
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design of bimetallic photocatalysts based on plasmonically-derived hot carriers
Jorge U.
Salmón-Gamboa
 *,
Mayela
Romero-Gómez
,
Diane J.
Roth
,
Alexey V.
Krasavin
,
Pan
Wang
,
Wayne
Dickson
and
Anatoly V.
Zayats
*
*,
Mayela
Romero-Gómez
,
Diane J.
Roth
,
Alexey V.
Krasavin
,
Pan
Wang
,
Wayne
Dickson
and
Anatoly V.
Zayats
*
Department of Physics and London Centre for Nanotechnology, King's College London, Strand, London WC2R 2LS, UK. E-mail: jorge.salmon@kcl.ac.uk
First published on 18th December 2020
Abstract
Hot carriers generated by plasmonic excitations have recently opened up new avenues in photocatalysis. The transfer of these energetic carriers to adjacent molecules can promote chemical transformations that are important for hydrogen generation by water splitting, CO2 reduction and degradation of organic pollutants. Here, we have developed and optimised a plasmonic hot-carrier catalytic system based on silica nanoparticles decorated with plasmonic gold nanoparticles as a source of hot carriers, equipped with platinum nanoclusters as co-catalyst for the enhancement of hot-carrier extraction. The latter plays a triple role by providing: a surface favourable for molecular adsorption; hot-electron generation near the nanoclusters due to field enhancement effects and electron momentum relaxation facilitating the electron transfer across the metal surface, exactly where molecules are adsorbed. The combination of plasmonic and catalytic metals in nano-heterostructured devices provides a new platform for photocatalytic processes and is of significant interest for future solar-based clean technologies.
Coherent collective oscillations of free electrons in metal nanoparticles are referred to as localised surface plasmons (LSPs) and can be excited under external light illumination. They may decay either radiatively into light or non-radiatively, producing electrons in a conduction band with energies above the Fermi level up to the LSP energy, termed hot carriers. These hot carriers can relax via electron–phonon coupling, locally heating the particle, but some can reach the nanoparticle surface and experience transitions into unoccupied levels of acceptor adsorbates, hence triggering chemical reactions.1–9 One of the interesting examples of plasmon-induced photochemistry is the dissociation of a hydrogen molecule H2 at room temperature and atmospheric pressure,10,11 a chemical process which is immensely difficult to achieve due to the high dissociation energy of ∼2.3 eV, and which had for long only been demonstrated at high temperatures. This proved that well-designed plasmonic nanostructures are able to perform difficult chemical reactions in ambient conditions, which is of paramount interest for catalytic applications. Other important examples of applications of hot carriers include hydrogen production by water splitting, ammonia production, and water purification from organic pollutants by semiconductor–noble metal hybrids.12,13 While for redox reactions the role of the electron transfer is clear, other types of reactions may benefit from both the carrier transfer and the local temperature increase mediated by the LSP excitation.14
In contrast to conventional catalysts, which accelerate chemical reactions by forming a temporary intermediate and/or modifying the activation energy upon adsorption, the efficient electron transfer takes place to the reacting agents when a reaction is catalysed with transient hot-electrons. Therefore, the ideal design of a plasmonic catalyst should provide (A) efficient light absorption in a plasmonic material, which is relatively easy to achieve near a plasmonic resonance; (B) a low radiative decay of the surface plasmons and their efficient non-radiative decay in hot carriers via Landau damping; (C) an easy route for the hot carriers to leave a plasmonic metal and interact with the species adsorbed on a surface; and therefore, (D) a surface with an appropriate surface energy for efficient adsorption. Conditions A–C can be optimised for a required illumination wavelength, which determines the energy distribution of hot carriers by the choice of a plasmonic metal, as well as the size and topology of the nanoparticles. Condition D is difficult to satisfy using plasmonic metals for which the electronic d-bands are far in energy from the Fermi level, where the bonding and anti-bonding states of an adsorbate are fully occupied, and, therefore, no metal–adsorbate interactions are likely to occur. At the same time, in typical catalytic metals, a d-band is close to the Fermi energy, promoting effective adsorption, but for the same reason these metals are poor plasmonic metals in the visible and near-infrared spectral ranges. Therefore, a combination of plasmonic and catalytic nanoparticles is needed to satisfy all conditions A–D. Moreover, such combination may also improve condition B by creating an additional electromagnetic field enhancement near the nanoparticle junctions, where hot-carriers are generated more efficiently. If nm-sized, such catalytic nanoparticles also provide good conditions for effective hot-carrier extraction by supplying an additional momentum to the electron through its scattering on the nanoparticle. In this way, plasmonically-derived hot carriers are most efficiently generated and extracted at the locations where the adsorbates are positioned. The other considerations for the photocatalytic efficiency are to provide large surface area, so that nanoparticles are preferable to a planar surface, and to avoid semiconductor materials as, for example, a substrate that could trap the excited hot carriers. As opposed to metal–semiconductor heterostructures exhibiting interfacial Schottky barriers through which hot carriers have to tunnel, no Schottky barrier is present in bi-metallic nanostructures, leading to the availability of more hot carriers for catalytic processes.
In this work, based on the above principles, we design an optimised topology of a catalytic platform and study the hot-carrier generation and extraction in gold nanoparticles decorated with platinum nanoclusters. SiO2–Au–Pt hetero-nanoparticles were used for efficient plasmon-induced photodegradation of methylene blue (MB) dye, outperforming other photocatalysts. The applications of the proposed plasmonic hetero-nanostructures are not limited to photodegradation of organic pollutants for water purification, but can also be used for other photocatalytic processes by tailoring their design and hot-carrier energy to maximise the targeted chemical reaction, such as hydrogen generation by water splitting,15,16 CO2 reduction17 or bacterial disinfection.18
Results
Particle morphology
The architecture and surface morphology of SiO2–Au(3)–Pt+++ hetero-nanoparticles is shown in Fig. 1 (see Table 2 in Methods for the nanoparticle nomenclature). Silica (SiO2) particles were used as an inert substrate to maximise the surface area and prevent nanoparticle agglomeration. Silica nanospheres with an average diameter of 150 nm are uniformly covered with Au nanoparticle with a diameter of 12 ± 1.2 nm. EDS measurements of the SiO2–Au(3)–Pt+++ hetero-nanoparticles (Fig. 1d–h) reveal an elemental composition of Pt and Au of 2.34% and 11.20%, respectively, which is in agreement with the nominal values from the fabrication conditions in Table 2. Pt metal selectively deposits on the surface of the Au nanoparticles, not accumulating elsewhere, and, if sufficient number of Pt ions are reduced, the Au nanoparticles can become completely coated with Pt.19 | ||
| Fig. 1 TEM images of synthesised nanoparticles. (a and b) A single nanoparticle of (a) SiO2–Au(3) and (b) SiO2–Au(3)–Pt. (c) Statistical analysis of SiO2–Au(3)–Pt+++ nanoparticles. Inset shows the size distribution of Au(3) nanoparticles with an average diameter of 12 nm. In order to obtain the images, the solution containing the nanostructures was dried on a TEM grid which led to their partial aggregation. (d–g) High resolution elemental maps and (h) EDS spectrum of SiO2–Au(3)–Pt+++ hetero-nanoparticles. | ||
Optical properties
The studied heterostructures feature a LSP resonance in the visible spectral range derived from the response of the Au nanoparticles, which can be tuned by varying their size. The LSP resonance width and position position also depends on the concentration of the Au nanoparticles on the surface of the SiO2 nanoparticles20 (Fig. 2a). The extinction of the naked silica cores, without Au, shows no resonance over the visible spectrum (Fig. 2a). The LSP resonance of Au-seeded SiO2 nanoparticles is located at around 500 nm. As the size of the Au nanoparticles is increased, the LSP extinction peak red-shifts and increases in magnitude. The LSP linewidth becomes broader, signifying the increase of the Au particle size distribution during the growth. When Pt nanoclusters are introduced, an additional red shift of the resonance develops, along with a pronounced broadening of the LSP linewidth and an increase of the extinction over a broad spectral range (Fig. 2b). Pt nanoparticles themselves do not support LSPs in the visible spectral range. The LSP broadening can be attributed to additional LSP dephasing mechanisms arising from the presence of Pt nanoclusters. For comparison, the extinction of the SiO2–Pt nanoparticles without Au does not present any LSP resonance in the visible spectral range (Fig. 2b) and increases towards UV wavelengths, where Pt may support surface plasmons.21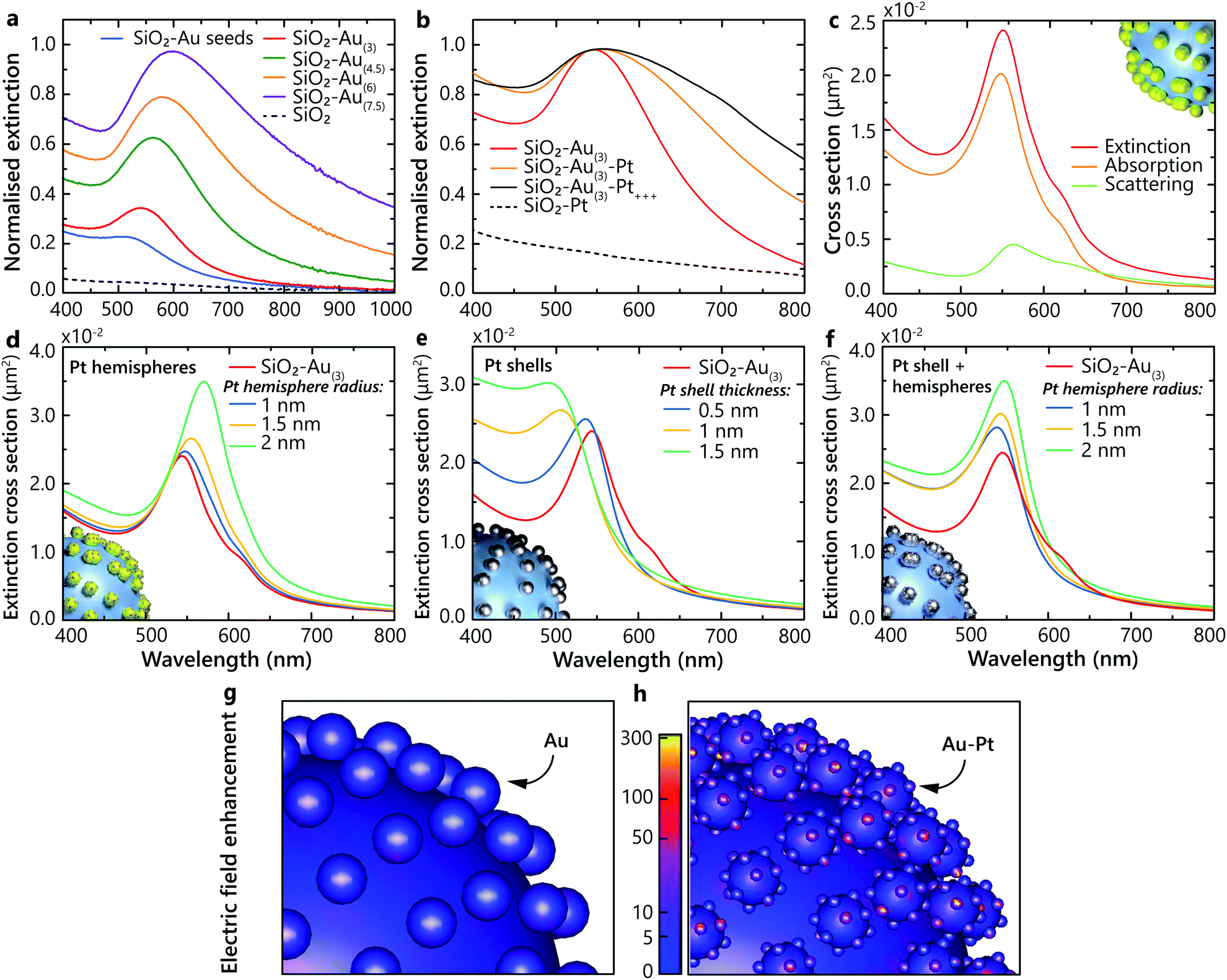 | ||
| Fig. 2 Optical properties of hetero-nanoparticles. (a) Experimentally measured extinction of SiO2–Au nanoparticles in water for different sizes of the Au nanoparticles together with the extinction of the SiO2 nanoparticles. (b) Extinction of SiO2–Au(3)–Pt nanoparticles in water for different Pt loading together with the extinction of SiO2–Pt nanoparticles. (c) Numerically simulated extinction, absorption and scattering cross-sections of a single SiO2–Au(3) nanoparticle in water (diameters of SiO2 and Au nanoparticles are 150 nm and 12 nm, respectively). (d–f) Extinction cross-section spectra of SiO2–Au(3)–Pt hetero-nanoparticles simulated with three different architectures: a SiO2–Au(3)–Pt nanoparticle with (d) Pt hemispheres of 1, 1.5 and 2 nm radii, (e) a smooth Pt shell with thicknesses of 0.5, 1 and 1.5 nm, and (f) combination of a Pt shell with thickness of 0.5 nm and Pt hemispheres of 1, 1.5 and 2 nm radii. All other parameters as in (c). (g and h) Electric field distribution for (g) SiO2–Au(3) and (h) SiO2–Au(3)–Pt+++ heterostructures simulated at their respective LSP resonances. Insets in (c and d) show schematics of the modelled architectures. The surface density of Au nanoparticles used in the simulations is the same as obtained from the TEM images in the experiment. | ||
To understand the observed optical properties, numerical simulations were performed on several architectures of hetero-nanoparticles. The simulated extinction spectra are in agreement with the experimental measurements, with the extinction maximum located around a wavelength of 550 nm for the SiO2–Au nanoparticles (Fig. 2a and c). As was mentioned above, the measured extinction spectra may exhibit a broad LSP resonance due to the nanoparticle size dispersion, whereas the simulations reproduce the homogeneous LSP linewidth with a narrower resonance peak. As expected for small plasmonic particles, the contribution of the scattering to the extinction is small compared to absorption (Fig. 2c).
Three different models were systematically studied in order to understand how the Pt nanoclusters form on the Au nanoparticles: (1) Pt hemispheres with a diameter from 2 to 4 nm on the surface of the Au nanoparticles, (2) a smooth and uniform Pt shell with a thickness from 0.5 to 1.5 nm on the surface of the Au nanoparticles and (3) the combination of the Pt shell of 0.5 nm in thickness and hemispheres with 1, 1.5 and 2 nm radii. In the case of Pt hemispheres formed on the surface of the Au nanoparticles, the plasmonic resonance red shifts as the Pt hemisphere size increases (Fig. 2d). This suggests a strong influence of the Pt nanocluster size on the optical properties of the nanoparticles. On the other hand, in the limit of the uniform Pt shell deposition, a strong blue shift of the resonance is observed with the increase of the shell thickness (Fig. 2e). Therefore, these two models cannot alone reproduce the experimentally observed behaviour. However, by combining these two models together, a satisfactory agreement between simulations and experiments can be achieved for the positions and trends of the observed LSP resonances (Fig. 2f). This suggests that, in addition to the thin layer of Pt formed after deposition on the Au nanoparticles, Pt nanoclusters are formed on the surface, providing a rough interface between Au and the surrounding aqueous environment.
Using the derived architecture of the hetero-nanoparticles, the electric field distributions excited at the LSP wavelength were calculated for the SiO2–Au(3) and SiO2–Au(3)–Pt+++ heterostructures. The local field intensity becomes stronger in the presence of Pt nanoclusters, compared to the bare Au nanoparticles excited at the resonance (Fig. 2g and h). Specifically, the electric field enhancement is strongest at the irregularities formed by the Pt nanoclusters on the surface. By tailoring the surface morphology of the plasmonic heterostructure, it is therefore possible to control the density of the field enhancement hot-spots. The field enhancement factor η, defined as η = |E|2/|E0|2, where E is the electric field at the surface of the nanostructure and E0 is the incident electric field, was calculated for both types of nanoparticles, at their corresponding LSP resonances. The enhancement at the surface of Au over the Au nanoparticle assembly on the SiO2–Au(3) nanoparticle is estimated to be approximately η = 45 ± 4. The corresponding enhancement factor at the surface of the Pt nanoclusters on the SiO2–Au(3)–Pt+++ heterostructure is significantly increased to approximately η = 276 ± 22. This field enhancement is associated with the sharp curvature of the nanoclusters and their nanosize scale. It has been recently demonstrated that spatial locations of the strongest electric field are responsible for the highest density of excited hot electrons and mark the areas where photochemical reaction rates are enhanced, triggering chemical transformations.22–24 Therefore, the Pt-decorated Au nanoparticles are beneficial compared to smooth Au nanoparticles in terms of the field enhancement and, thus, hot-carrier density. In the designed hetero-nanostructures, the hot carriers are generated near the Pt nanoclusters, exactly where they are needed to interact with adsorbates which preferentially attach to Pt.
Photocatalytic process
The hot carrier generation and extraction from the synthesised hetero-nanoparticles were investigated by observing the photocatalytic degradation of MB organic dye. During a photocatalytic processes with heterogeneous catalysts in an aqueous medium, reactive oxygen species (ROS) may be generated such as hydroxyl radical (·OH) and superoxide anion radical (·O2−). Both of them can chemically interact with organic molecules and break them down. The degradation of MB by heterogeneous photocatalysts was confirmed to be activated by generated ROS.25,26The formation of ·O2− and ·OH occurs due to the interaction of photogenerated charge carriers with the surrounding aqueous medium and requires electron transfer.27,28 The hot carriers interact with the adsorbed oxygen molecules and hydroxide ions as
| (O2)ads + e− → ·O2−, | (1) |
| (H2O ⇔ H+ + OH−)ads + h+ → H+ + ·OH. | (2) |
By monitoring the photocatalytic degradation process, the generation and extraction rates of hot-carriers can be evaluated. The MB degradation process can be described as
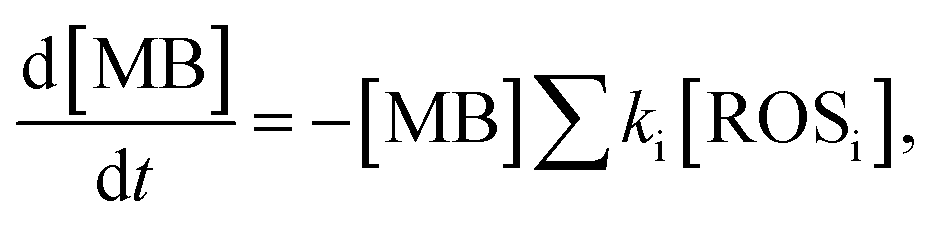 | (3) |
Assuming a dominant contribution of one type of ROS, its time dependent concentration, in addition to the right-hand term in eqn (3), includes a decay term related to a finite ROS lifetime τROS and the generation term R related to their creation by hot carriers:
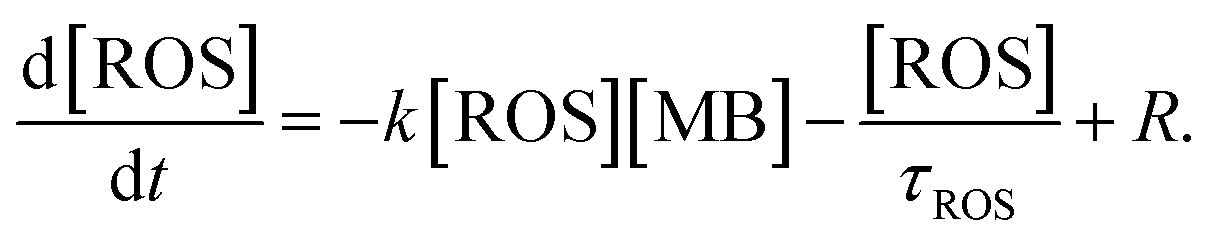 | (4) |
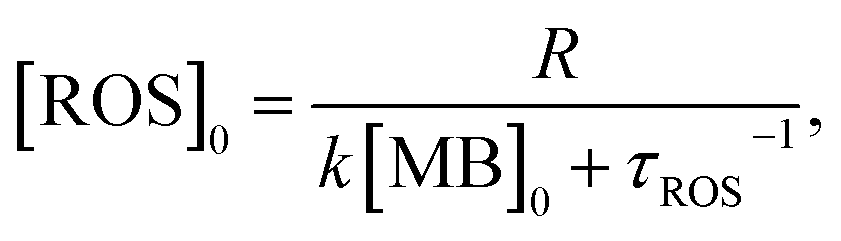 | (5) |
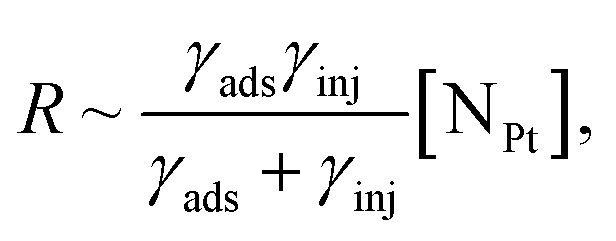 | (6) |
Finally, the hot-electron injection rate is determined by the light absorption rate γabs in the nanoparticles, the efficiency of the hot carrier generation ηhc, which depends on the illumination wavelength and is determined by the Landau damping, in the case of the LSP absorption, or by the interband absorption, and the efficiency of the hot-electron transfer to the adsorbate ηextr:
| γinj = γabsηhcηextr. | (7) |
Effect of metal loading
The experiments show that the hot-carriers excitation and their transfer to adsorbates strongly depend on the hetero-nanoparticle geometrical parameters, and the size of both Au nanoparticles and Pt nanoclusters affect the overall degradation process (Fig. 3). The Au nanoparticles efficiently absorb light and generate hot-carriers. Different Pt loading affects the population of hot carriers that are able to transfer from Pt into the adsorbates on the Pt surface. These processes were studied under illumination with light in a 556–566 nm spectral band, specifically selected to excite the LSP in all studied hetero-nanoparticles (Fig. 3a). The average power was set to avoid direct degradation of MB by photolysis and not to induce temperature-related effects. In this wavelength range, the absorption of all types of studied nanoparticles is similar and for the used pulsed laser power, the numerical estimates show that one laser pulse changes the temperature of the nanoparticles negligibly. The pulse repetition rate is low enough to ensure temperature relaxation between the pulses (50 ns interval between pulses, compared with a few ns nanoparticle-environment temperature relaxation time).30 While the local temperature can be higher, we do not expect that temperature effects significantly influence the studied processes, which require electron transfer. The advantage of the ultrashort pulsed illumination is a significant peak rate of hot-electrons, while maintaining low average power and energy transferred to water.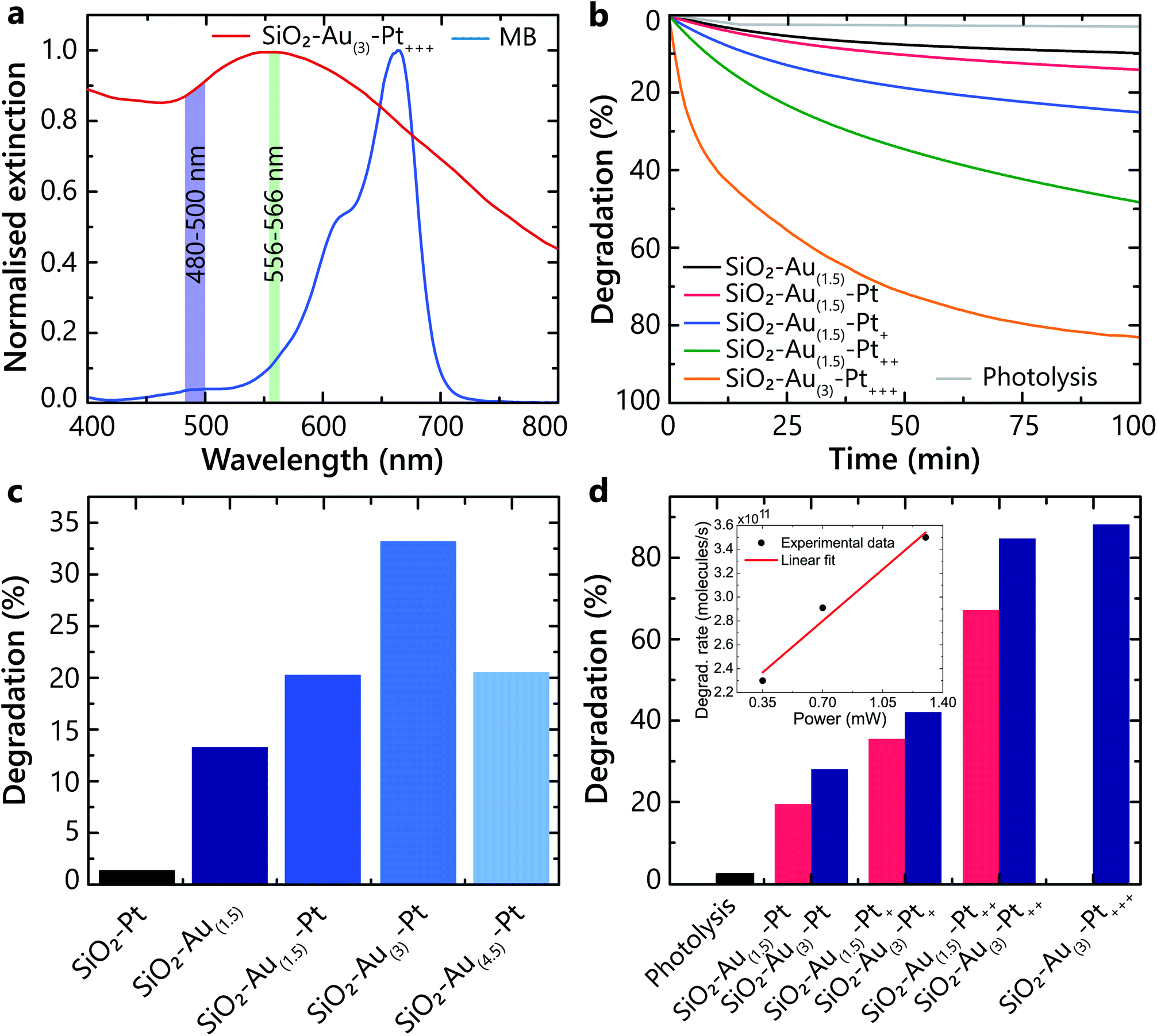 | ||
| Fig. 3 The effect of Au and Pt loading. (a) Normalised optical extinction of MB and the SiO2–Au(3)–Pt+++ heterostructures measured in water. (b) Time evolution of the MB absorbance during the illumination for hetero-nanoparticles with different Pt loading. (c and d) Photocatalytic degradation dependence on (c) Au and (d) Pt loading. For photodegradation, the pulsed laser illumination was used in a 556–566 nm spectral range with an average power of 0.35 mW. Inset in (d) shows the initial degradation rate dependence on the illuminating power for SiO2–Au(3)–Pt nanoparticles. | ||
Firstly, to demonstrate that the plasmonically generated hot carriers trigger the photochemical reaction (and it is not provoked solely by the intrinsic catalytic properties of Pt), a direct comparison of the photocatalytic performance between the SiO2–Pt and SiO2–Au–Pt nanoparticles was carried out. Even though the smaller (3–8 nm) Pt particles provide a larger surface area which is favourable for adsorption, the SiO2–Pt nanoparticles have a negligible effect on photodegradation compared to those containing Au (Fig. 3c). Hot carriers originate from Au, as a consequence of the non-radiative LSP decay, and are aided by a few nm size Pt clusters to efficiently transfer to the adsorbed molecules, triggering the chemical transformations.
On the other hand, it can be seen that the degradation rate increases as the Au nanoparticles size increases, but only up to a certain value (Fig. 3c). The optimal Au content was found to be that of the SiO2–Au(3)–Pt heterostructure (∼12 nm Au nanoparticle diameter, ∼550 nm LSP peak), and a further increase in the Au size was detrimental to the degradation process. This can be explained by the fact that for larger Au nanoparticles, the nonradiative relaxation would become less prominent and the hot-electron generation rate would decrease.31 Additionally, the hot electrons excited inside large nanoparticles cannot easily reach the surface and, instead, thermalise in the bulk, not contributing to the ROS generation. This is in agreement with the theoretical results for hot-carrier generation rates in spherical plasmonic nanoparticles predicting the optimal diameter within the range of 12–16 nm.32
To maximise the photocatalytic activity of the heterostructures, the Au nanoparticle size was chosen to be ∼12 nm, which corresponds to the sample SiO2–Au(3), and with this architecture, the effect of Pt loading was investigated under the same conditions as above. As a control experiment, photolysis (molecular dissociation due to photon absorption) was monitored and resulted in only 3.7% degradation of MB (Fig. 3d). An increase of the degradation rate was observed with the increase of the Pt loading (Fig. 3b and d). This results from an increased number of hot carriers generated and extracted through the Pt nanoclusters, enhancing the ROS generation. The highest performance was reached in the case of the SiO2–Au(3)–Pt+++ hetero-nanoparticles, which have optical properties similar to those in Fig. 2h. Even though the addition of Pt onto the Au nanoparticles is beneficial for the photocatalytic activity, a limit of performance is reached, as a thicker Pt layer hinders hot carriers from reaching the surface and prevent their interaction with the adsorbates (Fig. 3).
Improved photocatalytic performances of the plasmonic hetero-nanostructures come from a triple role of the Pt nanoclusters for the hot-electron-based photocatalyst efficiency, as they provide a surface favourable for molecular adsorption, enhance the hot-electron generation near the nanoclusters due to the field enhancement effects, and ensure the electron momentum relaxation facilitating the electron transfer across the metal surface, exactly where molecules are adsorbed.
Considering the MB degradation dynamics (Fig. 3b), the hot-carrier generation and extraction efficiency can be evaluated. The analysis shows that the degradation kinetics over the period studied (t = 100 min) are not described by a single exponential dependence as would be expected for a pseudo-first-order reactions, indicating the need for a full description of the second-order reactions (eqn (3) and (4)). This description includes in principle other ROS contributions to the degradation (e.g., the generation of ·OH via hot-holes) and a possible competition of O2 and the solvent for the same active sites (influencing γads); additionally, after significant MB depletion, the reaction may become diffusion limited.33 However, at the initial stages of the degradation (t < 10 min), the dependences can be approximated by a single exponential and the reaction rate can be determined for the dominant ROS. Taking into account the illumination conditions (the rate of photon absorption of approximately 4 × 1013 s−1), the initial number of MB molecules in the experiment (5.6 × 1015) and the degradation rate during this period (t < 10 min), the efficiency of the MB molecule degradation using optimised hetero-nanoparticles can be estimated of the order of 10% per photon absorbed by the nanoparticles. This is the lower bound of the hot-carrier generation and extraction efficiency ηhcηextr as it assumes that all extracted carriers generate ROS and all produced ROS live long enough to react with the MB molecules. It also assumes that one ROS is sufficient to initiate the degradation process. The observed reaction rates have a linear dependence on the intensity of the illuminating light in the studied power range (Fig. 3d, inset), which, together with eqn (5) and (6), shows that γinj, being much smaller than γads, is the major factor defining the photocatalytic process. It should be noted that this linear dependence should be expected only for sufficiently high intensities of the illuminating light to establish the equilibrium ROS concentration (eqn (4) and (5)), and its observation confirms that this has been achieved in the experiments.
For the considered Au nanoparticle size (∼12 nm), and the illumination wavelength at the LSP resonance, the nonradiative decay of the excited LSP is predominant and, through the Landau damping, generates hot-carriers. The detailed estimates of the competition between the Landau damping and other nonradiative LSP-decay processes34 in Au–Pt nanoparticles are beyond the scope of this work, and for the order of magnitude estimations of a lower bound of the hot-carrier extraction efficiency, the hot-carrier generation efficiency can be taken as ∼1. Under these assumptions, the lower bound of the hot-electron extraction efficiency is estimated to be of the order of 10% for the optimized configuration of the Au–Pt hetero-nanoparticles and 20 times lower for the least performing configuration in Fig. 2b. Theoretically, the extraction efficiency can be relatively easily evaluated considering a hot-carrier transport in Au–Pt core–shell nanoparticles, which for the Au and Pt effective electron masses and Fermi energies, gives approximately 50%,34,35 which is of the same order of magnitude as the estimates from the experiment. The measured efficiency – the ratio of a number of extracted hot electrons to a number of absorbed photons – is higher than those reported for other types of nanoparticle architectures36–38 and comparable to the hot-carrier injection efficiency in metal–semiconductor nanostructures.39
Wavelength dependence
In order to determine whether hot carriers derived from the LSP decay are legitimately causing the chemical reaction, two degradation experiments with SiO2–Au(3)–Pt+++ samples were carried out at two different irradiation wavelength ranges 480–500 nm and 556–566 nm (Fig. 3a). The light in the wavelength range of 480–500 nm mostly excites interband transitions from d- to sp-bands in Au. The illumination in the 556–566 nm spectral range corresponds to hot-carrier generation predominantly by the non-radiative LSP decay in the Au nanoparticles (Fig. 2a). Under the 480–500 nm illumination, only ∼25% of the MB degraded (Fig. 4a). In contrast, under the LSP excitation, more than 50% degradation was observed over the same time period (Fig. 4b). This confirms more effective hot-carrier generation and extraction through the LSP decay process. Through this pathway, hot electrons are excited in the conduction band of Au via the Landau damping and, after transferring to Pt, participate in the superoxide generation. In contrast, inter-band excitation in Au results also in hot-hole excitation,32 which may contribute to hydroxyl generation. Please note that in different plasmonic materials, different pathways of hot carries generation maybe more efficient.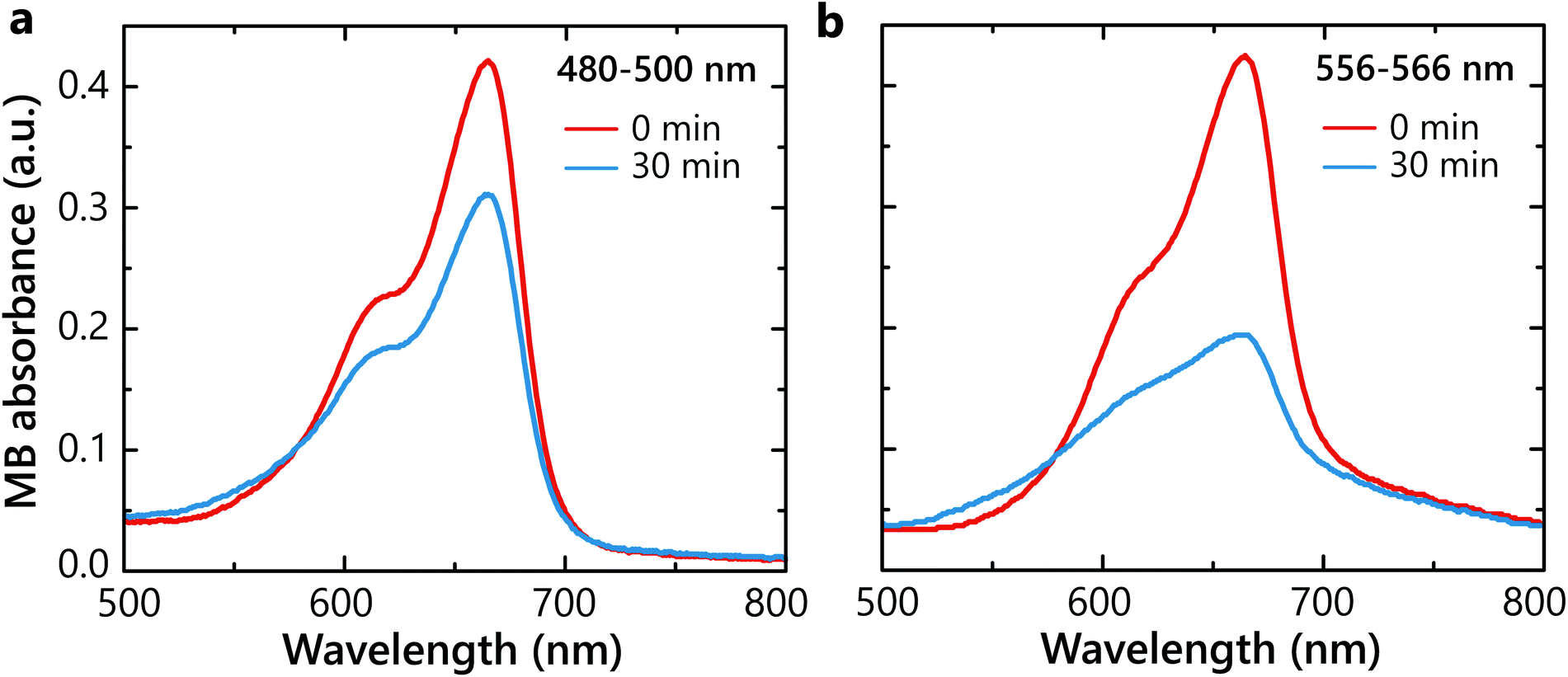 | ||
| Fig. 4 The effect of the illumination wavelength. MB absorbance spectra before and after 30 min photocatalytic degradation in water with SiO2–Au(3)–Pt+++ hetero-nanoparticles under the illumination in the spectral range of (a) 480–500 nm and (b) 556–566 nm with the same average absorbed power. | ||
Effect of electron and hole scavengers
In order to further understand the role of hot electrons and hot holes in the photocatalytic process, the degradation kinetics in the presence of hole and electron scavengers were studied. Scavengers are sacrificial reagents that interact and consume the generated hot-holes40 or hot-electrons.41 For this study, degradation experiments were carried out in the presence of methanol CH3OH (MeOH) as the hole scavenger or silver nitrate (AgNO3) as the electron scavenger, respectively. Methanol can be oxidised by a two step two-hole process, forming formaldehyde (CH2O) and formic acid (HCOOH).42 Silver nitrate is an ionic compound breaking into Ag+ and NO3− ions when dissolved in water. The silver cations will act as electron acceptors and will be reduced into Ag0. Even though molecular oxygen O2 itself is an electron acceptor, Ag+ cations possess a higher electronegativity due to their cationic nature and scavenge electrons more efficiently.Without the scavengers, the control experiment showed a degradation of MB of about 56% after 30 min (Fig. 5a). When hot holes were scavenged, the MB degradation decreased to a minor extent, resulting in the total degradation of 41% (Fig. 5b). In contrast, when hot electrons were scavenged, the reaction became completely deactivated (Fig. 5c). Thus, oxygen interaction with plasmonically-generated hot electrons constitute the dominating pathway for the ROS generation and the MB degradation. Specifically, O2 is reduced by one-electron reduction into a superoxide radical on the surface of Pt and, in turn, ·O2− species interact with the adjacent MB molecules, breaking them down.
 | ||
| Fig. 5 The effect of different environments. (a–c) MB absorbance spectra before and after 30 min photocatalytic degradation in water (a) without added scavengers and in the presence of (b) hole scavenger MeOH and (c) electron scavenger AgNO3. (d) Time evolution of the MB absorbance during the illumination for SiO2–Au(3)–Pt+++ hetero-nanoparticles in glycerol (red line) and water (blue line). The photodegradation experiments were performed under the 556–566 nm illumination. | ||
Solvent effect
The impact of solvents with different oxygen solubilities was studied in order to confirm the role of ROS derived from O2 molecules in the solvent. Glycerol, known for having a substantially low oxygen solubility43 and DI water, with a higher oxygen solubility,44 were compared as solvents in the photocatalytic mixtures. No photodegradation effect was observed in glycerol for up to 60 min of illumination (Fig. 5d). At the same time, more than 70% degradation was observed with water. With these results, it was concluded that MB molecules were degraded in their vast majority by the generated ·O2− species, with hot electrons as intermediaries. This result is in agreement with other studies on photocatalytic degradation of MB, where generated ·O2− species were considered as the reason for degradation of organic molecules.Conclusions
Based on the formulated requirements for plasmon-enhanced photocatalysts, Au-decorated silica nanoparticles covered with Pt nanoclusters were synthesised and optimised for the enhancement of hot carrier generation and extraction at the active catalytic sites for plasmonically enhanced photocatalysis. The design of the heterostructured architecture was guided by considerations of the specifics of hot-carrier excitation and extraction, and conditions for facilitating their interaction with adsorbates on a surface. The size of the Au nanoparticles was chosen to promote the LSP-enabled light absorption and hot-electron generation due to the non-radiative Landau damping of the LSPs. The design provides an opportunity to tune the energy of the required hot-carriers by controlling the Au nanoparticle size. The Pt nanoclusters produce the field enhancement hot spots, leading to higher population of hot carriers than in other regions of the surface, and deliver the efficient electron scattering, facilitating the electron extraction process. At the same time, the Pt surface is advantageous for adsorbing molecules to be affected by the hot carriers. These key factors responsible for the enhanced photocatalytic action were experimentally demonstrated by studying the impact of Pt loading on MB photodegradation in the presence of hetero-nanoparticles. It was confirmed from the TEM and EDS measurements, as well as the optical characterisation of the best performing photocatalytic heterostructures, that small Pt clusters or irregularities on the surface of the Au nanoparticles are crucial for the enhanced performance. The optimal design included SiO2–Au(3)–Pt+++ hetero-nanoparticles with ∼12 nm Au nanoparticles and ∼3 nm Pt nanoclusters, enabling the most efficient hot-carrier generation through the Landau damping of the Au-related LSPs and their most efficient extraction through Pt nanoclusters, where oxygen molecules are adsorbed.The developed photocatalyst outperforms some of the most recent and popular photocatalysts used for MB degradation (Table 1). For fair comparison of the photocatalysts of different nature, the metrics considered is the number of degraded molecules per gram of catalyst. Since not all the papers report the illumination conditions, it was not always possible to compare the performance in terms of the illumination power used. The plasmonic catalyst developed in this work (SiO2–Au(3)–Pt+++) seems to outperform other nanocatalysts, with a degradation rate of 1.8 × 1017 molecules per g per s achieved with the Au(3)–Pt+++ hetero-nanoparticles under 4.5 × 10−4 W cm−2 illumination (the estimate considers the degradation up to 40%, before significant molecule depletion when diffusion starts limiting the degradation rate (Fig. 3)). The lowest power density in the list with the narrowest illumination range of only 10 nm was used. The bimetallic Au–Pt nanostars system shows a performance close to our non-optimised nanoparticles in terms of the considered metrics, but it was not possible to further compare the efficiency because the power density was not reported in ref. 25.
| Material | Reference | Light source | Degraded MB, molecules per g per s |
|---|---|---|---|
| Au–Pt nanostars | 25 | Xe lamp, white light | 6.2 × 1016 |
| NiWO4–ZnO–NRGO | 36 | 250 W Hg lamp, 200–400 nm | 2.5 × 1016 |
| ZnO/Ag/Ag2WO4 | 37 | 50 W LED lamp, white light | 5.2 × 1014 |
| TiO2/ZnO | 38 | 9.5 × 10−2 W cm−2, white light | 6.0 × 1014 |
| SiO2–Au(3)–Pt+++ | This work | 4.5 × 10−4 W cm−2, 556–566 nm | 1.8 × 1017 |
Not only MB, but all organic pollutants sensitive to superoxide anion species can also be degraded by the designed photocatalyst. The proposed plasmonic heterostructures are not limited to water purification from organic dyes only, but could also be used for bacterial disinfection since superoxide reactive species kill bacteria.
The combination of plasmonic and catalytic metals provides important impact on hot-carrier dynamics and extraction processes. The bimetallic character of the nanoparticles, their size and the condition of Pt-coated surface are important factors for optimisation of the photocatalyst. By engineering an appropriate plasmonic nanostructure in conjunction with the a choice of catalytic metal suitable for a given reaction, it is possible to maximise a reaction rate due to hot-electron excitation and transfer. The developed approach allows for optimisation and exploitation of plasmonically generated hot carriers in a wide range of photocatalytic applications.
Methods
Chemicals
The following chemicals were used to produce the precursor solutions needed to synthesise the heterostructures: 3-aminopropyltriethoxysilane (APTES, 99%), tetraethyl orthosilicate (TEOS, 98%), ammonium hydroxide solution (NH3 in H2O, 28%), 200 proof ethanol, Au(III) chloride trihydrate (HAuCl4·3H2O, 99.9%), potassium carbonate (K2CO3, 99.995%), tetrakis(hydroxymethyl)phosphonium chloride (THPC, 80% in water), hexachloroplatinic(IV) acid hexahydrate (H2PtCl6·6H2O, 99%), sodium hydroxide (NaOH, 98%), formaldehyde solution (CH2O, 37 wt% in H2O), sodium chloride (NaCl, 99%), L-ascorbic acid (C6H8O6, 99%). The chemicals were used as received without further purification. All the aqueous solutions were made with deionised (DI) water with a resistivity of 18.2 MΩ.Precursor solutions
The procedure followed for the making of the precursor solutions necessary for the synthesis of the SiO2–Au heterostructures was based on a protocol outlined in ref. 45. All synthesis procedures were performed in air at atmospheric pressure, and all glassware was previously washed and thoroughly rinsed with DI water to avoid impurities. All the subsequently prepared solutions were aqueous (i.e., DI water based), unless specified otherwise for specific experiments, where ethanol was used.Au plating solution (K–Au)
The Au ions needed for the growth of the Au seeds into larger Au particles are provided by a K–Au solution. First, 12.42 mg of K2CO3 were weighed and added into 50 mL of DI water, followed by the addition of 750 μL of 1 wt% HAuCl4 solution. Afterwards, the mixture was stirred in the dark at 900 rpm for 30 min. Finally, the solution was stored at 4 °C overnight before use.Au seeds suspension
The method to produce this colloidal suspension was originally described by Duff et al.,46 where Au seeds were formed by reacting with THPC in an alkaline aqueous environment. The series of steps and quantities of reagents used in this work are as follows: firstly, 500 μL of 1 M NaOH was added into 45 mL of DI water under vigorous stirring, followed by the addition of 1 mL of a previously prepared 68 mM THPC aqueous solution. After 5 min of continuous stirring, 2 mL of a 1 wt% HAuCl4 solution was rapidly added into the mixture. The solution immediately turned dark brown, indicating the quick formation of Au nanoparticles, and was left to stir in the dark for further 15 min. Afterwards, the mixture was stored at 4 °C for a period of 15 days before use. The result of this process was a colloidal suspension of spherical Au nanoparticles with diameters of 1–2 nm. The THPC capping of the Au seeds endows them with a negative charge, having zeta potentials from −40 to −45 mV,47 which prevents particle aggregation and provides a good stability of the seeds in the suspension over a large period of time after being synthesised.SiO2 nanoparticles
A colloidal suspension of SiO2 nanospheres in ethanol was synthesized following a method described in ref. 48, wherein TEOS is hydrolyzed and condensed under alkaline conditions to form monodisperse nanoparticles. First, 167.5 μL of TEOS was mixed with 1.33 mL of ethanol, while 38.1 μL of 28% ammonium hydroxide was mixed with 407.8 μL of DI water and 1.05 mL of ethanol. Subsequently, the two solutions were mixed under continuous stirring at 300 rpm for 3.5 hours. The mixture was centrifuged 3 times, at 900 rpm for 15 min, and redispersed in 5 mL of ethanol. This process results in monodisperse silica nanoparticles of ∼150 nm in diameter. Silica nanoparticles produced with this method have been reported to have zeta potentials between −70 to −80 mV,49 due to the silanol groups at their surfaces.Functionalisation of SiO2 nanoparticles
APTES molecules are required to be adsorbed on the SiO2 surface in order for the Au seeds to be able to attach onto the SiO2 cores and grow in size. The functionalisation process was originally described in ref. 50. The steps followed in this work are as follows: firstly, 500 μL of the colloidal solution of SiO2 nanoparticles was mixed with 5 mL of ethanol and, subsequently, after being stirred for 10 min at 650 rpm, 10 μL of APTES was added. The mixture was left to stir at 100 rpm for 24 hours. Afterwards, the solution was heated up to 90 °C whilst being stirred for one hour, with the periodical addition of ethanol during the synthesis which prevented total evaporation of the solvent. Finally, in order to wash them, the nanoparticles were centrifuged three times in ethanol for 15 min at 8500 rpm and redispersed in 2 mL of ethanol.SiO2–Au nanoparticles
The process for decorating SiO2 cores with discrete Au nanoparticles is as follows: firstly, the pH of 3 mL of Au seeds solution was adjusted to 3 by the addition of HCl. At low pH, positive H+ protons interact with the APTES functionalised silica nanoparticles, making them positively charged. The adjustment of pH is to ensure the electrostatic interaction between the positively charged silica nanoparticle and the negatively charged Au seeds, allowing the Au seeds to strongly attach to the SiO2 surface. For this, 60 μL of 1 M NaCl and 200 μL of amino-terminated SiO2 cores were added into the mixture, sonicated for 5 min and left to rest overnight. Subsequently, the mixture was centrifuged three times at 7500 rpm for 15 min and redispersed in 1 mL of DI water. The result of this process is a colloidal suspension of SiO2 cores with attached Au seeds of 1–2 nm in size. To keep it fresh, this suspension has to be refrigerated at 4 °C, and can be used immediately without ageing.In order to increase the amount of Au loading on the surface of the SiO2 cores, the Au seeds will be used as the nucleation sites for the deposition of Au. When formaldehyde is introduced into the K–Au solution, it donates electrons to the Au ions, until equilibrium between the participating species is reached. The presence of carbonate in the plating solution (K–Au) is necessary to raise the pH of the solution. In this condition, pH between 7 and 9, the Au ions have less tendency to reduce and form independent Au clusters in the solution, reducing only over the Au seeds adsorbed on the surface of the silica nanoparticles.51
Implementing this recipe, 3 mL of K–Au was added into 4 mL of the previously prepared seeded silica nanoparticles whilst stirring, followed by the addition of 50 μL of formaldehyde, the reducing agent. Then, the mixture was maintained whilst stirring for 15 more minutes and a change in colour, from red to violet, was observed. This change in colour is due to the change of optical properties of the nanostructure due to the growth of the Au nanoparticles in size. The nanoparticles were immediately centrifuged at 3000 rpm for 15 min and redispersed in 8 mL of DI water to remove the formaldehyde. Failure to do so results in irreversible particle aggregation. The outcome of this synthesis procedure is SiO2–Au(3) nanoparticles with the LSP wavelength of around 550 nm and Au nanoparticles of around 12 nm in diameter (measured by TEM). The resultant colloidal solution can be stored at room temperature when not in use.
SiO2–Au–Pt nanoparticles
The deposition of Pt on the nanoparticles requires Pt ions, provided by H2PtCl6, reduced by L-ascorbic acid; an environmentally friendly reducing agent (also known as vitamin C). Firstly, 200 μL of aqueous 0.1 M L-ascorbic acid and 6.5 μL of 10 mM H2PtCl6 solution were mixed with 4 mL of SiO2–Au solution. The mixture was kept in a water bath at 70 °C for 3 hours and allowed to rest overnight at room temperature. Finally, the mixture was centrifuged at 2900 rpm for 15 min and redispersed in 4 mL of DI water. The final concentration of heterostructures was estimated to be ∼0.15 nM. Additional samples with different amounts of HAuCl4 and H2PtCl6 were synthesised in order to study the effect of Au and Pt loading on the catalytic performance of the heterostructures.The nomenclature and volume of the metal precursors of the studied samples are summarised in Table 2. The number of moles of Au atoms per nanoparticle was calculated by considering the concentration of K–Au used (number of Au ions), the concentration of the SiO2 cores and assuming that all Au ions reduce to form small Au nanoparticles distributed over all the silica cores. With this information, and taking into account the volume of the unit cell of Au which contains 4 Au atoms, it was possible to estimate the number of Au particles per silica core, approximately 135, and their size as a function of the Au precursor is in agreement with the TEM measurements. The average Au nanoparticle size of the different samples is also summarised in Table 2. Similarly, the number of Pt moles was likewise estimated by considering the number of ions reduced and given in Table 2. The estimated ratio of number of the number of moles of Au and Pt is in agreement with the ratio of elemental percentages obtained by electron dispersive X-ray spectroscopy (EDS) measurements.
| Sample label | Vol. of HAuCl4 | Au NPs diameter |
|---|---|---|
| SiO2–Au(1.5) | 1.5 mL | 9.5 nm |
| SiO2–Au(3) | 3.0 mL | 12.0 nm |
| SiO2–Au(4.5) | 4.5 mL | 13.7 nm |
| SiO2–Au(6) | 6.0 mL | 15.1 nm |
| SiO2–Au(7.5) | 7.5 mL | 16.3 nm |
| Sample label | Vol. of H2PtCl6 | Moles of Pt per particle |
|---|---|---|
| SiO2–Au(3)–Pt | 6.5 μL | 3.60 × 10−19 |
| SiO2–Au(3)–Pt+ | 13 μL | 7.22 × 10−19 |
| SiO2–Au(3)–Pt++ | 26 μL | 1.44 × 10−18 |
| SiO2–Au(3)–Pt+++ | 39 μL | 2.16 × 10−18 |
SiO2–Pt nanoparticles
In order to investigate the effect of Pt on the photocatalytic degradation of MB without being paired with Au, purely Pt decorated silica nanoparticles (SiO2–Pt) were also fabricated. For this, Pt nanoparticles were synthesised by wet chemistry with a Pt precursor H2PtCl6 and then were attached onto APTES functionalised silica cores. Citrate capped Pt nanoparticles with diameters of approximately 3–8 nm were synthesised following the method in ref. 52. First, 3 mL of 0.2% H2PtCl6 was added into 39 mL of boiling DI water. Then, 920 μL of a solution containing 0.05% of citric acid and 1% of sodium citrate was poured into the boiling mixture. After 30 s, 460 μL of a reducing solution consisting of 0.08% sodium borohydrate, 0.05% citric acid and 1% sodium citrate was added. The reaction was allowed to continue for 10 min, and then left to cool down to room temperature. The result of this process is a suspension of brown colour. To obtain Pt decorated silica composites SiO2–Pt, the functionalised SiO2 nanoparticles simply need to contact the suspension of the Pt nanoparticles.53 The citrate capping allows for interaction with the APTES functionalised silica surface, leading to attachment of the Pt nanoparticles onto the silica cores. The procedure was performed as follows: first 800 μL of the solution containing the Pt nanoparticles was introduced dropwise into 200 μL of the solution of APTES-functionalised SiO2 cores, and stirred vigorously for 1 hour. Then the mixture was centrifuged at 7500 rpm three times and redispersed in 1 mL of DI water.Characterisation of hot-electron extraction via photocatalytic experiments
The hot carrier extraction from the synthesised heterostructures was investigated with a series of experiments by photocatalytic degradation of MB. During the degradation measurements, the absorbance spectra of MB or the absorbance maximum at 664 nm (Fig. 3a), were monitored as a function of time allowing for the reaction kinetics to be studied. All photocatalytic studies were performed in a standard 1 cm × 1 cm fluorescence cuvette (Optical Glass 340–2500 nm) at ambient temperature. A supercontinuum laser (400 fs pulse duration, 20 MHz repetition rate), with collimated beam having a diameter of 1 cm, was used as a light source for the nanoparticle excitation, while the optical absorption spectrum of MB was recorded to monitor the MB degradation (Ocean Optics QEPro spectrometer). Optical filters were used to selectively control the excitation wavelength range, and a neutral density filter was used to control the illumination power.For the degradation measurements, unless specified otherwise, mixtures composed of 467 μL of the MB molecules with a concentration of 20 μM, 333 μL of the chosen catalyst (SiO2 nanoparticles concentration of ∼0.15 nM) and 200 μL of DI water were used. Control experiments were carried in similar conditions without nanoparticles, with solutions containing 467 μL of 20 μM MB mixed with 533 μL of DI water.
For the experiments with scavengers, a volume of 200 μL of each of the scavengers (a concentration of 0.01 M) was mixed with 333 μL of the hetero-nanoparticle solution and 467 μL of 20 μM MB.
Numerical modelling
Numerical simulations of extinction, scattering and absorption cross-sections and the field distributions were performed with the Finite-Difference Time-Domain method (FDTD) using Lumerical FDTD software and Finite Integration Technique (FIT) using CST Microwave Studio software. A simulation domain size of 2 μm and mesh size of 1 nm were used, sufficient for convergence of the solution. The geometrical parameters and metal coverage of SiO2–Au(3) were derived from the TEM images and material analysis (see above), so the structures for this composition were modelled considering a uniform distribution of 135 Au nanoparticles with a diameter of 12 nm, attached onto the surface of a SiO2 nanosphere with a diameter of 150 nm. Since the average distance between the individual SiO2 nanoparticles estimated from the concentration of the solution used in the experiments was found to be ∼2 μm, the particles were considered to be optically isolated and, therefore, the absorption and scattering of an individual heterostructure was numerically studied. The permittivity of Au was taken from the experimentally measured values by Johnson and Christy,54 with a corrected 5 nm mean free path of the conduction electrons imposed by the size of the object.55,56 The permittivity of Pt was taken from the values reported in ref. 57. Dispersionless refractive index values of 1.46 and 1.33 were used for SiO2 and water, respectively. The studied nanostructure was excited by a linearly polarised plane wavefront pulse with a broad spectrum from 400 to 800 nm.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by EPSRC (UK) under the Reactive Plasmonics Programme grant (EP/M013812/1). J. U. S.-G. acknowledges a studentship from CONACYT-Mexico. M. R.-G. acknowledges a studentship from SENER-CONACYT-Mexico. We are grateful to S. M. Fairclough for the help with the energy-dispersive X-ray spectroscopy. All the data supporting this research are provided in full in the Results section and available from the corresponding author upon reasonable request.References
- M. L. Brongersma, N. J. Halas and P. Nordlander, Plasmon-induced hot carrier science and technology, Nat. Nanotechnol., 2015, 10, 25–34 CrossRef CAS.
- N. Jiang, X. Zhuo and J. Wang, Active plasmonics: principles, structures, and applications, Chem. Rev., 2018, 118, 3054–3099 CrossRef CAS.
- P. Wang, et al., Plasmonic metamaterials for nanochemistry and sensing, Acc. Chem. Res., 2019, 52, 3018–3028 CrossRef CAS.
- X. Zhang, Y. L. Chen, R.-S. Liu and D. P. Tsai, Plasmonic photocatalysis, Rep. Prog. Phys., 2013, 76, 046401 CrossRef.
- S. Linic, U. Aslam, C. Boerigter and M. Morabito, Photochemical transformations on plasmonic metal nanoparticles, Nat. Mater., 2015, 14, 567–576 CrossRef CAS.
- P. Wang, A. V. Krasavin, M. E. Nasir, W. Dickson and A. V. Zayats, Reactive tunnel junctions in electrically-driven plasmonic nanorod metamaterials, Nat. Nanotechnol., 2018, 13, 159–164 CrossRef CAS.
- T. P. Araujo, J. Quiroz, E. C. Barbosa and P. H. Camargo, Understanding plasmonic catalysis with controlled nanomaterials based on catalytic and plasmonic metals, Curr. Opin. Colloid Interface Sci., 2019, 39, 110–122 CrossRef CAS.
- M. Kim, M. Lin, J. Son, H. Xu and J.-M. Nam, Hot-electron-mediated photochemical reactions: principles, recent advances, and challenges, Adv. Opt. Mater., 2017, 5, 1700004 CrossRef.
- Z. Zhang, C. Zhang, H. Zheng and H. Xu, Plasmon-driven catalysis on molecules and nanomate rials, Acc. Chem. Res., 2019, 52, 2506–2515 CrossRef CAS.
- S. Mukherjee, et al., Hot-electron-induced dissociation of H2 on gold nanoparticles supported on SiO2, J. Am. Chem. Soc., 2014, 136, 64–67 CrossRef CAS.
- L. Zhou, et al., Aluminum nanocrystals as a plasmonic photocatalyst for hydrogen dissociation, Nano Lett., 2016, 16, 1478–1484 CrossRef CAS.
- S. Dong, et al., Recent developments in heterogeneous photocatalytic water treatment using visible light-responsive photocatalysts: a review, RSC Adv., 2015, 5, 14610–14630 RSC.
- R. A. Rather, S. Singh and B. Pal, Photocatalytic degradation of methylene blue by plasmonic metal-TiO2 nanocatalysts under visible light irradiation, J. Nanosci. Nanotechnol., 2017, 17, 1210–1216 CrossRef CAS.
- Y. Sivan, I. W. Un and Y. Dubi, Assistance of metal nanoparticles in photocatalysis – nothing more than a classical heat source, Faraday Discuss., 2019, 214, 215–233 RSC.
- G. Kawamura and A. Matsuda, Synthesis of plasmonic photocatalysts for water splitting, Catalysts, 2019, 9(12), 982 CrossRef CAS.
- H. Tada, S. i. Naya and M. Fujishima, Water splitting by plasmonic photocatalysts with a gold nanoparticle/cadmium sulfide heteroepitaxial junction: a mini review, Electrochem. Commun., 2018, 97, 22–26 CrossRef CAS.
- J. S. DuChene, G. Tagliabue, A. J. Welch, W.-H. Cheng and H. A. Atwater, Hot hole collection and photoelectrochemical CO2 reduction with plasmonic Au/p-GaN photocathodes, Nano Lett., 2018, 18, 2545–2550 CrossRef CAS.
- C. Liu, et al., Rapid water disinfection using vertically aligned MoS2 nanofilms and visible light, Nat. Nanotechnol., 2016, 11, 1098–1104 CrossRef CAS.
- J. U. Salmón-Gamboa, et al., Optimizing hot carrier effects in pt-decorated plasmonic heterostructures, Faraday Discuss., 2019, 214, 387–397 RSC.
- B. E. Brinson, et al., Nanoshells made easy: improving Au layer growth on nanoparticle surfaces, Langmuir, 2008, 24, 14166–14171 CrossRef CAS.
- N. C. Bigall, et al., Monodisperse platinum nanospheres with adjustable diameters from 10 to 100 nm: synthesis and distinct optical properties, Nano Lett., 2008, 8, 4588–4592 CrossRef CAS.
- E. Cortes, et al., Plasmonic hot electron transport drives nano-localized chemistry, Nat. Commun., 2017, 8, 1–10 CrossRef.
- A. Sousa-Castillo, et al., Boosting hot electron-driven photocatalysis through anisotropic plasmonic nanoparticles with hot spots in Au–TiO2 nanoarchitectures, J. Phys. Chem. C, 2016, 120, 11690–11699 CrossRef CAS.
- V.-Q. Nguyen, Y. Ai, P. Martin and J.-C. Lacroix, Plasmon-induced nanolocalized reduction of diazonium salts, ACS Omega, 2017, 2, 1947–1955 CrossRef CAS.
- D. Kumar, S. B. Lee, C. H. Park and C. S. Kim, Impact of ultrasmall platinum nanoparticle coating on different morphologies of gold nanostructures for multiple one-pot photocatalytic environment protection reactions, ACS Appl. Mater. Interfaces, 2018, 10, 389–399 CrossRef CAS.
- A. Houas, et al., Photocatalytic degradation pathway of methylene blue in water, Appl. Catal., B, 2001, 31, 145–157 CrossRef CAS.
- T. L. Thompson and J. T. Yates, Surface science studies of the photoactivation of TiO2 new photochemical processes, Chem. Rev., 2006, 106, 4428–4453 CrossRef CAS.
- M. Hayyan, M. A. Hashim and I. M. AlNashef, Superoxide ion: generation and chemical implications, Chem. Rev., 2016, 116, 3029–3085 CrossRef CAS.
- K. Das and A. Roychoudhury, Reactive oxygen species (ROS) and response of antioxidants as ROS-scavengers during environmental stress in plants, Front. Environ. Sci., 2014, 2, 53 Search PubMed.
- C. Kuppe, K. R. Rusimova, L. Ohnoutek, D. Slavov and V. K. Valev, “Hot” in plasmonics: temperature-related concepts and applications of metal nanostructures, Adv. Opt. Mater., 2020, 8, 1901166 CrossRef CAS.
- C. Sönnichsen, et al., Drastic reduction of plasmon damping in gold nanorods, Phys. Rev. Lett., 2002, 88, 077402 CrossRef.
- S. Dal Forno, L. Ranno and J. Lischner, Material, size, and environment dependence of plasmon induced hot carriers in metallic nanoparticles, J. Phys. Chem. C, 2018, 122, 8517–8527 CrossRef CAS.
- H. Al-Ekabi and N. Serpone, Kinetics studies in heterogeneous photocatalysis. I. photocatalytic degradation of chlorinated phenols in aerated aqueous solutions over titania supported on a glass matrix, J. Phys. Chem., 1988, 92, 5726–5731 CrossRef CAS.
- J. B. Khurgin, Hot carriers generated by plasmons: where are they generated and where do they go from there?, Faraday Discuss., 2019, 214, 35–58 RSC.
- X. Xu, et al., TiN@TiO2 core–shell nanoparticles as plasmon-enhanced photosensitizers: the role of hot electron injection, Laser Photonics Rev., 2020, 14, 1900376 CrossRef CAS.
- M. M. J. Sadiq, U. S. Shenoy and D. K. Bhat, NiWO4-ZnO-NRGO ternary nanocomposite as an efficient photocatalyst for degradation of methylene blue and reduction of 4-nitro phenol, J. Phys. Chem. Solids, 2017, 109, 124–133 CrossRef CAS.
- M. Pirhashemi and A. Habibi-Yangjeh, Ultrasonic-assisted preparation of plasmonic ZnO/Ag/Ag2WO4 nanocomposites with high visible-light photocatalytic performance for degradation of organic pollutants, J. Colloid Interface Sci., 2017, 491, 216–229 CrossRef CAS.
- P. Prasannalakshmi and N. Shanmugam, Fabrication of TiO2/ZnO nanocomposites for solar energy driven photocatalysis, Mater. Sci. Semicond. Process., 2017, 61, 114–124 CrossRef CAS.
- K. Wu, J. Chen, J. R. McBride and T. Lian, Efficient hot-electron transfer by a plasmon-induced interfacial charge-transfer transition, Science, 2015, 349, 632–635 CrossRef CAS.
- M. Shen and M. A. Henderson, Identification of the active species in photochemical hole scavenging reactions of methanol on TiO2, J. Phys. Chem. Lett., 2011, 2, 2707–2710 CrossRef CAS.
- M. F. R. Samsudin, et al., Exploring the role of electron-hole scavengers on optimizing the photocatalytic performance of BiVO4, Mater. Today: Proc., 2018, 5, 21703–21709 Search PubMed.
- S. Mubeen, J. Lee, D. Liu, G. D. Stucky and M. Moskovits, Panchromatic photoproduction of H2 with surface plasmons, Nano Lett., 2015, 15, 2132–2136 CrossRef CAS.
- I. Kutsche, G. Gildehaus, D. Schuller and A. Schumpe, Oxygen solubilities in aqueous alcohol solutions, J. Chem. Eng. Data, 1984, 29, 286–287 CrossRef CAS.
- W. Xing, et al., Oxygen solubility, diffusion coefficient, and solution viscosity, in Rotating Electrode Methods and Oxygen Reduction Electrocatalysts, ed. W. Xing, G. Yin and J. Zhang, Elsevier, Amsterdam, 2014, ch. 1, pp. 1–31 Search PubMed.
- P. Wang, et al., Metaparticles: dressing nano-objects with a hyperbolic coating, Laser Photonics Rev., 2018, 12, 1800179 CrossRef.
- D. G. Duff, A. Baiker and P. P. Edwards, A new hydrosol of gold clusters. 1. Formation and particle size variation, Langmuir, 1993, 9, 2301–2309 CrossRef CAS.
- S. Park, M. Park, P. Han and S. Lee, The effect of pH-adjusted gold colloids on the formation of gold clusters over APTMS-coated silica cores, Bull. Korean Chem. Soc., 2006, 27, 1341–1345 CrossRef CAS.
- W. Stöber, A. Fink and E. Bohn, Controlled growth of monodisperse silica spheres in the micron size range, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- S. Park, M. Park, P. Han and S. Lee, Relative contributions of experimental parameters to NIR-absorption spectra of gold nanoshells, J. Ind. Eng. Chem., 2007, 13, 65–70 CAS.
- S. Oldenburg, R. Averitt, S. Westcott and N. Halas, Nanoengineering of optical resonances, Chem. Phys. Lett., 1998, 288, 243–247 CrossRef CAS.
- M. J. García-Soto and O. González-Ortega, Synthesis of silica-core gold nanoshells and some modifications/variations, Gold Bull., 2016, 49, 111–131 CrossRef.
- M. Cueto, et al., Platinum nanoparticles as photoactive substrates for mass spectrometry and spectroscopy sensors, J. Phys. Chem. C, 2014, 118, 11432–11439 CrossRef CAS.
- N. Zhang, et al., Near-field dielectric scattering promotes optical absorption by platinum nanoparticles, Nat. Photonics, 2016, 10, 473–482 CrossRef CAS.
- P. B. Johnson and R. W. Christy, Optical constants of the noble metals, Phys. Rev. B: Solid State, 1972, 6, 4370–4379 CrossRef CAS.
- P. Lissberger and R. Nelson, Optical properties of thin film Au-MgF2 cermets, Thin Solid Films, 1974, 21, 159–172 CrossRef CAS.
- R. Atkinson, et al., Anisotropic optical properties of arrays of gold nanorods embedded in alumina, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 235402 CrossRef.
- W. S. M. Werner, K. Glantschnig and C. Ambrosch-Draxl, Optical constants and inelastic electron scattering data for 17 elemental metals, J. Phys. Chem. Ref. Data, 2009, 38, 1013–1092 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |