
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDonor-free oligothiophene based dyes with di-anchor architecture for dye-sensitized solar cells†
John
Marques dos Santos‡
a,
Ellie
Tanaka‡
 b,
Alan A.
Wiles
a,
Graeme
Cooke
*a and
Neil
Robertson
*b
b,
Alan A.
Wiles
a,
Graeme
Cooke
*a and
Neil
Robertson
*b
aSchool of Chemistry, Joseph Black Building, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: Graeme.Cooke@glasgow.ac.uk
bSchool of Chemistry, EaStCHEM, University of Edinburgh, King's Buildings, David Brewster Road, Edinburgh, Scotland EH9 3FJ, UK
First published on 17th March 2021
Abstract
Traditionally, metal free DSSCs are fabricated from sensitizers designed using a donor–pi–acceptor (D–π–A) architecture. More recently, non-conventional dyes without strong donor units have emerged and have provided competitive power conversion efficiencies (PCEs) with respect to their (D–π–A)-based counterparts. Here, we report the synthesis and DSSC fabrication of oligothiophene molecules featuring two anchoring groups that do not possess strong donor moieties and are able to adopt V- and U-shape conformations. PCEs of 3.70% were obtained with the smaller 5-thiophene dyes (5T2A, 5T2A-E) possessing higher PCEs than their larger 8-thiophene counterparts containing diacetylene units (8T4A, 8T4A-E). Comparison to the DSSC properties of the analogous linear dye 5T, indicates that the architecture and structure of this series of dyes are likely responsible for their lower performance.
Design, System, ApplicationWe have designed and synthesised two generations of oligothiophene dyes that do not possess strong donor units and incorporate two peripheral anchoring groups to investigate this type of functionalisation for inexpensive sensitisers for dye-sensitised solar cells (DSSCs). The molecules can adopt V- and U-shape conformations, which is desirable for a di-anchoring process, and benefit from slight bathochromic shifts upon ethylenedioxythiophene (EDOT) functionalisation. We find that the larger 8-thiophene dyes display slightly blue-shifted spectra compared with their 5-thiophene counterparts due to the presence of diacetylene residues, and poorer performances likely due to dye architecture as will be discussed in depth. This work provides valuable guidance towards improving the design and performance of inexpensive “donor-free” thiophene-based dyes for DSSCs. |
1. Introduction
Dye-sensitized solar cells (DSSCs) continue to attract significant attention in endeavours to harness the considerable potential of solar radiation to provide renewable energy. It has been shown that the correct molecular engineering of sensitizers can greatly improve the performance of these solar energy conversion devices.1,2 The overall power conversion efficiency (PCE, η) has been raised from about 7% to circa 14% since this technology emerged, and DSSCs have also proven to be a very promising technology for low light scenarios such as indoor applications.3–5 Significant effort continues to be directed towards improving the PCE of DSSCs by modifying the electrolyte and sensitizers,6–8 and structural modification of the latter continues to be an attractive way to increase this parameter.6,9–11As the central component of DSSCs, the sensitizer harvests sunlight which leads to the production of photon-excited electrons. It is noteworthy that efficient electron injection from the excited dye to the conduction band of the TiO2 through the anchoring group is achieved using dyes with a D–π–A (D = electron donor, A = electron acceptor/anchoring group) archictecture.12 However, it has been shown in recent reports that outstanding efficiencies can be reached with dyes containing only thiophene π-bridges as relatively weak donors (referred to as “donor-free” dyes since they are absent of strong electron-donating moieties) with a general structure of πn–A.13,14 This indicated that these simple building blocks can be functionalised in simple ways to provide low-cost and efficient DSSCs. On the other hand, the choice of the anchoring group determines the electron injection rate in DSSCs devices.15–18 It is also partially influential on the arrangement of the dyes in the monolayer anchored on the surface of the semiconductor.19 Due to its strong electron-accepting character, strong binding interaction with the oxide and efficient electron injection, cyanoacetic acid is the most commonly used.10,16,20–27
In spite of these observations, single D–π–A dyes usually have a rod-shaped structure that facilitate dye loading; however, this may contribute to aggregation and charge recombination due to undesirable π–π stacking. Hence, considerable attention has been paid to dyes containing multiple anchoring groups,26 since they show limited aggregation as well as increased electron extraction channels, enhanced binding strength, lower optical-gap and enhanced extinction coefficient thus enhancing the device performance.2,28–31 In this regard, Eiamprasert et al. recently compared triphenylamine-based dyes with various anchoring groups and found the highest efficiency for the di-anchored dyes in comparison to the mono and the tri-anchored ones. The performance was also improved when an additional donor was incorporated into the semi-circular system.32
Here, to gain further insight into the design of ‘donor-free’ thiophene-based sensitizers, we present two generations of dyes featuring two anchoring cyanoacrylate acceptor units (Fig. 1). Despite the important advantages that the di-anchoring architecture can offer, it is so far not clear in the literature whether this structural feature would benefit “donor-free” thiophene-based dyes. Moreover, it is well-known that adjacent thiophenes preferably adopt an anti-conformation that usually leads oligothiophenes to adopt a linear conformation.33,34 This suggests that to effectively di-anchor at the TiO2 surface, these dyes would possibly have to overcome this rotational barrier.
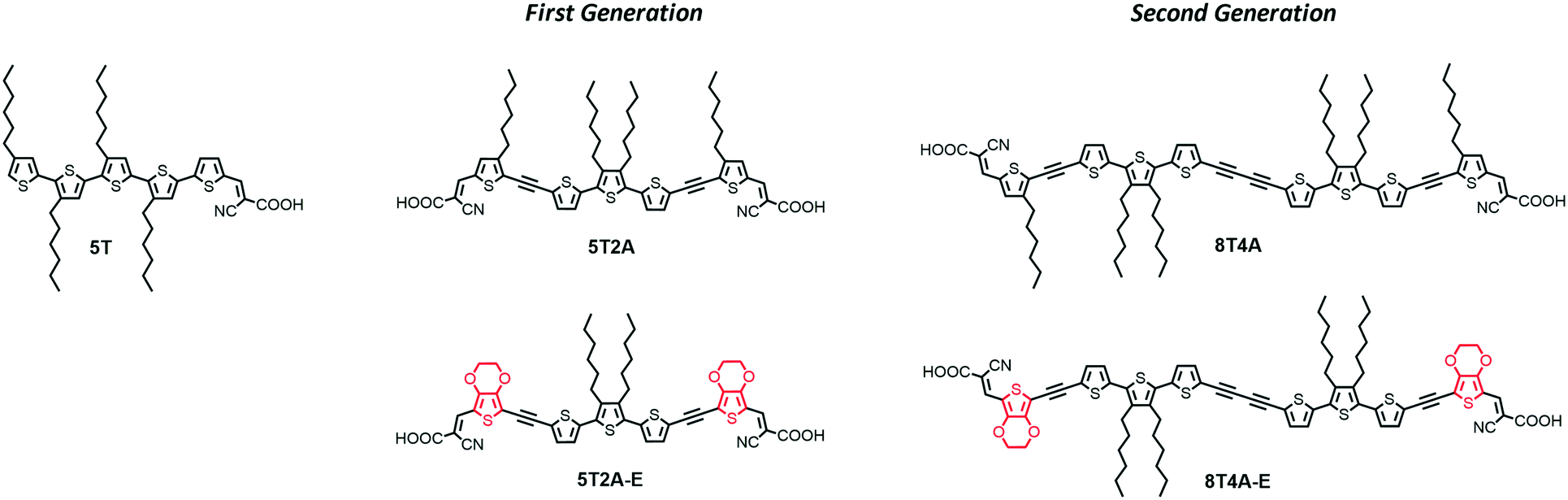 | ||
| Fig. 1 Chemical structures of dye 5T and the first- and second-generation dyes used in this study. | ||
The first-generation dyes were designed to be V-shaped if di-anchored (5T2A and 5T2A-E) whereas the larger second-generation dyes (8T4A and 8T4A-E), formed from precursors that were side products from the synthesis of the first-generation dyes, would adopt a more U-shaped conformation and feature diacetylene residues. Acetylenes in these molecules were designed to mitigate steric hindrance between moieties. These residues can compromise the intramolecular charge transfer (ICT) effect,35 however they seem not to be detrimental for dye-sensitizers, since structures are sought after with permanent molecular dipole and several devices with high PCE include this architectural feature.5,15,35–38 Furthermore, we have incorporated ethylenedioxythiophene (EDOT) residues into two of the dyes to increase the donor ability of one of the thiophenes units. We performed density functional theory (DFT) calculations to investigate the possibility of di-anchoring by accessing the number and distribution of possible molecular conformations, and investigated the DSSC properties of these sensitizers while comparing them to related linear donor-free dye 5T.13,14
2. Results and discussion
2.1. Molecular design and synthesis
The first-generation of di-anchoring dyes (5T2A, 5T2A-E) were designed to feature a conjugated system composed of 5 thiophene units, similar to dye 5T. However, unlike 5T, two acetylene residues were introduced to help to alleviate conformational strain and favour a di-anchoring conformation at the TiO2 surface. To investigate the effect of additional thiophenes and additional acetylenes, the second-generation dyes (8T4A and 8T4A-E) were designed to feature a conjugated system composed of 8 thiophene units and an extra diacetylene residue. The thiophene units closest to the acceptor moieties were substituted for EDOT moieties in both generations of dyes (identified by the suffix E) to aid with ICT. The synthesis of dyes 5T2A, 5T2A-E, 8T4A and 8T4A-E was accomplished as described in the ESI† and depicted in Scheme S1. The compound characterisation data are also provided in the ESI.†2.2. Optical properties
The UV-vis absorption spectra of 5T2A, 5T2A-E, 8T4A and 8T4A-E are presented in Fig. 2 and were recorded as THF solutions (1 × 10−5 M). It is noteworthy that 5T2A-E shows a bathochromic shift (λmax = 470 nm) in comparison with 5T2A (λmax = 455 nm), and 8T4A-E demonstrates similar behavior (λmax = 464 nm) in comparison with its counterpart 8T4A (λmax = 446 nm, see Fig. 2). The π-extended dyes (featuring 8 thiophene units) showed slightly blue-shifted spectra in comparison with the 5 thiophene analogues. In addition, in comparison with the mono-anchoring dye 5T that possesses an oligothiophene backbone featuring five thiophene units (λmax = 478 nm),13,14 the second anchoring/acceptor moieties did not produce a red-shift of the λmax. This can be attributed to the presence of the alkyne residues which, due to their electro-withdrawing nature, compromise ICT as they weaken the comparative electron accepting ability of the terminal acceptor.35 The bands shown in the spectra are attributed to S0–S1 type transition due to the ICT between the electron rich units and the cyanoacrylate terminal acceptor moieties. All dyes present extinction coefficients within the same order of magnitude but with slight differences, ranging from 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 680 M−1 cm−1 (5T2A-E) to 31630 M−1 cm−1 (8T4A-E). The optical gaps (Eopt) were estimated based on the onset values of absorption (λonset), and little difference was found with values ranging from ca. 2.09 eV (5T2A-E) to 2.14 eV (5T2A). All the optical and photophysical properties are presented in Table 1. In summary, the first- generation dyes displayed red-shifted spectra with a concomitant decrease in Eopt with respect to their larger second-generation counterparts, while both generations showed slightly red-shifted λmax values for the dyes containing the EDOT units.
680 M−1 cm−1 (5T2A-E) to 31630 M−1 cm−1 (8T4A-E). The optical gaps (Eopt) were estimated based on the onset values of absorption (λonset), and little difference was found with values ranging from ca. 2.09 eV (5T2A-E) to 2.14 eV (5T2A). All the optical and photophysical properties are presented in Table 1. In summary, the first- generation dyes displayed red-shifted spectra with a concomitant decrease in Eopt with respect to their larger second-generation counterparts, while both generations showed slightly red-shifted λmax values for the dyes containing the EDOT units.
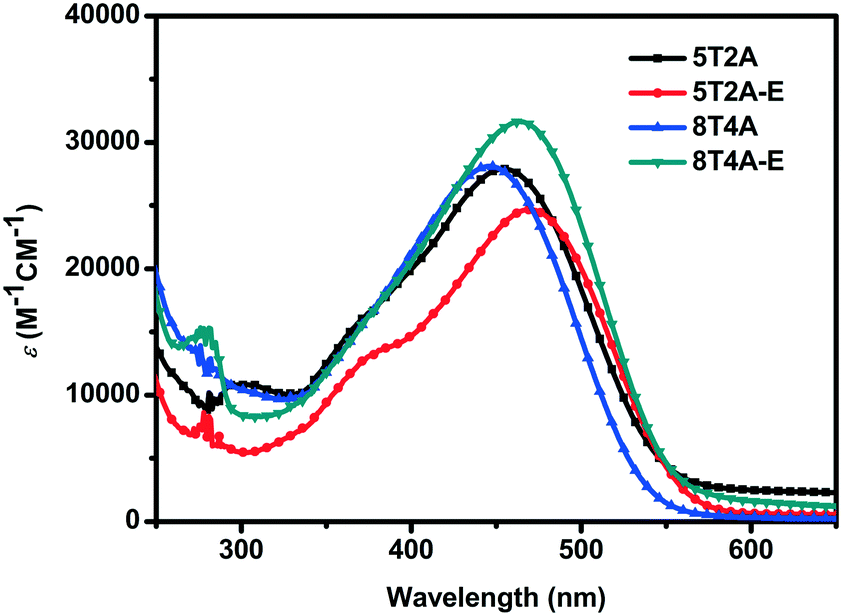 | ||
| Fig. 2 Absorption spectra of 5T2A, 5T2A-E, 8T4A and 8T4A-E recorded in THF solution (1 × 10−5 M). | ||
| Dye | λ max [nm] | ε max [M−1 cm−1] | λ onset [nm] | E opt [eV] | E 1/2ox [V] | E 1/2red [V] |
|---|---|---|---|---|---|---|
| a Measured in tetrahydrofuran (THF, 1 × 10−5 M). b Calculated using the formula Eopt = 1240/λonset. c Measured in THF (1 × 10−3 M) and calibrated versus the ferrocene/ferrocenium (Fc/Fc+) redox couple. | ||||||
| 5T2A | 455 | 27890 |
578 | 2.14 | 0.90 | −1.34 |
| 5T2A-E | 470 | 24680 |
591 | 2.09 | 0.98 | −1.71 |
| 8T4A | 446 | 28120 |
565 | 2.19 | 0.84 | −1.76 |
| 8T4A-E | 464 | 31630 |
586 | 2.11 | 0.82 | −1.73 |
2.3. Electrochemical properties
To investigate the electrochemical properties of the dyes and determine the position of the energy levels relative to the conduction band of TiO2 and the redox shuttles, square wave voltammetry measurements were performed in THF solution (1 × 10−3 M) versus the ferrocene/ferrocenium (Fc/Fc+) redox couple. Evaluation of chemical/electrochemical reversibility was unfortunately not possible via cyclic voltammetry due to the low peak current intensity observed for this series (Fig. S10†). In the square wave voltammograms presented in Fig. 3, it is possible to observe one reduction (Ered) and one oxidation (Eox) wave for each compound. Values of Eox and Ered are presented in Table 1. The first-generation dyes show slightly more positive oxidation Eox (0.90 V for 5T2A and 0.98 V for 5T2A-E) than their second-generation counterparts (0.84 V for 8T4A and 0.82 V for 8T4A-E), with 5T2A-E reaching the highest value of the series. All dyes display similar Ered (circa −1.7 V) except for 5T2A, which shows the least negative value (−1.34 eV). On the other hand, comparison of dyes with the same number of π-spacer units (5T2A and 5T2A-E) shows a slightly positive shift in the Eox value for the EDOT containing 5T2A-E. This minor difference in Eox is unexpected since 5T2A-E should have a more electron-rich nature due to the EDOT unit, however, a similar result has been shown in the literature.39 The Eox is an important parameter since it modulates the voltage loss in devices caused by excess driving force in the dye recovery process by redox mediators.8,40 The electrochemical study shows that although these dyes have Eox positive enough to allow regeneration by the redox shuttle I−/I3, they are perhaps too positive and may induce recombination. The Ered values are negative enough to ensure efficient electron injection into the conduction band of TiO2.12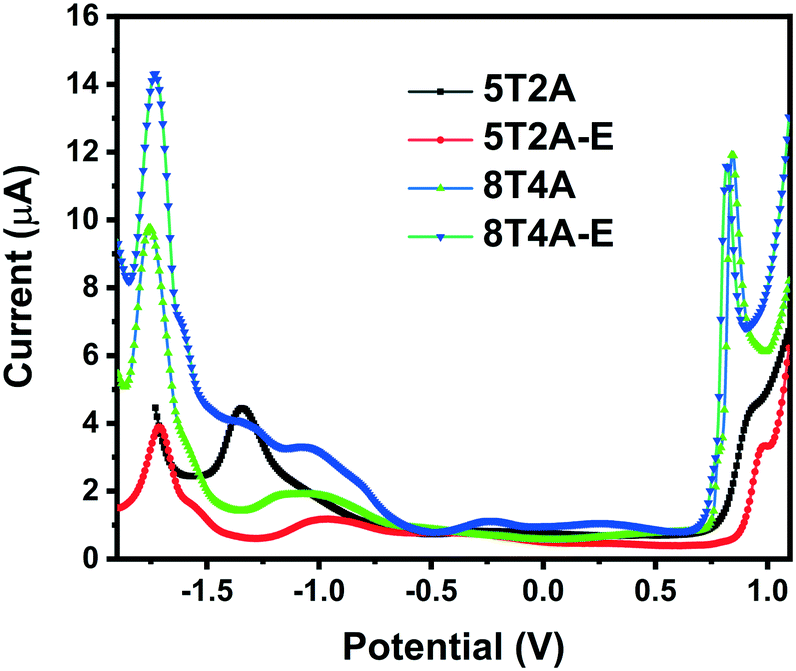 | ||
| Fig. 3 Square wave voltammograms of 5T2A, 5T2A-E, 8T4A and 8T4A-E, recorded in THF (1 × 10−3 M), calibrated versus the ferrocene/ferrocenium (Fc/Fc+) redox couple. | ||
2.4. Computational studies
Density functional theory (DFT) calculations are applied in these molecular systems to gain more insight into their electronic properties. Firstly, theoretical studies were performed at the B3LYP/6-31G(d,p) level of theory in the gas phase to obtain a representation of molecular orbital distribution of 5T2A, 5T2A-E, 8T4A and 8T4A-E, which is provided in Fig. 4. All molecules display a linear optimised structure in the gas phase. The electron density plot of the highest occupied molecular orbital (HOMO) for 5T2A and 5T2A-E is localised over the whole molecule according to the calculations, whereas for 8T4A and 8T4A-E it is not found to extend to the acceptor groups and is mainly located on the oligothiophene residue. In the case of lowest unoccupied molecular orbital (LUMO), the electron density distribution is also localised over the whole molecule for 5T2A and 5T2A-E, resulting in a complete overlap between frontier orbitals. However, for 8T4A and 8T4A-E, the distribution is mostly localised towards the cyanoacrylate acceptor moieties not reaching the centre of the molecule, thus in these dyes the overlap between HOMO and LUMO happens to a lower extent than in the first-generation dyes.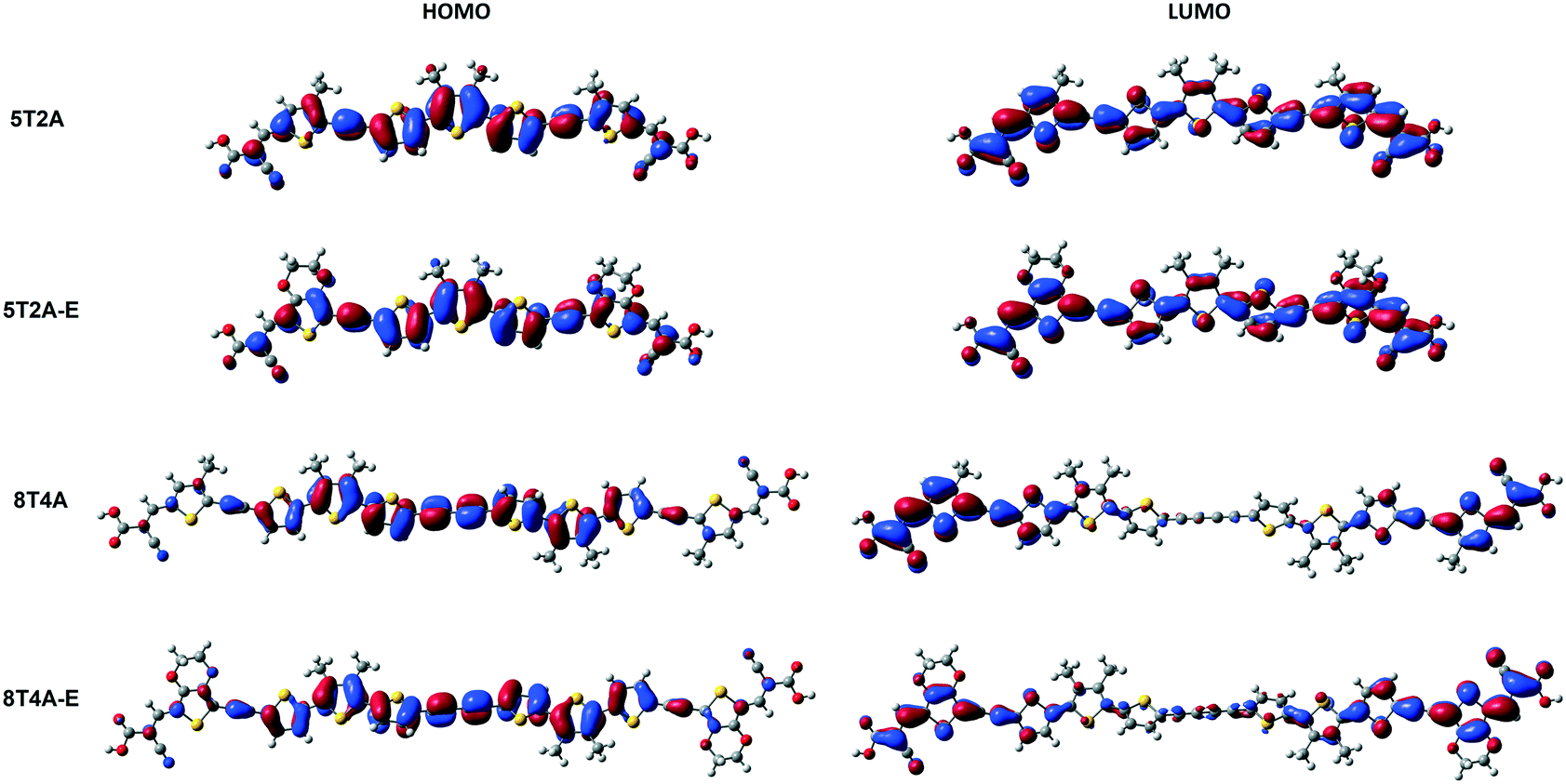 | ||
| Fig. 4 Molecular orbital distribution of 5T2A, 5T2A-E, 8T4A and 8T4A-E computed at the B3LYP/6-31G(d,p) level. | ||
To further understand the unimolecular properties and study the self-assembly tendencies of these dyes for a di-anchoring process, we used a computational method to estimate the number, distribution and properties of possible conformers of 5T2A. The structural variation from 5T to the first and second-generation dyes is expected to influence their structural conformation and self-assembly on the surface of the TiO2 semiconductor. For instance, adjacent thiophenes usually adopt an anti-conformation due to non-covalent intramolecular CH–S interactions, and EDOT moieties usually cause a similar effect due to RO–S interactions.33,34 Hence, oligothiophenes preferably adopt a linear arrangement that we consider non-ideal here for a di-anchoring process to happen, although evidences show that these rotational barriers can be overcome in a fluctuating environment at room temperature.41,42 The acetylene residues in this work are thus expected to help to circumvent such conformational lock.43–48
The calculations were carried out using DFT at the ωB97X-D/6-31+G(d,p) level of theory, a method successfully applied in a similar study,42 in the solvent state (in toluene and in THF at 300 K). A distribution analysis was performed to estimate the most likely distribution of conformer in solution. THF and toluene were chosen as their relative polarities are significantly different (polarity ε values of 7.4257 and 2.3741 respectively) and therefore should show greater difference in the effect of solvent polarisation of the conformers. The diffuse and polarisation functions were used because it better accounts for the solvent effects.49 Considering that 5T2A has six points of rotation in total (omitting the hydroxy group of the carboxylic acid moiety), the number of possible conformers is 26 (= 64) conformers (Fig. 5(a)). However due to symmetry a number of these conformers are energetically identical, therefore there are only 36 unique conformers (28 of them with a degeneracy of 2). Out of these conformers we identified 17 that adopt a linear shape, 14 that adopt a V shape and 5 that adopt a Z shape. Firstly, according to the calculations, the solvent polarity plays an important role in the distribution of accessible conformers at a given temperature. At 300 K, the most accessible conformer in toluene shows a Z-like shape (conformer 33), whereas in THF a linear (L) conformer (4) is the most favoured (see Fig. 5(b)). Furthermore, in THF, the relative population of other accessible conformers are considerably lower than in toluene. In toluene a total of five conformers were found at above 0.1 relative population to conformer 33. The third most populated conformer (26) has a V-shape. In THF, the next most abundant conformer is predicted to have a population of 0.05 relative to 4.
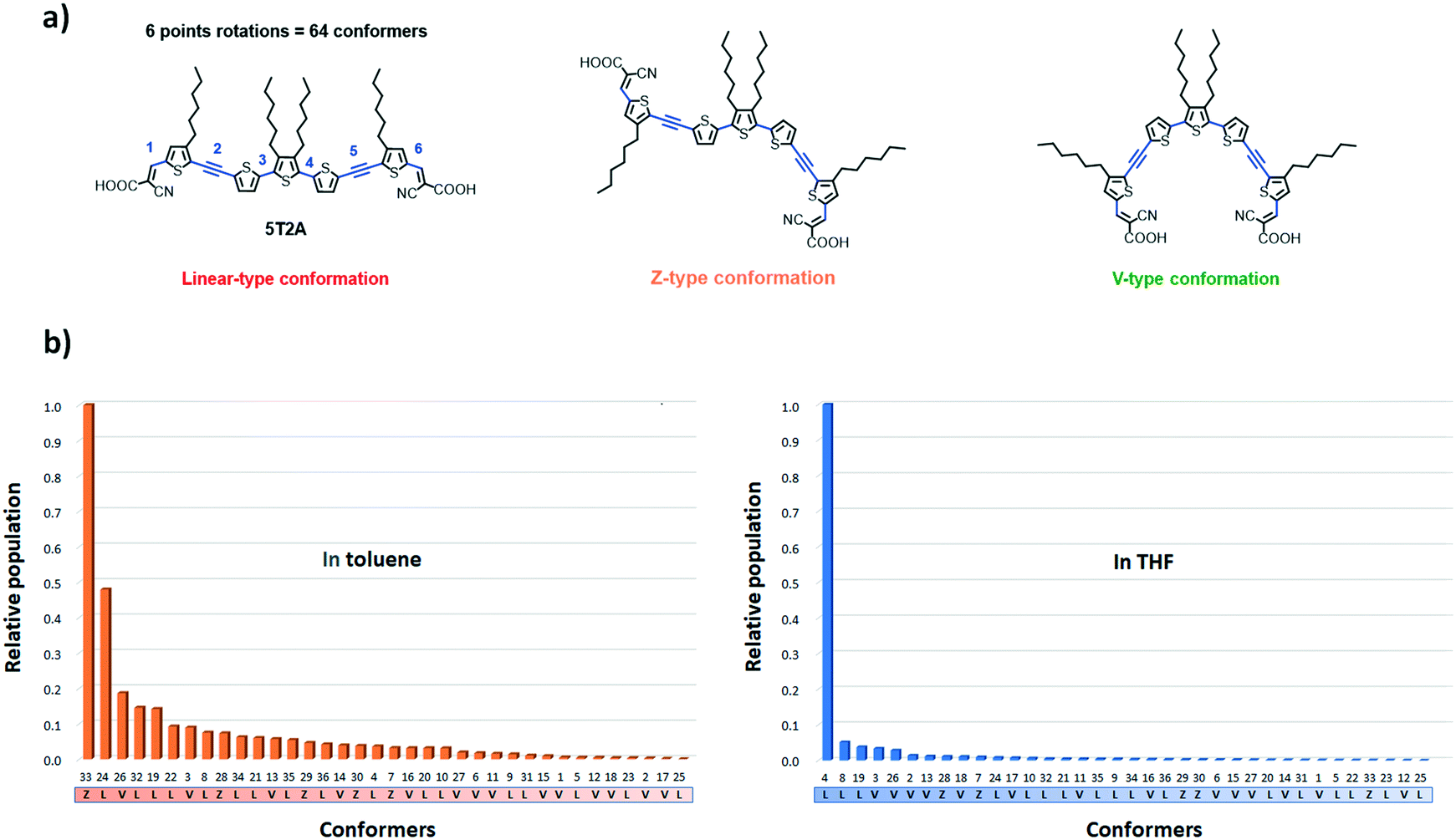 | ||
| Fig. 5 (a) Schematic representation of 5T2A in a linear (L), Z and V-type conformation. The points of rotations are represented in blue. (b) Relative population of independent conformers of 5T2A in toluene (left) and in THF (right) at 300 K computed at the ωB97X-D/6-31+G(d,p) level of theory. | ||
The geometry of each conformer was optimised using the ωB97X-D/6-31+G(d,p) method (Table S3†). Importantly, in both solvents, the conformers are not significantly different in energy from one another. This suggests that the desirable V-shaped conformers are accessible at room temperature since the rotational cost is low.41 Thus, the study shows that the V-shaped conformers are considerably lower in population, which could retard their uptake at the surface of TiO2via di-anchor and favour the mono-anchoring of dyes. The polarisation effect of the solvent suggests that toluene would favour a greater number of V-shaped conformers. Unfortunately, these dyes show considerably poorer solubility in toluene than in THF which can limit their study in the device.
2.5. Device performance
The four dyes, 5T2A, 5T2A-E, 8T4A and 8T4A-E, were tested in dye-sensitized solar cells (DSSCs) with the structure Fluorine-doped SnO2 (FTO)/compact TiO2/mesoporous TiO2/dye/I−/I3− electrolyte/Pt/FTO. 5 T dye was used as a reference. The current density–voltage (J–V) curves of the champion cells are shown in Fig. 6, with the values summarised in Table 2. The statistics are shown in Fig. S11.† All dyes in the series exhibited a PCE of around 2–3%, compared to the reference 5T dye at around 5–6%. The maximum PCEs achieved for the best cells were 3.70% with 5T2A or 5T2A-E, and 2.82% with 8T4A or 8T4A-E. Overall, all A–πn–A dyes showed lower performance in respect to the πn–A 5T dye. The relatively low VOC range (circa 0.5–0.6 V) suggests that recombination is facilitated in these systems. One possible cause for this is the highly positive Eox of the new dyes, which could act as a driving force for the TiO2 conduction band electrons to recombine with the oxidised dye. Within the series, the first-generation dyes, 5T2A and 5T2A-E, achieved higher PCE compared to the second generation, mainly attributed to larger current generation. Larger current can often arise from higher extinction coefficient of the dyes, however in our case this appears to be irrelevant. It is therefore likely that the molecular size or/and anchoring of the dyes is contributing to the performance trends.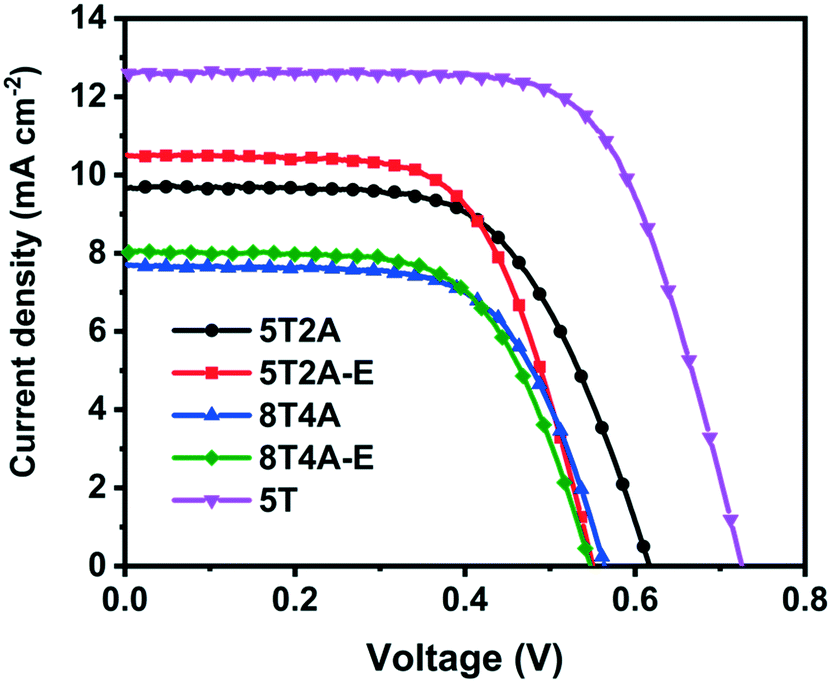 | ||
| Fig. 6 J–V curves of the best DSSCs employing 5T2A, 5T2A-E, 8T4A and 8T4A-E as light absorbers, with 5T dye as a reference. | ||
| Dye | J SC (mA cm−2) | V OC (V) | ff | PCE (%) |
|---|---|---|---|---|
| 5T2A | 9.68 (8.8 ± 1.3) | 0.62 (0.60 ± 0.07) | 0.62 (0.64 ± 0.06) | 3.70 (3.4 ± 0.3) |
| 5T2A-E | 10.5 (9.3 ± 1.3) | 0.55 (0.53 ± 0.03) | 0.64 (0.62 ± 0.02) | 3.70 (2.8 ± 0.7) |
| 8T4A | 7.69 (6.8 ± 1.4) | 0.56 (0.54 ± 0.02) | 0.65 (0.62 ± 0.06) | 2.82 (2.4 ± 0.5) |
| 8T4A-E | 8.03 (8.0 ± 0.7) | 0.55 (0.51 ± 0.02) | 0.64 (0.59 ± 0.06) | 2.82 (2.4 ± 0.4) |
| 5T | 12.6 (12.1 ± 0.6) | 0.72 (0.70 ± 0.03) | 0.69 (0.66 ± 0.03) | 6.24 (5.6 ± 0.4) |
Computational studies suggest that the dyes in the series occupy circa 4 nm2 when mono-anchored and 9 nm2 when di-anchored to the TiO2. These values will lead to estimated dye amounts of 6.64 10−8 mol cm−2 for mono-anchoring and 2.95 10−8 mol cm−2 for di-anchoring. Dye uptake for 5T2A, 5T2A-E, 8T4A and 8T4A-E was measured from UV-vis absorption and derived as 9.60 10−8 mol cm−2, 8.38 10−8 mol cm−2, 6.01 10−8 mol cm−2 and 2.68 10−8 mol cm−2, respectively. Comparing these values with the estimated amount in each case indicate that 5T2A, 5T2A-E and 8T4A are mono-anchored, while 8T4A-E may or may not be di-anchored. This follows the suggestion from the computational studies, where the A–πn–A dyes prefer linear binding to the TiO2 over di-anchoring. In the case of mono-anchoring of the A–πn–A dye, the electron pathway within the dye is deviated into two opposite directions, with one end receding from the TiO2 network. The remote acceptor residue would then work as an unwanted electron sink and recombination site, leading to lower current and voltage. However, as indicated above, di-anchoring may occur for 8T4A-E, due to the rapidly fluctuating conformation of the dye molecules at the liquid/solid interface during dye loading. However, the poor current and voltage trends displayed by the larger 8T4A and 8T4A-E dyes suggest that the larger structures lead to low dye uptake and recombination at the TiO2/electrolyte interface, due to the creation of large voids when di-anchored.
Poor solubility of the dyes is also detrimental to attaining higher performance. The incident photon-to-current efficiency (IPCE) data of DSSCs employing the most efficient A–πn–A dye, 5T2A (Fig. S12†), indicates that the use of THF leads to lower IPCE, hence lower current. However, in our case, the low solubility of the four dyes, especially 8T4A and 8T4A-E in most organic solvents left us with the sole choice to employ a THF-based binary dye bath system for sufficient dye uptake. This could also be one of the causes of their low performance. Dye bath solvent effects have been studied by Tian et al.,50 where our findings are relatively consistent with their suggestions.
The EDOT-substituted 5T2A-E and 8T4A-E displayed slightly higher current compared to their counterparts, which is expected due to the improved electron donating ability by incorporation of the EDOT residues. It is interesting to note that the dye uptake is lower for these dyes. On the other hand, the VOC appeared to decrease slightly, resulting in a lower average PCE in the case of 5T2A-E and similar in the case of 8T4A-E.
3. Conclusions
The oligothiophene based dyes composed of 5 and 8 thiophenes and possessing di-anchoring architecture were synthesized and compared with 5T in terms of photophysical and DSSC properties. Within this series, slightly red-shifted spectra were observed for the dyes containing EDOT residues, whereas the larger second-generation dyes showed slightly blue shifted spectra in comparison with their 5-thiophene analogues. The blue-shifted spectra of this series with respect to 5T were attributed to the presence of acetylene residues. By comparing the first-generation (5T2A and 5T2A-E) and second-generation (8T4A and 8T4A-E) systems, the former shows better performance in general. The trend is irrelevant to the absorption properties and is likely to come from the dye architecture and solubility.The computational studies implied that although the V- and U-shaped conformers are perfectly accessible at room temperature, this series likely show a considerable population of mono-anchored dyes at the surface of the TiO2 adopting a more linear conformation. However, as demonstrated for 5T2A, solvents with higher polarity may have a detrimental effect in allowing molecular motion by stabilising linear conformations. This is problematic due to the low solubility of the dyes in less polar solvents. The dye uptake analysis suggests that 5T2A, 5T2A-E and 8T4A have mono-anchored, while 8T4A-E is possibly di-anchored. Mono-anchored A–πn–A dyes are detrimental to the extraction of charge and Jsc, since the remote acceptor residue works as an electron sink and recombination site.
Within the series, 5T2A exhibits the highest current density, voltage and PCE, presumably due to the higher dye uptake and higher solubility. The optical/electrochemical properties of this series have been demonstrated to be favourable as a DSSC dye sensitizer, however the overall photovoltaic performance does not appear to be advantageous over the reference πn–A 5T dye. Future designing of such dyes should involve further consideration on compactness, rigidity, and solubility. Overall, ‘donor-free’ oligothiophene based A–πn–A dyes were introduced for the first time, and their ability as DSSC sensitizers were demonstrated. We believe that our study will serve as pathways towards improved designing of such dye families.
4. Experimental
4.1. Synthesis
Reagents were purchased from Sigma Aldrich®, Fluorochem®, TCI®, Alfa Aesar®, Acros® or Fisher Scientific® and used as received. Dry solvents were obtained either from Innovative Technology inc. Pure Solv 400-5-MD solvent purification system (activated alumina columns) or Sigma Aldrich®. Column chromatography was carried out using silica gel (Sigma-Aldrich) 40–63 nm particle size, 60 Å pore size. The solvent system is specified in each experiment. Thin-layer chromatography (TLC) was performed using Merck silica gel 60 covered aluminium plates F254. NMR spectra were obtained with either a Bruker Avance III 400 or a Bruker Avance III 500 spectrometers. Mass spectra were obtained from the mass spectrometry service at the University of Glasgow and from the EPSRC UK National Mass Spectrometry Facility at Swansea University. All the spectroscopic (except NMR) and electrochemical data were processed using Origin Pro 8.5 software suite.4.2. Optical and electrochemical properties
UV-vis spectra were recorded on a Perkin Elmer 25 UV/VIS Spectrometer at room temperature. Voltammograms were recorded on a CH Instrument Electrochemical Workstation (CHI 440a), Austin, TX, USA. Solutions were prepared in THF (1 × 10−3 M) containing TBA.PF6 (0.1 M) as electrolyte. The scan rate of 0.07 V s−1 was utilized. The redox couples are reported versus the ferrocene/ferrocenium (Fc/Fc+) redox couple, using a 1.6 mm diameter platinum working electrode, a platinum wire counter electrode and a silver wire pseudo reference electrode.4.3. Computational studies
Density functional theory (DFT) calculations were performed using Gaussian 09 software suite.51 In the gas phase, molecular geometries were initially optimised semi-empirically (PM6) and then re-optimised by DFT (B3LYP/6-31G(d,p)). In the conformational studies ωB97X-D/6-31+G(d,p) (solution state) was used. Some conformers were found to have negative frequency which were identified as transition states between two more stable conformers in energy. To facilitate the convergence of the geometry optimisations, the hexyl chains were replaced by methyl groups.4.4. Solar cell fabrication
Fluorine-doped tin oxide (FTO) glass (TCO22-7/Li, Solaronix) substrates were cleaned by sonication in 2% Decon 90 (Decon Laboratories) aq., deionized water, acetone and ethanol for 15 min each, followed by UV/O3 treatment for 20 min to make the surface hydrophilic. The blocking TiO2 layer was formed by immersing the substrates in 40 mM TiCl4 aq. at 80 °C for 30 min and rinsing with water. The mesoporous TiO2 layer was formed by screen-printing Dyesol 18NR-D (transparent layer, thickness 4 μm) and 18NR-AO (scattering layer, 4 μm) paste. The photoanodes were gradually heated to 500 °C and annealed for 15 min in a programmable furnace. Upon cooling, a second TiCl4 treatment was applied and the films were annealed at 500 °C for 30 min. Dye loading was initiated while the films were warm (>80 °C) and was performed overnight. 5T dye was used as a reference. The solvent system and concentration were optimised for each dye (full details in Table S4†). The cathodes were fabricated by doctor-blading Platisol T/SP (Solaronix) on cleaned FTO substrates and annealing at 400 °C for 15 min to obtain a Pt-coated FTO glass. The anode and cathode were assembled using a 25 μm Surlyn film (Du Pont) as spacer. The I−/I3−electrolyte was injected under vacuum through a pre-drilled hole in the cathode. The composition of the electrolyte used in the study was 0.1 M LiI, 0.05 M I2, 0.6 M 1,2-dimethyl-3-propylimidazolium iodide (DMPII) and 0.5 M 4-tert-butylpyridine in acetonitrile. The hole was finally sealed with a piece of Surlyn and cover glass slip.4.5. Solar cell characterization
The current–voltage (J–V) curves were recorded on an Autolab PGSTAT30 potentiostat. The cells were irradiated with an AM 1.5G simulated sunlight at 100 mW cm−2, generated by a Sciencetech SLB-300A compact solar simulator class AAA. The light intensity was calibrated by a Si reference cell before each measurement. A black mask with an aperture size of 0.2827 cm2 was used to define the active area.The incident photon-to-current conversion efficiency (IPCE) curves were recorded on a Bentham PVE300 EQE/IQE system in DC mode. The incident light was calibrated with a reference silicon photodiode within the range of 300–1000 nm.
The dyes were desorbed from the TiO2 by immersion in 0.1 M tetrabutylammonium hydroxide ethanol/methanol 9:1 (v/v) solution. Dye uptake was derived from the UV/Vis absorption peak of the resulting solutions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
JMS acknowledges the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) - Finance Code 001 for PhD funding. JMS thanks EPSRC UK National Mass Spectrometry Facility at Swansea University. ET thanks the Japan Student Services Organization (JASSO) for a PhD studentship. GC acknowledges the EPSRC for funding (EP/E036244/1). Open data available at: http://dx.doi.org/10.5525/gla.researchdata.1128. We acknowledge the Centre for Plastic Electronics (CPE), Imperial College London for access to their IPCE facility.Notes and references
- A. Yella, C. Mai, S. M. Zakeeruddin, S. Chang, C. Hsieh, Y. Yeh and M. Grätzel, Angew. Chem., 2014, 126, 3017–3021 CrossRef.
- N. Manfredi, B. Cecconi and A. Abbotto, Eur. J. Org. Chem., 2014, 7069–7086 CrossRef CAS.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- S. Mathew, A. Yella, P. Gao, R. Humphry-baker, B. F. E. Curchod, N. Ashari-astani, I. Tavernelli, U. Rothlisberger, K. Nazeeruddin and M. Gra, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed.
- J. Gong, K. Sumathy, Q. Qiao and Z. Zhou, Renewable Sustainable Energy Rev., 2017, 68, 234–246 CrossRef CAS.
- P. P. Kumavat, P. Sonar and D. S. Dalal, Renewable Sustainable Energy Rev., 2017, 78, 1262–1287 CrossRef.
- W. Zhang, Y. Wu, H. W. Bahng, Y. Cao, C. Yi, Y. Saygili, J. Luo, Y. Liu, L. Kavan, J. Moser, A. Hagfeldt, H. Tian, S. M. Zakeeruddin, W. Zhu and M. Gr, Energy Environ. Sci., 2018, 10, 8–10 Search PubMed.
- X. Zhang, A. Bolag, W. Yun, C. Eerdun, Y. Du, J. Ning and H. Alata, Chem. Lett., 2018, 48, 204–207 CrossRef.
- P. Kumaresan, S. Vegiraju, Y. Ezhumalai, S. L. Yau, C. Kim, W. H. Lee and M. C. Chen, Polymer, 2014, 6, 2645–2669 Search PubMed.
- X. Dai, H. Feng, W. Chen, Y. Yang, L. Nie, L. Wang, D. Kuang, H. Meier and D. Cao, Dyes Pigm., 2015, 122, 13–21 CrossRef CAS.
- H. L. Jia, M. D. Zhang, W. Yan, X. H. Ju and H. G. Zheng, J. Mater. Chem. A, 2016, 4, 11782–11788 RSC.
- Y. Hu, A. Ivaturi, M. Planells, C. L. Boldrini, A. O. Biroli and N. Robertson, J. Mater. Chem. A, 2016, 4, 2509–2516 RSC.
- A. Abate, M. Planells, D. J. Hollman, S. D. Stranks, A. Petrozza, A. R. S. Kandada, Y. Vaynzof, S. K. Pathak, N. Robertson and H. J. Snaith, Adv. Energy Mater., 2014, 4, 1–7 Search PubMed.
- A. Listorti, B. O'Regan and J. R. Durrant, Chem. Mater., 2011, 23, 3381–3399 CrossRef CAS.
- N. T. Hai, L. Q. Bao, S. Thogiti, R. Cheruku, K. S. Ahn and J. H. Kim, J. Nanosci. Nanotechnol., 2017, 17, 3181–3187 CrossRef.
- W. C. Chen, S. Nachimuthu and J. C. Jiang, Sci. Rep., 2017, 7, 1–13 CrossRef PubMed.
- I. Arbouch, D. Cornil, Y. Karzazi, B. Hammouti, R. Lazzaroni and J. Cornil, Phys. Chem. Chem. Phys., 2017, 19, 29389–29401 RSC.
- L. Zhang and J. M. Cole, ACS Appl. Mater. Interfaces, 2015, 7, 3427–3455 CrossRef CAS PubMed.
- R. Kesavan, F. Attia, R. Su, P. Anees, A. El-Shafei and A. V. Adhikari, J. Phys. Chem. C, 2019, 123, 24383–24395 CrossRef CAS.
- Ö. Birel, S. Nadeem and H. Duman, J. Fluoresc., 2017, 27, 1075–1085 CrossRef PubMed.
- S. S. M. Fernandes, M. C. R. Castro, A. I. Pereira, A. Mendes, C. Serpa, J. Pina, L. L. G. Justino, H. D. Burrows and M. M. M. Raposo, ACS Omega, 2017, 2, 9268–9279 CrossRef CAS PubMed.
- G. Koyyada, R. K. Chitumalla, S. Thogiti, J. H. Kim, J. Jang, M. Chandrasekharam and J. H. Jung, Molecules, 2019, 24, 1–18 CrossRef PubMed.
- P. Naik, R. Su, M. R. Elmorsy, A. El-Shafei and A. V. Adhikari, Photochem. Photobiol. Sci., 2018, 17, 302–314 CrossRef CAS PubMed.
- C. Qin, A. Mirloup, N. Leclerc, A. Islam and A. El-shafei, Adv. Energy Mater., 2014, 2, 2–7 Search PubMed.
- Y. C. Chen and J. T. Lin, Sustainable Energy Fuels, 2017, 1, 969–985 RSC.
- H. Klfout, A. Stewart, M. Elkhalifa and H. He, ACS Appl. Mater. Interfaces, 2017, 9, 39873–39889 CrossRef CAS PubMed.
- G. Wu, F. Kong, Y. Zhang, X. Zhang, J. Li, W. Chen, W. Liu, Y. Ding, C. Zhang, B. Zhang, J. Yao and S. Dai, J. Phys. Chem., 2014, 118, 8756–8765 CrossRef CAS PubMed.
- Y. S. Yang, H. Do Kim, J. H. Ryu, K. K. Kim, S. S. Park, K. S. Ahn and J. H. Kim, Synth. Met., 2011, 161, 850–855 CrossRef CAS.
- Y. H. Lee, H. J. Yun, S. K. Choi, Y. S. Yang, T. Park, K. S. Ahn, T. Suresh and J. H. Kim, Synth. Met., 2016, 217, 248–255 CrossRef CAS.
- H. Wu, Z. Huang, T. Hua, C. Liao, H. Meier, H. Tang, L. Wang and D. Cao, Dyes Pigm., 2019, 165, 103–111 CrossRef CAS.
- U. Eiamprasert, J. Sudchanham and P. Surawatanawong, J. Photochem. Photobiol., A, 2018, 352, 86–97 CrossRef CAS.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed.
- Y. Wang, Q. Liao, G. Wang, H. Guo, X. Zhang, M. A. Uddin and X. Guo, Chem. Mater., 2017, 29, 4109–4121 CrossRef CAS.
- C. Teng, X. Yang, C. Yang, H. Tian, S. Li, X. Wang, A. Hagfeldt and L. Sun, J. Phys. Chem. C, 2010, 114, 11305–11313 CrossRef CAS.
- H. Jia, K. Shen, X. Ju, M. Zhang and H. Zheng, New J. Chem., 2016, 40, 2799–2805 RSC.
- A. F. Buene, E. E. Ose, A. G. Zakariassen, A. Hagfeldt and B. H. Hoff, J. Mater. Chem. A, 2019, 7, 7581–7590 RSC.
- R. Tarsang, V. Promarak, T. Sudyoadsuk, S. Namuangruk, N. Kungwan, P. Khongpracha and S. Jungsuttiwong, RSC Adv., 2015, 5, 38130–38140 RSC.
- Z. Shen, X. Zhang, F. Giordano, Y. Hu and J. Hua, Mater. Chem. Front., 2016, 1, 181–189 RSC.
- A. Baheti, P. Singh, C. P. Lee, K. R. J. Thomas and K. C. Ho, J. Org. Chem., 2011, 76, 4910–4920 CrossRef CAS PubMed.
- N. E. Jackson, B. M. Savoie, K. L. Kohlstedt, M. Olvera De La Cruz, G. C. Schatz, L. X. Chen and M. A. Ratner, J. Am. Chem. Soc., 2013, 135, 10475–10483 CrossRef CAS PubMed.
- C. Mcdowell, K. Narayanaswamy, B. Yadagiri, M. C. Heifner, V. Gupta, C. Risko and P. Singh, J. Mater. Chem. A, 2018, 6, 383–394 RSC.
- K. Nakao, M. Nishimura, T. Tamachi, Y. Kuwatani, H. Miyasaka, T. Nishinaga and M. Iyoda, J. Am. Chem. Soc., 2006, 128, 16740–16747 CrossRef CAS PubMed.
- D. K. Frantz, A. Linden, K. K. Baldridge and J. S. Siegel, J. Am. Chem. Soc., 2012, 134, 1528–1535 CrossRef CAS PubMed.
- M. El Garah, A. Dianat, A. Cadeddu, R. Gutierrez, M. Cecchini, T. R. Cook, A. Ciesielski, P. J. Stang, G. Cuniberti and P. Samorì, Small, 2016, 12, 343–350 CrossRef CAS PubMed.
- M. D. Peeks, P. Neuhaus and H. L. Anderson, Phys. Chem. Chem. Phys., 2016, 18, 5264–5274 RSC.
- S. Q. Zhang, Z. Y. Liu, W. F. Fu, F. Liu, C. M. Wang, C. Q. Sheng, Y. F. Wang, K. Deng, Q. D. Zeng, L. J. Shu, J. H. Wan, H. Z. Chen and T. P. Russell, ACS Nano, 2017, 11, 11701–11713 CrossRef CAS PubMed.
- I. Gru, H. J. Eggimann, L. M. Herz and H. L. Anderson, J. Am. Chem. Soc., 2019, 141, 7965–7971 CrossRef PubMed.
- D. J. Carmona, D. R. Contreras, O. A. Douglas-Gallardo, S. Vogt-Geisse, P. Jaque and E. Vöhringer-Martinez, Theor. Chem. Acc., 2018, 137, 1–11 Search PubMed.
- H. Tian, X. Yang, R. Chen, R. Zhang, A. Hagfeldt and L. Sun, J. Phys. Chem. C, 2008, 112, 11023–11033 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. J. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, 2009 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1me00009h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |