
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-omics data integration considerations and study design for biological systems and disease
Stefan
Graw
a,
Kevin
Chappell
a,
Charity L.
Washam
ab,
Allen
Gies
a,
Jordan
Bird
a,
Michael S.
Robeson
II
 *c and
Stephanie D.
Byrum
*ab
*c and
Stephanie D.
Byrum
*ab
aDepartment of Biochemistry and Molecular Biology, University of Arkansas for Medical Sciences, 4301 West Markham Street (slot 516), Little Rock, AR 72205-7199, USA. E-mail: sbyrum@uams.edu; Fax: +1 501 526 7008; Tel: +1 501 686 5783
bArkansas Children's Research Institute, 13 Children's Way, Little Rock, AR 72202, USA
cDepartment of Biomedical Informatics, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA. E-mail: mrobeson@uams.edu; Fax: +1 501 526 5964; Tel: +1 501 526 4242
First published on 21st December 2020
Abstract
With the advancement of next-generation sequencing and mass spectrometry, there is a growing need for the ability to merge biological features in order to study a system as a whole. Features such as the transcriptome, methylome, proteome, histone post-translational modifications and the microbiome all influence the host response to various diseases and cancers. Each of these platforms have technological limitations due to sample preparation steps, amount of material needed for sequencing, and sequencing depth requirements. These features provide a snapshot of one level of regulation in a system. The obvious next step is to integrate this information and learn how genes, proteins, and/or epigenetic factors influence the phenotype of a disease in context of the system. In recent years, there has been a push for the development of data integration methods. Each method specifically integrates a subset of omics data using approaches such as conceptual integration, statistical integration, model-based integration, networks, and pathway data integration. In this review, we discuss considerations of the study design for each data feature, the limitations in gene and protein abundance and their rate of expression, the current data integration methods, and microbiome influences on gene and protein expression. The considerations discussed in this review should be regarded when developing new algorithms for integrating multi-omics data.
Introduction
The biological system is complex with many regulatory features such as DNA, mRNA, proteins, metabolites, and epigenetic features such as DNA methylation and histone post-translational modifications (PTMs). Each of these features can be influenced by a disease and cause changes in cell signaling cascades and phenotypes. In addition to the host regulatory mechanisms response to disease, the microbiome can make changes to the expression of the host features such as their genes, proteins, and/or PTMs. In order to gain insight into mechanisms of disease, we need to investigate each of these features and their interplay. For instance, cancers such as melanoma, lung, and thyroid cancers are driven by the BRAF oncogene.1 However, when patients are treated with therapies that inhibit BRAF, they often develop resistance. Recent multi-omics studies have revealed the heterogeneity and complexity of tumor features such as their genetic mutations, transcriptome, proteins, and signaling pathways. It is now appreciated that tumors can bypass the therapy and give rise to resistance programs.1,2Proper integration of multi-omics approaches has allowed deeper insights into disease etiology, such as unveiling the myriad ways in which the microbiome may play a part in mitigating or enhancing disease risk. This case can be exemplified in regard to incomplete breakdown of bisphenol A (BPA), a mass-produced chemical that is widely used in food packaging, plastics, and resins. BPA has become a growing public health concern as BPA is an endocrine disruptor (as reviewed in Yu 20193). Thus, research into the fast and complete degradation of BPA, and other compounds via microbial means is of great interest. Yu and colleagues (2019)3 were able to effectively combine multi-omics data to analyze a microbial community's ability to break down bisphenol A (BPA) products. Though prior research had already discovered the microbes’ ability to break down BPA, the interactions that allowed this reaction were yet unknown. Through a clever multi-omics design, the authors were able to use three major types of integrated analyses to identify differences in encoded and expressed microbial functions that were involved in the BPA-degrading microbial community.3
Another example, Poore et al. (2020) leveraged multi-omics and machine learning tools, to detect microbial biomarkers from blood and tissues, serving as a great example of microbiome-informed oncology.4 Here the research team was able to discriminate among healthy and cancer-free individuals as well as between multiple cancer types using plasma-derived, cell-free microbial nucleic acids. Finally, we refer the reader to other reviews about the importance of integrating microbes into multi-omics studies.5–10
There is a growing appreciation for multi-omics studies in context of therapeutic treatments. However, the methodologies are challenging for a variety of reasons. Each biological regulatory feature has technical hurdles to overcome due to sample preparation, sequencing platforms and depth, limits in instrumentation, and dynamic range.7,11 New data integration algorithms are being developed at a rapid pace. In this review, we discuss the background of cellular processes, current data integration methodologies, the considerations for multi-omics study design, and future directions.
Understanding cellular processes in context of ‘omics’
Biological systems are complex organisms with many various regulatory features. For instance, the human genome is composed of approximately 3.2 billion nucleotides that give rise to 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 25000 protein coding genes, and through alternative splicing events lead to over 1 million proteins (Fig. 1). Epigenetic modifications, as well as the microbiome, can influence the expression of both genes and proteins within the biological system under various conditions. In addition to varying numbers of genes and proteins within the biological system, there is also a large dynamic range of high and low abundant molecules within each feature. On top of biological complexity, there are limitations in each of the omic sequencing platforms. These factors should be considered when developing novel data integration methods and are discussed below.
000 to 25000 protein coding genes, and through alternative splicing events lead to over 1 million proteins (Fig. 1). Epigenetic modifications, as well as the microbiome, can influence the expression of both genes and proteins within the biological system under various conditions. In addition to varying numbers of genes and proteins within the biological system, there is also a large dynamic range of high and low abundant molecules within each feature. On top of biological complexity, there are limitations in each of the omic sequencing platforms. These factors should be considered when developing novel data integration methods and are discussed below.
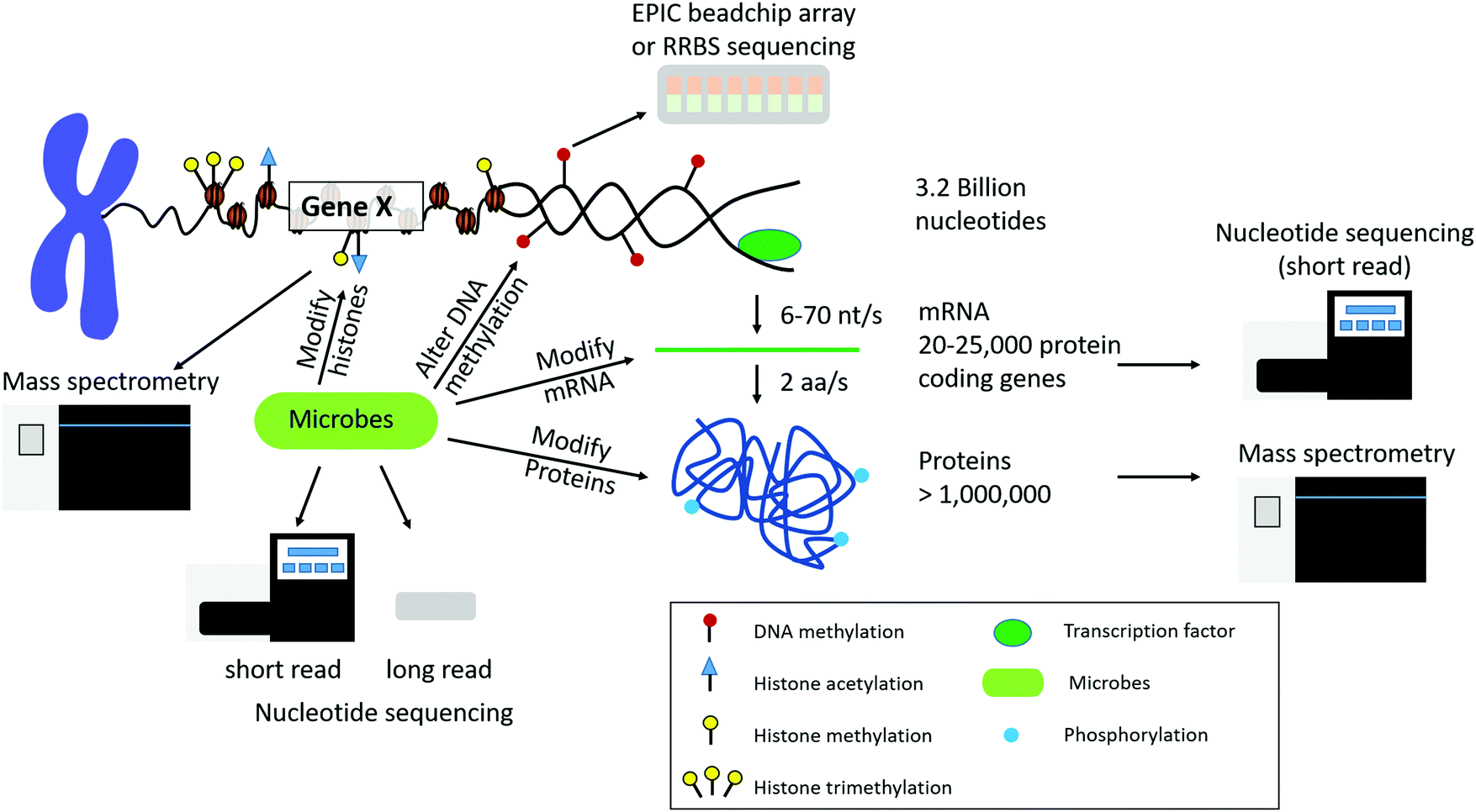 | ||
| Fig. 1 Overview of chromatin structure and gene/protein regulation. DNA access is regulated by DNA methylation and histone post-translational modifications (PTMs). There are approximately 3.2 billion nucleotides in the human genome transcribed to approximately 20000–25000 protein coding genes, which are translated to over 1 million proteins due to alternative splicing events. Each layer of regulation can also be modified by microbes that are present in the environment and host organism. Each level of biological regulation can be sequenced by using various nucleotide and protein/peptide sequencing technologies. | ||
Different organisms have varying numbers of genes and proteins. For instance, there are approximately 4300, 6000, and 25000 genes in the E. coli, S. cerevisiae, and H. sapiens genomes, respectively.12 This leads to approximately 2400 to 7800, 15000, and 300000 mRNA molecules per cell for E. coli,13S. cerevisiae,14 and H. sapiens,15 respectively. Mitochondrial transcripts can account for approximately 20% of polyadenylated RNA. Other high abundant transcripts include those that encode for ribosomal proteins and proteins involved in energy metabolism.16 It is important to note in sequencing platforms that only a fraction of all transcripts in a sample are actually sequenced and the potentially large number of transcript isoforms generated by alternative splicing events presents another challenge when integrating gene and protein level expression.17 The transcript isoforms may also change across biological conditions.18 An overview of the complexity of DNA, DNA methylation, histone post-translational modifications, mRNA, and proteins in humans is depicted in Fig. 1.
The estimated number of proteins in a cell is around 2.36 × 106, in E. coli and about 2.3 × 109 in H. sapiens HeLa cells.19 Within the vast number of total proteins in a cell, the most abundant proteins can make up 5–10% of protein content and consist of ribosomal proteins, acyl carrier protein (ACP) (functions in fatty acid biosynthesis), chaperones and folding catalysts, proteins of glycolysis (backbone of energy and carbon metabolism), and structural proteins such as actin. Transcription factors are low abundant proteins and range from 1–103 copies per cell in bacteria and 103–106 in mammalian cells. The most abundant proteins usually have many thousands of copies in bacteria and many millions in mammalian cells. The number of genes regulated by a transcription factor depends on its concentration.19 The protein content depends on the growth conditions and gene induction. Finally, this can become more complicated given the ratio of microbial-to-host cell count, which can depend on host cell type, and other factors.20
Sequencing technologies for various omics platforms only capture a snapshot of what is happening in a population of cells at one point in time due to limitations in instrument detection, dynamic range, and the lifetime expression of the molecules. For instance, the lifetime expression of mRNA transcripts and proteins are vastly different. The median lifetime of an mRNA in E. coli is 5 min, 20 min in budding yeast, and 600 min for H. sapiens.19 However, the lifetime of proteins is approximately 1–2 days. The rate of transcription and translation varies among organisms (E. coli: 10–100 nucleotides (nt) per second (s) and 10–20 amino acids (aa) per s. H. sapiens: 6–70 nt per s and 2 aa per s; rate of transcription and translation, respectively) (Fig. 1). For E. coli a single mRNA transcript can give rise to 10–100 proteins before being degraded. Given this information we can see that there will be an increased chance of detection of proteins with a longer life span, conflating our choice of omics platforms and the resultant interpretations of cellular processes.
It is important to recognize the biological complexity of organisms, dynamic range of molecules, sequencing limitations, as well as the lifetime of expression of those molecules when considering a data integration study design, developing a new algorithm, and when interpreting the results.
Microbiome influences on genes and proteins
In recent years, the importance of the microbiome in host health has been recognized. The idea of the holobiont and the hologenome has had profound implications in how we view the microbiome,21,22 especially in regard to therapeutics. The idea is that the interactions of the host's own genome and its “second genome”,23 collectively called the hologenome, work together to provide an “insurance policy” against a variety of perturbations24,25 that affect host health. This close relationship of microbe–host interactions can be more explicitly termed the “microbiota–nutrient metabolism–host epigenetic-axis”.26Microbiota and their metabolites can affect the host epigenetic landscape, by directly modifying histones, altering DNA methylation profiles, and influencing the nature of noncoding RNAs (Fig. 1). For example, histones can be modified by microbiota by altering the activity of histone modification enzymes, and the levels of the enzymes substrates.27–29
Microbiota can also affect the therapeutic nature of drugs. Many prodrugs, i.e. a drug that must metabolically converted in order to become pharmacologically useful, may remain inactive (i.e. the microbiota that mediate the conversion of the prodrug to its active form are not present), or the drug/prodrug, may not become bioavailable to the host, as a result of degradation by the hosts microbiota.30 Moreover, patients taking NSAIDs (non-steroidal anti-inflammatory drugs), may promote the preponderance of antibiotic resistant bacteria as 24% of tested over-the-counter NSAIDs inhibited the growth of at least one microbe in vitro.31 These metabolomic effects, raises concerns about potential side-effects of therapeutic drugs, or other diet and treatment regimens, intended to be used on humans and agricultural systems. For example, antibiotics can eliminate histone deacetylase (HDAC) inhibitor-producing microbes. These microbes, when present, can augment regulatory T (Treg) cells, which aids in anti-inflammatory processes.32
This means that the diversity of microbial metabolic pathways, and their impact on drug pharmacokinetics and pharmacodynamics,33,34 may partly explain the variation to drug responses between individuals and populations. Therefore, therapeutic treatments that involve the microbiome, may have to be regionally tailored.30,35,36
Histones can undergo both variant replacement and post-translational modification (PTM), together these form the “histone code”. These local arrangements can affect chromatin structure in such a way that leads to the activation or repression of transcriptional activity.37,38 Thus microbes, through diet, have the ability to modify methylation and PTM profiles of the host, and can also affect the generation of short-chain fatty acids (SCFAs) through the fermentation of dietary carbohydrates. SCFAs, such as butyrate and acetate, can inhibit deacetylase levels. Meaning that chromatin structure becomes increasingly relaxed due to acetylation driving increased transcriptional activity.26 In fact, it has been shown that microbes can affect host tissue acetylated and methylated chromatin states in a site-specific and combinatorial fashion and even impact host developmental and metabolic phenotypes.37–39
Modelling the development of the microbiome and its commensurate ontogenetic changes of the host, are increasingly being considered when trying to interrogate host health and therapeutics.40 Many microbial ecological principles such as community assembly are being brought to bear to investigate these processes.41,42 These changes can be exemplified through host immune maturation, considering that the host immune system must not only be able to recognize “self” antigens, but also those of symbiotic microbes. How microbes influence the expression of the major histocompatibility complex (MHC), or how host heterozygosity in turn affects the diversity of the microbiota through MHC, is largely unknown and is an active area of study.26,43 Finally, the role of microbes as they relate to cancer and immune treatments are increasingly becoming targets for the development of therapeutic strategies.44,45
Proteomics, in combination with other omics strategies have been used to interrogate disease processes. However, if we do not take into account the effects of microbiota (i.e. the entirety of the holobiont), then we may miss meaningful insights to develop potentially therapeutic treatments. Particularly those related to metabolic disorders (e.g. obesity), or the systemic effect of metabolites (e.g. bile acids) on organ systems.46 There is far more variation of our “second genome” that can be leveraged for human benefit compared to our own.47
Advances in microbial ecology
With heavy emphasis on understanding the effect of the microbiome it has become common practice for biomedical researchers to include methods to investigate the diversity of bacteria and archaea in their samples. The history of microbial ecology centers around the sequencing and alignment of appropriate phylogenetic marker genes. The 16S rRNA gene, first purposed as a marker by Woese and Fox (1977),48 is by far the most commonly used marker gene with massive databases of full length gene isolate from environmental and culture-derived sources (e.g. SILVA, RDP, Greengenes).49–51 (Table 1). New microbial taxonomy databases, such as the Genome Taxonomy Database (GTDB), not only curate a 16S rRNA gene reference database, but are also leveraging phylogenomic information52,53 to provide a consistent framework for determining the phylogenetic context partial or complete genomes derived from metagenomes.54| Resource | Data type | Link | Ref. |
|---|---|---|---|
| SILVA is a resource of databases of aligned ribosomal RNA (rRNA) gene sequences from the bacteria, archaea and eukaryota domains. | Gene sequences of 16S for prokaryotes and 18S for eukarya | https://www.arb-silva.de/ | 121 |
| Ribosomal database project: aligned and annotated rRNA gene sequence data | 16S rRNA sequences | http://rdp.cme.msu.edu/ | 122 |
| Greengenes is a dedicated full-length 16S rRNA gene database that provides users with a curated taxonomy based on de novo tree inference. | Taxonomy based on the 16S rRNA gene | https://greengenes.secondgenome.com/ | 123 |
| Genome Taxonomy Database is an initiative to establish a standardized microbial taxonomy based on genome phylogeny. The genomes used to construct the phylogeny are obtained from RefSeq and Genbank. | A comprehensive and phylogenomic-based taxonomy for bacterial and archaeal taxa | https://gtdb.ecogenomic.org/ | 52 and 53 |
| Universal Protein Resource (UniProt) is a comprehensive resource for protein sequence and annotation data | Protein sequence and annotation database | https://www.uniprot.org/ | 124 |
| NIH National Center for Biotechnology Information (NCBI) GenBank is an annotated collection of all publically available DNA sequences. Complete bimonthly release updates are available. Data is exchanged daily with the DNA DataBank of Japan and the European Nucleotide Archive. | Genomic sequence and annotation | https://www.ncbi.nlm.nih.gov/genbank/ | 125 |
| NIH/NCBI Reference Sequence (RefSeq) collection provides a comprehensive, integrated, non-redundant, well-annotated set of sequences, including genomic DNA, transcripts, and proteins | Genomic, transcriptomics, and proteomic sequence and annotation | https://www.ncbi.nlm.nih.gov/refseq/ | 126 |
| University of California Santa Cruz (UCSC) Genome Browser for exploring genome sequences and annotation. GenBank updates for mRNA, RefSeq, and EST data occur on a semi-quarterly basis. | Genome sequence and annotation database | http://genome.ucsc.edu/ | 127 |
| NIH National Human Genome Research Institute Encyclopedia of DNA Elements (ENCODE) Consortium project uses Reference Genomes from NCBI or UCSC | DNA methylation, and immunoprecipitation (IP) of proteins that interact with DNA and RNA, modified histones, transcription factors, chromatin regulators, and RNA-binding proteins. Genome sequence and annotation database. | https://www.encodeproject.org/ | 128 |
| Ensembl is a genome browser for vertebrate genomes that supports research in comparative genomics, evolution, sequence variation and transcriptional regulation. Updates are released every 2–3 months. | Genome sequence and annotation, gene models, transcriptional data, genetic variation and comparative analysis | http://ensembl.org/ | 129 |
| The Cancer Genome Atlas (TCGA) is a landmark cancer genomics program that molecularly characterized over 20000 primary cancer and matched normal samples spanning 33 cancer types. This a joint effort between the National Cancer Institute and the National Human Genome Research Institute. |
Individual patient tumor samples: DNA, RNA, protein, epigenetic changes | https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga | 130 |
| Cancer Cell Line Encyclopedia (CCLE) is a collaboration between the Broad Institute, and the Novartis Institutes for Biomedical Research and its Genomics Institute of the Novartis Research Foundation to conduct a detailed genetic and pharmacologic characterization of a large panel of human cancer models. CCLE contains genomics data and visualization for over 1400 cell lines. | Copy number, mRNA expression (Affy), RPPA, RRBS, and mRNA expression (RNAseq) | https://portals.broadinstitute.org/ccle | 131 |
| Therapeutically Applicable Research to Generate Effective Treatments (TARGET) is a community resource project. TARGET is organized into a collaborative network of disease-specific project teams with the goal of identifying molecular changes that drive childhood cancers. | Clinical information, gene expression, miRNA expression, copy number, sequencing data for cancers | https://ocg.cancer.gov/programs/target | Initiative phs000218 |
| Omics Discovery Index (OmicsDI) an open-source platform that enables access, discovery and dissemination of omics data sets. | Genomics, transcriptomics, proteomics, metabolomics | https://www.omicsdi.org/ | 132 |
| Multi-Omics Profiling Expression Database (MOPED) is a repository for multi-omics data of human and model organisms. | Transcriptomics and proteomics data and visualization | https://omictools.com/moped-tool | 133 |
| ProteomeXchange (PX) Consortium consists of PRIDE, PeptideAtlas, PASSEL, MassIVE and jPOST. Devoted to mass spectrometry (MS)-based proteomics data. | Proteomics data sets | http://www.proteomexchange.org/ | 134 and 135 |
Apart from selecting a marker gene and appropriate database, researchers also have a choice between sequencing methods and platforms. Due to limitations of short-read platforms such as Illumina and Ion Torrent, researchers must select between variable regions of the ∼1500 bp 16S rRNA gene. Each variable region provides a different level of sensitivity and specificity depending on microbial community composition. This is why preliminary amplicon surveys often compare a collection of primer sets and variable regions. The combination of the primer set and the amplicon region that best differentiates among the common taxa in the study, is then chosen. Alternative long-read platforms have recently been adapted to deliver high-throughput full-length 16S rRNA for researchers that need taxonomic resolution beyond the genus to family level typically provided by short-read technologies.55
Current metagenomic analysis techniques have allowed researchers to obtain partial and complete draft genomes from environmental/host-derived samples given sufficient sequence coverage. This coverage factor is highly dependent on the species evenness and richness. Researchers using these techniques can investigate potential functional differences of a collection of metagenome assembled (draft) genomes. However, often they have to use concentrated universal proteins to place these genomes in a phylogenetic context because of the difficulty of assembling and correctly binning highly conserved genes like the ribosomal subunit genes. Combined universal marker genes are used to construct the phylogeny from genomes assembled from environmental and host-derived sequences along with a minority of familiar microbial genomes from culture collections. The sudden rush of sequencing microbial genomes has necessitated the construction of easy-to-use wrappers and pipelines to aid biologists in learning how to approach the analysis of their metagenome data, either in whole or in part. Some great examples of such tools are, QIIME 2,56 metaWRAP,57 Sunbeam,58 SqueezeMeta,59 metAMOS,60 mg-RAST,61 IMG/M,62 Anvi’o,63 MicrobiomeAnalyst,64 and the variety of tools within the biobakery65 collection (e.g. MetaPhlan2,66 PhyloPhlan,67 HUMAnN,68 LEfSe69), among others (see ref. 70 for a review these and many other meta’omics tools). Biomedical researchers wading into the depths of microbial ecology looking to integrate disease metrics, host proteomics, and microbial diversity should be aware of the various databases, curatorial rigor, and the limitations of the sequencing platform they choose.
Sequencing technologies
Depending on the biological question, there are many types of omics technologies targeting DNA, total RNA, mRNA, miRNA, DNA methylation, proteins, protein modifications, histone post-translational modifications, metagenomics, metaproteomics, etc. Sequencing platforms have improved over the years and now allow for the sequencing of large complex human samples within a few days from small amounts of material (Table 2). Several workflows have been developed to sequence the whole genome, the whole exome (protein-coding portion of DNA), and transcriptome (mRNA), and arrays for specific cancer or immune-related genes. In addition, we can profile modifications, such as DNA methylation using either whole genome bisulfite sequencing or Illumina's MethylationEPIC BeadChip arrays. The detection of such modifications can also be determined through the direct sequencing of long read DNA and RNA via the Oxford Nanopore Technologies (ONT) MinION platform,71–74 and PacBio instrumentation.| Application | Recommended coverage (×) or reads (millions) | Ref. | ||
|---|---|---|---|---|
| Illumina | PacBio | Nanopore | ||
| Whole genome sequencing | >15× | >35× | >40× | 75 and 137 |
| Whole exome sequencing | >15× | >35× | >40× | 75 and 137 |
| Transcriptome sequencing (mRNA; differential expression analysis) | 10–30m | >30m | >30m | 138 and 139 |
| Transcriptome sequencing (alternative splicing; allele specific expression) | 50–100m | 50–100m | 50–100m | 139 |
| miRNA sequencing | >30m | >30m | >30m | 138 |
| 16S metagenomics | >100× | >100× | >100× | |
| Shotgun metagenomics | >80m | >80m | >80m | 140 |
| Histone ChIP-seq | >20m for narrow peak, >45m for broadpeak | >20m for narrow peak, >45m for broadpeak | >20m for narrow peak, >45m for broadpeak | 141 |
| Transcription factor ChIP-seq | >20m for narrow peak, >45m for broadpeak | >20m for narrow peak, >45m for broadpeak | >20m for narrow peak, >45m for broadpeak | 141 |
| ATAC-seq | >25m | >25m | >25m | 141 |
| DNA methylation sequencing (RRBS per strand) | >15× | >15× | >15× | 142 |
Error rates and read lengths vary between DNA sequencing technologies. Illumina short read sequencing (i.e. Hiseq, Miniseq, etc.) typically have very low error rates, at about 0.25% per base, but are sensitive to low diversity libraries, as is the case with applications such as 16S metagenomics and targeted gene approaches. Long read technologies have higher error rates, ranging from 13–15% for PacBio and 5–20% for Oxford Nanopore instruments.75,76 Read length for Illumina platforms have a maximum length of 600 bases but long read technologies commonly achieve 10–30 kb for a single read.77 Optimal read length is also dependent on the application. Where most sequencing experiments can collect suitable information with 150–300 base pair read lengths, there are exceptions. Illumina's 16S Metagenomics protocol requires 2× 300 base pairs. For whole genome sequencing (WGS), the longest read possible is optimal but with long read technologies, the error rate increases with the length. Many researchers have combined long read and short read sequencing to “fill gaps” with WGS. Due to the fairly recent advent of long read sequencing technology, information on optimal long read lengths for applications other than WGS is sorely lacking but Illumina short read sequencing is rich in optimal read length recommendations.78
Long read sequencing technologies such as ONT and PacBio have already ushered in significant improvements in both the amplicon and metagenomic sequencing space. From high resolution analysis of the full length 16S gene,55 the entire rRNA operon,79 to improving the ability to close entire microbial genomes.80 For an in-depth overview on these long-read sequencing technologies, see Amarasinghe et al.77
Mass spectrometers have also improved by increasing sequencing depth capabilities over the past 5–10 years. The technology has advanced from sequencing roughly 3000 proteins in a cell line experiment using older LTQ mass spectrometers to routinely sequencing 8000–10000 proteins using newer Orbitrap Lumos and Orbitrap Eclipse mass spectrometers. Most proteomics experiments are performed using data dependent acquisition (DDA) mode. In this method, the top 20 most abundant peptides in the MS1 scan that are eluted from a liquid chromatography (LC) column are selected for fragmentation in the orbitrap in order to generate the peptide sequence MS2 scan. The complexity of the sample mixture highly influences the sequencing depth and how many proteins will be identified. Understanding the protein abundance and make-up of the samples is critical. If transcription factors are the target molecules, then some method of removing highly abundant proteins prior to mass spectrometry may be necessary. This is especially critical for serum and plasma samples that have high abundant molecules, such as albumin and hemoglobin. Otherwise, the mass spectrometer will sequence thousands of molecules of albumin and miss the most interesting low abundant proteins.81
The latest mass spectrometry technology utilizes data independent acquisition (DIA) to sequence all of the peptides from the MS1 scan as they elute from the LC column as opposed to DDA methods that only sequence the top most abundant peaks. DIA methods are beneficial over DDA for complex mixtures, such as in the serum example above. This method helps to overcome complex mixtures that are highly influenced by high abundant proteins.82–84
In addition to shotgun sequencing for the host genes and/or proteins, we can also utilize shotgun sequencing for the microbiome. Shotgun metagenomics/metaproteomics may only sample the dominant microbiota when the sequencing depth is very shallow. A major challenge of shotgun sequencing the microbiome is the difficulty in assembling genome fragments due to under sampling, it is also just as difficult to piece together peptides for robust protein and taxa identification.
Despite these potential issues, it is possible to sample the microbial proteome in depth from a variety of human body sites and diseases, such as saliva, gut/feces, cervicovaginal, or chronic kidney disease.40,85–87 However, the study/sampling design and analytical approaches one must consider can differ greatly between each study. Several sampling preparation approaches have been shown to enrich microbial biomass ranging from differential centrifugation through double-filtering differential separation. These approaches are often followed by a variety of optimized microbial lysis protocols, typically involving mechanical disruption (e.g. bead beating, sonications), complemented with enzymes (e.g. trypsin) and detergents. Upon successful lysis, it is just as important that remaining enzymes, detergents and salts be removed. For more details see the review by Issa Isaac et al. and Lin et al.7,40 and the references therein.
Another complication for metaproteomics experiments is due to the fact that proteins within the same organism have shared peptide sequences. In order to have confidence in the protein identification, a unique peptide match for the protein should be identified with high confidence. This is made even more complicated when mapping peptide sequences to hundreds of different species that have conserved protein sequences. Mass spectrometry does not sequence proteins, but rather measures the mass-to-charge of peptides and relies on mass spectra matches to a database of protein sequences for protein identification. However, there is hope to make sense of these (Tables 1 and 2).7,40
Curated databases are critical to properly analyze nucleotide and protein sequencing data generated from these various sequencing platforms. The ability to align reads to a reference genome is only as good as the sequence and annotation information present in the reference genome. There are several resources that continually curate and update nucleotide sequence information and annotation including University of California Santa Cruz (UCSC) Genomics Institute genomes, National Center for Biotechnology Information (NCBI) GenBank and RefSeq, Encyclopedia of DNA Elements (ENCODE), and Ensembl to name a few. The Universal Protein Resource (UniProt) contains both Swiss-Prot (manually annotated and reviewed) and TrEMBL (automatically annotated and not reviewed) databases for protein sequence information (Table 1).
Data integration and current methodologies
Several data integration methodologies have been developed to integrate certain types of omics data. In addition, large data repositories have been created to house data from sequencing experiments for various diseases. These resources provide valuable building blocks and large amounts of biological samples that can be utilized to push data integration methods forward. Currently, data integration tools implement a variety of methods but generally fall under two categories: multi-staged analysis and meta-dimensional analysis.88 Multi-staged integration models are constructed using only two numerical or categorical features of the data. For example, gene counts from an RNA-seq experiment are combined with protein information from a mass spectrophotometry run. Meta-dimensional analysis attempts to incorporate all the types of data of interest by concatenation or transformation into a simultaneous matrix or “metadata” set that can be analyzed simultaneously. The latter method has more statistical power but can be challenging when attempting to combine data from different types of datasets. Yet, how does a researcher decide which tool or method is most appropriate? As stated above, the biological question is the driving force in the type of analysis method chosen and factors such as sampling, the type of platform, and quality of the data are important. How were the samples collected and prepared? Can the data be effectively analyzed if sequencing depth or quality is low? Are the data types compatible? How much signal is lost after normalization and filtering? These are all questions that should be considered before choosing the appropriate tools.Unfortunately, data integration and analysis are very complicated and there currently do not exist many user-friendly tools for researchers with limited bioinformatics backgrounds. Many tools utilize the statistical language R, which requires programming expertise in addition to strong biostatistical knowledge. For example, the R package integrOmics, which combines proteomics, transcriptomics, and pathway analysis on two data sets uses correlation analysis and partial least squares regression.89 The R package mixOmics uses multivariate analysis for data exploration, dimension reduction and visualization.90 Micrographite integrates miRNA and gene expression with pathway analysis91 and iClusterplus92 and LRACluster93 use clustering to integrate methylation and gene expression data (Table 3).
| Tool | Purpose | Types of omics data | Pros | Cons | Ref. | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Metabolomics | Proteomics | Transcriptomics | Pathway analysis | miRNA | SNP analysis | Microbiome | DNA methylation | Copy number variants (CNV) | Genomics | Visualization | |||||
| Metaboanalyst 4.0 | Metabolomics data analysis, interpretation, and integration with other omics data | × | × | × | × | Relatively easy to use. Has a web interface. Basic computer skills. | File size limit (50 Mb), installed version requires Linux and programming experience. | 143 | |||||||
| Paintomics 3.0 (web based) | Joint visualization of transcriptomics and metabolomics data | × | × | × | × | Relatively easy to use. Automatic feature name conversion. Has a web interface. Basic computer skills. | Most web applications have a file size limit. | 97 | |||||||
| integrOmics (R package) | Integrative analysis of two types of omics datasets | × | × | × | × | Customizable | Requires programming skills in R | 89 | |||||||
| Omics Integrator | Maps protein data to other data sets | × | × | × | × | × | × | Easy to use web interface. Basic computer skills. | Most web applications have a file size limit. Local installation requires advanced computer skills | 144 | |||||
| mixOmics (R package) | Data exploration, dimension reduction, and visualization | × | × | × | × | × | Customizable | Requires advanced programming skills, programming in R | 90 | ||||||
| PARADIGM | Extraction of disease-perturbed sub pathways within pathway networks | × | × | × | × | Uses a combination of algorithms to improve accuracy | Requires advanced programming skills, command line interface, programming in R. Pathways are measured independently, and interactions among pathways are not considered. | 145 | |||||||
| Micrographite (R Package) | Pathway analysis of miRNA and gene expression profiles | × | × | × | Customizable, integrates pathway information with predicted and validated miRNA-target interactions. | Requires advanced computer skills, programming in R. | 91 | ||||||||
| iClusterplus | Integrative clustering of multiple data sets | × | × | × | × | Customizable. Incorporates flexible modeling of the associations between different data types | Requires advanced computer skills, computationally intensive, limitations in statistical inference, programming skills in R | 92 | |||||||
| LRAcluster | Integrative clustering of multiple data sets | × | × | × | × | Fast and efficient unsupervised clustering | Command line interface, requires advanced computer skills. | 93 | |||||||
| GENEASE | Disease ontology exploration, analysis, and visualization of multiple databases | × | × | × | × | × | × | × | Web based interface. Uses multiple databases in real time. | Most web applications have a file size limit. | 146 | ||||
| ProteoClade | Annotate taxa to proteomics data | × | × | × | Customizable. Can work with large data sets. Targeted and de novo database searches. Good tutorials. | Requires advanced computer skills, programming in Python. | 147 | ||||||||
| Qiime2 (q2-micom) | Metabolic modeling | × | × | Customizable, Highly versatile. Good tutorials. | Steep learning curve. Requires advanced computer skills | 148 | |||||||||
| Qiime2 (q2-mmvec) | Learning microbiome/metabolic interactions | × | × | Customizable, highly versatile. Good tutorials. | Steep learning curve. Requires advanced computer skills | 149 | |||||||||
| Qiime2 (q2-metabolomics) | Tool to import metabolomic data into Qiime2 | × | × | Customizable, highly versatile. Good tutorials. | Steep learning curve. Requires advanced computer skills | 150 | |||||||||
For both multi-state and meta-dimensional methods, many different algorithms are used, but the most common ones are clustering, network analysis, data reduction (PCA), and Bayesian analysis.94 Ray et al. (2014) used Bayesian analysis to analyze gene expression and methylation data in ovarian cancer using data collected from the Cancer Genome Atlas Project and detected a gene, SPON1, which appears to be regulated by methylation of its CpG site.95 Correlation based analysis can be useful when prior knowledge of biochemical interactions is lacking.96 Regardless of the methodology, appropriate normalization and data filtering is very important as data is being incorporated from multiple sources.
There also exist some web-based tools such as Paintomics97 that attempt to make the data analysis easier but can still be difficult for the inexperienced user and the researcher must have a good working knowledge of their data.98 Further, there are databases that are commonly used in integrated omics analysis, such as the Cancer Cell Line Encyclopedia (CCLE), The Cancer Genome Atlas Program (TCGA), Tumor Alterations Relevant for Genomics-driven Therapy (TARGET), and Omics Discovery Index (OmicsDI)94 (Table 1). CCLE and TCGA have characterized thousands of cancer data sets and can be used for data mining and visualization. TARGET utilizes clinical information and has resources for analytical tools on their websites. OmicsDI provides a platform for searching public and protected data for a large variety of organisms.
Considerations for study design and power evaluation
As for any high-quality study, conducting a multi-omics study should always begin with identifying the scope and restrictions of a study. Careful planning and execution will improve a study's robustness and reproducibility and are especially crucial in multi-omics studies, as they involve a large number of comparisons, tailored statistical analysis, substantial financial and timely investments.10,98 Involving a statistician from the very beginning of a study is critically important to assist the researcher to identify the research question, define clear a priori hypotheses, proper experimental design, study analysis and interpretation, drawing conclusions and much more.99,100Once research hypotheses are clearly defined, a suitable study design is selected that addresses the research hypotheses best. Therefore, several questions need to be evaluated, such as: are one or more intervention groups compared to a control (or themselves), or is an effect evaluated in the same samples before and after intervention? Is an intervention effect over one period of time or will samples be measured at several different time points? Will biological samples be pooled or analyzed individually and what is the scientific justification for it101? Which types of omic platforms will provide the most value101 and how are the multi-omics data going to be integrated? Are samples from the same biological source available for all multi-omics platforms of interest? Ideally, samples for all omic platforms would be collected from the same source. However, this is not always possible due to sample-specific limitations or accessibility and amount of the material.101 For instance, generating multi-omics data from formalin-fixed paraffin-embedded (FFPE) tissue might not be possible for certain omic platforms.101 While there are many questions to be considered during the selection of the experimental design, the deciding factor for the choice of a study design is usually its feasibility and financial limitations.10,102
Following the selection of a study design, available resources need to be allocated between the individual omic platforms.101 This allocation should be guided by the cost and contribution of each individual omic platforms to the multi-omics study as well as the statistical power of each individual omic platforms. Omic platforms with a substantial signal-to-background noise ratio will require less samples and allow for an allocation of more resources to omic platforms with a small(er) signal-to-background noise ratio, as these platforms require more samples to achieve (similar) adequate statistical power. In addition, some omic platforms will also require some internal distribution of resources. For example, when designing an RNA-Seq study the trade-off between the number of samples and sequencing depth will need to be balanced.10
The sample and data collection should be guided by the data analysis to reduce confounding and technical artifacts, such as batch effects.10 These effects can be introduced during steps of the sample collection, preparation and storing (e.g. multiplexing).10,101 While some ad hoc methods attempt to reduce such biases introduced by technical artifacts, they are inferior to a randomized design.98 However, some technical artifacts cannot be avoided, and in these cases it is important to identify and understand such limitations early in experimental design to mitigate and recognize their impact on the results and conclusions.10,101
Due to the complexity and large volume of data associated with multi-omics studies it is crucially important to tailor the statistical analysis to a specific research project.10 A variety of methods for integrating multi-omics data have been proposed and categorized as either supervised, semi-supervised, or unsupervised;103 as well as, conceptual, statistical, correlation, network, and model-based integration.102 The integration and statistical analysis of a multi-omics study depend on the selection of omic platforms and their associated types of data (e.g. count values, percentages). Nevertheless, each analysis and method have its underlying assumptions that need to be verified.98
As in any well-designed study, an initial power calculation is increasingly crucial to evaluate and estimate a sufficient number of samples and avoid a potential waste of resources, especially in such large-scale studies.10 Power is defined as the probability of correctly rejecting the null hypothesis, which is the likelihood of detecting a true signal or effect. A mathematical power calculation is usually impossible due to the complex nature of the study design and data; however, estimating statistical power using simulation is a valuable alternative. The evaluation of statistical power involves four major steps. First, data needs to be simulated and a pilot study, prior knowledge, literature or experts can be beneficial for the generation of realistic data. Next, a true signal needs to be introduced and should be guided by the expected effect size. The data can then be analyzed, and the statistical power can be estimated empirically. In the context of a case-control study, the statistical power is the percentage of correctly identified features out of all features with an introduced effect size.
The statistical power of a study depends on several factors (Fig. 2) of which some can be controlled, while others are fixed due to the study and its design. First, the choice of statistical method used for the analysis. While some tests are more powerful than others, it is important that their assumptions are verified and met. Another factor that influences the statistical power of study is the number of variables measured by the individual omic platform, which is usually dictated by the omic platform.101 For example, genomics typically measures millions of variants,104,105 transcriptomics quantifies tens of thousands of molecules,106 and proteomics107 and metabolomics108,109 profile thousands of molecules. Further, statistical power is affected by the magnitude and prevalence of the effect of the phenotype or exposure (effect size). How distinct is the effect? How substantial is the signal difference between groups? And how many measured variables are affected? Information about the effect size might be available from previous literature or expert knowledge but is often unknown.101 In such cases, a pilot study can assist with estimating the effect size, but these estimates need to be handled with caution due to instability.101,110 Yet another power influencing factor is the homogeneity of the measured values, describing the natural variance of the sample, the precision of the measurement instrument and detection limits. With an increasing variance the statistical power will be reduced. The variance of the samples can be the result of many aspects, such as the sample population selection, choice of tissue type or confounding factors.111 In addition to sample variance inflation, confounding factors can also introduce biases in the data, and therefore it is important to collect sample meta-data to mitigate some effects of confounding.101 Because most of the factors affecting the statistical power of a study are fixed or dictated by the study design, the factor that is most commonly used to adjust the statistical power of a study is the sample size.
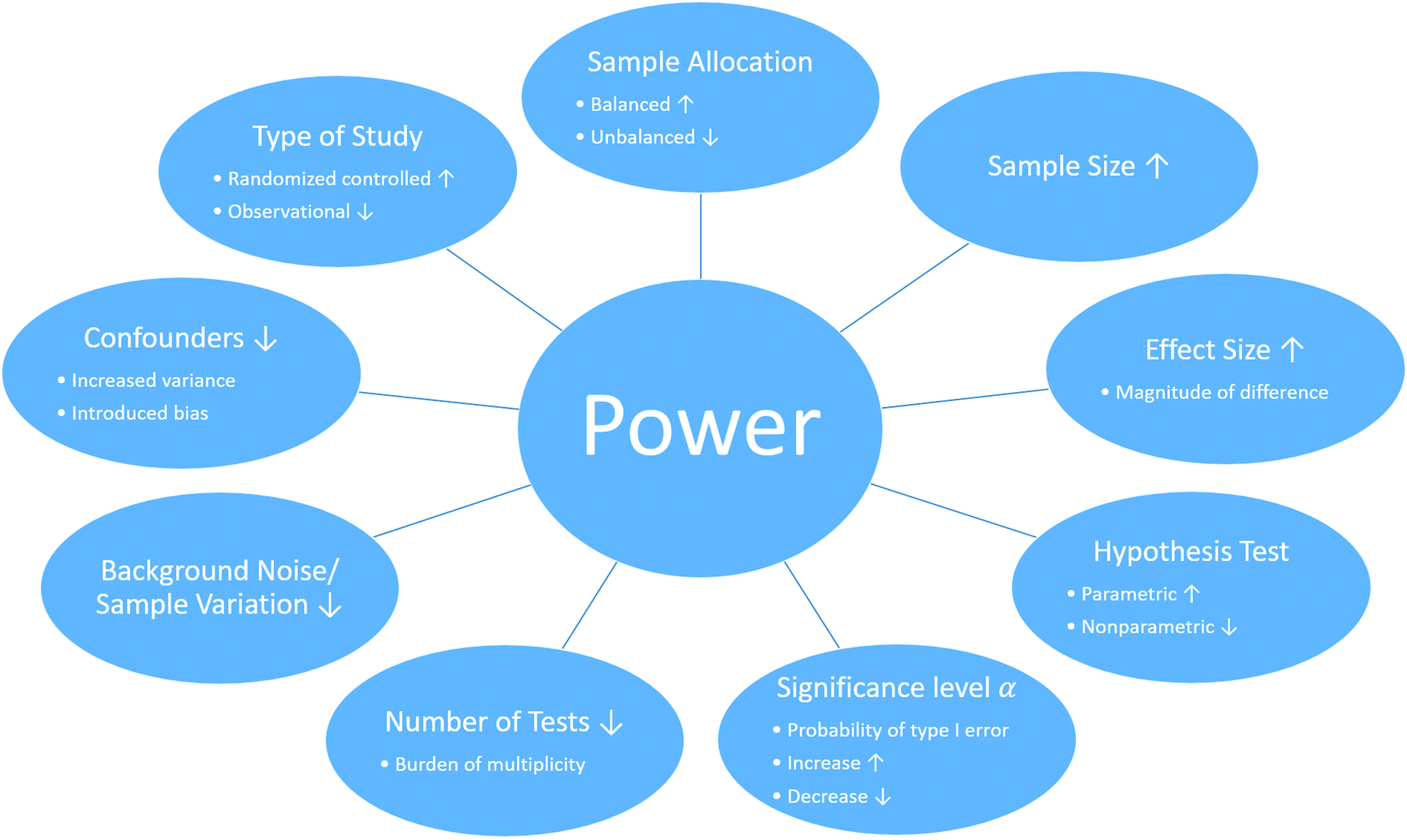 | ||
| Fig. 2 Factors that influence the statistical power in multi-omics studies. The statistical power of a multi-omics study can be effected by several factors including and must be considered at the beginning of the study. Such factors include, but are not limited to (the effect of the following factors on power are under the assumption that the remaining factors remain constant): (1) the type of the study. While randomized controlled studies are generally more powerful than observational studies due to controlling unwanted effects, limitations can prohibit this application of a randomized controlled study. (2) The sample allocation. In general, a balanced study, where samples are equally distributed among group, is more powerful unbalanced study. (3) Sample size. As the number of samples in a study increases the statistical power improves. (4) Effect size. The greater the true differences between groups, the greater the statistical power of a study. (5) Hypothesis test. While parametric tests are in general more powerful than nonparametric test, parametric tests are not applicable if there assumptions are not met. (6) Significance level α. The significant level represents the probability of type I errors, the probability of rejecting the null hypothesis given that the null hypothesis is true. As the numerical value of α increases, the probability of type I errors increases as well as the statistical power (probability of rejecting the null hypothesis given that the null hypothesis is true). (7) Number of tests. Testing multiple hypotheses requires a correction and reduces the statistical power. (8) Background noise and sample variation increase the variance and complicate the detection of a true signal and therefore decrease the statistical power. (9) Confounders can increase variance and/or introduce a bias, which decreases the statistical power. | ||
Applying power analyses for microbiome data is still a burgeoning field of inquiry and is replete with difficulties.112–115 The types of power analyses, like those outlined above, differ based on the questions being asked of microbiome data. Typically, power analyses of microbiome data center on measures of alpha and beta diversity, and differences in compositional abundances of taxa.114,116,117 Which of these measures to use will depend on the question at hand. How to integrate these into a multi-omics study is still underdeveloped.115
Power and sample size evaluation is a valuable technique during the experimental design of a study to ensure adequate power and sample size. While under- and overpower studies unnecessarily deplete resources, the risk of failure of a study is especially prevalent in underpowered studies. Underpowered studies and studies with improper experimental design are more likely to miss true signals, produce bias results, false positive (type I error) and false negative (type II error) results, which will lead to misinterpretations.10,101,111 Such incorrect inferences will impact the reproducibility, scientific progress and the cost of science.98,118
Conclusion and future directions
It is important to consider the context of the disease or research question that is under investigation and what types of data will provide valuable insight when integrated together. Depending on the biological question, type of material (fresh tissue, FFPE tissue, serum/plasma, and cell lines), amount of DNA/RNA/protein, number of biological replicates, and the number of confounding effects in a study, these factors will determine the best sample preparation and sequencing methods needed for data acquisition. Sample preparation methods including the day each sample is prepared, the type of DNA, RNA, and/or protein that is extracted, library generation for genomics, protein digestion and peptide labeling methods for mass spectrometry, and the sequencing platform/instrumentation are all key factors in the study design and the interpretation of the final results. If one sample is prepared on a different day than the other biological replicates, this will introduce variance and/or bias and reduce the statistical power of the analysis. If proteomics samples are multiplexed using multiple TMT-10plex batches, this will introduce a batch effect across sequencing runs. These factors should be discussed prior to sample preparation.It is also critical to know what population of regulatory features were captured for sequencing and can be integrated. For example, membrane bound proteins cannot be integrated with gene expression data if membrane proteins were not solubilized during sample preparation prior to performing mass spectrometry. A caveat with mass spectrometry data is the fact that a missing value does not necessarily mean a protein is not expressed, but only that the protein is below the detection limits of the mass spectrometer. The biological questions should be a driving force in the methodology used for multi-omics data integration.
Though multi-omics datasets can provide an individual with a greater depth of understanding in certain scenarios, this is not without cost. Omics studies often rely on large numbers of comparisons, the correct data type, appropriate statistical analyses, and a considerable investment of time, skilled personnel, and money. When constructing an experiment one must be weary of what types of omics data can and should be integrated to achieve the greatest understanding of the system being studied.98 High throughput omics platforms are not always necessary to answer the research question. Traditional techniques, such as enzyme-linked immunosorbent assay (ELISA) assays, immunohistochemistry (IHC), and quantitative polymerase chain reaction (qPCR), may be all that is necessary to validate a particular biological mechanism. In fact, these techniques are often required to validate the findings from a larger omics study in order to verify the significant molecule identified from omics data is a true positive result.
For the most part, current tools utilize clustering, networking, data reduction and Bayesian analysis. Because of ever increasing acquisition of data, resulting in large datasets and increasing numbers of them, machine learning will become more and more necessary for effective analysis and data mining. There is a need for accessible and well documented methods, tools and algorithms.96 As with all scientific endeavors, the easy questions will be answered first and “low hanging fruit” will be become less prevalent. Thus, there is a need for more effective algorithms and computing resources.88 Because of the variety of platforms used to generate multi-omics data, standardization of data formats would make integration easier.94
Future multi-omics data integration algorithms should take advantage of the “big data” resources (Table 1) and the advent of machine learning and artificial intelligence algorithms.10,119,120 Machine Learning has played an increasingly important role in allowing scientists to integrate multi-omics datasets. By utilizing a machines ability to compare and identify patterns in large quantities of biological data, we allow for far more accurate and efficient methods of elucidating complex cellular mechanisms and in some cases providing predictions to clinical outcomes. This is achieved through the computer's unique ability to observe multiple layers of omics data simultaneously providing a more holistic view of the systems at play, rather than observing each omic system individually and drawing simple conclusions based on visible correlations.4,120
New data integration methods should include variables related to each omic platform's weaknesses and limitations. Each method is limited by its statistical power, sample size, technical variables, batch effects, sequencing depths, sample preparation, and a multitude of other factors. These factors are important to keep in mind when designing, conducting and analyzing a study and interpreting the results. Therefore, it is highly recommended to involve a biostatistician/bioinformatician from the very beginning of any study. Their expert knowledge can be valuable at any stage of a study to prevent errors, wasting resources and optimize the study. The need for trainings program in this rapidly evolving field has been recognized by many institutes, such as Jackson Laboratory, Bioinformatics.org, UC Davis and Johns Hopkins, and many bioinformatics training programs are available online for free or with costs. Lastly, researchers should always remember to validate significant findings using other traditional wet lab techniques to unmask false positive results.
Conflicts of interest
No potential conflict of interest was reported by the authors.Acknowledgements
This study was supported by the Arkansas Children's Research Institute, the Arkansas Biosciences Institute, and the Center for Translational Pediatric Research funded under the National Institutes of Health grant P20GM121293.References
- A. Zaman, W. Wu and T. G. Bivona, Targeting Oncogenic BRAF: Past, Present, and Future, Cancers, 2019, 11(8), 1197 CrossRef CAS PubMed.
- A. Alvarez-Arenas, et al., Interplay of Darwinian Selection, Lamarckian Induction and Microvesicle Transfer on Drug Resistance in Cancer, Sci. Rep., 2019, 9(1), 9332 CrossRef PubMed.
- K. Yu, et al., An integrated meta-omics approach reveals substrates involved in synergistic interactions in a bisphenol A (BPA)-degrading microbial community, Microbiome, 2019, 7(1), 16 CrossRef PubMed.
- G. D. Poore, et al., Microbiome analyses of blood and tissues suggest cancer diagnostic approach, Nature, 2020, 579(7800), 567–574 CrossRef CAS PubMed.
- A. Gonzalez, et al., Characterizing microbial communities through space and time, Curr. Opin. Biotechnol., 2012, 23(3), 431–436 CrossRef CAS PubMed.
- D. Gurwitz, The Gut Microbiome: Insights for Personalized Medicine, Drug Dev. Res., 2013, 74(6), 341–343 CrossRef CAS.
- N. Issa Isaac, et al., Metaproteomics of the human gut microbiota: Challenges and contributions to other OMICS, Clin. Mass Spectrom., 2019, 14, 18–30 CrossRef.
- R. Mariam Reyad, A. Rama Saad and R. Karam, The Human Microbiome Project, Personalized Medicine and the Birth of Pharmacomicrobiomics, Curr. Pharmacogenomics Pers. Med., 2010, 8(3), 182–193 CrossRef.
- R. Saad, M. R. Rizkallah and R. K. Aziz, Gut Pharmacomicrobiomics: the tip of an iceberg of complex interactions between drugs and gut-associated microbes, Gut Pathog., 2012, 4(1), 16 CrossRef CAS PubMed.
- Y. Hasin, M. Seldin and A. Lusis, Multi-omics approaches to disease, Genome Biol., 2017, 18(1), 83 CrossRef PubMed.
- H. Lin, et al., Proteomics and the microbiome: pitfalls and potential, Expert Rev. Proteomics, 2018, 16(6), 501–511 CrossRef PubMed.
- B. Alberts, et al., Molecular Biology of the Cell, Garland Science, New York, 3rd edn, 1994 Search PubMed.
- A. Bartholomäus, et al., Bacteria differently regulate mRNA abundance to specifically respond to various stresses, Philos. Trans. R. Soc., A, 2016, 374(2063), 20150069 CrossRef PubMed.
- S. P. Gygi, et al., Correlation between protein and mRNA abundance in yeast, Mol. Cell. Biol., 1999, 19(3), 1720–1730 CrossRef CAS PubMed.
- V. E. Velculescu, et al., Analysis of human transcriptomes, Nat. Genet., 1999, 23(4), 387–388 CrossRef CAS PubMed.
- S. Welle, K. Bhatt and C. A. Thornton, Inventory of High-Abundance mRNAs in Skeletal Muscle of Normal Men, Genome Res., 1999, 9(5), 506–513 CAS.
- L. Liu, et al., The human microbiome: a hot spot of microbial horizontal gene transfer, Genomics, 2012, 100(5), 265–270 CrossRef CAS PubMed.
- G. A. Brar, et al., High-resolution view of the yeast meiotic program revealed by ribosome profiling, Science, 2012, 335(6068), 552–557 CrossRef CAS PubMed.
- R. Milo, R. Phillips and N. Orme, Cell Biology by the numbers, Garland Science, Taylor and Francis Group, LLC, 2016 Search PubMed.
- R. Sender, S. Fuchs and R. Milo, Revised Estimates for the number of human and bacteria cells in the body, PLoS Biol., 2016, 14(8), e1002533 CrossRef PubMed.
- R. M. Brucker and S. R. Bordenstein, The capacious hologenome, Zoology, 2013, 116(5), 260–261 CrossRef PubMed.
- I. Zilber-Rosenberg and E. Rosenberg, Role of microorganisms in the evolution of animals and plants: the hologenome theory of evolution, FEMS Microbiol. Rev., 2008, 32(5), 723–735 CrossRef CAS PubMed.
- A. M. O'Hara and F. Shanahan, The gut flora as a forgotten organ, EMBO Rep., 2006, 7(7), 688–693 CrossRef PubMed.
- S. Yachi and M. Loreau, Biodiversity and ecosystem productivity in a fluctuating environment: the insurance hypothesis, Proc. Natl. Acad. Sci. U. S. A., 1999, 96(4), 1463–1468 CrossRef CAS PubMed.
- E. Rosenberg and I. Zilber-Rosenberg, Role of Microorganisms in Adaptation, Development, and Evolution of Animals and Plants: The Hologenome Concept, in The Prokaryotes: Prokaryotic Biology and Symbiotic Associations, ed. E. Rosenberg, et al., Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 347–358 Search PubMed.
- J. Miro-Blanch and O. Yanes, Epigenetic Regulation at the Interplay Between Gut Microbiota and Host Metabolism, Front. Genet., 2019, 10, 638 CrossRef CAS PubMed.
- Y. Qin and P. A. Wade, Crosstalk between the microbiome and epigenome: messages from bugs, J. Biochem., 2017, 163(2), 105–112 CrossRef PubMed.
- M. A. J. Hullar, A. N. Burnett-Hartman and J. W. Lampe, Gut microbes, diet, and cancer, Cancer Treat. Res., 2014, 159, 377–399 CrossRef CAS PubMed.
- E.-S. Lee, E.-J. Song and Y.-D. Nam, Dysbiosis of Gut Microbiome and Its Impact on Epigenetic Regulation, J. Clin. Epigenetics, 2017, 3(2) DOI:10.21767/2472-1158.100048.
- Y. Vazquez-Baeza, et al., Impacts of the Human Gut Microbiome on Therapeutics, Annu. Rev. Pharmacol. Toxicol., 2018, 58, 253–270 CrossRef CAS PubMed.
- L. Maier, et al., Extensive impact of non-antibiotic drugs on human gut bacteria, Nature, 2018, 555(7698), 623–628 CrossRef CAS PubMed.
- A. D. Lieber, et al., Loss of HDAC6 alters gut microbiota and worsens obesity, FASEB J., 2019, 33(1), 1098–1109 CrossRef CAS PubMed.
- R. Mariam Reyad, S. Rama and A. R. Karam, The Human Microbiome Project, Personalized Medicine and the Birth of Pharmacomicrobiomics, Curr. Pharmacogenomics Pers. Med., 2010, 8(3), 182–193 CrossRef.
- R. Saad, M. R. Rizkallah and R. K. Aziz, Gut Pharmacomicrobiomics: the tip of an iceberg of complex interactions between drugs and gut-associated microbes, Gut Pathog., 2012, 4(1), 16 CrossRef CAS PubMed.
- B. Foxman and E. T. Martin, Use of the Microbiome in the Practice of Epidemiology: A Primer on -Omic Technologies, Am. J. Epidemiol., 2015, 182(1), 1–8 CrossRef PubMed.
- B. M. Hanson and G. M. Weinstock, The importance of the microbiome in epidemiologic research, Ann. Epidemiol., 2016, 26(5), 301–305 CrossRef PubMed.
- K. A. Krautkramer, F. E. Rey and J. M. Denu, Chemical signaling between gut microbiota and host chromatin: What is your gut really saying?, J. Biol. Chem., 2017, 292(21), 8582–8593 CrossRef CAS PubMed.
- K. A. Krautkramer, et al., Diet-Microbiota Interactions Mediate Global Epigenetic Programming in Multiple Host Tissues, Mol. Cell, 2016, 64(5), 982–992 CrossRef CAS PubMed.
- F. Sommer, et al., Site-specific programming of the host epithelial transcriptome by the gut microbiota, Genome Biol., 2015, 16, 62 CrossRef PubMed.
- H. Lin, et al., Proteomics and the microbiome: pitfalls and potential, Expert Rev. Proteomics, 2019, 16(6), 501–511 CrossRef CAS PubMed.
- J. L. Darcy, et al., A phylogenetic model for the recruitment of species into microbial communities and application to studies of the human microbiome, ISME J., 2020, 14, 1359–1368 CrossRef PubMed.
- L. Feng, et al., Identifying determinants of bacterial fitness in a model of human gut microbial succession, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(5), 2622 CrossRef CAS PubMed.
- M. A. W. Khan, et al., Does MHC heterozygosity influence microbiota form and function?, PLoS One, 2019, 14(5), e0215946 CrossRef CAS PubMed.
- V. Gopalakrishnan, et al., Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients, Science, 2018, 359(6371), 97–103 CrossRef CAS PubMed.
- G. Kroemer and L. Zitvogel, Cancer immunotherapy in 2017: The breakthrough of the microbiota, Nat. Rev. Immunol., 2018, 18(2), 87–88 CrossRef CAS PubMed.
- R. A. Quinn, et al., Global chemical effects of the microbiome include new bile-acid conjugations, Nature, 2020, 579(7797), 123–129 CrossRef CAS PubMed.
- K. Califf, et al., The human microbiome: getting personal, Microbe, 2014, 9(10), 410–415 Search PubMed.
- C. R. Woese and G. E. Fox, Phylogenetic structure of the prokaryotic domain: The primary kingdoms, Proc. Natl. Acad. Sci. U. S. A., 1977, 74(11), 5088–5090 CrossRef CAS PubMed.
- E. Pruesse, et al., SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB, Nucleic Acids Res., 2007, 35(21), 7188–7196 CrossRef CAS PubMed.
- J. R. Cole, et al., The Ribosomal Database Project: improved alignments and new tools for rRNA analysis, Nucleic Acids Res., 2009, 37, D141–D145 CrossRef CAS PubMed.
- T. Z. DeSantis, et al., Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB, Appl. Environ. Microbiol., 2006, 72(7), 5069–5072 CrossRef CAS PubMed.
- D. H. Parks, et al., A complete domain-to-species taxonomy for Bacteria and Archaea (vol 58, pg 561, 2020), Nat. Biotechnol., 2020 DOI:10.1038/s41587-020-0501-8.
- D. H. Parks, et al., A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life, Nat. Biotechnol., 2018, 36(10), 996–1004 CrossRef CAS PubMed.
- S. H. Yoon, et al., A large-scale evaluation of algorithms to calculate average nucleotide identity, Antonie van Leeuwenhoek, 2017, 110(10), 1281–1286 CrossRef CAS PubMed.
- B. J. Callahan, et al., High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution, Nucleic Acids Res., 2019, 47(18), e103 CrossRef CAS PubMed.
- E. Bolyen, et al., Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2, Nat. Biotechnol., 2019, 37(8), 852–857 CrossRef CAS PubMed.
- G. V. Uritskiy, J. DiRuggiero and J. Taylor, MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis, Microbiome, 2018, 6, 158 CrossRef PubMed.
- E. L. Clarke, et al., Sunbeam: an extensible pipeline for analyzing metagenomic sequencing experiments, Microbiome, 2019, 7(1), 46 CrossRef PubMed.
- J. Tamames, F. Puente-Sanchez and S. Meta, A Highly Portable, Fully Automatic Metagenomic Analysis Pipeline, Front. Microbiol., 2019, 9 DOI:10.3389/fmicb.2018.03349.
- T. J. Treangen, et al., MetAMOS: a modular and open source metagenomic assembly and analysis pipeline, Genome Biol., 2013, 14, R2 CrossRef PubMed.
- F. Meyer, et al., The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes, BMC Bioinf., 2008, 9, 386 CrossRef CAS PubMed.
- I. M. A. Chen, et al., IMG/M v.5.0: an integrated data management and comparative analysis system for microbial genomes and microbiomes, Nucleic Acids Res., 2019, 47(D1), D666–D677 CrossRef CAS PubMed.
- A. M. Eren, et al., Anvi'o: an advanced analysis and visualization platform for 'omics data, PeerJ, 2015, 3, e1319 CrossRef PubMed.
- A. Dhariwal, et al., MicrobiomeAnalyst: a web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data, Nucleic Acids Res., 2017, 45(W1), W180–W188 CrossRef CAS PubMed.
- L. J. McIver, et al., bioBakery: a meta'omic analysis environment, Bioinformatics, 2018, 34(7), 1235–1237 CrossRef CAS PubMed.
- D. T. Truong, et al., MetaPhlAn2 for enhanced metagenomic taxonomic profiling (vol 12, pg 902, 2015), Nat. Methods, 2016, 13(1), 101 CrossRef CAS.
- F. Asnicar, et al., Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0, Nat. Commun., 2020, 11(1), 2500 CrossRef CAS PubMed.
- E. A. Franzosa, et al., Species-level functional profiling of metagenomes and metatranscriptomes, Nat. Methods, 2018, 15(11), 962–968 CrossRef CAS PubMed.
- N. Segata, et al., Metagenomic biomarker discovery and explanation, Genome Biol., 2011, 12, R60 CrossRef PubMed.
- N. Segata, et al., Computational meta’omics for microbial community studies, Mol. Syst. Biol., 2013, 9, 666 CrossRef PubMed.
- T. Wongsurawat, et al., Decoding the Epitranscriptional Landscape from Native RNA Sequences, bioRxiv, 2018, 487819 Search PubMed.
- P. Jenjaroenpun, et al., Complete genomic and transcriptional landscape analysis using third-generation sequencing: a case study of Saccharomyces cerevisiae CEN.PK113-7D, Nucleic Acids Res., 2018, 46(7), e38 CrossRef CAS PubMed.
- S. Gigante, et al., Using long-read sequencing to detect imprinted DNA methylation, bioRxiv, 2019, 445924 Search PubMed.
- J. T. Simpson, et al., Detecting DNA cytosine methylation using nanopore sequencing, Nat. Methods, 2017, 14(4), 407–410 CrossRef CAS PubMed.
- M. Ferrarini, et al., An evaluation of the PacBio RS platform for sequencing and de novo assembly of a chloroplast genome, BMC Genomics, 2013, 14, 670 CrossRef CAS PubMed.
- Illumina, 16S Metagenomic Sequencing Library Preparation [Internet]. Available from: https://support.illumina.com/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf.
- S. L. Amarasinghe, et al., Opportunities and challenges in long-read sequencing data analysis, Genome Biol., 2020, 21, 30 CrossRef PubMed.
- Illumina, Illumina sequencing platforms [Internet]. Available from: https://www.illumina.com/systems/sequencing-platforms.html.
- L. de Oliveira Martins, et al., Taxonomic resolution of the ribosomal RNA operon in bacteria: implications for its use with long-read sequencing, NAR Genomics Bioinf., 2020, 2(1), lqz016 CrossRef PubMed.
- E. L. Moss, D. G. Maghini and A. S. Bhatt, Complete, closed bacterial genomes from microbiomes using nanopore sequencing, Nat. Biotechnol., 2020, 38, 701–707 CrossRef CAS PubMed.
- J. M. Heather and B. Chain, The sequence of sequencers: The history of sequencing DNA, Genomics, 2016, 107(1), 1–8 CrossRef CAS PubMed.
- N. L. Anderson and N. G. Anderson, The Human Plasma Proteome. History, Character, and Diagnostic Prospects, Mol. Cell. Proteomics, 2002, 1(11), 845–867 CrossRef CAS PubMed.
- S. A. Gerber, et al., Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS, Proc. Natl. Acad. Sci. U. S. A., 2003, 100(12), 6940–6945 CrossRef CAS PubMed.
- J. J. Porter, et al., Absolute Quantification of the Lower Abundance Proteome Through Immunoaffinity Depletion of the Twenty Most Abundant Proteins in Human Serum, 2006 Search PubMed.
- N. Grassl, et al., Ultra-deep and quantitative saliva proteome reveals dynamics of the oral microbiome, Genome Med., 2016, 8(1), 44 CrossRef PubMed.
- S. Afiuni-Zadeh, et al., Evaluating the potential of residual Pap test fluid as a resource for the metaproteomic analysis of the cervical-vaginal microbiome, Sci. Rep., 2018, 8(1), 10868 CrossRef PubMed.
- G. P. Hobby, et al., Chronic kidney disease and the gut microbiome, Am. J. Physiol. Renal. Physiol., 2019, 316(6), F1211–F1217 CrossRef CAS PubMed.
- M. D. Ritchie, et al., Methods of integrating data to uncover genotype–phenotype interactions, Nat. Rev. Genet., 2015, 16(2), 85–97 CrossRef CAS PubMed.
- K. A. Le Cao, I. Gonzalez and S. Dejean, integrOmics: an R package to unravel relationships between two omics datasets, Bioinformatics, 2009, 25(21), 2855–2856 CrossRef CAS PubMed.
- F. Rohart, et al., mixOmics: An R package for 'omics feature selection and multiple data integration, PLoS Comput. Biol., 2017, 13(11), e1005752 CrossRef PubMed.
- E. Calura, et al., Wiring miRNAs to pathways: a topological approach to integrate miRNA and mRNA expression profiles, Nucleic Acids Res., 2014, 42(11), e96 CrossRef CAS PubMed.
- Q. Mo and R. Shen, iClusterPlus: Integrative clustering of multi-type genomic data, 2019 Search PubMed.
- D. Wu, et al., Fast dimension reduction and integrative clustering of multi-omics data using low-rank approximation: application to cancer molecular classification, BMC Genomics, 2015, 16(1), 1022 CrossRef PubMed.
- I. Subramanian, et al., Multi-omics Data Integration, Interpretation, and Its Application, Bioinf. Biol. Insights, 2020, 14, 1177932219899051 CrossRef PubMed.
- P. Ray, et al., Bayesian joint analysis of heterogeneous genomics data, Bioinformatics, 2014, 30(10), 1370–1376 CrossRef CAS PubMed.
- K. Wanichthanarak, J. F. Fahrmann and D. Grapov, Genomic, Proteomic, and Metabolomic Data Integration Strategies, Biomarker Insights, 2015, 10s4, BMI.S29511 CrossRef PubMed.
- F. Garcia-Alcalde, et al., Paintomics: a web based tool for the joint visualization of transcriptomics and metabolomics data, Bioinformatics, 2011, 27(1), 137–139 CrossRef CAS PubMed.
- B. B. Misra, et al., Integrated Omics: Tools, Advances, and Future Approaches, J. Mol. Endocrinol., 2018, R21–R45 Search PubMed.
- S. Nakagawa and I. C. Cuthill, Effect size, confidence interval and statistical significance: a practical guide for biologists, Biol. Rev. Cambridge Philos. Soc., 2007, 82(4), 591–605 CrossRef PubMed.
- S. Holmes and W. Huber, Modern Statistics for Modern Biology, 2018 Search PubMed.
- F. R. Pinu, et al., Systems Biology and Multi-Omics Integration: Viewpoints from the Metabolomics Research Community, Metabolites, 2019, 9(4), 76 CrossRef CAS.
- R. Cavill, et al., Transcriptomic and metabolomic data integration, Briefings Bioinf., 2016, 17(5), 891–901 CrossRef PubMed.
- C. Wu, et al., A Selective Review of Multi-Level Omics Data Integration Using Variable Selection, High-Throughput, 2019, 8(1), 4 CrossRef PubMed.
- A. Auton, et al., A global reference for human genetic variation, Nature, 2015, 526(7571), 68–74 CrossRef PubMed.
- W. S. Bush and J. H. Moore, Chapter 11: Genome-wide association studies, PLoS Comput. Biol., 2012, 8(12), e1002822 CrossRef CAS.
- K.-H. Liang, in 3-Transcriptomics, in Bioinformatics for Biomedical Science and Clinical Applications, ed. K.-H. Liang, Woodhead Publishing, 2013, pp. 49–82 Search PubMed.
- L.-R. Yu, N. A. Stewart and T. D. Veenstra, Chapter 8 – Proteomics: The Deciphering of the Functional Genome, in Essentials of Genomic and Personalized Medicine, ed. G. S. Ginsburg and H. F. Willard, Academic Press, San Diego, 2010, pp. 89–96 Search PubMed.
- X. Liu and J. W. Locasale, Metabolomics: A Primer, Trends Biochem. Sci., 2017, 42(4), 274–284 CrossRef CAS PubMed.
- M. Mussap, M. Zaffanello and V. Fanos, Metabolomics: a challenge for detecting and monitoring inborn errors of metabolism, Ann. Transl. Med., 2018, 6(17), 338 CrossRef PubMed.
- M. A. Pourhoseingholi, M. Vahedi and M. Rahimzadeh, Sample size calculation in medical studies, Gastroenterol. Hepatol. Bed Bench., 2013, 6(1), 14–17 Search PubMed.
- C. Manzoni, et al., Genome, transcriptome and proteome: the rise of omics data and their integration in biomedical sciences, Briefings Bioinf., 2018, 19(2), 286–302 CrossRef CAS PubMed.
- J. Debelius, et al., Tiny microbes, enormous impacts: what matters in gut microbiome studies?, Genome Biol., 2016, 17(1), 217 CrossRef.
- Y. Xia and J. Sun, Hypothesis Testing and Statistical Analysis of Microbiome, Genes Dis., 2017, 4(3), 138–148 CrossRef PubMed.
- M. A. Sze and P. D. Schloss, Looking for a Signal in the Noise: Revisiting Obesity and the Microbiome, mBio, 2016, 7(4), e01018 CrossRef PubMed.
- C. Casals-Pascual, et al., Microbial Diversity in Clinical Microbiome Studies: Sample Size and Statistical Power Considerations: Statistical Power for Microbiome Studies, Gastroenterology, 2020, 158(6), 1524–1528 CrossRef PubMed.
- B. J. Kelly, et al., Power and sample-size estimation for microbiome studies using pairwise distances and PERMANOVA, Bioinformatics, 2015, 31(15), 2461–2468 CrossRef CAS PubMed.
- P. S. La Rosa, et al., Hypothesis Testing and Power Calculations for Taxonomic-Based Human Microbiome Data, PLoS One, 2012, 7(12), e52078 CrossRef CAS PubMed.
- P. D. Schloss, Identifying and Overcoming Threats to Reproducibility, Replicability, Robustness, and Generalizability in Microbiome Research, mBio, 2018, 9(3), e00525 CrossRef PubMed.
- Y. V. Sun and Y. J. Hu, Integrative Analysis of Multi-omics Data for Discovery and Functional Studies of Complex Human Diseases, Adv. Genet., 2016, 93, 147–190 CrossRef CAS PubMed.
- B. Mirza, et al., Machine Learning and Integrative Analysis of Biomedical Big Data, Genes, 2019, 10(2), 87 CrossRef CAS PubMed.
- P. Yilmaz, et al., The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks, Nucleic Acids Res., 2014, 42(D1), D643–D648 CrossRef CAS PubMed.
- J. R. Cole, et al., Ribosomal Database Project: data and tools for high throughput rRNA analysis, Nucleic Acids Res., 2014, 42(D1), D633–D642 CrossRef CAS.
- D. McDonald, et al., An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea, ISME J., 2012, 6(3), 610–618 CrossRef CAS.
- A. Bateman, et al., UniProt: a worldwide hub of protein knowledge, Nucleic Acids Res., 2019, 47(D1), D506–D515 CrossRef PubMed.
- D. A. Benson, et al., GenBank, Nucleic Acids Res., 2012, 40(D1), D48–D53 CrossRef CAS PubMed.
- J. O. Jo McEntyre, The NCBI Handbook [Internet], National Center for Biotechnology Information (US), Bethesda (MD), 2002. Available from: https://www.ncbi.nlm.nih.gov/books/NBK21101/ Search PubMed.
- M. Haeussler, et al., The UCSC Genome Browser database: 2019 update, Nucleic Acids Res., 2019, 47(D1), D853–D858 CrossRef CAS PubMed.
- I. Dunham, et al., An integrated encyclopedia of DNA elements in the human genome, Nature, 2012, 489(7414), 57–74 CrossRef CAS PubMed.
- K. L. Howe, et al., Ensembl Genomes 2020—enabling non-vertebrate genomic research, Nucleic Acids Res., 2019, 48(D1), D689–D695 CrossRef PubMed.
- Program, T.C.G.A., The Cancer Genome Atlas Program [Internet]. Available from: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga.
- M. Ghandi, et al., Next-generation characterization of the Cancer Cell Line Encyclopedia, Nature, 2019, 569(7757), 503–508 CrossRef CAS.
- Y. Perez-Riverol, et al., Quantifying the impact of public omics data, Nat. Commun., 2019, 10, 3512 CrossRef PubMed.
- E. Montague, et al., MOPED 2.5-An Integrated Multi-Omics Resource: Multi-Omics Profiling Expression Database Now Includes Transcriptomics Data, OMICS, 2014, 18(6), 335–343 CrossRef CAS PubMed.
- J. A. Vizcaino, et al., ProteomeXchange provides globally coordinated proteomics data submission and dissemination, Nat. Biotechnol., 2014, 32(3), 223–226 CrossRef CAS PubMed.
- E. W. Deutsch, et al., The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition, Nucleic Acids Res., 2017, 45(D1), D1100–D1106 CrossRef CAS PubMed.
- Genohub, Recommended Coverage and Read Depth for NGS Applications.
- R. Bowden, et al., Sequencing of human genomes with nanopore technology, Nat. Commun., 2019, 10, 1869 CrossRef PubMed.
- A. Byrne, et al., Realizing the potential of full-length transcriptome sequencing, Philos. Trans. R. Soc., B, 2019, 374(1786), 20190097 CrossRef CAS PubMed.
- Y. Liu, et al., Evaluating the impact of sequencing depth on transcriptome profiling in human adipose, PLoS One, 2013, 8(6), e66883 CrossRef CAS PubMed.
- H. S. Gweon, et al., The impact of sequencing depth on the inferred taxonomic composition and AMR gene content of metagenomic samples, Environ. Microbiome, 2019, 14, 7 CrossRef PubMed.
- ENCODE, ENCODE [Internet]. Available from: https://www.encodeproject.org/help/citing-encode/.
- Elements, E.E.o.D., Standards and Guidelines for Whole Genome Shotgun Bisulfite Sequencing (WGBS) [Internet]. Available from: https://www.encodeproject.org/documents/108d2515-c053-4b18-bc65-27e8f26d62c5/@@download/attachment/MethylC-SeqStandards_ENCODE3_EM.pdf. 2015.
- J. Chong, D. S. Wishart and J. Xia, Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis, Curr. Protoc. Bioinformatics, 2019, 68(1), e86 CrossRef PubMed.
- N. Tuncbag, et al., Network-Based Interpretation of Diverse High-Throughput Datasets through the Omics Integrator Software Package, PLoS Comput. Biol., 2016, 12(4), e1004879 CrossRef PubMed.
- C. J. Vaske, et al., Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM, Bioinformatics, 2010, 26(12), i237–i245 CrossRef CAS PubMed.
- S. Ghandikota, G. K. K. Hershey and T. B. Mersha, GENEASE: real time bioinformatics tool for multi-omics and disease ontology exploration, analysis and visualization, Bioinformatics, 2018, 34(18), 3160–3168 CrossRef CAS PubMed.
- A. D. Mooradian, et al., ProteoClade: A taxonomic toolkit for multi-species and metaproteomic analysis, PLoS Comput. Biol., 2020, 16(3), e1007741 CrossRef PubMed.
- C. Diener, S. M. Gibbons and O. Resendis-Antonio, MICOM: Metagenome-Scale Modeling To Infer Metabolic Interactions in the Gut Microbiota, Msystems, 2020, 5, e00606–19 CrossRef CAS PubMed.
- J. T. Morton, et al., Learning representations of microbe-metabolite interactions, Nat. Methods, 2019, 16(12), 1306–1314 CrossRef CAS PubMed.
- T. Pluskal, et al., MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data, BMC Bioinf., 2010, 11, 395 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2021 |