
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceScalable nanoporous carbon films allow line-of-sight 3D atomic layer deposition of Pt: towards a new generation catalyst layer for PEM fuel cells†
Marwa
Atwa
 ab,
Xiaoan
Li‡
a,
Zhaoxuan
Wang
c,
Samuel
Dull
d,
Shicheng
Xu
e,
Xia
Tong
a,
Rui
Tang
f,
Hirotomo
Nishihara
f,
Fritz
Prinz
ceg and
Viola
Birss
*a
ab,
Xiaoan
Li‡
a,
Zhaoxuan
Wang
c,
Samuel
Dull
d,
Shicheng
Xu
e,
Xia
Tong
a,
Rui
Tang
f,
Hirotomo
Nishihara
f,
Fritz
Prinz
ceg and
Viola
Birss
*a
aDepartment of Chemistry, University of Calgary, 2500 University Dr NW, Calgary, AB T2N 1N4, Canada. E-mail: Birss@ucalgary.ca
bDepartment of Chemistry, Suez Canal University, El Salam District, Ismailia, 41522, Egypt
cDepartment of Material Science and Engineering, Stanford University, 496 Lomita Mall, Stanford, CA 94305, USA
dDepartment of Chemical Engineering, Stanford University, 443 Via Ortega, Stanford, CA 94305, USA
eDepartment of Mechanical Engineering, Stanford University, Building 530, 440 Escondido Mall, Stanford, CA 94305, USA
fAdvanced Institute for Materials Research/Institute of Multidisciplinary Research for Advanced Materials, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai, 980-8577, Japan
gDepartment of Mechanical and Industrial Engineering, Norwegian University of Science and Technology, Høgskoleringen 1, Trondheim, 7491, Norway
First published on 20th August 2021
Abstract
Although nanoporous carbons are ubiquitous materials that are used in many clean energy and environmental applications, most are in powder form, thus requiring binders to hold particles together. This results in uncontrolled and complex pathways between particles, potentially exacerbating mass transport issues. To overcome these problems, we have developed an unprecedented binderless, self-supported, nanoporous carbon scaffold (NCS) with tunable and monodisperse pores (5–100+ nm), high surface area (ca. 200–575 m2 g−1), and 3-dimensional scalability (1–150+ cm2, 1–1000 μm thickness). Here, it is shown that NCS85 membranes (85 nm pores) are particularly promising as a host for the homogeneous and efficient 3-D atomic layer deposition (ALD) of Pt nanoparticles, due to the facile penetration of gas phase Pt precursor throughout the homogeneous, low tortuosity internal structure. Furthermore, the high density of surface defects of the as-synthesized NCS promotes uniform Pt nucleation with minimal agglomeration. These advantageous features are key to the rapid oxygen reduction kinetics observed under polymer electrolyte membrane (PEM) fuel cell MEA testing conditions. Cells constructed with an optimal ALD Pt loading of 30 cycles are shown to exhibit a specific activity of ≥0.4 mA cm−2Pt which is exemplary when compared to two commercial catalyst layers with comparable Pt mass loadings and tested under the same conditions. Furthermore, a maximum power density of 1230 mW cm−2 (IR-corrected) is obtained, with the limiting current densities approaching a very respectable 3 A cm−2.
New conceptsWe have developed a paradigm-shifting self-supported and nanoporous carbon scaffold (NCS), a 100% carbon membrane with fully tunable pore sizes, ultra-low tortuosity, and exceptional scalability. We demonstrate that the NCS is ideally suited for the uniform (in 3D) atomic layer deposition (ALD) of Pt nanoparticles (NPs), exhibiting exemplary performance as a catalyst layer in proton exchange membrane (PEM) fuel cells. Currently, only carbon powders are available for this purpose, as no self-supported carbon sheets with tunable nanoporosity exist. Also, after Pt NP deposition onto the powder, they must be mixed with a binder, sprayed, and often hot-pressed, involving multiple preparation steps. This generates catalyst layers with uncontrolled/tortuous pathways that cause catalyst utilization limitations. Previous efforts to ALD Pt into conventional carbon powders or even monoliths gave an anisotropic distribution of Pt, poor deposition efficiency, and a shallow penetration depth. The remarkably ordered nanoporosity of the NCS combined with its rich surface density of defect sites results in uniform Pt NP distribution, facilitating gas/ion transport during oxygen reduction in membrane-electrode-assembly testing. This work represents the unprecedented use of a pre-prepared nanoporous carbon sheet in a PEM fuel cell, showing great promise for future applications in batteries, capacitors, and electrolysis cells. |
1. Introduction
Carbonaceous and graphitic materials having nanoscale features are highly desirable for many clean energy applications, e.g., in lithium ion and redox flow batteries, capacitors, fuel cells, electrolysis cells, etc.,1–4 as well as in filtration and separation science. This is due to their low specific gravity, high electrical conductivity, controllable porosity and surface areas, modifiable surfaces, and ease of large-scale production.Most common porous carbons are in powder form, where the random orientation of the particles as well as particle agglomeration can create tortuous pathways for reactants and products. For most electrochemical applications, the carbon particles are held together with polymeric binders. However, binders typically lower electrical conductivity by interfering with carbon/carbon particle connectivity and may also add further tortuosity for the transport of solution and gaseous species. These issues have led to increasing interest in the development of robust and low cost porous carbon membranes that are binder-free, self-supported, scalable, and preferably have an ordered porous structure. Some successes include the preparation of gels or monoliths,5 as well as tape-cased carbon films,6,7 although the pores are typically either too small (micropores <2 nm in dia) or overly large (macroporous).7 Micropores make fluid access challenging, leading to difficulties in accessing the full internal surface area, while macroporous carbons have low surface areas, which limit the attainable power densities in electrochemical applications.
While several binderless and self-supported carbon materials have been reported, Table S1 (ESI†) shows that each of these has limitations, such as a limited thickness, non-tunable and broad pore size distributions, pores that are predominantly macro-scale or discontinuous in nature, or the absence of a 3D pore network. Table S1 (ESI†) includes two scalable porous carbon films.6,7 However, the pore size of the graphene film is not controllable, and full internal accessibility is questionable.6 In terms of the polyimide porous film,7 the pores are neither tunable nor 3D-percolating, the conductivity is low (<0.1 S cm−1), and the preparation method is non-green (using N-methyl-2-pyrrolidone).
To address these deficiencies, we have developed a nanoporous carbon scaffold (NCS) with a fully tunable and monodisperse pore size in the range of 5–100 nm in diameter.8 These NCS sheets, formed using a hard-templating method,8 contain no binders and are 100% carbon, and are also remarkably scalable in both area (1–150+ cm2) and thickness (1–1000 μm). Together with their high porosity (∼90%), ultra-low tortuosity, exceptional chemical and physical stability, the NCS materials are ideally suited for a wide variety of applications.
To date, we have reported on NCS membranes with pore sizes of either 22 or 85 nm in two different applications. In one of these studies,9 the internal surface NCS85 was modified to control its wettability, revealing that a combination of small hydrophobic pores and larger hydrophilic cracks facilitate oxygen and water transport, respectively. In our other work,10 NCS22 and NCS85 were used as a model of an ideal nanoporous material to study the dynamics of nanoliter droplet imbibition and capillary rise for application in natural porous media, e.g., shale rocks. To further expand the applications of these NCS materials, we have become interested in depositing a second or even third phase into their internal structure, starting with electrocatalytic metallic nanoparticles (NPs), especially for the oxygen reduction reaction (ORR), one of the major bottlenecks of PEM fuel cells. We began by investigating the performance of the NCS85 as a catalyst layer after loading it with Pt NPs using conventional wet impregnation methods.11 While promising, our interest has gravitated towards use of the NCS as an accessible, low tortuosity, porous support material for the atomic layer deposition (ALD) of Pt NPs. This is because ALD is known to generate metallic NPs with well distributed and reproducibly uniform sizes. The many ALD operating variables, including the ALD cycle number and temperature, have been shown to enable the fine tuning of NP size and distribution, ultimately dictating the activity, selectivity and stability of the deposited metals.12
Previous work involving ALD of Pt on high surface area carbons has been aimed primarily at producing ultra-high activity catalysts for use in electrochemistry. Liu et al. reported the ALD of Pt on onion-like carbon particles to catalyze the hydrogen evolution reaction (HER).13 While the performance was very good, testing was only carried out in 0.5 M sulfuric acid, not in a membrane-electrode-assembly (MEA). Also, as the support was in powder form, and the deposition required a specialized viscous flow reactor and an ALD cycle time of up to 400 seconds.13 In addition, these carbons required thermal annealing and chemical oxidation in order to produce a sufficiently high density of surface oxygen functional groups to aid in Pt loading and distribution. King and co-workers carried out Pt ALD on a 500 μm thick carbon aerogel monolith having a 1 cm2 geometric surface area but a real surface area of ∼480 m2 g−1.14 However, Rutherford backscattering depth analysis showed that the deposition was non-uniform and a penetration depth of only 10 μm could be achieved, even after many ALD cycles, due to the high tortuosity and the large percentage of surface area arising from micropores.14 Prior work in the Prinz group involved ALD preparation of highly active Pt catalysts for the ORR, using either glassy carbon or low surface area carbon black as the support.15 However, incorporation of these carbons or thick carbon monoliths into a PEM MEA has yet to be accomplished. As there have been no previous reports of the successful use of a thin porous carbon sheet or membrane for gas phase ALD of Pt precursors, the NCS material was considered to be ideally suited for the controlled deposition of catalytic Pt NPs at the atomic scale.
Here, we show that ALD of Pt into our NCS sheets (85 nm pores) produces uniformly small (ca. 3–5 nm dia) Pt NPs through the full NCS thickness, with no NP agglomeration seen even after 30 deposition cycles. Combined with the very good electronic conductivity and rapid ion injection/expulsion characteristics of the NCS85 backbone, this has resulted in excellent performance of the ALD Pt/NCS material as the cathode catalyst layer in an MEA configuration. A specific activity of 0.40 mA cm−2Pt was obtained for the best performing 30 ALD cycle NCS85 catalyst. This was shown to surpass the performance of two types of commercial Pt/C benchmark catalysts, a gas diffusion electrode (GDE) and a catalyst coated membrane (CCM) configuration, both tested under the same conditions in this study.
2. Results and discussion
2.1 NCS preparation and physical properties
As explained in our recent work,8–10 the NCS is fabricated by a combination of sonication and low energy ball-milling of mixtures of mesophase pitch (MP)/n-butanol (BtOH), polyvinyl alcohol (PVA)/water and silica nanoparticles/1,3-propanediol (PD)/water (Fig. 1 and Fig. S1, ESI†), producing a homogeneous slurry that is then tape-casted onto glass. Here, PD serves as a plasticizer for PVA, allowing the film to be easily removed from the glass substrate (Video S1, ESI†). PVA serves as a surfactant and also decomposes, itself forming a pitch-like material at 250–280 °C,16 improving the connectivity between the MP and silica. Without PVA added, only carbon powder forms as the final product after carbonization, with Fig. S2–S4 (ESI†) showing the thermal behavior of the as-cast precursor film.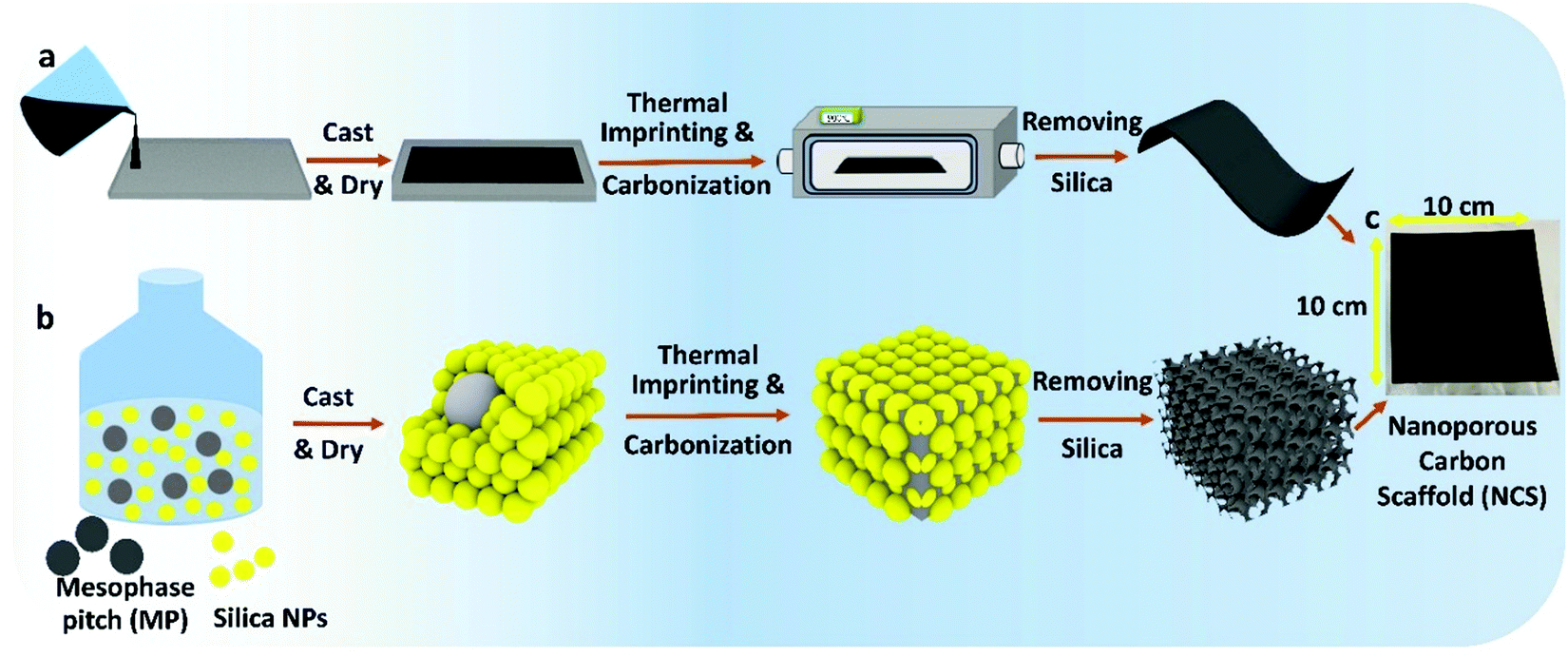 | ||
| Fig. 1 (a) Schematic showing nanoporous carbon scaffold (NCS) fabrication. (b) 3D cartoons of corresponding nano/microstructure of the NCS at each step in (a). Yellow represents silica colloid and grey represents pitch and resulting carbon phase. (c) Photograph of self-supported NCS film as final product with 100 cm2 area. | ||
The precursor membrane is then heated at 900 °C in nitrogen to remove PD and PVA, imprinted with SiO2, and then carbonized, followed by removal of the silica in strong base. Fig. S5 (ESI†) shows FESEM images of the identical site in the NCS, before and after silica removal. This confirms that the silica can be extracted while retaining the templated morphology and that no damage occurs to the carbon backbone by the NaOH treatment. The final NCS product is flexible (Fig. S6 and Video S2, ESI†), an advantage in bendable and cylindrical energy storage devices.
Top-down FESEM and TEM images of three NCS membranes (nominal pore diameters of 22, 50 and 85 nm) are seen in Fig. 2a–c, respectively, showing the excellent tunability of the nanostructure, controlled entirely by the silica template particle size (Fig. 2d). Cross-sectional analysis (Fig. S7, ESI†) reveals that the NCS nanostructure is organized and uniform throughout its thickness, resulting in its excellent permeability towards a wide range of fluids, with the air and glass sides of the tape-casted NCS being very similar (Fig. S8, ESI†). Twelve necks are seen within each NCS pore (Fig. 2e), with the pore neck diameter increasing proportionally with pore size (Table S2, ESI†).
 | ||
| Fig. 2 (a–c) FESEM (upper) and TEM (lower) images of NCS22, NCS50 and NCS85, respectively, with corresponding average pore sizes of 22, 50 and 85 nm. (d) Pore and neck diameter as a function of size of silica colloid used in NCS preparation. (e) High magnification FESEM image (left) of NCS85 showing 12 pore necks within each pore (reproduced with permission from ref. 47), as indicated in the 3D-model (right) of NCS85, with the pore necks highlighted in orange. | ||
Gas sorption data (Fig. S9, ESI†) show the hysteresis typical of mesoporous materials, where Table S2 (ESI†) demonstrates that the NCS surface area increases from 200–580 m2 g−1 for the NCS85 to NCS8 series, all being logically smaller than for microporous carbon materials. Table S3 (ESI†) shows that all of the NCS membranes have a similarly high porosity of ∼90%, making them ideal for enhanced mass transport and infiltration of precursors, such as in ALD of Pt NPs.
The NCS is also found here to be an excellent conductor, with the conductivity of the NCS backbone determined to be ≥2 S cm−1, much higher than for typical well-pressed carbon powders (3–30 mS cm−1).17,18 This is not surprising, as the NCS sheets are 100% carbon in composition. In terms of solution accessibility into the NCS pores, Fig. 3a shows the cyclic voltammograms (CVs) of a series of NCS materials in 0.5 M sulfuric acid, with the currents given per NCS mass to facilitate comparison between the various NCS samples and literature results. Table S4 (ESI†) shows that the electrochemically active surface area (ECSA in m2 g−1, calculated by dividing the gravimetric capacitance by 0.15 F m−2, a commonly used area-specific capacitance) ranges from 200–500 m2 g−1 in 0.5 M H2SO4.19 As these specific areas are very similar to the BET-determined surface areas (Fig. 3b), this indicates that essentially 100% of the NCS surface area is electrochemically accessible in this medium, attributed to its open, ordered and 3-D interconnected nanostructure.
 | ||
| Fig. 3 (a) CVs (10 mV s−1) of various NCS materials, with (b) showing a plot of their ECSA vs. BET-determined surface area. (c) shows the capacitance retention as a function of the scan rate, with (d) showing the Nyquist plot (inset: normalized complex capacitance as a function of frequency) for NCS22 as an example. All electrochemical experiments were carried out in N2-saturated, room temperature 0.5 M H2SO4. | ||
The majority of the currents seen in Fig. 3a represents double layer charging, with some additional charge arising from the pronounced pseudocapacitive quinone/hydroquinone redox peaks (at ∼0.6 V, Fig. 3a),20 which forecast a high surface oxygen group density. The specific capacitances are quite high, ranging from 30 to 120 F/g for NCS85 to NCS8, compared to 35–40 F g−1 for most other mesoporous carbons,21 consistent with the increasing surface area in this same sequence (Table S2, ESI†).
To confirm rapid ion transport rates through the NCS pores, we measured the capacity retention at high scan rates in 0.5 M sulfuric acid, a common approach in the literature. Excellent capacity retention is observed (loss of only 20%, Fig. 3c), indicative of fast ion transport rates inside the NCS pores, also confirmed by the small charge/discharge time constant obtained from electrochemical impedance spectroscopy (EIS). Fig. 3d shows that the full ∼40 μm NCS22 film can be charged/discharged in ≤1 s, while for most commercial carbon-based devices, the same process requires 10–100 s.22
Taken together, Fig. 2 and 3 show that the NCS membranes have an ideal ultra-low tortuosity microstructure without any binder present, possess the needed conductivity properties, and have exquisite tunability in terms of their pore size and the scalability of the sheets in all three dimensions. The NCS was therefore considered to be an ideal material for use as a substrate for the ALD of Pt NPs, with this combination of nano-engineering shown below to produce a paradigm-shifting catalyst layer for use in PEMFCs.
2.2 ALD of Pt into self-supported nanoporous carbon scaffold
As stated above, the literature has shown that it is challenging to attain a uniform deposition of a range of materials onto carbon powder surfaces via atomic layer deposition (ALD), especially of catalytic metal NPs,23 partly as the tortuous microstructures found in packed carbon powders do not offer the required line-of-sight. Because of this problem, the ALD deposit is far heavier at the deposition side of the carbon powder and lighter towards the back side. In the case of ALD of metals such as Pt, this causes agglomeration and non-uniform Pt nanoparticle (NP) size, leading to compromised deposition efficiency and sub-optimal performance.For these pioneering experiments, we placed a primary focus on NCS85, with a nominal 85 nm pore size, because its relatively large and fully 3D interconnected pores and ultra-low tortuosity (close to 1) was expected to best facilitate full penetration of the gas phase Pt precursor throughout its internal pore structure, thus forecasting the uniform deposition of Pt within the NCS-85 sheet. Also, the absence of any binders was expected to be advantageous in the subsequent infiltration of the Nafion ionomer, required for the transport of protons in the MEA work described below. A 12 μm NCS85 thickness was chosen in this work as this is comparable to standard catalyst layer thicknesses used in both fundamental PEMFC research and in commercial cells (5 to ≥15 μm).24,25 Note that an 18 μm thick NCS22 membrane was also ALD-loaded with Pt and MEA-tested for comparison with Ptx/NCS85. While the Pt NPs were found to be distributed uniformly throughout the NCS22 structure, the small neck sizes caused problems for Nafion infiltration. Thus, the NCS85 is the primary focus of this work.
20, 30 and 40 ALD cycles were investigated, producing Pt20/NCS85, Pt30/NCS85, and Pt40/NCS85, respectively. The Pt ALD process was demonstrated to be very efficient when using these open-structured NCS85 materials, requiring only ca. 165 s per ALD cycle (i.e., 9, 100, 5 and 50 s for exposure to the Pt precursor ((methylcyclopentadienyl)-trimethylplatinum (MeCpPtMe3)), a first Ar purge, air exposure time, and a final Ar purge, respectively).
Fig. 4 shows the TEM (a–c) and FESEM (d–f) images of the 20, 30 and 40 ALD cycle samples. It is clear that the higher Pt loading obtained with more ALD cycle numbers increases the average Pt NP size, from 3 nm for Pt20/NCS85 to 5 and 6.4 nm for Pt30/NCS85 and Pt40/NCS85, respectively (Fig. S10, ESI†). This is expected, as higher Pt loadings per real area, obtained by conventional solution infiltration methods, are also known to generate larger Pt NP sizes, due to the finite number of nucleation sites present. Importantly, Pt NP agglomeration is seen only for the Pt40/NCS85 catalyst, establishing 40 deposition cycles as the upper limit for the total carbon surface area available in the 12 μm thick NCS85 materials.
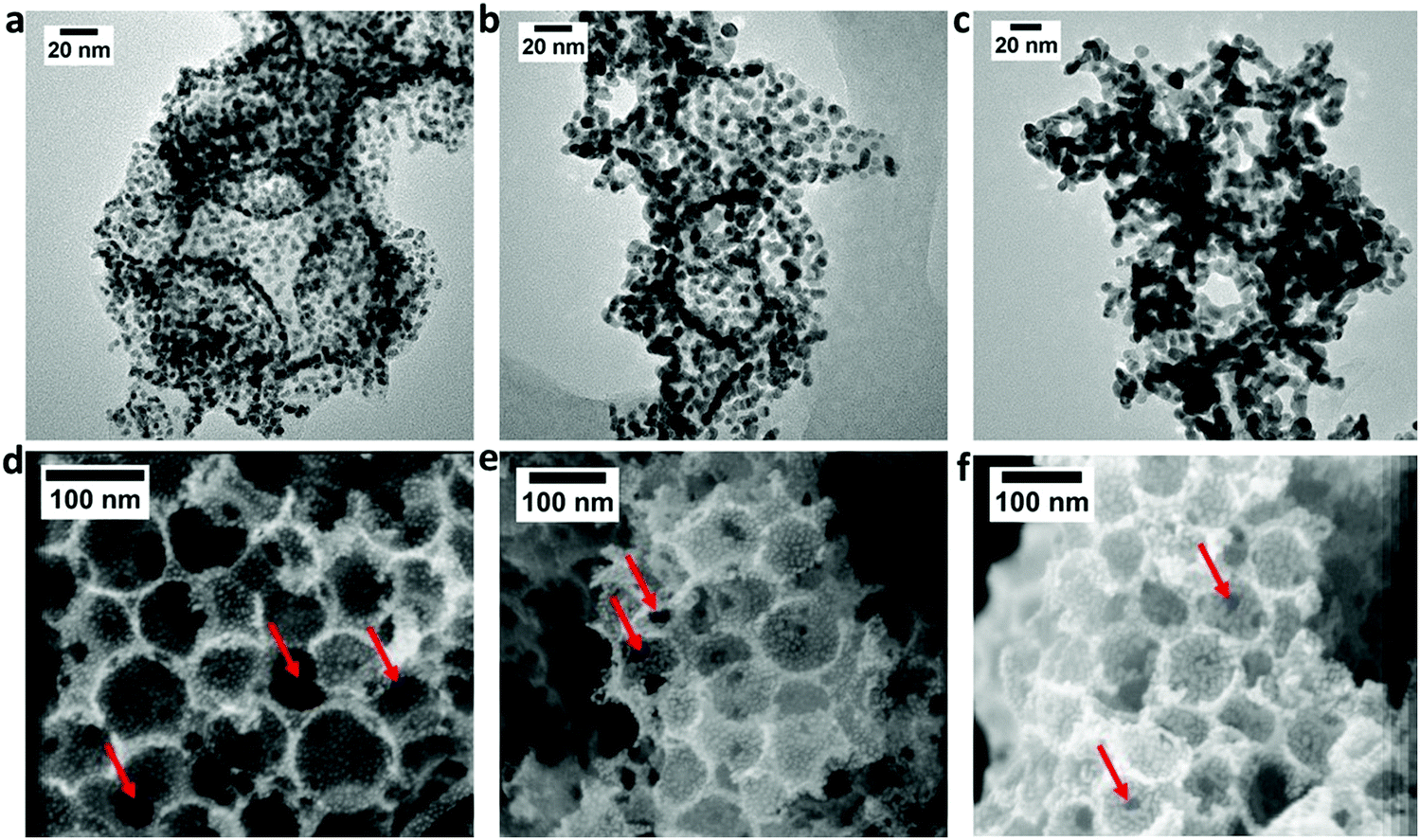 | ||
| Fig. 4 (a–c) HR-TEM images and (d–f) FESEM images in the central region of the sample cross-sections of Pt20/NCS85, Pt30/NCS85 and Pt40/NCS85, respectively. Red arrows indicate the pore necks within the Ptx/NCS85 pore structure. | ||
Fig. 4(d–f) also show that the ca. 85 nm pore sizes, as well as the 12 pore necks per pore of the NCS85 material, have been retained fully after the ALD process. The average neck diameter is 16 nm in the case of both Pt20/NCS85 (Fig. S11, ESI†) and Pt30/NCS85, the same as for the bare NCS85 (Fig. 2e), with minimal blockage of the pore necks caused by the Pt NPs. For Pt40/NCS85, however, the average neck diameter has decreased to 14 nm (as determined by SEM), likely due to the presence of the larger Pt NPs in the regions of the pore necks. This will be shown below to have a deleterious effect on mass transport at high current densities during PEMFC performance testing of these Pt40/NCS85 catalysts.
Fig. S12 (ESI†) shows the uniform Pt distribution across the full 12 μm thickness of the Ptx/NCS85 scaffolds. Also, Fig. S13 (ESI†) shows a selection of FESEM images collected from the Pt30/NCS85 cross-section, one from the outer (top) NCS surface and the other from the bottom, showing the same Pt content through the thickness and at both surfaces. These results are direct evidence of the ultra-low tortuosity of the NCS and its 3D interconnected pore structure, providing a minimal path length (clear line of sight) for the gas phase Pt precursor, allowing it to diffuse freely and uniformly throughout the NCS85 thickness. In contrast, powder substrates contain long and tortuous path lengths because of the random orientation and packing of the particles. Another impressive outcome of this work is that, for all Pt loadings, ALD has produced a narrow Pt particle size distribution on the NCS85 substrate (Fig. S10, ESI†), contrary to previous work using pristine carbons, such as carbon nanotubes (CNTs).26
As discussed earlier, the contribution of the pseudo-capacitance is ∼13% of the total capacitance of the NCS85 films obtained in the CV experiments (Fig. 3a and Table S4, ESI†), attributed to the presence of a significant concentration of surface oxygen groups on the NCS85 surface. ALD is known to occur by nucleation of the gas phase species onto the specimen surface, and in the case of carbon substrates, oxygen functional groups serve as ideal active sites for the anchoring of the ALD precursor.26 However, most carbons (e.g., CNTs, carbon black, graphene) contain very few surface oxygen sites, thus being weakly hydrophilic, or more commonly, hydrophobic.27–29 This low density of surface defects could be responsible for the non-uniformity of the thin metallic films and nanoparticles grown in other work using ALD on carbon, as precursor adsorption occurs primarily on edge sites. For this reason, recent efforts with carbon substrates have included a pre-treatment step to create a large number of homogenously distributed active sites. For instance, Yan et al. acid-treated graphene to produce a high concentration of surface oxygen functional groups, such as –COOH and –OH, on the graphene, thus enhancing Pd deposition via ALD.30
The NCS materials are all intrinsically highly wettable materials, giving a DI water contact angle of ∼30° (Fig. S14, ESI†). The hydrophilicity of the NCS arises from the high coverage of surface oxygen groups, originating from its imprinting with silica colloids and potentially also from the last step of NCS fabrication when the silica NPs are removed by treatment in base.10 For these reasons, the NCS surface characteristics were expected at the outset to be uniquely beneficial for the tethering of the Pt precursor during ALD, subsequent Pt NP nucleation and growth, and for stabilization of the NPs during their operation as an ORR catalyst.
To identify the surface oxygen functionalities on the NCS85 surface that are likely nucleating sites for the ALD Pt precursor, we performed temperature programmed desorption (TPD) in vacuum with quadrupole mass spectrometry analysis of the products (Fig. S15, ESI†). CO2 is produced from surface lactone, acid anhydride and carboxyl groups at 200–1000 °C, while CO, the primary product, forms from surface hydroxyl, carbonyl, acid anhydride and ether group oxidation at 100–1200 °C.31 The stability of these groups at high temperatures is also highly advantageous, as these sites need to survive the thermal conditions used to convert metal precursors to stable metal nanoparticles at 190 °C and potentially also handle annealing to still higher temperatures if a greater extent of NCS crystallinity was desired. Fig. S16 (ESI†) shows that a substantial amount of H2 (∼5× the CO or CO2 yield) is released during TPD, arising mostly from hydrogen terminated edge sites (C–H), but also from COH, CHOH, and COOH surface groups located at graphene sheet edge sites.32 These H2-generating edge sites (e.g., C–H groups) could also serve as useful nucleation sites for the Pt ALD precursor and then subsequent growth of the Pt NPs.33,34 The as-prepared NCS therefore has a high percentage of active/edge sites (H-terminated and oxygen functional groups) that are likely responsible for the observed uniform distribution of ALD Pt NPs throughout the full thickness of the NCS85 membranes.
HR-TEM analysis (Fig. S17, ESI†) of the Ptx/NCS85 catalyst shows that rather few of the Pt nanoparticles are spherical in shape and that more are elongated, also revealing a monocrystalline structure. Although the XRD pattern of the Pt NPs shows Pt(111), Pt(200), Pt(220), Pt(311), and Pt(222) peaks (Fig. S18, ESI†), TEM analysis also reveals that the individual Pt particles are monocrystalline in nature.
Overall, the uniform distribution of non-agglomerated Pt NPs achieved by ALD of the Pt precursor into the NCS85 membrane augers very well in terms of its promise as a catalyst layer in PEMFC MEA testing. Furthermore, the small size of the Pt NPs, combined with the rich density of edge sites and oxygen surface functionalities, predicts a high performance, especially in the kinetic region, under these realistic testing conditions.
2.3 MEA-based performance of ALD Ptx/NCS cathode catalyst layers
Following the atomic layer deposition of Pt, Nafion was deposited into the Pt-loaded NCS85 materials using a drop-casting method. The resulting Ptx/NCS85 membranes were then assembled into an MEA as the cathode, using a standard anode-coated membrane, with the details given in the Experimental section. Initial MEA testing was then carried out in a H2/N2 flow at 20 mV s−1 to determine the electrochemical characteristics of the Pt NPs.Fig. 5a shows the cyclic voltammetry of the three Ptx/NCS85 catalyst layers in the MEA under H2/N2 flow. Typical of Pt NPs, two pairs of peaks are seen for Pt30/NCS85 and Pt40/NCS85 in the HUPD (hydrogen underpotential deposition and removal) region, one centered at ca. 0.1 V and the second at 0.25 V. These peaks are broader in the case of Pt20/NCS85, perhaps suggesting the presence of a wider range of surface sites than for the other two catalysts. Fig. 5a also shows that the double layer capacitance between 0.4 and 0.6 V is slightly higher in the case of Pt40/NCS85, attributed to the higher Pt loading.
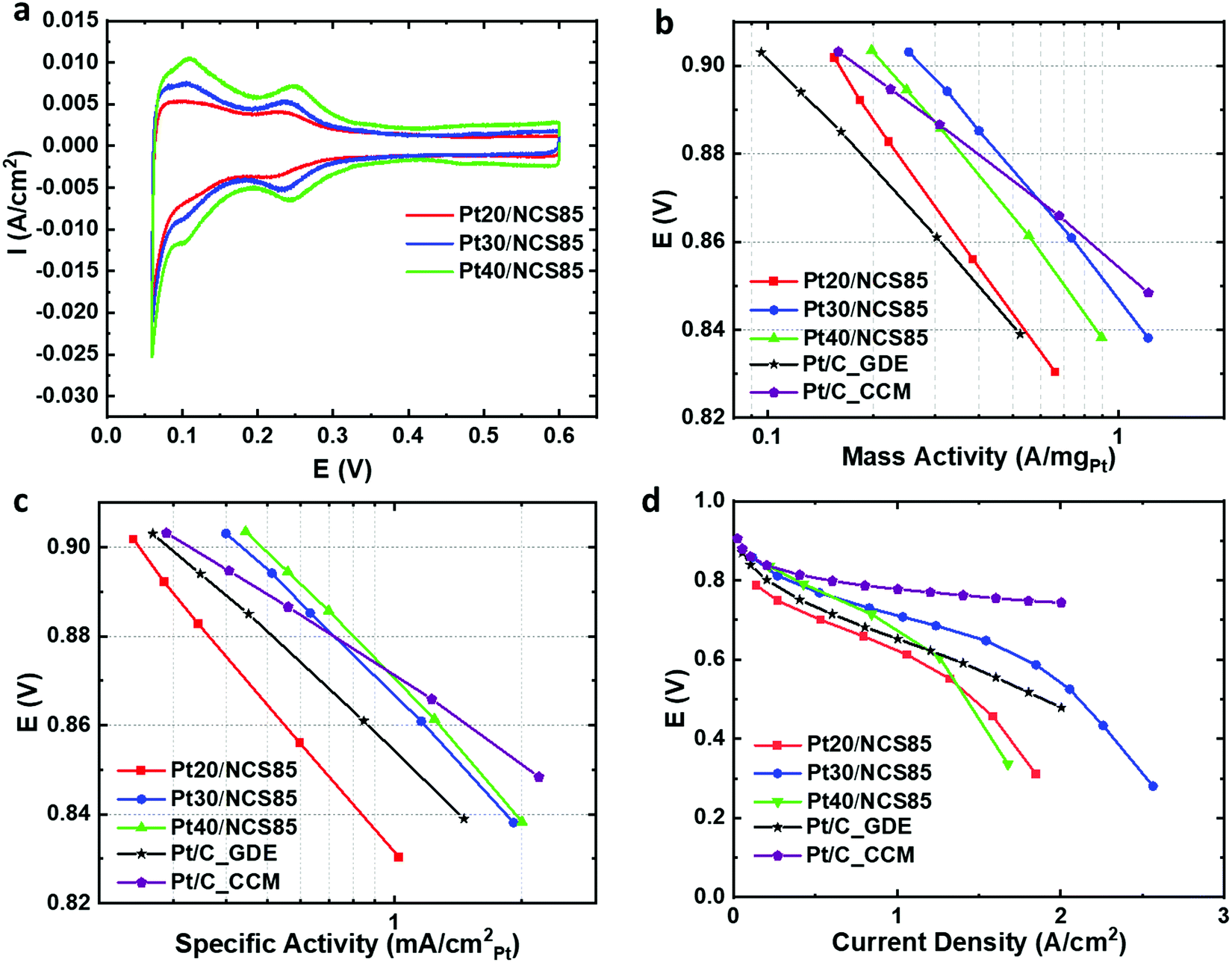 | ||
| Fig. 5 (a) Cyclic voltammetry of Ptx/NCS85 catalyst layers in an MEA in H2/N2 at 80 °C. (b) and (c) show the IR-corrected mass activity and specific activity, respectively, of the Ptx/NCS85 cathode layers and the commercial Pt/C cathode layers (CCM and GDE) as a function of current density. (b) ORR mass activity in A mgPt−1 and (c) shows the ORR specific activity in mA cm−2Pt. (d) H2-air fuel cell polarization curves of Ptx/NCS85 and commercial MEAs, with (b) to (d) collected at 80 °C, 100% RH and 150 kPaabs. Pt loadings for Pt20/NCS85, Pt30/NCS85 and Pt40/NCS85 were 0.26, 0.44 and 0.60 mgPt cm−2, respectively, while the Pt loading was 0.5 mgPt cm−2 for the commercial GDE and 0.4 mgPt cm−2 for the commercial CCM. | ||
Fig. S19 and Table S5 (ESI†) give the electrochemically active surface area (ECSA) of Pt in the ALD Ptx/NCS catalyst layers within the MEA. The ECSA values of Pt30/NCS85 and Pt40/NCS85 are similar (5.0 ± 0.7 and 6.4 ± 1.1 nm, respectively) as their Pt NPs are similar in size, while the Pt20/NCS85 material has the largest ECSA (64.5 m2 gPt−1), related to its smaller NP size (Fig. 5a). The Pt utilization of the Ptx/NCS85 catalyst layers was determined from the ratio of the experimental ECSA, calculated from the hydrogen underpotential deposition (HUPD) charge obtained in the MEA, to the maximum surface area of Pt estimated from the average Pt particle size of Pt obtained from TEM analyses, assuming a spherical NP shape. The results reveal high utilization values in the range of ca. 80–100%, which is as high as the catalyst layers reported by Harzer et al.35 The Pt20/NCS85 CL has the maximum possible ECSA (80 m2 gPt−1), based on the TEM-observed Pt NP size. This is as expected, as this CL has the lowest Pt loading and the smallest Pt NP size (3.5 ± 0.3 nm) of all of the ALD Ptx/NCS85 CLs studied in this work.
MEA testing was then carried out in 100% RH H2/O2 at 80 °C, with Fig. 5b showing the IR-corrected mass activity data in the kinetic region. In this mode of comparison, Pt30/NCS85 is the best performer relative to Pt20/NCS85 and Pt40/NCS85, giving the highest mass activity (0.25 A mgPt−1). This is attributed to the relatively large Pt NP size36,37 and the non-blocked pore necks of the Pt30/NCS85 catalyst, which would also help in facilitating the uniform and inter-connected distribution of Nafion. A similar volcano trend (Fig. 5b) has been observed in previous studies,36,37 indicating that, for NCS85, the 5 nm Pt NP size is optimal for these first generation ALD Ptx/NCS catalyst layers.
Most notably, the mass activity of the ALD Pt30/NCS85 and Pt40/NCS85 catalyst layers is higher at 0.9 V than reported for the two benchmark commercial Pt/C catalysts examined here, one being a GDE (gas diffusion electrode) and the other a CCM (cathode coated membrane) configuration. Importantly, the Pt loadings for these two commercial benchmarks are 0.4 and 0.5 mgPt cm−2 for the CCM and GDE MEAs, respectively (Fig. 5b and Table S6, ESI†), which is very comparable to the 0.44 mgPt cm−2 loading of our ALD Pt30/NCS85 catalysts. This may be attributed to the use of ALD Pt loading as ALD, which is known to produce particularly active Pt NPs compared to conventional Pt loading methods. It is also possible that the ordered and hydrophilic NCS85 structure is contributing to these very high mass activities.
Fig. 5c reveals the IR-corrected specific activity of the Ptx/NCS catalyst layers in the kinetic region in the H2/O2-fed MEA. Both the Pt30/NCS85 and Pt40/NCS85 CLs are also better performing by this measure as compared to the commercial CCM and GDE Pt/C catalysts, making our new ALD-Ptx/NCS85 membranes highly promising for future PEM fuel cell applications (Fig. 5c). In terms of optimal Pt loading, both the Pt30/NCS85 and Pt40/NCS85 catalysts give the highest specific activities, roughly 0.4 mA cm−2Pt at 0.9 V, while the Pt20/NCS85 is significantly less active. Several previous studies have reported that the specific activity of Pt NPs can be increased by increasing their diameter, argued to be because this would give a larger number of high-coordination surface sites, which may be the case here too.36,37 Similarly, Xu et al. reported an increase in the specific activity with increasing Pt mass loading and Pt NP size, with Pt prepared by ALD on glassy carbon.15
The performance of both the Pt30/NCS85 and Pt40/NCS85 CLs is better by this measure as compared to both of the commercial CCM and GDE Pt/C benchmark catalysts (Fig. 5b and c). Furthermore, the performance of our ALD Pt/NCS85 catalysts approaches that of the best specific and mass activities reported for several conventional ink-based Pt/C catalysts.35,38–40 For example, Yarlagadda et al.40 used testing conditions that were quite similar to ours, facilitating a direct comparison. This shows that, while a significantly lower Pt loading was used, a similar mass activity (ca. 0.3 A mgPt−1) but a higher specific activity (ca. 0.6 mA cmPt−2) is obtained. While the Yarlagadda Pt/C catalyst does out-perform our ALD Pt/NCS85 materials in the kinetic region,40 it remains clear that our new catalyst layers are very promising for future PEM fuel cell applications, especially as they have not yet been fully optimized.
The Tafel slopes were also obtained from Fig. 5c (Table S5, ESI†), assuming that they are dominated by the ORR at the ALD-Ptx/NCS cathodes (the HOR is known to be very rapid and thus contributes insignificantly to the observed losses in the kinetic region). The ORR at Pt30/NCS85 and Pt40/NCS85 gives a similar Tafel slope of ca. 90 mV dec−1. However, the ORR at the Pt20/NCS85 catalyst layer exhibited a significantly higher Tafel slope of 115 mV dec−1. This high Tafel slope value may reveal an inhomogeneous ionomer distribution, as the ionomer:Pt ratio was kept constant for all of the ALD Ptx/NCS85 catalysts and the ionomer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :carbon ratio was therefore the lowest for Pt20/NCS85. Indeed, lower Tafel slopes should be obtained for the catalysts that give the highest mass activity, which is the case also in our work (Tables S5 and S6, ESI†). It is worth noting that a normal ∼70 mV dec−1 Tafel slope value was obtained here by correcting the slopes for the Pt30/NS85 and Pt40/NCS85 materials for the protonic resistance (RH+,catalyst layer) using a value of 0.5 Ohm cm2, reported for non-ALD Pt deposited Pt/NCS85 catalyst layers.41 This is consistent with the fact that Nafion infiltration and loading was not optimized in the present work.
:carbon ratio was therefore the lowest for Pt20/NCS85. Indeed, lower Tafel slopes should be obtained for the catalysts that give the highest mass activity, which is the case also in our work (Tables S5 and S6, ESI†). It is worth noting that a normal ∼70 mV dec−1 Tafel slope value was obtained here by correcting the slopes for the Pt30/NS85 and Pt40/NCS85 materials for the protonic resistance (RH+,catalyst layer) using a value of 0.5 Ohm cm2, reported for non-ALD Pt deposited Pt/NCS85 catalyst layers.41 This is consistent with the fact that Nafion infiltration and loading was not optimized in the present work.
Fig. 5d shows the full performance plot for the Ptx/NCS85 catalysts in the H2/air-fed MEA in comparison to a commercial Pt/C with a Pt loading of 0.5 mgPt cm−2. Again, the Pt30/NCS85 catalyst exhibits a better performance than the other three catalysts in both the high and low current density regions, with full mass transport limitations projected to be reached only at a very high current density of 3 A cm−2 (Fig. 5d). In contrast, it is clear that Pt20/NCS85 has some problems with its Pt activity, arising from its low Pt loading and small Pt NP size, as seen also in the low current kinetic region (Fig. 5b and c). However, at high current densities, limitations related to local oxygen transport are likely the problem.42 For Pt40/NCS85, this catalyst is likely experiencing some blockage by Pt NPs at its pore necks (Fig. 4f), resulting in mass transport limitations at Pt40/NCS85, as was also predicted from Fig. 4c. Furthermore, there may also be problems due to flooding as a result of the high Pt loading (0.6 mgPt cm−2) employed.
We also tested a Pt10/NCS22 (with a 22 nm nominal pore size and 7 nm neck diameter, Table S2, ESI†) catalyst in an MEA configuration. Although ALD of Pt on the internal surface of the NCS22 film was successful, giving a very uniform Pt NP distribution of location, the performance of the Pt10/NCS22 catalyst layer when incorporated in the MEA was quite poor. Due to its small pore and neck sizes, it was difficult to infiltrate Nafion into the NCS22 membrane, resulting in poor performance in both the kinetic and diffusion regions (Fig. S20, ESI†). Thus the mass transport performance of the ALD Px/NCS85 CLs is not as good as seen for either the commercial CCM benchmark or as reported for several leading ink-based Pt/C catalysts. Even so, the present study clearly demonstrates that our unique ALD Ptx/NCS85 films are very promising for use as a highly active catalyst layer for the ORR in PEM fuel cells.
Overall, it is clear that Pt30/NCS85 is an excellent catalyst material for the ORR in MEA testing, thus opening the door towards a new generation of catalyst layers, involving the pre-construction of a carbon framework with the desired nanostructure, followed by atomic layer deposition of catalytic particles. A rigorous Nafion optimization study using these highly nano-engineered materials is underway and may lead to still better performances and propel this approach to the forefront of PEMFC fabrication. Future experiments will also focus on establishing the durability of the ALD Ptx/NCS catalysts under MEA testing conditions, as well as ALD of Pt into NCS materials with completely different nanostructures.
3. Conclusions
A new generation nanoporous carbon scaffold (NCS), containing monodisperse pores ranging in diameter from 5–100+ nm, is shown here to be ideal for application as a support material for the atomic layer deposition (ALD) of Pt NPs and then for implementation as the cathode layer of PEM fuel cells. Key advantages of the NCS include its preparation using a straightforward synthesis, it is 100% carbon (no binder) and is remarkably tunable in three-dimensions, and it is also fully scalable, from a few mm2 to 150+ cm2 in area. It is also shown that these NCS materials are very good electronic conductors, as they consist of a single carbon sheet, while their ordered pore structure facilitates fluid transport, thus opening new doors for the design of high-performance catalyst layers for PEM fuel cells.Here we show that a 12 μm thick NCS85 membrane (85 nm nominal pore size) can be efficiently and rapidly loaded with Pt in 20–40 ALD cycles, giving a narrow Pt NP size distribution through the full thickness of the NCS, attributed in part to the high surface density of edge sites and oxygen-based functionalities that serve to nucleate Pt from its gas-phase precursor. It is also shown that the best performing catalyst, the 30 ALD cycle Pt30/NCS85 material, gives Pt NPs that are ca. 5 nm in diameter and have a mixture of surface facets exposed.
After Nafion infiltration, these Pt30/NCS85 materials were tested in a membrane-electrode-assembly (MEA), generating specific and mass activities in the kinetic region of 0.40 mA cm−2Pt and 0.25 A mgPt−1, respectively, under H2/O2 gas flow at 80 °C. Full mass transport limitations were not reached even at current densities as high as 2.5 A cm−2, projecting towards a noteworthy limiting current density of 3 A cm−2, thus giving a maximum power density of ca. 1230 mW cm−2 (IR-corrected). The ORR activity of the Pt30/NCS catalyst layer is shown to be significantly better than seen for two commercial Pt/C benchmark catalyst layers (GDE and CCM), both having a comparable Pt loading and tested under the identical conditions.
For an analogous NCS22 film (22 nm nominal pore size) loaded with Pt in 10 ALD cycles, the Pt NPs were also found to be distributed uniformly throughout the internal NCS22 film structure. However, the ALD Pt10/NCS22 was shown to perform less well as a cathode catalyst in the MEA than either the ALD Ptx/NCS85 or the commercial Pt/C materials. This is proposed to be due to challenges in infiltrating Nafion within the narrow channels and necks of the NCS22 membranes.
4. Experimental section
4.1 NCS slurry precursor preparation8
0.1 g of mesophase pitch (MP, Momentum Materials Solutions (https://momentummaterials.ca/mesophase-pitch), softening point: 250 °C, ash <150 ppm, coking value ≥90%, toluene insolubles: 65 ± 3% and quinoline insolubles: 42 ± 3%), 0.2 g n-butanol and 5.0 g 10% polyvinyl alcohol (PVA, Alfa Aesar, 86–89% hydrolyzed, low molecular weight)/water were ball-milled together, producing a uniform MP/PVA ink. A colloidal silica suspension containing 0.5 g silica (Ludox SM-30, Ludox AS-40, NexSil-8040, or NexSil-125-40 with an average particle size of 8, 22, 50 and 85 nm, respectively) was added to 1.0 g of 1,3-propanediol (PD):water (mass ratio 1:1) to produce a silica suspension. The silica suspensions were added to the MP/PVA ink and then ball-milled to obtain the MP/PVA/PD/silica ink (or slurry), which was then degassed for 15 min to remove any bubbles.
4.2 NCS preparation
The slurries were normally tape-cast on glass with a 0.010 inch gap between the doctor blade and the substrate. After drying overnight, a pristine composite MP/PVA/PD/silica film was obtained. These films were imprinted in an alumina tubular furnace at 400 °C for 2 h in nitrogen, heated at 900 °C for 2 h to achieve carbonization, soaked in 3 M NaOH at 80 °C for 2 days to remove the silica template, washed successively with deionized water, 1 M HCl, and deionized water, and then dried at 80 °C overnight.4.3 Atomic layer deposition of Pt
Trimethyl(methylcyclo-pentadienyl) platinum(IV) (MeCpPtMe3, Strem Chemicals) was used as the Pt ALD precursor and air was used as the oxidant. Pt ALD was conducted using the NCS22 and NCS85 substrates at a reactor temperature of 190 °C and the Pt precursor cylinder was heated to 78 °C. The exposure time to the Pt precursor and air were optimized as 9 s and 5 s, respectively. Ar was used as the inert gas to remove any excess reactant. The Ar purge time after Pt precursor exposure was 100 s and the purge time after air exposure was 50 s.4.4 Physical characterization methods
A 3Flex Version 3.01 gas sorption instrument (Alberta Sulfur Research Ltd) was used to run nitrogen gas sorption analysis and determine the specific surface area and porosity of the NCS samples. Data were collected at 77 K with prior degassing in N2 at 150 °C for 4 h. Porosity values were determined using the grain and bulk volume/density values, where grain density values were measured by helium pycnometry using an AccuPyc II 1340 (Micromeritics®). The bulk density of the NCS films was determined by weighing a large piece of NCS film of a precise area (e.g., 10 cm2) with the thickness determined using FESEM.Thermogravimetric analysis of the pristine MP/PVA/PD/silica film and NCS components was conducted using a Mettler-Toledo TGA/DSC HT1600, heating to 900 °C at 2 °C min−1 under a nitrogen atmosphere. High-temperature TPD was performed using an in-house high vacuum apparatus,43 allowing quantitative detection of gases evolved during heating of samples to 1800 °C at 10 °C min−1. At these high temperatures, most of the carbon edge sites terminated by H or O were decomposed, forming H2, H2O, CO, and CO2, thus allowing the number of edge sites to be estimated. X-ray diffraction (XRD) patterns of Ptx/NCS85 films were collected using a Bruker D8 Advance ECO instrument equipped with Cu Kα source (λ = 1.54178 Å, kV = 40, mA = 25) and a Lynxeye XE detector.
In situ mass spectrometry analysis (Dycor ProLine Process Mass Spectrometer from Ametek Process Instruments, with Alicat Scientific mass flowmeters used to control gas flows) of the NCS precursor film (MP, silica, PD and PVA) was carried out on 100 mg samples in order to determine the most dominant gas evolution process that occurs during thermal treatment under He. The gas lines from the reactor to the mass spectrometer were heated to 140 °C to prevent condensation of the expected decomposition/reaction products (water, acetaldehyde and acetone). The ramp rate was 10 °C min−1 with 1 h soaks at 350, 500 and 900 °C.
Field emission scanning electron microscopy (FESEM) analysis of the NCS samples was carried out using a Zeiss Sigma VP instrument at an accelerating voltage of 8 kV (Microscopy and Imaging Facility, University of Calgary). A FEI Magellan 400 XHR scanning electron microscope with an accelerating voltage of 5 kV and a beam current of 50 pA was used for SEM analysis of the Ptx/NCS85 samples. The platinum elemental mapping of the Ptx/NCS85 and Pt10/NCS22 samples was carried out using an FEI Quanta 250 FEG environmental field emission scanning electron microscope equipped with a Bruker Quantax energy-dispersive X-ray spectroscopy (EDS) system with an accelerating voltage of 15 kV. JEOL JEM-ARM200cFS/TEM (nanoFAB Center, University of Alberta), equipped with a cold Field-Emission Gun (cFEG) and a probe Cs corrector and operating at an acceleration voltage of 200 kV, was used to conduct Bright-Field (BF) transmission electron microscopy (TEM) analysis of the NCS and Ptx/NCS films. The samples were prepared by suspending the NCS powder in ethanol and then adding one drop of this liquid onto a carbon-coated Cu TEM grid.
The van der Pauw method was used to measure the conductivity of a 1 × 1 cm2 piece of NCS film.44 Each corner was connected to a Ni wire using Ag paste, with the wires having the same diameter and length. Current was applied between two of the contacts and the voltage was measured between the other two contacts. As a reference, the conductivity of Vulcan carbon (VC) powder was measured by loading it into a ceramic tube and pressing it between two stainless steel rods at 4.6 MPa.
The Wilhelmy plate method was used to measure the water contact angle of NCS under ambient conditions (25 °C and 0.1 MPa).45 A 1 × 2 cm2 piece of the NCS was dispensed on the top of an empty 10 cm diameter Petri dish. DI water was then gradually added until the base of the NCS contacted the surface of the DI water. Due to the hydrophilicity of the NCS, a DI water meniscus formed spontaneously and stabilized within seconds. A Canon EOS Rebel SL2 camera was used to record the process at 25 frames per second with a resolution of 18–30 μm per pixel. The stabilized water contact angle was measured using ImageJ software.
4.5 Electrochemical studies of NCS films in sulfuric acid solution
Cyclic voltammetry (2–100 mV s−1), and electrochemical impedance spectroscopy (10 mV rms amplitude; 0.4 vs. RHE) were carried out with a Bio-Logic SP-300 potentiostat in 3-electrode cells containing deaerated 0.5 M H2SO4, a platinized Pt mesh as the counter electrode, and a reversible hydrogen (RHE) reference electrode. Generally, a piece of the NCS film (1 × 1 cm2) was sandwiched between two microscopic slides that had been sputter-coated with Au on their inner surfaces. A small circular hole was cut on the Au-coated slide, exposing a 0.8 cm2 geometric area of the NCS for electrochemical studies.4.6 MEA construction and testing
Ptx/NCS85 films were placed on a GDL/MPL (Freudenberg H23C8, Fuel Cell Store) followed by drop casting 4 wt% of Nafion solution D2021 in isopropyl alcohol to form the gas diffusion electrodes (GDE). After air drying, the cathode GDE was hot pressed onto a commercial anode-coated membrane ((ACM), where the membrane thickness was 18 μm and the Pt/C anode had a Pt loading of 0.1 mgPt cm−2) at 120 °C and 500 lbf cm−2. Importantly, the ACM was kept unchanged in all of our tested MEAs to ensure that any changes in performance could be ascribed fully to changes in the activity of the cathode catalyst layer. The membrane-electrode-assembly (MEA) was assembled with Freudenberg H23C8 gas diffusion layers and loaded into a Greenlight research cell fixture with a serpentine flow pattern. The cell was compressed to 4 bar.For MEA testing, a Scribner 840 fuel cell testing system was used. Conditions were set to at 80 °C, 100% RH, and 150 kPa absolute backpressure. After a standard activation procedure, a cathode recovery step was employed.46 Cyclic voltammograms (20 mV s−1) were collected with the cathode purged with Ar (99.999%, Praxair). Activity measurements were carried out in O2 (99.993%, Praxair) and H2 (99.999%, Praxair) fed at 5000 sccm and 500 sccm, respectively, with potentials held for 5 min. Polarization curves were collected in house air at 5000 sccm with currents held constant for 3 min. The high-frequency resistance was ascertained from an electrochemical impedance spectroscopy (EIS) Nyquist plot, collected at 0.25 A in oxygen. A commercial gas diffusion electrode (GDE, 60% Pt (0.5 mgPt cm−2) on Vulcan Carbon Paper, Fuel Cell Store) and a commercial catalyst-caoted membrane (CCM, 0.1 mgPt cm−2 anode loading and 0.4 mgPt cm−2 cathode loading) were tested to compare with the Ptx/NCS85 and Pt10/NCS22 catalyst layers.
Author contributions
All authors have read and approved the final version of the manuscript. In addition, M. A. prepared and carried out the characterization and electrochemical testing of the NCS material, X. L. optimized the slurry used in NCS preparation and carried out experiments to better understand the process, R. T. and H. N. carried out the TPD analyses and interpreted the results, Z. W. and S. X. conducted the Pt ALD on the NCS substrate, M. A. and S. D. carried out the characterization of the Pt/NCS films, S. D. and S. X. tested the performance of the ALD Pt/NCS films in a membrane-electrode assembly PEM fuel cell test station, and M. A., S. D., F. P. and S. X. analyzed the fuel cell data. M. A., X. L., S. D., Z. W., S. X, X. T. and V. B. wrote the overall manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
V. I. B. acknowledges the Natural Sciences and Engineering Research Council of Canada (NSERC) for financial support, as well as support from the Canada Research Chairs program. H. N. acknowledges partial support from the Cooperative Research Program of Five-star Alliance and NJRC Mater. & Dev. of Japan. M. A. extends her acknowledgements to the Egyptian Cultural Affairs and Missions Sector and NSERC for scholarship support. We also thank Drs. Robert Marriott and Ruohong Sui for their help with the gas sorption measurements. We also thank Dr Scott Paulson for useful discussions, Bin Pan for contact angle measurements and Arlene Ai for a SEM image. Z. W., S. D., S. X., and F. P. thank the Volkswagen Group of America for financial support and extend their acknowledgments to the Stanford Nano-Shared Facilities (SNSF) where part of the work was performed, supported by the National Science Foundation under award ECCS-1542152. Finally, V. I. B. and M. A. would like to thank Drs. Haoyang Yu and Peng Li from the nanoFAB Center at the University of Alberta for collecting the HR-TEM images.References
- Y. K. Sun, Z. Chen, H. J. Noh, D. J. Lee, H. G. Jung, Y. Ren, S. Wang, C. S. Yoon, S. T. Myung and K. Amine, Nat. Mater., 2012, 11, 942–947 CrossRef CAS PubMed.
- J. Ni and Y. Li, Adv. Energy Mater., 2016, 6, 1600278 CrossRef.
- Z. Zhao, M. Li, L. Zhang, L. Dai and Z. Xia, Adv. Mater., 2015, 27, 6834–6840 CrossRef CAS PubMed.
- M. Zhou, H. L. Wang and S. Guo, Chem. Soc. Rev., 2016, 45, 1273–1307 RSC.
- B. H. Han, W. Zhou and A. Sayari, J. Am. Chem. Soc., 2003, 125, 3444–3445 CrossRef CAS PubMed.
- S. Korkut, J. D. Roy-Mayhew, D. M. Dabbs, D. L. Milius and I. A. Aksay, ACS Nano, 2011, 5, 5214–5222 CrossRef CAS PubMed.
- S. Ohya, Y. Fujii, S. Yao, Y. Asano, K. Nakayama and K. Fukunaga, US Pat., 6565962, 2003 Search PubMed.
- V. Birss, X. Li, D. Banham and D. Y. Kwok, WO2015135069A1PCT/CA2015/000516, 2015.
- M. N. Islam, U. Shrivastava, M. Atwa, X. Li, V. Birss and K. Karan, ACS Appl. Mater. Interfaces, 2020, 12, 39215–39226 CrossRef CAS PubMed.
- B. Pan, C. R. Clarkson, M. Atwa, C. Debuhr, A. Ghanizadeh and V. I. Birss, J. Colloid Interface Sci., 2021, 489, 411–423 CrossRef PubMed.
- M. Atwa, X. Li, F. Forouzandeh, S. Luong, E. Bertin, M. J. Parnian, K. Karan and V. Birss, in Low-Temp. fuel cells, electrolysers & H2 processing, ed. A. Gasteiger and H. Bandarenka, Lucerne, 2019, pp. 112–120 Search PubMed.
- B. J. O’Neil, D. H. K. Jackson, J. Lee, C. Canlas, P. C. Stair, C. L. Marshall, J. W. Elam, T. F. Kuech, J. A. Dumesic and G. W. Huber, ACS Catal., 2015, 5, 1804–1825 CrossRef.
- D. Liu, X. Li, S. Chen, H. Yan, C. Wang, C. Wu, Y. A. Haleem, S. Duan, J. Lu, B. Ge, P. M. Ajayan, Y. Luo, J. Jiang and L. Song, Nat. Energy, 2019, 4, 512–518 CrossRef CAS.
- J. S. King, A. Wittstock, J. Biener, S. O. Kucheyev, Y. M. Wang, T. F. Baumann, S. K. Giri, A. V. Hamza, M. Baeumer and S. F. Bent, Nano Lett., 2008, 8, 2405–2409 CrossRef CAS PubMed.
- S. Xu, Y. Kim, J. Park, D. Higgins, S. J. Shen, P. Schindler, D. Thian, J. Provine, J. Torgersen, T. Graf, T. D. Schladt, M. Orazov, B. H. Liu, T. F. Jaramillo and F. B. Prinz, Nat. Catal., 2018, 1, 624–630 CrossRef CAS.
- H. Yang, S. Xu, L. Jiang and Y. Dan, J. Macromol. Sci., Part B: Phys., 2012, 51, 464–480 CrossRef CAS.
- F. Liu, A. E. Rotaru, P. M. Shrestha, N. S. Malvankar, K. P. Nevin and D. R. Lovley, Energy Environ. Sci., 2012, 5, 8982–8989 RSC.
- J. Zou, J. Liu, A. S. Karakoti, A. Kumar, D. Joung, Q. Li, S. I. Khondaker, S. Seal and L. Zhai, ACS Nano, 2010, 4, 7293–7302 CrossRef CAS PubMed.
- D. Banham, F. Feng, J. Burt, E. Alsrayheen and V. Birss, Carbon, 2010, 48, 1056–1063 CrossRef CAS.
- K. Kinoshita, Carbon: Electrochemical and Physicochemical Properties, Wiley, 1988 Search PubMed.
- Z. Wu, W. Li, Y. Xia, P. Webley and D. Zhao, J. Mater. Chem., 2012, 22, 8835–8845 RSC.
- L. Wei, M. Sevilla, A. B. Fuertes, R. Mokaya and G. Yushin, Adv. Energy Mater., 2011, 1, 356–361 CrossRef CAS.
- T. Onn, R. Küngas, P. Fornasiero, K. Huang and R. Gorte, Inorganics, 2018, 6, 34 CrossRef.
- J. P. Owejan, J. E. Owejan, W. Gu, T. A. Trabold, T. W. Tighe and M. F. Mathias, J. Electrochem. Soc., 2010, 157, B1456 CrossRef CAS.
- M. Chen, C. Zhao, F. Sun, J. Fan, H. Li and H. Wang, eTransportation, 2020, 5, 100075 CrossRef.
- G. Wang, L. Zou, Q. Huang, Z. Zou and H. Yang, J. Mater. Chem. A, 2019, 7, 9447–9477 RSC.
- A. I. Aria and M. Gharib, Langmuir, 2011, 27, 9005–9011 CrossRef CAS PubMed.
- B. Yao, J. Chen, Y. Li, Y. Wen, M. Wu and G. Shi, Adv. Mater. Interfaces, 2016, 3, 3–8 Search PubMed.
- Q. Shen and L. H. Jiang, ChemistrySelect, 2016, 1, 3132–3135 CrossRef CAS.
- H. Yan, H. Cheng, H. Yi, Y. Lin, T. Yao, C. Wang, J. Li, S. Wei and J. Lu, J. Am. Chem. Soc., 2015, 137, 10484–10487 CrossRef CAS PubMed.
- R. Tang, K. Taguchi, H. Nishihara, T. Ishii, E. Morallón, D. Cazorla-Amorós, T. Asada, N. Kobayashi, Y. Muramatsu and T. Kyotani, J. Mater. Chem. A, 2019, 7, 7480–7488 RSC.
- K. Nomura, H. Nishihara, N. Kobayashi, T. Asada and T. Kyotani, Energy Environ. Sci., 2019, 12, 1542–1549 RSC.
- H. Itoi, H. Nishihara, S. Kobayashi, S. Ittisanronnachai, T. Ishii, R. Berenguer, M. Ito, D. Matsumura and T. Kyotani, J. Phys. Chem. C, 2017, 121, 7892–7902 CrossRef.
- H. Nishihara, F. Ohtake, A. Castro-Muñiz, H. Itoi, M. Ito, Y. Hayasaka, J. Maruyama, J. N. Kondo, R. Osuga and T. Kyotani, J. Mater. Chem. A, 2018, 6, 12523–12531 RSC.
- G. S. Harzer, A. Orfanidi, H. El-Sayed, P. Madkikar and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165, F770–F779 CrossRef CAS.
- D. Li, C. Wang, D. S. Strmcnik, D. V. Tripkovic, X. Sun, Y. Kang, M. Chi, J. D. Snyder, D. Van Der Vliet, Y. Tsai, V. R. Stamenkovic, S. Sun and N. M. Markovic, Energy Environ. Sci., 2014, 7, 4061–4069 RSC.
- M. Shao, A. Peles and K. Shoemaker, Nano Lett., 2011, 11, 3714–3719 CrossRef CAS PubMed.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- S. Ott, A. Orfanidi, H. Schmies, B. Anke, H. N. Nong, J. Hübner, U. Gernert, M. Gliech, M. Lerch and P. Strasser, Nat. Mater., 2020, 19, 77–85 CrossRef CAS PubMed.
- V. Yarlagadda, M. K. Carpenter, T. E. Moylan, R. S. Kukreja, R. Koestner, W. Gu, L. Thompson and A. Kongkanand, ACS Energy Lett., 2018, 3, 618–621 CrossRef CAS.
- M. N. Islam, PhD thesis, University of Calgary, 2020.
- A. Kongkanand and M. F. Mathias, J. Phys. Chem. Lett., 2016, 7, 1127–1137 CrossRef CAS PubMed.
- T. Ishii, S. Kashihara, Y. Hoshikawa, J.-I. Ozaki, N. Kannari, K. Takai, T. Enoki and T. Kyotani, Carbon, 2014, 80, 135–145 CrossRef CAS.
- O. Philips’Gloeilampenfabrieken, Philips Res. Rep., 1958, 13, 1–9 Search PubMed.
- A. W. Adamson, Physical Chemistry of Surfaces, John Wiley and Sons, Inc., 1990 Search PubMed.
- J. Zhang, B. Litteer, F. Coms and R. R. Makharia, ECS Trans., 2019, 41, 1471–1485 CrossRef.
- K. Karan, Curr. Opin. Electrochem., 2017, 5(1), 27–35 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1mh00268f |
| ‡ Current address: Nanjing Momentum Materials Technologies Ltd Co., 368 East Zhe’ning Road, Lishui, Nanjing, Jiangsu, 211215, China. |
| This journal is © The Royal Society of Chemistry 2021 |