses.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank RTI International for support of this work and thank RTI International team members for support to characterize NPs: Brenda Fletcher (FT-IR), Dr Jean Kim (endotoxin testing), Rodney Snyder (UV-Vis), Randy Ruthowske (NMR), Randi Carter (GC/MS), and Sarah Harrison (TEM). FESEM, XPS and Raman work was performed at the Joint School of Nanoscience and Nanoengineering (JSNN), a member of the Southeastern Nanotechnology Infrastructure Corridor (SENIC) and National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the National Science Foundation (Grant ECCS-1542174). We thank also JSNN team members for their support: Klinton Davis (FESEM, XPS) and Matthew Craps (Raman).References
- M. Gilbert, Chapter 1 - Plastics Materials: Introduction and Historical Development, in Brydson's Plastics Materials, ed. M. Gilbert, Butterworth-Heinemann, eighth edn, 2017, pp. 1–18 Search PubMed.
- C. M. Rochman, Microplastics research—from sink to source, Science, 2018, 360(6384), 28 CrossRef CAS PubMed.
- C. M. Rochman, The Complex Mixture, Fate and Toxicity of Chemicals Associated with Plastic Debris in the Marine Environment, in Marine Anthropogenic Litter, ed. M. Bergmann, L. Gutow and M. Klages, Springer International Publishing, Cham, 2015, pp. 117–140 Search PubMed.
- A. Cózar, F. Echevarría, J. I. González-Gordillo, X. Irigoien, B. Úbeda, S. Hernández-León, Á. T. Palma, S. Navarro, J. García-de-Lomas, A. Ruiz, M. L. Fernández-de-Puelles and C. M. Duarte, Plastic debris in the open ocean, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(28), 10239 CrossRef PubMed.
- J. A. Ivar do Sul and M. F. Costa, The present and future of microplastic pollution in the marine environment, Environ. Pollut., 2014, 185, 352–364 CrossRef CAS PubMed.
- M. Eriksen, L. C. M. Lebreton, H. S. Carson, M. Thiel, C. J. Moore, J. C. Borerro, F. Galgani, P. G. Ryan and J. Reisser, Plastic Pollution in the World's Oceans: More than 5 Trillion Plastic Pieces Weighing over 250,000 Tons Afloat at Sea, PLoS One, 2014, 9(12), e111913 CrossRef PubMed.
- A. A. Horton, C. Svendsen, R. J. Williams, D. J. Spurgeon and E. Lahive, Large microplastic particles in sediments of tributaries of the River Thames, UK - Abundance, sources and methods for effective quantification, Mar. Pollut. Bull., 2017, 114(1), 218–226 CrossRef CAS PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Production, use, and fate of all plastics ever made, Sci. Adv., 2017, 3(7), e1700782 CrossRef PubMed.
- H. C. A. Lim, Chapter 20 - Thermoplastic Polyesters, in Brydson's Plastics Materials, ed. M. Gilbert, Butterworth-Heinemann, eighth edn, 2017, pp. 527–543 Search PubMed.
- https://www.plasticsinsight.com/resin-intelligence/resin-prices/polyethylene-terephthalate/ .
- K. Dutt and R. K. Soni, A review on synthesis of value added products from polyethylene terephthalate (PET) waste, Polym. Sci., Ser. B, 2013, 55(7), 430–452 CrossRef CAS.
- G. P. Karayannidis and D. S. Achilias, Chemical Recycling of Poly(ethylene terephthalate), Macromol. Mater. Eng., 2007, 292(2), 128–146 CrossRef CAS.
- B. Singh and N. Sharma, Mechanistic implications of plastic degradation, Polym. Degrad. Stab., 2008, 93(3), 561–584 CrossRef CAS.
- M. Day and D. M. Wiles, Photochemical decomposition mechanism of poly(ethylene terephthalate), J. Polym. Sci., Part B: Polym. Lett., 1971, 9(9), 665–669 CrossRef CAS.
- M. Day and D. M. Wiles, Photochemical degradation of poly(ethylene terephthalate). II. Effect of wavelength and environment on the decomposition process, J. Appl. Polym. Sci., 1972, 16(1), 191–202 CrossRef CAS.
- M. Day and D. M. Wiles, Photochemical degradation of poly(ethylene terephthalate). III. Determination of decomposition products and reaction mechanism, J. Appl. Polym. Sci., 1972, 16(1), 203–215 CrossRef CAS.
- A. Launay, F. Thominette and J. Verdu, Hydrolysis of poly(ethylene terephthalate): a kinetic study, Polym. Degrad. Stab., 1994, 46(3), 319–324 CrossRef CAS.
- S. Venkatachalam, S. G. Nayak, J. V. Labde, P. R. Gharal, K. Rao and A. K. Kelkar, Degradation and Recyclability of Poly (Ethylene Terephthalate), in Polyester, ed. H. E.-D. M. Saleh, IntechOpen, 2012, DOI:10.5772/48612.
- B. E. Oßmann, G. Sarau, H. Holtmannspötter, M. Pischetsrieder, S. H. Christiansen and W. Dicke, Small-sized microplastics and pigmented particles in bottled mineral water, Water Res., 2018, 141, 307–316 CrossRef PubMed.
- M. Pivokonsky, L. Cermakova, K. Novotna, P. Peer, T. Cajthaml and V. Janda, Occurrence of microplastics in raw and treated drinking water, Sci. Total Environ., 2018, 643, 1644–1651 CrossRef CAS PubMed.
- D. Schymanski, C. Goldbeck, H.-U. Humpf and P. Fürst, Analysis of microplastics in water by micro-Raman spectroscopy: release of plastic particles from different packaging into mineral water, Water Res., 2018, 129, 154–162 CrossRef CAS PubMed.
- G. Liebezeit and E. Liebezeit, Synthetic particles as contaminants in German beers, Food Addit. Contam., Part A, 2014, 31(9), 1574–1578 CrossRef CAS PubMed.
- A. A. Koelmans, N. H. Mohamed Nor, E. Hermsen, M. Kooi, S. M. Mintenig and J. De France, Microplastics in freshwaters and drinking water: critical review and assessment of data quality, Water Res., 2019, 155, 410–422 CrossRef CAS PubMed.
- M. E. Iñiguez, J. A. Conesa and A. Fullana, Microplastics in Spanish Table Salt, Sci. Rep., 2017, 7(1), 8620 CrossRef PubMed.
- M. Fischer, I. Goßmann and B. M. Scholz-Böttcher, Fleur de Sel—An interregional monitor for microplastics mass load and composition in European coastal waters?, J. Anal. Appl. Pyrolysis, 2019, 144, 104711 CrossRef CAS.
- L. Van Cauwenberghe and C. R. Janssen, Microplastics in bivalves cultured for human consumption, Environ. Pollut., 2014, 193, 65–70 CrossRef CAS PubMed.
- L. G. A. Barboza, C. Lopes, P. Oliveira, F. Bessa, V. Otero, B. Henriques, J. Raimundo, M. Caetano, C. Vale and L. Guilhermino, Microplastics in wild fish from North East Atlantic Ocean and its potential for causing neurotoxic effects, lipid oxidative damage, and human health risks associated with ingestion exposure, Sci. Total Environ., 2020, 717, 134625 CrossRef CAS PubMed.
- M. Eriksen, N. Maximenko, M. Thiel, A. Cummins, G. Lattin, S. Wilson, J. Hafner, A. Zellers and S. Rifman, Plastic pollution in the South Pacific subtropical gyre, Mar. Pollut. Bull., 2013, 68(1), 71–76 CrossRef CAS PubMed.
- X. Guo and J. Wang, The chemical behaviors of microplastics in marine environment: a review, Mar. Pollut. Bull., 2019, 142, 1–14 CrossRef CAS PubMed.
- M. A. Browne, P. Crump, S. J. Niven, E. Teuten, A. Tonkin, T. Galloway and R. Thompson, Accumulation of Microplastic on Shorelines Worldwide: Sources and Sinks, Environ. Sci. Technol., 2011, 45(21), 9175–9179 CrossRef CAS PubMed.
- P. J. Kole, A. J. Löhr, F. G. A. J. Van Belleghem and A. M. J. Ragas, Wear and Tear of Tyres: A Stealthy Source of Microplastics in the Environment, Int. J. Environ. Res. Public Health, 2017, 14(10), 1265 CrossRef PubMed.
- A. L. Andrady, Microplastics in the marine environment, Mar. Pollut. Bull., 2011, 62(8), 1596–1605 CrossRef CAS PubMed.
- N. B. Hartmann, T. Hüffer, R. C. Thompson, M. Hassellöv, A. Verschoor, A. E. Daugaard, S. Rist, T. Karlsson, N. Brennholt, M. Cole, M. P. Herrling, M. C. Hess, N. P. Ivleva, A. L. Lusher and M. Wagner, Are We Speaking the Same Language? Recommendations for a Definition and Categorization Framework for Plastic Debris, Environ. Sci. Technol., 2019, 53(3), 1039–1047 CrossRef CAS PubMed.
- A. J. Verschoor, Towards a definition of microplastics: Considerations for the specification of physico-chemical properties, National Institute for Public Health and the Environment, Bilthoven: RIVM, 2015 Search PubMed.
- C. Arthur, J. Baker and H. Bamford, in Proceedings of the International Research Workshop on Microplastic Marine Debris, NOAA Technical Memorandum, NOS-OR&R-30, 2009 Search PubMed.
- H. Bouwmeester, P. C. H. Hollman and R. J. B. Peters, Potential Health Impact of Environmentally Released Micro- and Nanoplastics in the Human Food Production Chain: Experiences from Nanotoxicology, Environ. Sci. Technol., 2015, 49(15), 8932–8947 CrossRef CAS PubMed.
- A. Nel, T. Xia, L. Mädler and N. Li, Toxic Potential of Materials at the Nanolevel, Science, 2006, 311(5761), 622 CrossRef CAS PubMed.
- A. Nel, T. Xia, H. Meng, X. Wang, S. Lin, Z. Ji and H. Zhang, Nanomaterial Toxicity Testing in the 21st Century: Use of a Predictive Toxicological Approach and High-Throughput Screening, Acc. Chem. Res., 2013, 46(3), 607–621 CrossRef CAS PubMed.
- J. Jeevanandam, A. Barhoum, Y. S. Chan, A. Dufresne and M. K. Danquah, Review on nanoparticles and nanostructured materials: history, sources, toxicity and regulations, Beilstein J. Nanotechnol., 2018, 9, 1050–1074 CrossRef CAS PubMed.
- N. P. Mortensen, L. M. Johnson, K. D. Grieger, J. L. Ambroso and T. R. Fennell, Biological interactions between nanomaterials and placental development and function following oral exposure, Reprod. Toxicol., 2019, 90, 150–165 CrossRef CAS.
- T. R. Fennell, N. P. Mortensen, S. R. Black, R. W. Snyder, K. E. Levine, E. Poitras, J. M. Harrington, C. J. Wingard, N. A. Holland, W. Pathmasiri and S. C. J. Sumner, Disposition of intravenously or orally administered silver nanoparticles in pregnant rats and the effect on the biochemical profile in urine, J. Appl. Toxicol., 2017, 37(5), 530–544 CrossRef CAS PubMed.
- T. Hüffer, A.-K. Weniger and T. Hofmann, Sorption of organic compounds by aged polystyrene microplastic particles, Environ. Pollut., 2018, 236, 218–225 CrossRef PubMed.
- T. Xia, M. Kovochich, M. Liong, J. I. Zink and A. E. Nel, Cationic Polystyrene Nanosphere Toxicity Depends on Cell-Specific Endocytic and Mitochondrial Injury Pathways, ACS Nano, 2008, 2(1), 85–96 CrossRef CAS PubMed.
- S. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Cellular uptake of nanoparticles: journey inside the cell, Chem. Soc. Rev., 2017, 46(14), 4218–4244 RSC.
- D. Magrì, P. Sánchez-Moreno, G. Caputo, F. Gatto, M. Veronesi, G. Bardi, T. Catelani, D. Guarnieri, A. Athanassiou, P. P. Pompa and D. Fragouli, Laser Ablation as a Versatile Tool To Mimic Polyethylene Terephthalate Nanoplastic Pollutants: Characterization and Toxicology Assessment, ACS Nano, 2018, 12(8), 7690–7700 CrossRef PubMed.
- F. M. Bauers, R. Thomann and S. Mecking, Submicron Polyethylene Particles from Catalytic Emulsion Polymerization, J. Am. Chem. Soc., 2003, 125(29), 8838–8840 CrossRef CAS PubMed.
- A. G. Rodríguez-Hernández, J. A. Muñoz-Tabares, J. C. Aguilar-Guzmán and R. Vazquez-Duhalt, A novel and simple method for polyethylene terephthalate (PET) nanoparticle production, Environ. Sci.: Nano, 2019, 6(7), 2031–2036 RSC.
- M. Kokkinopoulou, J. Simon, K. Landfester, V. Mailänder and I. Lieberwirth, Visualization of the protein corona: towards a biomolecular understanding of nanoparticle-cell-interactions, Nanoscale, 2017, 9(25), 8858–8870 RSC.
- P. Eaton, P. Quaresma, C. Soares, C. Neves, M. P. de Almeida, E. Pereira and P. West, A direct comparison of experimental methods to measure dimensions of synthetic nanoparticles, Ultramicroscopy, 2017, 182, 179–190 CrossRef CAS PubMed.
- M. Edge, R. Wiles, N. S. Allen, W. A. McDonald and S. V. Mortlock, Characterisation of the species responsible for yellowing in melt degraded aromatic polyesters—I: Yellowing of poly(ethylene terephthalate), Polym. Degrad. Stab., 1996, 53(2), 141–151 CrossRef CAS.
- A. P. d. S. Pereira, M. H. P. d. Silva, É. P. Lima Júnior, A. d. S. Paula and F. J. Tommasini, Processing and Characterization of PET Composites Reinforced With Geopolymer Concrete Waste, Mater. Res., 2017, 20, 411–420 CrossRef.
- I. Donelli, G. Freddi, V. A. Nierstrasz and P. Taddei, Surface structure and properties of poly-(ethylene terephthalate) hydrolyzed by alkali and cutinase, Polym. Degrad. Stab., 2010, 95(9), 1542–1550 CrossRef CAS.
- R. Dukali, I. Radović, D. Stojanović, D. Sevic, V. Radojevic, D. Jocic and R. Aleksic, Electrospinning of laser dye rhodamine B-doped poly(methyl methacrylate) nanofibers, J. Serb. Chem. Soc., 2014, 79, 867 CrossRef CAS.
- M. B. Gorbet and M. V. Sefton, Endotoxin: the uninvited guest, Biomaterials, 2005, 26(34), 6811–6817 CrossRef CAS PubMed.
- Services U. S. D. o. H. a. H., Guidance for Industry: Pyrogen and Endotoxins Testing: Questions and Answers, Administration F. a. D., 2012 Search PubMed.
- M. Olson and L. Julian, Apoptotic membrane dynamics in health and disease, Cell Health Cytoskeleton, 2015, 7, 133 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00888e |
| This journal is © The Royal Society of Chemistry 2021 |