
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of molecular flexibility and head group repulsion on aramid amphiphile self-assembly†
Samuel J.
Kaser‡
a,
Andrew J.
Lew‡
 a,
Dae-Yoon
Kim
b,
Ty
Christoff-Tempesta
c,
Yukio
Cho
c and
Julia H.
Ortony
*c
a,
Dae-Yoon
Kim
b,
Ty
Christoff-Tempesta
c,
Yukio
Cho
c and
Julia H.
Ortony
*c
aDepartment of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Ave, Cambridge, MA 02139, USA
bInstitute of Advanced Composite Materials, Korea Institute of Science and Technology (KIST), 92 Chudong-Ro, Bongdong-Eup, Wanju-Gun, Jeonbuk 55324, Republic of Korea
cDepartment of Materials Science and Engineering, Massachusetts Institute of Technology, 77 Massachusetts Ave, Cambridge, MA 02139, USA. E-mail: ortony@mit.edu
First published on 28th October 2021
Abstract
The self-assembly of amphiphilic molecules in water has led to a wide variety of nanostructures with diverse applications. Many nanostructures are stabilized by strong interactions between monomer units, such as hydrogen bonding and π–π stacking. However, the morphological implications of these strong, anisotropic interactions can be difficult to predict. In this study, we investigate the relationships between molecular flexibility, head group repulsion, and supramolecular geometry in an aramid amphiphile nanostructure that is known to exhibit extensive hydrogen bonding and π–π stacking – features that give rise to their unusual stability. We find by electron microscopy that increasing backbone flexibility disrupts molecular packing into high aspect-ratio nanoribbons, and at the highest degree of flexibility long-range ordering is lost. Even when backbone rigidity favors tight packing, increasing head group charge through pH-modulation leads to intermolecular electrostatic repulsion that also disrupts close packing. Spectroscopic measurements suggest that these changes are accompanied by disruption of π–π stacking but not hydrogen bonding. Backbone rigidity and head group repulsion are thus important design considerations for controlling internal stability and nanostructure curvature in supramolecular assemblies stabilized by π–π stacking interactions.
Design, System, ApplicationControl over nanomorphology is important for biomedical, energy, and environmental applications of self-assembled small molecules in water, but there is a dearth of knowledge about how intermolecular interactions interplay with molecular features to determine nanostructure. Aramid amphiphiles were investigated in this study as an example of a self-assembling molecular system containing strong and anisotropic intermolecular forces, i.e. hydrogen bonding and π–π stacking. We synthesized aramid amphiphiles with varying backbone flexibility and adjusted their head group repulsion by modulating pH. Through electron microscopy, spectrophotometric studies, and acid–base titrations, we provide evidence that rigidity of individual molecules corresponds to nanostructures of lower curvature and that at the highest molecular flexibility, long-range ordering is lost. We conclude that π–π stacking occurs most effectively when the constituent molecules exhibit rigid backbones, and that the nanostructures of rigid amphiphiles are also more sensitive to electrostatic interactions of the head groups. These findings suggest that backbone rigidity could play a significant role in other self-assembled systems with built-in strong intermolecular interactions, and offer design parameters to achieve pH-sensitive nanostructures with a range of architectures. |
1. Background
Self-assembly of amphiphilic small molecules in water can generate highly-ordered nanoscale structures.1–3 Such nanostructures have become important materials in regenerative medicine and drug delivery due to their modular design, ease of synthesis, and biocompatibility.4 Traditionally, self-assembly is driven by the hydrophobic effect, with the hydrophilic domains providing solubility and a low-energy interface of defined geometry.5 Equilibrium nanoarchitecture is determined by the structure of both the hydrophobic and hydrophilic domains. In these assemblies, isotropic intermolecular interactions impose predictable nanoscale geometries. However, the weakness of these interactions also leads to dynamic instabilities, wherein complete monomer exchange occurs within minutes to hours.6–8 As a consequence of this dynamic instability, amphiphile nanostructures generally dissociate upon drying, sometimes limiting their application scope.9 The introduction of stronger, anisotropic intermolecular interactions into these supramolecular ensembles is thus a key strategy for stabilizing their nanoarchitectures and expanding the utility of this class of materials.10A prevalent strategy to increase intermolecular cohesion in small molecule self-assembly is to incorporate hydrogen bonding, π–π interactions, or both into the amphiphile design.11–13 One such design motif is the aramid amphiphile (AA), which produces unusually stable self-assembled nanostructures by incorporating aromatic amide (aramid) domains into the molecular structure.14–16 The prototypical AA is composed of a charged head group, a triaramid structural domain, and a short aliphatic tail (Fig. 1).14 When added to water, these AAs spontaneously self-assemble to form high aspect-ratio nanoribbons with 4 nm thicknesses, 5 nm widths, and 20+ μm lengths. The aramid structural domain imparts strong and collective hydrogen bonding down the long axis of the nanoribbon, while π–π stacking is responsible for lateral cohesion. These intermolecular interactions endow self-assembled AA nanoribbons with undetectably low molecular exchange rates, enhanced mechanical properties (with strength and Young's modulus on the order of GPa), and air stability.14
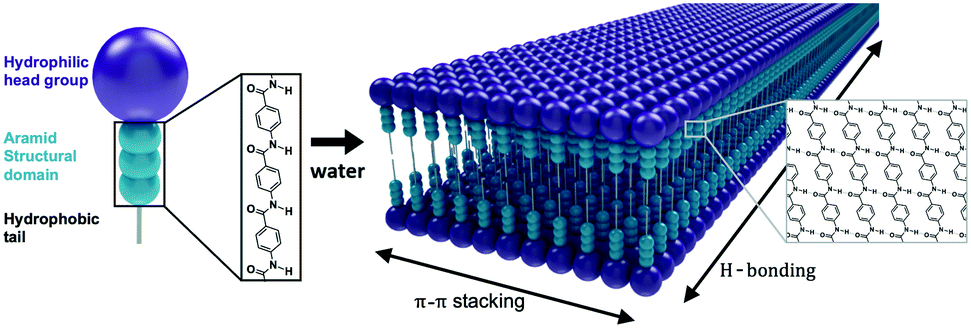 | ||
| Fig. 1 Aramid amphiphiles contain an aromatic amide “aramid” structural domain (turquoise), a charged head group (purple), and an aliphatic tail. Aramid amphiphiles spontaneously self-assemble upon addition of water. Anisotropic π–π stacking and hydrogen bonding interactions occur between molecules, producing stable high aspect-ratio nanoribbons with 4 nm thicknesses, 5 nm widths, and 20+ μm lengths. These graphics are not to scale, and a distribution of nanoribbon widths is expected. | ||
The relationship between molecular design and nanostructure morphology of simple surfactant self-assembly is conveniently described by the classical Israelachvili model.5 This model defines a critical packing parameter, p, that describes the respective cross-sectional areas of the hydrophilic head and hydrophobic tail of the constituent molecules. As the critical packing parameter approaches zero, molecular cone angle increases, and the overall nanoarchitecture has a greater degree of curvature – favoring spheres, as opposed to cylinders or lamellae.
While the Israelachvili packing parameter model is valuable for its elegance and simplicity, the introduction of strong and directional intermolecular interactions can give rise to geometries that the model cannot predict. As a result, rational design of self-assembling nanoarchitectures of amphiphiles with strong hydrogen bonding requires advanced computational molecular dynamics simulations.17 In fact, the introduction of hydrogen bonding and π–π stacking can produce exotic and unpredictable geometries like helices,18–20 twisted nanoribbons,19,21 and 3D cages.22
An outstanding objective is to better understand the relationships between molecular design and ultimate morphological outcomes. This goal is important because subtle differences in nanoarchitecture can have substantial consequences to applications. For example, the degree of curvature of lipid nanoparticles regulates cytotoxicity in drug delivery applications,23 high aspect-ratios are important for control over nanostructure gelation,24,25 and shape control regulates the self-assembly and conductivity of nanoscale organic optoelectronic devices.26–28
AAs were studied as an example of a nanostructure whose ordering relies on strong intermolecular forces - specifically, π–π stacking and hydrogen bonding interactions. While the AA platform produces nanostructures with a high degree of internal cohesion, there is so far no systematic description of how AA backbone design features influence nanoarchitecture. Here, we introduce varying degrees of flexibility into the molecular structure of an AA through the incorporation of alternating alkyl spacers of variable lengths and we modify the electrostatic interactions of the head groups by adjusting pH. We demonstrate self-assembly of these molecules in water and investigate the effects of these systematically varied molecular features on nanostructure morphology. Finally, changes in the intermolecular interactions underlying morphological changes with pH modulation were investigated through UV-vis and ATR-FTIR spectroscopy.
2. Results and discussion
We synthesized aramid amphiphiles (AAs) with structural domains containing an ABABA motif, where A is the aramid moiety and B is a flexible aliphatic group. This molecular design strategy was implemented to modify the flexibility of AA molecules. Fig. 2 shows the chemical structures of compounds 1, 2, and 3, where the head and tail groups remain unchanged, but the flexibility of the structural domain varies. In compound 2, propanamido groups in the structural domain introduce additional rotational freedom compared to compound 1. In compound 3, butanamido groups are incorporated into the chemical structure, leading to the greatest molecular flexibility of the three compounds.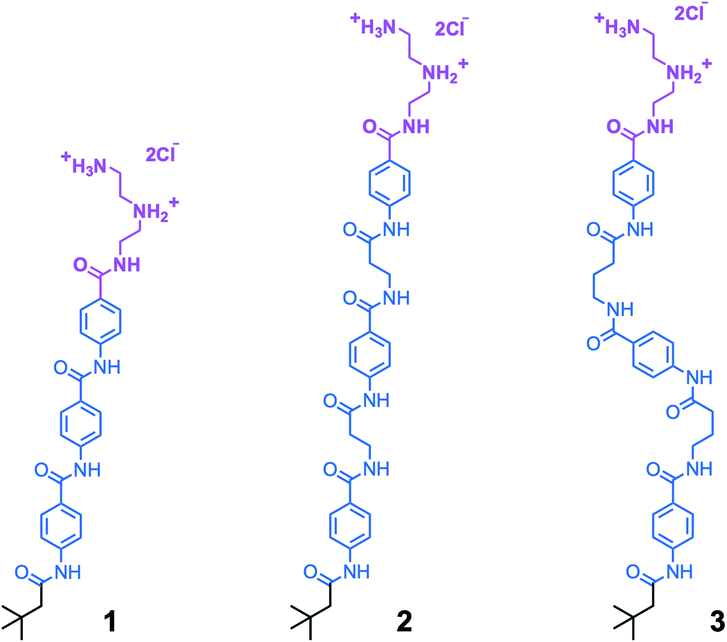 | ||
| Fig. 2 The aramid amphiphiles with variable backbone flexibility investigated in this study, where compound 1 is the most rigid, 2 is more flexible, and 3 is the most flexible. Each comprises a cationic triazaheptane head group (magenta), an aramid structural domain (blue), and a short, branched alkyl tail (black). | ||
The introduction of sp3 hybridized carbon atoms into the aramid backbone has implications for molecular structure besides greater flexibility. Namely, the aliphatic groups introduce the possibility of orientational disruption to the hydrogen bonding network or changes to the planarity of the molecules within the nanostructures. However, it should be possible to resolve the relative contribution of molecular flexibility to nanomorphological outcomes despite these effects: the number of carbon atoms in compound 2 should preserve the good intermolecular amide bond alignment observed previously in compound 1,29 and the alkyl groups of the head and tail groups preclude a perfectly coplanar molecular conformation for all three compounds.
Transmission electron microscopy (TEM) was used to image compounds 1–3 dried from water at neutral pH to study their self-assembly behavior. Acidic and basic suspensions were examined as well to modulate head group electrostatic repulsion and probe the contribution of head group charge to overall nanomorphology.
Fig. 3 illustrates the self-assembly of compound 1, and its sensitivity to changes in pH. At pH 3, when the amphiphiles are expected to exhibit the highest degree of head group protonation, we observe that compound 1 forms tightly coiled structures in water after brief bath sonication, as shown in Fig. 3a. Upon further sonication, these coils fuse along the edges to form nanotubes, shown in Fig. 3b, via a previously reported mechanism.16 At neutral pH, as shown in Fig. 3c and described previously,14 compound 1 forms nanoribbons, and at high pH, where electrostatic repulsion is minimized, disordered planar aggregates are observed (Fig. 3d). No fundamental morphological changes were observed with longer sonication at pH 7 or 11 (ESI† Fig. S1a and b).
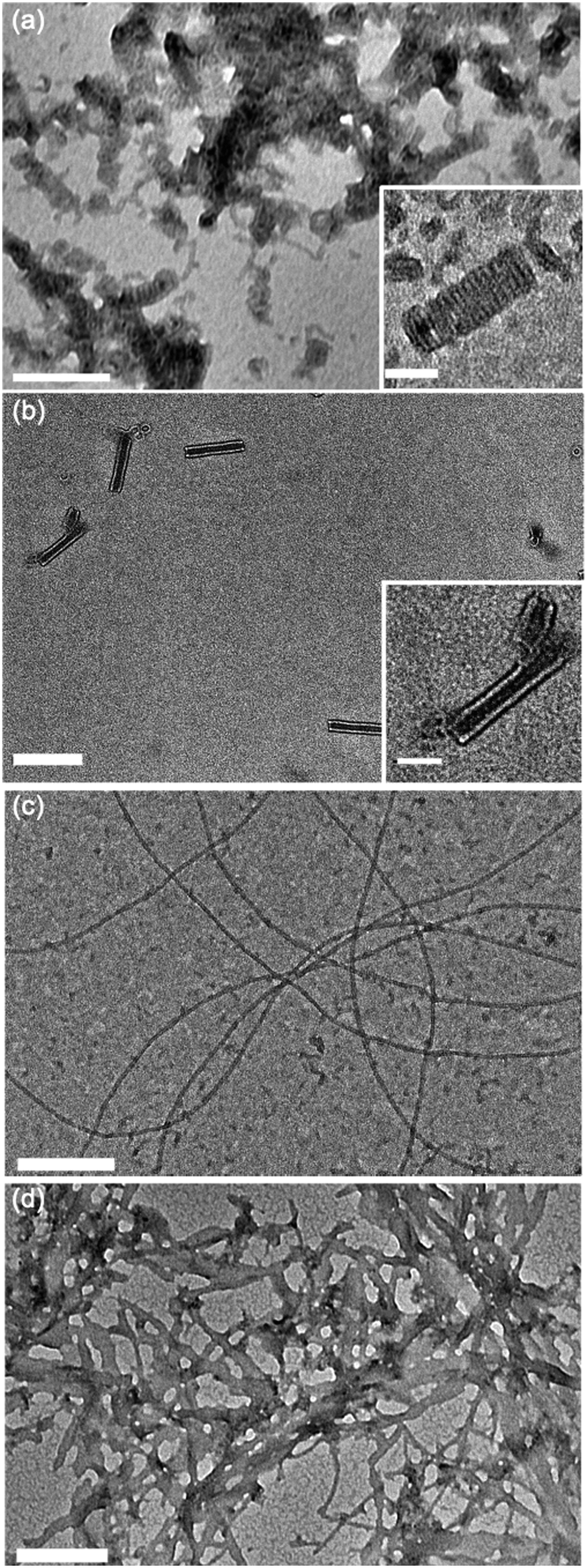 | ||
| Fig. 3 Transmission electron micrographs of compound 1 dried from water under various aqueous self-assembly conditions. (a) pH 3, (b) pH 3, sonicated for 24 hours for full equilibration, (c) pH 7, (d) pH 11. Main scale bar = 200 nm, inset scale bar = 50 nm. | ||
Aqueous suspensions of compound 2 were prepared under the same conditions as compound 1 to investigate the effects of introducing propanamido groups into the AA chemical structure. Nanotube formation was observed for compound 2 under all pH values investigated (Fig. 4). In contrast with compound 1 at pH 3, compound 2 forms short tube fragments immediately upon self-assembly rather than passing through an intermediate coil architecture. This observation suggests that when propanamido groups are introduced between the aramids, nanotube formation occurs via different pathways compared to assembly of the triaramid amphiphile, compound 1. Longer sonication of compound 2 did not induce deviation from the observed tube geometry under any pH condition (ESI† Fig. S1c–e).
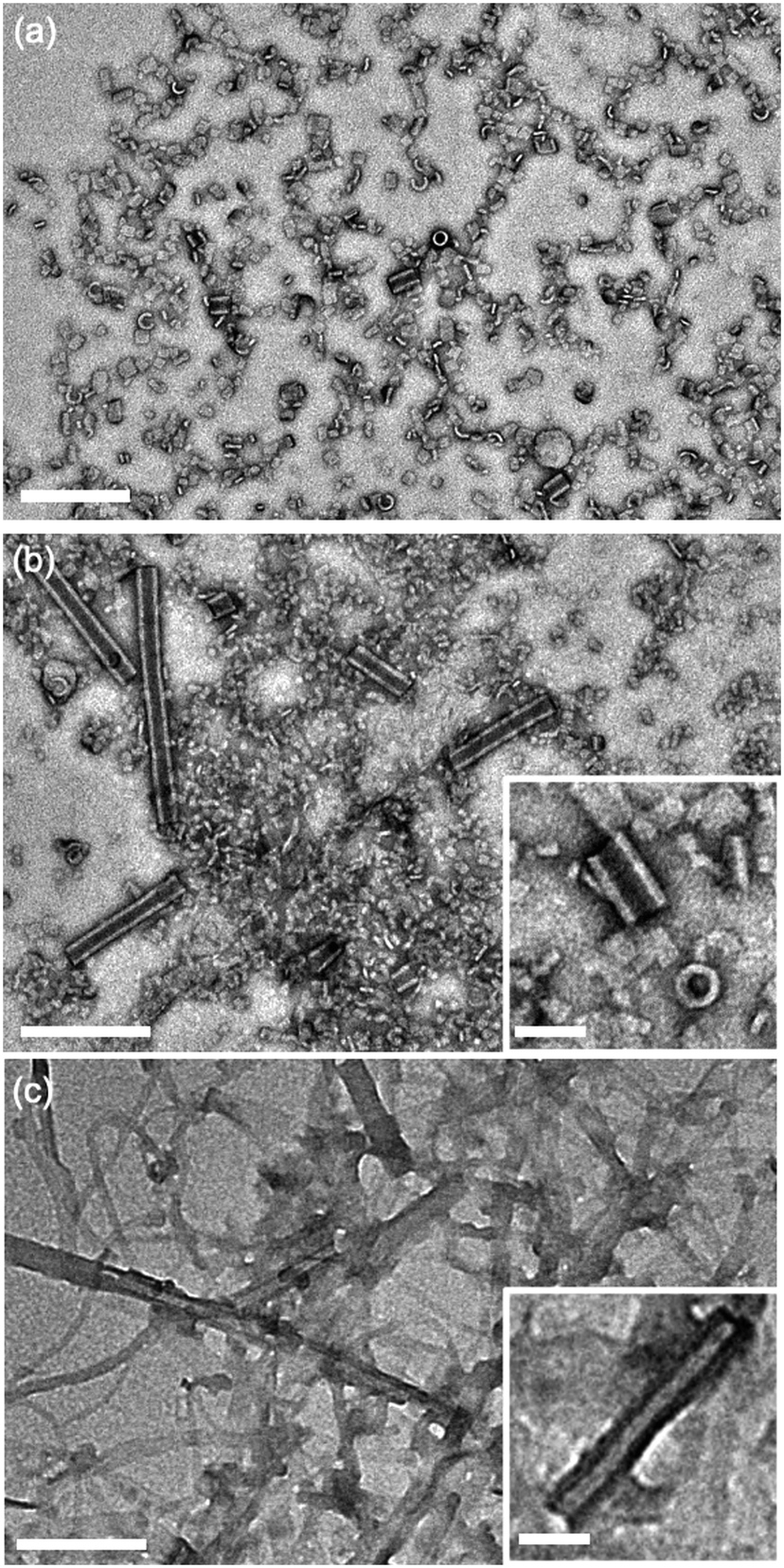 | ||
| Fig. 4 Transmission electron micrographs of compound 2 dried from water following self-assembly at varying pH values. (a) pH 3, (b) pH 7, (c) pH 11. Main scale bar = 200 nm, inset scale bar = 50 nm. | ||
TEM images were also recorded for compound 3 at pH 3, 7, and 11 to investigate the role of an even more flexible aramid amphiphile, with two flexible butanamido groups integrated into the AA chemical structure. Under acidic conditions, disordered aggregates were observed (Fig. 5a), while lamellar plaques appear to be visible under neutral and basic conditions (Fig. 5b and c). The observation of less-well-defined nanoscale geometries for suspensions of compound 3 at each pH is attributed to the high flexibility of its AA backbone. Specifically, the disorder imposed by backbone flexibility disrupts the anisotropic nature of AA hydrogen bonding and π–π stacking interactions and therefore reduces their order-imposing effects. As with compound 2, no noteworthy changes in morphology occurred after 24 hours of sonication in any pH condition studied (ESI† Fig. S1f–h).
 | ||
| Fig. 5 Transmission electron micrographs of compound 3 dried from water following self-assembly at varying pH values. (a) pH 3, (b) pH 7, (c) pH 11. Main scale bar = 200 nm, inset scale bar = 50 nm. | ||
The difference in self-assembly behavior between compounds 1–3 motivated further investigations into the underlying driving forces producing these discrepancies. Previous studies have demonstrated aggregation-induced changes in pKa triggered by energetic penalties from electrostatic repulsion.30,31 Titrations were therefore carried out to investigate the role of pH-modulated head group repulsion on the nanostructures formed by each AA, and a titration of the free head group triazaheptane (TAH) was carried out as a control.
Titration curves were acquired by acidifying aqueous suspensions of compounds 1–3 to a pH of about 3.3, then adding aliquots of an NaOH solution until a final pH of 10.5 was reached. These titration data were used to determine pKa values for each AA through visual analysis of horizontal inflection points.
Fig. 6a illustrates the titration curves of nanostructure suspensions of compounds 1–3 in water, with the curves offset along the x-axis for clarity. Single buffer regions are observed for compounds 1 and 2, with pKa values of 6.4 and 6.1, respectively. In contrast, no obvious buffer region appears in the titration of compound 3, likely due to lower effective AA concentration resulting from poor solubilization of the nanostructure head groups. This observation is consistent with the aggregation behavior of compound 3, especially at low pH, where disordered structures are observed by TEM. Replicates of these titrations for compounds 1 and 2 were carried out and produced the same pKa values (ESI† Fig. S2).
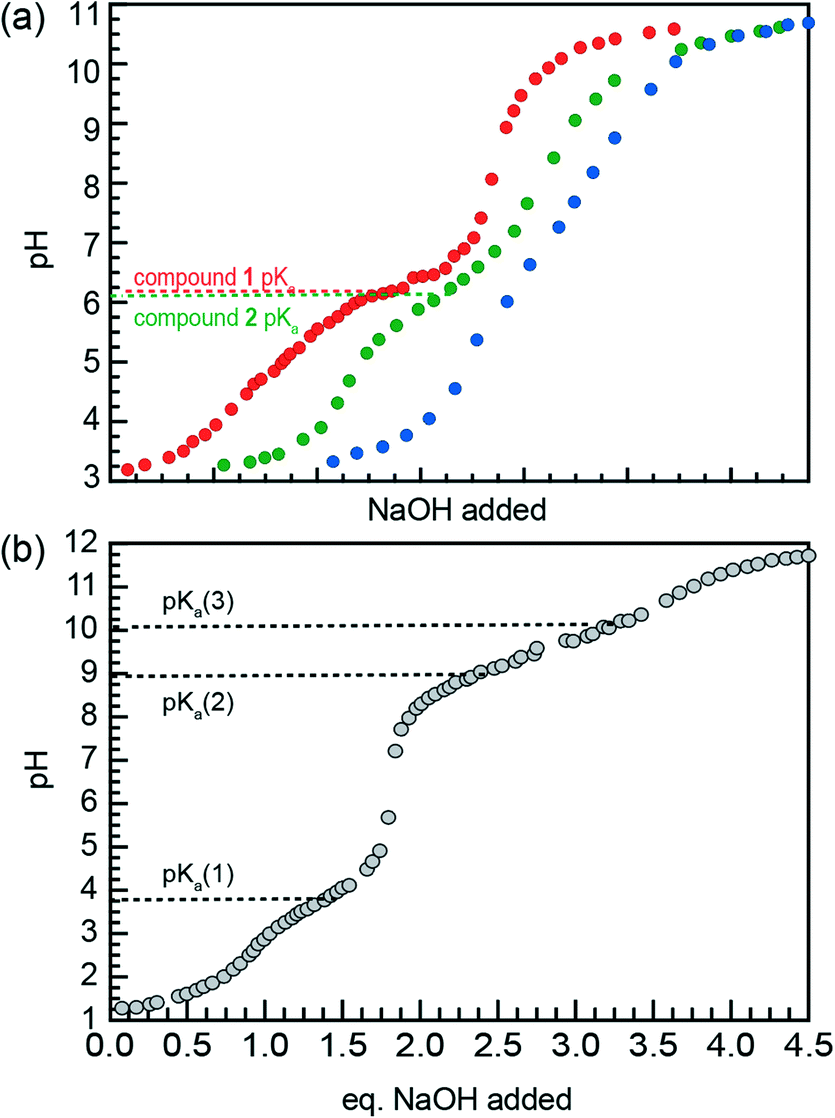 | ||
| Fig. 6 (a) Titration of acidified suspensions of compounds 1 (red), 2 (green), and 3 (blue) with 5.0 mM NaOH (aq). Curves are offset along the x-axis for clarity. Each titration was carried out until 3 equivalents of NaOH were added. (b) Titration of acidified triazaheptane with 45 mM NaOH serves as a control for the head groups of compounds 1–3. pKa values were approximated by visual analysis of horizontal inflection points. | ||
Titration of the fully solubilized TAH control (Fig. 6b) shows pKa values of 4.2, 8.9, and 10.1. While fully acidified TAH is triprotic, the head groups of compounds 1–3 are diprotic due to their attachment to the AA backbone via amide bond. Thus, the acid–base equilibria described by the pKa values of AA nanostructures are comparable to pKa(2) of TAH, as shown in Fig. 6b. Therefore, Fig. 6 illustrates that the pKa value of AA nanostructures is substantially less than the comparable pKa of the TAH control.
The observed decrease in pKa for AA nanostructures compared to free TAH reflects a destabilization of head group protons after self-assembly due to electrostatic repulsion from enforced close proximity. The pKa values observed for compounds 1 and 2 are both 6.1. Therefore, the more flexible molecular structure of compound 2 offers no additional head group stabilization compared to that of 1.
Based on this result, we hypothesize that both AAs 1 and 2 are forced into the tube architecture when efficient packing promoted by π–π stacking is impossible due to head group repulsion (compound 1, pH 3), molecular flexibility (compound 2, pH 7), or both (compound 2, pH 3). These results indicate that a rigid backbone is crucial for the imposition of order by π–π stacking interactions. This conclusion is consistent with the comparison of pre-equilibration TEM images of compound 1 at pH 3 (Fig. 3a) and compound 2 at neutral pH (Fig. 4b), with the coil-to-tube transition observed for compound 1 suggesting π–π stacking along the short axis of the nanostructure can occur but a tube geometry is ultimately thermodynamically favorable. Meanwhile, compound 2 forms tube fragments after brief sonication, suggesting that efficient π–π stacking to form nanoribbons is never achieved.
To test this hypothesis, we measured UV-vis absorbance spectra for each compound at pH 3, 7, and 11 to compare π–π stacking behavior between compounds 1–3 (Fig. 7). Aromatic moieties absorb strongly between wavelengths of 240 and 340 nm and absorption spectroscopy is thus a valuable tool for studying perturbations to the electronic environment of aramids.32 In particular, shifts in the wavelength of peak intensity (λmax) allow us to make inferences about π–π stacking, with closer and more optimized π–π stacking giving rise to hypsochromic (blue) shifts in the absorbance maxima.33
 | ||
| Fig. 7 UV-visible light absorbance spectra of AA assemblies in water. Spectra of compounds 1–3 (a–c, respectively) are shown at three pH values: red = pH 3, purple = pH 7, blue = pH 11. | ||
Fig. 7 shows absorption spectra for compounds 1–3 at pH 3, 7, and 11. For compound 1 (Fig. 7a), a hypsochromic shift of 17 nm in λmax is observed from pH 3 to pH 7, corresponding to the nanotube to nanoribbon transition observed in TEM. In contrast, the λmax of compound 1 remains the same between neutral and basic conditions (Fig. 7a), when nanoribbons and lamellar aggregates are observed, respectively. For compound 2, where the presence of nanotubes is insensitive to pH, a modest 5 nm shift in λmax is observed between acidic and neutral conditions. A subtle (1 nm) bathochromic shift was observed from neutral to basic conditions (Fig. 7b). Finally, no hypsochromic shift occurs from acidic to neutral conditions for compound 3 (Fig. 7c), where disordered aggregates transition to lamellar plaques. This observation suggests that π–π interactions are weak in both pH conditions, and thus that any changes in their strength with modulation of head group repulsion over this range are too slight to be detected by UV–vis absorption spectroscopy. Basic conditions, however, produced a roughly 8 nm hypsochromic shift from pH 7. This shift may correspond to the increase in planarity seen for compound 3 nanostructures in basic aqueous suspension (Fig. 5c). In addition, the absorption spectrum for compound 3 at pH 11 is very broad, suggesting that packing is not uniform throughout the nanostructures. This reflects poor ordering for compound 3 nanostructures under these conditions.
We observe that increasing pH generally leads to a hypsochromic shift in the absorbance spectra of nanostructures of all three compounds, 1, 2, and 3, reflecting the competition between head group electrostatic repulsion and backbone π–π stacking efficiency. Compound 1 shows a particularly prominent hypsochromic shift from pH 3 to pH 7 (see Fig. 7a), indicating that an acidic environment, and the tube formation it induces, dramatically disrupts π–π stacking.
In contrast to compound 1, the absorbance spectrum of compound 2 shows minor shifts across the pH range investigated, reflecting its persistent nanotube morphology. The much larger hypsochromic shift shown between acidic and neutral pH for compound 1 compared to compound 2 is consistent with the hypothesis. Namely, a nanoribbon morphology is produced by effective π–π stacking and a tube morphology occurs upon the disruption of this stacking.
Finally, attenuated total reflection Fourier transformed infrared (ATR-FTIR) transmittance spectra were measured to probe the roles of backbone flexibility and head group repulsion on hydrogen bonding. Infrared spectroscopy can be used to elucidate information about bond strength, with changes in the strength of hydrogen bonds to amide moieties leading to predictable changes in C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond stretch absorption frequency.34 Samples were sonicated for 24 hours in heavy water, then diluted to acidic, neutral, and basic pH values. IR spectra for each compound in dimethyl sulfoxide (DMSO) were also collected as a control condition where no self-assembly occurs.
O bond stretch absorption frequency.34 Samples were sonicated for 24 hours in heavy water, then diluted to acidic, neutral, and basic pH values. IR spectra for each compound in dimethyl sulfoxide (DMSO) were also collected as a control condition where no self-assembly occurs.
Fig. 8 shows ATR-FTIR spectra of compounds 1–3 dissolved in DMSO (black) and following overnight sonication in D2O and subsequent pH adjustment (red, purple, and blue). For compound 1 (Fig. 8a), the aqueous solutions show narrowing of the broad amide CO peak in the DMSO spectrum to produce defined peaks at 1638 and 1673 cm−1, suggesting increased uniformity of hydrogen bonding conditions through molecular self-assembly. This assembly-based spectral transformation has been observed previously for compound 1.14 Moreover, the peak height ratios at 1673 and 1638 cm−1 and amide region lineshapes in general are very similar between the aqueous samples at all three pH values, indicating that changes in head group repulsion have little effect on the number or strength of intermolecular hydrogen bonds.
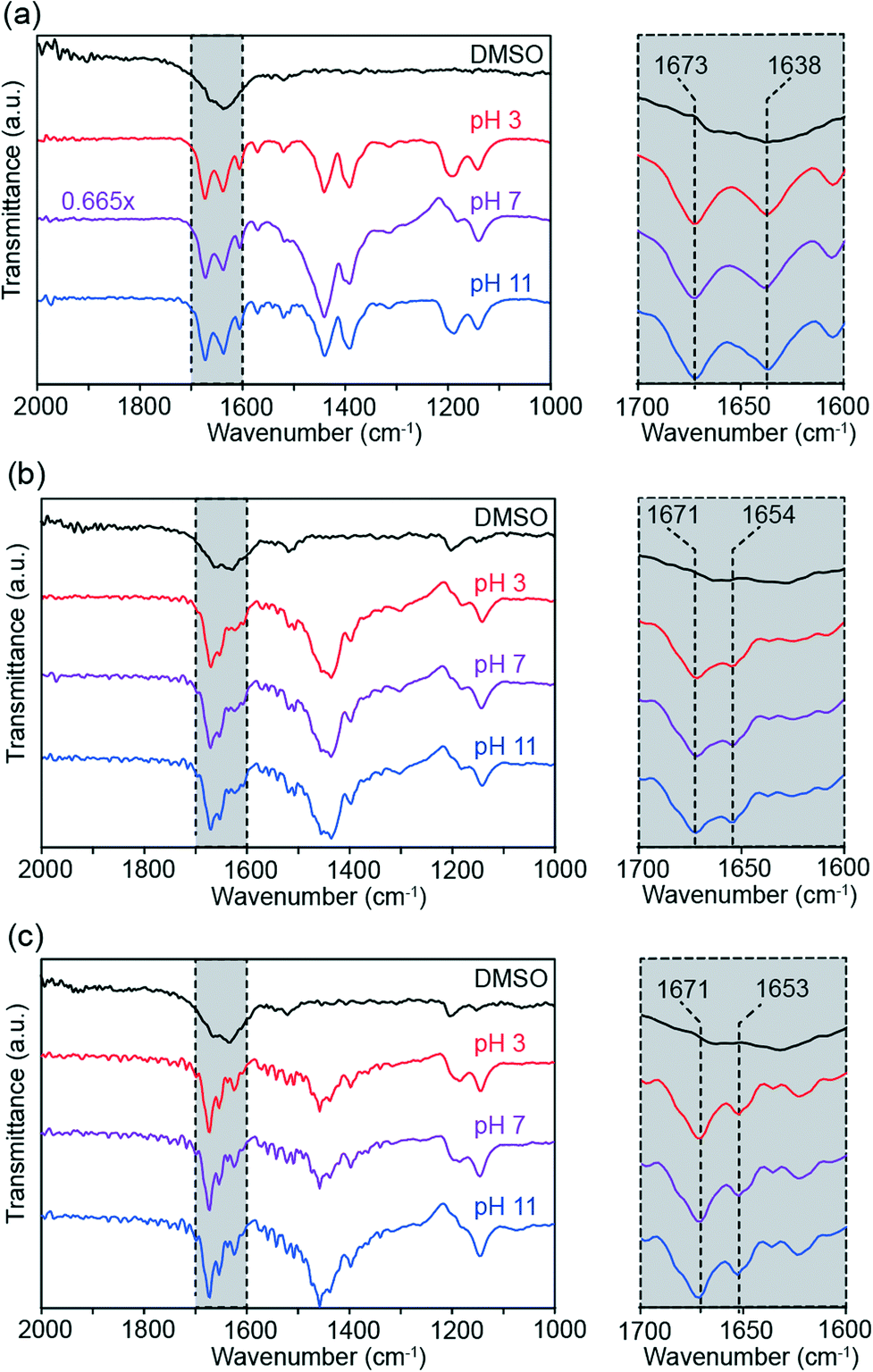 | ||
| Fig. 8 Solution-phase ATR-FTIR spectra of compounds 1–3 (a–c, respectively). Each compound was assembled in D2O at pH 3 (red), pH 7 (purple), and pH 11 (blue). The black curves represent absorption spectra for each compound dissolved in DMSO. Scaling factors for spectra are reported on the left, otherwise the spectra are unscaled. Insets of the amide bond stretch region are shown in grey. | ||
Just as seen for compound 1, self-assembly sharpens the broad amide peak for compound 2 in DMSO into well-defined peaks (Fig. 8b). The IR absorbance maxima for compound 2 occur at 1654 and 1671 cm−1, in contrast with compound 1. These shifts in peak position between compounds are attributed to the change in molecular electronic environment with the introduction of the aliphatic linkers, rather than differences in hydrogen bonding. Fig. 8b shows effectively identical amide CO stretch lineshapes across pH conditions, again suggesting that head group repulsion changes do not affect internal hydrogen bonding. The highly uniform ATR-FTIR spectra between compounds are also consistent with the persistence of tube morphology across all pH conditions.
As seen in the other compounds, aqueous self-assembly of compound 3 produces narrow, well-defined peaks in the amide CO stretch region that are consistent across pH values (Fig. 8c), indicating the formation of an organized network of intermolecular hydrogen bonds within compound 3 nanostructures. These sharp, pH-independent peaks are observed despite changes to hydrogen bond orientation compared to compound 2 and the lack of defined nanoscale geometries observed in TEM images of compound 3 (Fig. 5). Transmittance minima occur at 1653 and 1671 cm−1. While the nanostructures of compound 3 differ significantly from those formed by both compounds 1 and 2, compound 3's CO stretch frequencies closely resemble those of 2. This observation reaffirms that the discrepancy between CO stretch frequencies between compounds 1 and 2 is primarily a consequence of changes in electronic structure, rather than nanomorphology.
3. Conclusions
This study offers that (1) the degree of molecular flexibility of amphiphiles and (2) head group repulsion are parameters that may be varied for tuning morphological outcomes of small molecule self-assembly in water. The results of this study indicate that increasing molecular flexibility in the aramid amphiphile backbone inhibits efficient packing into planar nanoribbons through disruption of π–π stacking interactions. Notably, the introduction of propanamido spacers into a triaramid amphiphile leads to the formation of nanotubes in water at neutral pH, whereas a rigid backbone gives rise to nanoribbons. This result defies the Israelachvili critical packing parameter model, which does not predict that an increase in backbone length will favor geometries of greater curvature. We also observe a greater pH sensitivity when the backbone is rigid, as a consequence of close packing of the head groups. This relationship between pH sensitivity and backbone rigidity provides design parameters for future pH-modulated nanostructures.We further increased the length of the spacers between aramid groups in order to impose even greater molecular flexibility within the nanostructures. This modification results in the disruption of order-imposing anisotropic intermolecular forces and leads to nanostructures with less well-defined geometries.
Spectroscopic analysis (UV-Vis and ATR-FTIR) demonstrated that the more dramatic morphological changes observed through pH adjustment were accompanied by significant changes in the strength of π–π stacking interactions, but not in the strength or extent of hydrogen bonding interactions. This result implies that π–π stacking is more sensitive to increased head group repulsion than is hydrogen bonding.
TEM characterization, in combination with acid–base titrations and spectrophotometric studies, provides insights into the structural implications of molecular flexibility in an amphiphile nanostructure with higher-degree ordering produced by anisotropic intermolecular forces.
Experimental
Detailed synthetic procedures and chemical characterization are provided in the ESI.†Transmission electron microscopy (TEM)
TEM micrographs were captured on a FEI Tecnai G2 Spirit TWIN microscope at an accelerating voltage of 120 kV. 10 mL of 1 mg mL−1 aqueous suspensions of compounds 1–3 were deposited on a continuous carbon grid (Electron Microscopy Sciences, 200 mesh, copper) for 20 s, then blotted to remove excess solution. Grids were stained with 10 mL 0.1% phosphotungstic acid (aq), blotted to remove excess stain, then dried for 10 min.Acid–base titrations
10 mg mL−1 solutions of compounds 1–3 in distilled water were sonicated for 24 h and diluted to 1 mg mL−1. Each dilution was adjusted to pH of approx. 3.30 with HCl, then titrated through dropwise addition of 5.00 mM NaOH (aq). A 10 mM solution of triazaheptane (TAH) in 10 mL H2O was acidified to pH 2.0 through addition of 1.0 M HCl, then subsequently titrated with 45.82 mM NaOH. Change in pH following each addition was monitored by Mettler Toledo FP20-Bio benchtop meter, which was calibrated using commercially available Millipore Certipur standardized buffer solutions at pH 4.00, 7.00, and 12.00.UV-vis absorption spectroscopy
UV-visible light absorption spectra were obtained with a Perkin-Elmer LAMBDA 850 spectrophotometer. 50 μM aqueous suspensions of compounds 1–3 were prepared and their absorbance spectra were measured between 230–400 nm in a 1 cm quartz cuvette against a distilled water blank. Spectra are normalized by maximum absorbance.Fourier-transformed infrared spectroscopy
Compounds 1–3 were dissolved in either DMSO or D2O for an overall concentration of 20 mg mL−1. Following overnight sonication, the aqueous samples were diluted to 15 mg mL−1 through the addition of DCl (aq), D2O, or NaOD (aq) to respectively achieve pH values of 3, 7, and 11. Spectra were measured on a Bruker Alpha II FT-IR spectrometer with a monolithic diamond crystal. For each sample condition, 256 scans were recorded against background solvent.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the National Science Foundation under grant no. CHE-1945500. This work was supported in part by the Professor Amar G. Bose Research Grant Program and the Abdul Latif Jameel Water and Food Systems Lab. A. J. L and T. C.-T. acknowledge the support of the National Science Foundation Graduate Research Fellowship Program under Grant No. 1122374. T. C.-T. acknowledges the support of the Martin Family Society of Fellows for Sustainability and the Hugh Hampton Young Fellowship. This work made use of the MRSEC Shared Experimental Facilities at MIT supported by the National Science Foundation under award number DMR-14-19807 and the MIT Department of Chemistry Instrumentation Facility (DCIF).References
- T. Aida, E. W. Meijer and S. I. Stupp, Functional supramolecular polymers, Science, 2012, 335(6070), 813–817 CrossRef CAS PubMed.
- J.-M. Lehn, Supramolecular chemistry: receptors, catalysts, and carriers, Science, 1985, 227(4689), 849 CrossRef CAS PubMed.
- G. M. Whitesides, J. P. Mathias and C. T. Seto, Molecular self-assembly and nanochemistry: a chemical strategy for the synthesis of nanostructures, Science, 1991, 254(5036), 1312 CrossRef CAS.
- H. Cui, M. J. Webber and S. I. Stupp, Self-assembly of peptide amphiphiles: from molecules to nanostructures to biomaterials, Biopolymers, 2010, 94(1), 1–18 CrossRef CAS.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, Theory of self-assembly of hydrocarbon amphiphiles into micelles and bilayers, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- R. M. Da Silva, D. Van Der Zwaag, L. Albertazzi, S. S. Lee, E. Meijer and S. I. Stupp, Super-resolution microscopy reveals structural diversity in molecular exchange among peptide amphiphile nanofibres, Nat. Commun., 2016, 7, 11561 CrossRef CAS PubMed.
- W. C. Wimley and T. E. Thompson, Transbilayer and interbilayer phospholipid exchange in dimyristoylphosphatidylcholine/dimyristoylphosphatidylethanolamine large unilamellar vesicles, Biochemistry, 1991, 30(6), 1702–1709 CrossRef CAS PubMed.
- J. H. Ortony, C. J. Newcomb, J. B. Matson, L. C. Palmer, P. E. Doan and B. M. Hoffman, et al., Internal dynamics of a supramolecular nanofibre, Nat. Mater., 2014, 13(8), 812 CrossRef CAS PubMed.
- P. K. Hashim, J. Bergueiro, E. W. Meijer and T. Aida, Supramolecular polymerization: a conceptual expansion for innovative materials, Prog. Polym. Sci., 2020, 105, 101250 CrossRef CAS.
- S. I. Stupp and L. C. Palmer, Supramolecular chemistry and self-assembly in organic materials design, Chem. Mater., 2014, 26(1), 507–518 CrossRef CAS.
- R. P. Sijbesma and E. W. Meijer, Self-assembly of well-defined structures by hydrogen bonding, Curr. Opin. Colloid Interface Sci., 1999, 4(1), 24–32 CrossRef CAS.
- M. Lübtow, I. Helmers, V. Stepanenko, R. Q. Albuquerque, T. B. Marder and G. Fernández, Self-assembly of 9,10-bis(phenylethynyl) anthracene (BPEA) derivatives: influence of π-π and hydrogen-bonding interactions on aggregate morphology and self-assembly mechanism, Chemistry, 2017, 23(25), 6198–6205 CrossRef.
- C. J. Newcomb, S. Sur, J. H. Ortony, O.-S. Lee, J. B. Matson and J. Boekhoven, et al., Cell death versus cell survival instructed by supramolecular cohesion of nanostructures, Nat. Commun., 2014, 5(1), 3321 CrossRef.
- T. Christoff-Tempesta, Y. Cho, M. Geri, G. Lamour, A. J. Lew, X. Zho, W. R. Zuo and J. H. Ortony, Self-assembly of aramid amphiphiles into ultrastable nanoribbons and aligned nanoribbon threads, Nat. Nanotechnol., 2021, 16, 447–454 CrossRef CAS PubMed.
- W. R. Lindemann, T. Christoff-Tempesta and J. H. Ortony, A global minimization toolkit for batch-fitting and χ2 cluster analysis of CW-EPR spectra, Biophys. J., 2020, 119(10), 1937–1945 CrossRef CAS PubMed.
- D.-Y. Kim, T. Christoff-Tempesta, G. Lamour, X. Zuo, K.-H. Ryu and J. H. Ortony, Morphological transitions of a photoswitchable aramid amphiphile nanostructure, Nano Lett., 2021, 21(7), 2912–2918 CrossRef CAS PubMed.
- Y. S. Velichko, S. I. Stupp and M. O. de la Cruz, Molecular simulation study of peptide amphiphile self-assembly, J. Phys. Chem. B, 2008, 112(8), 2326–2334 CrossRef CAS.
- S. Ahmed, J. H. Mondal, N. Behera and D. Das, Self-assembly of peptide amphiphile-forming helical nanofibers and in situ template synthesis of uniform mesoporous single wall silica nanotubes, Langmuir, 2013, 29(46), 14274–14283 CrossRef CAS PubMed.
- Y. Wan, Z. Wang, J. Sun and Z. Li, Extremely stable supramolecular hydrogels assembled from nonionic peptide amphiphiles, Langmuir, 2016, 32(30), 7512–7518 CrossRef CAS PubMed.
- A. K. Awasthi, S. D. Bhagat, R. Ramakrishnan and A. Srivastava, Chirally twisted ultrathin polydopamine nanoribbons: synthesis and spontaneous assembly of silver nanoparticles on them, Chem. – Eur. J., 2019, 25(56), 12905–12910 CrossRef CAS.
- S. Song, J. Wang, N. Song, H. Di, D. Liu and Z. Yu, Peptide interdigitation-induced twisted nanoribbons as chiral scaffolds for supramolecular nanozymes, Nanoscale, 2020, 12(4), 2422–2433 RSC.
- P. Chidchob, T. G. W. Edwardson, C. J. Serpell and H. F. Sleiman, Synergy of two assembly languages in DNA nanostructures: self-assembly of sequence-defined polymers on DNA cages, J. Am. Chem. Soc., 2016, 138(13), 4416–4425 CrossRef CAS PubMed.
- E. Villarreal, G. G. Li, Q. Zhang, X. Fu and H. Wang, Nanoscale surface curvature effects on ligand–nanoparticle interactions: A plasmon-enhanced spectroscopic study of thiolated ligand adsorption, desorption, and exchange on gold nanoparticles, Nano Lett., 2017, 17(7), 4443–4452 CrossRef CAS PubMed.
- T. Christoff-Tempesta, A. J. Lew and J. H. Ortony, Beyond covalent crosslinks: applications of supramolecular gels, Gels, 2018, 4(2), 40 CrossRef.
- A. S. Weingarten, R. V. Kazantsev, L. C. Palmer, M. McClendon, A. R. Koltonow and A. P. S. Samuel, et al., Self-assembling hydrogel scaffolds for photocatalytic hydrogen production, Nat. Chem., 2014, 6(11), 964–970 CrossRef CAS PubMed.
- L. C. Palmer and S. I. Stupp, Molecular self-assembly into one-dimensional nanostructures, Acc. Chem. Res., 2008, 41(12), 1674–1684 CrossRef CAS.
- A. Patra, C. Chandaluri and T. P. Radhakrishnan, Optical materials based on molecular nanoparticles, Nanoscale, 2011, 4, 343–359 RSC.
- F. Di Maria, P. Olivelli, M. Gazzano, A. Zanelli, M. Biasiucci and G. Gigli, et al., A successful chemical strategy to induce oligothiophene self-assembly into fibers with tunable shape and function, J. Am. Chem. Soc., 2011, 133(22), 8654–8661 CrossRef CAS.
- T. Fort, The wettability of a homologous series of Nylon polymers, in Contact angle, wettability, and adhesion, ed. F. M. Foulkes, American Chemical Society, Washington DC, 1964, vol. 43, pp. 302–309 Search PubMed.
- J. R. Kanicky and D. O. Shah, Effect of premicellar aggregation on the pKa of fatty acid soap solutions, Langmuir, 2003, 19(6), 2034–2038 CrossRef CAS.
- E. S. Kartashynska, Y. B. Vysotsky, D. Vollhardt and V. B. Fainerman, Relationship between the bulk and surface basicity of aliphatic amines: a quantum chemical approach, ACS Omega, 2020, 5(49), 32032–32039 CrossRef CAS PubMed.
- J. H. Ortony, T. Chatterjee, L. E. Garner, A. Chworos, A. Mikhailovsky and E. J. Kramer, et al., Self-assembly of an optically active conjugated oligoelectrolyte, J. Am. Chem. Soc., 2011, 133(21), 8380–8387 CrossRef CAS.
- Z. Tang, M. S. Johal, P. Scudder, N. Caculitan, R. J. Magyar and S. Tretiak, et al., Study of the non-covalent interactions in Langmuir–Blodgett films: An interplay between π−π and dipole–dipole interactions, Thin Solid Films, 2007, 516(1), 58–66 CrossRef CAS.
- A. Barth, Infrared spectroscopy of proteins, Biochim. Biophys. Acta, Bioenerg., 2007, 1767(9), 1073–1101 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic details and chemical characterization are included. See DOI: 10.1039/d1me00120e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |