
Synaptic vesicle-inspired nanoparticles with spatiotemporally controlled release ability as a “nanoguard” for synergistic treatment of synucleinopathies†
Weihong
Ji‡
ab,
Yan
Li‡
a,
Ruiyuan
Liu
ab,
Zhiguo
Lu
ab,
Linying
Liu
ab,
Zhuyan
Shi
ab,
Jie
Shen
ab and
Xin
Zhang
 *a
*a
aState Key Laboratory of Biochemical Engineering, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: xzhang@ipe.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 9th March 2021
Abstract
Synaptic vesicle-inspired nanoparticles (RT-PPB NPs) as a “nanoguard” were designed for clearing the toxic α-synuclein aggregates in diseased neurons and preventing the culprits from escaping to affect other normal cells. The NPs could overcome a series of tissue and cellular barriers and controllably release drugs in the diseased neurons, which ensured the optimization of synergistic treatment. This study indicates that the synaptic vesicle-inspired NPs may have the potential to open up a new avenue for the treatment of synucleinopathies, as well as other neurodegenerative diseases.
New conceptsThe low intracellular drug concentration caused by tissue and cellular barriers has been a major obstacle limiting the efficacy of treating neurodegenerative diseases. Herein, evolved synaptic vesicle (SV)-mimicking nanoparticles (RT-PPB NPs) are constructed for achieving intracellular drug enrichment in synucleinopathies. The NPs can efficiently cross the blood–brain barrier and target/dock to the neurons. The lipid shell of the NPs then fuses with the cell membrane and directly releases the drug core into the cytoplasm, which can avoid lysosomal trapping and exocytosis. Most importantly, the drug core with dual reactive oxygen species-responsive ability can controllably release rapamycin and small interfering RNA for reducing the intracellular α-synuclein (α-syn) aggregate level and neuron-to-neuron transmission of α-syn aggregates via the aggregates themselves and exosomes. The RT-PPB NPs play the role of a guard to clear the toxic α-syn aggregates in the diseased neurons and prevent the culprits from escaping to affect other normal cells. These “nanoguards” can significantly rescue the dopaminergic neurons and recover the motor behaviour of Parkinson's disease model mice. This study indicates that the SVs-inspired NPs may have the potential to open up a new avenue for the treatment of synucleinopathies, as well as other neurodegenerative diseases. |
Synucleinopathies, such as Parkinson's disease (PD), multiple system atrophy and dementia with Lewy bodies, are a group of neurodegenerative diseases that are characterised by the pathological deposition of α-synuclein (α-syn).1,2 The α-syn aggregates as the culprits are not only toxic to host neurons, but also have prion-like activities. Neuronal exosomes can facilitate α-syn aggregate transmission, resulting in more neuron degeneration and the development of the disease.3–7 Both pathologies are devastating to neurons, and treatment for only one aspect is not comprehensive. Therefore, it is necessary to employ synergistic therapy for clearing intracellular α-syn aggregates and reducing neuron-to-neuron transmission of α-syn aggregates simultaneously.
In synucleinopathies, autophagy goes awry, which results in intracellular α-syn aggregates not being degraded in time.8,9 Rapamycin (RAPA) can increase lysosome-mediated autophagic degradation activity, thereby reducing intracellular and transmitted α-syn aggregates.10,11 Studies have shown that inhibition of neutral sphingomyelinase-2 (nSMase2) can reduce the synthesis of exosomes.12,13 Inspired by the abovementioned, small interfering RNA targeting nSMase2 (sinSMase2) will be used to halt exosome-mediated α-syn aggregate release and transmission.12,13 Therefore, RAPA and sinSMase2 can achieve a synergistic treatment effect by reducing intracellular α-syn aggregates and intercellular transmission simultaneously. However, there are a series of barriers in drug delivery for neurodegenerative disease treatment. The most common problem is the poor ability to target the blood–brain barrier (BBB) and neurons.14,15 In recent years, various brain-targeting nanomaterials have been developed for synucleinopathies and other neurodegenerative diseases.16–20 These nanomaterials can enhance drug accumulation in the brain and have good therapeutic effects. However, the primary uptake pathway, cellular endocytosis, is another immediate problem, which can result in lysosomal trapping and exocytosis, and cause a significant decrease in intracellular drug concentration.21,22 Therefore, it is necessary to design a reasonable drug carrier to deliver the two drugs directly into the cytoplasm of diseased neurons.
Synaptic vesicles (SVs), approximately 40–100 nm in diameter, are made of a lipid bilayer inserted with soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) synaptobrevin II (syb) proteins.23,24 And neurotransmitters are trapped in their hydrophilic core. After docking to the plasma membrane, the SNARE proteins drive SVs to fuse with the cell membrane to release neurotransmitters directly.25,26 We noted that the unique structure and action process of SVs were worth learning for drug delivery and the big hydrophilic lumen was conducive to drug loading. The docking and fusion process can help drugs overcome the cell membrane barrier and skip the lysosomal trapping to release drugs into the cytoplasm of the target cell. However, RAPA and sinSMase2 have almost no similar features, and cannot be directly encapsulated in the lumen like neurotransmitters. Therefore, it is urgent to design a suitable drug core with spatiotemporally controlled ability to load these two drugs and release them in a controlled manner.
Herein, inspired by the structure and action process of SVs, we constructed evolved SV-mimicking nanoparticles (NPs) with spatiotemporally controlled release ability as a “nanoguard” for clearing toxic α-synuclein aggregates and halting their pathology propagation. The SV-mimicking NPs consisted of a lipid envelope with targeting and fusogenic function, and a dual-reactive oxygen species (ROS) responsive drug core (Scheme 1a). RVG29, a 29 amino acid peptide derived from rabies virus glycoprotein (RVG), was conjugated to facilitate the NPs across the BBB and docking to neurons via its recognition of acetylcholine receptor (AchR).27–30 Transmembrane segment syb (TMS-syb), derived from SNARE syb proteins of SVs, was inserted in the lipid envelope, which could drive the membrane fusion between NPs and the cell membrane (Scheme 1b).31 And the lipid envelope with similar components to the SV lipid layer was coated on the drug core. Using the characteristic of high ROS level in diseased neurons, a dual-ROS responsive micelle was designed.1,2 Dual-ROS responsive amphiphilic polymer poly(propylene sulfide)-b-poly[(2-acryloyl)ethyl(p-boronic acid benzyl) dimethylammonium bromide] (PPS–PDMAEA-B, PPB) was synthesized and self-assembled into micelles, which encapsulated RAPA and superparamagnetic iron oxide NPs (SPIONs) in the hydrophobic region and absorbed sinSMase2 via electrostatic interaction to form the dual-ROS responsive ![[P with combining low line]](https://www.rsc.org/images/entities/char_0050_0332.gif)
![[B with combining low line]](https://www.rsc.org/images/entities/char_0042_0332.gif) /RAPA/SPIONs@sinSMase2 (PPB) core. In the presence of ROS, the thioether group of hydrophobic block PPS could be oxidized to form a hydrophilic sulfone group to release RAPA.32,33 Meanwhile, positive to negative charge reversal occurred for the hydrophilic block PDMAEA-B thanks to the production of poly(acrylic acid) upon oxidation to release sinSMase2.34,35 Therefore, the PPB core could load two drugs and release drugs more rapidly as far as possible compared with single-ROS responsive or non-ROS responsive micelles.
/RAPA/SPIONs@sinSMase2 (PPB) core. In the presence of ROS, the thioether group of hydrophobic block PPS could be oxidized to form a hydrophilic sulfone group to release RAPA.32,33 Meanwhile, positive to negative charge reversal occurred for the hydrophilic block PDMAEA-B thanks to the production of poly(acrylic acid) upon oxidation to release sinSMase2.34,35 Therefore, the PPB core could load two drugs and release drugs more rapidly as far as possible compared with single-ROS responsive or non-ROS responsive micelles.
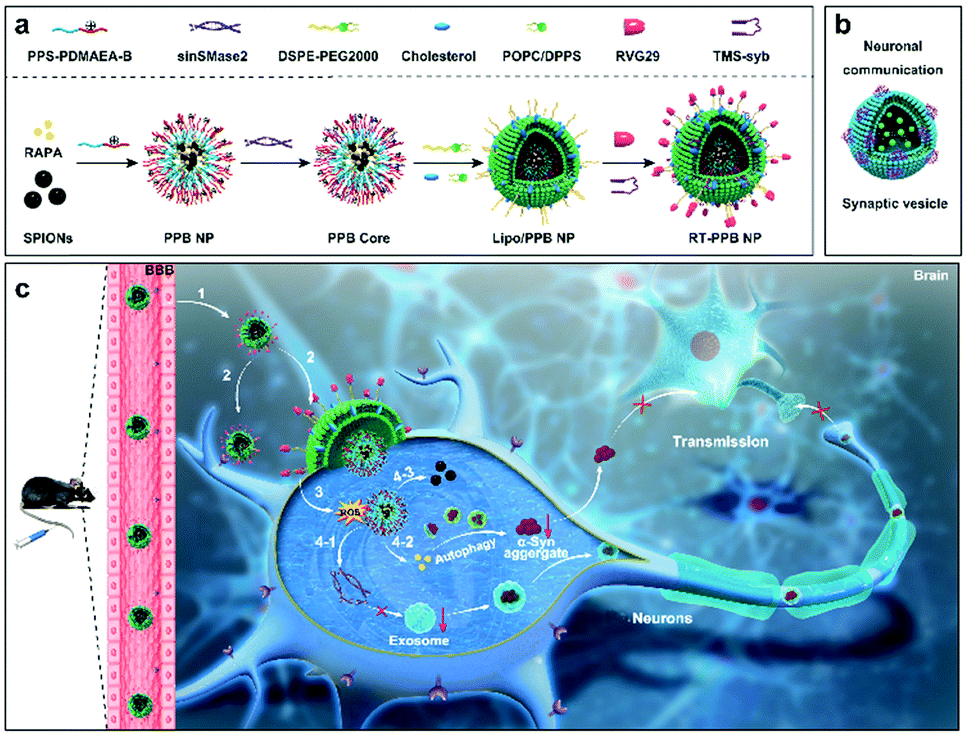 | ||
| Scheme 1 Schematic illustration of the evolved SV-mimicking RT-PPB NPs. (a) Structural composition and preparation of the RT-PPB NPs. (b) Structure of the SVs. The purple clusters represented SNAREs synaptobrevin II (syb) protein and the green intravesicular spheres represented neurotransmitters. (c) Schematic diagram of the mechanism of the RT-PPB NPs for synergistic therapy of synucleinopathies. (1) The NPs crossed the BBB and (2) docked/targeted to the cell body of neurons and the presynapses. (3) TMS-syb peptides drove the membrane fusion and the PPB core was directly delivered into the cytoplasm. (4-1) The sinSMase2, and (4-2) RAPA were rapidly released under the ROS environment. The RAPA and sinSMase-2 could synergistically reduce intracellular α-syn aggregates and intercellular α-syn aggregates transmission. (4-3) The accumulation of NPs in the brain was tracked by SPIONs via MRI. | ||
As shown in Scheme 1c, with the help of RVG29, the “nanoguard”, SV-mimicking ![[R with combining low line]](https://www.rsc.org/images/entities/char_0052_0332.gif) VG-
VG-![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) MS-Liposome (/RAPA/SPIONs@sinSMase2) (RT-PPB) NPs, crossed the BBB and targeted/docked to the neurons. TMS-syb peptides drove the membrane fusion and delivered the PPB core directly into the cytoplasm. Both drugs were triggered to be released from the dual-ROS responsive core due to the intracellular high ROS level caused by α-syn aggregates in diseased neurons. The released RAPA and sinSMase2 could clear intracellular α-syn aggregates and inhibit the synthesis of exosomes, which could synergistically reduce intercellular α-syn aggregate transmission for the treatment of synucleinopathies. The accumulation of NPs could be tracked by SPIONs via magnetic resonance imaging (MRI) due to their high r2 relaxivity.30
MS-Liposome (/RAPA/SPIONs@sinSMase2) (RT-PPB) NPs, crossed the BBB and targeted/docked to the neurons. TMS-syb peptides drove the membrane fusion and delivered the PPB core directly into the cytoplasm. Both drugs were triggered to be released from the dual-ROS responsive core due to the intracellular high ROS level caused by α-syn aggregates in diseased neurons. The released RAPA and sinSMase2 could clear intracellular α-syn aggregates and inhibit the synthesis of exosomes, which could synergistically reduce intercellular α-syn aggregate transmission for the treatment of synucleinopathies. The accumulation of NPs could be tracked by SPIONs via magnetic resonance imaging (MRI) due to their high r2 relaxivity.30
Dual-ROS responsive amphiphilic polymer PPB and control polymers hydrophobic block single-ROS responsive PPS–PDMAEA-Bn (PPBn), hydrophilic block single-ROS responsive C-PDMAEA-B (CPB), and non-ROS responsive C-PDMAEA-Bn (CPBn) were synthesized by similar routes (Fig. S1–S3, ESI†).32–35 The structures of the intermediates and end products were confirmed by gel permeation chromatography and 1H NMR, which indicated the successful synthesis (Fig. S4–S10, ESI†). The change of the 1.2–2.2 ppm peaks in 1H NMR indicated the conversion of the thioether group in PPS, and the shifting of the peaks from 7.5–8.0 ppm to 6.8–7.5 ppm confirmed the change of PDMAEA-B after H2O2 treatment (Fig. S11 and S12, ESI†). The DSPE–PEG–RVG29 was synthesized and the structure was confirmed by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (Fig. S13 and S14, ESI†). SPIONs were synthesized via high-temperature thermal decomposition.36,37 The monodispersed spherical SPIONs with 15 nm diameter were observed by transmission electron microscopy (TEM) (Fig. S15, ESI†).
Four NPs were self-assembled from the amphiphilic polymers with RAPA and SPIONs in the hydrophobic region, and had comparable diameters of approximate 100 nm and zeta potentials of about 30 mV (Fig. S16, ESI†). The NPs also showed a low polydispersity index (PDI), indicating the uniformity in size (Table S2, ESI†). Gel electrophoresis assays demonstrated that all four NPs could completely adsorb siRNA at an N/P ratio of 8 (Fig. S17, ESI†). Considering that excess materials could cause metabolic burden, NPs with an N/P ratio of 8 were selected and utilized for the following experiments. At a N/P ratio of 8, the diameter of the PPB core was 118 nm with a zeta potential of 8.3 mV (Fig. 1a) and the other three cores had comparable sizes and zeta potentials (Fig. S18, ESI†).
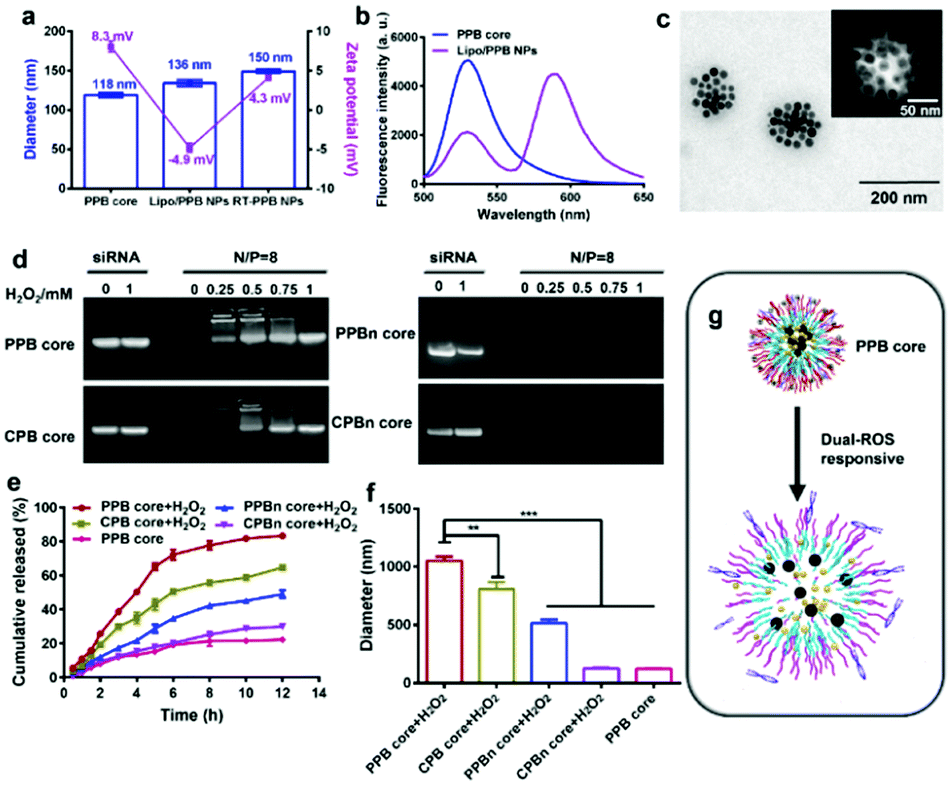 | ||
| Fig. 1 Characterization of the NPs. (a) Diameter and zeta potential of the PPB core, Lipo/PPB NPs, and RT-PPB NPs detected by DLS (n = 3). (b) Fluorescence emission scans (490 nm excitation) of the PPB core and Lipo/PPB NPs. (c) TEM image of RT-PPB NPs. (d–g) Dual-ROS responsive ability of the PPB core. (d) SiRNA released from four cores following H2O2 treatment as determined by gel electrophoresis. (e) The cumulative release of RAPA from the cores at 37 °C in 0.5% Tween-80 with or without 0.5 mM H2O2 detected by high performance liquid chromatography (n = 3). (f) The diameter changes of four cores after being treated with 0.5 mM H2O2 for 3 h at 37 °C (n = 3). (g) Schematic illustration of drugs released from the PPB core following H2O2 treatment. Data are presented as the mean ± SD. **P < 0.01, ***P < 0.001, Student's t-test. | ||
The liposomes were then enveloped on the core using the thin film method. The diameter of the Liposome (/RAPA/SPIONs@sinSMase2) (Lipo/PPB) NPs increased to around 136 nm and the zeta potential decreased to −4.9 mV due to the negatively charged POPC lipid (Fig. 1a). To confirm the core–shell structure, a fluorescence resonance energy transfer (FRET) assay was utilized.38,39 The core and shell were separately labeled with a pair of FRET dyes, carboxyfluorescein (FAM-sinSMase) and rhodamine-B (DSPE–PEG–RhoB). At an excitation wavelength of 490 nm, strong emission intensity of FAM at 525 nm was detected in the PPB core, while strong emission intensity of RhoB at 580 nm with fading emission intensity of FAM at 525 nm was detected in the Lipo/PPB NPs, indicating that strong FRET occurred (Fig. 1b). The distance (R) between two dyes was calculated to be about 5.1 nm. These results suggested the formation of shell–core Lipo/PPB NPs.
We further utilized RVG29 and TMS-syb decoration to construct the SV-mimicking RT-PPB NPs. RT-PPB NPs were approximately 150 nm in diameter with a zeta potential of 4.3 mV (Fig. 1a). The modification increased the zeta potential due to the positively charged TMS-syb (Fig. S19, ESI†). The conjugation efficiency of the TMS-syb peptide was estimated as 62.4 ± 1.6%, and the average density of the peptide on the NP surface was approximately 375 peptide ligands per particle. TEM images showed that RT-PPB NPs presented monodispersed spherical structures with SPIONs in the core (Fig. 1c). The magnetic properties of the NPs were evaluated with r2 values. RT-PPB NPs had an r2 value of 164.54 mM−1 s−1 and the other NPs had comparable values, which were high enough for in vivo application (Fig. S20, ESI†).
In theory, both drugs were proposed to be released from the PPB core after entering into the neurons with a high level of ROS via membrane fusion. Therefore, we detected the ability of the core NPs to release drugs after H2O2 treatment.34,40 The PPB core exhibited an active ROS response and siRNA was released upon treatment with H2O2 for 3 h due to the charge reversal, as well as the CPB core, whereas no siRNA was released from the PPBn core and CPBn core (Fig. 1d). Meanwhile, 40.22% RAPA was released from the PPB core with H2O2 treatment at 3 h and up to 83.14% at 12 h. Under the same treatment for 3 h, the RAPA cumulative release quantities of the CPB core, PPBn core and CPBn core were only 29.75%, 17.30% and 12.46%, respectively (Fig. 1e). The size of the PPB core after incubation with H2O2 for 3 h increased to 1051 nm with the largest change (Fig. 1f). As anticipated, the results revealed that the PPB core with dual-ROS responsive ability could lead to more complete disintegration of the core, which resulted in rapid release of two drugs (Fig. 1g and Fig. S11, ESI†).
Before in vivo application, the biocompatibility of the NPs was studied using the MTT assay. Cell viability of SH-SY5Y was above 90% for the RT-PPB NPs at sinSMase2 concentration of 1 μg mL−1 and RAPA concentration of 100 nM (Fig. S21, ESI†). And an alamar blue assay was used to observe changes in cell metabolism (Fig. S22, ESI†).41 Besides, the serum stability test indicated that the RT-PPB NPs could be stable under physiological conditions (Fig. S23, ESI†). These results indicated that the NPs had no obvious cytotoxicity and were suitable for in vivo application.
The BBB is a major challenge in the treatment of most brain disorders.14,15 Additionally, NPs have to recognize the neurons for the treatment of synucleinopathies. Firstly, the endothelial barrier of bEnd.3 cells was formed after 7 days of culture and the transepithelial electric resistances before and after RT-PPB NP incubation were over 200 Ω cm2 (Fig. S24a and b, ESI†). And the permeability of the NPs across the BBB was thoroughly examined, and particularly the permeability coefficients were calculated (Fig. S24c and d, ESI†).42–44 The permeability coefficient of RT-PPB NPs was 59.33 × 10−4 cm h−1, which was nearly 3 times that of T-PPB NPs (Fig. S24c, ESI†). Namely, RT-PPB NPs exhibit nearly 2 times better BBB transcytosis than T-PPB NPs (Fig. S24d, ESI†). To assess the targeting ability of RT-PPB NPs, the transwell was established as the BBB model in vitro (Fig. 2a).42–45 Compared with the T-PPB NPs without RVG29, we found that the RT-PPB NPs group had an apparent enhancement of the mean fluorescence intensity of Cy5 in the upper bEnd.3 cells and bottom SH-SY5Y cells (Fig. 2b, c and Fig. S25, ESI†). As a comparison, the addition of free RVG peptide inhibited the promoting effect (Fig. S25, ESI†). These results indicated that RVG29 modification significantly promoted the RT-PPB NPs translocation across the BBB and accumulation in SH-SY5Y cells. Furthermore, in order to prove that the RT-PPB NPs had the ability to target the neurons, the uptake ability of diverse cells toward RT-PPB NPs was compared. The Cy5 mean fluorescence intensity was detected after incubating RT-PPB NPs with the murine microglial cell line BV2, the mouse hepatic stellate cell HSC and SH-SY5Y cells for 3 h, respectively (Fig. S26, ESI†). Compared with the other two types of cells without AchRs, SH-SY5Y cells had the most uptake. This result indicated that RT-PPB NPs could effectively target neurons.
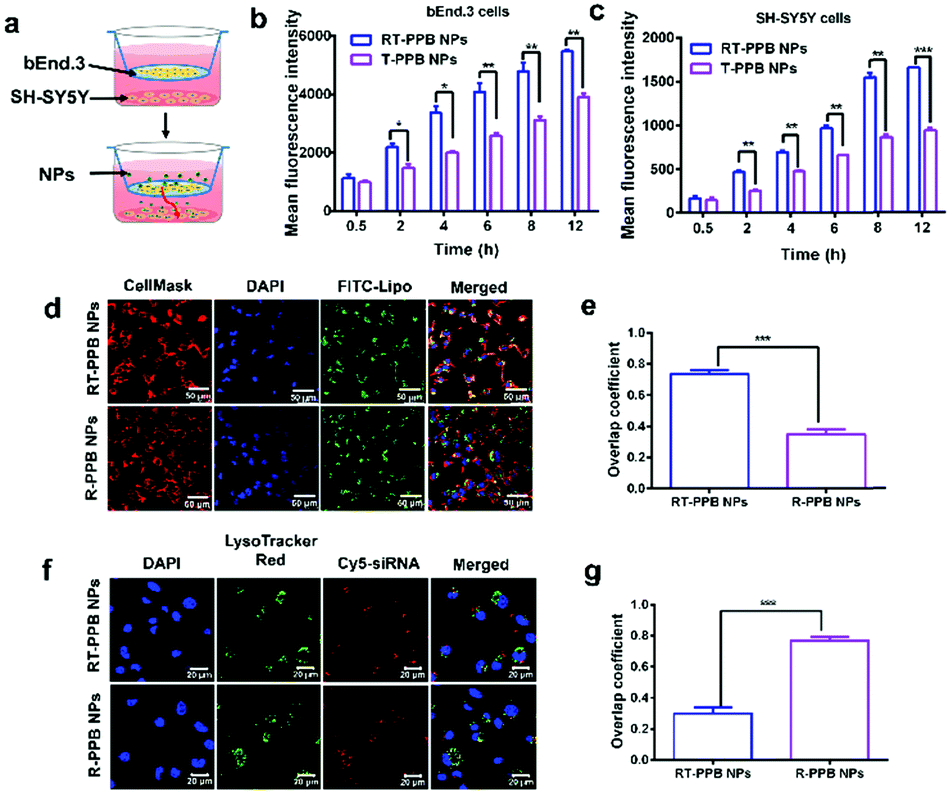 | ||
| Fig. 2 Blood brain barrier permeability and cellular internalization pathway of NPs in vitro. (a) The schematic illustration of the transwell model. (b and c) The cellular uptake of NPs in bEnd.3 cells (b) and SH-SY5Y cells (c) in a transwell model (n = 3). (d–g) The cellular internalization of NPs with 1 μg FITC-Lipo or 1 μg Cy5-siRNA assessed by confocal laser scanning microscopy (CLSM) after 3 h of incubation. (d) The cellular internalization via membrane fusion and (e) the overlap coefficient quantified results. (f) The cellular internalization by avoiding endosomal trapping and (g) the overlap coefficient quantified results (n = 3). More than 100 cells were analyzed for the quantification. Data are presented as the mean ± SD. **P < 0.01, ***P < 0.001, Student's t-test. | ||
Subsequently, we explored how the RT-PPB NPs entered cells. The green FITC fluorescence of RT-PPB NPs was almost co-localized with the cellular plasma membrane with an overlap coefficient of 0.75, while the R-PPB NPs were mainly distributed in the cell (Fig. 2d and e). Furthermore, the cores of RT-PPB NPs and R-PPB NPs were labeled with Cy5-siRNA to track their intracellular fate. The red Cy5 fluorescence of RT-PPB NPs was barely co-localized with the green fluorescence of endosomes/lysosomes, whereas the R-PPB NPs were mainly co-localized with endosomes/lysosomes as yellow dots with an overlap coefficient of 0.77 (Fig. 2f and g). Then we observed the co-localization of the RT-PPB NPs and the cytoplasm. The overlap coefficient of the RT-PPB NPs and the cytoplasm was 0.71, which was significantly higher than the overlap coefficient of R-PPB NPs and the cytoplasm (Fig. S27, ESI†). Besides, endocytosis inhibitors were adopted to study the cellular uptake pathway. Chlorpromazine inhibited the clathrin-mediated endocytosis by triggering the dissociation of the clathrin lattice. Genistein and mβCD could inhibit caveolae by inhibiting tyrosine kinase and depleting cholesterol, respectively. Dynasore inhibited both clathrin-mediated endocytosis and caveolae by inhibiting dynamin. Wortmannin inhibited macropinocytosis by inhibiting phosphatidyl inositol-3-phosphate.46,47 As shown in Fig. S28 (ESI†), the cellular uptake efficiencies of RT-PPB NPs were seldom affected by these five inhibitors, indicating that RT-PPB NPs might enter cells predominantly via a membrane fusion mechanism. And the cellular uptake efficiencies of R-PPB NPs were significantly inhibited. These results demonstrated that TMS-syb could mediate membrane fusion, which made the drug core mostly bypass lysosomal trapping.
To more fully verify the ROS-responsive drug release ability of the PPB core in vitro, α-syn-mCherry over-expression SH-SY5Y (α-syn-mCherrySH-SY5Y) cell line was used as the synucleinopathy pathological model cell with high intracellular ROS level (Fig. S29, ESI†). RT-PPB NPs loaded Cy5-siRNA and hydrophobic FITC representing RAPA. After co-culture for 3 h, the fluorescence of Cy5-siRNA and FITC was separated in α-syn-mCherrySH-SY5Y cells, but not in SH-SY5Y cells (Fig. S30, ESI†). This result further demonstrated that the PPB core could quickly release two drugs in diseased neurons. Moreover, Fig. S31 and S32 (ESI†) showed that the level of nSMase2 protein in SH-SY5Y cells after treatment with RT-PPB NPs was not significantly reduced. Therefore, RT-PPB NPs had little or no negative effect on normal neurons, and would not adversely affect normal physiological functions.
Next, we studied the effect of SV-mimicking RT-PPB NPs in vitro. The western blot results showed that the RT-PPB NPs had the lowest nSMase2 protein level compared to free sinSMase2 and other NP groups (Fig. 3a, b and Fig. S33, ESI†). The inhibition of nSMase2 reduces the synthesis of exosomes as it can decrease the synthesis of ceramide lipid, which is an essential lipid for a subtype of exosome biogenesis in multivesicular bodies.48 Exosomes extracted from cell culture medium had a diameter of around 100 nm (Fig. 3c), which were consistent with reports by other researchers.49 The concentration of exosomes secreted by the cells with RT-PPB NPs treatment was significantly reduced by 57.6%, which was more effective than other treatment groups (Fig. S34, ESI† and Fig. 3d). We further examined the α-syn aggregate levels by ELISA assays (Fig. 3e) and using CLSM after co-culture with the different groups for 48 h (Fig. 3f). The SV-mimicking RT-PPB NPs decreased α-syn aggregates most compared to other groups. These results indicated that RT-PPB NPs could synergistically reduce the secretion of exosomes and the intracellular α-syn aggregate level.
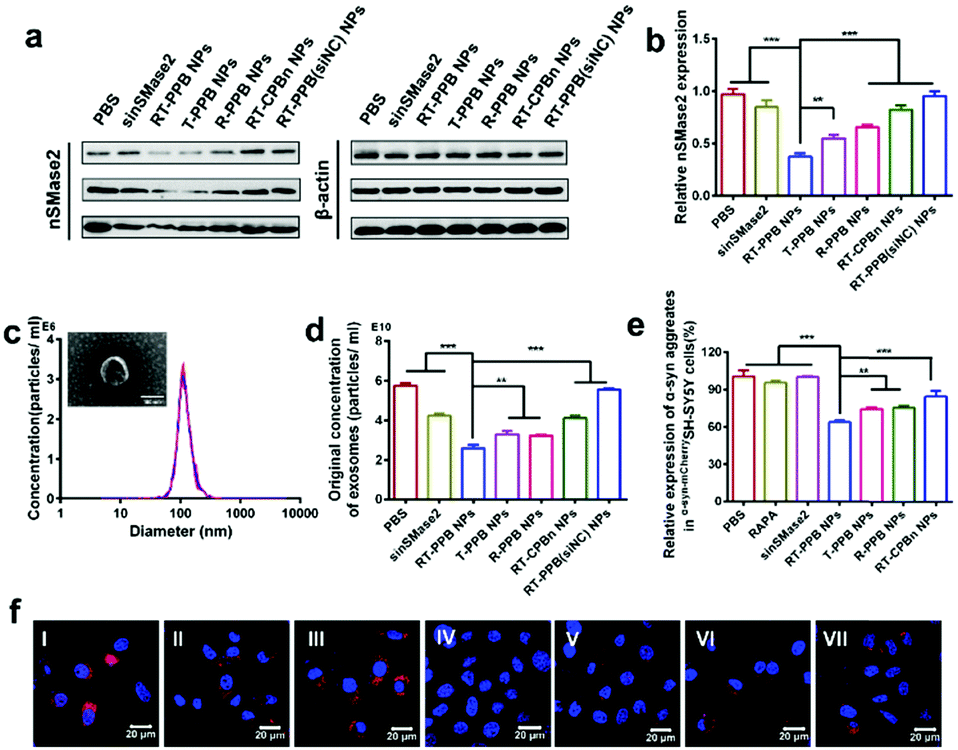 | ||
| Fig. 3 In vitro gene silencing effects of NPs, release of exosomes and levels of intracellular α-syn aggregates. (a) Western blot analysis of nSMase2 protein level under the different formulations at 37 °C for 48 h. The concentration of siRNA was 3 μg per well in 6-well plates. (b) Quantified results of western blot using ImageJ (n = 3). (c) The average size of exosomes (the inset image is the TEM result of exosomes, scale bar: 100 nm). (d) The concentrations of exosomes measured by Nanoparticle Tracking Analysis (n = 3). (e) The relative expression of α-syn aggregates in α-syn-mCherrySH-SY5Y cells detected by ELISA assays under NP treatment with sinSMase2 concentration of 1 μg mL−1 and RAPA concentration of 100 nM for 48 h (n = 5). (f) The α-syn aggregates in α-syn-mCherrySH-SY5Y cells. Samples: (I) PBS, (II) RAPA, (III) sinSMase2, (IV) RT-PPB NPs, (V) T-PPB NPs, (VI) R-PPB NPs, and (VII) RT-CPBn NPs. Data are presented as the mean ± SD. **P < 0.01, ***P < 0.001, Student's t-test. | ||
To confirm that diseased neurons could affect normal neurons through neuronal communication, we constructed the transwell model (Fig. 4a).50 The culture medium and recipient cells (SH-SY5Y) were collected and the α-syn aggregate levels were detected by ELISA assays (Fig. 4b and c). The exosomes were extracted from cell culture medium and the exosome-loaded α-syn aggregate levels of the RT-PPB NP group were the lowest. After co-culture with RT-PPB NP-treated donor cells (α-syn-mCherrySH-SY5Y), the α-syn aggregate levels in SH-SY5Y cells were significantly reduced. In addition, fluorescence images were taken with CLSM to more directly observe the protein distribution in the recipient cells (Fig. 4d). There were obvious α-syn-mCherry signals in the recipient SH-SY5Y cells after co-culture with donor α-syn-mCherrySH-SY5Y cells for 24 h (Fig. 4d(I)), which indicated that α-syn aggregates could indeed be transmitted from diseased neurons to healthy neurons. Compared with co-culture with untreated or other donor cells, the α-syn-mcherry signals in the recipient cells almost disappeared after co-culture with RT-PPB NP-treated donor cells (Fig. 4d). These results supported that the “nanoguard” RT-PPB NPs could effectively inhibit exosome-mediated transmission of the culprit α-syn aggregates between neurons (Fig. 4e). In fact, exosome transmission not only exists between neurons, but also between different cell types, such as between neurons and glial cells.51–54 Therefore, the NPs may play a broader role in vivo.
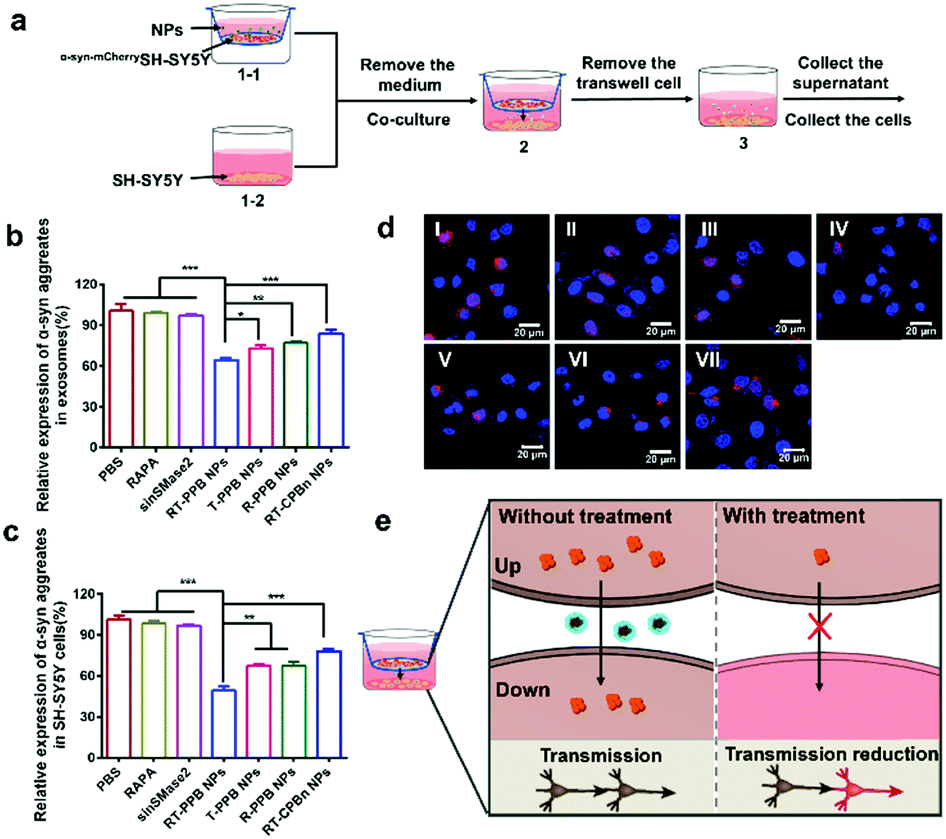 | ||
Fig. 4
In vitro neuronal transmission of α-syn aggregates. (a) The schematic illustration of the transwell model. (1-1) The donor cells (α-syn-mCherrySH-SY5Y) were treated with different formulations for 12 h in the upper chambers. (1-2) Meanwhile, SH-SY5Y cells were seeded in 6-well plates for 24 h. (2) The α-syn-mCherrySH-SY5Y cells were placed on top of the recipient SH-SY5Y cells, and subsequently co-cultured in medium supplemented with 10% exosome-depleted FBS for 24 h. (3) Removal of the donor α-syn-mCherrySH-SY5Y cells. (b) The α-syn aggregate levels of exosomes in culture medium and (c) the α-syn aggregates levels in recipient cells (SH-SY5Y cells) were detected by ELISA assays (n = 5). (d) The α-syn aggregates in SH-SY5Y cells in the transwell model. Samples: (I) PBS, (II) RAPA, (III) sinSMase2, (IV) RT-PPB NPs, (V) T-PPB NPs, (VI) R-PPB NPs, (VII) RT-CPBn NPs. (e) The schematic illustration of α-syn aggregate transmission. Up: α-syn-mCherrySH-SY5Y cells. Down: SH-SY5Y cells.  : α-syn aggregates. : α-syn aggregates.  : α-syn aggregates-loaded exosomes. Data are presented as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, Student's t-test. : α-syn aggregates-loaded exosomes. Data are presented as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, Student's t-test. | ||
Subsequently, the PD was taken as an example to evaluate the therapeutic effect of the “nanoguard” on synucleinopathies in vivo. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD model mice were successfully constructed (Fig. S35, ESI†).55–58 We then studied the BBB permeability of RT-PPB NPs. Compared with T-PPB NPs, RT-PPB NPs could significantly enhance the accumulation of NPs in the brain at 12 h post injection (Fig. S36a, ESI†), demonstrating the important role of RVG29 in achieving good brain-specific enrichment. The T2* MRI images also revealed that more RT-PPB NPs were accumulated in the brain (Fig. S36b, ESI†). Moreover, the RAPA concentrations in the brain were measured by LC-MS. The amount of drug accumulation in the brain of the RT-PPB group and the T-PPB group was 6.8% and 1.2% of the initial dose injected, respectively (Fig. S36c, ESI†). The immunofluorescence staining showed that the majority of RT-PPB NPs signal was co-localized with TH-labeled neurons (Fig. S36d, ESI†). The results indicated that RVG29 peptide could mediate the effective delivery of the RT-PPB NPs into the brain and target the neurons.
Next, the PD model mice were treated with the different groups by i.v. injection twice a week. The mice were monitored for 5 weeks and their behavior was evaluated weekly (Fig. 5a). The open field test and pole test were utilized to investigate the changes in exercise behavior. Weekly behavioral data reflected that treatment effects were time-dependent. Specifically, the increase of the total distance indicated that the RT-PPB NPs could significantly improve the exercise ability of the PD mice (Fig. 5b–d). The coordination ability of the PD mice treated with RT-PPB NPs was also obviously improved in the pole test (Fig. 5e and f). Taken together, these behavioral tests indicated the treatment effect of RT-PPB NPs in the MPTP-induced PD model mice.
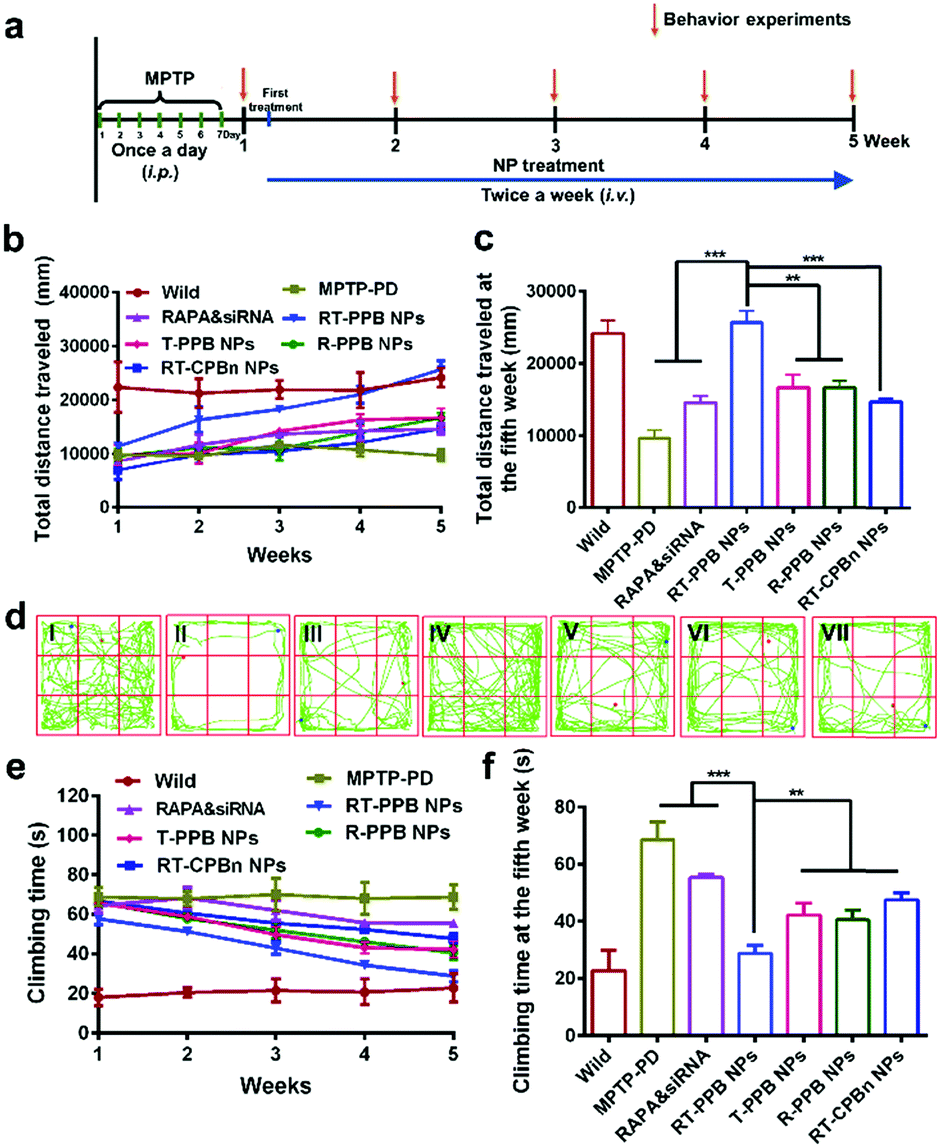 | ||
| Fig. 5 Behavior improvement of the MPTP-induced PD model mice (n = 5). (a) Experimental timeline for the construction of the MPTP-induced PD model mice and the administration of formulations. The 8-week-old male C57BL/6 mice were treated intraperitoneally with MPTP (30 mg kg−1) for 7 consecutive days and the exercise behaviour was tested. The MPTP-induced PD model mice were randomly grouped (n = 5) and treated with the different formulations (1 mg kg−1 siRNA and 3 mg kg−1 RAPA) by i.v. injection twice a week. (b) Changes in the total distance traveled in the open-field experiment within 5 weeks. (c and d) Total distance traveled in the open-field experiment at the fifth week. (e) Changes of the pole test score of the treated mice within 5 weeks. (f) Pole test score of the treated mice at the fifth week. (I) Wild control, (II) MPTP-PD control, (III) RAPA & sinSMase2, (IV) RT-PPB NPs, (V) T-PPB NPs, (VI) R-PPB NPs, and (VII) RT-CPBn NPs. Data are presented as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, Student's t-test. | ||
Western blot was employed to test the gene silencing performance in vivo, and the nSMase2 level with RT-PPB NP treatment was reduced most (Fig. 6a, b and Fig. S37, ESI†). We then detected the decrease of α-syn (red) in tyrosine hydroxylase (TH)-positive dopaminergic neurons (green) at the brain substantia nigra region of the mice treated with RT-PPB NPs. The overlap coefficient between the α-syn (red) and TH-positive neurons (green) was much lower than that for others (Fig. 6c and e). Additionally, the number of TH-positive neurons increased most at the brain substantia nigra region of the mice treated with RT-PPB NPs (Fig. 6d and f). Fig. 6g shows that the α-syn aggregate levels of brain tissues in all treatment groups were detected by ELISA assays. The RT-PPB NP group had the lowest level of α-syn aggregates.
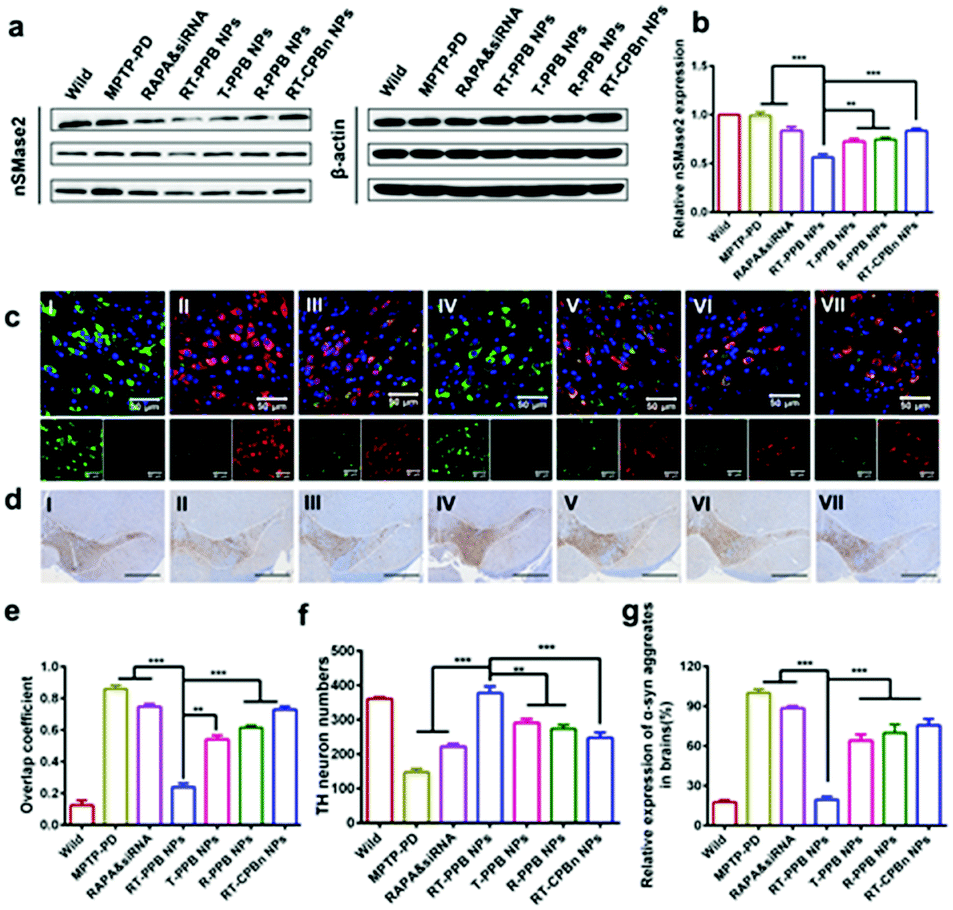 | ||
| Fig. 6 In vivo treatment effect of RT-PPB NPs. (a) Western blot and (b) quantified results of nSMase2 in the brains of mice with different treatment (n = 3). (c) Immunofluorescence staining of the treated mice brain substantia nigra region: nucleic (DAPI, blue), TH (green), and α-syn (red). (d) TH immunohistochemistry of brain slides in the treated mice substantia nigra region. Scale bar: 100 μm. (e) Overlap coefficient quantification of tyrosine hydroxylase (green) and α-syn (red) in Fig. 6c (n = 5). (f) TH-neuron numbers quantified from Fig. 6d by using Image-Pro Plus 6.0 software (n = 5). (g) The relative expression of α-syn aggregates in the brain was detected by ELISA (n = 5). Samples: (I) wild control, (II) MPTP-PD control, (III) RAPA & sinSMase2, (IV) RT-PPB NPs, (V) T-PPB NPs, (VI) R-PPB NPs, and (VII) RT-CPBn NPs. Data are presented as the mean ± SD. **P < 0.01, ***P < 0.001, Student's t-test. | ||
One of the important pathological features of synucleinopathies is that microglia would be activated.12,59 Fig. S38 (ESI†) shows that the number of microglia in the striatum area of the PD mice brain decreased after four weeks of treatment with RT-PPB NPs. The possible reason was that RT-PPB NPs could effectively reduce neuron-derived exosome transfer and alleviate PD pathology, resulting in depletion of microglia. These results indicated that the RT-PPB NPs indeed had the best synergy effect. The RT-PPB NPs could reduce the level of α-syn aggregates and their propagation, which significantly rescued the neurons and depleted the microglia. The biosafety of NPs was confirmed by hematoxylin and eosin staining. The major organs showed normal morphology and no lesions (Fig. S39, ESI†). The synergistic treatment not only took the diseased cells as the target, but also took into account the dysfunctional communication between the cells which played an important role in the disease development process.59,60
Conclusions
In this study, we designed evolved synaptic vesicle-mimicking RT-PPB NPs as the “nanoguard” to rescue the diseased neurons and guard the healthy cells. The RT-PPB NPs had the ability to overcome the drug delivery obstacles. Firstly, the NPs could permeate the BBB and dock to neurons to achieve drug enrichment. Secondly, the TMS-syb peptide, derived from SNARE syb proteins of SVs, drove the membrane fusion between NPs and the cell membrane, which could effectively avoid the lysosomal trapping, and achieve direct and effective enrichment of drugs in the cytoplasm. Thirdly, the drug core could simultaneously load rapamycin and sinSMase2 with completely different characteristics, and achieve controlled release in diseased neurons. The “nanoguard” significantly alleviated the synucleinopathy pathological features by simultaneously reducing the toxic aggregates in the diseased neurons and preventing them from escaping to affect other normal cells, and improved motor performance of the MPTP-induced PD mice. Actually, due to cell-to-cell transmission of neurotoxic proteins, the intercellular miscommunication, not just the neuronal miscommunication, is a common mechanism of the onset and progression for various neurodegenerative diseases.61,62 Hence, we provided an advanced platform for the treatment of synucleinopathies and other neurodegenerative diseases at different stages.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the Beijing Natural Science Foundation (L172046, 2192057), the National High Technology Research and Development Program (2016YFA0200303), the National Natural Science Foundation of China (21905283, 31771095, and 21875254), and the Beijing Nova Program (Z201100006820140).Notes and references
- K. Vekrellis, M. Xilouri, E. Emmanouilidou, H. J. Rideout and L. Stefanis, Lancet Neurol., 2011, 10, 1015 CrossRef CAS.
- E. Angot, J. A. Steiner, C. Hansen, J.-Y. Li and P. Brundin, Lancet Neurol., 2010, 9, 1128 CrossRef.
- Y. C. Wong and D. Krainc, Nat. Med., 2017, 23, 1 CrossRef CAS PubMed.
- G. A. Garden and A. R. La Spada, Neuron, 2012, 73, 886 CrossRef CAS PubMed.
- K. C. Luk, V. Kehm, J. Carroll, B. Zhang, P. O'Brien, J. Q. Trojanowski and V. M. Y. Lee, Science, 2012, 338, 949 CrossRef CAS PubMed.
- P. Desplats, H. J. Lee, E. J. Bae, C. Patrick, E. Rockenstein, L. Crews, B. Spencer, E. Masliah and S. J. Lee, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13010 CrossRef CAS PubMed.
- M. Danzer, L. R. Kranich, W. P. Ruf, O. Cagsal-Getkin, A. R. Winslow, L. Zhu, C. R. Vanderburg and P. J. McLean, Mol. Neurodegener., 2012, 7, 42 CrossRef PubMed.
- R. A. Nixon, Nat. Med., 2013, 19, 983 CrossRef CAS PubMed.
- D. C. Rubinsztein, P. Codogno and B. Levine, Nat. Rev. Drug Discovery, 2012, 11, 9 CrossRef PubMed.
- J. Bové, M. Martínez-Vicente and M. Vila, Nat. Rev. Neurosci., 2011, 12, 437 CrossRef PubMed.
- D. C. Rubinsztein, J. E. Gestwicki, L. O. Murphy and D. J. Klionsky, Nat. Rev. Drug Discovery, 2007, 6, 304 CrossRef CAS PubMed.
- H. Asai, S. Ikezu, S. Tsunoda, M. Medalla, J. Luebke, T. Haydar, B. Wolozin, O. Butovsky, S. Kügler and T. Ikezu, Nat. Neurosci., 2015, 18, 1584 CrossRef CAS PubMed.
- M. B. Dinkins, J. Enasko, C. Hernandez, G. Wang, J. Kong, I. Helwa, Y. Liu, A. V. Terry Jr and E. Bieberich, J. Neurosci., 2016, 36, 8653 CrossRef CAS PubMed.
- W. A. Banks, Nat. Rev. Drug Discovery, 2016, 15, 275 CrossRef CAS PubMed.
- D. Furtado, M. Björnmalm, S. Ayton, A. I. Bush, K. Kempe and F. Caruso, Adv. Mater., 2018, 30, 1801362 CrossRef PubMed.
- Y. Zhao, J. Cai, Z. Liu, Y. Li, C. Zheng, Y. Zheng, Q. Chen, H. Chen, F. Ma, Y. An, L. Xiao, C. Jiang, L. Shi, C. Kang and Y. Liu, Nano Lett., 2019, 19, 674 CrossRef.
- Q. Chen, Y. Du, K. Zhang, Z. Liang, J. Li, H. Yu, R. Ren, J. Feng, Z. Jin, F. Li, J. Sun, M. Zhou, Q. He, X. Sun, H. Zhang, M. Tian and D. Ling, ACS Nano, 2018, 12, 1321 CrossRef CAS.
- D. Kim, J. M. Yoo, H. Hwang, J. Lee, S. H. Lee, S. P. Yun, M. J. Park, M. Lee, S. Choi, S. H. Kwon, S. Lee, S. H. Kwon, S. Kim, Y. J. Park, M. Kinoshita, Y. H. Lee, S. Shin, S. R. Paik, S. J. Lee, S. Lee, B. H. Hong and H. S. Ko, Nat. Nanotechnol., 2018, 13, 812 CrossRef CAS PubMed.
- I. Javed, G. Peng, Y. Xing, T. Yu, M. Zhao, A. Kakinen, A. Faridi, C. L. Parish, F. Ding, T. P. Davis, P. C. Ke and S. Lin, Nat. Commun., 2019, 10, 3780 CrossRef PubMed.
- P. Chen, F. Ding, R. Cai, I. Javed, W. Yang, Z. Zhang, Y. Li, T. P. Davis, P. C. Ke and C. Chen, Nano Today, 2020, 35, 100937 CrossRef CAS PubMed.
- G. Sahay, W. Querbes, C. Alabi, A. Eltoukhy, S. Sarkar, C. Zurenko, E. Karagiannis, K. Love, D. Chen, R. Zoncu, Y. Buganim, A. Schroeder, R. Langer and D. G. Anderson, Nat. Biotechnol., 2013, 31, 653 CrossRef CAS PubMed.
- N. D. Donahue, H. Acar and S. Wilhelm, Adv. Drug Delivery Rev., 2019, 143, 68 CrossRef CAS PubMed.
- M. M. Holm, J. Kaiser and M. E. Schwab, Trends Neurosci., 2018, 41, 360 CrossRef CAS PubMed.
- R. B. Kelly, Cell, 1993, 72, 43 CrossRef.
- R. Jahn and R. H. Scheller, Nat. Rev. Mol. Cell Biol., 2006, 7, 631 CrossRef CAS PubMed.
- A. V. Pobbati, A. Stein and D. Fasshauer, Science, 2006, 313, 673 CrossRef CAS PubMed.
- M. J. Schnell, J. P. McGettigan, C. Wirblich and A. Papaneri, Nat. Rev. Microbiol., 2010, 8, 51 CrossRef CAS PubMed.
- P. Kumar, H. Wu, J. L. McBride, K. E. Jung, M. H. Kim, B. L. Davidson, S. K. Lee, P. Shankar and N. Manjunath, Nature, 2007, 448, 39 CrossRef CAS PubMed.
- L. Alvarez-Erviti, Y. Seow, H. Yin, C. Betts, S. Lakhal and M. J. Wood, Nat. Biotechnol., 2011, 29, 341 CrossRef CAS PubMed.
- J. A. Dani, D. Y. Ji and F. M. Zhou, Neuron, 2001, 31, 349 CrossRef CAS PubMed.
- D. Langosch, J. M. Crane, B. Brosig, A. Hellwig, L. K. Tamm and J. Reed, J. Mol. Biol., 2001, 311, 709 CrossRef CAS PubMed.
- J. P. Bearinger, S. Terrettaz, R. Michel, N. Tirelli, H. Vogel, M. Textor and J. A. Hubbell, Nat. Mater., 2003, 2, 259 CrossRef CAS.
- M. K. Gupta, T. A. Meyer, C. E. Nelson and C. L. Duvall, J. Controlled Release, 2012, 162, 591 CrossRef CAS PubMed.
- X. Liu, J. Xiang, D. Zhu, L. Jiang, Z. Zhou, J. Tang, X. Liu, Y. Huang and Y. Shen, Adv. Mater., 2016, 28, 1743 CrossRef CAS PubMed.
- X. C. Jiang, J. J. Xiang, H. H. Wu, T. Y. Zhang, D. P. Zhang, Q. H. Xu, X. L. Huang, X. L. Kong, J. H. Sun, Y. L. Hu, K. Li, Y. Tabata, Y. Q. Shen and J. Q. Gao, Adv. Mater., 2019, 31, 1807591 CrossRef PubMed.
- J. Park, K. An, Y. Hwang, J. G. Park, H. J. Noh, J. Y. Kim, J. H. Park, N. M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891 CrossRef CAS PubMed.
- B. Hu, F. Dai, Z. Fan, G. Ma, Q. Tang and X. Zhang, Adv. Mater., 2015, 17, 5499 CrossRef PubMed.
- X. Sun, G. Wang, H. Zhang, S. Hu, X. Liu, J. Tang and Y. Shen, ACS Nano, 2018, 12, 6179 CrossRef CAS PubMed.
- W. Shan, X. Zhu, M. Liu, L. Li, J. Zhong, W. Sun, Z. Zhang and Y. Huang, ACS Nano, 2015, 9, 2345 CrossRef CAS PubMed.
- R. Liu, J. Yang, L. Liu, Z. Lu, Z. Shi, W. Ji, J. Shen and X. Zhang, Adv. Sci., 2019, 7, 1901555 CrossRef PubMed.
- J. Y. Liu, C. Liu, J. F. Zhang, Y. M. Zhang, K. Y. Liu, J. X. Song, S. G. Sreenivasmurthy, Z. Y. Wang, Y. S. Shi, C. C. Chu, Y. Zhang, C. S. Wu, X. H. Deng, X. Y. Liu, J. Song, R. Q. Zhuang, S. G. Huang, P. F. Zhang, M. Li, L. Wen, Y. W. Zhang and G. Liu, ACS Nano, 2020, 14, 1533 CrossRef CAS PubMed.
- Y. Liu, R. Huang, L. Han, W. Ke, K. Shao, L. Ye, J. Lou and C. Jiang, Biomaterials, 2009, 30, 4195 CrossRef CAS PubMed.
- Y. Jiang, J. Zhang, F. H. Meng and Z. Y. Zhong, ACS Nano, 2018, 12, 11070 CrossRef CAS PubMed.
- Y. Omidi, L. Campbell, J. Barar, D. Connell, S. Akhtar and M. Gumbleton, Brain Res., 2003, 990, 95 CrossRef CAS PubMed.
- Z. G. Lu, Y. Li, Y. J. Shi, Y. H. Li, Z. B. Xiao and X. Zhang, Adv. Funct. Mater., 2017, 27, 1703967 CrossRef.
- L. Yin, Z. Song, K. H. Kim, N. Zheng, H. Tang, H. Lu, N. Gabrielson and J. Cheng, Biomaterials, 2013, 34, 2340 CrossRef CAS PubMed.
- D. Nie, Z. Dai, J. L. Li, Y. W. Yang, Z. Y. Xi, J. Wang, W. Zhang, K. Qian, S. Y. Guo, C. L. Zhu, R. Wang, Y. M. Li, M. R. Yu, X. X. Zhang, X. H. Shi and Y. Gan, Nano Lett., 2020, 20, 936 CrossRef CAS PubMed.
- K. Trajkovic, C. Hsu, S. Chiantia, L. Rajendran, D. Wenzel, F. Wieland, P. Schwille, B. Brügger and M. Simons, Science, 2008, 319, 1244 CrossRef CAS.
- C. Théry, L. Zitvogel and S. Amigorena, Nat. Rev. Immunol., 2002, 2, 569 CrossRef.
- M. S. Sinha, A. Ansell-Schultz, L. Civitelli, C. Hildesjö, M. Larsson, L. Lannfelt, M. Ingelsson and M. Hallbeck, Acta Neuropathol., 2018, 136, 41 CrossRef.
- C. Peng, J. Q. Trojanowski and V. M. Y. Lee, Nat. Rev. Neurol., 2020, 16, 199 CrossRef CAS.
- R. E. Musgrove, M. Helwig, E. J. Bae, H. Aboutalebi, S. J. Lee, A. Ulusoy and D. A. Di Monte, J. Clin. Invest., 2019, 129, 3738 CrossRef.
- N. Uemura, M. T. Uemura, K. C. Luk, V. M. Lee and J. Q. Trojanowski, Trends Mol. Med., 2020, 26, 936 CrossRef CAS PubMed.
- M. Beekes, A. Thomzig, W. Schulz-Schaeffer and R. Burger, Acta Neuropathol., 2014, 128, 463 CrossRef CAS PubMed.
- J. B. Koprich, L. V. Kalia and J. M. Brotchie, Nat. Rev. Neurosci., 2017, 18, 515 CrossRef CAS.
- H. J. Kwon, D. Kim, K. Seo, Y. G. Kim, S. I. Han, T. Kang, M. Soh and T. Hyeon, Angew. Chem., Int. Ed., 2018, 57, 9408 CrossRef CAS.
- M. Beekes, A. Thomzig, W. Schulz-Schaeffer and R. Burger, Behav. Brain Res., 2010, 211, 1 CrossRef.
- M. F. Chesselet and F. Richter, Lancet Neurol., 2011, 10, 1108 CrossRef.
- M. Guo, J. Wang, Y. Zhao, Y. Feng, S. Han, Q. Dong, M. Cui and K. Tieu, Brain, 2020, 143, 1476 CrossRef.
- J. L. Guo and V. M. Y. Lee, Nat. Med., 2014, 20, 130 CrossRef CAS.
- J. Hardy and T. Revesz, N. Engl. J. Med., 2012, 366, 2126 CrossRef CAS PubMed.
- S. J. Lee, P. Desplats, C. Sigurdson, I. Tsigelny and E. Masliah, Nat. Rev. Neurol., 2010, 6, 702 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, experimental details, and additional data. See DOI: 10.1039/d0mh01542c |
| ‡ W. J. and Y. L. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |