
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComprehensive analysis of epigenetic signatures of human transcription control†
Guillaume
Devailly
 *a and
Anagha
Joshi
*b
*a and
Anagha
Joshi
*b
aGenPhySE, Université de Toulouse, INRAE, ENVT, 31326, Castanet Tolosan, France. E-mail: guillaume.devailly@inrae.fr
bComputational Biology Unit, Department of Clinical Science, University of Bergen, 5021, Bergen, Norway. E-mail: Anagha.Joshi@uib.no
First published on 21st June 2021
Abstract
Advances in sequencing technologies have enabled exploration of epigenetic and transcriptional profiles at a genome-wide level. The epigenetic and transcriptional landscapes are now available in hundreds of mammalian cell and tissue contexts. Many studies have performed multi-omics analyses using these datasets to enhance our understanding of relationships between epigenetic modifications and transcription regulation. Nevertheless, most studies so far have focused on the promoters/enhancers and transcription start sites, and other features of transcription control including exons, introns and transcription termination remain underexplored. We investigated the interplay between epigenetic modifications and diverse transcription features using the data generated by the Roadmap Epigenomics project. A comprehensive analysis of histone modifications, DNA methylation, and RNA-seq data of thirty-three human cell lines and tissue types allowed us to confirm the generality of previously described relationships, as well as to generate new hypotheses about the interplay between epigenetic modifications and transcription features. Importantly, our analysis included previously under-explored features of transcription control, namely, transcription termination sites, exon–intron boundaries, and the exon inclusion ratio. We have made the analyses freely available to the scientific community at http://joshiapps.cbu.uib.no/perepigenomics_app/ for easy exploration, validation and hypothesis generation.
Background
Epigenetic modifications of the DNA sequence and DNA-associated proteins along with transcriptional machinery are thought to be the main driver shaping mammalian genomes during development and disease.1 Epigenetic modifications include DNA methylation, histone variants and histone post-translational modifications (such as acetylations and methylations), and facilitate tissue specific expression.2 The advent and maturation of sequencing technologies have facilitated large scale generation of epigenomic data across diverse organisms in multiple cell and tissue types. Accordingly, consortia were established to generate large epigenomic datasets, including ENCODE,3 Roadmap Epigenomics,2 and Blueprint epigenome4 for humans, modENCODE5 for model organisms, and FAANG6,7 for farm animal species. The International Human Epigenome Consortium (IHEC) was set up to gather reference maps of human epigenomes.8 The data from these efforts have generated new findings through integrated analyses. Such analyses are facilitated by consortia data portals8,9 as well as portals gathering data from multiple sources,10–14 which allow easy browsing as well as downloading of both sequences and processed data. In addition, many data portals include (or link to) genome browsers to allow online solutions for data exploration.15Several online tools have been developed to explore publicly available epigenomic data16–20 to gain insights into mammalian epigenetic control. These tools used diverse computational frameworks ranging from data integration and visualisation (e.g. ChIP-Atlas allows visualisation of multiple histone modifications and transcription factor binding sites at any given genomic locus by using public ChIP-seq and DNase-seq data21) to semi-automated genome annotation (e.g. Segway performed genomic segmentation of human chromatin by integrating histone modifications, transcription-factor binding and open chromatin22). Though identification of functional elements from epigenetic data2,22 has been highly effective in annotating the enhancer and promoter regions of a genome, they failed to capture other transcription regulation features such as exon–intron boundaries and transcription termination features. For example, the Roadmap Epigenomics project mapped about 30 epigenetic modifications across human cell lines and tissues to gather a representative set of “complete” epigenomes.2 Using these data, Kundaje et al. built a hidden Markov model based classifier to define 15 distinct chromatin states, including active or inactive promoters, active or inactive enhancers, and condensed and quiescent states. Notably, this unsupervised approach did not lead to the definition of “exon” states, and even less to “exon-included” and “exon-excluded” states. This might be because epigenetic modifications enriched at enhancers and promoters have strong signals (or peaks), while the ones abundant at gene bodies (DNA methylation, H3K36me3) are wide and diffuse. The promoter and enhancer features therefore dominate in epigenetic data analyses, hindering recovery of associations between epigenetic modifications and other transcription control events such as splicing (constitutive or alternative). Moreover, some transcription features might not have a strong correlation with any chromatin modification studied. For example, Curado et al.23 estimated that only 4% of the differentially included exons were associated with changes in H3K9ac, H3K27ac, and/or H3K4me3 across 5 different cell lines. A gap therefore remains in genome-wide computational analyses towards getting a comprehensive overview of the associations between epigenetic modifications and transcription control features.
On the other hand, individual targeted studies have provided evidence for the interplay between epigenetics and other transcriptional features. DNA methylation at gene bodies has been positively correlated with the gene expression level.24,25 Maunakea et al.26 observed that DNA methylation was positively correlated with splicing at alternatively spliced exons, and proposed a mechanism involving DNA methylation reader MECP2. Lev Maor et al.27 observed that DNA methylation at exons can be either positively or negatively correlated with splicing depending on the exons, through a mechanism involving CTCF and MECP2. A causal role of DNA methylation in alternative splicing was established by drug-induced de-methylation,28 as well as by targeted DNA methylations and de-methylations by Shayevitch et al.29 Xu et al.30,31 identified H3K36me3 epigenetic modification associated with alternative splicing. There is some evidence for epigenetic control at transcription termination as well; the loss of gene body DNA methylation was found to favour the use of a proximal alternative poly-adenylation site by unmasking CTCF binding sites.32
In summary, many large data integration approaches only allow extraction of epigenetic signatures for dominant features of transcription control (e.g. enhancer, promoter, transcription start site (TSS)), missing many other transcription features (e.g. exon, intron, transcription termination site (TTS)). We therefore performed a systematic analysis of associations between epigenetic modifications and diverse transcription features. Using the Roadmap Epigenomics project data for 30 epigenetic modifications in 33 cell and tissue contexts, we explored links between epigenetic modifications and transcription control. We confirmed previously known associations as well as generated novel observations. We have provided our analyses along with thousands of visualisations freely through a companion web application available at http://joshiapps.cbu.uib.no/perepigenomics_app/, allowing researchers to browse the results and generate working hypotheses.
Results
Exploration of epigenetic signatures of transcription control using the Roadmap Epigenomics project data
To explore the epigenetic signatures at transcription control sites, we first extracted three gene features: transcription start site (TSS), transcription termination site (TTS) and middle exons from GENCODE annotation version 2933 (Table 1). We classified genes in two different ways. Firstly, we partitioned all genes based on the gene length into “long” (>3 kb), “short” (≤1 kb), and “intermediate” length genes. This classification allows an investigation of epigenetic modifications at the TTSs while excluding any spurious signals from the TSSs, observable in short but not in long genes. We also classified genes based on simplified GENCODE gene types, namely: protein coding genes, RNA genes, pseudogenes and other genes (see the Methods section).| Mark | TSS | TTS | Exons | Exon expression | Exon inclusion ratio | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Centered at TSS | Within cell types | Across cell types | Centered at TTS | Within cell types | Across cell types | Centered at exon | Within cell types | Across cell types | Within cell types | Across cell types | ||
| DNA methylation | CpG density | ✓ | +++ | ✓ | + | ✓ | + | − | ||||
| mCpG ratio | ✓ | −−− | ✗ | · | · | ✗ | + | − | − | + | ||
| mCpG density | ✓ | −−− | ✓ | ++ | ✓ | + | · | |||||
| Chromatin accessibility | DNAsel | ✓ | +++ | ++ | ✓ | · | ++ | ✓ | ++ | − | − | |
| Histone variant | H2A.Z variant | ✓ | ++ | ✗ | · | · | ✗ | · | · | − | · | |
| Histone methylations | H3K4mel | ✓ | + | + | ✗ | + | ++ | ✓ | + | ++ | − | − |
| H3K4me2 | ✓ | +++ | + | ✗ | ✓ | + | + | − | · | |||
| H3K4me3 | ✓ | +++ | ++ | ✗ | + | ✗ | ++ | ++ | · | |||
| H3K9me3 | ✗ | · | · | ✗ | − | + | ✓ | − | + | · | ||
| H3K23me2 | ✓ | ++ | ✗ | ✗ | · | · | ||||||
| H3K27me3 | ✓ | −− | · | ✗ | ✓ | + | − | |||||
| H3K36me3 | ✗ | +++ | ++ | ✓ | +++ | +++ | ✓ | +++ | +++ | · | ||
| H3K79mel | ✓ | ++ | · | ✗ | ++ | ++ | ✓ | ++ | ++ | · | ||
| H3K79me2 | ✓ | ++ | · | ✗ | + | + | ✓ | ++ | + | · | ||
| H4K20mel | ✓ | + | ✗ | + | ✓ | + | ||||||
| Histone acetylations | H2AK5ac | ✓ | + | + | ✗ | ✗ | + | · | ||||
| H2BK5ac | ✓ | ++ | ++ | ✗ | + | ✗ | + | · | ||||
| H2BK12ac | ✓ | + | + | ✗ | + | ✓ | + | · | ||||
| H2BK15ac | ✓ | + | · | ✗ | ✓ | − | · | |||||
| H2BK2.ac | ✗ | · | ✗ | ✗ | ||||||||
| H2BK12.ac | ✓ | + | + | ✗ | + | ✗ | + | · | ||||
| H3K4ac | ✓ | + | ++ | ✗ | + | ✗ | + | − | · | |||
| H3K9ac | ✓ | +++ | ++ | ✗ | + | ✗ | + | − | − | |||
| H3K14ac | ✓ | + | + | ✗ | ✗ | · | ||||||
| H3K18ac | ✓ | + | ++ | ✗ | + | ✗ | + | − | · | |||
| H3K23ac | ✓ | + | + | ✗ | ✗ | + | · | |||||
| H3K27ac | ✓ | +++ | ++ | ✗ | ++ | ++ | ✗ | ++ | ++ | · | ||
| H3K56ac | ✓ | + | ✗ | ✗ | · | · | ||||||
| H4K5ac | ✓ | ++ | ✗ | ✗ | ||||||||
| H4K8ac | ✓ | + | + | ✗ | + | + | ✗ | + | + | − | − | |
| H4K12ac | ✓ | + | ✗ | + | ✗ | + | − | |||||
| H4K91ac | ✓ | + | + | ✗ | + | ✗ | − | |||||
We obtained RNA sequencing data for 33 cell or tissue types from the Roadmap data portal.2 Gene and exon normalised expression levels (transcripts per million, TPM) were calculated by pseudo-mapping of the reads to the human transcriptome using Salmon34 in each sample. Moreover, for every middle exon in each cell type, the exon inclusion ratio (ψ) was calculated (see Methods), ranging from 0 to 1 (0 for exons not included, and 1 for exons included in all the transcripts).
The genome-wide histone modification and DNAseI profiles for the cell types corresponding to the transcriptome data were obtained from the Roadmap Epigenomics consortium. For the Whole Genome Bisulfite Sequencing (WGBS) data, we computed three new tracks (Fig. S1, ESI†): CpG nucleotide density (consistent across cell and tissue types), CpG methylation ratio (average ratio of methylation at CpG sites in the window), and CpG methylation density (number of methylated CpG sites in each window), for each sample, using the WGBS CpG coverage track as a control. The mCpG density is the number of methylated sites in a given genomic region, calculated as a product of the CpG density and the average mCpG ratio.
For each pair of transcription feature and epigenetic modification, the associations were explored at two levels: (1) cell or tissue level – for all genes (or exons) in each cell or tissue type and (2) gene level – for each gene (or exon) across all cell or tissue types. Cell or tissue level analysis allows within-assay comparison of highly and weakly expressed genes (or exons), but is sequence and genomic context (unique for each gene or exon) dependent. On the other hand, gene level analysis fixes the sequence and genomic contexts, but is more sensitive to technical variability across experiments.
The main observations from these analyses are summarised in Table 1 and elaborated in the sections below. We have developed a companion web application to allow exploration of analyses at http://joshiapps.cbu.uib.no/perepigenomics_app/. Unless explicitly specified, the associations described in this manuscript were common to all cell types and tissues. The main figures focus mostly on the gastric tissue but readers are encouraged to explore other cell types and tissues using the companion web application.
Transcription activity and epigenetic modifications near transcription start sites
Many epigenetic modifications are enriched around the TSS of expressed genes, a region containing gene promoters. To investigate the link between the transcription level and epigenetic modifications at the TSS, we generated stack profiles of each epigenetic modification around the TSS. When epigenetic modifications were sorted according to gene expression (Fig. 1A), most histone modifications studied were more abundant at highly expressed genes than at weakly expressed ones. Specifically, only one histone acetylation (H2BK20ac, Fig. S2, ESI†) and three histone methylations (H3K9me3, H3K27me3, and H3K36me3, Fig. S3–S5, ESI†) did not show a positive correlation with the gene expression level. We further noted that only H3K27me3 was more abundant at the TSSs of weakly or non-expressed protein coding genes than at the TSSs of highly expressed protein coding genes. H3K27me3 was not present at the TSSs of non-expressed, non-protein coding genes in any of the cell or tissue types, highlighting the fact that the associations between an epigenetic modification and the transcriptional level may be gene-type specific (see the companion web application).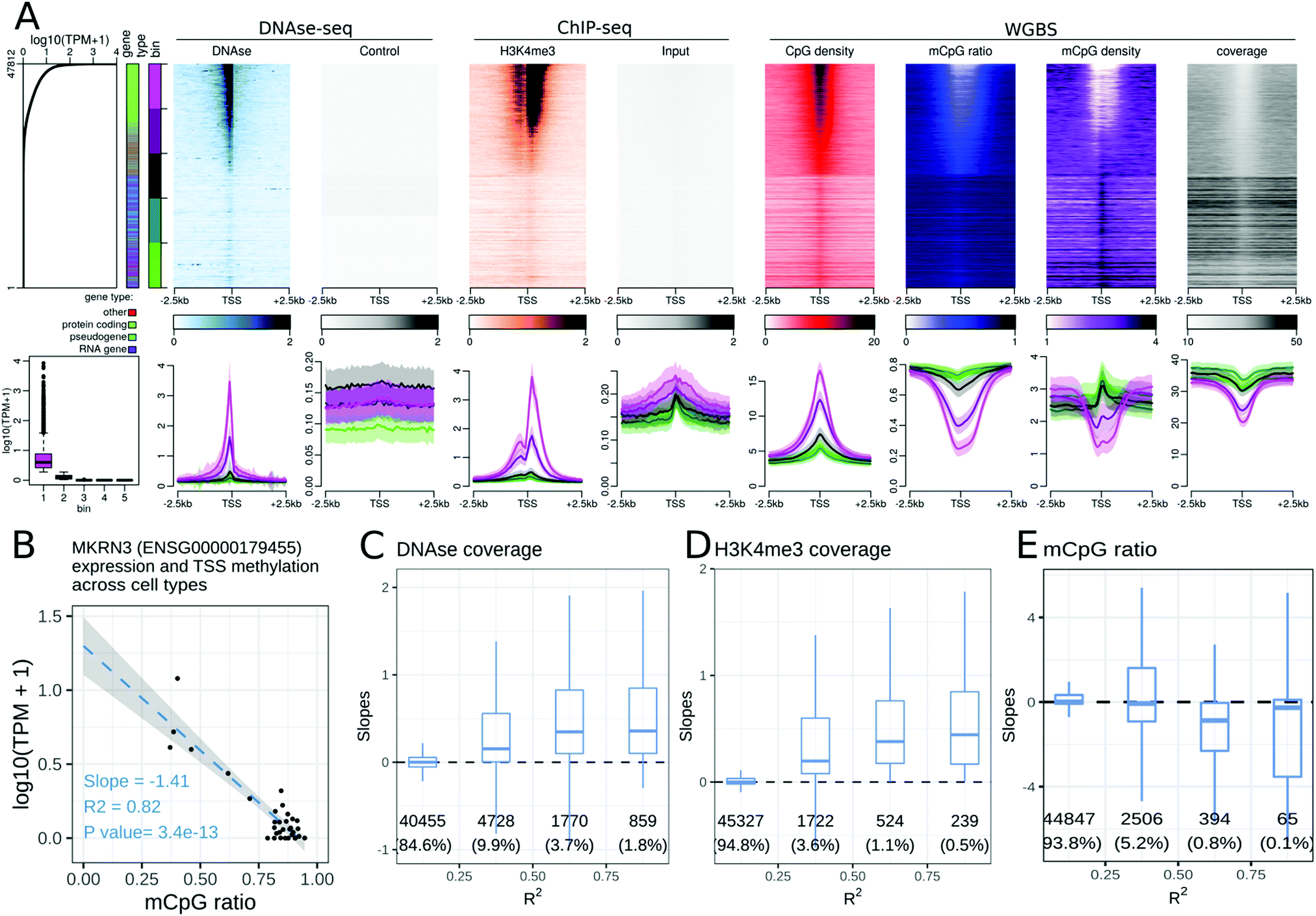 | ||
Fig. 1 Relationships between epigenetic modifications near the Transcription Start Sites (TSSs) and gene expression levels. (A) Association between epigenetic modifications near the TSSs and gene expression levels in the gastric tissue. Upper part, from left to right: gene expression levels in all 47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 812 autosomal genes annotated using GENCODE. The first side bar indicates the gene type (green: protein coding genes, blue: pseudogenes, purple: RNA genes, red: other types of genes), and the second side bar indicates the genes sorted according to their expression level (lower panels of A, 5 bins in total, purple: highly expressed genes, green: weakly expressed genes). Stacked profiles of (i) DNAse-seq and the respective control and (ii) H3K4me3 ChIP-seq and the respective input control, and (iii) the CpG density, mCpG ratio (mCpG/CpG), mCpG density, and WGBS coverage near the TSS, sorted according to the corresponding gene expression level. Lower part, from left to right: Boxplot of gene expression levels in each of the 5 expression bins defined in the upper part. Average profiles of DNAse-seq and the respective control, H3K4me3 ChIP-seq and the respective input control, and the CpG density, mCpG ratio, mCpG density and WGBS coverage, ± SEM (Standard Error of the Mean) for each bin of promoters. (B–E) Association between epigenetic modifications near the TSSs and gene expression levels across cell types. (B) Regression of the expression level (in log10(TPM + 1)) of the MKRN3 gene and the mean DNA methylation ratio at CpG sites 500 bp around the TSS of the MKRN3 gene. Each dot corresponds to a cell type. The slope is negative and the correlation coefficient (R2) is greater than 0.75 for MKRN3. Similar regressions were generated for each gene and epigenetic modification pair. (C–E) Distributions of the slopes from the gene regressions (as in (B)) according to the R2 correlation coefficients for the DNAse-seq signals (C), H3K4me3 ChIP-seq signals (D) and the mCpG ratios (E) near the TSSs of the corresponding genes. The number of genes and percentage of genes in each category are displayed below each box. 812 autosomal genes annotated using GENCODE. The first side bar indicates the gene type (green: protein coding genes, blue: pseudogenes, purple: RNA genes, red: other types of genes), and the second side bar indicates the genes sorted according to their expression level (lower panels of A, 5 bins in total, purple: highly expressed genes, green: weakly expressed genes). Stacked profiles of (i) DNAse-seq and the respective control and (ii) H3K4me3 ChIP-seq and the respective input control, and (iii) the CpG density, mCpG ratio (mCpG/CpG), mCpG density, and WGBS coverage near the TSS, sorted according to the corresponding gene expression level. Lower part, from left to right: Boxplot of gene expression levels in each of the 5 expression bins defined in the upper part. Average profiles of DNAse-seq and the respective control, H3K4me3 ChIP-seq and the respective input control, and the CpG density, mCpG ratio, mCpG density and WGBS coverage, ± SEM (Standard Error of the Mean) for each bin of promoters. (B–E) Association between epigenetic modifications near the TSSs and gene expression levels across cell types. (B) Regression of the expression level (in log10(TPM + 1)) of the MKRN3 gene and the mean DNA methylation ratio at CpG sites 500 bp around the TSS of the MKRN3 gene. Each dot corresponds to a cell type. The slope is negative and the correlation coefficient (R2) is greater than 0.75 for MKRN3. Similar regressions were generated for each gene and epigenetic modification pair. (C–E) Distributions of the slopes from the gene regressions (as in (B)) according to the R2 correlation coefficients for the DNAse-seq signals (C), H3K4me3 ChIP-seq signals (D) and the mCpG ratios (E) near the TSSs of the corresponding genes. The number of genes and percentage of genes in each category are displayed below each box. | ||
We explored the trends in peak shapes and noted that a ‘double hill’ (or an ‘M’ shape) with a gap at the exact location of the TSS was the most common shape. The ‘gap’ of the ChIP-seq signal between the ‘hills’ was located exactly at a sharp peak of the DNAseI signal around the TSS (Fig. 1A), indicating very high DNA accessibility at the TSS. This suggests the presence of a nucleosome-free region at the promoter terminating at the TSS, with the first nucleosome positioned near the +1 of transcription. This double hill pattern was either symmetric or asymmetric depending on the mark and cell or tissue type (e.g. a stronger peak on the downstream hill than the upstream hill in H3K4me3 in the gastric tissue, Fig. 1A). The H3K79me2 profile was particularly strongly asymmetric (Fig. S6, ESI†), with a strong peak at around ±500 bp downstream of the TSS across 2 of the 3 cell lines for which the mark was studied in the Roadmap Epigenomics dataset.
The CpG density around the TSS was positively correlated with gene expression in all cell and tissue types in the dataset. The CpG methylation ratio and CpG methylation density near the gene TSS were negatively correlated with gene expression levels. While the CpG methylation ratio showed a flat profile near the TSS of non-expressed genes, the CpG methylation density transitioned from a gap at highly expressed genes to a peak at non-expressed genes at the TSS (Fig. 1A).
We further explored these trends for individual gene types. As protein-coding genes formed the majority of all genes, the above observations for all gene types were preserved when the analyses were restricted to protein coding genes only. Separating genes according to gene type highlighted differences in epigenetic profiles at lincRNAs and unprocessed pseudogenes compared to protein coding genes. Processed pseudogenes often showed a different relationship between their expression level and the epigenetic status of their promoter. For example, expressed processed pseudogenes showed neither a DNAseI accessibility peak at the TSS, nor an enrichment of active epigenetic modifications at the TSS. While the CpG density near the TSS was correlated with the processed pseudogene expression level, their promoters did not show any decrease in DNA methylation ratio, resulting in a positive relation between the DNA methylation density and processed pseudogene expression (see the companion web application).
Expressed genes of the ‘antisense’ gene type often mirrored the epigenetic signature of the ‘protein coding’ gene type. For example, the H3K79me2 peak was pronounced 500 bp before the TSS of ‘antisense’ genes, whereas it peaked 500 bp after the TSS of protein coding genes (Fig. S7, ESI†). However, it is likely that the observed epigenetic signal at antisense genes might be due to the corresponding ‘sense’ gene.
Altogether, the gene expression level was positively or negatively correlated with many epigenetic modifications at gene promoters when comparing expressed and non-expressed genes within a cell type. The associations between epigenetic modifications and gene expression in a cell or tissue type are promoter sequence and gene context dependent. For example, the CpG density at the TSS is a good predictor of both the gene expression level and the CpG methylation ratio and density (Fig. 1A). To study the association between epigenetic modifications and transcription across different cell and tissue types, we calculated linear regression between each epigenetic modification and gene expression level across cell or tissue types. Specifically, the epigenetic signals in the ±500 bp window around the TSS and the gene expression level (log10(TPM + 1)) for each gene (Fig. 1B) were linearly regressed to obtain a slope and a linear correlation coefficient (R2). For example, the linear regression between the MKRN3 gene expression level and the average CpG methylation ratio near the TSS of the MKRN3 gene resulted in a slope of −1.41, an R2 of 0.82, and a p-value of 3.4 × 10−13. As expected, some of these associations were highly tissue specific (Fig. S8, ESI†): for example CLDN18 in the gastric tissue, GCG and INS in the pancreas, MYOD1 in the skeletal muscle and NPPB in the heart showed a positive correlation between DNAse, H3K27ac and H3K4me3 signals and gene expression levels, and a negative correlation between H3k27me3 and WGBS signals and gene expression levels. For each epigenetic modification, the distribution of slopes across all genes was plotted against their R2 (Fig. 1C–E and the companion web application). The epigenetic modifications with positive slopes across cell or tissue types were: H3K4me3 (Fig. 1D), H3K36me3, H2BK5ac, H3K4ac, H3K9ac, H3K18ac, H3K27ac (Fig. S8A–F, ESI†) and chromatin accessibility measured by DNAseI digestion assay (Fig. 1C). DNA methylation showed a negative slope for the R2 greater than 0.5 (Fig. 1E). For each epigenetic modification, only a minority of genes displayed high correlation values. This is in part due to the fact that most genes did not show high expression variability across the datasets. For each epigenetic modification, gene ontology enrichment analysis for the top 1000 genes with the highest R2 value was performed. Highly correlated genes were often enriched for the biological processes involved in development or muscle biology (Fig. S9, ESI†). These top 1000 correlated genes were often enriched for genes with a CpG island near the promoter (Fig. S10, ESI†), and in protein coding and lincRNA genes (Fig. S11, ESI†).
Transcription activity and epigenetic modifications near transcription termination sites
We repeated the analyses described above at transcription termination sites (TTSs). We noted that highly expressed genes tend to be longer than non-expressed genes. In short genes, it is difficult to distinguish the effect of epigenetic modifications at the TSS from that at the TTS. We therefore defined three classes of genes: short (≤1 kb), long (>3 kb), and intermediate length genes, to mitigate this gene-length effect. The number of genes in each category for each GENECODE gene type is presented in Fig. S13 (ESI†).While many epigenetic modifications showed a peak (or a gap) centred at the gene TSS, only two modifications were enriched at the gene TTS. First, H3K36me3 displayed a broad hill-shape profile, with a peak at the TTS (Fig. 2A). Levels of H3K36me3 at TTSs were positively correlated with gene expression levels for different genes within a cell type (Fig. 2A), and also for gene expression levels of the same gene across cell types (Fig. 2C). In some samples, the H3K36me3 profile was slightly asymmetric at the TTS, with more signal in the gene body than after the TTS (see the companion web application).
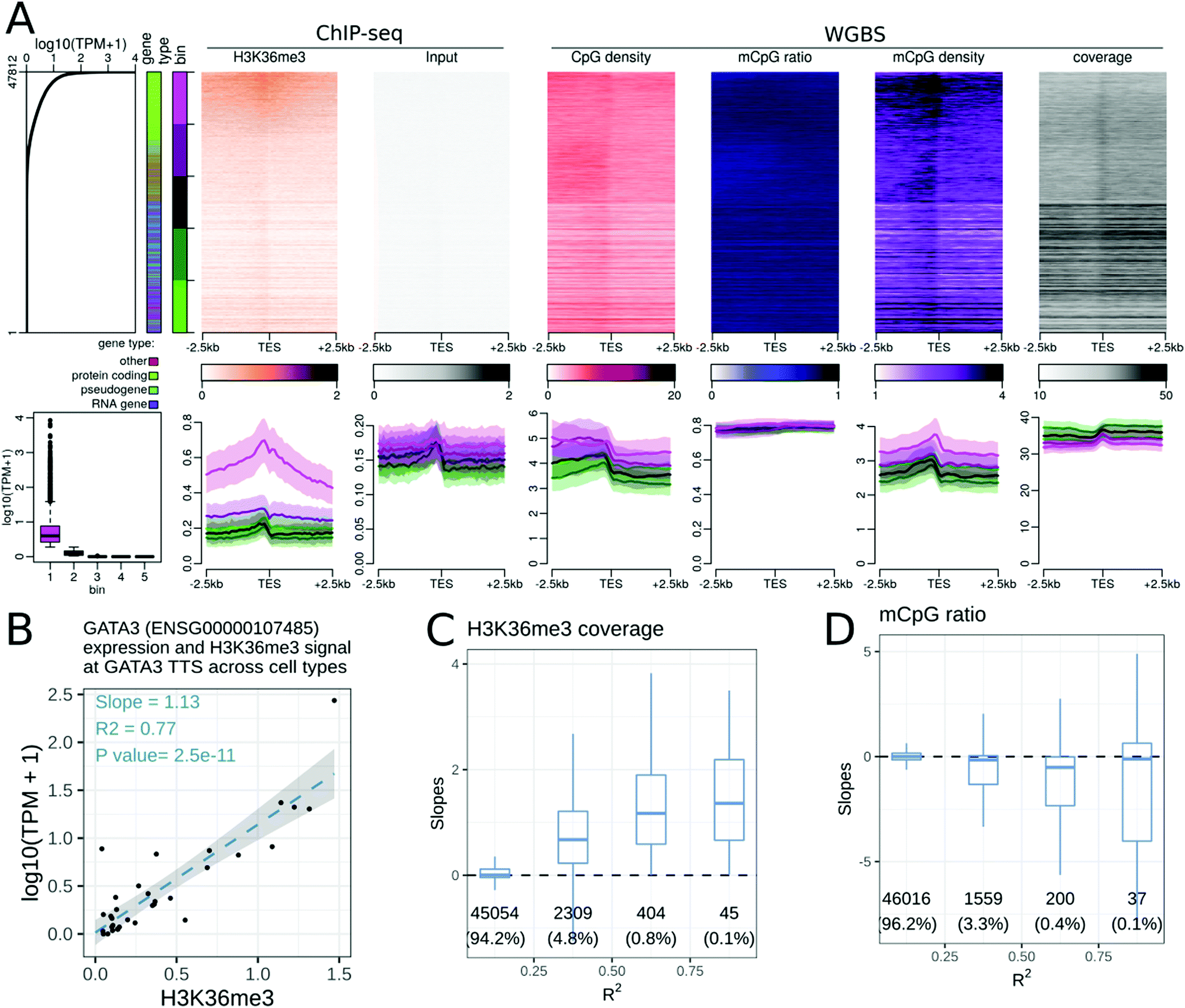 | ||
| Fig. 2 Relationships between epigenetic modifications near the transcription termination sites (TTSs) and expression levels. (A) Association between epigenetic modifications near the TTS and gene expression levels in the gastric tissue. Upper part, from left to right: Gene expression levels in all 47812 genes annotated using GENCODE. The first side bar indicates the gene type (green: protein coding genes, blue: pseudogenes, purple: RNA genes, red: other types of genes), and the second side bar indicates the 5 bins used in the lower panels of A (purple: highly expressed genes, green: weakly expressed genes). Stacked profiles of (i) H3K36me3 ChIP-seq and the respective input control, and (ii) the CpG density, mCpG ratio (mCpG/CpG), mCpG density, and WGBS coverage near the TTS, sorted according to the corresponding gene expression level. Lower part, from left to right: Boxplot of the gene expression level in each of the 5 bins defined in the upper part. Then, average profiles of DNAse-seq and the respective control, H3K4me3 ChIP-seq and the respective input control, and the CpG density, mCpG ratio, mCpG density and WGBS coverage ± SEM (Standard Error of the Mean) for each bin of promoters. (B–D) Association between epigenetic modifications near the TTS and gene expression levels across cell types. (B) Regression of the expression level (in log10(TPM + 1)) of the GATA3 gene with the mean H3L36me3 ChIP-seq signal 500 bp around the TTS of the GATA3 gene, where each dot corresponds to a cell or tissue type. The slope was positive and the correlation coefficient (R2) was greater than 0.75 in this case. Similar regressions were conducted for each gene and epigenetic modification pair. (C and D) Distribution of the slopes from the gene regressions (as in B) according to the R-squared correlation coefficients for H3K36me3 modification (C), and the mCpG ratios (D) near the TTSs of the corresponding genes. The number of genes and percentage of genes in each category are displayed below each box. | ||
The DNA methylation density increased at the TTS (Fig. 2A). As the DNA methylation ratio was nearly constant, the increase of DNA methylation density was mostly due to the increase of CpG density at the TTS. The DNA methylation density at the TTS was positively correlated with the gene expression level when comparing different genes within a cell type. A weak negative correlation between DNA methylation and the gene expression level at the TTS was observed for a subset of genes when comparing the same gene across cell and tissue types (Fig. 2D). This is in agreement with recent findings.32 The negative correlation between DNA methylation and gene expression was evident only in ‘long’ genes, and in protein coding genes (see the companion web application).
Though most epigenetic modifications did not show enrichment at TTSs, four epigenetic modifications (DNAseI accessibility, H3K4me1, H3K79me1, H3K27ac, Fig. S10A–D, ESI†), were positively correlated with the expression level of a gene across cell and tissue types. These epigenetic modifications showed no enrichment at TTSs, yet the change in expression level was associated with the change in epigenetic modification strength. These modifications could be reflecting broader chromatin organisational features such as topologically associating domains and A/B chromatin domains.35
Exon transcription and epigenetic modifications at middle exons
Several studies have found a correlation between DNA methylation26,27,36 or histone modifications23,30,31 and exon and splicing events, in either single or a few cell and tissue contexts. We explored whether these observations hold true in the Roadmap Epigenomics dataset. We focused on the middle exons of protein coding genes and excluded the first and last exons. A total of 16811 middle exons were expressed in at least one cell or tissue type in the Roadmap dataset. The expression level of an exon was defined as the sum of the TPM of the transcripts including that exon. Similar to TSS and TTS analysis, we performed epigenetic modification enrichment analysis at middle exons. H3K36me3 showed enrichment at middle exons, correlated with the exon expression level within a cell or tissue type (Fig. 3A). Changes in H3K36me3 at exons were also strongly associated with exon expression levels across cell and tissue types (Fig. 3D). Though some other epigenetic modifications showed a weak enrichment at exons, the same observation can be made for input samples used as negative controls, thus likely reflecting a technical artefact. Epigenetic modifications, including H3K4me1, H3K4me3 (Fig. 3C), H3K27ac, H3K79me1, H3K79me2, H3K9ac, and H3K8ac, were correlated with exon expression levels across cell and tissue types, but did not show any enrichment at the middle exons (see the companion web application). The H3K4me3 signal, peaked at the gene TSS, and terminated around the start of the first internal exon (Fig. 3A), resulting in a transition from H3K4me3 marked chromatin to H3K36me3 marked chromatin near the beginning of the second exon of genes.
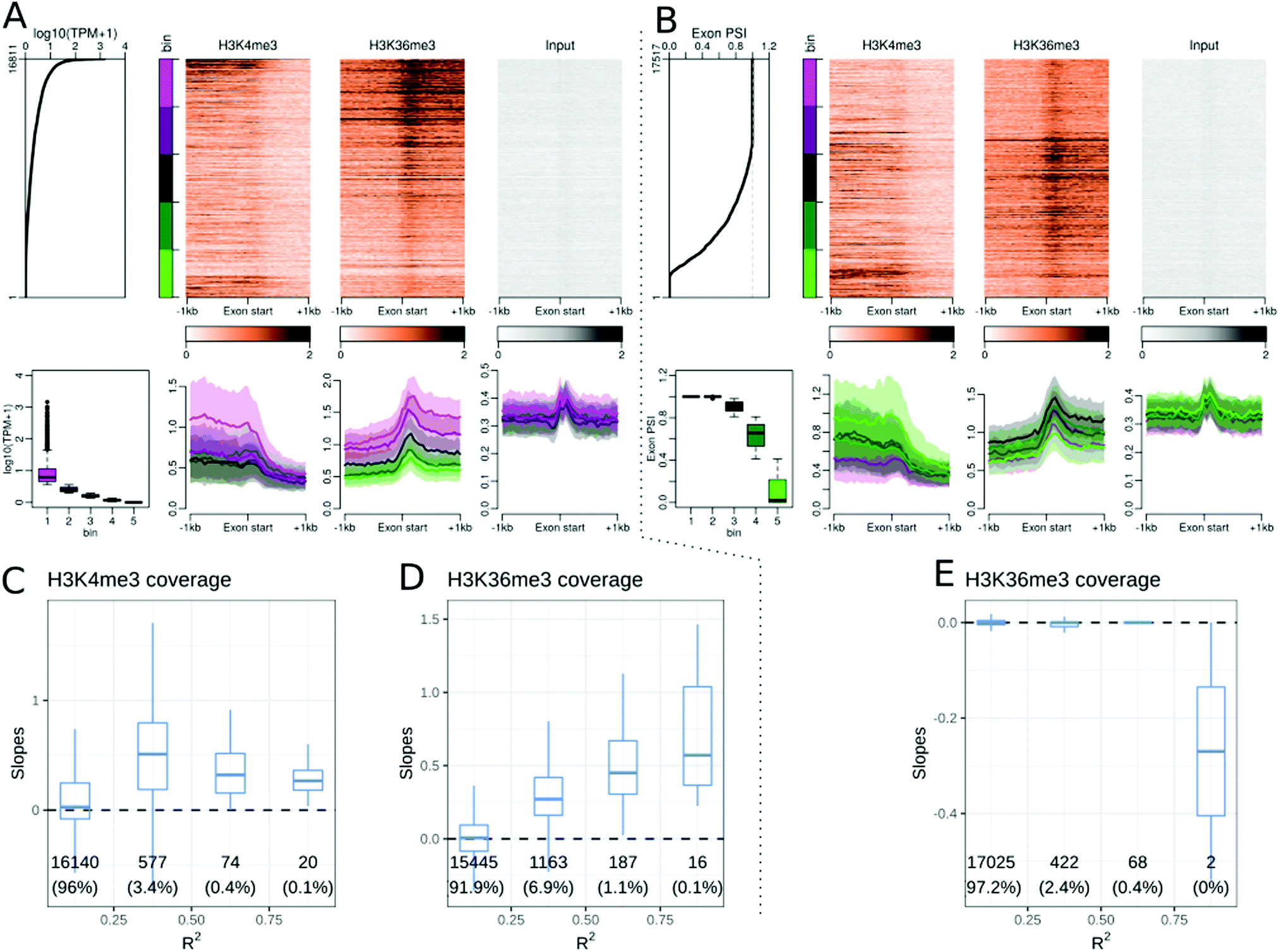 | ||
| Fig. 3 Relationships between epigenetic modifications near middle exon starts and exon expression levels or exon inclusion ratios. (A) Association between epigenetic modifications near middle exon start sites and exon expression levels in the gastric tissue. Upper part, from left to right: middle exon expression levels in 16811 middle exons annotated using GENCODE. The side bar indicates 5 bins used in the lower panels of A (purple: highly expressed exons, green: weakly expressed exons). Then: Stacked profiles of H3K4me3 ChIP-seq, H3K36me3, and input control, sorted according to the exon expression levels. Lower part, from left to right: boxplot of exon expression levels in each of the 5 bins defined in the upper part. Then, the average profiles of H3K4me3 ChIP-seq, H3K36me3, and the respective input control ± SEM (Standard Error of the Mean) for each bin of exons. (B) Association between epigenetic modifications near middle exon start sites and exon inclusion ratios in the gastric tissue. Upper part, from left to right: middle exon inclusion ratios in 17517 middle exons annotated using GENCODE. The side bar indicates 5 bins used in the lower panels of B (purple: included exons, green: excluded expressed exons). Then: stacked profiles of H3K4me3 ChIP-seq and H3K36me3, and the respective input controls, sorted according to the corresponding exon inclusion ratio. Lower part, from left to right: boxplot of exon inclusion ratios in each of the 5 bins defined in the upper part. Then, average profiles of H3K4me3 ChIP-seq, H3K36me3, and the respective input control ± SEM (standard error of the mean) for each bin of exons. (C and D) Distribution of the slopes from exon expression level regressions and epigenetic marks present at the exon start (±100 bp) according to the R2 correlation coefficients for H3K4me3 signals (C), and H3K36me3 (D) near the start of the corresponding middle exons. (E) Distribution of the slopes from exon inclusion ratio regressions and epigenetic modification at the exon start (±100 bp) grouped by the R2 correlation coefficients for H3K36me3 modification. The number of exons and percentage of exons in each category are displayed below each box. | ||
There was no difference between the DNA methylation ratios at exons and introns, but exons had overall more CpG sites than introns, resulting in a higher DNA methylation density at middle exons than at the surrounding introns (see the companion web application). The DNAseI accessibility further decreased at the exon start (Fig. S15, ESI†), from the already low surrounding accessibility, suggesting that the splicing acceptor site has even lower accessibility than the surrounding regions.
Exon inclusion ratios and epigenetic modifications at middle exons
The exon expression level in a cell type consists of both constitutive and alternative splicing events. To study the association between epigenetic modifications and alternate splicing events, we calculated the exon inclusion ratio for each exon. The exon inclusion ratio was calculated for all transcripts of a given gene (see Methods) in each cell or tissue type and sorted using the inclusion ratio (ψ). Accordingly, we obtained ψ values for 17517 exons, including 706 exons that were never included in the Roadmap datasets, but were part of genes that were expressed in this dataset. We checked whether epigenetic modifications were correlated with the exon inclusion ratio and noted that only a few modifications, namely, H3K27ac, H3K4me3, and H3K36me3, were associated with the exon inclusion ratio within a cell type (Fig. 3B). DNA methylation was also associated with the exon inclusion ratio. No epigenetic modification showed a strong association with the changes in inclusion ratio at the alternatively included exons (Fig. 3E and Table 1). There were nevertheless very weak associations for the mCpG ratio, DNAseI, H3K4me1, H3K27me3, H3K9ac and H4K8ac.
A linear model for gene expression
So far, we have analysed the associations between epigenetic modifications and transcription control in a pair-wise manner. In order to better model the combinatorial effect of modifications, we selected 6 epigenetic modifications (DNA methylation, H3K4me1, H3K4me3, H3K9me3, H3K27me3 and H3K36me3) for which epigenetic and transcriptome data were available for 27 cell types in the Roadmap dataset, and regressed a linear model at four transcriptional features for all types of genes: (i) epigenetic modifications around TSSs and gene expression levels (Fig. 4A), (ii) epigenetic modifications around TTSs and gene expression levels (Fig. 4B), (iii) epigenetic modifications around the start of middle exons and exon expression levels (Fig. 4C), and (iv) epigenetic modifications around the start of middle exons and exon inclusion ratios (Fig. 4D).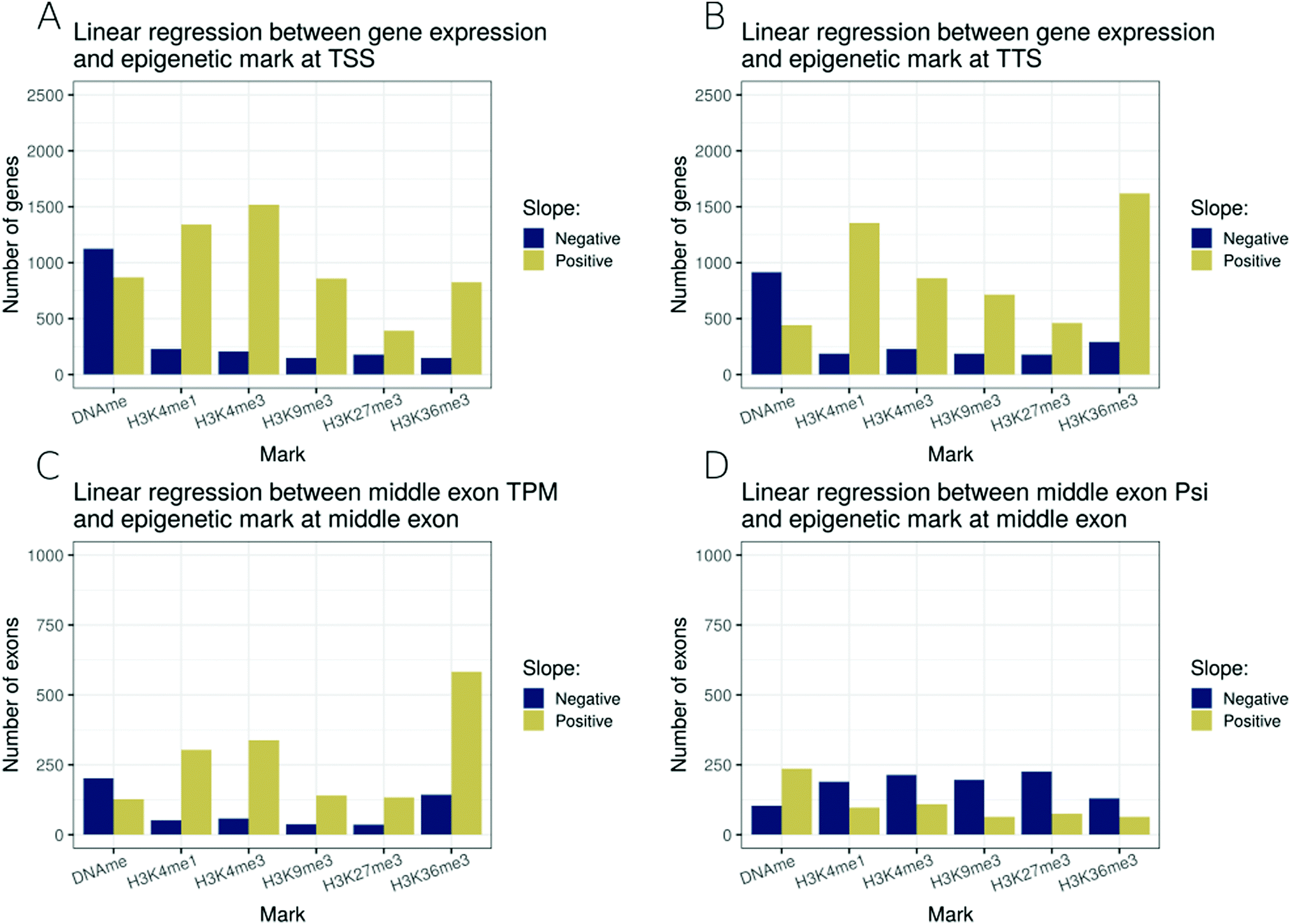 | ||
| Fig. 4 Linear regression models including six epigenetic modifications characterised in 27 cell types. Each bar represents the number of genes or exons with a statistically significant slope (p ≤ 0.01), either positive (golden) or negative (deep blue). (A) Linear regression model of gene expression levels and the levels of 6 epigenetic modifications near their respective TSSs (±500 bp). (B) Linear regression model of gene expression levels and the levels of 6 epigenetic modifications near their respective TTSs (±500 bp). (C) Linear regression model of middle exon expression levels and the levels of 6 epigenetic modifications near their respective starts (±100 bp). (D) Linear regression model of the middle exon inclusion ratio and the levels of 6 epigenetic modifications near their respective starts (±100 bp). | ||
At TSSs, amongst the 6 marks studied, DNA methylation was associated with gene repression, while all studied histone methylations were associated with the activation of the expression level (Fig. 4A). At TTSs, we noted a similar pattern to that at TSSs, with a difference that H3K36me3 at the TTS was the modification most strongly associated with an increase in expression level, followed by H3K4me1 as the second most positively associated (Fig. 4B). We noted that restricting the analysis to long genes only, TTS associations with epigenetic modifications were much weaker (Fig. S4, ESI†), highlighting that an important fraction of the TTS associations were due to short genes for which the promoter marks might confound the TTS signal. H3K36me3 at middle exons was also strongly associated with exon expression levels (Fig. 4C). Finally, no strong associations were detected for epigenetic modifications (of the 6 selected ones) at exon inclusion ratios (Fig. 4D).
PEREpigenomics, a web resource to explore associations between epigenetic modifications and transcription features
Using the Roadmap dataset, we unravelled a range of associations between epigenetic modifications and transcription control. We generated a total of 9024 stacked profiles of epigenetic modifications near the TSS, TTS and middle exons, sorted according to the gene expression level, exon expression level or exon inclusion ratio, in 33 different cell and tissue types. We also provided stacked profiles for the TSS and TTS for each gene type (protein-coding genes, RNA genes, pseudogenes, other types), as well as for short (≤1 kb), long (>3 kb) and intermediate length genes. Users furthermore can generate regressions between an epigenetic modification at the TSS or TTS and gene expression levels across cell and tissue types for a gene of interest, and the same feature is available for middle exons and exon expression levels or exon inclusion ratios as well.We have made the analyses and visualisations available to users through a web-application at http://joshiapps.cbu.uib.no/perepigenomics_app/.
This allows users to explore relationships between epigenetic modifications and transcription across many tissues, including their tissue of interest.
Discussion
In summary, this multi-faceted analysis of the Roadmap Epigenomics data, freely available through a web application, has enabled the confirmation of known (or previously observed in only one of few cell types) associations between epigenetic modifications and transcription on a large data set and further formulation of new hypotheses. We specifically discuss epigenetic associations of previously under-explored transcription features below grouped according to the epigenetic modification.Histone modifications
Of the 28 studied histone modifications near the TSS, 25 were positively correlated with gene expression across genes within a cell type, and a subset of them (16) were also correlated with changes in gene expression when comparing the same gene across cell and tissue types (Table 1). H3K27me3 was the only mark that was more abundant at the TSSs of weakly or non-expressed genes than at those of highly expressed protein coding genes. H3K27me3 modification was not present at non-protein coding genes, highlighting that the negative correlation between H3K27me3 and gene expression is a gene type specific association. H3K9me3 has been associated with closed chromatin,37,38 and was not enriched at the TSSs of non-expressed genes in the Roadmap Epigenomics data set. While many active histone marks showed a TSS asymmetry, with a higher signal downstream of the TSS than that upstream of the TSS. H3K79me1 and H3K79me2 were enriched downstream of the TSS of expressed genes (Fig. S6, ESI†), suggesting an association with the transcription direction. Indeed, H3K79 methylations are catalysed by the DOT1L enzyme during transcription elongation.39 It should be noted that their profile was only available in 5 and 3 cell lines, respectively, all derived from the embryonic stem cell line H1. H3K79me1 and H3K79me2 asymmetries were less marked in the H1-derived trophoblast, which could either reflect a relevant biological difference or be due to experimental issues.Among the histone modifications, H3K36me3 displayed a unique profile across all studied transcription features. The H3K36me3 modification is positively correlated with gene expression at the TTS across genes within a cell type, and also when comparing the same gene across cell and tissue types. While largely absent from the TSS region, it is enriched at all middle exons and on the last exon. We noted that the gene body H3K36me3 begins at the start of the second exon, where the H3K4me3 peak decreases. We further explored a potential link between exonic H3K36me3 and alternative splicing. Our approach using the exon inclusion ratio revealed that any such association was either weak or restricted to only a few exons. Similar observations have been made by others: Xu et al.31 noted that from about 3000 alternative splicing events, 800 were positively correlated with changes in H3K36me3 and 700 were negatively correlated with changes in H3K36me3. It should be noted that H3K36me3 modified genomic regions (wide peaks) tend to be an order of magnitude larger than an average exon size (1000 bp vs. 100 bp). Altogether, though 10 histone modifications showed some enrichment at exons, the associations between epigenetic modifications and changes in exon inclusion ratio were weak and/or limited to a small subset of genes.
DNA methylation
The CpG density is highly variable in the human genome, with CpG depleted or CpG poor regions spanning most of the genome. We noted that the CpG density at the TSS was strongly associated with gene expression across genes within a cell type, where most expressed genes had a CGI centred at their TSS. Accordingly, at the TSS, the mCpG ratio, or the average methylation at CpG sites, was negatively associated with gene expression across genes within a cell type. This trend overlaps with the CpG density where most of the CpG deserts are heavily methylated, and most CGIs are unmethylated40 (Fig. S1, ESI†). An increase in mCpG ratio at TSSs was also associated with the down-regulation of gene expression in the gene level analysis. Non-promoter regions were methylated with mCpG ratios around 85%; this ratio decreased to around 30% at the TSSs of the expressed protein-coding genes. We noted that this is not the case for expressed pseudogenes, whose TSSs had higher CpG density than the surrounding regions, but remained methylated.The mCpG density is the number of methylated sites in a given genomic region, calculated as a product of the CpG density and the average mCpG ratio in a given genomic region. It has been shown that the mCpG density, but not the mCpG ratio, was the main driver of the binding of DNA methylation readers of the MBD family.41 While many publications focus solely on the mCpG ratio as a metric to evaluate DNA methylation, we argue that multiple metrics provide complementary information. For example, while the mCpG ratio remains constant across the promoters of repressed genes, the mCpG density peaks near the TSSs of repressed genes suggest that these regions might preferentially recruit repressive DNA binding proteins (e.g. MBP). The DNA methylation density also peaks at the TSSs of expressed processed pseudogenes, as their CGIs remain methylated. At exons, the mCpG ratio is as high as that at introns, but the CpG density is higher at exons than that at introns, resulting in a higher mCpG density.
It has been observed that GC rich regions might be more difficult to sequence using some Illumina sequencing protocols, resulting in lower coverage at CGIs.42 We noted this bias in about half of the WGBS samples, where the WGBS coverage at the TSS was anti-correlated with gene expression across genes within a cell type. Some WGBS samples were less affected by this bias, while a few showed even an inverse trend, with higher coverage in GC rich regions. These biases leave the mCpG ratio and mCpG density profiles largely unaffected, thus preserving the validity of the analysis.
DNA accessibility and nucleosome positioning
The DNAseI assay showed a narrow (<100 bp) peak before and at the TSS of expressed genes. This narrow peak of DNA accessibility matched the location of a dip in the bi-modal signal present in many histone modifications positively correlated with the expression level (e.g. H3K4me3 and many histone acetylations). These observations suggest that there is a short nucleosome-free region before the TSS of expressed genes, with a nucleosome positioned just after the +1 of transcription. Such a nucleosome positioning effect is well described in yeast43 and in mammals.44 Intriguingly, middle exon starts and TTS positions appear to be depleted of DNAseI signals, even more so than the surrounding regions (Fig. S3, ESI†). This suggests that middle and last exon starts are particularly inaccessible regions, maybe due to nucleosome positioning45 or the presence of the splicing machinery at acceptor sites.Analysis across cell and tissue types
For each gene and middle exon, we correlated the level of epigenetic modification with the expression level or exon inclusion ratio across the different cell and tissue types in the Roadmap dataset. 459 genes had a linear regression coefficient R2 ≥ 0.5 (0.9% of all genes) between promoter DNA methylation and gene expression levels, including 209 protein coding genes (1.1% of the protein coding genes), and the slope was negative, indicating that for these genes, a higher level of promoter DNA methylation is associated with a lower gene expression level. Conversely, 763 genes had a linear regression coefficient R2 ≥ 0.5 (1.6% of all genes) when regressing H3K4me3 levels at the promoter and gene expression levels, including 492 protein coding genes (2.6% of the protein coding genes). The slope in this case was positive: a higher level of promoter H3K4me3 was associated with a higher gene expression level. Nonetheless, most genes or exons did not have regression coefficients of ≥0.25. This might be because many genes and exons might not have large enough epigenetic or expression variability in this dataset. Indeed, correlated genes were often enriched for gene ontologies related to development, genes with likely variable expressions across different tissues (Fig. S9, ESI†). ChIP-seq and DNAse-seq peak heights can also be biased by the epigenetic modifications (or accessibility) in the dominant fraction of alleles and cells, as well as biased by changes in ChIP efficiency due to hard-to-control experimental variations. Importantly, many transcriptional control mechanisms might not show any correlation at the epigenetic level, e.g. transcription factor activities, and binding with co-factors as well as variable rates of post-transcriptional degradation of mRNAs. The dataset itself poses many limitations in the analysis. For example, the Roadmap dataset provides only one sample per tissue for each modification. Despite the richness of these data, the absence of replicates hinders the ability to perform a more in depth analysis of tissue specific associations between epigenetic modifications and transcription control.Conclusions
In summary, we performed a comprehensive analysis to study links between epigenetic modifications and transcription control using the Roadmap Epigenomics data. The Roadmap Epigenomics histone modifications, whole genome bisulfite sequencing, and RNA-seq data, across diverse human cell and tissue types, allowed us to confirm the generality of previously described relationships between epigenetic modifications and transcription control in one or few cell types, as well as to generate new hypotheses about the interplay between epigenetic modifications and transcript diversity. Importantly, our analysis focused on previously under-explored features of transcription control including transcription termination sites, exon–intron boundaries, middle exons and the exon inclusion ratio. We have produced thousands of stack profile plots of epigenetic modifications around gene features sorted according to their gene expression level, exon expression level or exon inclusion ratio, and filterable by gene type. These plots are made freely available through a web application, http://joshiapps.cbu.uib.no/perepigenomics_app/. We hope this web application will serve the community (i) as a resource to validate known or previously described epigenetic modifications associated with transcription features as well as (ii) an interactive tool for allowing exploration of data as a novel hypothesis generator of epigenetic and transcriptional control.Methods
Data retrieval
GENCODE human annotation version 29 (main annotation file) was downloaded from the GENCODE as gff3 files. Reads from RNA-seq data were retrieved from the European Nucleotide Archive using the Roadmap sample table as a reference. Whole genome bisulphite sequencing (WGBS) data sets (bigwig files of fractional methylation and read coverage) were downloaded from the Roadmap Epigenomics data portal.Histone modifications and DNAseI data were downloaded as consolidated, not subsampled, tagAlign files from the Roadmap data portal.
Altogether, we retrieved 27 RNA-seq and the corresponding WGBS datasets, 13 DNAseI profiles (with matching controls), and 242 histone ChIP-seq datasets in 27 human cell lines or tissues (with 27 matching controls).
RNA sequencing analysis
RNA-sequencing reads were quantified using Salmon34 v12.0 by pseudoalignment to the human reference genome hg38 and annotations v29 provided using GENCODE.33 The parameters validateMappings, seqBias, gcBias were on, with biasSpeedSamp equals to 5, and libType equals to A were selected. For samples with biological replicates, the median expression value in TPM samples was used for genes and transcripts. For each exon, the exon expression level was calculated as the sum of the TPM of the transcripts including this exon. Exon inclusion ratios are computed as the sum of the TPM of transcripts including the exon divided by the TPM value of the gene.We considered each gene uniquely, by selecting a representative TSS (or TTS) per gene, from all annotated TSSs (or TTSs). We sorted all transcripts of a gene according to their TSS (or TTS) genomic coordinates and selected the TSS (or TTS) of the transcript at the middle of the list, i.e. the median TSS (or TTS). The list of middle exons was obtained by taking the shortest transcript of each protein coding gene, then selecting genes with 3 or more exons, and excluding the first and last exons of each transcript. The shortest known transcript isoform was used to ensure that the list of middle exons could not contain the first or last exons of other isoforms. Most annotated non-protein coding genes were monoexonic, and were excluded from the exonic analyses.
Epigenetic modification data processing
For WGBS, three different tracks were generated from the FractionalMethylation.bigwig files using bedtools46 and rtracklayer:47 number of CpG sites per window, mean DNA methylation ratio per window, and density of mCpG sites per window, using windows of 250 base pair width, sliding by 100 base pairs. The WGBS coverage file was processed similarly to produce a fourth track serving as a control. No post-processing was done for DNAseI and Histone tagAlign files.Heatmap generation
Gene types were derived from GENCODE,33 where the pseudogene category contained all genes with the word “pseudogene”, and the “other” types of genes were defined as neither protein coding genes, nor pseudogenes, nor RNA genes. Genes were binned in 5 groups of equal sizes according to their expression values, and exons in 5 groups according to their expression values or exon inclusion ratios.For genes with multiple GENCODE annotations for TSSs (or TTSs), we sorted all TSSs (or TTSs) according to their genomic coordinates (5′ to 3′, taking into account their orientation) and took the TSS (or TTS) of the transcript in the middle of the list. The list of middle exons was obtained using protein coding genes. For genes with several annotated transcripts, the transcript with the smallest length was selected, the first and last exons were filtered, and the remaining exons were kept. Stacked profiles of tracks centred at TSSs or TTSs were generated using a region of ±2.5 kb around the TSS or TTS, using windows of 100 bases every 100 bases. Stacked profiles of tracks centred at middle exons were drawn using a region of ±1 kb around middle exon starts, with windows of 50 bases every 50 bases. For histone modifications and DNAseI data, and the corresponding input controls, the coverage was expressed as FPKM values. For CpG density and mCpG density, it was defined as the number of (methylated) sites per window (250 bp for TSSs or TTSs, 100 bp for exons). The mCpG ratio ranged from 0 (all the CpG sites in the window fully unmethylated) to 1 (all the CpG sites in the window fully methylated), and the coverage was expressed as the number of reads covering a region. For each of the five gene (or exon) bins, we displayed the value distributions as boxplots, and the average profiles ± standard error of the mean (SEM) for each bin. Heatmaps were drawn with a custom script using the following packages: seqplots,48 Repitools,49 GenomicRanges,50 rtracklayer,47 and plotrix.51
Regression analysis
At each gene TSS (or TTS), the epigenetic modification level in a sample was averaged in the ±500 bp region around the TSS (or TTS) and a linear regression was calculated for each gene using expression values in log10(TPM + 1). Epigenetic modifications in the ±100 bp region around middle exon starts were regressed with either log10(TPM + 1) of the exon or the exon inclusion ratio. For each regression the slope and regression coefficient (R2) were obtained, using dplyr,52 purrr,53 and broom.54 From each of the regressions of epigenetic modification near the TSS, the top 1000 genes with the highest R2 were extracted and submitted to PantherDB v1655 using the API to obtain the list of enriched biological process ontology terms. CpG islands were obtained from the UCSC Table Browser.56
Data availability
PEREpigenomics is developed in Shiny.57 Source code and data of the application are available at http://forgemia.inra.fr/guillaume.devailly/perepigenomics_app. Scripts used to process the data and generate the plots can be found at: http://github.com/gdevailly/perepigenomicsAnalysis.Author contributions
GD and AJ designed the analyses and the web applications, and wrote this manuscript. GD performed the analysis and developed the web application.Abbreviations
| TSSs | Transcription start sites |
| TTSs | Transcription termination sites |
| TESs | Transcription end sites |
| TPM | Transcripts per million reads |
| CpG | Cytosine–guanine dinucleotide |
| CGI | CpG island |
| ψ | Exon inclusion ratio |
| WGBS | Whole genome bisulfite sequencing |
| H3K9me3 | Tri-methylation of lysine 9 of histone 3 |
| H4K5ac | Acetylation of lysine 5 of histone 4 |
Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
GD was funded for this work by the People Program (Marie Curie Actions FP7/2007-2013) under REA grant agreement no. PCOFUND-GA-2012-600181. AJ was supported by the Bergen Research Foundation Grant No. BFS2017TMT01. The authors would like to thank Olaf Sarnow, Kjell Petersen and Stanislav Oltu for their help with the web server configuration, and Anna Mantsoki and Barry Horne for their help at the beginning of the project.Notes and references
- J. Romanowska and A. Joshi, Genes, 2019, 10, 76 CrossRef CAS PubMed.
- A. Kundaje, W. Meuleman, J. Ernst, M. Bilenky, A. Yen, A. Heravi-Moussavi, P. Kheradpour, Z. Zhang, J. Wang, M. J. Ziller, V. Amin, J. W. Whitaker, M. D. Schultz, L. D. Ward, A. Sarkar, G. Quon, R. S. Sandstrom, M. L. Eaton, Y.-C. Wu, A. R. Pfenning, X. Wang, M. Claussnitzer, Y. Liu, C. Coarfa, R. A. Harris, N. Shoresh, C. B. Epstein, E. Gjoneska, D. Leung, W. Xie, R. D. Hawkins, R. Lister, C. Hong, P. Gascard, A. J. Mungall, R. Moore, E. Chuah, A. Tam, T. K. Canfield, R. S. Hansen, R. Kaul, P. J. Sabo, M. S. Bansal, A. Carles, J. R. Dixon, K.-H. Farh, S. Feizi, R. Karlic, A.-R. Kim, A. Kulkarni, D. Li, R. Lowdon, G. Elliott, T. R. Mercer, S. J. Neph, V. Onuchic, P. Polak, N. Rajagopal, P. Ray, R. C. Sallari, K. T. Siebenthall, N. A. Sinnott-Armstrong, M. Stevens, R. E. Thurman, J. Wu, B. Zhang, X. Zhou, A. E. Beaudet, L. A. Boyer, P. L. D. Jager, P. J. Farnham, S. J. Fisher, D. Haussler, S. J. M. Jones, W. Li, M. A. Marra, M. T. McManus, S. Sunyaev, J. A. Thomson, T. D. Tlsty, L.-H. Tsai, W. Wang, R. A. Waterland, M. Q. Zhang, L. H. Chadwick, B. E. Bernstein, J. F. Costello, J. R. Ecker, M. Hirst, A. Meissner, A. Milosavljevic, B. Ren, J. A. Stamatoyannopoulos, T. Wang and M. Kellis, Nature, 2015, 518, 317–330 CrossRef CAS PubMed.
- ENCODE Project Consortium, Nature, 2012, 489, 57–74, DOI:10.1038/nature11247.
- D. Adams, L. Altucci, S. E. Antonarakis, J. Ballesteros, S. Beck, A. Bird, C. Bock, B. Boehm, E. Campo, A. Caricasole, F. Dahl, E. T. Dermitzakis, T. Enver, M. Esteller, X. Estivill, A. Ferguson-Smith, J. Fitzgibbon, P. Flicek, C. Giehl, T. Graf, F. Grosveld, R. Guigo, I. Gut, K. Helin, J. Jarvius, R. Küppers, H. Lehrach, T. Lengauer, Å. Lernmark, D. Leslie, M. Loeffler, E. Macintyre, A. Mai, J. H. Martens, S. Minucci, W. H. Ouwehand, P. G. Pelicci, H. Pendeville, B. Porse, V. Rakyan, W. Reik, M. Schrappe, D. Schübeler, M. Seifert, R. Siebert, D. Simmons, N. Soranzo, S. Spicuglia, M. Stratton, H. G. Stunnenberg, A. Tanay, D. Torrents, A. Valencia, E. Vellenga, M. Vingron, J. Walter and S. Willcocks, Nat. Biotechnol., 2012, 30, 224–226 CrossRef CAS PubMed.
- S. E. Celniker, L. A. L. Dillon, M. B. Gerstein, K. C. Gunsalus, S. Henikoff, G. H. Karpen, M. Kellis, E. C. Lai, J. D. Lieb, D. M. MacAlpine, G. Micklem, F. Piano, M. Snyder, L. Stein, K. P. White and R. H. Waterston, Nature, 2009, 459, 927–930 CrossRef CAS PubMed.
- L. Andersson, A. L. Archibald, C. D. Bottema, R. Brauning, S. C. Burgess, D. W. Burt, E. Casas, H. H. Cheng, L. Clarke, C. Couldrey, B. P. Dalrymple, C. G. Elsik, S. Foissac, E. Giuffra, M. A. Groenen, B. J. Hayes, L. S. Huang, H. Khatib, J. W. Kijas, H. Kim, J. K. Lunney, F. M. McCarthy, J. C. McEwan, S. Moore, B. Nanduri, C. Notredame, Y. Palti, G. S. Plastow, J. M. Reecy, G. A. Rohrer, E. Sarropoulou, C. J. Schmidt, J. Silverstein, R. L. Tellam, M. Tixier-Boichard, G. Tosser-Klopp, C. K. Tuggle, J. Vilkki, S. N. White, S. Zhao and H. Zhou, Genome Biol., 2015, 16, 57 CrossRef PubMed.
- S. Foissac, S. Djebali, K. Munyard, N. Vialaneix, A. Rau, K. Muret, D. Esquerré, M. Zytnicki, T. Derrien, P. Bardou, F. Blanc, C. Cabau, E. Crisci, S. Dhorne-Pollet, F. Drouet, T. Faraut, I. Gonzalez, A. Goubil, S. Lacroix-Lamandé, F. Laurent, S. Marthey, M. Marti-Marimon, R. Momal-Leisenring, F. Mompart, P. Quéré, D. Robelin, M. S. Cristobal, G. Tosser-Klopp, S. Vincent-Naulleau, S. Fabre, M.-H. P.-V. der Laan, C. Klopp, M. Tixier-Boichard, H. Acloque, S. Lagarrigue and E. Giuffra, BMC Biol., 2019, 17, 108 CrossRef CAS PubMed.
- D. Bujold, D. A. de Lima Morais, C. Gauthier, C. Côté, M. Caron, T. Kwan, K. C. Chen, J. Laperle, A. N. Markovits, T. Pastinen, B. Caron, A. Veilleux, P.-É. Jacques and G. Bourque, Cell Syst., 2016, 3, 496–499 CrossRef CAS PubMed.
- C. A. Davis, B. C. Hitz, C. A. Sloan, E. T. Chan, J. M. Davidson, I. Gabdank, J. A. Hilton, K. Jain, U. K. Baymuradov, A. K. Narayanan, K. C. Onate, K. Graham, S. R. Miyasato, T. R. Dreszer, J. S. Strattan, O. Jolanki, F. Y. Tanaka and J. M. Cherry, Nucleic Acids Res., 2017, 46, D794–D801 CrossRef PubMed.
- M. Sánchez-Castillo, D. Ruau, A. C. Wilkinson, F. S. Ng, R. Hannah, E. Diamanti, P. Lombard, N. K. Wilson and B. Gottgens, Nucleic Acids Res., 2014, 43, D1117–D1123 CrossRef PubMed.
- H. G. Stunnenberg, M. Hirst, S. Abrignani, D. Adams, M. de Almeida, L. Altucci, V. Amin, I. Amit, S. E. Antonarakis, S. Aparicio, T. Arima, L. Arrigoni, R. Arts, V. Asnafi, M. Esteller, J.-B. Bae, K. Bassler, S. Beck, B. Berkman, B. E. Bernstein, M. Bilenky, A. Bird, C. Bock, B. Boehm, G. Bourque, C. E. Breeze, B. Brors, D. Bujold, O. Burren, M. J. Bussemakers, A. Butterworth, E. Campo, E. C. de Santa-Pau, L. Chadwick, K. M. Chan, W. Chen, T. H. Cheung, L. Chiapperino, N. H. Choi, H.-R. Chung, L. Clarke, J. M. Connors, P. Cronet, J. Danesh, M. Dermitzakis, G. Drewes, P. Durek, S. Dyke, T. Dylag, C. J. Eaves, P. Ebert, R. Eils, J. Eils, C. A. Ennis, T. Enver, E. A. Feingold, B. Felder, A. Ferguson-Smith, J. Fitzgibbon, P. Flicek, R. S.-Y. Foo, P. Fraser, M. Frontini, E. Furlong, S. Gakkhar, N. Gasparoni, G. Gasparoni, D. H. Geschwind, P. Glažar, T. Graf, F. Grosveld, X.-Y. Guan, R. Guigo, I. G. Gut, A. Hamann, B.-G. Han, R. A. Harris, S. Heath, K. Helin, J. G. Hengstler, A. Heravi-Moussavi, K. Herrup, S. Hill, J. A. Hilton, B. C. Hitz, B. Horsthemke, M. Hu, J.-Y. Hwang, N. Y. Ip, T. Ito, B.-M. Javierre, S. Jenko, T. Jenuwein, Y. Joly, S. J. Jones, Y. Kanai, H. G. Kang, A. Karsan, A. K. Kiemer, S. C. Kim, B.-J. Kim, H.-H. Kim, H. Kimura, S. Kinkley, F. Klironomos, I.-U. Koh, M. Kostadima, C. Kressler, R. Kreuzhuber, A. Kundaje, R. Küppers, C. Larabell, P. Lasko, M. Lathrop, D. H. Lee, S. Lee, H. Lehrach, E. Leitão, T. Lengauer, Å. Lernmark, R. D. Leslie, G. K. Leung, D. Leung, M. Loeffler, Y. Ma, A. Mai, T. Manke, E. R. Marcotte, M. A. Marra, J. H. Martens, J. I. Martin-Subero, K. Maschke, C. Merten, A. Milosavljevic, S. Minucci, T. Mitsuyama, R. A. Moore, F. Müller, A. J. Mungall, M. G. Netea, K. Nordström, I. Norstedt, H. Okae, V. Onuchic, F. Ouellette, W. Ouwehand, M. Pagani, V. Pancaldi, T. Pap, T. Pastinen, R. Patel, D. S. Paul, M. J. Pazin, P. G. Pelicci, A. G. Phillips, J. Polansky, B. Porse, J. A. Pospisilik, S. Prabhakar, D. C. Procaccini, A. Radbruch, N. Rajewsky, V. Rakyan, W. Reik, B. Ren, D. Richardson, A. Richter, D. Rico, D. J. Roberts, P. Rosenstiel, M. Rothstein, A. Salhab, H. Sasaki, J. S. Satterlee, S. Sauer, C. Schacht, F. Schmidt, G. Schmitz, S. Schreiber, C. Schröder, D. Schübeler, J. L. Schultze, R. P. Schulyer, M. Schulz, M. Seifert, K. Shirahige, R. Siebert, T. Sierocinski, L. Siminoff, A. Sinha, N. Soranzo, S. Spicuglia, M. Spivakov, C. Steidl, J. S. Strattan, M. Stratton, P. Südbeck, H. Sun, N. Suzuki, Y. Suzuki, A. Tanay, D. Torrents, F. L. Tyson, T. Ulas, S. Ullrich, T. Ushijima, A. Valencia, E. Vellenga, M. Vingron, C. Wallace, S. Wallner, J. Walter, H. Wang, S. Weber, N. Weiler, A. Weller, A. Weng, S. Wilder, S. M. Wiseman, A. R. Wu, Z. Wu, J. Xiong, Y. Yamashita, X. Yang, D. Y. Yap, K. Y. Yip, S. Yip, J.-I. Yoo, D. Zerbino and G. Zipprich, Cell, 2016, 167, 1145–1149 CrossRef CAS PubMed.
- S. J. Marygold, M. A. Crosby and J. L. Goodman, Methods in Molecular Biology, Springer, New York, 2016, pp. 1–31 Search PubMed.
- J. Chèneby, M. Gheorghe, M. Artufel, A. Mathelier and B. Ballester, Nucleic Acids Res., 2017, 46, D267–D275 CrossRef PubMed.
- J. Chèneby, Z. Ménétrier, M. Mestdagh, T. Rosnet, A. Douida, W. Rhalloussi, A. Bergon, F. Lopez and B. Ballester, Nucleic Acids Res., 2020, 48(D1), D180–D188 Search PubMed.
- D. Li, S. Hsu, D. Purushotham, R. L. Sears and T. Wang, Nucleic Acids Res., 2019, 47, W158–W165 CrossRef CAS PubMed.
- C. Coarfa, C. S. Pichot, A. Jackson, A. Tandon, V. Amin, S. Raghuraman, S. Paithankar, A. V. Lee, S. E. McGuire and A. Milosavljevic, BMC Bioinf., 2014, 15, S2 CrossRef PubMed.
- F. Albrecht, M. List, C. Bock and T. Lengauer, Nucleic Acids Res., 2016, 44, W581–W586 CrossRef CAS PubMed.
- G. Devailly, A. Mantsoki and A. Joshi, Bioinformatics, 2016, 32, 3354–3356 CrossRef CAS PubMed.
- Y. He and T. Wang, Bioinformatics, 2017, 33, 3268–3275 CrossRef CAS PubMed.
- M. G. Dozmorov, Bioinformatics, 2017, 33, 3323–3330 CrossRef CAS PubMed.
- S. Oki, T. Ohta, G. Shioi, H. Hatanaka, O. Ogasawara, Y. Okuda, H. Kawaji, R. Nakaki, J. Sese and C. Meno, EMBO Rep., 2018, 19, 12 CrossRef PubMed.
- M. M. Hoffman, O. J. Buske, J. Wang, Z. Weng, J. A. Bilmes and W. S. Noble, Nat. Methods, 2012, 9, 473–476 CrossRef CAS PubMed.
- J. Curado, C. Iannone, H. Tilgner, J. Valcárcel and R. Guigó, Genome Biol., 2015, 16, 236 CrossRef PubMed.
- Y. Li, J. Zhu, G. Tian, N. Li, Q. Li, M. Ye, H. Zheng, J. Yu, H. Wu, J. Sun, H. Zhang, Q. Chen, R. Luo, M. Chen, Y. He, X. Jin, Q. Zhang, C. Yu, G. Zhou, J. Sun, Y. Huang, H. Zheng, H. Cao, X. Zhou, S. Guo, X. Hu, X. Li, K. Kristiansen, L. Bolund, J. Xu, W. Wang, H. Yang, J. Wang, R. Li, S. Beck, J. Wang and X. Zhang, PLoS Biol., 2010, 8, e1000533 CrossRef PubMed.
- M. B. Stadler, R. Murr, L. Burger, R. Ivanek, F. Lienert, A. Schöler, C. Wirbelauer, E. J. Oakeley, D. Gaidatzis, V. K. Tiwari and D. Schübeler, Nature, 2011, 480, 490–495 CrossRef CAS PubMed.
- A. K. Maunakea, I. Chepelev, K. Cui and K. Zhao, Cell Res., 2013, 23, 1256–1269 CrossRef CAS PubMed.
- G. L. Maor, A. Yearim and G. Ast, Trends Genet., 2015, 31, 274–280 CrossRef PubMed.
- X.-L. Ding, X. Yang, G. Liang and K. Wang, Sci. Rep., 2016, 6, 24545 CrossRef CAS PubMed.
- R. Shayevitch, D. Askayo, I. Keydar and G. Ast, RNA, 2018, 24, 1351–1362 CrossRef CAS PubMed.
- Y. Xu, Y. Wang, J. Luo, W. Zhao and X. Zhou, Nucleic Acids Res., 2017, 45, 12100–12112 CrossRef CAS PubMed.
- Y. Xu, W. Zhao, S. D. Olson, K. S. Prabhakara and X. Zhou, Genome Biol., 2018, 19, 133 CrossRef PubMed.
- V. Nanavaty, E. W. Abrash, C. Hong, S. Park, E. E. Fink, Z. Li, T. J. Sweet, J. M. Bhasin, S. Singuri, B. H. Lee, T. H. Hwang and A. H. Ting, Mol. Cell, 2020, 78, 752–764 CrossRef CAS PubMed.
- A. Frankish, M. Diekhans, A.-M. Ferreira, R. Johnson, I. Jungreis, J. Loveland, J. M. Mudge, C. Sisu, J. Wright, J. Armstrong, I. Barnes, A. Berry, A. Bignell, S. C. Sala, J. Chrast, F. Cunningham, T. D. Domenico, S. Donaldson, I. T. Fiddes, C. G. Girón, J. M. Gonzalez, T. Grego, M. Hardy, T. Hourlier, T. Hunt, O. G. Izuogu, J. Lagarde, F. J. Martin, L. Martínez, S. Mohanan, P. Muir, F. C. P. Navarro, A. Parker, B. Pei, F. Pozo, M. Ruffier, B. M. Schmitt, E. Stapleton, M.-M. Suner, I. Sycheva, B. Uszczynska-Ratajczak, J. Xu, A. Yates, D. Zerbino, Y. Zhang, B. Aken, J. S. Choudhary, M. Gerstein, R. Guigó, T. J. P. Hubbard, M. Kellis, B. Paten, A. Reymond, M. L. Tress and P. Flicek, Nucleic Acids Res., 2018, 47, D766–D773 CrossRef PubMed.
- R. Patro, G. Duggal, M. I. Love, R. A. Irizarry and C. Kingsford, Nat. Methods, 2017, 14, 417–419 CrossRef CAS PubMed.
- E. Lieberman-Aiden, N. L. van Berkum, L. Williams, M. Imakaev, T. Ragoczy, A. Telling, I. Amit, B. R. Lajoie, P. J. Sabo, M. O. Dorschner, R. Sandstrom, B. Bernstein, M. A. Bender, M. Groudine, A. Gnirke, J. Stamatoyannopoulos, L. A. Mirny, E. S. Lander and J. Dekker, Science, 2009, 326, 289–293 CrossRef CAS PubMed.
- S. Shukla, E. Kavak, M. Gregory, M. Imashimizu, B. Shutinoski, M. Kashlev, P. Oberdoerffer, R. Sandberg and S. Oberdoerffer, Nature, 2011, 479, 74–79 CrossRef CAS PubMed.
- T. Kouzarides, Cell, 2007, 128, 693–705 CrossRef CAS PubMed.
- I. A. Tchasovnikarova, R. T. Timms, N. J. Matheson, K. Wals, R. Antrobus, B. Gottgens, G. Dougan, M. A. Dawson and P. J. Lehner, Science, 2015, 348, 1481–1485 CrossRef CAS PubMed.
- K. Wood, M. Tellier and S. Murphy, Biomolecules, 2018, 8, 11 CrossRef PubMed.
- A. M. Deaton and A. Bird, Genes Dev., 2011, 25, 1010–1022 CrossRef CAS PubMed.
- T. Baubec, R. Ivánek, F. Lienert and D. Schübeler, Cell, 2013, 153, 480–492 CrossRef CAS PubMed.
- J. C. Dohm, C. Lottaz, T. Borodina and H. Himmelbauer, Nucleic Acids Res., 2008, 36, e105 CrossRef PubMed.
- K. Struhl and E. Segal, Nat. Struct. Mol. Biol., 2013, 20, 267–273 CrossRef CAS PubMed.
- Y. Li, C. Li, S. Li, Q. Peng, N. A. An, A. He and C.-Y. Li, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8817–8822 CrossRef CAS PubMed.
- H. Guo, B. Hu, L. Yan, J. Yong, Y. Wu, Y. Gao, F. Guo, Y. Hou, X. Fan, J. Dong, X. Wang, X. Zhu, J. Yan, Y. Wei, H. Jin, W. Zhang, L. Wen, F. Tang and J. Qiao, Cell Res., 2016, 27, 165–183 CrossRef PubMed.
- A. R. Quinlan and I. M. Hall, Bioinformatics, 2010, 26, 841–842 CrossRef CAS PubMed.
- M. Lawrence, R. Gentleman and V. Carey, Bioinformatics, 2009, 25, 1841–1842 CrossRef CAS PubMed.
- P. Stempor and J. Ahringer, Wellcome Open Res., 2016, 1, 14 Search PubMed.
- A. L. Statham, D. Strbenac, M. W. Coolen, C. Stirzaker, S. J. Clark and M. D. Robinson, Bioinformatics, 2010, 26, 1662–1663 CrossRef CAS PubMed.
- M. Lawrence, W. Huber, H. Pagès, P. Aboyoun, M. Carlson, R. Gentleman, M. T. Morgan and V. J. Carey, PLoS Comput. Biol., 2013, 9, e1003118 CrossRef CAS PubMed.
- J. Lemon, R-News, 2006, 6(4), 8–12 Search PubMed.
- H. Wickham, R. François, L. Henry and K. Müller, dplyr: A Grammar of Data Manipulation, 2020 Search PubMed.
- L. Henry and H. Wickham, purrr: Functional Programming Tools, 2019 Search PubMed.
- D. Robinson and A. Hayes, broom: Convert Statistical Analysis Objects into Tidy Tibbles, 2019 Search PubMed.
- H. Mi, A. Muruganujan and P. D. Thomas, Nucleic Acids Res., 2012, 41, D377–D386 CrossRef PubMed.
- D. Karolchik, Nucleic Acids Res., 2004, 32, 493D–496D CrossRef PubMed.
- W. Chang, J. Cheng, J. Allaire, Y. Xie and J. McPherson, shiny: Web Application Framework for R, 2018 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0mo00130a |
| This journal is © The Royal Society of Chemistry 2021 |