
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTrafficking of JC virus-like particles across the blood–brain barrier†
Dong
Ye‡
*a,
Tina
Zimmermann‡§
b,
Victoria
Demina
c,
Sergey
Sotnikov
c,
Christian L.
Ried
d,
Harri
Rahn
d,
Marcus
Stapf
c,
Christopher
Untucht
b,
Michael
Rohe¶
b,
Georg C.
Terstappen||
b,
Karsten
Wicke
b,
Mario
Mezler
a,
Heiko
Manninga
c and
Axel H.
Meyer
 *a
*a
aAbbVie Deutschland GmbH & Co. KG, DMPK, Bioanalytical Research, Knollstraße, 67061 Ludwigshafen, Germany. E-mail: dong.ye@abbvie.com; axel.meyer@abbvie.com
bAbbVie Deutschland GmbH & Co. KG, Neuroscience Discovery, Knollstraße, 67061 Ludwigshafen, Germany
cNEUWAY Pharma GmbH, Ludwig-Erhard-Allee 2, 53175 Bonn, Germany
dAbbVie Deutschland GmbH & Co. KG, Development Sciences NBE, Knollstraße, 67061 Ludwigshafen, Germany
First published on 9th February 2021
Abstract
Hollow viral vectors, such as John Cunningham virus-like particles (JC VLPs), provide a unique opportunity to deliver drug cargo into targeted cells and tissue. Current understanding of the entry of JC virus in brain cells has remained insufficient. In particular, interaction of JC VLPs with the blood–brain barrier (BBB) has not been analyzed in detail. Thus, JC VLPs were produced in this study for investigating the trafficking across the BBB. We performed a carotid artery injection procedure for mouse brain to qualitatively study JC VLPs' in vivo binding and distribution and used in vitro approaches to analyze their uptake and export kinetics in brain endothelial cells. Our results show that clathrin-dependent mechanisms contributed to the entry of VLPs into brain endothelial cells, and exocytosis or transcytosis of VLPs across the BBB was observed in vitro. VLPs were found to interact with sialic acid glycans in mouse brain endothelia. The ability of JC VLPs to cross the BBB can be useful in developing a delivery system for transport of genes and small molecule cargoes to the brain.
Introduction
Delivery of biologics to the brain remains a great challenge due to the hurdle imposed by the blood–brain barrier (BBB). Substances with molecular weight over 400 Da are generally impeded from paracellular transport due to extensive tight junctions formed between brain microvascular endothelial cells, which constitute the BBB and form a neurovascular unit together with astrocytes, pericytes and microglia.2 ATP-binding cassette (ABC) transporters expressed on the luminal side of the BBB also hinder the transcellular transport of exogenous substances because of active efflux from the endothelium to the blood.2,3 Compared to invasive approaches that mechanically or chemically disrupt the BBB for temporary drug access into the brain tissue, non-invasive delivery technologies avoid risks of damaging brain endothelia and neighboring neurons (∼8 μm away from a capillary) and offers unique benefits for overcoming the intact BBB4 and comprehensive drug delivery to CNS. Using natural concepts, viral vectors have demonstrated a great potential for delivery of therapeutics to the brain, owing to their specialized BBB-shuttling mechanisms. For example, adeno-associated virus (i.e., AAV9) was reported to cross the BBB via receptor-mediated transcytosis for gene transfer.5 Herpes simplex virus, vesicular stomatitis virus, retrovirus and adenovirus were also individually demonstrated to deliver therapeutics for the treatment of brain tumors.4 However, as a simplified form of a natural virus, the shuttling mechanism for VLPs in biological applications has yet to be defined.John Cunningham virus (JCV) is an etiological agent that can transmit from latent sites (i.e., kidney, bone marrow) to the brain, where it damages the white matter and causes a neurodegenerative disease called progressive multifocal leukoencephalopathy (PML).6–8 Similar to other polyomaviruses, JCV is a non-enveloped DNA virus, enclosed by three viral capsid proteins, VP1, VP2 and VP3. Individually, VP1 serves as a major capsid constituent to control targeting specificities,7 while minor capsid proteins VP2 and VP3 function in regulation of the viral genome.9,10 Pathogenesis of native JCV has shown its wide infectivity in human brain, gastrointestinal tract, kidney, liver or lung tissue, although the overall consequence from such infection is clinically harmless for healthy adults due to suppression by the immune system.8 The cell type-specific tropism of JCV has been connected with kidney epithelial cells, tonsillar stromal cells or bone marrow-derived cells (i.e., B lymphocyte),7,8 oligodendrocytes,11 astrocytes12 as well as brain endothelial cells.13 In light of observed PML-infections, the possibility of JCV being able to cross the BBB via JCV-infected B-cells migrating to the brain was proposed.14 However, it has not been shown that infected B-cells are indeed the critical vectors responsible for JCV infection of the central nervous system.
JCV capsid protein VP1 is known to bind to viral receptors, including α-(2,3)- and α-(2,6)-linked sialic acids15,16 or 5-hydroxytryptamine (5-HT) 2A receptors.17 Only few studies are available to investigate mechanistic interactions between VP1-assembled particles and the BBB, despite the fact that native JCV has been reported to infect brain microvascular endothelial cells, even in the absence of 5HT2A receptor in the BBB.13 The possibility to harness such pathological mechanisms as a method to deliver external biomolecules into the brain for therapeutic purposes is of great interest to explore. In fact, gene therapy via JC VLPs in treating brain tumors such as glioblastoma was already shown.18 However, such intratumoral exposure does not evidence their trafficking into the brain under healthy conditions across an intact BBB (without being compromised by tumor).
JCV VP1 can be conveniently generated by using baculovirus insect cells,19,20Escherichia coli21,22 or yeast cell23,24 expression systems. Recombinant VP1 proteins are able to self-assemble to form virus-like particles (VLPs), and thus it is possible to package drug cargo inside the capsules. For example, DNA can be loaded into JC VLPs by either osmotic shock or a dissociation–reassociation cycle, with the latter procedure reported to be more effective through adjusting chemical conditions for Ca2+ ions and disulfide bonds.19 The produced VLPs were thought to share some characteristics (i.e., morphology, cell binding) with native JCV,19 although it remains unclear whether these biological characteristics of native JCV can be matched by its derived VLP. Recombinant VP1 protein-assembled JC VLPs were successfully used for packaging of chemical substances in vitro, i.e., propidium iodide,25 suggesting suitability of the system for transport of small molecules. Also, JC VLPs were reported as efficient vehicles for delivery of genes or nucleic acids in vivo.21,22,26 Such delivery technology has been proposed for disease treatment in bone fractures26 and for vaccine generation.19
In this study, VLPs derived from JCV recombinant VP1 protein were manufactured with a defined quality, and physicochemical properties of each batch were precisely controlled. We applied JC VLPs to investigate their mechanistic interactions with the brain, specifically understanding trafficking of JC VLPs across the BBB in vitro and in vivo. In order to focus on illustrating mechanistic underpinnings of transport and providing evidence for JC VLPs crossing the BBB, we employed a range of technological approaches to conduct the BBB analysis. For some of these results this would be qualitative, instead of quantitative, owing to the remaining technical challenges. Therefore, here we aim to demonstrate that JC VLPs are able to mechanistically cross the BBB and clarify whether they can translocate to the brain.
Experimental
Preparation and characterization of VLPs
Virus-like particles (VLPs) were manufactured by protein expression using a Sf9 insect cell line derived from the fall armyworm (Spodoptera frugiperda) (Thermo Fischer Scientific). VLPs were produced by infecting the cells with recombinant Baculovirus containing a John Cunningham virus VP1-protein expression cassette (mad-1 strain). The recombinant Baculovirus was prepared by using the Bac-to-Bac® Baculovirus expression system (Thermo Fischer Scientific). VLPs were produced at pH 6.3 after 7 to 10 days in a 3.4 L bioreactor (INFORS HT Minifors). Air flow and temperature (26 °C) were controlled over the time. To remove cells and cell debris suspension was centrifuged at 4 °C, 5000g and the supernatant containing VLPs was harvested. After that VLPs were concentrated and purified using a ÄKTAcross flow™ system (GE Healthcare) equipped with a 300 kDa cut-off membrane (Sartorius). The flow ultrafiltration was carried out with a constant pressure of 1.5 bar and a factor of 8 (1 L supernatant against 8 L buffer). VLPs were further purified by size exclusion chromatography (SEC) using HiPrep™ Sephacryl® S-500 HR column (GE Healthcare). Finally, VLPs were concentrated in Vivaspin® concentrators (Sartorius) with 5 kDa cut-off membrane, followed by filtration through 0.45 μm Acrodisc® filter with Supor® membrane (Pall Corporation).Purified VLPs were suspended in a buffer (150 mM NaCl, 10 mM Tris–HCl, pH 7.5) and subsequently characterized for quality assurance as described in ESI Table 1.† Here, physicochemical characterization results for VLPs were shown by using three example instruments: (1) in Transmission Electron Microscopy (TEM), VLPs were mounted on carbon-coated copper grids (Plano GmbH) and stained with 2% uranyl acetate (Sigma Aldrich). Specimens were visualized with a Zeiss EM900 electron microscope, operating a voltage at 80 kV with magnifications up to 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×; (2) in Western Blotting (WB), VLPs of total 800, 400 and 200 ng were run in SDS-PAGE gel (4–20% Criterion TGX Precast Gels) (Bio-Rad). The blotting of VP1 protein was visualized by staining with primary mouse anti-human JCV VP1 antibody (1:5000, Abcam) and secondary goat anti-mouse IgG (H + L)-HRPO (1:5000, Dianova). The blots were developed with Pierce ECL Western Blotting Substrate (ThermoFischer Scientific) and analysed using a chemiluminescence imaging system (ChemiDoc touch, Bio-Rad); (3) Dynamic Light Scattering (DLS) measurements were performed for thirty repeated runs at 23 °C using a Zetasizer Nano ZS instrument (Malvern Inc.). The VLP sizes were reported in a volume distribution curve after analysed by Zetasizer Software (version 7.11, Malvern Inc.). For additional physicochemical characterization methods, please refer to the details described in the ESI.†
000×; (2) in Western Blotting (WB), VLPs of total 800, 400 and 200 ng were run in SDS-PAGE gel (4–20% Criterion TGX Precast Gels) (Bio-Rad). The blotting of VP1 protein was visualized by staining with primary mouse anti-human JCV VP1 antibody (1:5000, Abcam) and secondary goat anti-mouse IgG (H + L)-HRPO (1:5000, Dianova). The blots were developed with Pierce ECL Western Blotting Substrate (ThermoFischer Scientific) and analysed using a chemiluminescence imaging system (ChemiDoc touch, Bio-Rad); (3) Dynamic Light Scattering (DLS) measurements were performed for thirty repeated runs at 23 °C using a Zetasizer Nano ZS instrument (Malvern Inc.). The VLP sizes were reported in a volume distribution curve after analysed by Zetasizer Software (version 7.11, Malvern Inc.). For additional physicochemical characterization methods, please refer to the details described in the ESI.†
Animal experiments
Animals were group housed in a temperature- and humidity-controlled room with a 12 h/12 h light/dark cycle (lights on 5 am to 5 pm) and had access to food and water ad libitum. AbbVie is committed to the internationally-accepted standard of the 3Rs (Reduction, Refinement, Replacement) and adhering to the highest standards of animal welfare in the company's research and development programs. The animal care and use conditions comply with the Directive 2010/63/EU of the European Parliament and of the Council on the protection of animals used for scientific purposes (published in German Federal Gazette 2013, Part I, Nr. 47). All procedures in animals are approved by the state authority (Landesuntersuchungsamt (LUA), Koblenz).Injection and tissue preparation
The intra-artery carotid injection was performed on 12 weeks old male CD-1 mice (Charles River). As in schematic diagram shown in Fig. 2a, mice were anesthetized with sevoflurane (AbbVie) and the external carotid artery (ECA) was catheterized to the bifurcation of internal carotid artery (ICA) without ligation of pterygopalatine artery (PPA). VLPs were dosed as a bolus (10 μL s−1), with a dose volume of 0.2 mL per animal. Animals were perfused transcardially with PBS 15 min and 120 min later. From every animal, brain and liver were carefully isolated for further analysis.Immunohistochemistry
Organs were post-fixed in 10% formalin (w/v) (Sigma-Aldrich) at RT for 24 h and dehydrated, freed from lipids and embedded in paraffin. After solidification of paraffin, the embedded tissue was cut into slices of 5 μm thickness using a microtome. Tissue slices were transferred to microscope slides. Samples were subjected to deparaffinization and rehydration in descending alcohol concentration series. Antigen retrieval was performed in a steamer containing pre-heated antigen retrieval buffer solution (10 mM sodium citrate buffer, pH 6.0) for 2 min. Following washes with Tris-buffered saline with Tween 20 (TBS-T, Dako), endogenous peroxidase activity was blocked by incubation of samples in methanol–30% hydrogen peroxide–water (7:1:2 volume ratio) for 10 min. Unspecific protein binding was prevented prior to immunostaining by 1 h incubation in normal donkey serum (NDS, Abcam) in TBS-T. Sections were incubated in 1% NDS overnight at 4 °C with primary antibodies, including rabbit anti-JCV VP1 (1:500, Neuway Pharma), rabbit anti-SV40 VP1 (1:500, Abcam), biotinylated Maackia amurensis lectin II (MAL, Vector Laboratories), biotinylated Sambucus nigra lectin (SNA, Vector Laboratories), and goat Mannose Receptor antibody (MR, 1:20, R&D Systems). After washing three times in TBS-T, a secondary antibody in a 1:500 dilution was introduced for 2 h incubation at RT, including donkey anti-rabbit IgG Biotin (Jackson ImmunoResearch), donkey anti-rabbit IgG Alexa Fluor 488 (1:500, Thermo Fisher), donkey anti-goat IgG Alexa Fluor 546 (1:500, Thermo Fisher), streptavidin Alexa Fluor 555 conjugate (1:200, Thermo Fisher).
For biotinylated antibody staining, sections were washed three times in TBS-T and biotinylated horseradish peroxidase was preincubated with avidin to form avidin–biotin–enzyme complexes using a Vectastain Elite ABC Kit (Peroxidase Standard) (Vector Laboratories). These complexes were transferred to the antibody-treated tissue slices for binding to biotinylated secondary antibodies for 30 min. After washing three times in TBS-T, detection of antigen was performed by adding hydrogen peroxide and 3,3′-diaminobenzidine using a DAB Peroxidase Substrate Kit (Vector Laboratories). Respective DAB times: brain, 11 min; liver, 2 min. Samples were washed in water for 5 min after immunodetection and counterstained with eosin and hematoxylin. Samples were dehydrated and coverslipped using PERTEX® medium (Histolab).
For fluorescent antibody staining, sections were washed in TBS-T, counterstained with Hoechst (Thermo Fisher) and washed with PBS. Samples were then mounted with Mowiol using a cover slip.
For image analysis, stained tissue samples were analyzed and imaged using a slide scanner Axio Scan.Z1 (Zeiss). Fluorescent signals were visualized with a Zeiss Axiovert LSM 700 (Carl Zeiss, Oberkochen, Germany) laser scanning confocal microscope, and z-stacks with optical sections of 1 μm were also performed.
Pre-embedding immunogold labeling electron microscopy
Organs were post-fixed at 4 °C with 4% paraformaldehyde (w/v) (Sigma-Aldrich) and 0.25% glutaraldehyde (v/v) (Science Services) and cut into small blocks. Tissue blocks were sectioned at a thickness of 10 μm and collected on Superfrost Plus Gold Slides (Science Services) before further labeling with 1.4 nm IgG-nanogold probes (Nanoprobes). The immunogold labeling procedure was operated according to a previous protocol.27 Briefly, the organ sections were fixed in a periodate-lysine-paraformaldehyde (PLP) fixative and permeabilized. Rabbit anti-human VP1 polyclonal antibody (Neuway Pharma) with a 1:500 dilution was incubated with the samples for 1 h. Later, a secondary nanogold-conjugated anti-rabbit IgG in 1:50 dilution (Nanoprobes) was introduced for 1 h. After post-fixed in 1% glutaraldehyde, the samples underwent the silver enhancement using a HQ-Silver kit according to the manufacturer's instructions (Nanoprobes). In order to stabilize the silver–immunogold complexes from subsequent treatment of osmium tetroxide, we applied an additional procedure with sodium thiosulfate, according to a previous report.28 After serial steps in fixation, osmication and dehydration as reported,27 the sections were flat-embedded using the BEEM® capsules (Plano EM) filled with epon resin. Finally, the epon blocks were cut into ultrathin (80 nm) sections on an ultramicrotome (Leica EM UC7) and then negatively stained with 2% uranyl acetate and 3% lead citrate before the imaging analysis at a voltage of 80 kV, using a Zeiss EM900 electron microscope.
Cell culture
hCMEC/D3 cells were obtained from INSERM France and used between passage 29 to 35 in a culture density of 40000 cells per cm2 according to previous reports.29,30 In our experiments, all culturewares were coated with low viscosity rat collagen I (R&D Systems) at a concentration of 34 μg mL−1. For cell propagation, hCMEC/D3 cells were grown in EBM-2 basal media (Lonza), supplemented with 5% FBS, 5 μg mL−1 ascorbic acid and 1 ng mL−1 bFGF (Sigma Aldrich), 1% chemically defined lipid concentrate and 1% β-mercaptoethanol (Gibco), 1% HEPES and 1% penicillin–streptomycin (Life Technologies). For cell monolayer formation, 1.4 μM hydrocortisone (Sigma Aldrich) was supplemented in addition to the above recipe, and the cells were cultured for minimal 7 days before use.
To evaluate expression profile of 5HT2A receptor, human iPSC-derived astrocytes were prepared via differentiation of human neural stem cells, using internally developed protocols in AbbVie. In order to validate the cell identity, the obtained iPSC astrocytes were characterized by immunocytochemistry using astrocyte markers (GFAP, CD44, S100, EEAT1 or ALDH1L1). For the experiment, the iPSC astrocytes were plated on laminin-coated 96-well plates and cultured in astrocyte differentiated media 1 (ADM1), which was prepared with advanced DMEM/F12 and Neurobasal™ media (Life Technologies), supplemented with BSA (Sigma), 5 ng mL−1 CNTF and 10 ng mL−1 BMP2 (Peprotech), 1× penicillin–streptomycin, 0.5× N-2 Supplement, 0.5× B-27-Supplement and 1 mM L-glutamine (all from Life Technologies).
Uptake and export studies
hCMEC/D3 cells were plated on 96-well μ-clear plates (Greiner Bio-One) and cultured for 48 h prior to exposure of VLPs. Depending on experimental requirements, the cells were treated as following: for concentration-dependent experiments, the cells were incubated with 0, 3.8, 7.9, 15.8 and 31.5 μg mL−1 VLPs along with the media for 24 h; for time-dependent experiments, the cells were exposed to VLPs at a concentration of 31.5 μg mL−1 along with the media for 0, 1, 2, 3, 4, 5 and 6 h; in a pulse-and-chase study, the cells were exposed to 31.5 μg mL−1 VLPs for 24 h (pulse). After removal of VLPs, the cells were rinsed with PBS for three times and cultivated in the fresh media for 0, 1, 2 and 4 h (chase). After various treatments described above, the immunofluorescence staining procedures were applied for VP1 protein. In brief, the cells were fixed in 4% paraformaldehyde for 10 min, and then permeabilized in 0.1% Triton X-100 for 10 min. After 1 h blocking in 1% BSA and 0.05% Tween 20, mouse anti-human VP1 monoclonal antibody (1:1500, Abcam) was incubated with the cells for 1 h, rinsed by PBS and then replaced with secondary Cy3- or Alexa Fluor 488-conjugated AffiniPure anti-mouse IgG (1:800, Jackson ImmunoResearch) for 1 h. In order to evaluate any lysosomal degradation in the pulse-and-chase study, the cells from 0 and 4 h chase were co-stained by using anti-VP1 mouse antibody (1:1500, Abcam) and anti-Lysosomal Associated Membrane Protein 1 (LAMP1) rabbit antibody (1:1000, Abcam). Secondary staining was performed using Alexa Fluor 488-conjugated anti-mouse IgG (1:800) and Cy5-conjugated anti-rabbit IgG (1:400) (Jackson ImmunoResearch). After stained with Hoechst 33342 (Invitrogen), the cells were imaged with a 20× or 40× lens on Arrayscan High Content Analysis (HCA) studio (Thermo Fisher Scientific) and analysed using a Spot Detector BioApplication program. The quantification was performed at well-levels with program-assisted determination of cell border. Valid individual spots (or punctate anti-VP1 staining objects) were identified in between cell border and cell perinuclear area (exclusion of nuclear area) to calculate total intensity of spots captured from cell population in each well. The relative cell uptake was normalized through dividing the well-level total spot intensity with the well-level cell count, as of the following equation:
Inhibition study
hCMEC/D3 cells and iPSC-derived astrocytes were seeded with an equal density of 40000 cells per cm2 on 96-well plates in order to evaluate the influence of 5HT2A receptor (5HT2AR) on VLP uptake. Each of them was first pre-treated for 1 h with rabbit anti-human 5HT2AR polyclonal antibody (1:200, Abcam) at 0.185 and 1.85 μg mL−1, or without the antibody. After removal of the antibody, the cells were washed with PBS and treated with 31.5 μg mL−1 VLPs in culture media for 24 h. For inhibition of clathrin-mediated endocytosis, chlorpromazine (CPZ) (Sigma-Aldrich) or Pitstop2 inhibitor (Abcam) was used respectively. CPZ was prepared with cell media before exposed to the cells for 1 h at concentrations of 1 and 100 μM, or without CPZ; in parallel, Pitstop2 inhibitor was dissolved in DMSO before diluted in cell media for cell treatment of 30 min at 30 μM, similar to a previous report.31 Pitstop2 negative control (Abcam) featured by its highly related structure to Pitstop2 inhibitor was also applied at 30 μM, together with DMSO only and untreated controls. After the pre-treatment, CPZ and Pitstop2 were removed and rinsed. The cells were subsequently incubated with 31.5 μg mL−1 VLPs for 1.5 h and stained for anti-VP1 immunocytochemistry. The analysis was performed on Arrayscan HCA studio as described above.
Confocal and electron microscopy
For confocal analysis, hCMEC/D3 cells were seeded on a collagen-coated chamber slide (Lab-Tek™ II, Thermo Fisher Scientific) and cultivated for 7 days to form the cell monolayer. The BBB monolayer was incubated with 31.5 μg mL−1 VLPs for 10 min and rinsed with PBS before immediately transferred to fresh media for incubation at 37 °C up to 110 min. VP1 immunocytochemistry was carried out as described above, with co-staining of anti-Early Endosome Antigen 1 (EEA1) rabbit antibody (1:900, Abcam). The cell monolayer was scanned in a basal-to-apical direction with a 20× lens in order to generate z-stacks with optical intervals of 1 μm. The images were acquired and processed using a Zeiss Axiovert LSM 700 laser scanning confocal microscope (Carl Zeiss, Oberkochen, Germany), as described above.
For transmission electron microscopy (TEM), hCMEC/D3 monolayer was formed on a transwell filter (0.4 μm polyester, Corning Costar) and treated with 31.5 or 126 μg mL−1 VLPs for up to 4 h. The cells were fixed with 2.5% glutaraldehyde at room temperature for 1 hour in Sorensen's phosphate buffer, and postfixed in 1% osmium tetroxide for 1 hour. The cells were dehydrated by serial washes of 30%, 50%, 70%, 90% and 100% ethanol, before cells were embedded in epon resin. Ultrathin sections (80 nm) were cut with a diamond knife (Diatome, US) using a Leica EM UC7 ultramicrotome. Negative staining was performed with 2% uranyl acetate and 3% lead citrate, same as already described. TEM images were analysed under 80 kV electron beam using a Zeiss EM900 electron microscope (Zeiss Germany).
Statistical analysis
Unless otherwise stated, reported data represent the mean ± standard deviation. Statistical analysis was performed with one-way ANOVA, using GraphPad Prism 7 (GraphPad Software Inc., La Jolla, CA, USA). p < 0.05 was considered statistically significant.Results
Production and physicochemical characterization of VLPs
In order to ensure material quality, VLPs underwent a range of bioanalytical assessments using the technical methods outlined in ESI Table S1.† Details in applying these technologies can be found in the Experimental and ESI sections.† To select qualified VLPs, certain threshold values were defined as target parameters. For in vitro and in vivo studies, only materials fulfilling the requirements for those parameters were considered as qualified. In addition, osmolality and endotoxin levels were also measured before using the VLPs in our studies. By performing these stringent analytical tests, we aimed to obtain VLPs with improved quality and defined physicochemical characteristics.Physicochemical characterization of VLPs was performed using three bioanalytical methods: Transmission Electron Microscopy (TEM), Western Blotting (WB) and Dynamic Light Scattering (DLS). In TEM (Fig. 1a and b), VLPs maintained optimal dispersion without apparent aggregation. The VLP shape was overall spherical and similar to natural JCV capsids. The particles were measured to have a mean diameter of 40–50 nm, consistent with a previous report.25 In Fig. 1b, a magnified feature (250000×) of a single VLP is shown. The major constituent of VLPs was identified as approx. 40 kDa monomeric VP1 protein by WB as seen in Fig. 1c. The pattern of protein bands was consistent across each lane after loading of VLP dilutions (by 2 fold). In DLS, VP1-assembled VLPs showed a single peak in both intensity and volume distribution graphs, where the homogenous particle population was measured without presence of aggregation (Fig. 1d). Here, a volume distribution was included to avoid underestimation of smaller particles and to compare with an intensity distribution to show dispersity features of VLPs. The average hydrodynamic diameter of VLPs was approx. 53 nm, close to the size determined via TEM. With a low polydispersity index value (PDI = 0.07), VLPs showed superior monodispersity. A buffer formulation prepared with 10 mM Tris–HCl, 150 mM NaCl, pH 7.5 likely contributed to the VLPs' dispersion, which was used with VLPs in the following studies.
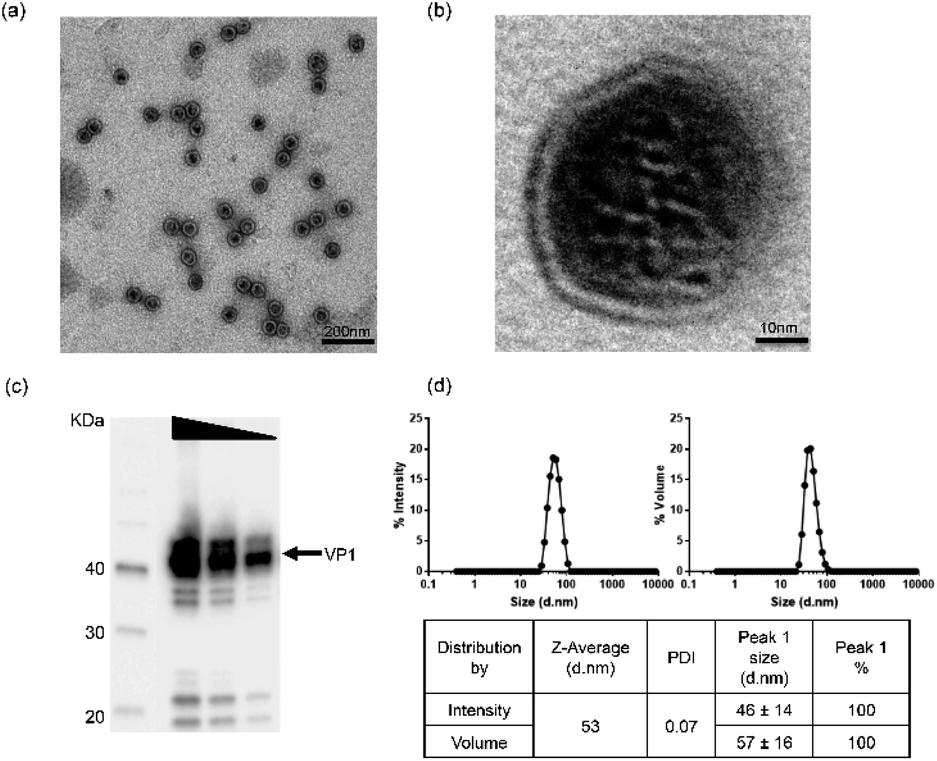 | ||
| Fig. 1 Characterization of virus-like particles (VLPs) assembled from recombinant JCV VP1 protein. (a) VLP stock samples were mounted on grids and visualized by transmission electron microscopy following a negative staining. (b) A single VLP particle magnified at 250000× is shown. (c) A 2-fold dilution series of VLP stock solution (800, 400 and 200 ng of VLPs were loaded in lane 2–4, respectively) was electrophoretically separated by SDS-PAGE and analyzed by western blotting using a mouse anti-human VP1 primary antibody for detection of monomeric VP1 protein (approx. 40 kDa, black arrow). The black arrow indicates the prominent monomeric VP1 protein. (d) In DLS, VLP particles were dispersed in 10 mM Tris–HCl, 150 mM NaCl, pH 7.5 and measured at 23 °C. The VLP stock solution was monodisperse as evident from both intensity and volume size distribution (data provided in the table). | ||
Mouse carotid artery administration
Before carotid artery injection study, we had performed an intravenous (IV) injection to validate VLP distribution to the mouse brain. After VLPs were intravenously administrated via mouse tail veins, we observed strong VP1 presence in mouse liver or kidney but little in brain (data not shown). This finding was consistent with an earlier study.32 After that, we were interested to further examine whether the low brain delivery observed in the IV study was caused by rapid peripheral clearance of VLPs or the inability of VLPs to engage the endothelial cells and reach beyond the BBB. We therefore altered our injection route by administrating VLPs into mouse internal carotid arteries (ICA). That approach allows testing the binding of VLPs to the mouse brain endothelium, before they further circulate through peripheral organs and undergo clearance.As shown in the schematic diagram (Fig. 2a), mouse external carotid artery (ECA) was surgically catheterized prior to injection of VLPs (50 μg) through ICA. After 15 and 120 min, the animals were euthanized and perfused. Brain and liver were then harvested for analysis of presence of VP1 protein using both IHC (at 15 and 120 min) and immunogold-labeling electron microscopy (IG-EM) (at 15 min). In Fig. 2b, presence of VP1 protein at brain microvascular endothelial cells was detected by anti-VP1 IHC at both time points, suggesting VLPs indeed bind to brain endothelia. As shown in Fig. 2c, extended brain exposure (from 15 min to 120 min) led to the clear distribution of anti-VP1 immunofluorescence staining from centralized brain vessel regions to the surrounding brain tissue, suggesting potential VLP transport after ICA injection. In Fig. 2d, distributed VP1 protein was co-stained with Maackia amurensis lectin II (MAL) or Sambucus nigra lectin (SNA), which binds to carbohydrate structures with α-(2,3)- or α-(2,6)-linked sialic acid, respectively. Co-localized fluorescence signals between VP1 protein and two sialic acids were observed, suggesting that both α-(2,3)- and α-(2,6)-linked sialic acids interact with VLPs during binding to the brain endothelia. Sialic acids were previously reported to mediate cell infection for native JCV.7,15,16 In that regard, our results suggest similarities between native JCV and the derived VLP.
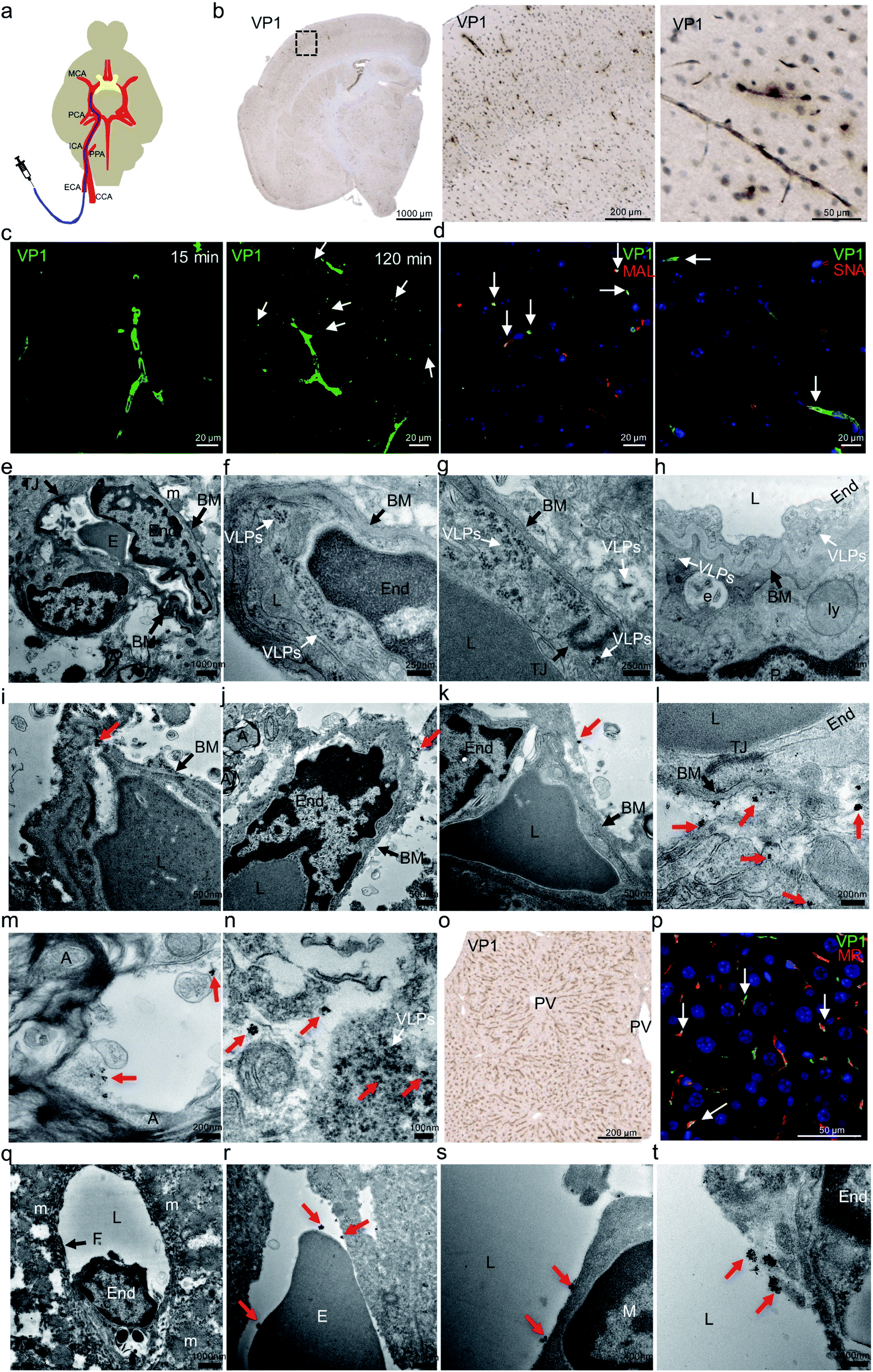 | ||
| Fig. 2 In vivo distribution of JC VLPs following carotid artery administration to mouse brain. (a) Schematic representation of catheterization of the external carotid artery (ECA) and injection of VLPs to the brain. The diagram shown here was produced with modification from a previous report.1 (b) After 15 min mouse brain sections were stained for VP1 protein by IHC. (c) With the same sections, Alex Fluor 488 fluorescence (green) was used for VP1 labeling. VP1 positive staining was observed to distribute from central brain microvascular capillary regions to surrounding cellular space (white arrows) from 15 min to 120 min after the injection. (d) Lectin histochemistry for SNA and MAL was applied to the mouse brain sections, where VP1 (green) and the lectins (red) were co-localized in the endothelial cells (blue, nuclei), as shown by the white arrows. (e) Employing electron microscopy imaging, mouse BBB presented with details for endothelial cell in close contact with its surrounding cellular entities. (f–h) A large number of intact JC VLPs (white arrows) were found to accumulate inside mouse brain endothelial cells and subsequently traverse the BBB from its lumen to the basal membrane (BM). (i–k) With pre-embedding immunogold (IG)-labeling electron microscopy, IG-labeled VP1 protein or VLPs (red arrows) were identified near or beyond the BM area of the BBB. A few magnified images showed that IG particles were clearly identified beyond the BBB (l) and inside the neuronal space (m), where some were accompanied by intact VLPs found nearby (n) (white arrow). (o) Similar to the brain, IHC of liver tissue was performed for detection of VP1 protein. (p) VP1 protein (green) was internalized by liver sinusoidal endothelial cells, as evidenced by its co-localization (white arrows) with mannose receptor (MR, red). As before, EM revealed IG-labeled VP1 or VLPs in close contact with erythrocyte (r), macrophage (s) and endothelial cell (t) in one exemplified fenestrated liver sinusoidal vessel (q). All red arrows indicate the location of IG particles. ECA, external carotid artery; CCA, common carotid artery; PPA, pterygopalatina artery; ICA, internal carotid artery; PCA, posterior cerebral artery; MCA, middle cerebral artery; VP1, JCV capsid protein; MAL, Maackia amurensis lectin II; SNA, Sambucus nigra lectin; PV, portal vein; MR, mannose receptor; E, erythrocyte; End, endothelial cell; TJ, tight junction; BM, basal membrane; P, pericyte; L, lumen; A, myelinated axon; PV, pulmonary veins; e, endosome; ly, lysosome; m, mitochondrion; F, fenestrae; M, macrophage. | ||
Due to the detection limit of IHC, we further used electron microscopy (EM) imaging to determine JC VLPs' distribution in the brain tissue. As shown in Fig. 2e, mouse BBB structure comprises brain endothelial cells interconnecting each other via tight junctions and vicinity support from pericytes and neurons. Erythrocytes visually occupied the partial space of the capillary lumen. As seen in Fig. 2f, a large number of intact JC VLPs were clearly visible in the lumen area of mouse brain microvascular endothelium, where the cell membrane movement was seen to endocytose the VLPs inside the cytoplasmic space. In Fig. 2g and h, VLPs were observed in the vicinity of the vascular basal membrane (BM), to penetrate the BBB and to approach its underneath area, as indicated by the presence of a pericyte. This demonstrates that JC VLPs were able to cross the BBB layer in vivo and remained intact following this transcytosis.
Due to a similar electron density of VLPs as the mouse tissue under the electron beam, it was challenging to identify distribution of intact VLPs in high tissue volumes without immunolabeling. We therefore applied a pre-embedding immunogold electron microscopy (IG-EM) approach to further detect VLPs throughout the parenchyma. Since VP1 monomers serve as building blocks for VLP particles, labeling with VP1 mouse primary antibody and 1.4 nm nanogold conjugated to anti-mouse IgG would likely lead to a unique pattern of gold particles clustered around VLPs, which therefore enables identification of VLPs and their distribution. IG-decorated VLPs were found to be distributed beyond the basal membrane (BM) of brain endothelial cells (Fig. 2i–k), confirming that VLPs had crossed the BBB. Furthermore, as shown in Fig. 2l, more IG particles were detected underneath the BM, where brain endothelial cells were still interconnected with tight junctions, indicating the BBB integrity and exclusion of paracellular transport. IG-labeled VLPs were also detected in the neuronal space, where myelinated axons were visible in vicinity (Fig. 2m). Intact VLPs also appeared alongside with IG labeling (Fig. 2n). In general, ultrasmall gold particles were clustered in an aggregation manner, mostly in rounded shapes with diameters of up to approx. 50–70 nm, dependent on silver enhancement. Individual colloid gold from those clustering could be still visible. The 1.4 nm colloid gold-labeling method enabled detection of a multi-VP1 epitope labeling feature and provided a unique way to identify JC VLPs.
On the other hand, JC VLPs were also detected in peripheral liver tissue (Fig. 2o). A co-staining against VP1 protein and mannose receptor (MR) showed a high degree of co-localization (Fig. 2p). Normally, MR is recruited as a scavenger receptor in liver sinusoidal endothelial cells (LSECs) for degradation of foreign materials from the blood.33 This result indicated that LSECs remained exerting an effect on mechanistic clearance after VLPs being injected via ICA. A cross section of liver sinusoidal blood vessel was visualized by EM imaging. The section featured fenestrated endothelial cells as demonstrated in Fig. 2q. Immunogold particles were found to accumulate near the vessel lumen as well as the plasma membrane of endothelial cells (Fig. 2t). Erythrocytes and macrophages were also seen in association with IGs (Fig. 2r and s). In liver sections IG-labeled VLPs were also detected.
Overall, our carotid artery injection demonstrated that VLPs reached the mouse brain, while the peripheral clearance of VLPs was associated with mouse liver and kidney (data not shown).
Uptake and export of VLPs in BBB cells
After successfully demonstrating the binding and transport of VLPs in the BBB in vivo, we further performed in vitro cell kinetic analysis using High Content Analysis (HCA). For that we employed hCMEC/D3 cells for human blood–brain barrier modeling, as they are often used to reproduce similar in vivo BBB phenotypic characteristics, useful for understanding trafficking outcome of VLPs.In Fig. 3, continuous uptake and export experiments are shown. Concentration-dependent kinetics are presented in Fig. 3a, where hCMEC/D3 cells were treated for 24 h with serial dilutions of VLPs. The uptake showed a progressive increase with VLP concentration in a linear fashion, implicating such uptake process was not saturable with the given concentrations. VLPs' effective exposure concentration (31.5 μg mL−1) was calculated based on our in vivo parameters, factored by mouse body weight (approx. 25 g), blood volume (approx. 58.5 mL kg−1) as well as the volume and dose of injected VLPs (approx. 50 μg per animal). With this concentration hCMEC/D3 cells were incubated with VLPs for 1–6 h. Again, a linear increase was shown, indicating a time-dependent VLP uptake (Fig. 3b). There was a three-fold uptake change observed in between 1 and 6 h. Finally, to validate VLP transport we performed a pulse-and-chase study over time. After 24 h of continuous exposure to VLPs (pulse time), hCMEC/D3 cells were re-cultured in fresh media for 0, 1, 2 or 4 h (chase time). As demonstrated in Fig. 3c, the cells pre-loaded with VLPs showed a steady decrease of fluorescence intensity from the cells. In particular, by 4 h chase time VLP content in the cells was reduced by approximately 50%, which was significantly different from 0 h chase. The diminishing quantity of cellular VLPs might be a result of the exocytosis of BBB cells. In addition, under fluorescence imaging (Fig. 3d), immunofluorescence from VP1 and Lysosomal Associated Membrane Protein 1 (LAMP1) staining was apparently separated, and lysosomal co-localization was hardly observed in between VLPs and lysosomes at chase of 0 and 4 h. This again indicates that VLPs were exocytosed from the BBB cells without involvement of lysosomal degradation.
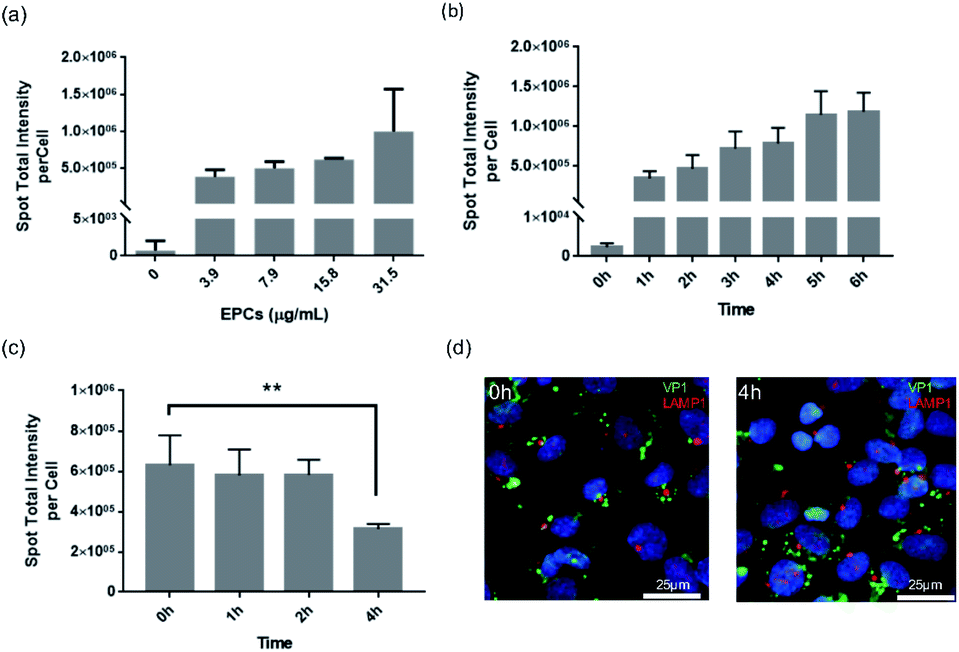 | ||
| Fig. 3 Cellular uptake and export profiles of VLP exposure to the BBB cells (hCMEC/D3). High content imaging analysis was used in conjunction with anti-VP1 immunocytochemistry for kinetic quantification. For uptake studies experiments were performed in concentration or time dependence, where the cells were treated either for 24 h with serial dilutions of VLPs (a) or up to 6 h with a fixed concentration of VLPs (31.5 μg mL−1) (b). For export, a pulse-and-chase study was performed to first expose the cells with VLPs for 24 hours and then re-culture them in fresh media (no VLPs) for 0, 1, 2 and 4 h (c). Additionally, (d) the co-staining of VP1 and LAMP1 was also performed to detect any VLP internalization in lysosomes after 0 or 4 h chase (green, VP1; red, LAMP1; blue, nuclei). All bar graphs show values for total spot intensity divided by total cell count, as described in the Experimental section. Error bars represent means ± SD from triplicate wells. One-way ANOVA analysis was applied to determine statistical significance (**p < 0.01). | ||
Subcellular transport process of VLPs across the BBB
In order to demonstrate VLP transport in a polarized BBB model, hCMEC/D3 cells were cultured for 7 days and then exposed to VLPs in a short pulse for 10 min, prior to a 4 hour chase. Immunofluorescence from anti-VP1 and anti-Early Endosome Antigen 1 (EEA1) staining was visualized by confocal microscopy at 0 min chase (Fig. 4a). Punctate signals from VLPs were found near the cell membrane and also in the intracellular space, suggesting an early cellular interaction. After a 110 min chase VLPs were found to accumulate in a perinuclear region and some of them were seen with increasing endosomal co-localization, as shown by the white arrows in Fig. 4b. Trafficking into early endosomes was previously reported as a contributing stage for transcytosis of biologics across the BBB,34 which may explain the absence of VLP sorting observed in lysosomes (Fig. 3d).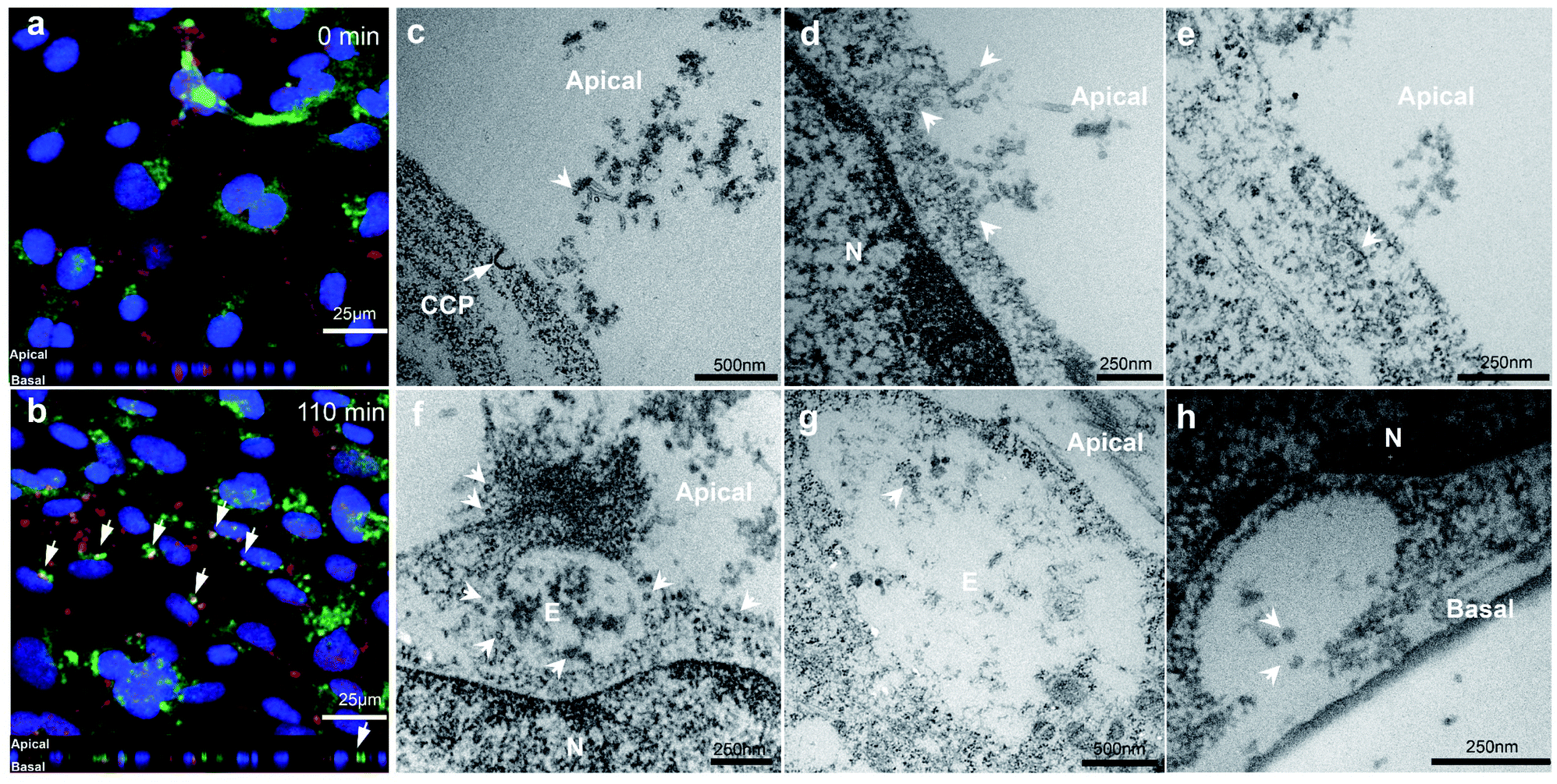 | ||
| Fig. 4 Confocal and electron microscopy imaging analysis for VLP transport across the BBB. For confocal microscopy, hCMEC/D3 cells were plated on a glass slide for 7 days prior to a pulse-and-chase study performed. The cells were then treated by VLPs (31.5 μg mL−1) as short as 10 min, then cultured in fresh media for 0 (a) and 110 min (b). A co-staining for VP1 and EEA1 showed VLPs were co-localized with early endosomes (indicated by white arrows) (red, VP1; green, EEA1; blue, nuclei). z-Scanning of the monolayer from a basal-to-apical direction was performed and displayed under the bottom of image (a) and (b), to illustrate the cross-section views of treated cell monolayer. For electron microscopy, hCMEC/D3 cell monolayer on a transwell filter were treated with 126 μg mL−1 VLPs for 4 h. Cross-section images were captured to illustrate directional transmigration of VLPs, and cell membrane interactions (c–e), intracellular internalization (f and g) and exocytosis (h) are individually shown. White arrow heads indicate the location of VLPs. CCP, clathrin-coated pits; N, nucleus; E, endosome. | ||
By electron microscopy cross sections of in vitro BBB monolayer were imaged to substantiate the overall transport process for VLPs. For this study, concentration- or time-dependent experiments were performed in order to obtain optimal imaging conditions for VLPs crossing the BBB (data not shown). We found that use of a higher concentration of 126 μg mL−1 (compared to 31.5 μg mL−1) was helpful to capture more cellular activities from transport of VLPs. We first observed that VLPs contacted cell membranes in large quantity (Fig. 4c). A clathrin-coated pit was found opening in the vicinity of VLPs while they were approaching the apical cell membrane. VLPs adhered to the cell surface like “pearls” and subsequently were engulfed via membrane movement (Fig. 4d). VLPs were also seen to traffic with a range of subcellular structures, which involved vesicular transport (Fig. 4e) or endosomal sorting (Fig. 4f and g). Importantly, VLPs seemed to remain intact in morphology during subcellular sorting. Lastly, as an evidence of transcytosis, an image of VLPs exiting the basal membrane is presented (Fig. 4h), where a vesicle was docking with opening cavity and VLPs of 40–50 nm in size were discharged towards the transwell membrane. VLP particles were visibly spherical in shape after traversing the BBB layer.
Inhibition of VLP endocytosis in BBB cells
The data presented in Fig. 2 and 4 demonstrated that VLPs were efficient in binding to or transporting across the BBB, indicating their high affinity for brain endothelium in vitro and in vivo. However, in literature one reported human BBB in vitro model was found not to express serotonin 5HT2A receptor (5HT2AR) in vitro,13 which was otherwise required for infectivity of native JCV in brain glial cells.17 Therefore, we performed an inhibition study to investigate the mechanism contributing to VLP uptake into human brain endothelial cells. We used hCMEC/D3 endothelial cells and iPSC-based astrocytes to evaluate roles of 5HT2AR in mediating VLP uptake (Fig. 5a and b). Using qPCR analysis, 5HT2AR gene expression was observed in iPSC-based astrocytes but not in hCMEC/D3 cells (data not shown), which is in agreement with literature.13,17 When pre-treated by 5HT2AR antibody, iPSC-based astrocytes showed a significant decrease of VLP uptake for 24 h. The reduction of VLP uptake was concentration-dependent and it changed 10-times after increasing the antibody concentration from 0.185 to 1.85 μg mL−1 for cell treatment. Statistical differences were found between the groups treated with or without antibody (Fig. 5b). On the contrary, the endothelial cells showed neither comparable VLP uptake as in astrocytes nor significant differences regarding dependence of 5HT2AR antibody exposure (Fig. 5a). This confirms that 5HT2A receptor may not play a role during VLP endocytosis.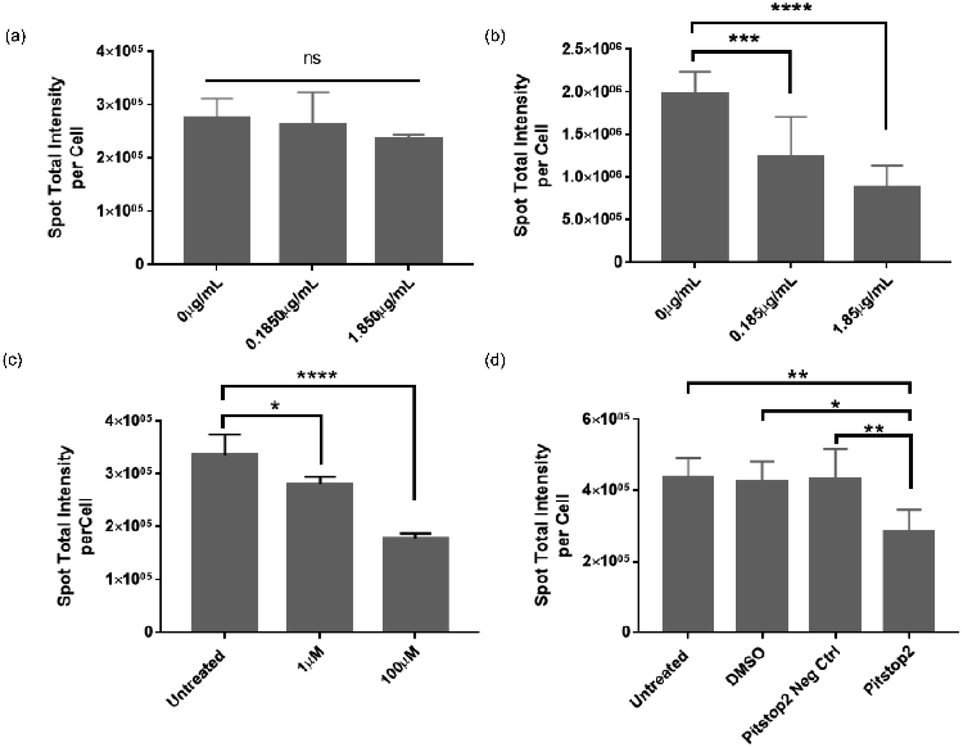 | ||
| Fig. 5 Roles of 5HT2A receptor (5HT2AR) and clathrin-mediated endocytosis (CME) in mediating VLP uptake into BBB cells. hCMEC/D3 cells (a) and human iPSC-derived astrocytes (b) were pre-treated by 5HT2AR antibody of 0, 0.185 and 1.85 μg mL−1 for 1 h and then exposed to 31.5 μg mL−1 VLPs for 24 h. The VLP uptake was assessed based on dependence of antibody treatment or cell type. For inhibition of CME, hCMEC/D3 cells were pre-treated by either chlorpromazine (CPZ) at 0, 1 or 100 μM for 1 h (c), or by Pitstop2 inhibitor and Pitstop2 negative control, both at 30 μM for 0.5 h (d). VLPs were subsequently applied for 1.5 h before cellular uptake was evaluated by HCA. Bar graphs were generated as described in the Experimental section. Error bars represent means ± SD from triplicate wells. One-way ANOVA analysis was applied to determine statistical significance (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 and no significance (ns) at p ≥ 0.05). | ||
On the other hand, clathrin-mediated endocytosis (CME) was also reportedly responsible for the infectivity of JCV in brain glial cells.35 Considering that clathrin-coated pits are the predominant type of vesicles at the BBB,36 we further investigated the involvement of CME during VLP uptake into BBB cells. Chlorpromazine (CPZ) and Pitstop2™ inhibitors were reported to deplete or block CME from cell plasma membrane or cytosol,37 therefore we applied them for inhibiting VLP endocytosis. As shown in Fig. 5c, hCMEC/D3 cells exhibited a concentration-dependent decrease in uptake of VLPs for 1.5 h, after 1 h pre-treatment with CPZ. VLP uptake was reduced by approx. 17% at 1 μM CPZ and 47% at 100 μM CPZ, both significantly different from the untreated control. It was observed that a higher CPZ dose at ∼300 μM affected cell viability, but not the concentrations used here (data not shown). On the other hand, we pre-treated hCMEC/D3 cells with Pitstop2 inhibitor for 30 min, and the VLP uptake for 1.5 h was assessed to be reduced by 36%, 32% and 34%, in comparison to the untreated, DMSO, Pitstop2 negative control, respectively. The differences between Pitstop2 and controls were statistically significant (Fig. 5d). In conclusion, we confirmed that clathrin-mediated endocytosis contributed to VLP endocytosis in human brain endothelial cells, similar to a previous report for native JCV.35
Discussion
Virus-based nanocarrier systems have been studied in recent years due to advantages of structural uniformity and biocompatibility.38 As a reduction form of native viral vectors, hollow virus-like particles (VLPs) are assembled by outer capsid protein VP1, with VP2, VP3 protein and viral DNA being absent, in order to facilitate receptor engagement as well as cargo incorporation. It therefore offers great flexibility and convenience in preparing VLPs for medical applications, via tuning buffer conditions of Ca2+ ions and disulfide bonds for VP1 dissociation and reassociation.25,39 Based on unique biology of the native virus, VLPs are interesting systems that bear promises to carry drug cargo for targeted brain delivery. As an example here we discuss JC VLPs, which are derived from JC virus that persists in healthy individuals without symptoms because of suppressed viral reproduction by the immune system. Upon reactivation, however, JCV is thought to infiltrate the BBB and infect brain cells using 5HT2A receptor or clathrin-mediated mechanisms.It was previously reported that liver sinusoid vessels served as a sink for clearance of blood-borne JC VLPs from intravenous injection via the tail vein.32 In our independent IV pilot study we also found a similar result for VLPs in peripheral organs. Moreover, production quality and bioanalytical control for these viral particles would improve VLPs' in vivo distribution outcomes. For example, an abnormal endotoxin level from a non-GMP production process may aggravate an innate immune response to VLPs and promote a peripheral clearance, which therefore needs to be carefully controlled.40 Also, elevated endotoxins could induce neuroinflammation and exert an effect on the disruption of the BBB.41 Therefore, it is necessary to implement a range of bioanalytical tests as proposed in ESI Table S1† to circumvent emerging stability and integrity issues during in vivo testing so that the results are comparable in follow-up studies.
As a proof of concept, we used in vivo carotid artery injection to demonstrate delivery outcome of JC VLPs into mice. Our qualitative analysis revealed modest translocation of JC VLPs into brain, although a more quantitative assessment of JC VLP delivery into brain will be required in future studies for understanding the brain entry efficiency. With carotid artery injection we found that if given the chance to bind, VLPs could be enriched in brain vasculature before liver clearance occurs. Anti-VP1 IHC and IF results together showed that VLPs were indeed binding to the endothelia throughout the whole brain. From 15 to 120 min post injection, the brain capillary binding persistently occurred, and IF staining showed increased signals spreading from central brain capillary regions to the nearby brain tissue. According to Simon-Santamaria, J. et al.,32 negligible brain distribution was observed in an IV study using radiolabeled JC VLPs with normal or mutant binding properties for sialic acids. However, it remains unclear whether the radioactivity fully represented characteristics of JC VLPs, particularly regarding their interacting nature with sialic acids from brain tissue. Based on evidence provided in our study VLPs are enriched on brain capillary walls, which seems to be the result of receptor interaction with sialic acids. Our IHC data showed that α-(2,3)- and α-(2,6)-linked sialic acids were co-localized with VP1 protein and indeed required for VLP binding to mouse brain, while they were reportedly not required for liver binding due to its scavenging mechanism.32 The exact nature of interactions between JC VLPs and sialic acids remains under debate in the literature. α-(2,6)-Linked sialic acid was reported as a primary binding site for recombinant JCV VP1 pentamer.42 However, α-(2,3)- and α-(2,6) linkages reportedly shared similar functions in mediating multivalent JCV infection to brain cells.15,16 In particular, lactoseries tetrasaccharide C (LSTc) was reported in literature as a specific JCV receptor motif, which was found to mediate JCV attachment and infectivity in human glial cells.42 In an attempt to validate the presence of LSTc-mediated VLP uptake mechanism in human BBB cells, we had tried to use LSTc as an inhibitor to block VLP binding and entry in hCMEC/D3 cells. However, no clear LSTc-dependent effect was observed to influence VLP uptake, suggesting such mechanism is not present with hCMEC/D3 cells (ESI Fig. S1†).
Despite VLP clearance was observed in mouse peripheral organs, employing EM imaging we showed evidence that VLPs were capable of crossing the BBB and entering the neuronal space in vivo. It is likely that the binding properties of VLPs to brain vasculature could be a prerequisite for their transmigration through the BBB.
With regard to viral transport, native JCV was postulated in literature to utilize its etiological pathway transmitting from its peripheral latency sites to the brain. During this stage, B lymphocytes reportedly served as a reservoir of viral genome and a dissemination vehicle for JCV crossing the BBB.14 The role of JC viral capsid during BBB transmigration remained unclear despite suggested infectivity of JCV to the brain endothelia.13 Moreover, the lack of 5HT2AR in the BBB in vitro suggested other potential mechanisms for VLP transport across the brain endothelium. Due to mechanistic inaccessibility from in vivo study, here we addressed this question by using in vitro approaches.
VLP uptake was investigated by employing hCMEC/D3 cells and quantified with anti-VP1 immunofluorescence staining. The overall uptake progressively increased with VLP exposure time and concentration, similar to kinetics of other forms of nanoparticles.43,44 Also, in a pulse-and-chase experiment we pre-loaded the BBB cells with VLPs for 24 h, and a steady export was observed over time as soon as the cells were re-cultured in fresh media. By fluorescence imaging we confirmed that the VLP export was not influenced by a “diminishing” factor from lysosomal degradation in hCMEC/D3 cells, a process otherwise commonly seen in case of polymeric nanoparticles.45
Further, we grew hCMEC/D3 cells into polarized cell monolayer in vitro to validate an apical-to-basal transport of VLPs by employing confocal and electron microscopy. After a pulse of 10 minutes only, VLPs already showed binding to the surface of the hCMEC/D3 monolayer without immediate localization in endosomes. Preceded by a 110 min chase the surface-bound VLPs were further translocated to the intracellular space, where endosomal co-localization concomitantly emerged. Thus, we clearly demonstrated the binding and transportation of VLPs throughout the BBB in vitro, consistent with the carotid injection study as discussed above. By electron microscopy we studied the VLP transport process in more detail. Overall, our EM data confirm that VLPs could be endocytosed by the hCMEC/D3 monolayer. Active cell membrane interactions were captured post VLP treatment, including the presence of clathrin-coated pits, membrane invagination or engulfment. Trafficking of VLPs was identified by their localization in vesicles or endosomes but rarely in lysosomes. We further observed that the transcytosis of VLPs indeed occurred in the BBB model in vitro, in accordance with IG-EM findings from the in vivo BBB study. Collectively, these results suggest that VLPs can transmigrate across the BBB using a pathway which does not involve lysosomal accumulation.
Our in vivo results suggested the involvement of sialic acids during JC VLP binding to brain endothelia, which were known to promote clathrin-mediated endocytosis (CME) of biologics across the BBB.46 Also, clathrin-coated pits (CCPs) are vesicles predominately present at the BBB,36 where their endocytosis is reportedly restricted by expression of Mfsd2a.47 According to Pho et al.35 JCV entered human glial cells via CME. Also, since 5HT2AR is not expressed in the BBB and hence cannot facilitate VLP transport, we further explored the involvement of CME during JC VLP transport across the BBB. CPZ was previously used to block infection of JCV in brain cells through CME.17,35,48 Pitstop2 was also reported to interfere with CME for viral uptake (i.e., swine fever virus,49 HIV31). Applying both inhibitors in our study resulted in a significant decrease of VLP uptake into BBB cells. One schematic diagram is provided to summarize the current understanding of JC VLP transport pathway across the BBB (Fig. 6). This also led to our conclusion that – although VLPs were only assembled with capsid protein VP1 and generally a simpler version compared to its native viral form – they preserved similar functionality to exploit CME for uptake and further transport across the BBB.
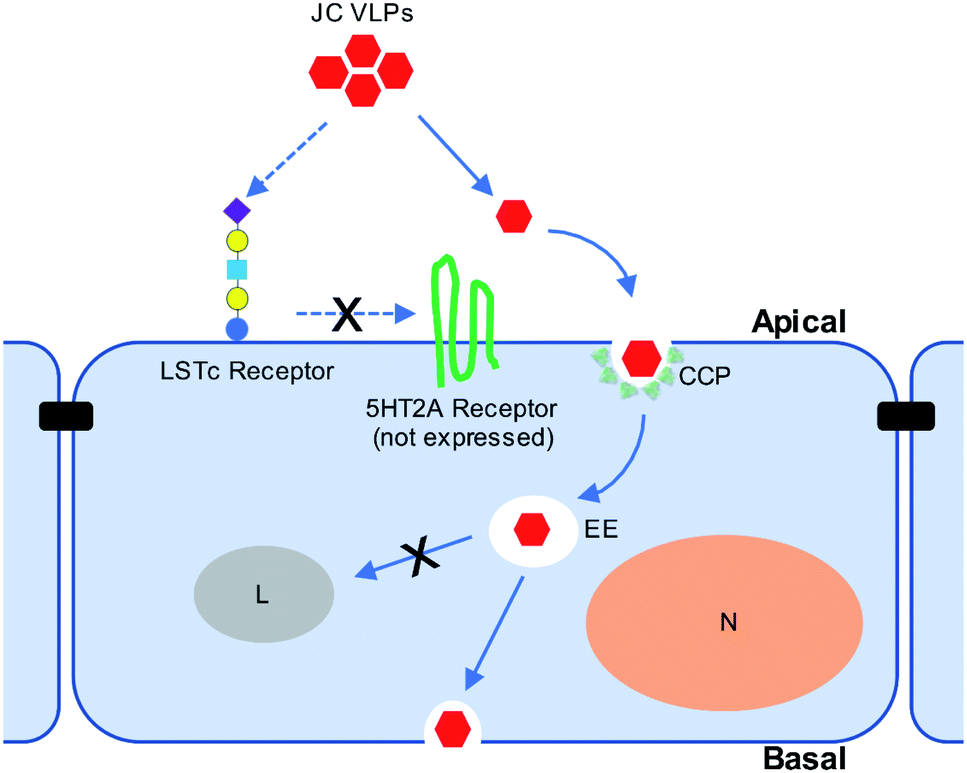 | ||
| Fig. 6 Schematic diagram for JC VLP transport pathway across the BBB using clathrin-mediated endocytosis as demonstrated in vitro and in vivo. CCP, clathrin-coated pit; EE, early endosome; L, lysosome; N, nucleus. | ||
Conclusion
In this study, we demonstrate that JC virus-like particles display similar physiochemical characteristics and cellular functionality as reported for native JCV in literature. Also, JC VLPs are able to transcytose across the blood–brain barrier as demonstrated in vitro and in vivo. From our study, JC VLPs remain functional under in vivo and in vitro conditions, where their binding or translocation across the BBB is mediated through combined interactions with sialic acids as well as clathrin-dependent mechanisms. Their uptake and translocation processes involve serial intracellular trafficking events through brain endothelial cells as demonstrated in mouse and human-derived BBB. In particular, different from the outcome observed from the IV injection study, JC VLPs can translocate into brain if injected to its close proximity, by such as a carotid injection, and magnitude of JC VLP delivery into brain needs a further investigation. Overall, JC VLPs are characterized by relatively lesser biological complexity and attractive versatility as drug carriers, as compared to native JCV. Delivery of JC VLPs packaged with suitable cargo types (i.e., plasmid DNA, siRNA) represents a promising technological solution for brain delivery across the BBB.Data availability
The raw/processed data required to reproduce these findings cannot be shared at this time due to legal or ethical reasons.Funding
The design, study conduct, and financial support for this research was provided by AbbVie and German Ministry of Education and Research (BMBF) under the program of “Efficient drug transport in biological systems – BioMatVital: BioTransporter”. All authors contributed to the study conception and design. The first draft of the manuscript was written by Dong Ye, and all authors actively contributed to revise and proofread the final version. AbbVie participated in the interpretation of data, review, and approval of the publication.Conflicts of interest
Dong Ye, Christian L. Ried, Harri Rahn, Christopher Untucht, Karsten Wicke, Mario Mezler and Axel H. Meyer are employees of AbbVie and may own AbbVie stock. Victoria Demina, Sergey Sotnikov, Marcus Stapf and Heiko Manninga are employees of NEUWAY Pharma GmbH. Tina Zimmermann, Georg C. Terstappen and Michael Rohe were employees of AbbVie at the time when they contributed to this work. Currently, Tina Zimmermann is an employee of Boehringer Ingelheim Pharma; Georg C. Terstappen is an employee of Cambrian Biopharma; Michael Rohe is an employee of H. Lundbeck A/S. All authors declare no competing financial interest in this work.Acknowledgements
We would like to acknowledge technical support by Aysel Gueler, Ina Mayrhofer, Juergen Korffmann, Gabriele Weber, Bettina Liebel, Lynda Nolan and Andreas Popp from AbbVie Deutschland GmbH & Co. KG, and by Andrea Tausch from NEUWAY Pharma GmbH. This study was funded by the German Ministry for Education and Research (BMBF) under the call “Efficient drug transport in biological systems - BioMatVital: BioTransporter”.References
- L. Zhang, R. L. Zhang, Q. Jiang, G. Ding, M. Chopp and Z. G. Zhang, Nat. Protoc., 2015, 10, 539–547 CrossRef CAS.
- N. J. Abbott, A. A. Patabendige, D. E. Dolman, S. R. Yusof and D. J. Begley, Neurobiol. Dis., 2010, 37, 13–25 CrossRef CAS.
- Z. Zhao, A. R. Nelson, C. Betsholtz and B. V. Zlokovic, Cell, 2015, 163, 1064–1078 CrossRef CAS.
- G. F. Woodworth, G. P. Dunn, E. A. Nance, J. Hanes and H. Brem, Front. Oncol., 2014, 4, 126 Search PubMed.
- H. Fu and D. M. McCarty, Curr. Opin. Virol., 2016, 21, 87–92 CrossRef CAS.
- E. Major, K. Amemiya, C. Tornatore, S. Houff and J. Berger, Clin. Microbiol. Rev., 1992, 5, 49–73 CrossRef CAS.
- M. W. Ferenczy, L. J. Marshall, C. D. Nelson, W. J. Atwood, A. Nath, K. Khalili and E. O. Major, Clin. Microbiol. Rev., 2012, 25, 471–506 CrossRef CAS.
- B. J. Brew, N. W. Davies, P. Cinque, D. B. Clifford and A. Nath, Nat. Rev. Neurol., 2010, 6, 667–679 CrossRef.
- M. L. Gasparovic, G. V. Gee and W. J. Atwood, J. Virol., 2006, 80, 10858–10861 CrossRef CAS.
- A. S. Saribas, S. Mun, J. Johnson, M. El-Hajmoussa, M. K. White and M. Safak, Virology, 2014, 449, 1–16 CrossRef CAS.
- C. Schaumburg, B. O'hara, T. Lane and W. Atwood, J. Virol., 2008, 82, 8896–8899 CrossRef CAS.
- B. F. Sabath and E. O. Major, J. Infect. Dis., 2002, 186(2), S180–S186 CrossRef.
- M. L. Chapagain, S. Verma, F. Mercier, R. Yanagihara and V. R. Nerurkar, Virology, 2007, 364, 55–63 CrossRef CAS.
- M. L. Chapagain and V. R. Nerurkar, J. Infect. Dis., 2010, 202, 184–191 CrossRef CAS.
- A. S. Dugan, M. L. Gasparovic and W. J. Atwood, J. Virol., 2008, 82, 2560–2564 CrossRef CAS.
- C. K. Liu, G. Wei and W. J. Atwood, J. Virol., 1998, 72, 4643–4649 CrossRef CAS.
- G. F. Elphick, W. Querbes, J. A. Jordan, G. V. Gee, S. Eash, K. Manley, A. Dugan, M. Stanifer, A. Bhatnagar, W. K. Kroeze, B. L. Roth and W. J. Atwood, Science, 2004, 306, 1380–1383 CrossRef CAS.
- C.-N. Chao, Y.-H. Yang, M.-S. Wu, M.-C. Chou, C.-Y. Fang, M.-C. Lin, C.-K. Tai, C.-H. Shen, P.-L. Chen and D. Chang, Sci. Rep., 2018, 8, 2213 CrossRef.
- C. Goldmann, H. Petry, S. Frye, O. Ast, S. Ebitsch, K. D. Jentsch, F. J. Kaup, F. Weber, C. Trebst, T. Nisslein, G. Hunsmann, T. Weber and W. Luke, J. Virol., 1999, 73, 4465–4469 CrossRef CAS.
- D. Chang, C. Y. Fung, W. C. Ou, P. C. Chao, S. Y. Li, M. Wang, Y. L. Huang, T. Y. Tzeng and R. T. Tsai, J. Gen. Virol., 1997, 78(6), 1435–1439 CrossRef CAS.
- L. Chen, M. Wang, W. Ou, C. Fung, P. Chen, C. Chang, W. Huang, J. Wang, P. Lin and D. Chang, Gene Ther., 2010, 17, 1033 CrossRef CAS.
- C.-F. Chang, M. Wang, W.-C. Ou, P.-L. Chen, C.-H. Shen, P. Y. Lin, C.-Y. Fang and D. Chang, Expert Opin. Biol. Ther., 2011, 11, 1169–1175 CrossRef CAS.
- A. D. Hale, D. Bartkeviciute, A. Dargeviciute, L. Jin, W. Knowles, J. Staniulis, D. W. Brown and K. Sasnauskas, J. Virol. Methods, 2002, 104, 93–98 CrossRef CAS.
- K. Sasnauskas, A. Bulavaite, A. Hale, L. Jin, W. A. Knowles, A. Gedvilaite, A. Dargeviciute, D. Bartkeviciute, A. Zvirbliene, J. Staniulis, D. W. Brown and R. Ulrich, Intervirology, 2002, 45, 308–317 CrossRef CAS.
- C. Goldmann, N. Stolte, T. Nisslein, G. Hunsmann, W. Luke and H. Petry, J. Virol. Methods, 2000, 90, 85–90 CrossRef CAS.
- D. B. Hoffmann, K. O. Böker, S. Schneider, E. Eckermann-Felkl, A. Schuder, M. Komrakova, S. Sehmisch and J. Gruber, Mol. Ther.--Nucleic Acids, 2016, 5(3), e298 CrossRef CAS.
- D. Ye, S. Anguissola, T. O'Neill and K. A. Dawson, Nanoscale, 2015, 7, 10050–10058 RSC.
- R. C. Melo, E. Morgan, R. Monahan-Earley, A. M. Dvorak and P. F. Weller, Nat. Protoc., 2014, 9, 2382 CrossRef CAS.
- B. Poller, H. Gutmann, S. Krähenbühl, B. Weksler, I. Romero, P. O. Couraud, G. Tuffin, J. Drewe and J. Huwyler, J. Neurochem., 2008, 107, 1358–1368 CrossRef CAS.
- B. B. Weksler, E. A. Subileau, N. Perriere, P. Charneau, K. Holloway, M. Leveque, H. Tricoire-Leignel, A. Nicotra, S. Bourdoulous, P. Turowski, D. K. Male, F. Roux, J. Greenwood, I. A. Romero and P. O. Couraud, FASEB J., 2005, 19, 1872–1874 CrossRef CAS.
- L. von Kleist, W. Stahlschmidt, H. Bulut, K. Gromova, D. Puchkov, M. J. Robertson, K. A. MacGregor, N. Tomilin, A. Pechstein, N. Chau, M. Chircop, J. Sakoff, J. P. von Kries, W. Saenger, H.-G. Kräusslich, O. Shupliakov, P. J. Robinson, A. McCluskey and V. Haucke, Cell, 2011, 146, 471–484 CrossRef CAS.
- J. Simon-Santamaria, C. H. Rinaldo, P. Kardas, R. Li, I. Malovic, K. Elvevold, P. McCourt, B. Smedsrød, H. H. Hirsch and K. K. Sørensen, PLoS One, 2014, 9, e111762 CrossRef.
- K. Elvevold, J. Simon-Santamaria, H. Hasvold, P. McCourt, B. Smedsrod and K. K. Sorensen, Hepatology, 2008, 48, 2007–2015 CrossRef CAS.
- A. S. Haqqani, C. E. Delaney, E. Brunette, E. Baumann, G. K. Farrington, W. Sisk, J. Eldredge, W. Ding, T.-L. Tremblay and D. B. Stanimirovic, J. Cereb. Blood Flow Metab., 2018, 38, 727–740 CrossRef CAS.
- M. T. Pho, A. Ashok and W. J. Atwood, J. Virol., 2000, 74, 2288–2292 CrossRef CAS.
- M. Simionescu, N. Ghinea, A. Fixman, M. Lasser, L. Kukes, N. Simionescu and G. Palade, J. Submicrosc. Cytol. Pathol., 1988, 20, 243 CAS.
- D. Dutta and J. G. Donaldson, Cell. Logist., 2012, 2, 203–208 CrossRef.
- Y. Ma, R. J. M. Nolte and J. J. L. M. Cornelissen, Adv. Drug Delivery Rev., 2012, 64, 811–825 CrossRef CAS.
- R. Noad and P. Roy, Trends Microbiol., 2003, 11, 438–444 CrossRef CAS.
- R. Cubas, S. Zhang, S. Kwon, E. M. Sevick-Muraca, M. Li, C. Chen and Q. Yao, J. Immunother., 2009, 32, 118–128 CrossRef.
- W. A. Banks, A. M. Gray, M. A. Erickson, T. S. Salameh, M. Damodarasamy, N. Sheibani, J. S. Meabon, E. E. Wing, Y. Morofuji and D. G. Cook, J. Neuroinflammation, 2015, 12, 223 CrossRef.
- U. Neu, M. S. Maginnis, A. S. Palma, L. J. Stroh, C. D. Nelson, T. Feizi, W. J. Atwood and T. Stehle, Cell Host Microbe, 2010, 8, 309–319 CrossRef CAS.
- F. Danhier, N. Lecouturier, B. Vroman, C. Jérôme, J. Marchand-Brynaert, O. Feron and V. Préat, J. Controlled Release, 2009, 133, 11–17 CrossRef CAS.
- J. Panyam and V. Labhasetwar, Adv. Drug Delivery Rev., 2003, 55, 329–347 CrossRef CAS.
- D. Ye, M. N. Raghnaill, M. Bramini, E. Mahon, C. Åberg, A. Salvati and K. A. Dawson, Nanoscale, 2013, 5, 11153–11165 RSC.
- A. Abulrob, H. Sprong, E. Henegouwen, P. Van Bergen and D. Stanimirovic, J. Neurochem., 2005, 95, 1201–1214 CrossRef CAS.
- A. Ben-Zvi, B. Lacoste, E. Kur, B. J. Andreone, Y. Mayshar, H. Yan and C. Gu, Nature, 2014, 509, 507–511 CrossRef CAS.
- S. Suzuki, H. Sawa, R. Komagome, Y. Orba, M. Yamada, Y. Okada, Y. Ishida, H. Nishihara, S. Tanaka and K. Nagashima, Virology, 2001, 286, 100–112 CrossRef CAS.
- B. Hernáez, M. Guerra, M. L. Salas and G. Andrés, PLoS Pathog., 2016, 12, e1005595 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00879f |
| ‡ Authors contributed equally. |
| § Current address: Boehringer Ingelheim Pharma GmbH & Co. KG, CardioMetabolic Diseases Research, 88397 Biberach an der Riß, Germany. |
| ¶ Current address: H. Lundbeck A/S, Ottiliavej 9, 2500 Valby, Denmark. |
| || Current address: Cambrian Biopharma, New York, USA. |
| This journal is © The Royal Society of Chemistry 2021 |