DOI:
10.1039/D0NA00730G
(Paper)
Nanoscale Adv., 2021,
3, 661-667
Phosphorescent MoS2 quantum dots as a temperature sensor and security ink†
Received
31st August 2020
, Accepted 9th November 2020
First published on 10th November 2020
Abstract
Currently, few phosphorescent materials (PMs) possess a long phosphorescence lasting time and have potential for application in chemical sensors. Herein, we disclose that the incorporation of few-layer molybdenum disulfide quantum dots (FL-MoS2 QDs) into poly(vinyl alcohol) (PVA) matrices leads to the emission of bright green phosphorescence with a long lasting time of 3.0 s and a phosphorescence quantum yield of 20%. This enhanced phosphorescence originates from the formation of O–H⋯S hydrogen bonding networks between the rich sulfur sites of the FL-MoS2 QDs and the hydroxyl groups of the PVA molecules, which not only rigidifies the vibration modes of the FL-MoS2 QDs but also provides an oxygen barrier. Further investigations reveal that the FL-MoS2 QD/PVA composites exhibit a longer phosphorescence lasting time than N,S-doped carbon dots, few layer tungsten disulfide quantum dots, Rhodamine 6G, and Rhodamine B in PVA matrices. Since heat efficiently induced the removal of water moisture from PVA matrices, the FL-MoS2 QD/PVA composites could be implemented for phosphorescence turn-on and naked-eye detection of temperature variations ranging from 30 to 70 °C. By contrast, the carbon dot/PVA composites were incapable of sensing environmental temperature due to their weak hydrogen bonding with the hydroxyl groups of PVA matrices. Additionally, this study reveals the potential of the FL-MoS2 QD/PVA composites as an advanced security ink for anti-counterfeiting and encryption applications. The given results could open a new direction for potential application of two-dimensional quantum dots in phosphorescence-based sensors and security inks.
Introduction
Phosphorescent materials (PMs) have received much attention over the last decade owing to their outstanding optical properties, including long-lived triplet excited states, massive Stokes shifts, and extremely low background.1,2 These characteristics make PMs attractive for a wide range of applications in chemosensors,3–5 security protection,6 optoelectronics,7,8 and imaging probes.9–11 In comparison with nanosecond-lifetime fluorescent materials, PMs possess a second-to-hour lifetime that is easily detected by the naked eye upon switching off the excitation source. However, the development of PMs is limited by the rapid nonradiative decay of triplet excitons12 and inefficient singlet-to-triplet intersystem crossing.13 For example, an oxygen molecule can induce triplet exciton quenching due to its triplet ground state.14 Additionally, a long phosphorescence lifetime makes the triplet excitons easy to be consumed via vibrational deactivation.15,16 Therefore, it can be expected that highly efficient phosphorescence will be obtained if the above events are suppressed. Two principal strategies have evolved to enhance the PMs' efficiency and lifetime through the promotion of singlet-to-triplet intersystem crossing and the suppression of nonradiative decay of triplet excitons.17 The first strategy is to introduce heavy metal atoms (e.g., Br, Cl, and I), heteroatoms (e.g., N and P), and aromatic carbonyls into PMs, boosting the spin–orbit coupling.17–19 The incorporation of heavy atoms and heteroatoms into PMs promotes the combination of the singlet and triplet states of the excitons, facilitating the intersystem crossing.19 Moreover, the aromatic carbonyl in PMs induces the generation of intrinsic triplet excitons which is beneficial to the formation of spin–orbit coupling.20 The other strategy involves embedding fluorophores into a solid matrix which provides an oxygen barrier to restrict the nonradioactive transitions from triplet excitons to oxygen molecules.21 Examples of the embedding approaches include host–guest doping (e.g., doped in the polymer matrix, steroids, and organic frameworks),5,22 crystal engineering (single crystal and co-crystal methods),23,24 and H-aggregation.25 The latter is particularly appealing to generate long-lifetime PMs due to the simplicity, metal-free composition, and environmentally friendly processes.
Previous studies on PMs mainly included organometallic complexes and heavy metal-based inorganics.26,27 Obstacles to these materials' applicability come from complicated synthetic routes, high-cost precursors, and high toxicity. In an effort to circumvent these drawbacks, organic luminogens are introduced instead of doping metals into PMs. However, organic luminogens rarely have ultralong-lived phosphorescence due to the rapid quenching of triplet excitons. Recently, nanomaterials are emerging as an alternative for PMs since they offer the distinct advantages of good water solubility, biocompatibility, simple synthetic routes, and low cost. Examples of nanomaterial-related PMs include Mn2+-doped semiconductor quantum dots,28 phosphorescent dye-loaded silica nanoparticles,29 gold nanoclusters,30 and carbon dots (CDs).31 Although exhibiting ultralong-lived phosphorescence in a solid matrix or in the presence of heavy metal ions, these nanomaterials rarely serve as a temperature sensor due to temperature-induced quenching of phosphorescence. As an example, Chen et al. prepared nitrogen-doped CDs through the hydrothermal treatment of polyvinyl alcohol (PVA) and ethylene diamine and showed a double-exponential decay in their phosphorescence with raising the temperature.32 Therefore, it is urgent to develop nanomaterial-based PMs for the phosphorescence detection of an increase in temperature, which can be easily monitored by the naked eye.
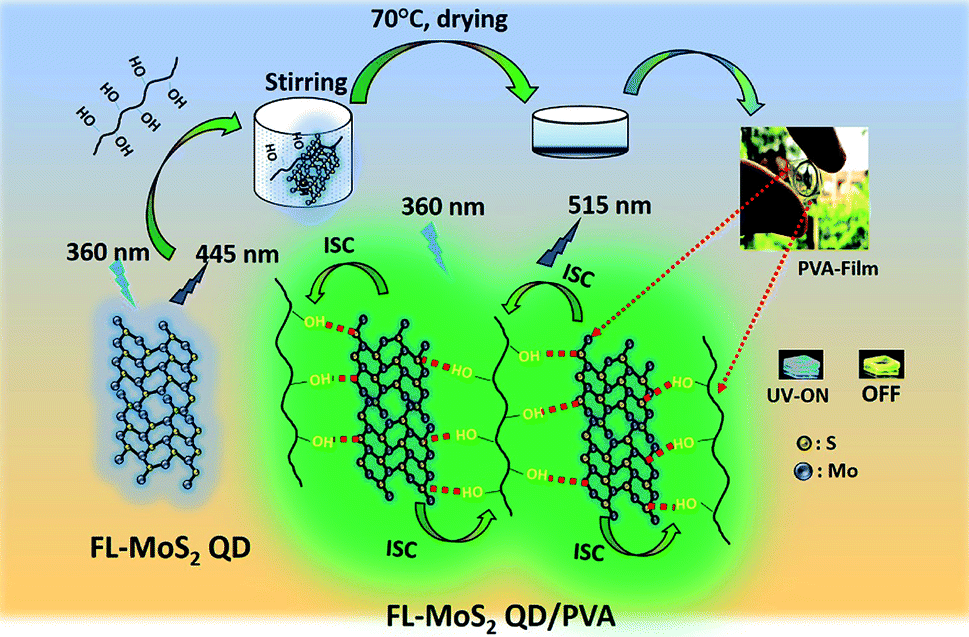
Previous studies have revealed that the loading of nanomaterial-related PMs into a PVA matrix is one of the most effective strategies to suppress oxygen-induced triplet–triplet quenching due to strong hydrogen bonding between them.32–34 Additionally, MoS2 nanosheets have been found to strongly bind to PVA through the formation of O–H⋯S hydrogen bonding.35–37 Inspired by these observations, we proposed that atomically thin two-dimensional (2D) QDs, an example being few-layer molybdenum disulfide (FL-MoS2) QDs, could behave as efficient PMs in PVA matrices through the formation of multiple hydrogen-bonding interactions; this might expand the applicability of 2D QDs in the field of phosphorescence. Herein, we report a straightforward method to fabricate 2D QD-based PMs by incorporating FL-MoS2 QDs into PVA matrices (Fig. 1). Upon the excitation with UV light (365 nm), the FL-MoS2 QD/PVA composites showed a green afterglow with an ultralong phosphorescence lasting time of 3.0 s and a phosphorescence quantum yield (PQY) of 20%. The phosphorescence lasting time of the FL-MoS2 QD/PVA composites is much longer than those of CDs (0.5 s), few-layer tungsten disulfide quantum dots (FL-WS2 QDs; 1.5 s), Rhodamine 6G (0.5 s), and Rhodamine B (1.5 s) in PVA matrices. Further studies found that an elevated temperature increased the phosphorescence lasting time of the FL-MoS2 QD/PVA composites in the range from room temperature to 70 °C due to heat-induced removal of water moisture from PVA matrices. This feature enables the FL-MoS2 QD/PVA composites to serve as a heat-induced phosphorescence “turn-on” sensor based on the naked-eye detection of their phosphorescence lasting time. Additionally, the FL-MoS2 QD/PVA composites integrated with the carbon dot (CD)/PVA composites or fluorescent organic dyes were applied as a security ink for anti-counterfeiting and encryption applications. This observation could pave a new route for expanding the scope of 2D QD-based PMs and their applications.
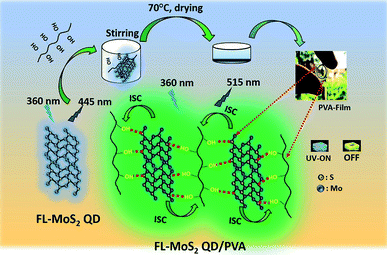 |
| | Fig. 1 Schematic illustration of the preparation of the FL-MoS2/PVA composites through hydrogen bonding between the FL-MoS2 QDs and PVA. | |
Results and discussion
Phosphorescent nanomaterials and organic dyes in PVA matrices
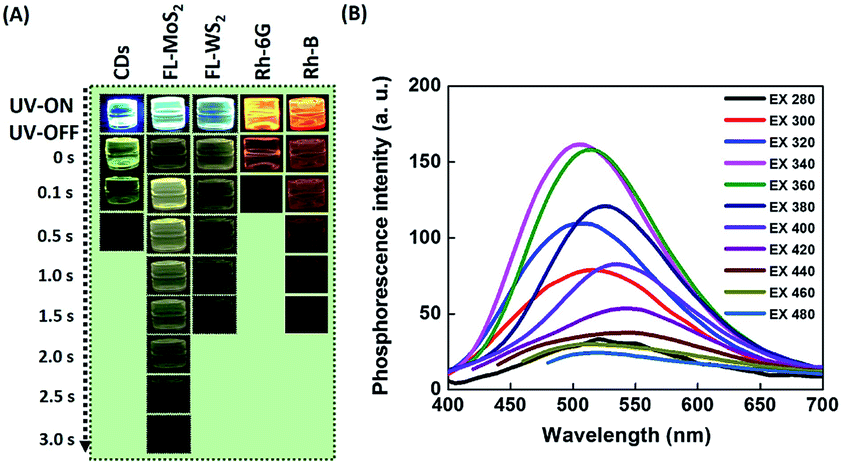
A series of PMs and organic dyes, including the FL-MoS2 QDs, FL-WS2 QDs, N,S-doped CDs, Rhodamine 6G, and Rhodamine B, were compared to investigate the differences in their phosphorescence lasting times in PVA matrices. Before the start of the test, we determined the morphology of the selected nanomaterials. Transmission electron microscopy (TEM) images display that the average sizes of the FL-MoS2 QDs, FL-WS2 QDs, and N,S-doped CDs were 3.64 ± 0.82, 3.90 ± 0.71 nm, and 3.51 ± 0.63 nm, respectively (Fig. S1, ESI†). The FL-MoS2 QDs and FL-WS2 QDs in TEM images appeared as dark spots, reflecting their few-layer thickness. Moreover, the topographic heights of the individual FL-MoS2 QDs, FL-WS2 QDs, and N,S-doped CDs, determined by AFM images, were approximately estimated to be 2.0, 1.8, and 2.8 nm, in sequence (Fig. S2, ESI†). Given that the heights of monolayer MoS2 and WS2 nanosheets were separately reported to be 0.82 and 0.93 nm, the thicknesses of FL-MoS2 and FL-WS2 QDs were suggested to be two or three layers. By contrast, the height of the N,S-doped CDs is almost the same as their diameter measured in the TEM image, reflecting that the N,S-doped CDs are spherical in shape. We next examined the optical properties of the three kinds of nanodots. The FL-MoS2 and FL-WS2 QDs separately have excitonic absorption peaks in the range of 270–340 nm accompanied by a long tail in the visible region (Fig. S3, ESI†), which is the characteristic of earlier published FL-MoS2 and FL-WS2 QDs.38,39 Moreover, the N,S-doped CDs exhibited two characteristic absorption peaks at 240 and 340 nm, which arise from π–π* and n–π* transitions at the CD core and edges, respectively (Fig. S3, ESI†). Fig. S4 (ESI†) reveals the fluorescence spectra of the FL-MoS2 QDs, FL-WS2 QDs and N,S-doped CDs at different excitation wavelengths. These three nanodots all exhibited excitation-dependent emission behavior, signifying that various degrees of edge sites and surface defects could occur on individual nanodots' surface.40,41 We rule out that this excitation-dependent emission behavior comes from different-sized nanodots due to their narrow size distribution. When excited at 360, 320, and 380 nm, the strongest fluorescence peaks of the FL-MoS2 QDs, FL-WS2 QDs, and N,S-doped CDs appeared at 445, 406, and 432 nm, respectively. At the same excitation wavelengths, the fluorescence quantum yields (QYs) of the FL-MoS2 QDs, FL-WS2 QDs, and N,S-doped CDs were separately determined to be 6.9, 3.46, and 72.2% with fluorescence lifetimes of 5.87, 1.9, and 8.9 ns.
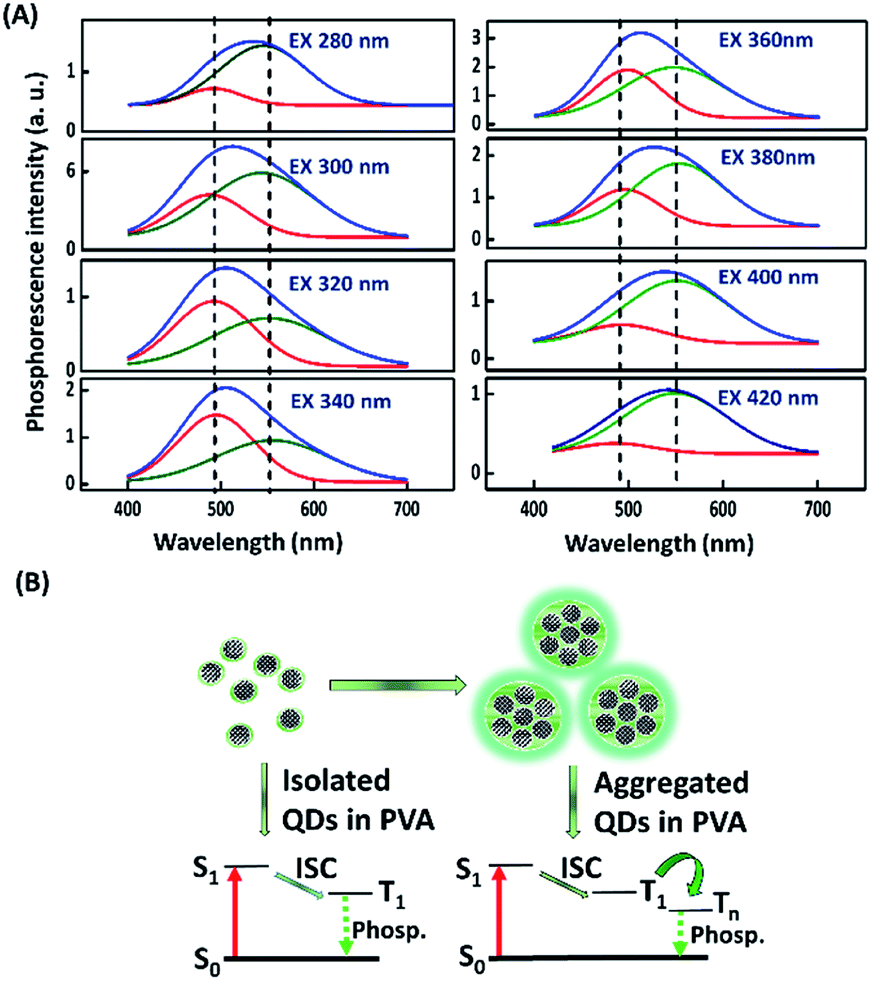
After switching off the excitation light (365 nm), the three present nanodots showed no phosphorescence in an aqueous solution which could be due to the oxygen-triggered triplet–triplet quenching and vibrational dissipation.42–44 Once prepared in PVA matrices, the three present nanodots emitted blue fluorescence under the excitation of UV light (365 nm) and green phosphorescence after removing the UV lamp (Fig. 2A). Moreover, Rhodamine B and Rhodamine 6G in PVA matrices were selected as reference standards to compare the three present nanodots' phosphorescence lasting time. The room-temperature phosphorescence of the FL-MoS2 QDs, FL-WS2 QDs, N,S-doped CDs, Rhodamine B, and Rhodamine 6G in PVA matrices lasted 3.0, 1.5, 0.5, 0.5, and 1.5 s, in sequence. It is suggested that the FL-MoS2 and FL-WS2 QDs offer sufficient sulfur sites to form intermolecular hydrogen bonds with the hydroxyl groups of PVA molecules. This hydrogen bond-induced fixation not only restrains the vibrational motion of the nanodots but also forms a protective layer against the phosphorescence quenchers of oxygen.20,32,34 To confirm the existence of hydrogen bonding between the FL-MoS2 QDs and PVA matrices, we examined the Fourier-transform infrared spectroscopy (FT-IR) spectra of the PVA polymers, the FL-MoS2 QDs, and the FL-MoS2 QD/PVA composites. Fig. S5 (ESI†) shows that incorporating FL-MoS2 QDs into PVA matrices led to a slight shift in the absorption band of hydroxyl groups from 3250 to 3275 cm−1 and an increase in their absorption intensity. This feature results from the formation of hydrogen bonding between the sulfur atoms of the FL-MoS2 QDs and the hydroxyl groups of the PVA matrices.36,45 Deng et al. showed that the phosphorescence lasting time of ethylenediaminetetraacetic acid-based CDs in PVA matrices was about 1 s.20 Gou et al. demonstrated that the embedding of 2,4-diaminotoluene, 2,6-diaminotoluene, 2,4-diaminobenzenesulfonic acid, and m-phenylenediamine-related CDs into PVA matrices resulted in the production of phosphorescent materials with a long lasting time (4 s).46 In comparison to our present N,S-doped CDs and previously reported CDs, it is concluded that the FL-MoS2 QD/PVA and FL-WS2 QD/PVA composites provided a comparable phosphorescence lasting time. Fig. 2B displays that the FL-MoS2 QD/PVA composites exhibited excitation-dependent phosphorescence from 280 to 480 nm. Under the excitation wavelength of 340 nm, the FL-MoS2 QD/PVA composites' strongest phosphorescence peak was located at 500 nm, and their PQY was determined to be 20%. This excitation-dependent phosphorescence was also observed in ethylenediamine-based CD/PVA47 and 1,2,4-triaminobenzene-related CD/molten salt composites.48 Table S1 (ESI†) shows that the PQY of the FL-MoS2 QD/PVA composites is comparable to that of the previously reported organic small molecules, CDs, and metal–organic frameworks. This excitation-dependent phosphorescence behaviour mechanism could be attributed to the mixing of the phosphorescence of isolated and aggregated FL-MoS2 QDs in PVA matrices. In confirmation of this assumption, the FL-MoS2 QD/PVA composites' phosphorescence spectra were deconvoluted using a multi-Gaussian function. At different excitation wavelengths, the FL-MoS2 QD/PVA composites' obtained phosphorescence bands were split into two Gaussian-shaped peaks at 490 and 550 nm which separately originate from the isolated and aggregated FL-MoS2 QDs (Fig. 3A). In contrast to the isolated FL-MoS2 QDs, the aggregated FL-MoS2 QDs exhibited relatively long-wavelength phosphorescence due to van der Waals stacking via inter-nanodot interaction in PVA matrices.49 Such a stacking structure could stabilize the triplet-excited state of the FL-MoS2 QDs and result in another triplet excited state (Fig. 3B). The TEM image of the FL-MoS2 QD/PVA composites confirms the aggregation of the FL-MoS2 QDs in PVA matrices (Fig. S6, ESI†). The air stability of the FL-MoS2 QD/PVA composites was tested by evaluating their phosphorescence lasting time in ambient air and a N2 gas environment. Fig. S7 (ESI†) reveals that the phosphorescence lasting time of the FL-MoS2 QD/PVA composites in ambient air resembled that in a N2 gas environment.
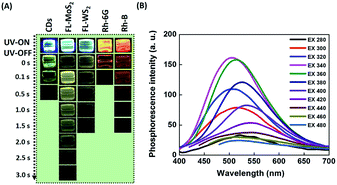 |
| | Fig. 2 Phosphorescence of nanomaterial- and organic dye-doped PVA composites. (A) Time-evolution photographs of PVA matrices incorporated with CDs, FL-MoS2 QDs, FL-WS2 QDs, Rhodamine 6G (Rh-6G), and Rhodamine B (Rh-B) before and after turning off a UV lamp (365 nm) at ambient temperature. (B) Phosphorescence spectra of the FL-MoS2/PVA composites as a function of excitation wavelength at ambient temperature. | |
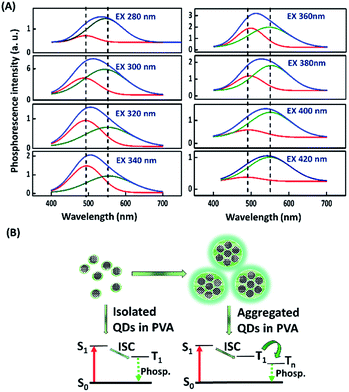 |
| | Fig. 3 Excitation-dependent phosphorescence of the FL-MoS2 QD/PVA composites. (A) Gaussian curve-fitting analysis of phosphorescence spectra (blue curve) of the FL-MoS2 QD/PVA composites at excitation wavelengths of 280, 300, 320, 340, 360, 380, 400, and 420 nm. Two Gaussian-shaped peaks represent isolated (red curve) and aggregated (green curve) FL-MoS2 QDs. (B) Proposed mechanism of excitation-dependent phosphorescence of the FL-MoS2 QD/PVA composites occurring from isolated and aggregated FL-MoS2 QDs in PVA matrices. “S”, “T”, “Phosp.”, and “ISC” correspond to “singlet state”, “triplet state”, “phosphorescence”, and “intersystem crossing”, in sequence. | |
Temperature sensor
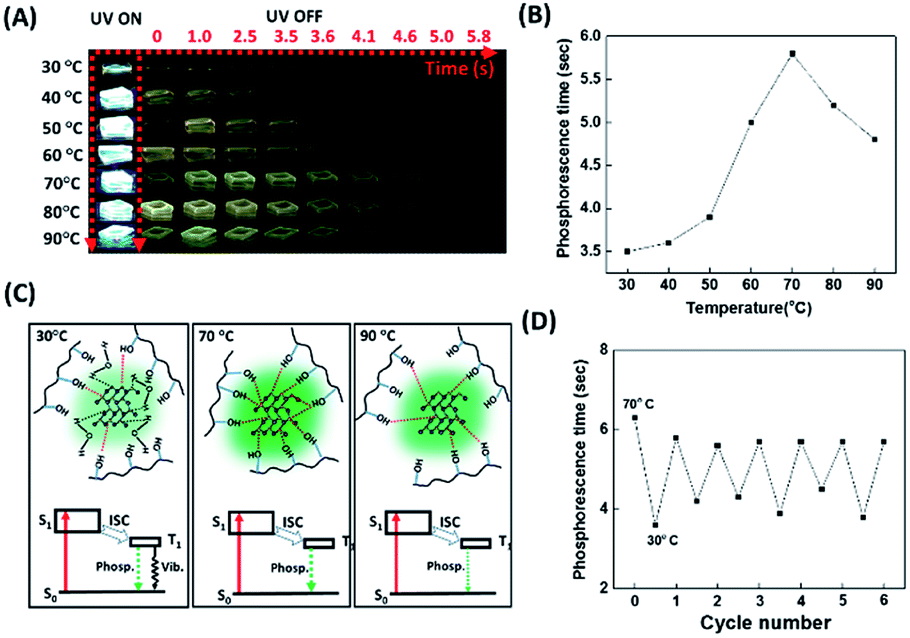
Bao et al. disclosed that water molecules dramatically quenched the phosphorescence of the CD/PVA composites due to the formation of hydrogen bonding between water molecules and the CD surface groups.50 Water molecules can interfere with the hydrogen bonding between PVA and the CDs, weakening the formation of a rigid structure that protects the CDs from the phosphorescence quencher of oxygen. Therefore, it is reasonable that the phosphorescence lasting time of the CD/PVA composites could be enhanced by increasing the temperature as a result of heat-induced dehydration of the composites. However, the increased temperature can also break the hydrogen bonding between PVA and the CDs. We point out that the competition between the heat-mediated breakdown of hydrogen bonding and heat-induced dehydration of water is a critical factor affecting the CD/PVA composites' phosphorescence lasting time. Considering that the hydrogen bonding also exists between the sulfur atoms of the FL-MoS2 QDs and the hydroxyl groups of PVA matrices, we suggest that the phosphorescence lasting time of the FL-MoS2 QD/PVA composites could be profoundly connected with the temperature variation. Fig. 4A reveals the photo images associated with the FL-MoS2 QD/PVA composites' phosphorescence lasting time as a function of temperature from 30 to 90 °C. Intriguingly, the FL-MoS2 QD/PVA composites' phosphorescence lasting time was exponentially increased as the temperature was varied from 30 to 70 °C (Fig. 4B). We propose that water from the air moisture could migrate into the FL-MoS2 QD/PVA composites at low temperatures, resulting in the creation of hydrogen bonding with the FL-MoS2 QDs together with the disruption of the rigid bonding between the FL-MoS2 QDs and PVA matrices. As a result, oxygen molecules easily penetrate the composites and then quench their phosphorescence. By contrast, the increased temperature can trigger water removal from the composites and rigidify their structure, leading to an increase in the phosphorescence lasting time. Thus, the phosphorescence lasting time of the FL-MoS2 QD/PVA composites at high temperatures is longer than that at low temperatures. However, heat above 70 °C can weaken or break the hydrogen bonding between the FL-MoS2 QDs and PVA matrices, facilitating oxygen molecules’ entry into the composites. The possible mechanism underlying the relationship between the heating temperature and phosphorescence lasting time of the composites is illustrated in Fig. 4C. An FT-IR technique was implemented to demonstrate the existence of water (from the air moisture) in the FL-MoS2 QD/PVA composites at low temperature (Fig. S5, ESI†). The apparent band at 1658 cm−1 of the FL-MoS2 QDs was identified to be the bending vibration of adsorbed water,36 reflecting the adsorption of water (from the air moisture) on the surface of the FL-MoS2 QDs. Although the absorption intensity of the same band was remarkably reduced in the FL-MoS2 QD/PVA composites, a few water molecules are still entrapped in the FL-MoS2 QD/PVA composites. This finding supports the notion mentioned above. Fig. 4D shows that the temperature sensing with the FL-MoS2 QD/PVA composites was completely reversible during 6 cycles of heating and cooling between 30 and 70 °C. In short, the FL-MoS2 QD/PVA composites can serve as a high-temperature sensor with remarkable advantages of naked-eye detection and good reusability. In confirmation of the FL-MoS2 QD/PVA composites' unique temperature sensing capabilities, the phosphorescence lasting time of the N,S-doped CD/PVA composites was also monitored at different temperatures. As indicated in Fig. S8 (ESI†), the phosphorescence lasting time of the N,S-doped CD/PVA composites at first was increased with a temperature increase from 30 to 40 °C as a result of the removal of water from the composites, and then it progressively decayed in the temperature range of 40–90 °C due to the breakdown of hydrogen bonding between CDs and PVA matrices. It is evident that the N,S-doped CD/PVA composites are poorly suitable for high-temperature sensing.
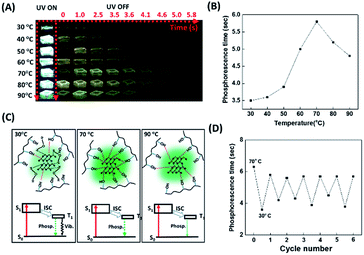 |
| | Fig. 4 Temperature-sensitive FL-MoS2 QD/PVA composites. (A) Time-evolution photographs of the FL-MoS2 QD/PVA composites before and after turning off the UV lamp at different temperatures. (B) Phosphorescence lasting time of the FL-MoS2 QD/PVA composites as a function of temperature from 30 to 90 °C. (C) Schematic illustration of the phosphorescence process occurring in the FL-MoS2 QD/PVA composites at different temperatures. “S”, “T”, “Phosp.”, “Vib.”, and “ISC” correspond to “singlet state”, “triplet state”, “phosphorescence”, “vibrational relaxation”, and “intersystem crossing”, in sequence. (D) Reversible phosphorescence response of the FL-MoS2 QD/PVA composites through the alternating change between 70 °C and 30 °C. | |
Anticounterfeiting and encryption
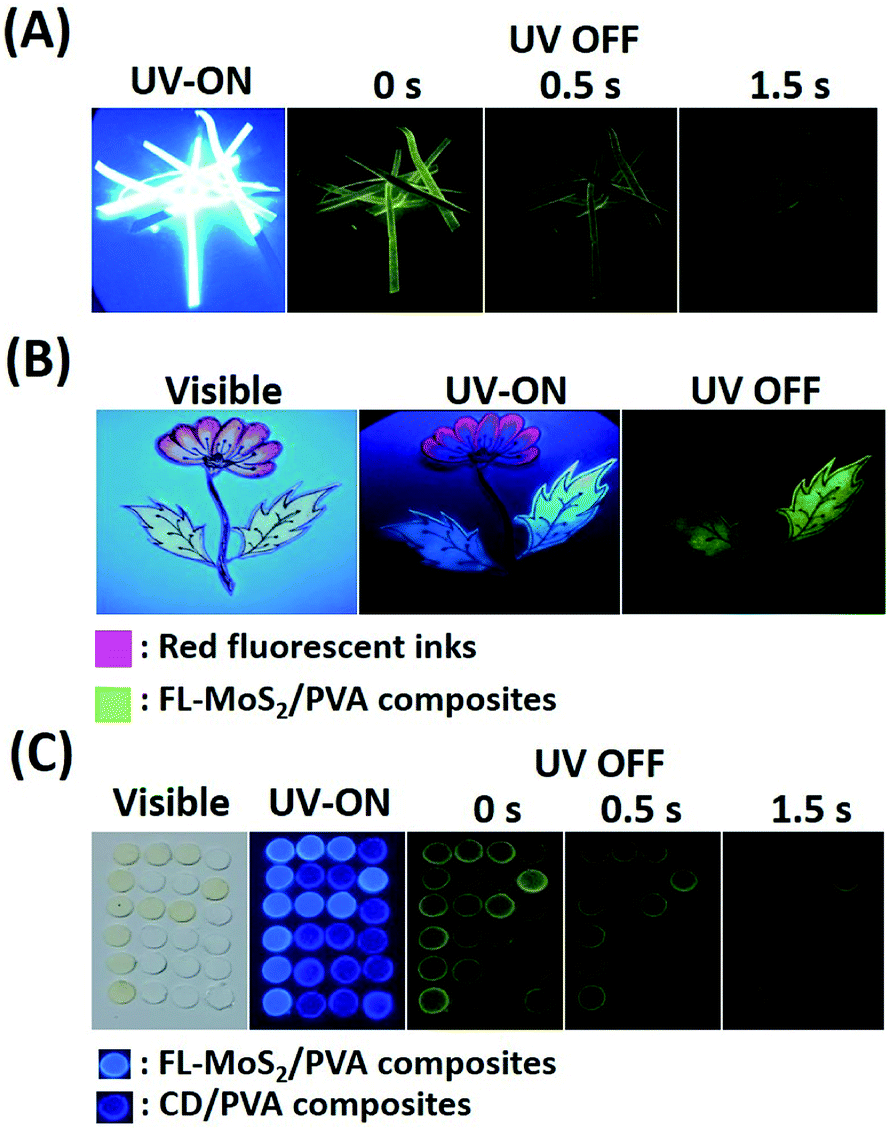
The long phosphorescence of the FL-MoS2 QD/PVA composites enables the preparation of unique anti-counterfeiting inks. In order to know whether the FL-MoS2 QD/PVA composites still emit phosphorescence on Whatman filter paper, paper strips were immersed in the composites for 2 min and then dried in an oven. Fig. 5A indicates that the FL-MoS2 QD/PVA composite-embedded paper strips exhibited bright blue color under 365 nm UV illumination and turned green after switching off the excitation light. This color change is easily visualized for a couple of seconds. It is evident that the Whatman filter paper rarely influences the fluorescence and phosphorescence properties of the FL-MoS2 QD/PVA composites. The flower and leaf parts were separately encoded with red fluorescent inks and the FL-MoS2 QD/PVA composites to fabricate the dual-mode security pattern of a flowering plant. Fig. 5B shows that the flower and leaves displayed red and blue colors under UV illumination, respectively. After turning off the UV light, only a green color was bright enough to be observed with the naked eye. This finding reflects that the FL-MoS2 QD/PVA composites can serve as a new anti-counterfeiting ink to fabricate dual-model security patterns. Since the phosphorescence lasting time of the CD/PVA composites differs from that of the FL-MoS2 QD/PVA composites, we expect this difference to be beneficial for time-dependent security protection. Four operation scenarios, including “light on”, “light off at the beginning, “light off after 0.5 s”, and “light off after 1 s”, were selected to conduct the encryption algorithm. Fig. 5C presents the fabrication of the letter “P” pattern with the FL-MoS2/PVA composites and other parts with the CD/PVA composites. The letter “P” and other parts appeared sky blue and indigo blue fluorescent colors, respectively, at an excitation wavelength of 365 nm. After switching off the excitation source at the beginning, the letter “P” with phosphorescence emission was not easily recognized by the naked eye due to the interference from the CD/PVA composites' phosphorescence. Since the phosphorescence of the FL-MoS2 QD/PVA composites is longer than that of the CD/PVA composites, the letter “P” was visualized only after 1 s. These findings point out that the FL-MoS2 QDs combined with fluorescent organic dyes and N,S-doped CDs have considerable potential to serve as a security ink for added information protection.
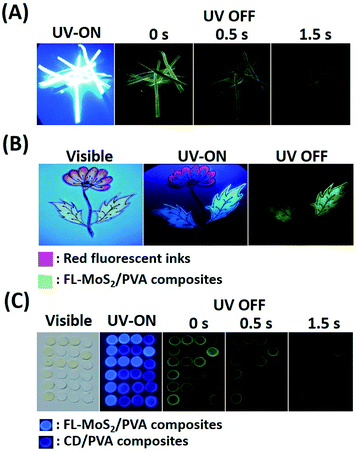 |
| | Fig. 5 (A) Time-evolution photographs of the FL-MoS2 QD/PVA composite-embedded paper strips before and after turning off the UV lamp at ambient temperature. (B) Photographs of a flowering plant pattern made from red fluorescent inks and the FL-MoS2 QD/PVA composites under visible light irradiation, under UV lamp excitation (365 nm), and after turning off the UV lamp. (C) Photographs of a rectangular array of small circular pieces made from the CD/PVA and FL-MoS2 QD/PVA composites under visible light irradiation, under UV lamp excitation (365 nm), and after turning off the UV lamp for 0, 0.5, and 1 s. | |
Conclusions
For the first time, we have demonstrated that the incorporation of the FL-MoS2 QDs into PVA matrices provides long phosphorescence with a satisfactory QY and they function as a phosphorescence platform for naked-eye and turn-on detection of temperature variations. Further studies reveal that the strong hydrogen-bond networks of the rich sulfur sites of the FL-MoS2 QDs with the hydroxyl groups of PVA molecules are the determining factors for generating long phosphorescence and fabricating phosphorescence-based temperature sensors. Importantly, the FL-MoS2 QD/PVA composites have been successfully utilized as a security ink for added information protection. In contrast to earlier published PVA-based PMs, the FL-MoS2 QD/PVA composites offer distinct advantages: (1) the FL-MoS2 QDs provide abundant sulfur sites and a flat surface to interact with PVA molecules, resulting in the formation of strong hydrogen-bond networks; (2) the phosphorescence lasting time and intensity of the FL-MoS2 QD/PVA composites were enhanced with increasing the temperature from 30 to 70 °C; (3) the FL-MoS2 QD/PVA composites exhibit a long phosphorescence lasting time and satisfactory PQY without the additional surface modification. We suggest that the concept associated with the phosphorescence of the FL-MoS2 QDs in PVA matrices is expected to apply to other 2D transition metal dichalcogenides.
Experimental section
Chemicals
Citric acid, L-cysteine, cetyltrimethylammonium bromide, N,N-dimethylformamide, and PVA (M.W. 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) were bought from Sigma-Aldrich (Louis, MO, USA). Rhodamine B and Rhodamine 6G were purchased from Acros (Geel, Belgium). MoS2 and WS2 powder were ordered from Alfa-Aesar (Ward Hill, MD, USA). Ultrapure water purified with a Milli-Q system (Millipore, Hamburg, Germany; 18.2 MΩ cm) was employed in all of the experiments.
000) were bought from Sigma-Aldrich (Louis, MO, USA). Rhodamine B and Rhodamine 6G were purchased from Acros (Geel, Belgium). MoS2 and WS2 powder were ordered from Alfa-Aesar (Ward Hill, MD, USA). Ultrapure water purified with a Milli-Q system (Millipore, Hamburg, Germany; 18.2 MΩ cm) was employed in all of the experiments.
Synthesis of the FL-WS2 QDs and FL-MoS2 QDs
The preparation of the FL-WS2 QDs was conducted according to a previously published procedure.39 Layered WS2 materials (0.53 g) and cetyltrimethylammonium bromide (0.25 g) are added to 50 mL of deionized water, followed by ultrasonication (Elmasonic E60H, Elma, Singen, Germany) in an ice bath (150 W) for 3 h. Subsequently, the resulting solution was centrifuged at 10 °C (3000 rpm, 15 min) to remove the unexfoliated layered WS2 materials. The surfactant-exfoliated nanosheets were centrifuged at 10 °C (12000 rpm, 45 min) and re-dispersed in deionized water three times. The homogeneous suspension of WS2 nanosheets in deionized water (10 mL) was transferred into a 60 mL Teflon autoclave, followed by thermal treatment at 180 °C for 6 h. After cooling to room temperature, the unreacted nanosheets were removed by centrifugation at 10 °C (12000 rpm, 30 min). The obtained supernatant corresponds to the FL-WS2 QDs. In the preparation of FL-MoS2 QDs,51,52 layered MoS2 materials (10 mg) were mixed with N,N-dimethylformamide (10 mL) in a serum bottle. The sealed bottle was incubated in an ice bath and treated with ultrasonication at 150 W for 36 h. Subsequently, the resultant solution was transferred to a tube and centrifuged at 10 °C (6000 rpm, 30 min). The collected supernatant was vigorously stirred in an oil bath at 145 °C for 8 h, followed by centrifugation at 10 °C (12000 rpm, 60 min). The obtained supernatant corresponding to the FL-MoS2 QDs was dried in an oven and then dispersed in deionized water. The resultant FL-WS2 and FL-MoS2 QDs were stored in a refrigerated box at 4 °C. The mass concentrations of the FL-WS2 and FL-MoS2 QDs were estimated to be 1.5 and 1 mg mL−1, respectively. The morphology and composition of the as-prepared QDs were characterized by TEM (JEM-2100, JEOL, Japan) integrated with energy-dispersive X-ray spectroscopy (EDS; Oxford, XMax 80T) attachment at an accelerating voltage of 200 keV and an FT-IR spectrometer (Nicolet 6700, Thermo Electron Corporation, Madison, WI). Moreover, their optical properties and color chromaticity were measured using a JASCO V-670 spectrophotometer (JASCO, Tokyo, Japan) and an Edinburgh FS5 spectrofluorometer (Edinburgh Instruments Ltd., UK).
Synthesis of the CDs
The CDs were obtained via hydrothermal pyrolysis treatment of L-cysteine and citric acid. L-Cysteine (300 mg) and citric acid (100 mg) were mixed in 500 μL of deionized water. The resultant solution was transferred to a silica crucible and subjected to heat in a muffle furnace at 260 °C for 3 min. After cooling to room temperature, the obtained ash (10 mg) was dispersed in 10 mL of ultrapure water, sonicated (150 W) at ambient temperature for 5 min, and then centrifuged at 12000 rpm for 10 min. The supernatant corresponding to the as-made CDs was stored in a glass container at 4 °C.
Preparation of PM-doped PVA composites
1 g of PVA powder was added to 10 mL of deionized water. The PVA powder was entirely dissolved by heating at 90 °C for 2 h under gentle stirring. After cooling to room temperature, a homogeneous PVA solution (100 mg mL−1, 200 μL) was diluted with deionized water (200 μL) and then blended with 50 μL of 0.1 mg mL−1 nanomaterials (CDs, FL-MoS2 QDs, and FL-WS2 QDs) and organic dyes (Rhodamine 6G and Rhodamine B). The resultant mixture (200 μL) was carefully cast into a mold and then cured at 70 °C in an oven until drying. The formed composites were exposed under UV light (365 nm), and their phosphorescence was imaged as a function of time at different temperatures after switching off the UV light.
Fabrication of the security pattern
An aqueous solution of the FL-MoS2 QDs (1 mg mL−1, 200 μL) was mixed with a PVA solution (100 mg mL−1, 400 μL). The resultant mixture was incubated with a filter paper strip (Whatman No 3, diameter 110 mm, Maidstone, England, UK) for 10 min at ambient temperature. Subsequently, the as-made strips were dried in an oven at 70 °C. Besides, the FL-MoS2 QD/PVA composites and red fluorescent inks were separately added into an empty pen to draw the dual-mode security pattern of a flowering plant. The maximum excitation and emission wavelengths of red fluorescent inks were found to be 290 and 565 nm, respectively. For obtaining the letter “P” pattern that emitted green phosphorescence, Whatman filter papers were cut into small circular pieces and then incorporated with either the MoS2 QD/PVA composites or the CD/PVA composites based on the above-discussed procedure. The CD/PVA and MoS2 QD/PVA composite-containing circular pieces were arranged in rows and columns to form a rectangular array.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
We would like to thank the Ministry of Science and Technology of Taiwan (MOST107-2113-M-110-013-MY3) and NSYSU-KMU Joint Research Project (NSYSUKMU 109-P002) for the financial support of this study.
Notes and references
- J. Yuan, R. Chen, X. Tang, Y. Tao, S. Xu, L. Jin, C. Chen, X. Zhou, C. Zheng and W. Huang, Chem. Sci., 2019, 10, 5031–5038 RSC.
- K. Kanosue and S. Ando, ACS Macro Lett., 2016, 5, 1301–1305 CrossRef CAS.
- Z. Chen, K. Y. Zhang, X. Tong, Y. Liu, C. Hu, S. Liu, Q. Yu, Q. Zhao and W. Huang, Adv. Funct. Mater., 2016, 26, 4386–4396 CrossRef CAS.
- X. Lin, J. Liu, M. Tian, Y. Bai, Y. Bao, T. Shu, L. Su and X. Zhang, Anal. Chem., 2020, 92, 6785–6791 CrossRef CAS.
- S. Jing, Y. Zhao, R.-C. Sun, L. Zhong and X. Peng, ACS Sustainable Chem. Eng., 2019, 7, 7833–7843 CrossRef CAS.
- R. Tian, S.-M. Xu, Q. Xu and C. Lu, Sci. Adv., 2020, 6, eaaz6107 CrossRef CAS.
- R. Kabe, N. Notsuka, K. Yoshida and C. Adachi, Adv. Mater., 2016, 28, 655–660 CrossRef CAS.
- X. Wang, H. Ma, M. Gu, C. Lin, N. Gan, Z. Xie, H. Wang, L. Bian, L. Fu and S. Cai, Chem. Mater., 2019, 31, 5584–5591 CrossRef CAS.
- Y. Tao, Z. An, P. Chen, C. Xu, R. Chen, W. Huang and K. Pu, Adv. Mater., 2017, 29, 1606665 CrossRef.
- H. Zhang, J. Jiang, P. Gao, T. Yang, K. Y. Zhang, Z. Chen, S. Liu, W. Huang and Q. Zhao, ACS Appl. Mater. Interfaces, 2018, 10, 17542–17550 CrossRef CAS.
- Y.-C. Liang, S.-S. Gou, K.-K. Liu, W.-J. Wu, C.-Z. Guo, S.-Y. Lu, J.-H. Zang, X.-Y. Wu, Q. Lou and L. Dong, Nano Today, 2020, 34, 100900 CrossRef CAS.
- M. S. Kwon, Y. Yu, C. Coburn, A. W. Phillips, K. Chung, A. Shanker, J. Jung, G. Kim, K. Pipe and S. R. Forrest, Nat. Commun., 2015, 6, 1–9 Search PubMed.
- S. M. Borisov, R. Pommer, J. Svec, S. Peters, V. Novakova and I. Klimant, J. Mater. Chem. C, 2018, 6, 8999–9009 RSC.
- C. Grewer and H.-D. Brauer, J. Phys. Chem., 1994, 98, 4230–4235 CrossRef CAS.
- D. Lee, O. Bolton, B. C. Kim, J. H. Youk, S. Takayama and J. Kim, J. Am. Chem. Soc., 2013, 135, 6325–6329 CrossRef CAS.
- W. Shi, J. Yao, L. Bai and C. Lu, Adv. Funct. Mater., 2018, 28, 1804961 CrossRef.
- K. Jiang, Y. Wang, X. Gao, C. Cai and H. Lin, Angew. Chem., Int. Ed., 2018, 57, 6216–6220 CrossRef CAS.
- L. Xiao, Y. Wu, J. Chen, Z. Yu, Y. Liu, J. Yao and H. Fu, J. Phys. Chem. A, 2017, 121, 8652–8658 CrossRef CAS.
- Z. Yang, Z. Mao, X. Zhang, D. Ou, Y. Mu, Y. Zhang, C. Zhao, S. Liu, Z. Chi and J. Xu, Angew. Chem., Int. Ed., 2016, 55, 2181–2185 CrossRef CAS.
- Y. Deng, D. Zhao, X. Chen, F. Wang, H. Song and D. Shen, Chem. Commun., 2013, 49, 5751–5753 RSC.
- Y. Gao, H. Zhang, Y. Jiao, W. Lu, Y. Liu, H. Han, X. Gong, S. Shuang and C. Dong, Chem. Mater., 2019, 31, 7979–7986 CrossRef CAS.
- X. Yang and D. Yan, Chem. Sci., 2016, 7, 4519–4526 RSC.
- J. Han, W. Feng, D. Y. Muleta, C. N. Bridgmohan, Y. Dang, G. Xie, H. Zhang, X. Zhou, W. Li and L. Wang, Adv. Funct. Mater., 2019, 29, 1902503 CrossRef.
- J. Wei, B. Liang, R. Duan, Z. Cheng, C. Li, T. Zhou, Y. Yi and Y. Wang, Angew. Chem., Int. Ed., 2016, 55, 15589–15593 CrossRef CAS.
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng and X. Liu, Nat. Mater., 2015, 14, 685–690 CrossRef CAS.
- S. Haneder, E. Da Como, J. Feldmann, J. M. Lupton, C. Lennartz, P. Erk, E. Fuchs, O. Molt, I. Münster and C. Schildknecht, Adv. Mater., 2008, 20, 3325–3330 CrossRef CAS.
- Q. Zhao, C. Huang and F. Li, Chem. Soc. Rev., 2011, 40, 2508–2524 RSC.
- X. Lu, J. Zhang, Y.-N. Xie, X. Zhang, X. Jiang, X. Hou and P. Wu, Anal. Chem., 2018, 90, 2939–2945 CrossRef CAS.
- B. Del Secco, L. Ravotto, T. V. Esipova, S. A. Vinogradov, D. Genovese, N. Zaccheroni, E. Rampazzo and L. Prodi, Photochem. Photobiol. Sci., 2019, 18, 2142–2149 RSC.
- M. Sugiuchi, J. Maeba, N. Okubo, M. Iwamura, K. Nozaki and K. Konishi, J. Am. Chem. Soc., 2017, 139, 17731–17734 CrossRef CAS.
- K. Jiang, Y. Wang, Z. Li and H. Lin, Mater. Chem. Front., 2020, 4, 386–399 RSC.
- Y. Chen, J. He, C. Hu, H. Zhang, B. Lei and Y. Liu, J. Mater. Chem. C, 2017, 5, 6243–6250 RSC.
- J. Li, B. Wang, H. Zhang and J. Yu, Small, 2019, 15, 1805504 CrossRef.
- K. Jiang, L. Zhang, J. Lu, C. Xu, C. Cai and H. Lin, Angew. Chem., Int. Ed., 2016, 55, 7231–7235 CrossRef CAS.
- S. Thakur, P. Bandyopadhyay, S. H. Kim, N. H. Kim and J. H. Lee, Composites, Part A, 2018, 110, 284–293 CrossRef CAS.
- S.-K. Kim, J. J. Wie, Q. Mahmood and H. S. Park, Nanoscale, 2014, 6, 7430–7435 RSC.
- Z. Lu, L. Sun, G. Xu, J. Zheng, Q. Zhang, J. Wang and L. Jiao, ACS Nano, 2016, 10, 5237–5242 CrossRef CAS.
- W. Gu, Y. Yan, C. Zhang, C. Ding and Y. Xian, ACS Appl. Mater. Interfaces, 2016, 8, 3107–3114 CrossRef.
- Y. Yan, C. Zhang, W. Gu, C. Ding, X. Li and Y. Xian, J. Phys. Chem. C, 2016, 120, 12170–12177 CrossRef CAS.
- X. Ding, F. Peng, J. Zhou, W. Gong, G. Slaven, K. P. Loh, C. T. Lim and D. T. Leong, Nat. Commun., 2019, 10, 41 CrossRef CAS.
- N. Dhenadhayalan, K.-C. Lin, R. Suresh and P. Ramamurthy, J. Phys. Chem. C, 2016, 120, 1252–1261 CrossRef CAS.
- K. Jiang, Y. Wang, C. Cai and H. Lin, Chem. Mater., 2017, 29, 4866–4873 CrossRef CAS.
- K. Jiang, L. Zhang, J. Lu, C. Xu, C. Cai and H. Lin, Angew. Chem., Int. Ed., 2016, 55, 7231–7235 CrossRef CAS.
- J. Tan, R. Zou, J. Zhang, W. Li, L. Zhang and D. Yue, Nanoscale, 2016, 8, 4742–4747 RSC.
- D. L. Howard and H. G. Kjaergaard, Phys. Chem. Chem. Phys., 2008, 10, 4113–4118 RSC.
- H. Gou, Y. Liu, G. Zhang, Q. Liao, X. Huang, F. Ning, C. Ke, Z. Meng and K. Xi, Nanoscale, 2019, 11, 18311–18319 RSC.
- K. Jiang, Y. Wang, C. Cai and H. Lin, Adv. Mater., 2018, 30, 1800783 CrossRef.
- C. Wang, Y. Chen, T. Hu, Y. Chang, G. Ran, M. Wang and Q. Song, Nanoscale, 2019, 11, 11967–11974 RSC.
- H. Jin, B. Baek, D. Kim, F. Wu, J. D. Batteas, J. Cheon and D. H. Son, Nano Lett., 2017, 17, 7471–7477 CrossRef CAS.
- X. Bao, E. V. Ushakova, E. Liu, Z. Zhou, D. Li, D. Zhou, S. Qu and A. L. Rogach, Nanoscale, 2019, 11, 14250–14255 RSC.
- D. Haldar, A. Ghosh, S. Bose, S. Mondal, U. K. Ghorai and S. K. Saha, Opt. Mater., 2018, 79, 12–20 CrossRef CAS.
- W. Gu, Y. Yan, C. Zhang, C. Ding and Y. Xian, ACS Appl. Mater. Interfaces, 2016, 8, 11272–11279 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00730g |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b and
Wei-Lung
Tseng
b and
Wei-Lung
Tseng