
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The pursuit of stability in halide perovskites: the monovalent cation and the key for surface and bulk self-healing†
D. R.
Ceratti
 *a,
A. V.
Cohen
a,
R.
Tenne
b,
Y.
Rakita
a,
L.
Snarski
a,
N. P.
Jasti
cd,
L.
Cremonesi
e,
R.
Cohen
c,
M.
Weitman
c,
I.
Rosenhek-Goldian
f,
I.
Kaplan-Ashiri
f,
T.
Bendikov
f,
V.
Kalchenko
g,
M.
Elbaum
h,
M. A. C.
Potenza
e,
L.
Kronik
*a,
G.
Hodes
*a and
D.
Cahen
*acd
*a,
A. V.
Cohen
a,
R.
Tenne
b,
Y.
Rakita
a,
L.
Snarski
a,
N. P.
Jasti
cd,
L.
Cremonesi
e,
R.
Cohen
c,
M.
Weitman
c,
I.
Rosenhek-Goldian
f,
I.
Kaplan-Ashiri
f,
T.
Bendikov
f,
V.
Kalchenko
g,
M.
Elbaum
h,
M. A. C.
Potenza
e,
L.
Kronik
*a,
G.
Hodes
*a and
D.
Cahen
*acd
aWeizmann Institute of Science, Department of Materials and Interfaces, 7610001, Rehovot, Israel. E-mail: davide-raffaele.ceratti@weizmann.ac.il; david.cahen@weizmann.ac.il; gary.hodes@weizmann.ac.il; leeor.kronik@weizmann.ac.il
bWeizmann Institute of Science, Department of Physics of Complex Systems, 7610001, Rehovot, Israel
cBar Ilan University, Department of Chemistry, 5290002, Ramat Gan, Israel
dBar Ilan University, Bar-Ilan Institute for Adv. Mater., BINA, 5290002, Ramat Gan, Israel
eDepartment of Physics and CIMAINA, University of Milan, via Celoria, 16, 20133, Milan, Italy
fWeizmann Institute of Science, Department of Chemical Research Support, 7610001, Rehovot, Israel
gWeizmann Institute of Science, Department of Veterinary Resources, 7610001, Rehovot, Israel
hWeizmann Institute of Science, Department of Chemical and Biological Physics, 7610001, Rehovot, Israel
First published on 12th March 2021
Abstract
We find significant differences between degradation and healing at the surface or in the bulk for each of the different APbBr3 single crystals (A = CH3NH3+, methylammonium (MA); HC(NH2)2+, formamidinium (FA); and cesium, Cs+). Using 1- and 2-photon microscopy and photobleaching we conclude that kinetics dominate the surface and thermodynamics the bulk stability. Fluorescence-lifetime imaging microscopy, as well as results from several other methods, relate the (damaged) state of the halide perovskite (HaP) after photobleaching to its modified optical and electronic properties. The A cation type strongly influences both the kinetics and the thermodynamics of recovery and degradation: FA heals best the bulk material with faster self-healing; Cs+ protects the surface best, being the least volatile of the A cations and possibly through O-passivation; MA passivates defects via methylamine from photo-dissociation, which binds to Pb2+. DFT simulations provide insight into the passivating role of MA, and also indicate the importance of the Br3− defect as well as predicts its stability. The occurrence and rate of self-healing are suggested to explain the low effective defect density in the HaPs and through this, their excellent performance. These results rationalize the use of mixed A-cation materials for optimizing both solar cell stability and overall performance of HaP-based devices, and provide a basis for designing new HaP variants.
New conceptsStudying photo-damage in and on halide perovskites, HaPs, we show how, after being damaged, the material heals much more readily in confined chemical environments than if products of the damage can escape, or can further react with the ambient. This finding is especially important because current encapsulation strategies of HaP-based devices do not take this into account and use materials that can react with damage products, thus interfering with self-healing. Building on our earlier discovery of HaP self-healing, we now define the reactions and conditions necessary for the process. We show and explain differences between the monovalent methylammonium, formamidinium and cesium cations for damaging and self-healing in-bulk and on-surface of these perovskites. This work underlines the relevance of the dynamic nature of HaPs, materials in a steady-state of damage and repair, determined by both the kinetics and thermodynamics of the critical reactions identified here. On this basis, we can now explain the contribution of each monovalent cation to the overall stability and quality of mixed cation perovskites in record efficiency solar cells. Finally we show that the materials’ self-healing is crucial for the properties of devices because self-healing is critical in determining the low defect densities of the HaPs. |
1 Introduction
1.1 Background
Halide Perovskites (HaPs) continue to have significant impact on the field of solar cells, thanks to their very low cost, ease of production, and competitive efficiencies that are comparable, if not superior, to those of established photovoltaic (PV) technologies.1–3 In the wake of these PV results, other applications, such as light-emission, radiation detection, and electronics are explored.4–7 Despite all these efforts, a critical issue hangs as a sword of Damocles over the entire field: stability. Various approaches for delaying or avoiding device degradation, due to external influences such as heat, light, or chemicals, have been tested.8 Encapsulation of the solar cell and perovskite passivation through long alkylamine molecules,9 as well as use of 2D variants of HaPs,10,11 show promise for avoiding degradation; still, the need for decades of stable operation (at least for use in solar cells) under realistic conditions makes the question of the intrinsic stability of the materials themselves crucial. Hereafter we use the term “intrinsic stability” for the stability of the material, irrespective of external (ambient, light, temperature) influence. This can be tested by stressing the material to different extents; intrinsically unstable (or metastable) materials will degrade already with low intensity stresses.Degradation pathways have been identified for the thermal and photo-decomposition of methylammonium (MA) lead halides using mass spectroscopy12 and STA-FTIR (simultaneous thermal analysis (STA) and Fourier Transform-Infrared (FT-IR) Spectroscopy).13 The pathways that were identified involve the release of methylamine, hydrogen halides, halomethanes, ammonia, and halogens and likely cause loss of photo-conversion efficiency. However, such mechanistic analysis is lacking for formamidinium (FA) and cesium (Cs) lead halides, which are critical building blocks of the currently most-efficient halide perovskite solar cells.2,14 Furthermore, an important question then follows: are decomposition mechanisms connected to an intrinsic material instability or are there ways to avoid or eliminate them, or heal the damage? For example, mechanisms involving the attack of the material by external chemicals are strongly inhibited by encapsulation and do not affect the intrinsic material stability. However, if there are readily accessible degradation pathways that do not involve external chemicals, HaPs would be practically unusable for long-term operation, unless…the material can self-heal. To address this question, we focus our study on the perovskites themselves. While studies on complete cells are the most relevant ones for applications, using multi-component samples makes determining the limits of stability of the HaP material, the one irreplaceable element of a perovskite solar cell, very difficult and practically impossible.
1.2 Approach
We assess the fundamental differences between the stability of the bulk (chemically inaccessible) and surface (chemically relatively accessible) of HaP single crystals (SI.1, ESI†), by monitoring healing from strong, supra-band-gap illumination-induced damage (SI.2 and SI.3, ESI†), focusing on the critical role of A cations in HaP stability.1.3 Summary of findings
(1) There are very significant differences between self-healing of perovskites15,16 in the bulk and that at surfaces, with major effects on the perceived stability of the material.(2) Upon illumination the optoelectronic properties of the materials change differently at the surface and in the bulk, including whether and how they revert to the original state after photo-damage, as shown through confocal, and fluorescence lifetime imaging microscopy (FLIM).
To pinpoint the chemical origin of these variations,
• We analyze the effect of photodamage at the surface and in the near-surface region.
• We use density functional theory (DFT) simulations to rationalize, at a molecular level, the A cation effect. In particular, we discuss how the self-healing directly controls the defect density in the HaPs.
We conclude that.
(3) the thermodynamically-determined stability and defect density in the bulk are the relevant quantities for solar energy applications.
(4) Surface degradation is more pronounced than bulk degradation, due to kinetic effects (material loss), which can be hindered with tailored encapsulation and judicious electron transporting layer (ETL) and hole transporting layer (HTL).17
(5) We identify and explain the mechanisms involved in the self-healing, an obvious step towards optimizing self-healing. This result constitutes a major step beyond the conclusion of our earlier work, i.e., that bromide perovskite can self-heal.15
Thus, our results also go well beyond earlier suggested, deduced, or assumed self-healing,18–22 and pave a way to reach long-term stable halide perovskite solar cells rationally, i.e., beyond doing so primarily empirically.
2 Results: PL effects of surface and bulk self-healing
2.1 Time dependence of photoluminescence (PL) intensity
For the sake of discussion, we can ask if self-healing of damage due to larger (than 1 K) temperature changes will occur. Because the halide perovskite chemistry is relatively simple (see Section 5 of this article), there is only a limited number of possible degradation products and, actually, all form whatever the origin of the damage. Even if the ratios of the amount of these will differ between light- and heat-induced damage, self-healing will occur, whatever the origin of the damage. Since damage can heal completely (cf. next paragraph) we deduce that each decomposition product will be part of the healing process, irrespective of the decomposition pathway that it originates from. The exception to this discussion is the case of PbBr2, which photolyzes27,28 to Pb0 and Br2, while thermally it does not decompose (rather, it evaporates). For the latter it was argued that temperature is critical for long-term PCE stability.29,30
 | ||
| Fig. 1 Background-normalized PL intensity of (a and b) MA, (c and d) FA, and (e and f) Cs lead tribromide perovskite samples, at time zero, t = 0, i.e., immediately after bleaching) (—) and after 12 h of recovery, t = 12 h (- - -) for the 1P (left) and 2P (right) experiments. The laser power (LP) is expressed in %, with 100% (1P) being 880 μW and (2P) 771 mW. For Cs, we see in the 1P experiment a large PL increase exceeding the initial level at >80% LP as marked by the gray shading. This corresponds to a new, blue-shifted phase discussed in the text. | ||
2.2 PL spectroscopy
PL spectroscopy can indicate modification of material composition and/or structure that is damage/healing-induced, beyond changes in PL intensity.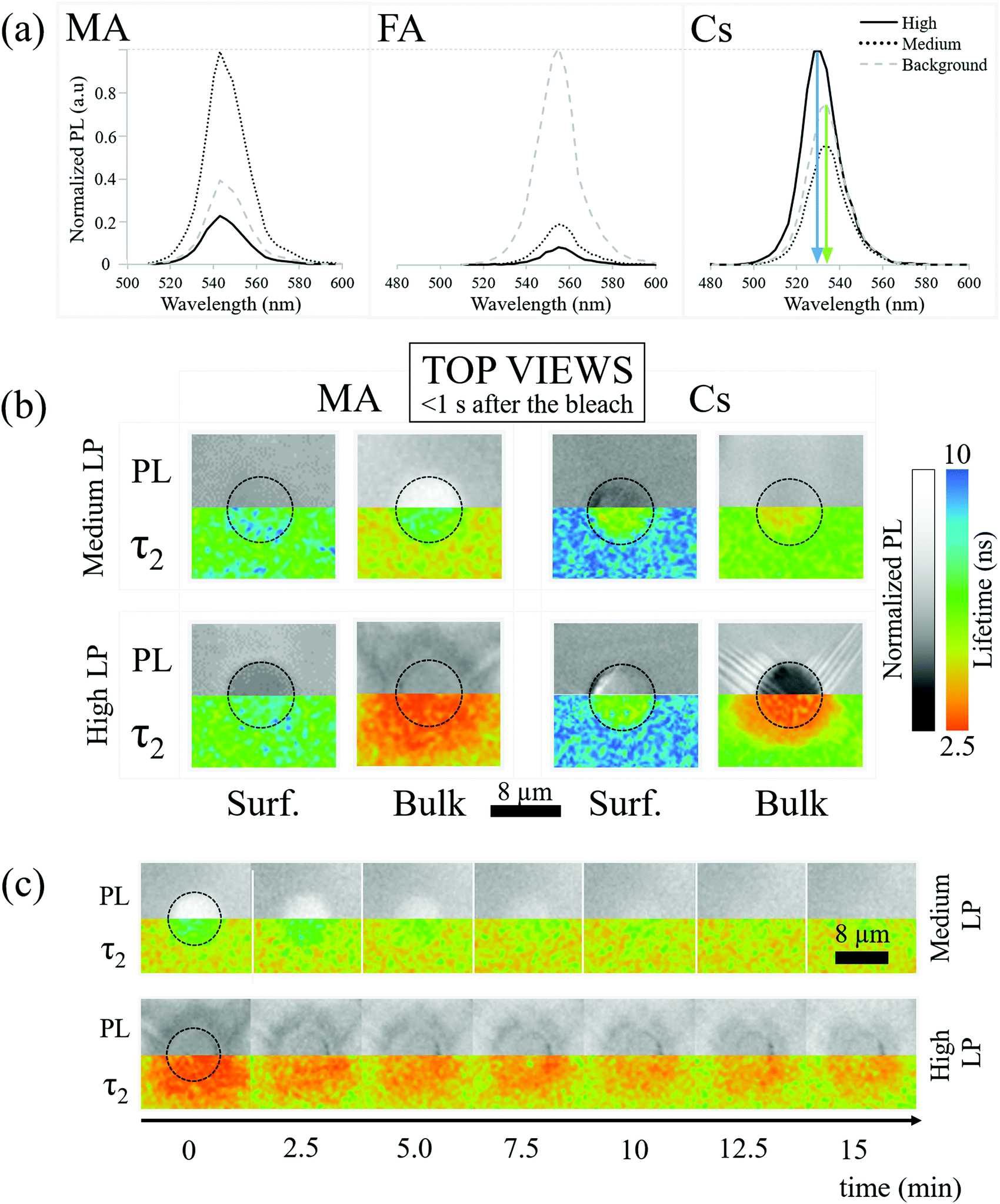 | ||
| Fig. 2 (a) Normalized PL spectra (the value of 1 is set for strongest PL after the bleaching cycle) from the surfaces of MA-, FA- and Cs-PbBr3. —: strongest bleaching. …: medium bleach. - - -: background (at 20 μm lateral distance from the ∼8 μm diameter bleached area). The arrows in the spectra of the Cs samples emphasize the blue shift of the PL peak for the high LP bleach (blue arrow), with respect to the PL peaks of the background or medium LP bleach (green arrow). The spectra are normalized to the maximum PL value of each experiment. Medium bleach in the MA case is recorded from slightly (1 μm) inside the single crystal. In all other cases it is recorded on the surface. (b) (Mirrored) images of the PL intensities (grayscale) and of PL lifetimes (color-coded – right-most legend) for the same areas/regions of the MA and Cs samples on the surface and in the bulk, just after (<1 s) medium and strong bleaching. (c) As (b) for only the MA sample and only in the bulk, showing the time evolution of the PL (grayscale) and PL lifetime after bleaching, at medium LP (top) and high LP (bottom). Each step corresponds to a delay of 2.5 minutes. As in (b) the PL and lifetime images are mirrored to facilitate comparison between them. | ||
2.3 PL lifetime imaging (fluorescence lifetime imaging microscopy, FLIM)
The absence of spectral shifts in the perovskite emission leaves the possibility that bleaching influences the material through thermal or photo-induced trap formation. To explore this, we map the PL lifetime of the perovskite crystals both at the surface and in the bulk.Bleaching-created traps should decrease the lifetime of the photo-generated carriers and decrease PL lifetime. Decreasing the lifetime also decreases the PL intensity. Specifically, in a trap-assisted recombination regime (indicated by exponential decay of the instantaneous PL intensity over time), the integrated PL intensity between pulses decreases if the lifetime decreases. In Fig. 2b we relate the PL intensities (top) and FLIM (bottom) measurements of the MA and Cs samples just after medium LP (30 seconds of solar illumination) and high LP (100–200 seconds of solar illumination) bleaching on the surface and in the bulk (110 μm). When discussing PL intensities we consider the PL “increased”/“decreased” if it is more/less intense than the background respectively.
The PL decay is found to be bi-exponential (details of the fitting are given in SI.7, ESI†). Following SI.7 (ESI†), the fastest decay is due to carrier trapping and the slowest to detrapping. We focus on the slowest decay because it is the kinetically limiting process. Additional information on the shorter lifetime results/processes is reported in SI.7 (ESI†).
Fig. 2b-MA-Bulk shows an increase of lifetime for medium LP compared to the pre-bleaching and background lifetime (Fig. 2b-MA-Bulk, top), where the PL increases, along with a decrease of the lifetime for high LP (Fig. 2b-MA-Bulk, bottom), where the PL intensity decreases. Note that the lifetime of the carriers emitted from unbleached parts of the material (background of image Fig. 2b-MA-Bulk) changes between the surface (∼6.5 ns) and the bulk (∼5 ns).
(1) Solvent molecules (γ-ButyroLactone (GBL), DiMethylFormamide (DMF) and/or DiMethyl SulfOxide (DMSO)) that can interact strongly38 with the Pb2+ ions are incorporated into the HaP crystals as defects and can act as recombination centers, thus reducing the lifetime. Mass spectroscopy shows that GBL, DMF and/or DMSO molecules are present in the single crystals (cf. SI.8, ESI†). Ref. 39 shows tail states in high-resolution photoemission spectroscopy, if DMSO is used in HaP film preparation. The assumption in this explanation is that solvent molecules are readily removed from the surface by evaporation, but remain trapped in the bulk.
(2) The density of shallow defects at/near the surface that can trap (minority) carriers is high enough so that less carriers actually recombine per unit time, increasing the measured carrier lifetime; the trapped carriers will eventually de-trap and recombine via band-to-band transitions. Such effects are known for other bulk semiconductors.40
(Dis)proving these hypotheses needs work that is beyond the scope of the present study.
For what concerns the effect of photobleaching, we interpret the observations showed in Fig. 2b-MA-Surface as a “shadowing” effect due to a non-emitting material formed on the surface. This material blocks the PL originating from the MAPbBr3 lying below it. In addition, the microscope assesses the material up to a depth of ∼1 μm, even if focused on the surface. If complete degradation happens at or very close to the surface (within ∼100 nm), the overall PL also decreases because the amount of emitting material decreases. Because the degraded material does not emit light, the photons emitted from deeper in the crystal will be the result of the recombination of carriers, with the lifetime typical for the undamaged material. Importantly, this result supports the observation that simply monitoring the lifetime is not sufficient for determining the quality of a material, a conclusion that we drew also earlier, based on other data.41
As to the bulk, what is observed in Fig. 2b-MA-Bulk follows what is expected from a decrease/increase of trap density which both increase/decrease the PL intensity and PL lifetime. The chemical origins of this effect cannot be simply extrapolated from these observations and constitute the subject of a subsequent section.
The effect of the bleaching follows what is expected at the surface for medium LP and in the bulk (Fig. 2b-Cs), i.e., bleaching reduces the lifetime. The PL intensity decreases because a larger number of carriers recombine through faster, non-radiative, processes. At the same time, the high LP Cs-Surface data show an opposite trend with an increase of PL intensity despite a reduction in lifetime compared to the pre-bleaching state. This is probably due to the fact that the blue-shifted material has a higher PL quantum efficiency than the background PL of the perovskite.
2.4 Interim conclusions
We showed that in nearly all cases changes in PL intensity directly relate to PL lifetime changes. This suggests that both have a common cause, which could be a change of defect density. However, we found that equivalent laser irradiation treatments affect the MA-, FA-, and Cs-perovskites differently. Moreover, we showed in Section 2.1 that the A cation influences self-healing of the perovskites (cf.Fig. 1). This indicates that the A cation is key in determining the stability of the perovskite. This conclusion points to a chemical basis for the differences, given the lack of a direct effect of the A cation on the electronic structure of the material.43,44 We proceed to study these differences and try to understand their chemical origins, analyzing the surface experimentally (Section 3) and the bulk through DFT simulation (Section 4).3 Results: characterization of surface damage
We used atomic force microscopy (AFM), scanning electron microscopy (SEM), energy-dispersive X-ray spectroscopy (EDS), and X-ray photoelectron spectroscopy (XPS) to characterize the (near-)surface damage and to investigate changes of morphology and composition after the bleaching cycles.3.1 AFM
Fig. 2b-MA-surface-high LP shows that MA samples exhibit a slightly more luminescent halo up to several μm from the strongly bleached area. Because no bleaching was performed in that region and the heat of the bleaching does not significantly increase the temperature in and around the bleached material (cf. SI.6, ESI†), the modification of the PL has to originate from chemicals released upon bleaching of the surface. Fig. 3(a) shows AFM images of a sample surface after 100 consecutive bleaching cycles (at high LP) taken in an area, equivalent to the one showing a halo in Fig. 2b-MA-Surface-High LP. We used 100 cycles to increase the effect of the halo formation sufficiently, to allow measuring the bleaching effect up to one day after bleaching. Because the time delay between bleach cycles is 10 seconds, they can be viewed as separate events that inflict cumulative damage (see SI.6, ESI† for details). In this way the amount of material, released from the bleached spot, increases with consequent stronger effect on the region around that spot.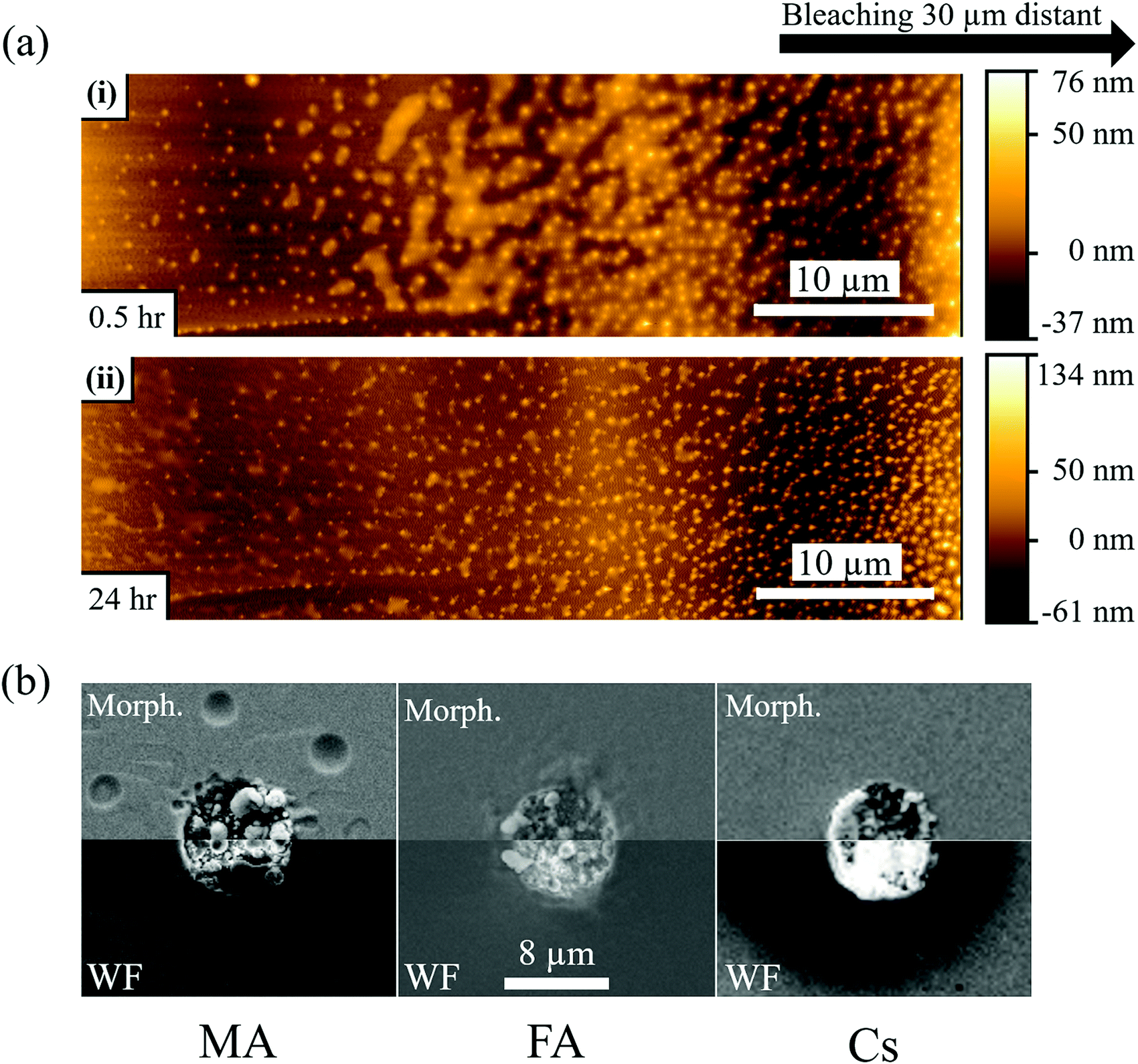 | ||
| Fig. 3 (a) AFM images of MAPbBr3 single crystal surface 30 min (i) and 24 hours (ii) after 100 cycles of 1P bleaching with a laser power of 880 μW (LP). The AFM image is taken 20–30 μm from the bleached area (see Fig. S6, ESI†), because the AFM tip could not make contact closer to the damaged area. The images show nanoparticles formed on the crystal surface and a semi-liquid phase between the particles that evaporates with time (compare top and bottom AFM images). (b) SEM images of MAPbBr3, FAPbBr3 and CsPbBr3 single crystals after 100 cycles of 1P bleaching with LP. The images were taken with the SE2 detector to show the morphology and with the InLens detector to map variations in work function (whiter for lower, darker for higher work function). The images are mirrored to facilitate comparison between them. For (a) and (b) 100 bleaching cycles were used to increase the amount of damage performed quantitatively. Qualitatively, though, there is no difference as the time delay between each cycle of 10 seconds is longer than any temperature or carrier concentration decay timescale. As is calculated in detail in the ESI† (SI.6), the temperature rise will be less than 1 K. | ||
AFM imaging reveals the formation of small, possibly HaP particles (as the PL spectrum, not shown, remains that of MAPbBr3). We suggest this is formed by the decomposition products, resulting from surface photochemical vaporization by the laser, that fall back on the sample (horizontally placed – laser is incident from the top) after cooling. However, we cannot be certain of their composition because the PL spectrum can originate from the underlying crystal. The AFM images in Fig. 3a were obtained 0.5 h (i) and 1 day (ii) after the 100 bleaching cycles of the area, around which a halo appeared. Increasing the number of bleaching cycles assists in locating the spot optically (Fig. S7, ESI†) and allows imaging of the halo area that formed away from the bleached area (details in SI.11, ESI†). Comparing Fig. 3a(i) and (ii), we infer that the material connecting the different particles, seen in (i), evaporates with time and is probably a viscous liquid, as suggested by its flattened shape and the issues with direct AFM imaging very near the bleached area (see SI.11, ESI†). We attribute the presence of this liquid to re-absorbed CH3NH2 and NH3, known to dissolve into the MAPbBr3 to form a viscous phase.45 The other products of decomposition (HBr, CH3Br) do not interact with the solid and would not form such a phase. Because the vapor pressure of NH3 at 25 °C (∼10 bar)46 is higher than that of CH3NH2 (∼2 bar),47 which is consistent with their boiling points (NH3: −33.3 °C;48 CH3NH2: 6 °C49), we expect CH3NH2 to dominate absorption after vaporization. This argument can explain why we do not find any PL halo around bleached areas on FA and Cs samples: their decomposition products cannot form such a liquid with the corresponding HaP.
3.2 SEM
Fig. 3b shows SEM images of the surfaces of MA, FA and Cs after 100 bleaching cycles. As in the case of Fig. 3a, the repetition of the bleaching increases the amount of damage (molecules lost) but does not change it qualitatively, because the 10 sec time between the bleaching cycles is longer than the time that any change in carrier concentration or temperature persists (cf. detailed calculation in SI.6 ESI,† that shows ΔT < 1 K during the illumination). Fig. 3b-MA shows SEM images of the surface of the MA crystal. Two modes were used: the SE2 detector detects secondary electrons from the (near)surface, which makes it sensitive to surface morphology. The higher surface sensitivity of the In-Lens electron detector makes its signal also sensitive to the electrical potential of the surface, and, thus, reflect the work function.50,51 Clearly the surface, where the material was bleached (with ∼870 μW laser power), is strongly affected by the treatment.The MA sample formed structures inside the bleached area. As noted above, some signs of the presumed liquid CH3NH2-rich phase are also detected by SEM outside the bleached zone (Fig. 3b-MA). However, because SEM is performed in vacuum, the phase that we proposed above as being the cause of the nanoparticles that were observed in the AFM images (Fig. 3a) and of the halo in Fig. 2b-MA-Surface-High LP, evaporates. The InLens image shows an increased signal relative to the background, indicating, as reported earlier,15,50,51 a decrease in work function. This can be explained by the formation of electron-rich Pb0 (work function of 4.0 eV,52 compared to 4.7 to 5.1 eV for the HaP single crystals) over electron-poorer PbBr2 (work function53 of 6.0 eV). The FA sample (Fig. 3b-FA), even if less stable for a single, defect-inducing bleaching cycle, seems to be more resistant to multiple cycles of bleaching/laser ablation, as seen from the SE2-morphology image. The Cs sample appears to be as affected by the strong laser pulses as the MA one, with an evident change in morphology. Noticeably, for the Cs sample the In-Lens image shows a very strong increase of the signal in the bleached area, indicating a more substantial decrease of the work function than for the MA and FA samples. At the same time, the area around the bleach shows a decrease of signal, corresponding to a work function increase.
3.3 EDS, XPS
To explain the above observations, we examined the surface through EDS and XPS, searching for variation in composition that can explain the above-mentioned observations. EDS analysis shows that photobleaching of the MA sample increases the Pb![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Br ratio in the bleached area by a factor of 1.4 compared to the bulk. The signal from a pure PbBr2 crystal would increase the Pb:Br signal ratio by a 1.5 factor. At first glance, one could conclude that PbBr2 is formed. However, it has to be taken into account that the EDS signal is collected down to a depth of ∼0.5 μm and that the real Pb:Br ratio on the surface is likely much higher. This supports the hint of Pb0 formation as concluded in the preceding paragraph. On the other hand, no difference in composition, within the sensitivity of EDS, is found between the background and the halo-area around the bleached area. EDS analysis of FA samples, on the other hand, showed only a factor of 1.15 increase in Pb:Br ratio. This is also consistent with Pb0 formation, although this does not exclude possible PbBr2 formation. Cs samples show no change in the Cs:Pb:Br ratio but a Pb:O ratio of 1.4:1 is found, indicating additional oxygen (see SI.13, ESI† – no O is found in the background by EDS).
:Br ratio in the bleached area by a factor of 1.4 compared to the bulk. The signal from a pure PbBr2 crystal would increase the Pb:Br signal ratio by a 1.5 factor. At first glance, one could conclude that PbBr2 is formed. However, it has to be taken into account that the EDS signal is collected down to a depth of ∼0.5 μm and that the real Pb:Br ratio on the surface is likely much higher. This supports the hint of Pb0 formation as concluded in the preceding paragraph. On the other hand, no difference in composition, within the sensitivity of EDS, is found between the background and the halo-area around the bleached area. EDS analysis of FA samples, on the other hand, showed only a factor of 1.15 increase in Pb:Br ratio. This is also consistent with Pb0 formation, although this does not exclude possible PbBr2 formation. Cs samples show no change in the Cs:Pb:Br ratio but a Pb:O ratio of 1.4:1 is found, indicating additional oxygen (see SI.13, ESI† – no O is found in the background by EDS).
The XPS analysis (see SI.14, ESI† for Experimental details) of a Cs sample, treated in the same way over a much larger (few mm2) area, did not show the presence of any Pb oxide, as the Pb spectrum did not differ from that of a non-treated one. XPS revealed, instead, a ∼15% increase of the Cs:Pb ratio on the surface. Since any sample is contaminated by oxygen and carbon and XPS cannot differentiate between traces of O coming from perovskite oxidation or from contamination, it is not possible to exploit it to determine O surface concentration. Nevertheless, as the depth analyzed by XPS is 1–2 orders of magnitude smaller than the EDS one, the two results are not contradictory. From the comparison of XPS and EDS, we conclude that Cs enrichment is limited to the surface region. The presence of oxygen is probably an indication of the formation of either lead or cesium oxides or, lead bromates. The formation of cesium oxides can explain the InLens-work function image of Fig. 3b-Cs, because Cs oxides are known low work-function (∼1 eV) materials.39,54 We note that neither lead oxide nor lead bromate are stable in acid environment. MAPbBr3 and FAPbBr3 are reactive towards oxides due to the acidity of the A+ cation and would transform to PbBr2. PbBr2 could then eventually transform to Pb0 under illumination,27,28 a photoreaction known also for PbI2.55 The possible formation of lead oxides or bromates would deplete the lead content of the original perovskite, leaving a Cs-rich volume that, as reported previously,36 has a stronger and blue-shifted photoluminescence.
3.4 Support from TGA-MS
Using Mass Spectroscopy (MS – see SI.6, ESI†) we analyzed the decomposition products that are released from MAPbBr3, FAPbBr3 and mixed FA-CsPbBr3 single crystals between 320 °C and 360 °C (during ThermoGravimetric Analysis, TGA). We could detect HBr and CH3Br from the decomposition of MAPbBr3, implying formation of both CH3NH2 and NH3 (not MS-detectable in our TG-MS because of their low molecular weight). FAPbBr3 decomposed to HBr and triazine. No Br2 formation was detected from any of these crystals. Therefore, we conclude that their thermal material degradation stops at PbBr2. However, PbBr2 is known to photolyze readily, which means that photobleaching will lead to Pb0 and Br2 formation. Unfortunately, illumination of the sample was not possible in those MS systems, that were suitable for our samples and available to us.4 Chemistry of self-healing in the bulk
In Section 2 of this article, we reported that the A cation strongly modifies both degradation and recovery of the three APbBr3 perovskites. This effect, at least in the bulk, should be explainable on the base of chemical instability of the species formed after photobleaching. One would indeed expect more stable species to heal slower than more unstable ones.Experimental identification of the chemical origin of such differences in the bulk is not possible given the low amount of defects that the bleaching produces (the material can be considered the same on a macroscopic level). No optical method (the bulk is not accessible otherwise) can indeed identify defects in traces (the effects on the PL intensity that we noticed are most probably produced by under ppb defect density). Because of the difficulty to use experimental methods to get insight into the defect chemistry and physics, we turned to state-of-the-art density functional theory (DFT) to assess the plausibility of pertinent chemical scenarios and to help understand the observed phenomena. Specifically, we focus on deformations caused in the minimum energy structure of the pristine material by introducing a defect, as well as on the degree of charge localization around the defect. For computational details, see SI.9 (ESI†).
4.1 DFT of bromide interstitials: general considerations
The A cation is known to have a minimal direct effect on the HaP electronic structure.43,44 However, it does influence the structure of the perovskite by changing the cell size and the symmetry, thereby indirectly affecting the defect chemistry of the material. Depending on the A cation, a similar defect can cause varying levels of distortion to the crystal, which can determine whether the defect will have long-term (meta)stability. The more distorted the structure, the less likely it is that the defect will persist, barring kinetic stabilization.A growing consensus points to halogen related defects, most notably interstitials, as possible traps and/or charge recombination centers in HaPs.56–59 For this reason, we focus our efforts on interstitial bromide defects in all three systems. In the point defect model, an interstitial defect can have different charge states, depending on the energy of its charge transition level relative to the Fermi level of the material. Specifically, a Br interstitial defect can be either positively charged (Bri+), neutral (Bri0), or negatively charged (Bri−). Experimentally, HaPs are commonly found to be slightly p-type,60,61i.e., with a Fermi level that is closer to the level of the valence band maximum (VBM) than the conduction band minimum (CBM) and lower than the defect level. This suggests that a positively charged defect is the most probable one.
4.2 Bri+: geometry in the three APbBr3 HaPs
Using DFT, we calculated the minimum energy structure of a Br interstitial in each of its possible charge states, in all three systems. The Bri+ structures are presented in Fig. 4a–c. For completeness, all other calculated minimum energy structures are reported in SI.8 (ESI†). In all three systems, the Bri+ defect produced a similar structure: the Bri+ relaxed to a position between two neighboring lattice Br anions, connecting two octahedral cages to create a rigid, linear Br3− structure with Br–Br distances of 0.255–0.259 nm. Br3− anions are known constituents of solids, e.g. CsBr3,62 and other organic cation equivalents63 or ionic liquids64 (e.g., MaBr3 and FABr3).15 Linear polyhalide anion species generated by interstitial Br have also been observed experimentally in some crystal structures, e.g. KCl,65,66 KBr,65,66 RbBr,67 and CsBr.68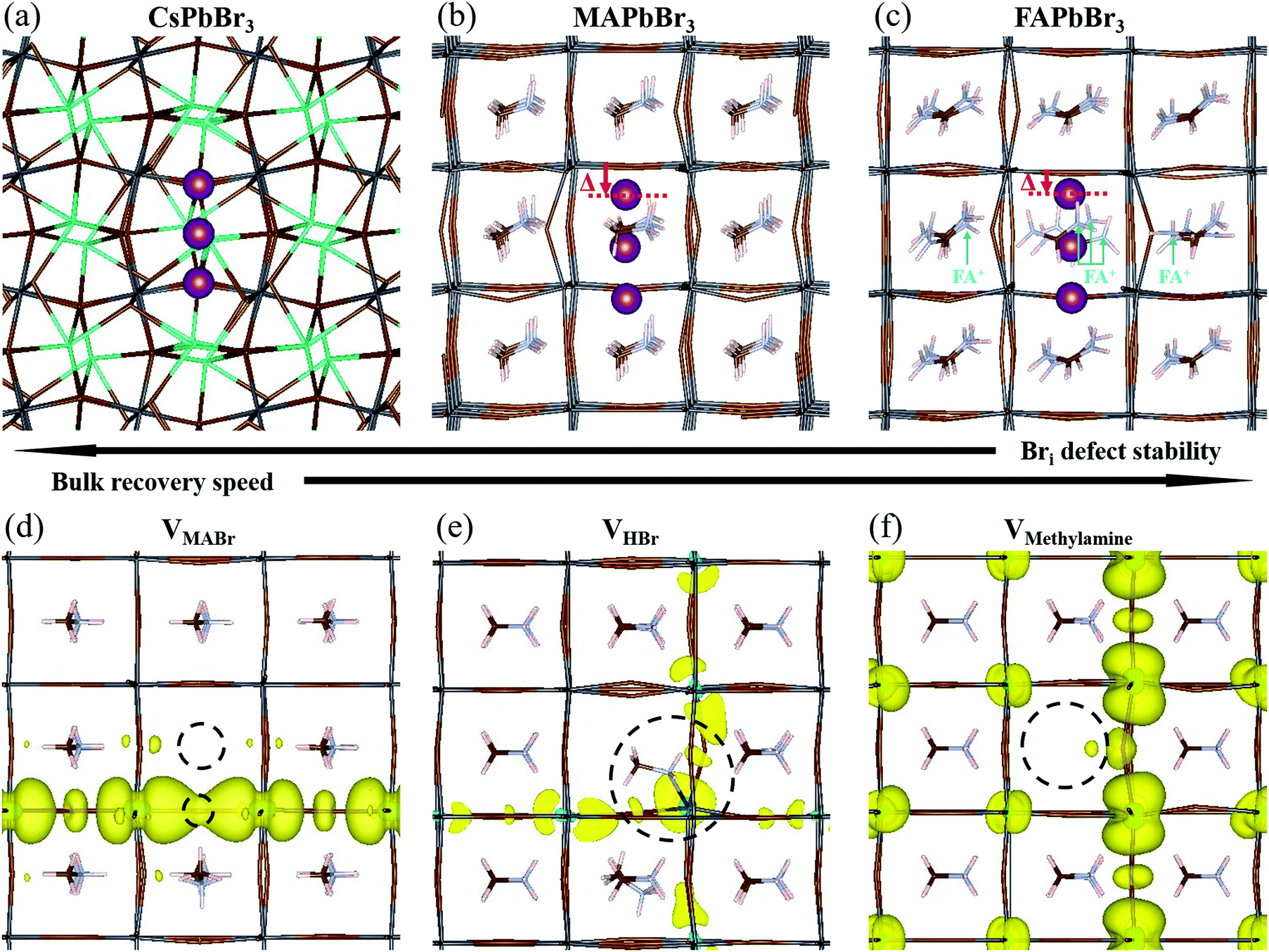 | ||
| Fig. 4 Minimum energy structures of (a) orthorhombic CsPbBr3, (b) cubic MAPbBr3, and (c) cubic FAPbBr3, containing a Bri+ defect, as well as minimum energy structures of MAPbBr3 containing (d) VMABr, (e) VHBr, and (f) VMeNH2. Bri+ defects are marked in purple and vacancies are marked by dashed circles. Cs atoms are marked in green, Pb atoms in dark grey, and Br atoms in brown. The red arrow indicates the displacement of the Br atoms from the Br–Pb–Br axis. The light blue arrows indicate the FA+ ions most affected by the defect. Yellow contours in (d)–(f) represent the partial charge density associated with the defect eigenvalue. | ||
We find that the room temperature (RT) orthorhombic CsPbBr3 structure remains almost undistorted around the defect, whereas the RT cubic MAPbBr3 and FAPbBr3 structures are strongly perturbed. In particular, in MAPbBr3 and FAPbBr3, one of the bromine atoms has to shift strongly off the axis formed by the two Pb ions to which it is bound, in order to bind to the Br interstitial ion (see the displacement (Δ) of the topmost Br atoms, marked in purple in Fig. 4b and c (red arrows)). As a consequence, the interstitial Br atom and the second structural Br atom are pushed downwards and away from the latter's equilibrium position. While for MAPbBr3 the MA+ orientations remain relatively unchanged, in the case of FA+ the presence of a Br interstitial defect causes a strong modification of the equilibrium positions of the nearby FA+ ions (see ions indicated by light blue arrows, Fig. 4c). In addition, reaching converged minimum energy geometries for the Br interstitial in FAPbBr3 is much more difficult than in MAPbBr3 and CsPbBr3. This, coupled to the large structural distortions in the FAPbBr3 system, indirectly indicates the improbability of a stable Br interstitial in that material. We can speculate that FA+, by way of its natural vibration, will “push” Bri+ out towards the next nearby equilibrium Br position (in the octahedral cage), with consequent displacement of the already-present Br ion that becomes an interstitial defect in the next unit cell. This process will repeat until the interstitial recombines with a vacancy or is otherwise annihilated.
4.3 Bri+: relation between geometry and stability
Our DFT calculations suggest that the self-healing in the bulk of the APbBr3 could be inversely related to the amount and localization of the deformation introduced by the Br interstitial.69 The stability of a Br interstitial defect is lowest for FAPbBr3, whereas in CsPbBr3 the defect can exhibit long term metastability. It is important to note that it has recently become clear that the perovskite structure is not static, but rather is subject to large, anharmonic, polar fluctuations.70–72 Moreover, these fluctuations may affect the defect energy landscape.73 Clearly, the static, relaxed structure can only represent an average picture. Because the static FA+-containing structure is already greatly distorted even in the absence of dynamic effects, much more so than the Cs+-containing one, it is reasonable to assume that these differences carry over to the dynamic, room temperature systems.4.4 The role of methylamine
In Section 2 we noted that MAPbBr3 exhibits increased PL brightness upon photo-bleaching in the bulk, whereas the two other bromide perovskites do not. This necessitates further examination of the consequences of an MA-related defect, also in the view that all reported cases of light soaking in HaPs contain MA in the composition (see ref. 74 for a recent review on the subject). Free methylamine possibly forms in the bulk after dissociation of the methylammonium to methylamine and HBr. We therefore look into three neutral defect complexes involving methylamine and bromine: (1) an MABr vacancy, VMABr; (2) an HBr vacancy, VHBr; (3) a methylamine vacancy, VMeNH2.In Fig. 4d, we show the structure and charge density associated with VMABr. In this case, the charge density is localized along the Pb–VBr–Pb axis. For the VHBr (Fig. 4e), the charge density spreads two dimensionally, while for the VMeNH2 (Fig. 4f) the defect induces a smaller effect with an almost unchanged geometry and a rather delocalized charge density in all three dimensions. This indicates a negligible effect on the electronic structure and a very low likelihood of trap formation. We stress that for the VHBr, we report a direct bond between the lone pair of the N atom of methylamine and the nearest Pb atom. When removing HBr (as well as MABr), two Pb–Br bonds are broken, creating a defect level in the gap. The additional electron density, due to the lone pair on the N atom in CH3NH2, partially screens the defect, making VHBr less harmful than VMABr. However, VHBr and VMeNH2 are not the thermodynamically favored structures. According to our calculations, the reaction:
| VHBr + VMeNH2 ↔ VMABr |
5 General discussion: pulling it all together
The presented data show the differences in degradation and self-healing between surface and bulk and between MAPbBr3, FAPbBr3, and CsPbBr3. These variations can be crucial in determining the stability of HaP materials and HaP-based devices.To understand the atomistic processes that lead to the reported results we consider:
• how the A cation influences self-healing on both the surface and in the bulk of HaPs;
• how the A cation influences the defect density in the HaPs;
• how “proper” encapsulation has to take into account all processes we have identified, rather than being limited to preventing H2O and O2 penetration in the HaP-based device.
5.1 Surface and bulk stability: the role of the A cation
In Fig. 5 and in Table 1 we summarize all the information on APbBr3 perovskites after photo-bleaching, provided by the current study. Closed circles represent degradation followed by self-healing processes. Red circles describe Bri-related ones and green circles describe methylamine-related processes. Thicker lines correspond to faster processes. Arrows pointing vertically represent material exchange with the environment. Bulk and surface processes are separated by the dotted line, showing that self-healing dominates in the bulk and material loss dominates at the surface. Each of the indicated processes is consistent by results from experiments and computations, as indicated by symbols.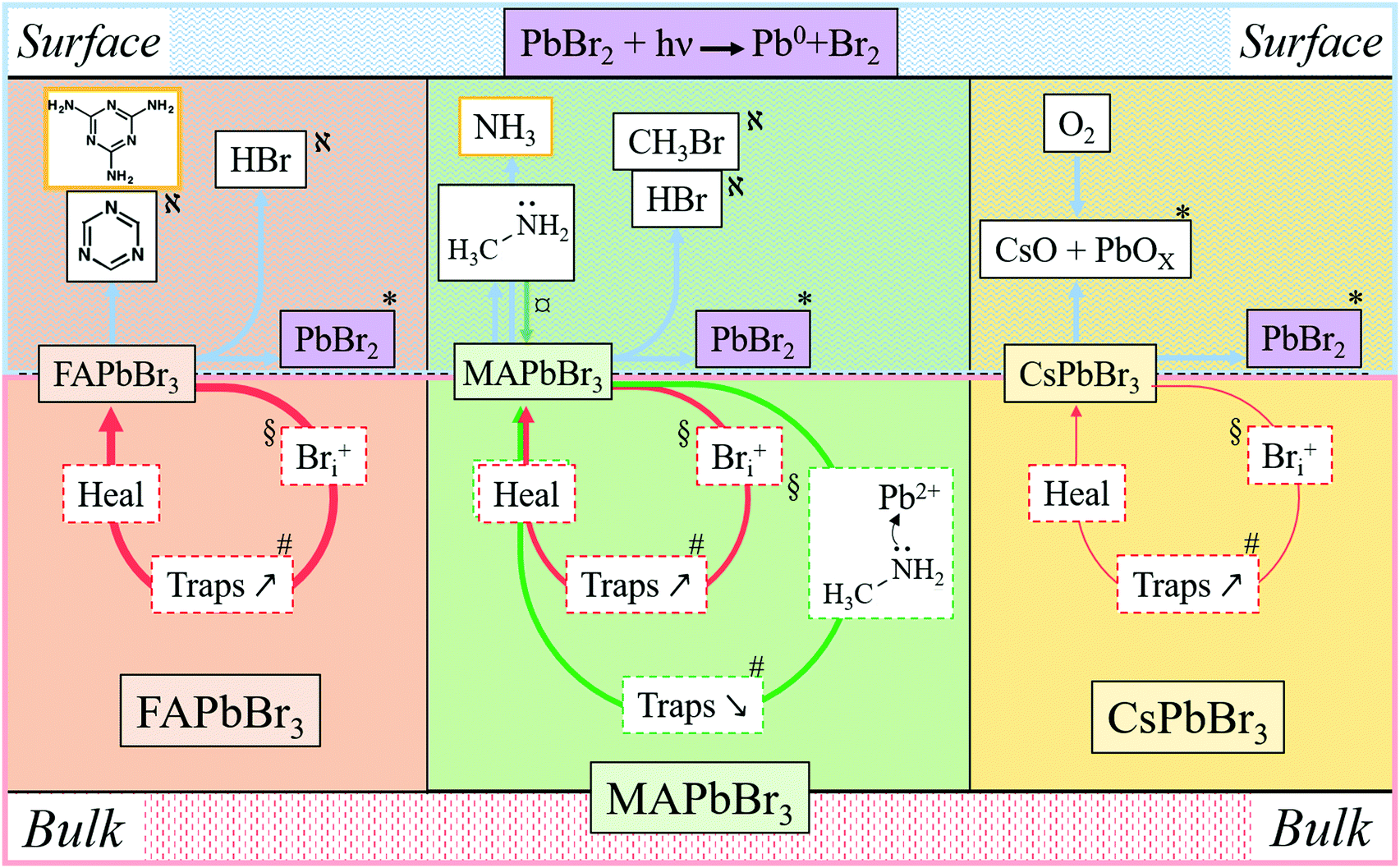 | ||
Fig. 5 Scheme of the chemical processes involved in the degradation and healing of APbBr3 perovskites on the surface and in the bulk. Information from the literature is contained in orange rectangles. Surface phenomena are reported in the upper row and bulk ones in the row below, under the dashed line. The thickness of the arrows (for the degradation on the surface and the self-healing in the bulk) corresponds qualitatively to the speed of the corresponding process. The symbols indicate observables, analyzed in the main text, and consistent with the indicated processes: § DFT; # 2P PL Lifetime and intensity;  1P PL Intensity and AFM; * InLens SEM, EDS and work function increase; 1P PL Intensity and AFM; * InLens SEM, EDS and work function increase;  TGA-MS. On MAPbBr3 and FAPbBr3 PbO will not be because it reacts with CH3NH3+, H2NCHNH2+ to give CH3NH2, HNCHNH2 and H2O. TGA-MS. On MAPbBr3 and FAPbBr3 PbO will not be because it reacts with CH3NH3+, H2NCHNH2+ to give CH3NH2, HNCHNH2 and H2O. | ||
| Cation | Location | Damage | Healing | Phenomenon | Supported by |
|---|---|---|---|---|---|
| MA | Surface | Threshold | Minimal | Halo – MA condensation, Pb/Br increase | AFM – SEM, EDS |
| Bulk | Progressive | Fast | Increase/decrease in PL | DFT | |
| FA | Surface | Threshold | Partial | Pb/Br increase | EDS |
| Bulk | Progressive | Very fast | Decrease in PL | DFT | |
| Cs | Surface | Threshold | Semi-complete | O-containing product(s), Cs/Pb increase | EDS, XPS |
| Bulk | Progressive | Slow | Decrease in PL | DFT |
| FAPbBr3 + hν ↔ FABr + PbBr2 | (a) |
| FABr ↔ (sym)-triazine + other decom. products | (b) |
Considering now the surface process, the MA perovskite decomposition proceeds, as indicated by our MS results and the AFM results, as follows:12,13
| MAPbBr3 + hν ↔ MABr + PbBr2 | (c) |
| MABr ↔ CH3NH2↑ + HBr↑ | (d1) |
| MABr ↔ CH3Br + NH3↑ | (d2) |
In the bulk material, the products of reaction (d1) can be absorbed by the crystal without forming a gas: methylamine, as discussed above (Section 4.3), can penetrate the crystal structure, HBr can dissociate into H+, which can penetrate into the structure due to its small size, while Br− can be accepted as part of an interstitial defect. CH3Br, however, is a hydrophobic molecule that does not readily react either with the perovskite or with NH3. Its formation would build up extremely high pressure inside the crystal. This reasoning rationalizes that reaction (d2) is inhibited in the bulk by the law of mass action. However, in contact with an organic HTL, ETL or encapsulating polymer, CH3Br will be readily absorbed, pushing the reaction to the right and leading to irreversible decomposition, even if the device is encapsulated.
Interestingly, only for CsPbBr3 do we find formation of a non-healable form with more intense PL inside the bleached area in the 1P-surface experiment at high LP (Fig. 1e, 2a and 3b). This result is explained by the reaction of CsPbBr3 with oxygen, as shown by EDS, which indicates formation of an O-containing product. XPS shows formation of a Cs-rich surface (Section 3). This decomposition pathway creates a PL increase only if the surface of the material is exposed to O2 (which is the case under our experimental conditions) and to light. Still, this strong bleaching-induced modification of the surface does not improve the PL lifetime, despite increasing its intensity (Fig. 2b-Cs-High LP).
Overall, surface decomposition of CsPbBr3, even if starting at lower energies than in the MA case, is less impactful than for FA and MA, probably because only a small amount of volatiles is actually created and most of the material can re-form (and, likely, quicker than in the bulk, as steric constraints are relaxed).
Summarizing, each A cation influences the chemistry and self-healing of the perovskite differently. When the A cations are mixed, each of them can synergistically contribute to the overall stability and performance of the material. This synergism is in addition to configurational entropy gains of the system, due to lack of order in A cation distribution.
5.2 Importance of self-healing for bulk defect density
The FA+ cation plays here the role of keeping the defect density low. As the defect density is crucial for HaP-based solar cells, we can understand why FA+ is the most abundant cation in MAxFAyCs1−x−yPbBr3 based solar cells.At equilibrium, a material's self-healing kinetics are connected to its defect density.85 For every defect, we consider that the defect density is determined by the equilibrium between formation and destruction (by healing) of the defect:
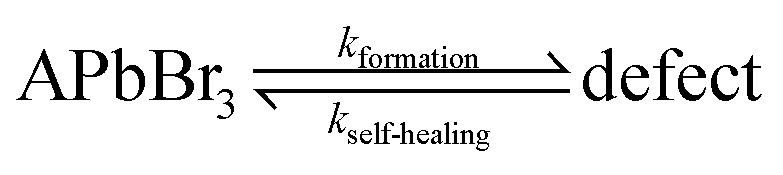 | (e) |
where [HaP] is the concentration of the perovskite, which is effectively constant for any relevant [defect]. From this we deduce that the [defect] is inversely proportional to the self-healing kinetic constant. We can then expect FAPbBr3 to have the lowest defect density, as its self-healing kinetics are fastest. The effect is particularly relevant during solar illumination. In this case, loss of efficiency is commonly attributed to the formation of defects that are continuously created by illumination.18,86 The self-healing process continuously repairs (i.e. eliminates) the defects, dictating a steady-state defect density that normally will be higher than the equilibrium one. The faster the recovery kinetics, the closer the steady-state defect density is to the equilibrium one as dictated by thermodynamics. Therefore, self-healing protects the material from damage, enhancing its chemical stability and decreasing the steady state density of performance-damaging defects.
5.3 Relation between self-healing and encapsulation
For self-healing to take place, any material loss has to be avoided to minimize the occurrence of reactions like (a) and (c). For this reason, it is now necessary to redefine what “proper” encapsulation is in the case of HaPs. There are many reports on how classical encapsulation significantly improves the stability of halide perovskite solar cells. This is not surprising, as the most obvious effect of encapsulation is protecting the material(s) against chemical attacks (i.e. H2O, O2) from the ambient. However, this is not all that encapsulation should do.The critical new insight from our study is that besides protecting from external chemical attack, and containing decomposition products, which will by the mass-action law, increase the probability of re-composition, this will allow self-healing to occur also at and near the surface. This insight shows that proper encapsulation can act beyond protection and containment by conveying also to the surface of the HaP (and its interfaces in devices) dynamic stabilization.
The self-healing that we demonstrated to occur in bulk perovskites requires any decomposition product to be readily available, to enable reversal of the decomposition reaction. This condition is clearly not fulfilled at a surface that is open to the ambient, with the consequence that self-healing will be limited. In most reported studies, the “escape” ways for decomposition products in HaP-based solar cells are not controlled. For example, an iodine (or bromine) molecule forming at the surface of a solar cell may well find its way between grain boundaries or through the larger than molecular spaces between the perovskite layer and the HTL (or in the porous, if not completely filled ETL as in case of mesoporous TiO2). Eventually the molecule can get away from the HaP surface and, for example, accumulate in open spaces. In such case, even if the product of the decomposition has not reacted with the ETL or HTL, but ends up spatially far from the decomposition site, it may not be available for self-healing.
The above issues point to one reason why hybrid 3D–2D perovskites can have enhanced stability and optoelectronic properties. The organic layers of these perovskites can prevent escape of any 3D decomposition products. Such an effect would be in addition to passivation of defects on the surfaces of the 3D perovskites, by adding the 2D layers as well as increased hydrophobicity to keep water away.
6 Conclusions
We compared and contrasted experimentally bulk and surface damage of APbBr3 halide perovskites and evaluated the results using thermodynamic, kinetic, chemical, and physical considerations. We demonstrated how the intrinsic stability of the perovskites, linked to thermodynamics, is significantly greater than their kinetic stability, thanks to self-healing. We performed PL, FLIM, AFM, EDS and XPS measurements, and DFT simulations to analyze and explain the differences in the damage that can be inflicted by a strong laser beam, and the self-healing processes for the three different A cation perovskites.FA provides superior healing properties in the bulk of the material and reduces the bulk defect density because of the instability of the Bri+ defect (in a Br3 complex) in its structure, differently from what happens in MA and Cs. When photoexcited in the bulk, MA decomposes to methylamine and HBr (eqn (c) and (d1)). Methylamine can migrate and passivate defects while H+ remains at the site of the MA+ ion without serious effect on the local electronic properties. This causes an increase of PL (and probably in photo-conversion) efficiency, increasing solar cell performance. Finally, Cs+ helps to stabilize the surface of the material.
In terms of fundamental understanding, our results lead us to hypothesize about the question of why HaPs have such good optoelectronic, and particularly PV properties, expressed by (a) relatively long charge carrier lifetimes and diffusions lengths. These qualities can be traced to (b) low densities of electronically and optically active defects, i.e., those that affect measurable properties. As (a) will be qualitatively inversely proportional to (b), if the defects are subject to self-healing, their density will be small. Taken to the extreme, self-healing becomes the fundamental cause for the excellent opto-electronic properties of the HaPs, because all derive from the self-healing. Even if self-healing is not the sole cause, it likely is a major reason for the HaPs’ excellent properties.
We stress how from our results we can infer that fulfilling the conditions for self-healing is necessary for long-lasting stability of devices made with HaP materials. In particular, apart from perfect encapsulation, the ETL and HTL have to be chemically unreactive towards the decomposition products of the perovskites in order to create “bulk-like” conditions for the HaP part of the interfaces. In the most stable devices that were reported, such an effect was likely an unintended consequence, in-depth analysis of which can help deciding on device architectures for commercialization.
Author contributions
D. R. Ceratti originated the idea of using a 2P/1P microscopy-based approach to assess perovskite stability, and using FRAP as reporting method; he synthetized the single crystals used in this study, collected and analyzed the data. G. Hodes* and D. Cahen* defined the problem and participated with DRC in analyzing and interpreting the data. DRC wrote the initial draft and with DC, GH and LK finalized the paper. A. V. Cohen* and L. Kronik* performed the DFT simulations and wrote that part of the text. R. Tenne,* M. Elbaum* and V. Kalchenko made possible the 1P and 2P microscopy experiments and helped DRC to acquire and analyze those data. Y. Rakita helped synthesize CsPbBr3 crystals and performed with DRC the SEM and EDS measurements. T. Bendikov performed and analyzed the XPS measurements. L. Snarski helped unravel the chemistry of methylamine. L. Cremonesi* and M. A. C. Potenza* performed the calculations on the focusing and aberration effects for the 1P and 2P laser beam experiments at the surface and inside the crystals. They participated with DRC in calculating the effective energy deposited inside the material and wrote the ESI† one these calculations. I. Rosenhek-Goldian acquired the AFM images with DRC. I. Kaplan-Ashiri performed the simulations of EDS penetration and emission profiles and wrote the relevant text. N. P. Jasti, R. Cohen and M. Weitman performed and analyzed the TGA measurements. The authors here marked with a * reviewed the final text before submission, helping to clarify and simplify the text.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dan Oron (Weizmann Inst.) for many fruitful and stimulating discussions. D. R. C. thanks the Weizmann Institute's SAERI, Sustainability And Energy Research Initiative, for a 2017–2020 PD fellowship in the first 2 years of which this work was done; D. R. C. and D. C. thank the Minerva Center for Self Repairing Systems for Energy and Sustainability for partial funding; D. C. thanks the Ullmann family foundation for partial 2017–2018 funding, when the research was done. Y. R. acknowledges a 2018 Perlman research grant for student-initiated research in chemistry for partial support. A. V. C. and L. K. acknowledge support by the Minerva Foundation. L. K. holds the Aryeh and Mintzi Katzman Professorial Chair. D. C. also thanks the Yotam project of the WIS’ SAERI, for partial support. D. R. C. held a SAERI PD fellowship. N. P. Jasti acknowledges funding from the European Union's Horizon 2020 MSCA Innovative Training Network under grant agreement no. 764787 (MAESTRO). D. R. C. thanks Ilario Gelmetti, Stefania Cacovich and Alexandre Py for fruitful discussions and clarity check of the text.Notes and references
- M. A. Green, Y. Hishikawa, E. D. Dunlop, D. H. Levi, J. Hohl-Ebinger, M. Yoshita and A. W. Y. Ho-Baillie, Prog. Photovolt. Res. Appl., 2019, 27, 3–12 CrossRef.
- Q. Jiang, Z. Chu, P. Wang, X. Yang, H. Liu, Y. Wang, Z. Yin, J. Wu, X. Zhang and J. You, Adv. Mater., 2017, 29, 1703852 CrossRef PubMed.
- T. M. Brenner, D. A. Egger, L. Kronik, G. Hodes and D. Cahen, Nat. Rev. Mater., 2016, 1, 1–16 Search PubMed.
- H. Kim, J. S. Han, J. Choi, S. Y. Kim and H. W. Jang, Small Methods, 2018, 2, 1700310 CrossRef.
- C. C. Stoumpos and M. G. Kanatzidis, Adv. Mater., 2016, 28, 5778–5793 CrossRef CAS PubMed.
- L. N. Quan, B. P. Rand, R. H. Friend, S. G. Mhaisalkar, T.-W. Lee and E. H. Sargent, Chem. Rev., 2019, 119, 7444–7477 CrossRef CAS PubMed.
- Y. He, L. Matei, H. J. Jung, K. M. McCall, M. Chen, C. C. Stoumpos, Z. Liu, J. A. Peters, D. Y. Chung, B. W. Wessels, M. R. Wasielewski, V. P. Dravid, A. Burger and M. G. Kanatzidis, Nat. Commun., 2018, 9, 1609 CrossRef PubMed.
- S. He, L. Qiu, L. K. Ono and Y. Qi, Mater. Sci. Eng., R, 2020, 140, 100545 CrossRef.
- B. Chen, P. N. Rudd, S. Yang, Y. Yuan and J. Huang, Chem. Soc. Rev., 2019, 48, 3842–3867 RSC.
- Y. Lin, Y. Bai, Y. Fang, Z. Chen, S. Yang, X. Zheng, S. Tang, Y. Liu, J. Zhao and J. Huang, J. Phys. Chem. Lett., 2018, 9, 654–658 CrossRef CAS PubMed.
- X. Zhang, X. Ren, B. Liu, R. Munir, X. Zhu, D. Yang, J. Li, Y. Liu, D.-M. Smilgies, R. Li, Z. Yang, T. Niu, X. Wang, A. Amassian, K. Zhao and S. (Frank) Liu, Energy Environ. Sci., 2017, 10, 2095–2102 RSC.
- E. J. Juarez-Perez, L. K. Ono, M. Maeda, Y. Jiang, Z. Hawash and Y. Qi, J. Mater. Chem. A, 2018, 6, 9604–9612 RSC.
- A. E. Williams, P. J. Holliman, M. J. Carnie, M. L. Davies, D. A. Worsley and T. M. Watson, J. Mater. Chem. A, 2014, 2, 19338–19346 RSC.
- D. Prochowicz, R. Runjhun, M. M. Tavakoli, P. Yadav, M. Saski, A. Q. Alanazi, D. J. Kubicki, Z. Kaszkur, S. M. Zakeeruddin, J. Lewiński and M. Grätzel, Chem. Mater., 2019, 31, 1620–1627 CrossRef CAS.
- D. R. Ceratti, Y. Rakita, L. Cremonesi, R. Tenne, V. Kalchenko, M. Elbaum, D. Oron, M. A. C. Potenza, G. Hodes and D. Cahen, Adv. Mater., 2018, 30, 1706273 CrossRef PubMed.
- Here we use self-healing to describe the series of chemical processes that undo damage in a material or system, without the involvement of any external factors/effects. We distinguish it from self-repair, which is used also to describe the ability to repair more complex systems, not by net reversal of the mechanisms that led to the damage, but through different actions that can involve other factors/effects, triggered without external intervention, to mend the damage.
- The system has to be made as closed as possible, i.e., limiting exposure to ambient and especially escape of eventual possible products as much as possible. These layers must not solubilize or react with the products of decompositions of the HaPs, so as to make the HaP thin film bulk-like.
- W. Nie, J.-C. Blancon, A. J. Neukirch, K. Appavoo, H. Tsai, M. Chhowalla, M. A. Alam, M. Y. Sfeir, C. Katan, J. Even, S. Tretiak, J. J. Crochet, G. Gupta and A. D. Mohite, Nat. Commun., 2016, 7, 11574 CrossRef CAS PubMed.
- J. Fan, Y. Ma, C. Zhang, C. Liu, W. Li, R. E. I. Schropp and Y. Mai, Adv. Energy Mater., 2018, 8, 1703421 CrossRef.
- F. Lang, N. H. Nickel, J. Bundesmann, S. Seidel, A. Denker, S. Albrecht, V. V. Brus, J. Rappich, B. Rech, G. Landi and H. C. Neitzert, Adv. Mater., 2016, 28, 8726–8731 CrossRef CAS PubMed.
- C. Ran, W. Gao, J. Li, J. Xi, L. Li, J. Dai, Y. Yang, X. Gao, H. Dong, B. Jiao, I. Spanopoulos, C. D. Malliakas, X. Hou, M. G. Kanatzidis and Z. Wu, Joule, 2019, 3, 3072–3087 CrossRef CAS.
- C. Wang, F. Gu, Z. Zhao, H. Rao, Y. Qiu, Z. Cai, G. Zhan, X. Li, B. Sun, X. Yu, B. Zhao, Z. Liu, Z. Bian and C. Huang, Adv. Mater., 2020, 32, 1907623 CrossRef CAS PubMed.
- B. Wenger, P. K. Nayak, X. Wen, S. V. Kesava, N. K. Noel and H. J. Snaith, Nat. Commun., 2017, 8, 590 CrossRef PubMed.
- Q. Dong, Y. Fang, Y. Shao, P. Mulligan, J. Qiu, L. Cao and J. Huang, Science, 2015, 347, 967–970 CrossRef CAS PubMed.
- R. Heiderhoff, T. Haeger, N. Pourdavoud, T. Hu, M. Al-Khafaji, A. Mayer, Y. Chen, H.-C. Scheer and T. Riedl, J. Phys. Chem. C, 2017, 121, 28306–28311 CrossRef CAS.
- N. Onoda-Yamamuro, T. Matsuo and H. Suga, J. Phys. Chem. Solids, 1990, 51, 1383–1395 CrossRef CAS.
- J. F. Verwey and J. Schoonman, Physica, 1967, 35, 386–394 CrossRef CAS.
- J. Schoonman and J. F. Verwey, Physica, 1968, 39, 244–250 CrossRef CAS.
- M. B. Islam, M. Yanagida, Y. Shirai, Y. Nabetani and K. Miyano, Sol. Energy Mater. Sol. Cells, 2019, 195, 323–329 CrossRef CAS.
- L. Shi, M. P. Bucknall, T. L. Young, M. Zhang, L. Hu, J. Bing, D. S. Lee, J. Kim, T. Wu, N. Takamure, D. R. McKenzie, S. Huang, M. A. Green and A. W. Y. Ho-Baillie, Science, 2020, 6497, eaba2412 CrossRef PubMed.
- D. Cahen and L. Chernyak, Adv. Mater., 1997, 9, 861–876 CrossRef CAS.
- K. T. Arulmozhi and N. Mythili, AIP Adv., 2013, 3, 122122 CrossRef.
- M. M. Amini, E. Najafi, A. Dehghani and S. W. Ng, J. Mol. Struct., 2015, 1083, 221–228 CrossRef CAS.
- I. A. Kamenskikh, A. Kirm, V. N. Kolobanov, V. V. Mikhailin, P. A. Orekhanov, I. N. Shpinkov, D. A. Spassky, A. N. Vasil’ev, B. I. Zadneprovsky and G. Zimmerer, IEEE Trans. Nucl. Sci., 2001, 48, 2324–2329 CAS.
- V. Gonzalez, D. Gourier, T. Calligaro, K. Toussaint, G. Wallez and M. Menu, Anal. Chem., 2017, 89, 2909–2918 CrossRef CAS PubMed.
- Y. Rakita, N. Kedem, S. Gupta, A. Sadhanala, V. Kalchenko, M. L. Böhm, M. Kulbak, R. H. Friend, D. Cahen and G. Hodes, Cryst. Growth Des., 2016, 16, 5717–5725 CrossRef CAS.
- C. Sun, Z. Gao, H. Liu, C. Geng, H. Wu, X. Zhang, C. Fan and W. Bi, Dalton Trans., 2018, 47, 16218–16224 RSC.
- J. C. Hamill, J. Schwartz and Y.-L. Loo, ACS Energy Lett., 2018, 3, 92–97 CrossRef CAS.
- F. Zhang, J. C. Hamill, Y.-L. Loo and A. Kahn, Adv. Mater., 2020, 32, 2003482 CrossRef CAS PubMed.
- D. Macdonald, R. A. Sinton and A. Cuevas, J. Appl. Phys., 2001, 89, 2772–2778 CrossRef CAS.
- I. Levine, S. Gupta, A. Bera, D. Ceratti, G. Hodes, D. Cahen, D. Guo, T. J. Savenije, J. Ávila, H. J. Bolink, O. Millo, D. Azulay and I. Balberg, J. Appl. Phys., 2018, 124, 103103 CrossRef.
- J. M. Harrowfield, B. W. Skelton and A. H. White, J. Chem. Soc., Dalton Trans., 1993, 2011–2016 RSC.
- Y. H. Chang, C. H. Park and K. Matsuishi, J. Korean Phys. Soc., 2004, 44, 889–893 CAS.
- J. Endres, D. A. Egger, M. Kulbak, R. A. Kerner, L. Zhao, S. H. Silver, G. Hodes, B. P. Rand, D. Cahen, L. Kronik and A. Kahn, J. Phys. Chem. Lett., 2016, 7, 2722–2729 CrossRef CAS PubMed.
- S. R. Raga, Y. Jiang, L. K. Ono and Y. Qi, Energy Technol., 2017, 5, 1750–1761 CrossRef CAS.
- Lange's Handbook of Chemistry, 17th Edition, https://www.accessengineeringlibrary.com/browse/langes-handbook-of-chemistry-seventeenth-edition, accessed March 31, 2019.
- Physical and thermodynamic properties of pure chemicals: data compilation (Continually updated resource, 1989) [WorldCat.org], https://www.worldcat.org/title/physical-and-thermodynamic-properties-of-pure-chemicals-data-compilation/oclc/19513328, accessed March 31, 2019.
- Ammonia (7664-41-7) MSDS Melting Point Boiling Point Density Storage Transport, https://www.chemicalbook.com/ProductMSDSDetailCB9854275_EN.htm, (accessed March 26, 2019).
- 74-89-5 CAS MSDS (Methylamine) Melting Point Boiling Point Density CAS Chemical Properties, https://www.chemicalbook.com/ChemicalProductProperty_US_CB4387750.aspx, (accessed March 26, 2019).
- J. Cazaux, Appl. Surf. Sci., 2010, 257, 1002–1009 CrossRef CAS.
- M. Nagoshi, T. Kawano and K. Sato, Surf. Interface Anal., 2016, 48, 470–473 CrossRef CAS.
- P. A. Anderson and A. L. Hunt, Phys. Rev., 1956, 102, 367–368 CrossRef CAS.
- H. P. Beck and G. Moretti, Solid State Sci., 2020, 110, 106359 CrossRef CAS.
- J. J. Uebbing and L. W. James, J. Appl. Phys., 1970, 41, 4505–4516 CrossRef CAS.
- M. R. Tubbs and A. J. Forty, Br. J. Appl. Phys., 1964, 15, 1553–1558 CrossRef CAS.
- D. Meggiolaro, S. G. Motti, E. Mosconi, A. J. Barker, J. Ball, C. A. R. Perini, F. Deschler, A. Petrozza and F. D. Angelis, Energy Environ. Sci., 2018, 11, 702–713 RSC.
- J. Wiktor, F. Ambrosio and A. Pasquarello, J. Mater. Chem. A, 2018, 6, 16863–16867 RSC.
- M. H. Du, J. Mater. Chem. A, 2014, 2, 9091–9098 RSC.
- M.-H. Du, J. Phys. Chem. Lett., 2015, 6, 1461–1466 CrossRef CAS PubMed.
- A. Zohar, M. Kulbak, I. Levine, G. Hodes, A. Kahn and D. Cahen, ACS Energy Lett., 2019, 4, 1–7 CrossRef CAS.
- I. Levine, O. G. Vera, M. Kulbak, D.-R. Ceratti, C. Rehermann, J. A. Márquez, S. Levcenko, T. Unold, G. Hodes, I. Balberg, D. Cahen and T. Dittrich, ACS Energy Lett., 2019, 4, 1150–1157 CrossRef CAS.
- G. L. Breneman and R. D. Willett, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1969, 25, 1073–1076 CrossRef CAS.
- P. K. Bakshi, M. A. James, T. S. Cameron and O. Knop, Can. J. Chem., 1996, 74, 559–573 CrossRef CAS.
- M. E. Easton, A. J. Ward, B. Chan, L. Radom, A. F. Masters and T. Maschmeyer, Phys. Chem. Chem. Phys., 2016, 18, 7251–7260 RSC.
- D. Schoemaker and A. Lagendijk, Phys. Rev. B: Solid State, 1977, 15, 5927–5937 CrossRef CAS.
- W. Känzig and T. O. Woodruff, J. Phys. Chem. Solids, 1959, 9, 70–92 CrossRef.
- A. Lushchik, Izv. Akad. Nauk Ehstonskoj SSR Fiz. Mat., 1980, 29, 173–180 Search PubMed.
- B. V. R. Chowdari and N. Itoh, J. Phys. Chem. Solids, 1972, 33, 1773–1783 CrossRef CAS.
- J.-H. Yang, W.-J. Yin, J.-S. Park and S.-H. Wei, J. Mater. Chem. A, 2016, 4, 13105–13112 RSC.
- O. Yaffe, Y. Guo, L. Z. Tan, D. A. Egger, T. Hull, C. C. Stoumpos, F. Zheng, T. F. Heinz, L. Kronik, M. G. Kanatzidis, J. S. Owen, A. M. Rappe, M. A. Pimenta and L. E. Brus, Phys. Rev. Lett., 2017, 118, 136001 CrossRef PubMed.
- A. Marronnier, G. Roma, S. Boyer-Richard, L. Pedesseau, J.-M. Jancu, Y. Bonnassieux, C. Katan, C. C. Stoumpos, M. G. Kanatzidis and J. Even, ACS Nano, 2018, 12, 3477–3486 CrossRef CAS PubMed.
- A. M. A. Leguy, A. R. Goñi, J. M. Frost, J. Skelton, F. Brivio, X. Rodríguez-Martínez, O. J. Weber, A. Pallipurath, M. I. Alonso, M. Campoy-Quiles, M. T. Weller, J. Nelson, A. Walsh and P. R. F. Barnes, Phys. Chem. Chem. Phys., 2016, 18, 27051–27066 RSC.
- A. V. Cohen, D. A. Egger, A. M. Rappe and L. Kronik, J. Phys. Chem. Lett., 2019, 10, 4490–4498 CrossRef CAS PubMed.
- Z. Andaji-Garmaroudi, M. Anaya, A. J. Pearson and S. D. Stranks, Adv. Energy Mater., 2020, 10, 1903109 CrossRef CAS.
- D. R. Ceratti, A. Zohar, R. Kozlov, H. Dong, G. Uraltsev, O. Girshevitz, I. Pinkas, L. Avram, G. Hodes and D. Cahen, Adv. Mater., 2020, 32(46), 2002467 CrossRef CAS PubMed.
- S. Sadhu, T. Buffeteau, S. Sandrez, L. Hirsch and D. M. Bassani, J. Am. Chem. Soc., 2020, 142(23), 10431–10437 CrossRef CAS PubMed.
- W. T. M. Van Gompel, R. Herckens, G. Reekmans, B. Ruttens, J. D’Haen, P. Adriaensens, L. Lutsen and D. Vanderzande, J. Phys. Chem. C, 2018, 122, 4117–4124 CrossRef CAS.
- F. C. Schaefer, I. Hechenbleikner, G. A. Peters and V. P. Wystrach, J. Am. Chem. Soc., 1959, 81, 1466–1470 CrossRef CAS.
- B. Conings, S. A. Bretschneider, A. Babayigit, N. Gauquelin, I. Cardinaletti, J. Manca, J. Verbeeck, H. J. Snaith and H.-G. Boyen, ACS Appl. Mater. Interfaces, 2017, 9, 8092–8099 CrossRef CAS PubMed.
- S. Shao, M. Abdu-Aguye, L. Qiu, L.-H. Lai, J. Liu, S. Adjokatse, F. Jahani, M. E. Kamminga, G. H. ten Brink, T. T. M. Palstra, B. J. Kooi, J. C. Hummelen and M. A. Loi, Energy Environ. Sci., 2016, 9, 2444–2452 RSC.
- C. Zhao, B. Chen, X. Qiao, L. Luan, K. Lu and B. Hu, Adv. Energy Mater., 2015, 5, 1500279 CrossRef.
- Z. Song, C. Wang, A. B. Phillips, C. R. Grice, D. Zhao, Y. Yu, C. Chen, C. Li, X. Yin, R. J. Ellingson, M. J. Heben and Y. Yan, Sustainable Energy Fuels, 2018, 2, 2460–2467 RSC.
- H. Syafutra, J.-H. Yun, Y. Yoshie, M. Lyu, S. N. Takeda, M. Nakamura, L. Wang and M.-C. Jung, Nanomaterials, 2020, 10, 1253 CrossRef CAS PubMed.
- D. H. Jung, J. H. Park, H. E. Lee, J. Byun, T. H. Im, G. Y. Lee, J. Y. Seok, T. Yun, K. J. Lee and S. O. Kim, Nano Energy, 2019, 61, 236–244 CrossRef CAS.
- Y. Rakita, I. Lubomirsky and D. Cahen, Mater. Horiz., 2019, 6, 1297–1305 RSC.
- H. Tan, A. Jain, O. Voznyy, X. Lan, F. P. G. de Arquer, J. Z. Fan, R. Quintero-Bermudez, M. Yuan, B. Zhang, Y. Zhao, F. Fan, P. Li, L. N. Quan, Y. Zhao, Z.-H. Lu, Z. Yang, S. Hoogland and E. H. Sargent, Science, 2017, 355, 722–726 CrossRef CAS PubMed.
- X. Tang, M. Brandl, B. May, I. Levchuk, Y. Hou, M. Richter, H. Chen, S. Chen, S. Kahmann, A. Osvet, F. Maier, H.-P. Steinrück, R. Hock, G. J. Matt and C. J. Brabec, J. Mater. Chem. A, 2016, 4, 15896–15903 RSC.
- F. Zu, T. Schultz, C. M. Wolff, D. Shin, L. Frohloff, D. Neher, P. Amsalem and N. Koch, RSC Adv., 2020, 10, 17534–17542 RSC.
- S. Kim, S. Bae, S.-W. Lee, K. Cho, K. D. Lee, H. Kim, S. Park, G. Kwon, S.-W. Ahn, H.-M. Lee, Y. Kang, H.-S. Lee and D. Kim, Sci. Rep., 2017, 7(1), 1–9 CrossRef PubMed.
- F. Ren, Z. Zhu, X. Qian, W. Liang, P. Mu, H. Sun, J. Liu and A. Li, Chem. Commun., 2016, 52, 9797–9800 RSC.
- K. Mahmood, S. Sarwar and M. Taqi Mehran, RSC Adv., 2017, 7, 17044–17062 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1mh00006c |
| This journal is © The Royal Society of Chemistry 2021 |