
Potency and metabolic stability: a molecular hybrid case in the design of novel PF74-like small molecules targeting HIV-1 capsid protein†
Rajkumar Lalji
Sahani‡
 a,
Thamina
Akther‡
a,
Maria E.
Cilento
b,
Andres Emanuelli
Castaner
b,
Huanchun
Zhang
b,
Karen A.
Kirby
bc,
Jiashu
Xie
a,
Stefan G.
Sarafianos
bc and
Zhengqiang
Wang
*a
a,
Thamina
Akther‡
a,
Maria E.
Cilento
b,
Andres Emanuelli
Castaner
b,
Huanchun
Zhang
b,
Karen A.
Kirby
bc,
Jiashu
Xie
a,
Stefan G.
Sarafianos
bc and
Zhengqiang
Wang
*a
aCenter for Drug Design, College of Pharmacy, University of Minnesota, Minneapolis, MN 55455, USA. E-mail: wangx472@umn.edu
bLaboratory of Biochemical Pharmacology, Department of Pediatrics, Emory University School of Medicine, Atlanta, GA 30322, USA
cChildren's Healthcare of Atlanta, Atlanta, GA 30322, USA
First published on 28th September 2021
Abstract
PF74 (1) is a potent and well-characterized prototypical small molecule targeting human immunodeficiency virus type 1 (HIV-1) capsid protein (CA), but not a viable antiviral lead due to the lack of metabolic stability. We report herein our molecular hybridization-based medicinal chemistry efforts toward potent and metabolically stable PF74-like small molecules. The design of the new sub-chemotype 4 rationally combines binding features of two recently reported PF74-like compounds 2 and 3. The subsequent confirmation and structure–activity relationship (SAR) of hit 4a entailed the chemical synthesis of 37 novel analogs, most of which showed modest but meaningful thermal shift, and low μM antiviral activity. The most potent analogs (4a, 4d, 4o, and 4r) all exhibited noticeably improved metabolic stability over PF74. Molecular modeling suggests that these new analogs bind to the PF74 binding site. Overall, our work demonstrated that the molecular hybridization approach is suitable for designing compounds with balanced potency and metabolic stability.
Introduction
A few classes of antiviral drugs have been approved for treating HIV-1 infection under combination antiretroviral therapy (cART) settings.1 However, HIV-1 remains incurable and the inevitable emergence of viral strains resistant to current drug classes continues to necessitate the development of mechanistically novel antivirals. HIV-1 capsid protein (CA) represents a new drug target of increasing interest.2 Functionally, CA and the capsid core play multiple roles in the HIV-1 replication cycle, including mainly uncoating, cytoplasmic trafficking, nuclear entry, and integration in the early stage and viral assembly in the late stage.3–5 On the molecular level, these functions are driven by CA–CA interactions and/or CA–host factor interactions. Research geared toward disrupting these molecular interactions has identified multiple small molecule prototypes binding to a few different sites of CA.1 Of these, PF74 (ref. 6 and 7) (1) is particularly well-characterized as a potent antiviral compound by binding to a pocket at the CA N-terminal domain (CANTD) and the adjacent CA C-terminal domain (CACTD) interfaces8 to confer both early stage and late stage antiviral activities. Significantly, this same binding pocket is used by host factors9 important for viral nuclear entry, such as NUP153 (ref. 10 and 11) and CPSF6,12,13 and targeted by GS-6207 (lenacapavir),14,15 a long-acting injectable antiviral candidate in advanced clinical development.The clinical validation of the PF74 binding site as a drug target has prompted notable medicinal chemistry efforts in search of novel PF74-like small molecules.7,16–22 Many of the efforts have aimed to address the major weakness of PF74 as an antiviral lead, the prohibitively low metabolic stability.19–21 When measured in human liver microsomes (HLMs), PF74 displayed a half-life (t1/2) of a round 1 min (0.7 min (ref. 21) and 1.3 min (ref. 17), Fig. 1, A), predicting excessive liver extraction and a complete lack of oral bioavailability. We have previously pursued medicinal chemistry to replace the indole moiety with various chemical moieties less electron-rich to mitigate the oxidative metabolism.20,21 A particularly interesting analog resulted from these efforts was compound 2 (ref. 21) which exhibited drastically improved metabolic stability (51-fold) while largely retaining the antiviral potency (Fig. 1, B). The design of compound 2 also featured the substitution of the para-H (right) with a chlorine atom (highlighted), which may have contributed to the observed metabolic stability, and may also benefit target binding via halogen bonding.7 In the meantime, Sun et al. recently reported another redesigned PF74-like compound 3 (ref. 17) with substantially improved antiviral activity (5.8-fold), conferred by the replacement of the indole moiety by a phenylsulfonyl oxopiperazine, and the substitution of the para-H with a methoxy group. However, the metabolic stability was only modestly (3.2-fold) enhanced, possibly due to the methoxy group as a metabolic soft spot.23 Nevertheless, the stronger potency of 3 and the enhanced metabolic stability of 2 make a strong case for molecular hybridization to identify novel PF74-like small molecules with balanced potency and metabolic stability. To that end, we further redesigned our compound 2 into a novel sub-chemotype 4 (Fig. 1, C) by using the design features from both 2 and 3. Specifically, we retained the para-Cl on the phenyl ring, and expanded the benzamide moiety into an aryl acyl oxopiperazine. We report herein the synthesis, the biophysical thermal shift assay (TSA), the antiviral and cytotoxicity results of 37 analogs. Selected compounds were also tested for microsomal stability in HLMs.
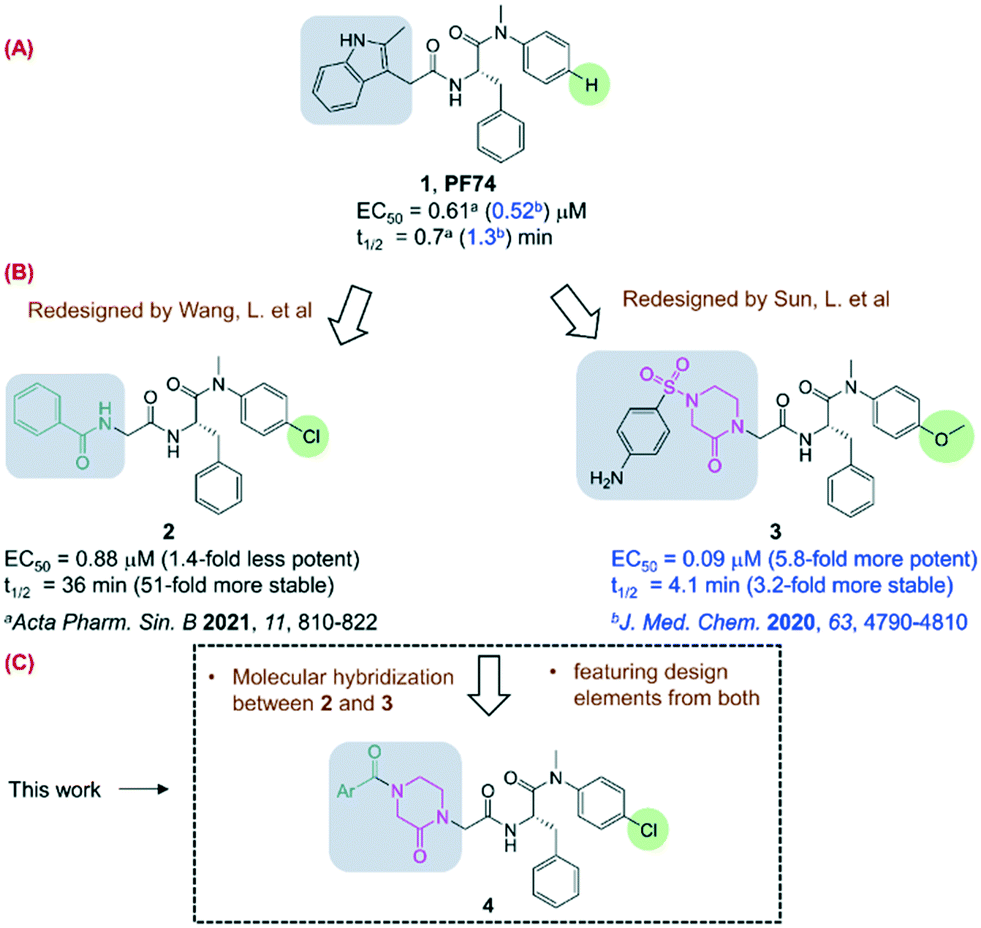 | ||
| Fig. 1 Design of a novel PF74-like chemotype for targeting the PF74 binding site of HIV-1 CA. (A) PF74 showed sub-μM potency and extremely poor microsomal stability; (B) previously PF74 was redesigned by replacing the para-H (right) and the indole ring (left) into 2 with 51-fold improvement in metabolic stability, and 3 with 5.8-fold improvement in antiviral potency; (C) the current work uses a molecular hybridization approach combining desirable features from both 2 and 3 to design a novel chemotype 4. | ||
Results and discussion
Chemistry
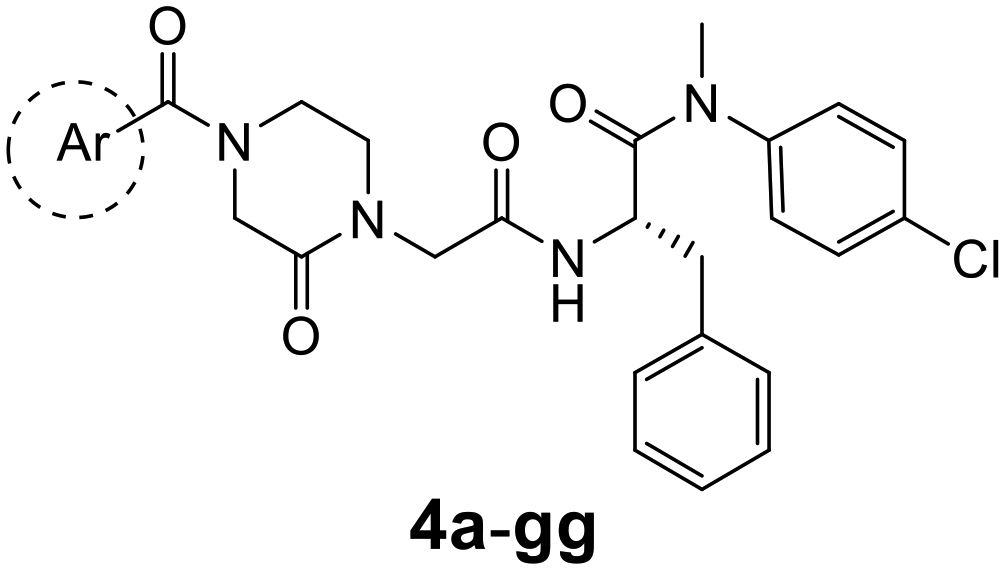
The synthetic route for the preparation of analogs 4a–4z, 4aa–4gg, and 13 is shown in Scheme 1. Coupling of 4-chloro-N-methylaniline (6) with Boc-protected phenylalanine (5) afforded 7. The Boc protecting group was removed using TFA to produce intermediate8,16 which was used for the subsequent amide coupling with the commercially available bromoacetic acid to produce 9. From this reaction, we also isolated compound 10 as a byproduct. To probe the SAR of the amide moiety (the indole equivalent), intermediates 11 and 14 were prepared by reacting 9 with 1-Boc-piperazine and 1-Boc-3-oxopiperazine, respectively, and were then treated with TFA for Boc deprotection to give 12 and 15. This generic scheme allowed the synthesis of 4a–4z, 4aa–4gg, and 13 upon the final amide coupling reaction. The amino analogs 4r–4t were prepared via the NO2 reduction from analogs 4o–4q.24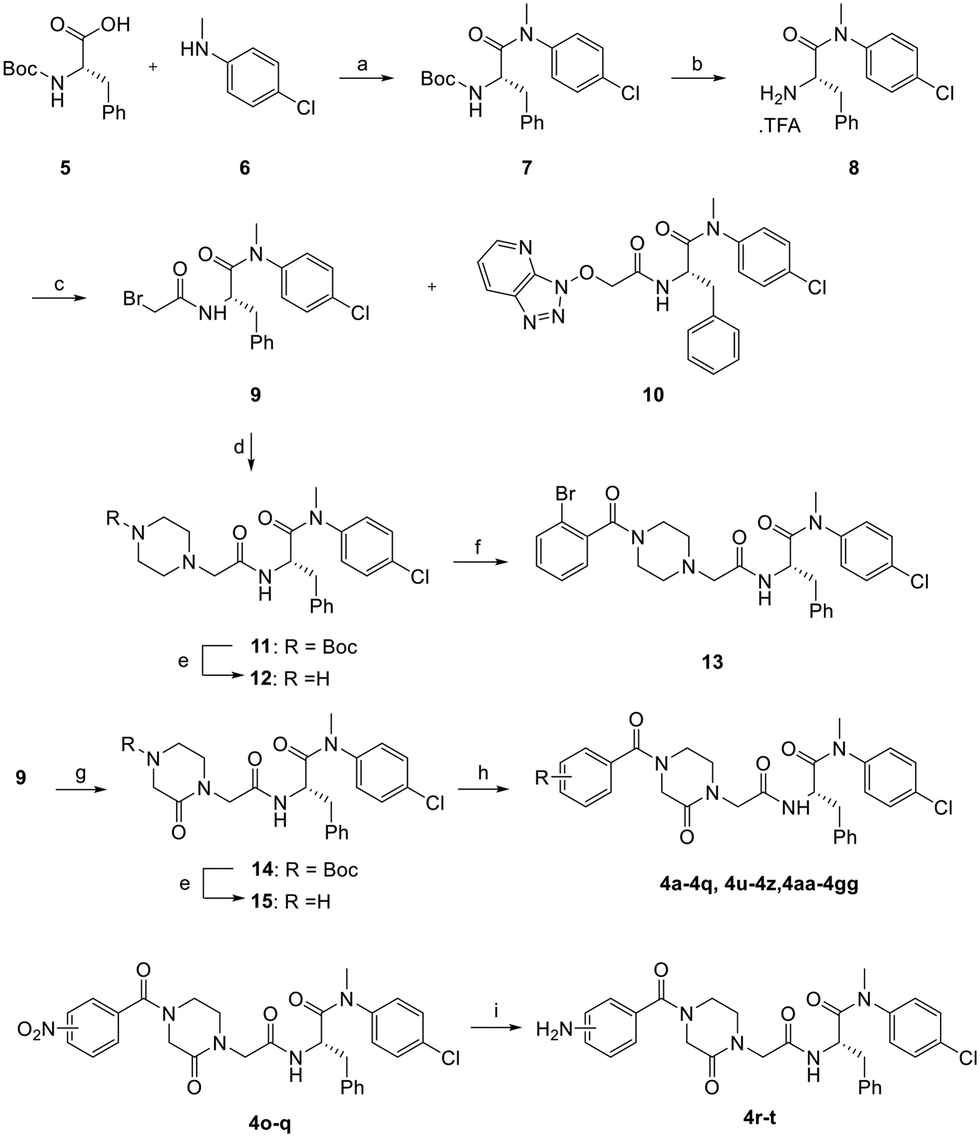 | ||
Scheme 1 Synthesis of analogs 4a–4z, 4aa–4gg, and 13. Reagents and conditions: a) HATU, DIPEA, DCM, rt, 12 h, 98%; b) TFA, DCM, 50 °C, 12 h, 98%; c) bromoacetic acid, HATU, DIPEA, DCM, rt, 12 h, 9 (61%), 10 (8%); d) 1-Boc-piperazine, K2CO3, DMF, 50 °C, 12 h, 91%; e) TFA, DCM, 50 °C, 12 h, 83%; f) 2-bromobenzoic acid, HATU, DIPEA, DMF, rt, 12 h, 73%; g) 1-Boc-3-oxopiperazine, K2CO3, DMF, 50 °C, 12 h, 55%; h) corresponding substituted benzoic acid, HATU, DIPEA, DMF, rt, 12 h or substituted benzoyl chloride, Et3N, DCM, 12 h, 4a–4q, 4u–4z, 4aa–4gg (50–98%), 13 (41%); i) Fe, CaCl2, EtOH/H2O = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 70 °C, 12 h, 39–62%. :1, 70 °C, 12 h, 39–62%. | ||
The preparation of analog 20 is described in Scheme 2. 3-Chloro-5-hydroxybenzonitrile 16 was treated with 2-chloro-3-fluoro-4-(trifluoromethyl)pyridine 17 (ref. 25) to afford ether intermediate 18 which was converted to carboxylic acid intermediate 19 upon cyanide hydrolysis in the presence of potassium hydroxide. Amide coupling between acid 19 and amine 9 using similar methods described in Scheme 1 yielded analog 20.
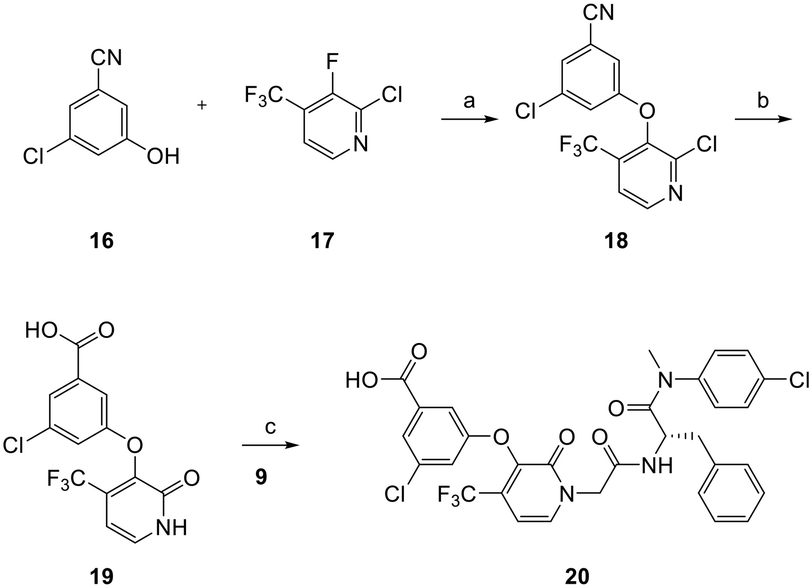 | ||
| Scheme 2 Synthesis of analog 20. Reagents and conditions: a) K2CO3, NMP, 80 °C, 12 h, 67%; b) KOH, t-BuOH, 100 °C, 12 h, dil. HCl, 76%; c) K2CO3, DMF, 50 °C, 12 h, 41%. | ||
Biological assays and SAR
We began the compound evaluation with a biophysical TSA which directly measures the melting point change (ΔTm) of the target protein upon compound binding. A positive ΔTm value (right shift) designates a stabilizing effect whereas a negative value (left shift) indicates a destabilizing effect of the compound on the protein. All 37 analogs were also tested in a cell-based antiviral assay against HIV-1 and a cytotoxicity assay. The values of the control compound PF74 in the current assays were consistent with previous reports.7,19–21 From these assays, designed hit 4a caused a modest but meaningful right shift in the TSA (ΔTm = 1.0 °C) and conferred low μM antiviral activity (EC50 = 3.8 μM, Table 1). Subsequently, a short series of compounds was synthesized to probe the SAR around the indole equivalent (highlighted in blue, Table 1). Not surprisingly, compound 15 with only the oxopiperazine moiety showed no significant shift in TSA or antiviral activity at concentrations up to 20 μM. In addition, substituting piperazine for the oxopiperazine resulted in a compound (13) with substantially reduced antiviral activity (EC50 = 20 μM for 13 vs. EC50 = 3.8 μM for 4a). Interestingly, when the indole equivalent was an aza-benzotriazole moiety (10: ΔTm = 1.0 °C, EC50 = 5.2 μM) or a diaryl ether moiety (20: ΔTm = 1.9 °C, EC50 > 20 μM), the resulting compounds conferred similar or better thermal shift (Table 1). However, compound 10 may lack chemical stability, whereas compound 20 did not show antiviral activity at concentrations up to 20 μM. In the cytotoxicity assay, none of these analogs showed significant cytotoxicity at concentrations up to 50 μM. Collectively, results from this concise SAR confirm compound 4a as a hit. The subsequent SAR efforts focused on the left aromatic moiety (Ar, Table 2) of hit 4a. The SAR was based on the synthesis of 33 analogs, of which most (4a–4z) feature an Ar moiety of a mono-substituted phenyl ring. The mono-substituent spans a range of functional groups of different electronic natures, including halogens (Br, Cl, F) and pseudohalogen (CN), SO2F, NO2, NH2, OH, and CF3. For each monosubstituent, three sets of compounds (ortho, meta, and para) were synthesized, except for the SO2F substituent with which the synthesis of the ortho analog was not successful. In addition, we also synthesized analogs featuring the unsubstituted phenyl, a pyridine of different substitution patterns, pyrazole, or indole as the Ar moiety (Table 2). Out of the 33 analogs in this SAR, 18 produced significant effect (ΔTm = 1.0–2.6 °C) in the TSA, and 20 inhibited HIV-1 in low μM range (EC50 = 2.7–15.6 μM). Overall, testing of these compounds in TSA and the antiviral assay revealed two prominent SAR trends. First of all, electron-withdrawing substituents in the Ar appeared to confer discernibly larger thermal shift in the TSA and overall better potency in the antiviral assay than electron-donating substituents (4a–4qvs.4r–4w, Table 2). The same trend was largely conformed to with analogs having a heteroaryl moiety (4bb–4gg) as electron-deficient chloropyridine (4ee: ΔTm = 1.1 °C, EC50 = 7.7 μM) produced a significant thermal shift and low μM antiviral activity, whereas the electron-rich indole (4gg: ΔTm = 0.3 °C, EC50 > 20 μM) did not confer noticeable effect in either assay. Second, the ortho substitution was favored over meta/para for both the thermal shift and the antiviral potency, which is particularly manifested by analogs substituted with CN (4jvs.4k/4l), NO2 (4ovs.4p/4q), NH2 (4rvs.4s/4t) and CF3 (4xvs.4y/4z). In the cytotoxicity assay, none of the 33 analogs showed toxicity at concentrations up to 50 μM.| Compd | Structure | TSA ΔTma (°C) | EC50b (μM) | CC50c (μM) |
|---|---|---|---|---|
| a TSA: thermal shift assay. ΔTm: change of CA hexamer melting point in presence of compound compared to DMSO control. b Concentration of compound inhibiting HIV-1 replication by 50%, expressed as the mean ± standard deviation from at least two independent experiments. c Concentration of compound causing 50% cell death, expressed as the mean ± standard deviation from at least two independent experiments. | ||||
| 4a |
|
1.1 ± 0.2 | 3.8 ± 0.5 | >50 |
| 15 |
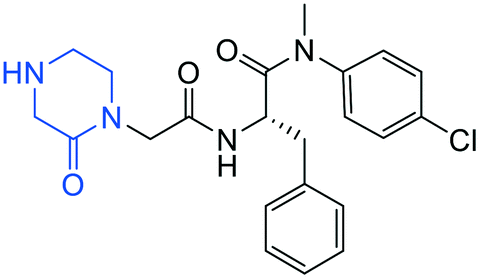
|
0.1 ± 0.1 | >20 | >50 |
| 13 |
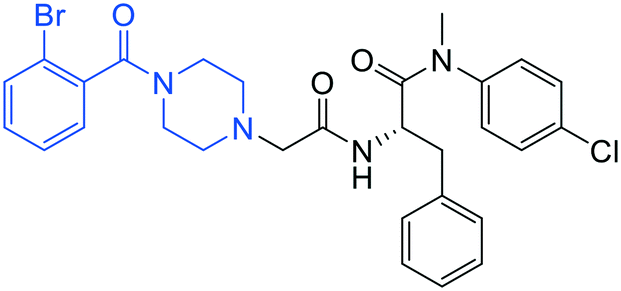
|
0.7 ± 0.1 | 20 ± 2 | >50 |
| 10 |
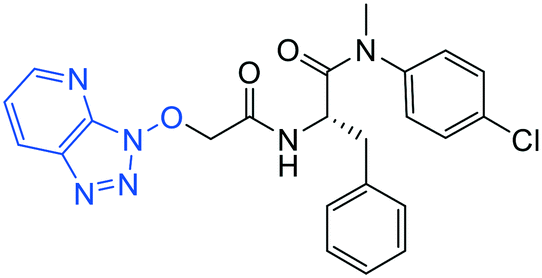
|
1.0 ± 0.3 | 5.2 ± 0.4 | >50 |
| 20 |
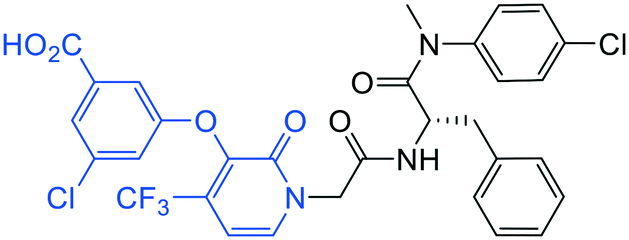
|
1.9 ± 0.4 | >20 | >50 |
| 1 |
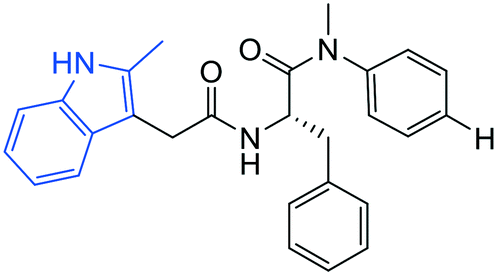
|
6.9 ± 0.3 | 0.6 ± 0.1 | >50 |
|
|
||||
|---|---|---|---|---|
| Compd | Ar | TSA ΔTma (°C) | EC50b (μM) | CC50c (μM) |
| a TSA: thermal shift assay. ΔTm: change of CA hexamer melting point in presence of compound compared to DMSO control. b Concentration of compound inhibiting HIV-1 replication by 50%, expressed as the mean ± standard deviation from at least two independent experiments. c Concentration of compound causing 50% cell death, expressed as the mean ± standard deviation from at least two independent experiments. | ||||
| 4a |
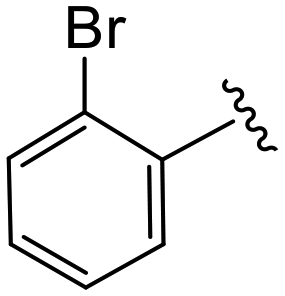
|
1.1 ± 0.2 | 3.8 ± 0.5 | >50 |
| 4b |
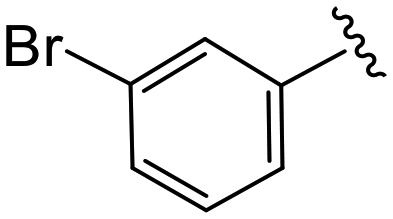
|
1.1 ± 0.4 | 9.0 ± 0.8 | >50 |
| 4c |
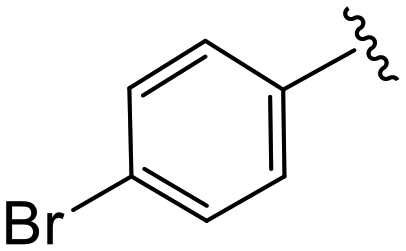
|
0.8 ± 0.2 | 10.2 ± 1.3 | >50 |
| 4d |
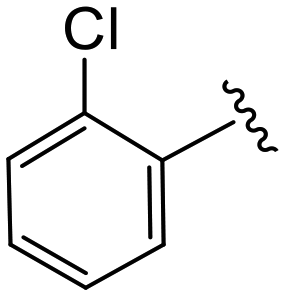
|
1.5 ± 0.4 | 6.4 ± 1.1 | >50 |
| 4e |
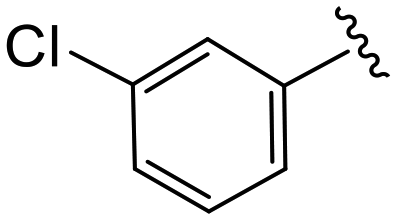
|
1.0 ± 0.4 | 10.2 ± 0.9 | >50 |
| 4f |
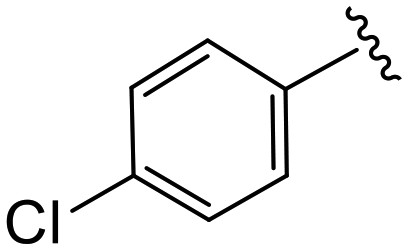
|
1.3 ± 0.4 | 9.2 ± 1.3 | >50 |
| 4g |
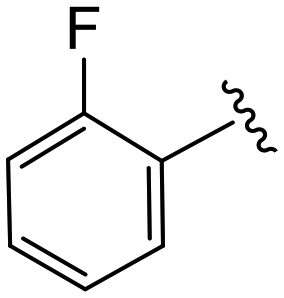
|
1.6 ± 0.3 | 9.5 ± 0.8 | >50 |
| 4h |
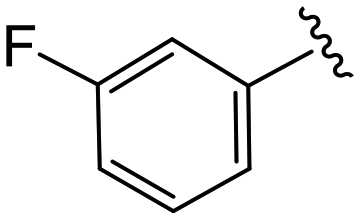
|
1.7 ± 0.3 | 13.3 ± 2.9 | >50 |
| 4i |
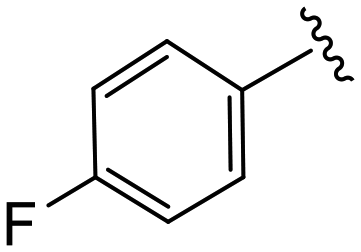
|
1.3 ± 0.5 | 11.5 ± 2.7 | >50 |
| 4j |

|
2.1 ± 0.1 | 7.5 ± 0.1 | >50 |
| 4k |

|
1.5 ± 0.4 | 12.9 ± 0.8 | >50 |
| 4l |
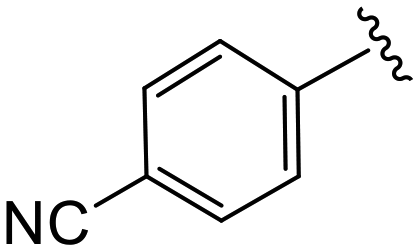
|
1.7 ± 0.3 | 11.0 ± 1.7 | >50 |
| 4m |
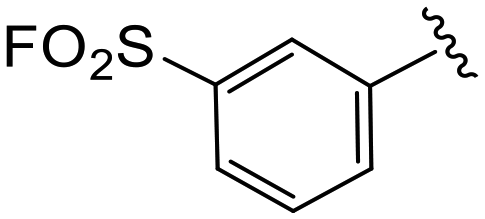
|
1.1 ± 0.2 | >20 | >50 |
| 4n |
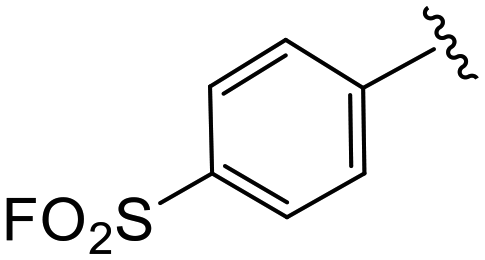
|
0.8 ± 0.4 | >20 | >50 |
| 4o |
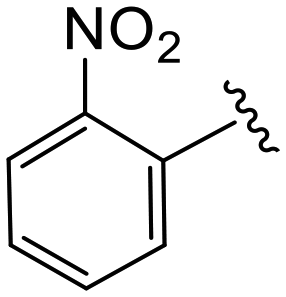
|
2.6 ± 0.2 | 2.7 ± 0.1 | >50 |
| 4p |
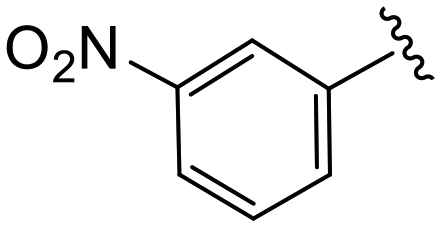
|
1.5 ± 0.3 | 9.3 ± 0.7 | >50 |
| 4q |
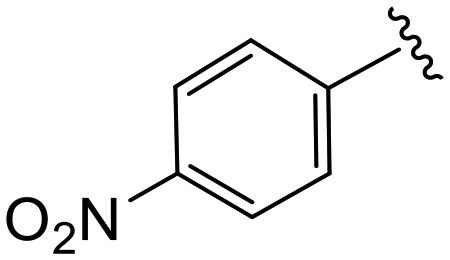
|
1.5 ± 0.1 | >20 | >50 |
| 4r |
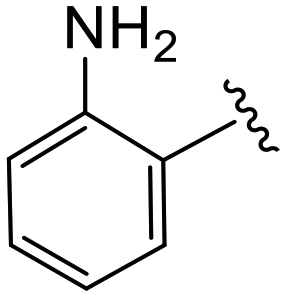
|
0.7 ± 0.1 | 4.5 ± 0.2 | >50 |
| 4s |
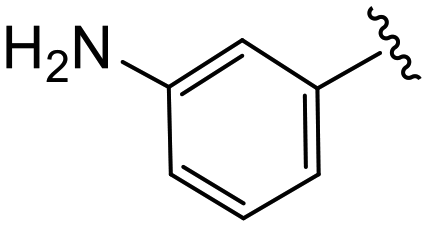
|
0.3 ± 0.1 | >20 | >50 |
| 4t |
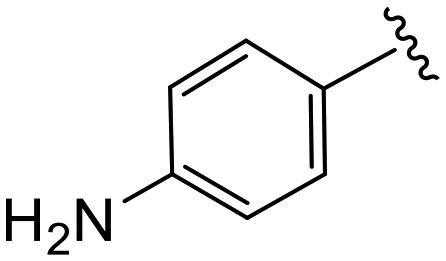
|
0.5 ± 0.1 | >20 | >50 |
| 4u |
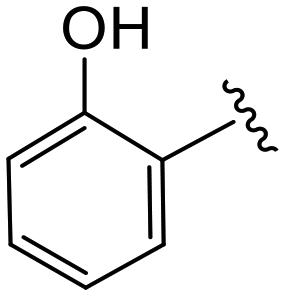
|
0.8 ± 0.2 | >20 | >50 |
| 4v |
|
0.8 ± 0.3 | >20 | >50 |
| 4w |
|
0.7 ± 0.3 | >20 | >50 |
| 4x |
|
1.4 ± 0.2 | 7.2 ± 3 | >50 |
| 4y |
|
0.3 ± 0.3 | >20 | >50 |
| 4z |
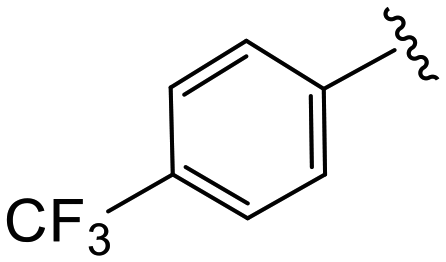
|
0.5 ± 0.2 | >20 | >50 |
| 4aa |
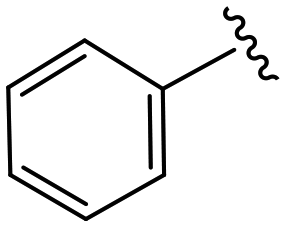
|
0.7 ± 0.1 | >20 | >50 |
| 4bb |
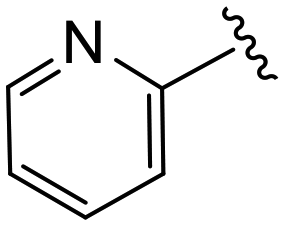
|
0.8 ± 0.2 | 12.7 ± 2.2 | >50 |
| 4cc |
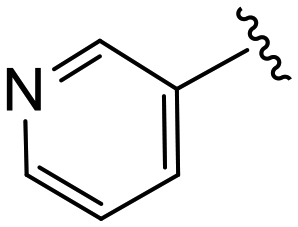
|
0.7 ± 0.2 | 12.5 ± 4.1 | >50 |
| 4dd |
|
0.7 ± 0.2 | 15.6 ± 2.1 | >50 |
| 4ee |
|
1.1 ± 0.3 | 7.7 ± 0.1 | >50 |
| 4ff |
|
1.3 ± 0.2 | >20 | >50 |
| 4gg |
|
0.3 ± 0.1 | >20 | >50 |
Metabolic stability in liver microsomes
As mentioned earlier, a main goal of designing new PF74-like small molecules is to improve metabolic stability. The metabolic liability of PF74 likely results from the cytochrome P450 (CYPs)-mediated oxidative metabolism, as it is known that peptidomimetics such as PF74 are good substrates for CYP3A4.26 To evaluate the metabolic stability of 4a and its analogs, we tested 10 selected compounds with low μM antiviral activity, along with PF74 and a control compound verapamil, in both HLMs and mouse liver microsomes (MLMs). Verapamil is known to be metabolized by liver microsomes and therefore was utilized as in house quality control to ensure the proper functionality of the incubation systems. The values of verapamil and PF74 are consistent with previous reports.7,19–21 As shown in Table 3, improved metabolic stability was observed with all analogs, with the exception of compound 10 which contains a highly labile N–O bond. The improvement is likely due to the introduction of a Cl atom on the right phenyl ring, and the replacement of the indole ring with less electron-rich moieties. Particularly, four of the most potent compounds in the antiviral assay, 4a (EC50 = 3.8 μM, t1/2 = 7.9 min, 11.3-fold over PF74), 4d (EC50 = 6.4 μM, t1/2 = 6.2 min, 8.9-fold over PF74), 4o (EC50 = 2.7 μM, t1/2 = 3.8 min, 5.4-fold over PF74), and 4r (EC50 = 4.5 μM, t1/2 = 4.4 min, 6.3-fold over PF74) all displayed substantially longer half-life (t1/2) than PF74 in HLMs.| Compd | Microsomal stability t1/2a (min) | |
|---|---|---|
| Human | Mouse | |
| a Data are represented as mean ± standard deviation (SD) (n = 2). b Values for PF74 were also reported in our previous publications.7,19–21 | ||
| 10 | 0.8 ± 0.03 | 0.5 ± 0.03 |
| 4a | 7.9 ± 0.3 | 1.8 ± 0.03 |
| 4b | 4.8 ± 0.1 | 1.4 ± 0.05 |
| 4d | 6.2 ± 0.3 | 1.1 ± 0.02 |
| 4f | 7.5 ± 0.1 | 1.3 ± 0.1 |
| 4g | 4.1 ± 0.2 | 0.9 ± 0.01 |
| 4o | 3.8 ± 0.04 | 0.9 ± 0.02 |
| 4p | 2.6 ± 0.1 | 0.8 ± 0.01 |
| 4r | 4.4 ± 0.004 | 1.3 ± 0.01 |
| 4ee | 3.6 ± 0.05 | 1.0 ± 0.01 |
| PF74 | 0.7 ± 0.02 | 0.6 ± 0.03 |
| Verapamil | 11.5 | 3.9 |
Molecular modeling
To computationally characterize the target binding of the newly designed analogs, we performed molecular docking for four selected compounds, 13, 4a, 4x and 4y using the co-crystal structure of PF74-bound HIV-1 CA (PDB code: 4XFZ8). A control docking was conducted with PF74 (docked pose shown in Fig. S1†). Overall, these analogs all docked reasonably well into the PF74 binding site (Fig. 2), with docking scores slightly better than or comparable to PF74 (Fig. 2, legend). Common molecular interactions for all ligands, including PF74, include two H-bonds with N57, and one with K70; and a π–cation interaction with K70. In addition, an inter-domain H-bond with K182 of the adjacent CACTD was observed with the ortho-substituted 4a (Fig. 2, A) and 4x (Fig. 2, C), but not with 13 (Fig. 2, B) or the meta-substituted 4y (Fig. 2, D). The loss of the inter-domain H-bond may account for the potency difference between the two pairs: 4y (ΔTm = 0.3 °C; EC50 > 20 μM) vs.4x (ΔTm = 1.4 °C; EC50 = 7.2 μM), and 13 (ΔTm = 0.7 °C; EC50 = 20 μM) vs.4a (ΔTm = 1.1 °C; EC50 = 3.8 μM).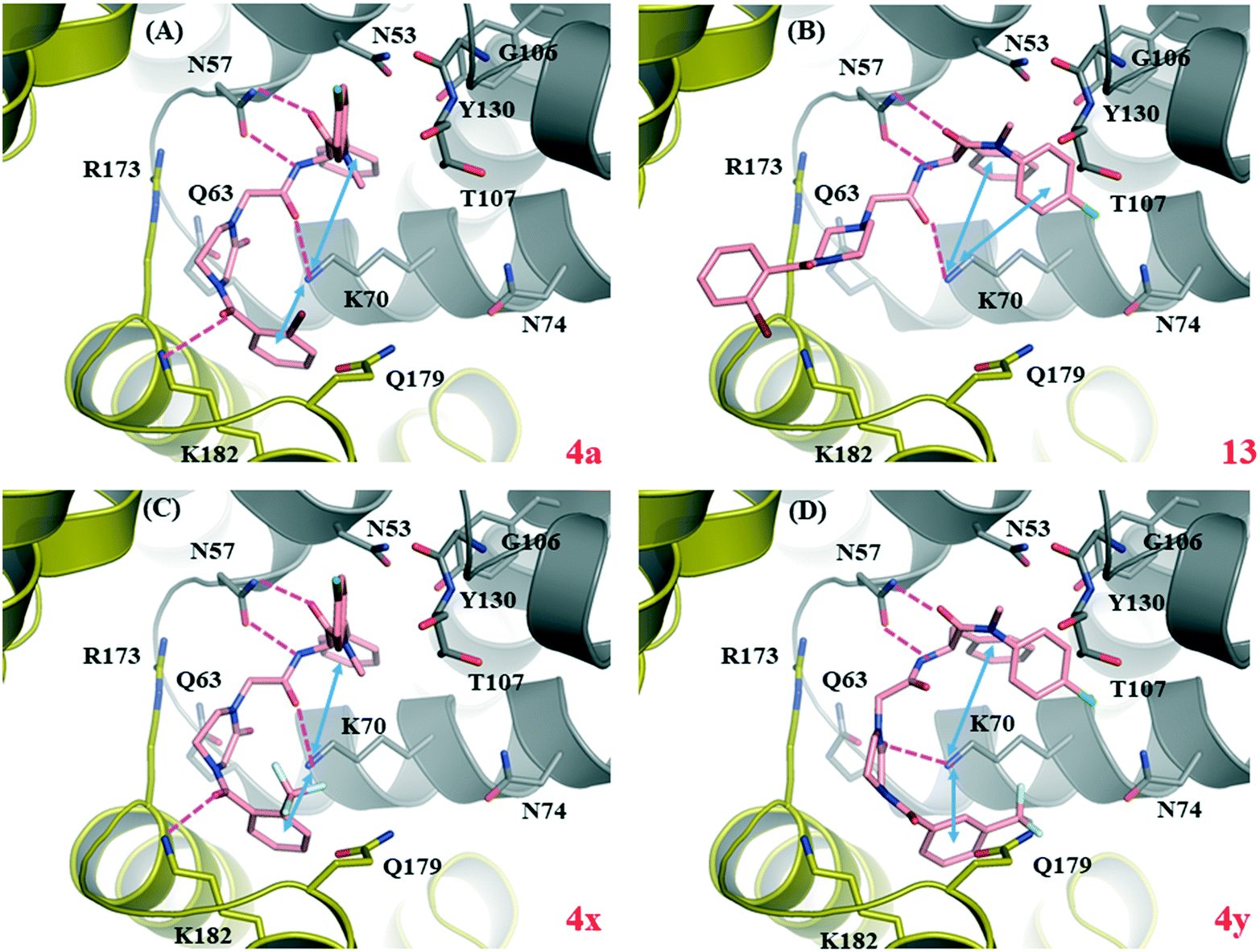 | ||
| Fig. 2 Docking of four selected analogs into PF74-bound HIV-1 CA (PDB ID: 4XFZ8). The Glide score for the PF74 control docking was −5.8 kcal mol−1 (Fig. S1†). Predicted binding modes of (A) compound 4a (Glide score = −6.5 kcal mol−1); (B) compound 13 (Glide score = −5.4 kcal mol−1); (C) compound 4x (Glide score = −6.5 kcal mol−1); and (D) compound 4y (Glide score = −6.1 kcal mol−1). Hydrogen-bonding and cation–π interactions are depicted as magenta dashed lines and blue double-headed arrows, respectively. CANTD is shown in grey cartoon and adjacent CACTD in gold cartoon, with key residues around binding site shown as pink sticks, and ligands shown as magenta sticks. The nitrogen, oxygen, fluorine and chlorine atoms are colored blue, red, light blue, and green, respectively. | ||
Conclusions
To discover potent and metabolically stable PF74-like small molecules, we used a molecular hybridization approach to design novel sub-chemotype 4 based on two recently reported compounds 2 and 3. Through the synthesis of 37 analogs, we confirmed the designed hit 4a and developed a comprehensive SAR around the Ar moiety. Importantly, most of the newly synthesized analogs produced a meaningful thermal shift, and low μM antiviral activity. SAR of the Ar moiety revealed that ortho-substituted, electron-withdrawing groups conferred the best potency. Metabolic stability of the most potent analogs (4a, 4d, 4o and 4r) improved substantially compared to PF74. From molecular docking results, these new analogs are predicted to bind to the PF74 binding site. Overall, although higher potency and stability are desired for PF74-like small molecules, our work proved that the molecular hybridization approach can be valuable in identifying PF74-like compounds with balanced potency and metabolic stability.Experimental
Chemistry
All commercial chemicals were used as supplied unless indicated otherwise. Compounds were purified via flash chromatography using a Combiflash RF-200 (Teledyne ISCO, Lincoln, NE, USA) with RediSep columns (silica) and indicated mobile phase. 1H and 13C NMR spectra were recorded on a Varian 600 MHz (Agilent Technologies, Santa Clara, CA, USA) or Bruker 400 spectrometer (Bruker, Billerica, MA, USA). Mass data were acquired using an Agilent 6230 TOF LC/MS spectrometer (Agilent Technologies). PF74 was synthesized according to reported procedures.7(S)-2-(2-(4-(2-Bromobenzoyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-chlorophenyl)-N-methyl-3-phenylpropanamide (4a). Yield 92%. 1H NMR (400 MHz, MeOD) δ 7.68 (dd, J = 8.2, 4.1 Hz, 1H), 7.49 (t, J = 7.5 Hz, 1H), 7.44–7.36 (m, 2H), 7.33 (d, J = 8.2 Hz, 2H), 7.28–7.14 (m, 3H), 6.96 (q, J = 5.0 Hz, 2H), 6.88 (s, 2H), 4.62 (q, J = 5.9, 4.3 Hz, 1H), 4.36–3.83 (m, 5H), 3.60–3.42 (m, 2H), 3.34 (t, J = 4.7 Hz, 1H), 3.15 (s, 3H), 2.97 (ddd, J = 13.3, 7.9, 2.3 Hz, 1H), 2.76 (dd, J = 13.4, 7.0 Hz, 1H). 13C NMR (100 MHz, MeOD) δ 172.3, 168.9, 167.1, 166.7, 141.9, 137.5, 137.3, 134.4, 133.4, 131.9, 130.2, 129.7, 129.6, 128.9, 128.8, 128.6, 127.5, 119.3, 52.6, 50.1, 45.9, 43.9, 39.4, 38.8, 37.4. HRMS (ESI) m/z calcd for C29H28BrClN4O4 [M − H]+ 611.1055, found: 611.1060.
(S)-2-(2-(4-(4-Bromobenzoyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-chlorophenyl)-N-methyl-3-phenylpropanamide (4c). Yield 98%. 1H NMR (400 MHz, MeOD) δ 7.65 (dd, J = 8.5, 2.2 Hz, 2H), 7.42 (d, J = 8.0 Hz, 2H), 7.36–7.29 (m, 2H), 7.28–7.20 (m, 3H), 6.95 (dd, J = 6.4, 2.9 Hz, 2H), 6.88 (s, 2H), 4.63 (t, J = 7.5 Hz, 1H), 4.43–3.87 (m, 5H), 3.69 (s, 1H), 3.37 (s, 2H), 3.15 (d, J = 1.2 Hz, 3H), 2.97 (dd, J = 13.2, 8.0 Hz, 1H), 2.76 (dd, J = 13.2, 7.2 Hz, 1H). 13C NMR (101 MHz, MeOD) δ 172.3, 170.5, 168.8, 167.1, 141.9, 137.3, 134.3, 132.4, 130.1, 129.7, 129.6, 128.9, 127.4, 52.6, 49.4, 44.8, 40.0, 38.8, 37.4. HRMS (ESI) m/z calcd for C29H28BrClN4O4 [M − H]+ 611.1055, found: 611.1050.
(S)-N-(4-Chlorophenyl)-2-(2-(4-(2-cyanobenzoyl)-2-oxopiperazin-1-yl)acetamido)-N-methyl-3-phenylpropanamide (4j). Yield 89%. 1H NMR (400 MHz, MeOD) δ 7.88 (d, J = 7.7 Hz, 1H), 7.80 (td, J = 7.7, 1.4 Hz, 1H), 7.72–7.60 (m, 2H), 7.36–7.30 (m, 2H), 7.27–7.20 (m, 3H), 6.95 (dd, J = 6.8, 2.8 Hz, 2H), 6.88 (s, 2H), 4.63 (t, J = 7.6 Hz, 1H), 4.42 (s, 1H), 4.19–3.97 (m, 4H), 3.61 (q, J = 5.6, 5.1 Hz, 1H), 3.40 (dq, J = 30.9, 5.6 Hz, 2H), 3.15 (s, 3H), 2.97 (dd, J = 13.2, 7.9 Hz, 1H), 2.77 (dd, J = 13.2, 7.2 Hz, 1H). 13C NMR (100 MHz, MeOD) δ 172.3, 168.9, 167.9, 166.9, 141.9, 139.4, 137.3, 134.4, 134.1, 131.0, 130.2, 129.7, 129.7, 128.9, 128.1, 127.4, 117.1, 110.6, 52.7, 49.4, 46.3, 44.3, 39.8, 38.8, 37.4. HRMS (ESI) m/z calcd for C30H28ClN5O4 [M − H]+ 558.1903, found 558.1900.
(S)-3-(4-(2-((1-((4-Chlorophenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-3-oxopiperazine-1-carbonyl)benzenesulfonyl fluoride (4m). Yield 61%. 1H NMR (400 MHz, CDCl3) δ 8.14–8.08 (m, 2H), 7.86 (dt, J = 7.7, 1.5 Hz, 1H), 7.74 (t, J = 7.9 Hz, 1H), 7.32–7.19 (m, 5H), 6.93 (p, J = 3.2, 2.5 Hz, 2H), 6.77 (d, J = 8.1 Hz, 2H), 4.74 (q, J = 7.5 Hz, 1H), 4.54–3.65 (m, 6H), 3.44 (s, 2H), 3.17 (s, 3H), 2.89 (dd, J = 13.3, 7.7 Hz, 1H), 2.73 (dd, J = 13.3, 6.9 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 171.4, 167.2, 167.0, 165.2, 140.8, 136.5, 135.9, 134.4, 134.3, 134.0 (d, J = 25.6 Hz), 130.4, 130.2, 130.1, 129.5, 128.8, 128.7, 127.6, 127.2, 53.2, 51.4, 50.4, 50.1, 47.2, 39.1, 37.9. HRMS (ESI) m/z calcd for C29H28ClFN4O6S [M − H]− 613.1329, found 613.1331.
(S)-4-(4-(2-((1-((4-Chlorophenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-3-oxopiperazine-1-carbonyl)benzenesulfonyl fluoride (4n). Yield 53%. 1H NMR (400 MHz, CDCl3) δ 8.10 (d, J = 8.1 Hz, 2H), 7.70 (d, J = 8.0 Hz, 2H), 7.32–7.20 (m, 5H), 6.92 (dd, J = 6.6, 2.9 Hz, 2H), 6.76 (s, 2H), 4.75 (q, J = 7.5 Hz, 1H), 4.43 (s, 1H), 4.22–3.86 (m, 4H), 3.59 (s, 1H), 3.44 (s, 2H), 3.17 (s, 3H), 2.89 (dd, J = 13.3, 7.7 Hz, 1H), 2.73 (dd, J = 13.2, 6.8 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 171.4, 167.5, 166.9, 165.4, 140.8, 135.9, 134.8 (d, J = 25.5 Hz), 134.3, 130.6, 130.1, 129.5, 129.1, 128.8, 128.7, 127.2, 124.3, 51.4, 50.0, 47.8, 39.1, 37.9. HRMS (ESI) m/z calcd for C29H28ClFN4O6S [M − H]− 613.1329, found 613.1326.
(S)-N-(4-Chlorophenyl)-2-(2-(4-(2-hydroxybenzoyl)-2-oxopiperazin-1-yl)acetamido)-N-methyl-3-phenylpropanamide (4u). Yield 64%. 1H NMR (400 MHz, MeOD) δ 7.37–7.20 (m, 7H), 6.98–6.83 (m, 6H), 4.63 (t, J = 7.5 Hz, 1H), 4.08 (s, 5H), 3.64 (d, J = 2.6 Hz, 1H), 3.37 (s, 2H), 3.16 (s, 3H), 2.97 (dd, J = 13.2, 7.9 Hz, 1H), 2.77 (dd, J = 13.2, 7.2 Hz, 1H). 13C NMR (100 MHz, MeOD) δ 172.4, 169.9, 168.9, 167.6, 154.5, 141.9, 137.3, 134.4, 131.9, 130.2, 129.7, 129.6, 128.9, 127.5, 122.8, 120.3, 116.3, 52.6, 49.5, 38.8, 37.4. HRMS (ESI) m/z calcd for C29H29ClN4O4 [M − H]− 547.1754, found 547.1759.
(S)-N-(4-Chlorophenyl)-2-(2-(4-(3-hydroxybenzoyl)-2-oxopiperazin-1-yl)acetamido)-N-methyl-3-phenylpropanamide (4v). Yield 50%. 1H NMR (400 MHz, MeOD) δ 7.50–7.18 (m, 6H), 6.99–6.83 (m, 7H), 4.63 (t, J = 7.5 Hz, 1H), 4.42–3.88 (m, 5H), 3.70 (s, 1H), 3.45–3.32 (m, 2H), 3.15 (s, 3H), 2.98 (dd, J = 13.2, 7.9 Hz, 1H), 2.77 (dd, J = 13.2, 7.2 Hz, 1H). 13C NMR (100 MHz, MeOD) δ 172.3, 171.7, 168.8, 167.6, 159.1, 141.9, 137.3, 136.4, 134.4, 130.4, 130.2, 129.7, 129.6, 128.9, 127.4, 118.2, 114.5, 52.6, 49.5, 38.8, 37.4. HRMS (ESI) m/z calcd for C29H29ClN4O4 [M − H]− 547.1754, found 547.1754.
(S)-N-(4-Chlorophenyl)-2-(2-(4-(4-hydroxybenzoyl)-2-oxopiperazin-1-yl)acetamido)-N-methyl-3-phenylpropanamide (4w). Yield 39%. 1H NMR (400 MHz, MeOD) δ 7.42–7.30 (m, 4H), 7.24 (dd, J = 5.0, 1.9 Hz, 3H), 6.95 (dd, J = 6.6, 2.9 Hz, 2H), 6.90–6.82 (m, 3H), 4.63 (t, J = 7.5 Hz, 1H), 4.28 (s, 2H), 4.09 (d, J = 2.2 Hz, 2H), 3.85 (s, 2H), 3.36 (dt, J = 13.3, 7.0 Hz, 2H), 3.15 (s, 3H), 2.98 (dd, J = 13.2, 7.9 Hz, 1H), 2.77 (dd, J = 13.2, 7.2 Hz, 1H). 13C NMR (100 MHz, MeOD) δ 172.3, 172.0, 168.9, 167.5, 160.6, 141.9, 137.3, 134.4, 130.2, 130.0, 129.7, 129.7, 128.9, 127.4, 125.7, 115.7, 52.6, 49.5, 38.8, 37.4. HRMS (ESI) m/z calcd for C29H29ClN4O5 [M − H]+ 549.1899, found: 549.1902.
(S)-N-(4-Chlorophenyl)-N-methyl-2-(2-(2-oxo-4-(2-(trifluoromethyl)benzoyl)piperazin-1-yl)acetamido)-3-phenylpropanamide (4x). Yield 70%. 1H NMR (400 MHz, MeOD) δ 7.85–7.76 (m, 1H), 7.74 (d, J = 7.5 Hz, 1H), 7.68 (t, J = 7.7 Hz, 1H), 7.54 (dd, J = 10.7, 6.6 Hz, 1H), 7.37–7.29 (m, 2H), 7.27–7.19 (m, 3H), 6.96 (q, J = 4.8 Hz, 2H), 6.88 (s, 2H), 4.74–4.60 (m, 1H), 4.55 (dd, J = 18.5, 5.7 Hz, 1H), 4.32–4.14 (m, 2H), 4.07–3.71 (m, 3H), 3.45 (p, J = 5.4 Hz, 2H), 3.15 (s, 3H), 2.97 (ddd, J = 13.4, 7.9, 2.2 Hz, 1H), 2.81–2.71 (m, 1H). 13C NMR (100 MHz, MeOD) δ 172.3, 168.6, 166.9, 166.4, 141.9, 137.3, 134.4, 133.4 (d, J = 9.6 Hz), 130.7, 130.6, 130.2, 129.7, 129.7, 128.9, 128.2 (d, J = 8.8 Hz), 127.4, 127.2 (q, J = 4.6 Hz), 125.8, 52.6, 50.6, 46.1, 44.2, 39.5, 38.8, 37.4. HRMS (ESI) m/z calcd for C30H28ClF3N4O4 [M − H]+ 601.1827, found: 601.1827.
(S)-2-(2-(4-Benzoyl-2-oxopiperazin-1-yl)acetamido)-N-(4-chlorophenyl)-N-methyl-3-phenylpropanamide (4aa). Yield 94%. 1H NMR (400 MHz, MeOD) δ 7.49 (d, J = 3.7 Hz, 5H), 7.37–7.30 (m, 2H), 7.23 (dd, J = 5.0, 1.9 Hz, 3H), 6.95 (dd, J = 6.6, 2.8 Hz, 2H), 6.88 (s, 2H), 4.63 (t, J = 7.5 Hz, 1H), 4.45–3.83 (m, 5H), 3.79–3.64 (m, 1H), 3.35 (d, J = 8.7 Hz, 2H), 3.15 (s, 3H), 2.97 (dd, J = 13.2, 7.9 Hz, 1H), 2.76 (dd, J = 13.2, 7.2 Hz, 1H). 13C NMR (100 MHz, MeOD) δ 172.3, 171.6, 168.8, 167.3, 141.9, 137.3, 135.3, 134.4, 131.0, 130.2, 129.7, 129.6, 129.2, 128.9, 127.6, 127.4, 52.6, 49.4, 44.5, 38.8, 37.3. HRMS (ESI) m/z calcd for C29H29ClN4O4 [M − H]− 531.1805, found 531.1811.
(S)-N-(4-Chlorophenyl)-N-methyl-2-(2-(2-oxo-4-picolinoylpiperazin-1-yl)acetamido)-3-phenylpropanamide (4bb). Yield 86%. 1H NMR (400 MHz, MeOD) δ 8.62 (dq, J = 4.8, 1.3 Hz, 1H), 7.97 (tt, J = 7.7, 1.8 Hz, 1H), 7.74 (dd, J = 18.3, 7.8 Hz, 1H), 7.52 (ddt, J = 7.7, 4.9, 1.4 Hz, 1H), 7.33 (d, J = 8.3 Hz, 2H), 7.23 (d, J = 5.5 Hz, 3H), 6.95 (dd, J = 6.6, 2.9 Hz, 2H), 6.88 (s, 2H), 4.63 (t, J = 7.5 Hz, 1H), 4.40 (s, 1H), 4.33 (s, 1H), 4.10 (d, J = 9.1 Hz, 2H), 4.02 (t, J = 5.5 Hz, 1H), 3.85 (t, J = 5.3 Hz, 1H), 3.42 (p, J = 6.5 Hz, 2H), 3.15 (d, J = 1.0 Hz, 3H), 2.97 (dd, J = 13.2, 7.9 Hz, 1H), 2.77 (dd, J = 13.2, 7.2 Hz, 1H). 13C NMR (100 MHz, MeOD) δ 172.3, 168.8, 168.2, 167.3, 153.4, 149.0, 141.9, 138.4, 137.3, 134.3, 130.2, 129.7, 128.9, 127.4, 126.0, 124.9, 124.5, 52.6, 50.9, 46.8, 44.4, 39.9, 38.8, 37.4. HRMS (ESI) m/z calcd for C28H28ClN5O4 [M − H]+ 534.1903, found: 534.1907.
(S)-N-(4-Chlorophenyl)-N-methyl-2-(2-(4-nicotinoyl-2-oxopiperazin-1-yl)acetamido)-3-phenylpropanamide (4cc). Yield 68%. 1H NMR (400 MHz, MeOD) δ 8.73–8.65 (m, 2H), 7.98 (d, J = 7.9 Hz, 1H), 7.55 (ddd, J = 7.9, 5.0, 0.9 Hz, 1H), 7.36–7.30 (m, 2H), 7.24 (dd, J = 4.9, 1.9 Hz, 3H), 6.95 (dd, J = 6.6, 2.9 Hz, 2H), 6.88 (s, 2H), 4.63 (t, J = 7.5 Hz, 1H), 4.37 (s, 1H), 4.23–3.93 (m, 4H), 3.70 (s, 1H), 3.39 (s, 2H), 3.15 (s, 3H), 2.98 (dd, J = 13.2, 7.9 Hz, 1H), 2.77 (dd, J = 13.2, 7.2 Hz, 1H). 13C NMR (100 MHz, MeOD) δ 172.3, 168.8, 168.6, 167.2, 151.3, 148.1, 141.9, 137.3, 136.4, 134.4, 132.0, 130.2, 129.7, 129.7, 128.9, 127.4, 124.6, 52.6, 49.5, 46.5, 44.8, 39.9, 38.8, 37.4. HRMS (ESI) m/z calcd for C28H28ClN5O4 [M − H]+ 534.1903, found: 534.1906.
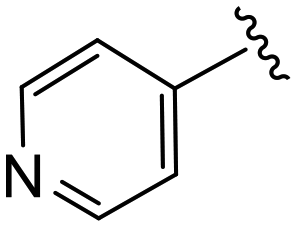
(S)-N-(4-Chlorophenyl)-2-(2-(4-isonicotinoyl-2-oxopiperazin-1-yl)acetamido)-N-methyl-3-phenylpropanamide (4dd). Yield 63%. 1H NMR (400 MHz, MeOD) δ 8.72–8.66 (m, 2H), 7.52 (d, J = 5.1 Hz, 2H), 7.33 (d, J = 8.4 Hz, 2H), 7.24 (d, J = 6.0 Hz, 3H), 6.95 (dd, J = 6.7, 2.7 Hz, 2H), 6.88 (s, 2H), 4.63 (t, J = 7.5 Hz, 1H), 4.37 (s, 1H), 4.25–3.85 (m, 4H), 3.63 (d, J = 6.0 Hz, 1H), 3.35 (d, J = 6.9 Hz, 2H), 3.15 (s, 3H), 2.98 (dd, J = 13.2, 7.9 Hz, 1H), 2.77 (dd, J = 13.2, 7.2 Hz, 1H). 13C NMR (100 MHz, MeOD) δ 172.3, 168.8, 168.5, 167.1, 150.4, 141.9, 137.3, 134.4, 130.2, 129.7, 129.7, 128.9, 127.4, 122.6, 122.3, 52.7, 49.0, 46.4, 44.5, 39.7, 38.8, 37.4. HRMS (ESI) m/z calcd for C28H28ClN5O4 [M − H]+ 534.1903, found: 534.1908.
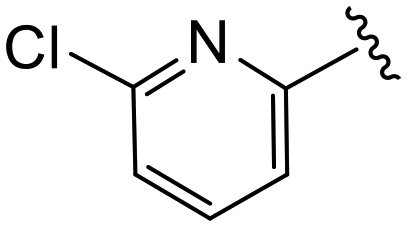
(S)-N-(4-Chlorophenyl)-2-(2-(4-(6-chloropicolinoyl)-2-oxopiperazin-1-yl)acetamido)-N-methyl-3-phenylpropanamide (4ee). Yield 83%. 1H NMR (400 MHz, MeOD) δ 8.01–7.92 (m, 1H), 7.71 (dd, J = 17.7, 7.6 Hz, 1H), 7.62–7.55 (m, 1H), 7.37–7.29 (m, 2H), 7.29–7.20 (m, 3H), 6.95 (dd, J = 6.6, 2.8 Hz, 2H), 6.88 (s, 2H), 4.63 (td, J = 7.5, 4.1 Hz, 1H), 4.38 (s, 2H), 4.10 (d, J = 9.2 Hz, 2H), 4.01 (t, J = 5.5 Hz, 1H), 3.88 (t, J = 5.5 Hz, 1H), 3.41 (ddt, J = 17.7, 11.9, 4.7 Hz, 2H), 3.15 (s, 3H), 2.98 (dd, J = 13.2, 7.9 Hz, 1H), 2.77 (ddd, J = 13.8, 7.2, 3.0 Hz, 1H). 13C NMR (100 MHz, MeOD) δ 172.3, 168.8, 167.2, 166.6, 153.6, 150.6, 141.9, 141.3, 137.3, 134.4, 130.2, 129.7, 129.7, 128.9, 127.4, 126.7, 123.8, 123.4, 52.6, 50.9, 46.9, 44.5, 40.1, 38.8, 37.4. HRMS (ESI) m/z calcd for C28H27Cl2N5O4 [M − H]+ 568.1513, found: 568.1518.
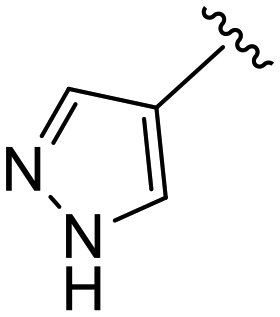
(S)-N-(4-Chlorophenyl)-N-methyl-2-(2-(2-oxo-4-(1H-pyrazole-4-carbonyl)piperazin-1-yl)acetamido)-3-phenylpropanamide (4ff). Yield 37%. 1H NMR (400 MHz, MeOD) δ 8.06 (s, 1H), 7.92 (s, 1H), 7.36–7.29 (m, 2H), 7.24 (p, J = 3.6 Hz, 3H), 6.99–6.92 (m, 2H), 6.88 (s, 2H), 4.63 (t, J = 7.5 Hz, 1H), 4.39 (s, 2H), 4.10 (s, 2H), 3.96 (t, J = 5.4 Hz, 2H), 3.41 (q, J = 6.2 Hz, 2H), 3.15 (s, 3H), 2.98 (dd, J = 13.2, 7.9 Hz, 1H), 2.77 (dd, J = 13.2, 7.2 Hz, 1H). 13C NMR (100 MHz, MeOD) δ 172.3, 168.9, 167.5, 165.1, 141.9, 139.9, 137.3, 134.4, 130.2, 129.7, 129.7, 128.9, 127.4, 115.9, 52.7, 49.5, 38.8, 37.4. HRMS (ESI) m/z calcd for C26H27ClN6O4 [M − H]+ 523.1855, found: 523.1857.
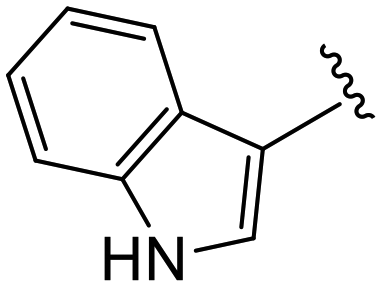
(S)-2-(2-(4-(1H-Indole-3-carbonyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-chlorophenyl)-N-methyl-3-phenylpropanamide (4gg). Yield 73%. 1H NMR (400 MHz, MeOD) δ1H NMR (400 MHz, MeOD) δ 7.97 (s, 0H), 7.65 (dt, J = 7.7, 1.0 Hz, 1H), 7.60 (s, 1H), 7.40–7.32 (m, 1H), 7.29–7.20 (m, 3H), 7.12 (tt, J = 8.1, 7.0, 2.9 Hz, 5H), 6.85 (dd, J = 6.8, 2.7 Hz, 3H), 4.53 (t, J = 7.5 Hz, 1H), 4.46 (s, 2H), 4.00 (s, 2H), 3.91 (t, J = 5.5 Hz, 2H), 3.57–3.51 (m, 1H), 3.05 (s, 4H), 2.93–2.74 (m, 1H), 2.74–2.54 (m, 2H). HRMS (ESI) m/z calcd for C31H30ClN5O4 [M − H]+ 570.1914, found: 570.1914.
(S)-2-(2-((3H-[1,2,3]Triazolo[4,5-b]pyridin-3-yl)oxy)acetamido)-N-(4-chlorophenyl)-N-methyl-3-phenylpropanamide (10). Yield 8%. 1H NMR (400 MHz, CDCl3) δ 8.78 (dd, J = 4.5, 1.4 Hz, 1H), 8.41 (dd, J = 8.4, 1.4 Hz, 1H), 8.28 (d, J = 7.9 Hz, 1H), 7.46 (dd, J = 8.4, 4.5 Hz, 1H), 7.32–7.13 (m, 5H), 7.00 (dd, J = 6.7, 2.8 Hz, 2H), 6.57 (s, 1H), 5.16–5.01 (m, 2H), 4.77 (ddd, J = 9.0, 7.9, 6.1 Hz, 1H), 3.14 (s, 3H), 3.12–3.07 (m, 1H), 2.87 (dd, J = 12.9, 6.1 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 170.7, 165.2, 151.5, 140.9, 139.5, 136.1, 135.1, 134.0, 129.9, 129.8, 129.6, 128.8, 128.7, 127.2, 121.3, 78.4, 51.6, 39.5, 37.7. HRMS (ESI) m/z calcd for C23H21ClN6O [M − H]+ 465.1463, found: 465.1460.
(S)-2-(2-(4-(2-Bromobenzoyl)piperazin-1-yl)acetamido)-N-(4-chlorophenyl)-N-methyl-3-phenylpropanamide (13). Yield 73%. 1H NMR (600 MHz, MeOD) δ 7.57 (dd, J = 8.1, 4.0 Hz, 1H), 7.38 (dt, J = 15.6, 7.3 Hz, 1H), 7.30–7.17 (m, 2H), 7.12 (d, J = 8.0 Hz, 2H), 6.96 (s, 2H), 6.83 (d, J = 6.9 Hz, 2H), 4.58 (d, J = 7.7 Hz, 1H), 3.62 (p, J = 6.5 Hz, 3H), 3.16–3.08 (m, 6H), 3.01 (s, 0H), 2.90 (ddd, J = 29.3, 15.0, 6.5 Hz, 2H), 2.47 (q, J = 26.9, 20.1 Hz, 3H), 2.36–2.20 (m, 1H). 13C NMR (100 MHz, MeOD) δ 171.65, 170.96, 169.48, 168.31, 141.80, 138.33, 137.29, 136.35, 132.58, 130.62, 129.54, 128.85, 128.24, 127.58, 127.35, 126.72, 126.22, 119.27, 59.94, 55.97, 54.44, 51.29, 47.15, 46.39, 43.76, 42.82, 41.12, 38.68, 36.65. HRMS (ESI) m/z calcd for C29H30BrClN4O3 [M + H]+ 597.1263, found: 597.1210.
(S)-2-(2-(4-(3-Bromobenzoyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-chlorophenyl)-N-methyl-3-phenylpropanamide (4b). Yield 79%. 1H NMR (400 MHz, MeOD) δ 7.71–7.64 (m, 2H), 7.46 (d, J = 7.7 Hz, 1H), 7.41 (td, J = 8.0, 1.3 Hz, 1H), 7.33 (d, J = 8.4 Hz, 2H), 7.23 (dd, J = 5.1, 1.8 Hz, 3H), 6.95 (dd, J = 6.6, 2.9 Hz, 2H), 6.88 (s, 2H), 4.63 (t, J = 7.5 Hz, 1H), 4.34 (s, 1H), 4.22–3.84 (m, 4H), 3.65 (d, J = 12.7 Hz, 1H), 3.36 (s, 2H), 3.15 (s, 3H), 2.97 (dd, J = 13.2, 7.9 Hz, 1H), 2.76 (dd, J = 13.2, 7.2 Hz, 1H). 13C NMR (100 MHz, MeOD) δ 172.3, 169.7, 168.8, 167.2, 141.9, 137.5, 137.3, 134.4, 133.9, 131.0, 130.6, 130.2, 129.7, 129.7, 128.9, 127.4, 126.3, 123.0, 52.6, 49.5, 46.5, 44.7, 38.8, 37.4. HRMS (ESI) m/z calcd for C29H28BrClN4O4 [M − H]+ 611.1055, found: 611.1053.
(S)-2-(2-(4-(2-Chlorobenzoyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-chlorophenyl)-N-methyl-3-phenylpropanamide (4d). Yield 81%. 1H NMR (400 MHz, MeOD) δ 7.47 (dddd, J = 25.5, 9.6, 7.9, 3.5 Hz, 4H), 7.36–7.29 (m, 2H), 7.28–7.19 (m, 3H), 6.95 (dq, J = 6.9, 3.1, 1.9 Hz, 2H), 6.90–6.85 (m, 2H), 4.63 (t, J = 7.6 Hz, 1H), 4.49 (dd, J = 18.6, 6.0 Hz, 1H), 4.30 (dd, J = 18.5, 5.9 Hz, 1H), 4.16 (dd, J = 15.6, 5.6 Hz, 1H), 4.05 (d, J = 17.1 Hz, 2H), 3.92 (d, J = 5.1 Hz, 1H), 3.58–3.42 (m, 2H), 3.15 (s, 3H), 2.97 (ddd, J = 13.2, 7.9, 2.2 Hz, 1H), 2.76 (dd, J = 13.3, 7.2 Hz, 1H). 13C NMR (100 MHz, MeOD) δ 172.3, 168.2, 167.1, 166.6, 141.9, 137.3, 135.3, 134.3, 131.7, 130.8, 130.2, 130.1, 129.7, 129.6, 128.9, 128.6, 128.1, 127.4, 52.6, 49.9, 45.9, 43.9, 39.4, 38.8, 37.4. HRMS (ESI) m/z calcd for C29H28Cl2N4O4 [M − H]+ 567.156, found 567.1561.
(S)-2-(2-(4-(3-Chlorobenzoyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-chlorophenyl)-N-methyl-3-phenylpropanamide (4e). Yield 90%. 1H NMR (400 MHz, MeOD) δ 7.56–7.52 (m, 2H), 7.48 (dd, J = 8.5, 7.4 Hz, 1H), 7.44–7.40 (m, 1H), 7.37–7.30 (m, 2H), 7.24 (dd, J = 5.0, 1.9 Hz, 3H), 6.95 (dd, J = 6.6, 2.8 Hz, 2H), 6.88 (s, 2H), 4.63 (t, J = 7.5 Hz, 1H), 4.34 (s, 1H), 4.22–3.85 (m, 4H), 3.67 (s, 1H), 3.36 (s, 2H), 3.15 (s, 3H), 2.98 (dd, J = 13.2, 7.9 Hz, 1H), 2.77 (dd, J = 13.2, 7.2 Hz, 1H). 13C NMR (100 MHz, MeOD) δ 172.3, 169.9, 168.8, 167.3, 141.9, 137.3, 135.2, 134.4, 130.9, 130.8, 130.2, 129.7, 129.6, 128.9, 127.4, 125.9, 52.7, 49.5, 44.7, 38.8, 37.4. HRMS (ESI) m/z calcd for C29H28Cl2N4O4 [M − H]+ 567.156, found 567.1555.
(S)-2-(2-(4-(4-Chlorobenzoyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-chlorophenyl)-N-methyl-3-phenylpropanamide (4f). Yield 98%. 1H NMR (400 MHz, MeOD) δ 7.50 (s, 4H), 7.36–7.29 (m, 2H), 7.23 (dd, J = 5.0, 1.9 Hz, 3H), 6.95 (dd, J = 6.6, 2.9 Hz, 2H), 6.88 (s, 2H), 4.63 (t, J = 7.5 Hz, 1H), 4.45–3.85 (m, 5H), 3.69 (s, 1H), 3.47–3.34 (m, 2H), 3.15 (s, 3H), 2.97 (dd, J = 13.2, 7.9 Hz, 1H), 2.76 (dd, J = 13.2, 7.2 Hz, 1H). 13C NMR (100 MHz, MeOD) δ 172.3, 170.5, 168.8, 167.4, 141.9, 137.3, 136.9, 134.4, 133.9, 130.2, 129.7, 129.7, 129.4, 128.9, 127.4, 52.6, 49.4, 46.7, 44.8, 38.8, 37.4. HRMS (ESI) m/z calcd for C29H28Cl2N4O4 [M − H]+ 567.156, found 567.1559.
(S)-N-(4-Chlorophenyl)-2-(2-(4-(2-fluorobenzoyl)-2-oxopiperazin-1-yl)acetamido)-N-methyl-3-phenylpropanamide (4g). Yield 98%. 1H NMR (400 MHz, MeOD) δ 7.59–7.49 (m, 1H), 7.53–7.42 (m, 1H), 7.42–7.29 (m, 3H), 7.23 (dd, J = 7.9, 2.7 Hz, 4H), 7.00–6.92 (m, 2H), 6.88 (s, 2H), 4.63 (t, J = 7.5 Hz, 1H), 4.39 (s, 1H), 4.19–3.91 (m, 4H), 3.61 (t, J = 5.3 Hz, 1H), 3.52–3.33 (m, 2H), 3.15 (s, 3H), 2.97 (dd, J = 13.2, 7.9 Hz, 1H), 2.76 (dd, J = 13.2, 7.2 Hz, 1H). 13C NMR (100 MHz, MeOD) δ 172.3, 168.8, 167.1, 166.5, 160.3, 157.9, 141.9, 137.3, 134.4, 132.8 (d, J = 8.1 Hz), 130.2, 129.7, 129.7, 128.9, 127.4, 125.5, 123.5 (d, J = 17.6 Hz), 116.4 (d, J = 21.3 Hz), 52.6, 49.4, 46.3, 44.2, 39.6, 38.8, 37.4. HRMS (ESI) m/z calcd for C29H28ClFN4O4 [M − H]+ 551.1856, found: 551.1861.
(S)-N-(4-Chlorophenyl)-2-(2-(4-(3-fluorobenzoyl)-2-oxopiperazin-1-yl)acetamido)-N-methyl-3-phenylpropanamide (4h). Yield 91%. 1H NMR (400 MHz, MeOD) δ 7.52 (td, J = 7.8, 5.4 Hz, 1H), 7.37–7.19 (m, 8H), 6.95 (dd, J = 6.6, 2.9 Hz, 2H), 6.89 (s, 2H), 4.63 (t, J = 7.5 Hz, 1H), 4.34 (s, 1H), 4.22–3.87 (m, 4H), 3.68 (s, 1H), 3.37 (s, 2H), 3.15 (s, 3H), 2.98 (dd, J = 13.2, 7.9 Hz, 1H), 2.77 (dd, J = 13.2, 7.2 Hz, 1H). 13C NMR (100 MHz, MeOD) δ 172.3, 170.0, 168.8, 167.2, 164.6, 162.1, 141.9, 137.3, 134.4, 131.3 (d, J = 7.9 Hz), 130.2, 129.7, 129.7, 128.9, 127.4, 123.5, 117.8 (d, J = 20.9 Hz), 114.8, 52.6, 49.4, 46.6, 44.7, 39.9, 38.8, 37.4. HRMS (ESI) m/z calcd for C29H28ClFN4O4 [M − H]+ 551.1856, found: 551.1852.
(S)-N-(4-Chlorophenyl)-2-(2-(4-(4-fluorobenzoyl)-2-oxopiperazin-1-yl)acetamido)-N-methyl-3-phenylpropanamide (4i). Yield 95%. 1H NMR (400 MHz, MeOD) δ 7.61–7.51 (m, 2H), 7.36–7.30 (m, 2H), 7.23 (m, 5H), 7.00–6.91 (m, 2H), 6.88 (s, 2H), 4.63 (t, J = 7.5 Hz, 1H), 4.36–3.65 (m, 6H), 3.37 (d, J = 6.7 Hz, 2H), 3.15 (s, 3H), 2.98 (dd, J = 13.2, 7.9 Hz, 1H), 2.76 (dd, J = 13.2, 7.2 Hz, 1H). 13C NMR (101 MHz, MeOD) δ 172.3, 170.6, 168.8, 167.2, 165.9, 163.4, 141.9, 137.3, 134.4, 131.5, 130.4, 130.2, 129.7, 129.7, 128.9, 127.4, 116.2, 115.9, 52.6, 49.4, 38.8, 37.3. HRMS (ESI) m/z calcd for C29H28ClFN4O4 [M − H]+ 551.1856, found: 551.1856.
(S)-N-(4-Chlorophenyl)-2-(2-(4-(3-cyanobenzoyl)-2-oxopiperazin-1-yl)acetamido)-N-methyl-3-phenylpropanamide (4k). Yield 89%. 1H NMR (400 MHz, MeOD) δ 7.92–7.84 (m, 2H), 7.80 (d, J = 7.9 Hz, 1H), 7.68 (ddd, J = 11.3, 5.6, 2.6 Hz, 1H), 7.37–7.29 (m, 2H), 7.29–7.20 (m, 3H), 6.98–6.92 (m, 2H), 6.89 (s, 2H), 4.63 (t, J = 7.5 Hz, 1H), 4.36 (s, 1H), 4.20–3.90 (m, 4H), 3.65 (d, J = 10.3 Hz, 1H), 3.37 (s, 2H), 3.15 (d, J = 1.4 Hz, 3H), 2.97 (dd, J = 13.2, 8.0 Hz, 1H), 2.76 (dd, J = 13.2, 7.2 Hz, 1H). 13C NMR (100 MHz, MeOD) δ 172.3, 169.2, 168.8, 167.1, 141.9, 137.2, 136.8, 134.4, 132.1, 131.3, 130.4, 130.1, 129.7, 129.6, 128.9, 127.4, 118.3, 113.4, 52.6, 49.4, 46.6, 44.7, 39.9, 38.8, 37.4. HRMS (ESI) m/z calcd for C30H28ClN5O4 [M − H]+ 558.1903, found 558.1900.
(S)-N-(4-Chlorophenyl)-2-(2-(4-(4-cyanobenzoyl)-2-oxopiperazin-1-yl)acetamido)-N-methyl-3-phenylpropanamide (4l). Yield 84%. 1H NMR (400 MHz, MeOD) δ 7.86 (d, J = 7.9 Hz, 2H), 7.67 (d, J = 7.8 Hz, 2H), 7.36–7.30 (m, 2H), 7.24 (dd, J = 4.9, 2.0 Hz, 3H), 6.95 (dd, J = 6.6, 2.9 Hz, 2H), 6.88 (s, 2H), 4.63 (t, J = 7.5 Hz, 1H), 4.37 (s, 1H), 4.21–3.95 (m, 4H), 3.78–3.54 (m, 1H), 3.36 (s, 2H), 3.15 (s, 3H), 2.97 (dd, J = 13.2, 7.9 Hz, 1H), 2.76 (dd, J = 13.2, 7.2 Hz, 1H). 13C NMR (101 MHz, MeOD) δ 172.3, 169.6, 168.8, 167.2, 141.9, 139.8, 137.3, 134.4, 133.1, 130.2, 129.7, 129.7, 128.9, 127.4, 118.4, 114.5, 52.7, 49.4, 46.5, 44.6, 39.8, 38.8, 37.3. HRMS (ESI) m/z calcd for C30H28ClN5O4 [M − H]+ 558.1903, found 558.1907.
(S)-N-(4-Chlorophenyl)-N-methyl-2-(2-(4-(2-nitrobenzoyl)-2-oxopiperazin-1-yl)acetamido)-3-phenylpropanamide (4o). Yield 95%. 1H NMR (400 MHz, MeOD) δ 8.26 (td, J = 7.9, 1.2 Hz, 1H), 7.86 (td, J = 7.5, 1.2 Hz, 1H), 7.78–7.69 (m, 1H), 7.58 (ddd, J = 12.5, 7.6, 1.5 Hz, 1H), 7.37–7.30 (m, 2H), 7.29–7.17 (m, 3H), 7.00–6.92 (m, 2H), 6.91–6.86 (m, 2H), 4.64 (q, J = 7.3 Hz, 1H), 4.40 (s, 1H), 4.01 (d, J = 63.3 Hz, 4H), 3.53 (q, J = 9.2, 7.3 Hz, 2H), 3.15 (s, 3H), 2.98 (ddd, J = 12.3, 7.8, 3.9 Hz, 1H), 2.77 (ddd, J = 12.8, 7.2, 4.8 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ13C NMR (100 MHz, MeOD) δ 172.3, 168.8, 168.0, 167.1, 146.2, 141.9, 137.3, 135.4, 134.4, 131.9, 131.3, 130.2, 129.72, 129.67, 128.9, 128.8, 127.4, 125.4, 52.6, 50.3, 46.2, 44.0, 39.7, 38.83, 38.79, 37.4. HRMS (ESI) m/z calcd for C29H28ClN5O6 [M − H]− 576.1655, found 576.1652.
(S)-N-(4-Chlorophenyl)-N-methyl-2-(2-(4-(3-nitrobenzoyl)-2-oxopiperazin-1-yl)acetamido)-3-phenylpropanamide (4p). Yield 88%. 1H NMR (400 MHz, CDCl3) δ 8.33 (dq, J = 6.5, 2.0 Hz, 2H), 7.79 (dt, J = 7.7, 1.4 Hz, 1H), 7.69–7.61 (m, 1H), 7.32–7.25 (m, 2H), 7.28–7.20 (m, 3H), 6.93 (dt, J = 6.0, 3.4 Hz, 2H), 6.77 (d, J = 9.2 Hz, 2H), 4.75 (q, J = 7.6 Hz, 1H), 4.49–3.67 (m, 6H), 3.52–3.41 (m, 2H), 3.17 (s, 3H), 2.90 (dd, J = 13.2, 7.7 Hz, 1H), 2.74 (dd, J = 13.3, 6.9 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 171.4, 167.6, 167.0, 165.2, 148.3, 140.9, 136.2, 135.9, 134.3, 133.4, 130.2, 130.1, 129.5, 128.8, 128.7, 127.2, 125.4, 122.8, 51.4, 50.1, 47.5, 39.4, 39.2, 37.9. HRMS (ESI) m/z calcd for C29H28ClN5O6 [M − H]− 576.1655, found 576.1669.
(S)-N-(4-Chlorophenyl)-N-methyl-2-(2-(4-(4-nitrobenzoyl)-2-oxopiperazin-1-yl)acetamido)-3-phenylpropanamide (4q). Yield 92%. 1H NMR (400 MHz, CDCl3) δ 8.34–8.26 (m, 2H), 7.63 (d, J = 8.5 Hz, 2H), 7.32–7.19 (m, 5H), 6.92 (dd, J = 6.6, 2.9 Hz, 2H), 6.76 (d, J = 7.8 Hz, 2H), 4.75 (q, J = 7.6 Hz, 1H), 4.42 (s, 1H), 4.23–3.84 (m, 4H), 3.60 (s, 1H), 3.49–3.42 (m, 2H), 3.17 (s, 3H), 2.90 (dd, J = 13.3, 7.7 Hz, 1H), 2.74 (dd, J = 13.3, 6.8 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 171.9, 167.9, 166.9, 164.9, 148.9, 140.9, 140.6, 135.9, 134.3, 130.1, 129.5, 128.8, 128.7, 127.6, 127.2, 124.2, 51.4, 50.1, 47.3, 44.4, 40.1, 39.2, 37.9. HRMS (ESI) m/z calcd for C29H28ClN5O6 [M − H]− 576.1655, found 576.1657.
(S)-N-(4-Chlorophenyl)-N-methyl-2-(2-(2-oxo-4-(3-(trifluoromethyl)benzoyl)piperazin-1-yl)acetamido)-3-phenylpropanamide (4y). Yield 91%. 1H NMR (400 MHz, MeOD) δ 7.83 (d, J = 8.0 Hz, 2H), 7.77 (d, J = 7.7 Hz, 1H), 7.70 (t, J = 7.7 Hz, 1H), 7.33 (d, J = 8.3 Hz, 2H), 7.23 (dd, J = 5.1, 1.9 Hz, 3H), 6.95 (dd, J = 6.6, 2.8 Hz, 2H), 6.88 (s, 2H), 4.63 (t, J = 7.5 Hz, 1H), 4.37 (s, 1H), 4.24–3.88 (m, 4H), 3.65 (d, J = 12.9 Hz, 1H), 3.38 (s, 2H), 3.15 (s, 3H), 2.98 (dd, J = 13.2, 7.9 Hz, 1H), 2.77 (dd, J = 13.2, 7.2 Hz, 1H). 13C NMR (100 MHz, MeOD) δ 172.3, 169.9, 168.8, 167.1, 141.9, 137.3, 136.5, 134.4, 131.7, 131.4, 130.2, 129.7, 129.6, 128.9, 127.4, 125.9, 124.7, 123.2, 52.7, 49.5, 44.8, 38.8, 37.3. HRMS (ESI) m/z calcd for C30H28ClF3N4O4 [M − H]− 599.1678, found 599.1684.
(S)-N-(4-Chlorophenyl)-N-methyl-2-(2-(2-oxo-4-(4-(trifluoromethyl)benzoyl)piperazin-1-yl)acetamido)-3-phenylpropanamide (4z). Yield 98%. 1H NMR (400 MHz, CDCl3) δ 7.71 (d, J = 8.0 Hz, 2H), 7.56 (d, J = 8.0 Hz, 2H), 7.32–7.19 (m, 5H), 6.93 (dd, J = 6.6, 2.9 Hz, 2H), 6.76 (d, J = 8.7 Hz, 2H), 4.75 (q, J = 7.6 Hz, 1H), 4.56–3.77 (m, 5H), 3.62 (s, 1H), 3.42 (s, 2H), 3.17 (s, 3H), 2.90 (dd, J = 13.3, 7.8 Hz, 1H), 2.74 (dd, J = 13.2, 6.8 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 171.3, 168.8, 167.0, 165.8, 140.9, 138.1, 135.9, 134.3, 132.55 (d, J = 32.9 Hz), 130.1, 129.5, 128.8, 128.7, 127.8 (d, J = 9.9 Hz), 127.2, 125.94 (q, J = 3.7 Hz), 125.00, 51.4, 50.2, 47.3, 39.7, 39.2, 37.9. HRMS (ESI) m/z calcd for C30H28ClF3N4O4 [M − H]− 599.1678, found 599.1684.
:1) stirred for 12 h at 70 °C in oil bath (monitored by TLC). The resulting mixture was filtered and extracted with ethyl acetate for three times. The organic layers were combined, washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure in vacuum. The crude mixture was purified using Combi-flash on silica gel (5% MeOH in DCM as eluent).
(S)-2-(2-(4-(2-Aminobenzoyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-chlorophenyl)-N-methyl-3-phenylpropanamide (4r). Yield 39%. 1H NMR (600 MHz, CD3OD) δ 7.33 (d, J = 8.3 Hz, 2H), 7.28–7.19 (m, 3H), 7.19 (t, J = 7.8 Hz, 1H), 7.16–7.04 (m, 1H), 6.98–6.93 (m, 2H), 6.88 (s, 2H), 6.79 (d, J = 8.1 Hz, 1H), 6.70 (t, J = 7.4 Hz, 1H), 4.63 (t, J = 7.6 Hz, 1H), 4.29–4.26 (m, 2H), 4.09 (d, J = 5.8 Hz, 2H), 3.78 (s, 2H), 3.42–3.33 (m, 2H), 3.15 (d, J = 1.7 Hz, 3H), 2.97 (dd, J = 13.4, 7.9 Hz, 1H), 2.77 (dd, J = 13.3, 7.1 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 171.6, 168.6, 167.4, 166.1, 152.3, 151.3, 144.8, 141.0, 136.3, 134.1, 131.7, 129.9, 129.5, 128.9, 128.7, 128.2, 127.1, 120.9, 51.8, 51.7, 50.5, 47.7, 38.8, 37.9. HRMS (ESI) m/z calcd for C29H30ClN5O4 [M − H]− 546.1914, found 546.1911.
(S)-2-(2-(4-(3-Aminobenzoyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-chlorophenyl)-N-methyl-3-phenylpropanamide (4s). Yield 53%. 1H NMR (600 MHz, CD3OD) δ 7.33 (d, J = 8.3 Hz, 2H), 7.23 (d, J = 5.6 Hz, 3H), 7.18 (t, J = 7.7 Hz, 1H), 6.97–6.93 (m, 2H), 6.88 (s, 2H), 6.83–6.78 (m, 1H), 6.75 (s, 1H), 6.71 (d, J = 7.4 Hz, 1H), 4.63 (t, J = 7.6 Hz, 1H), 4.33 (s, 1H), 4.07 (d, J = 16.5 Hz, 3H), 3.96 (s, 1H), 3.70 (s, 1H), 3.31 (s, 2H), 3.15 (d, J = 2.0 Hz, 3H), 2.97 (dd, J = 13.3, 7.8 Hz, 1H), 2.77 (dd, J = 13.3, 7.0 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 171.4, 169.9, 167.4, 166.1, 154.5, 151.8, 150.5, 140.9, 139.2, 136.1, 134.3, 130.1, 129.5, 128.9, 128.7, 127.2, 122.4, 51.9, 50.3, 47.9, 41.7, 38.8, 38.0. HRMS (ESI) m/z calcd for C29H30ClN5O4 [M − H]− 546.1914, found 546.1918.
(S)-2-(2-(4-(4-Aminobenzoyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-chlorophenyl)-N-methyl-3-phenylpropanamide (4t). Yield 62%. 1H NMR (400 MHz, MeOD) δ 7.37–7.31 (m, 2H), 7.31–7.20 (m, 5H), 6.95 (dd, J = 6.7, 2.8 Hz, 2H), 6.89 (s, 2H), 6.80–6.67 (m, 2H), 4.63 (t, J = 7.6 Hz, 1H), 4.28 (s, 2H), 4.08 (d, J = 1.6 Hz, 2H), 3.86 (t, J = 5.4 Hz, 2H), 3.37 (dt, J = 8.3, 5.6 Hz, 2H), 3.15 (s, 3H), 2.97 (dd, J = 13.2, 7.9 Hz, 1H), 2.77 (dd, J = 13.2, 7.3 Hz, 1H). 13C NMR (100 MHz, MeOD) δ 173.2, 172.9, 169.5, 168.2, 152.5, 142.6, 137.9, 135.0, 130.8, 130.6, 130.4, 130.3, 129.6, 128.1, 122.9, 114.9, 53.3, 50.1, 39.4, 37.9. HRMS (ESI) m/z calcd for C29H30ClN5O4 [M − H]− 546.1914, found 546.1914.
(S)-3-Chloro-5-((1-(2-((1-((4-chlorophenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-4-(trifluoromethyl)-1,2-dihydropyridin-3-yl)oxy)benzoic acid (20). Yield 41%. 1H NMR (400 MHz, MeOD) δ 7.71 (d, J = 1.5 Hz, 1H), 7.67 (d, J = 7.3 Hz, 1H), 7.47–7.41 (m, 1H), 7.30–7.20 (m, 5H), 7.17 (t, J = 2.1 Hz, 1H), 6.98 (dt, J = 7.5, 3.6 Hz, 2H), 6.77 (s, 2H), 6.61 (dd, J = 7.4, 1.5 Hz, 1H), 4.74 (d, J = 3.1 Hz, 2H), 4.59 (dd, J = 8.6, 6.5 Hz, 1H), 3.13 (s, 3H), 2.98 (dd, J = 13.1, 8.7 Hz, 1H), 2.79 (dd, J = 13.0, 6.5 Hz, 1H). 13C NMR (100 MHz, MeOD) δ 170.7, 165.5, 156.9, 156.7, 140.3, 135.6, 133.9, 132.8, 131.0, 130.7, 128.6, 128.3, 128.1, 127.5, 125.9, 123.1, 122.4, 119.7, 118.9, 114.3, 99.58 (d, J = 4.7 Hz), 51.3, 50.9, 37.5, 35.9. HRMS (ESI) m/z calcd for C31H24Cl2F3N3O6 [M − H]− 600.0921, found 600.0927.
Thermal shift assays (TSAs)
TSAs used purified covalently-crosslinked hexameric CAA14C/E45C/W184A/M185A (CA121). CA121 cloned in a pET11a expression plasmid was kindly provided by Dr. Owen Pornillos (University of Virginia, Charlottesville, VA, USA). CA121 was expressed in Escherichia coli BL21(DE3)RIL and purified according to reported protocols.27 The TSAs were conducted as previously described7,16,19–21 with each reaction containing 7.5 μM CA121 in 50 mM sodium phosphate buffer (pH 8.0), 1× Sypro Orange Protein Gel Stain (Life Technologies, Carlsbad, CA, USA), and either 1% DMSO (control) or 20 μM compound (1% DMSO final). The plate was heated from 25 to 95 °C with a heating rate of 0.2 °C every 10 s in the PikoReal Real-Time PCR system (Thermo Fisher Scientific, Waltham, MA, USA) or the QuantStudio 3 Real-Time PCR system (Thermo Fisher Scientific). The fluorescence intensity was measured with an Ex range of 475–500 nm and Em range of 520–590 nm. The difference in the melting temperature (ΔTm) of CA121 in DMSO (T0) vs. in the presence of compound (Tm) were calculated using the following eqn (1):| ΔTm (°C) = Tm − T0. | (1) |
Virus production
The wild-type laboratory HIV-1 strain, HIV-1NL4-3,28 was produced using a pNL4-3 vector (NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH, Bethesda, MD, USA). HIV-1NL4-3 was generated by transfecting HEK 293T/17 cells with 5 μg of pNL4-3 vector and X-tremeGENE HP DNA Transfection Reagent (Roche, Basel, Switzerland) in a 10 cm tissue culture dish. Supernatant was harvested after 48 h and concentrated overnight at 4 °C, with Lenti-X concentrator (Clontech, Mountain View, CA, USA) followed by centrifugation at 1500 × g for 45 min at 4 °C. The resulting viral-containing pellet was concentrated 10-fold by resuspension in DMEM with 10% FBS and stored at −80 °C.Anti-HIV-1 and cytotoxicity assays
Anti-HIV-1 activity of PF74 and related analogs was examined in TZM-GFP cells. The potency of HIV-1 inhibition was determined based on the inhibition of viral LTR-activated GFP expression in the presence of compounds compared to DMSO controls. Briefly, TZM-GFP cells were plated at density of 1 × 104 cells per well in a 96-well plate and media was also added that contained increasing concentrations of the compounds. Cells were exposed to HIV-1NL4-3 (MOI = 0.1) 24 h post treatment. After 48 h incubation, anti-HIV-1 activity was determined by counting the amount of GFP positive cells on a Cytation™ 5 Imaging Reader (BioTek, Winooski, VT, USA) and 50% effective concentration (EC50) values were determined.The cytotoxicity of each compound was also determined in TZM-GFP cells. Cells plated at a density of 1 × 104 cells per well in a 96-well plate were continuously exposed to increasing concentrations compounds over a period of 72 h. The number of viable cells in each well was determined using an XTT Cell Proliferation Kit (R&D Systems, Inc., Minneapolis, MN, USA), and 50% cytotoxicity concentration (CC50) values were determined. All cell-based assays were conducted in duplicate and in at least two independent experiments.
To obtain EC50 and CC50 dose response curves, values were plotted in GraphPad Prism 5 and analyzed with the loginhibitorvs. normalized response–variable slope equation as eqn (2):
| Y = 100/(1 + 10(((logIC50)−X)×HillSlope)) | (2) |
Microsomal stability assay
The in vitro microsomal stability assay was conducted in duplicate in commercially available mouse and human liver microsomes (Sekisui XenoTech, Kansas City, KS, USA), which were supplemented with nicotinamide adenine dinucleotide phosphate (NADPH) as a cofactor. Briefly, the test compound (1 μM final concentration) was spiked into the reaction mixture containing liver microsomal protein (0.5 mg mL−1 final concentration) and MgCl2 (1 mM final concentration) in 0.1 M potassium phosphate buffer (pH 7.4). The reaction was initiated by addition of 1 mM NADPH, followed by incubation at 37 °C. A negative control was performed in parallel in the absence of NADPH to reveal any chemical instability or non-NADPH dependent enzymatic degradation for each compound. A reaction with positive controls verapamil was also performed to confirm the proper functionality of the incubation systems. A 50 μL aliquot of reaction mixture was then taken at various time points (0, 5, and 15 min), and quenched with 150 μL of acetonitrile containing an appropriate internal standard and 0.1% formic acid. The quenched samples were vortexed and centrifuged at 15000 rpm for 5 min at 4 °C. The supernatants were collected and analyzed by LC-MS/MS to determine the in vitro metabolic half-life (t1/2).
Molecular modeling
Molecular modeling was performed using the Schrödinger small molecule drug discovery suite 2019-1 (ref. 29) (Schrödinger Inc., New York, USA) for PF74-bound full length native HIV-1 CA (PDB ID: 4XFZ).8 To analyze the crystal structure we used Maestro30 (Schrödinger Inc.). A standard docking protocol was followed step by step including protein preparation, receptor grid generation, ligand preparation, and molecular docking. Protein preparation wizard31 (Schrödinger Inc.) was used for the refinement of the above crystal structure for protein preparation, in which the missing hydrogen atoms, side chains, and loops were added using Prime. This was followed by minimization using the OPLS 3e force field,32 where hydrogen bonding network was optimized and the heavy atoms were converged to an rmsd of 0.3 Å. The receptor grid generation tool in Maestro (Schrödinger Inc.) was used to define the binding site around the native ligand PF74, surrounding all the key residues within the range of 12 Å. After sketching ligands in Maestro, different conformers were generated in LigPrep63 at pH of 7 ± 2 method to serve as input for docking process. As a final step, docking was performed by the Glide XP33 (Glide, version 8.2, New York, USA) with the van der Waals radii of nonpolar atoms for each of the ligands scaled by a factor of 0.8. After docking refinement and minimization, protein flexibility was also considered under implicit solvent. Numbering of residues of HIV-1 CA which used in this paper for description was based on the full length native HIV-1 CA.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This research was supported by the National Institute of Allergy and Infectious Diseases, the National Institutes of Health, grant R01AI120860 (to S. G. S. and Z. W.) and F31AI155158 (to M. E. C.). S. G. S. acknowledges funding from the Nahmias-Schinazi Distinguished Chair in Research. We thank the Minnesota Supercomputing Institute for molecular modeling resources.Notes and references
- Y. H. Shin, C. M. Park and C. H. Yoon, Infect. Chemother., 2021, 53, 29–45 CrossRef PubMed.
- S. K. Carnes, J. H. Sheehan and C. Aiken, Curr. Opin. HIV AIDS, 2018, 13, 359–365 CrossRef CAS PubMed.
- A. Fassati, Virus Res., 2012, 170, 15–24 CrossRef CAS PubMed.
- V. Le Sage, A. J. Mouland and F. Valiente-Echeverria, Virus Res., 2014, 193, 116–129 CrossRef CAS PubMed.
- M. Novikova, Y. Zhang, E. O. Freed and K. Peng, Virol. Sin., 2019, 34, 119–134 CrossRef CAS PubMed.
- W. S. Blair, C. Pickford, S. L. Irving, D. G. Brown, M. Anderson, R. Bazin, J. A. Cao, G. Ciaramella, J. Isaacson, L. Jackson, R. Hunt, A. Kjerrstrom, J. A. Nieman, A. K. Patick, M. Perros, A. D. Scott, K. Whitby, H. Wu and S. L. Butler, PLoS Pathog., 2010, 6, e1001220 CrossRef PubMed.
- L. Wang, M. C. Casey, S. K. V. Vernekar, H. T. Do, R. L. Sahani, K. A. Kirby, H. Du, A. Hachiya, H. Zhang, P. R. Tedbury, J. Xie, S. G. Sarafianos and Z. Wang, Eur. J. Med. Chem., 2020, 200, 112427 CrossRef CAS PubMed.
- A. T. Gres, K. A. Kirby, V. N. KewalRamani, J. J. Tanner, O. Pornillos and S. G. Sarafianos, Science, 2015, 349, 99–103 CrossRef CAS PubMed.
- A. J. Price, D. A. Jacques, W. A. McEwan, A. J. Fletcher, S. Essig, J. W. Chin, U. D. Halambage, C. Aiken and L. C. James, PLoS Pathog., 2014, 10, e1004459 CrossRef PubMed.
- C. Buffone, A. Martinez-Lopez, T. Fricke, S. Opp, M. Severgnini, I. Cifola, L. Petiti, S. Frabetti, K. Skorupka, K. K. Zadrozny, B. K. Ganser-Pornillos, O. Pornillos, F. Di Nunzio and F. Diaz-Griffero, J. Virol., 2018, 92, e00648-18 CrossRef PubMed.
- K. A. Matreyek, S. S. Yucel, X. Li and A. Engelman, PLoS Pathog., 2013, 9, e1003693 CrossRef PubMed.
- V. Achuthan, J. M. Perreira, G. A. Sowd, M. Puray-Chavez, W. M. McDougall, A. Paulucci-Holthauzen, X. Wu, H. J. Fadel, E. M. Poeschla, A. S. Multani, S. H. Hughes, S. G. Sarafianos, A. L. Brass and A. N. Engelman, Cell Host Microbe, 2018, 24, 392–404 CrossRef CAS PubMed , e398.
- D. A. Bejarano, K. Peng, V. Laketa, K. Borner, K. L. Jost, B. Lucic, B. Glass, M. Lusic, B. Mueller and H. G. Krausslich, eLife, 2019, 8, e41800, DOI:10.7554/eLife.41800.
- S. M. Bester, G. Wei, H. Zhao, D. Adu-Ampratwum, N. Iqbal, V. V. Courouble, A. C. Francis, A. S. Annamalai, P. K. Singh, N. Shkriabai, P. Van Blerkom, J. Morrison, E. M. Poeschla, A. N. Engelman, G. B. Melikyan, P. R. Griffin, J. R. Fuchs, F. J. Asturias and M. Kvaratskhelia, Science, 2020, 370, 360–364 CrossRef CAS PubMed.
- J. O. Link, M. S. Rhee, W. C. Tse, J. Zheng, J. R. Somoza, W. Rowe, R. Begley, A. Chiu, A. Mulato, D. Hansen, E. Singer, L. K. Tsai, R. A. Bam, C. H. Chou, E. Canales, G. Brizgys, J. R. Zhang, J. Li, M. Graupe, P. Morganelli, Q. Liu, Q. Wu, R. L. Halcomb, R. D. Saito, S. D. Schroeder, S. E. Lazerwith, S. Bondy, D. Jin, M. Hung, N. Novikov, X. Liu, A. G. Villasenor, C. E. Cannizzaro, E. Y. Hu, R. L. Anderson, T. C. Appleby, B. Lu, J. Mwangi, A. Liclican, A. Niedziela-Majka, G. A. Papalia, M. H. Wong, S. A. Leavitt, Y. Xu, D. Koditek, G. J. Stepan, H. Yu, N. Pagratis, S. Clancy, S. Ahmadyar, T. Z. Cai, S. Sellers, S. A. Wolckenhauer, J. Ling, C. Callebaut, N. Margot, R. R. Ram, Y. P. Liu, R. Hyland, G. I. Sinclair, P. J. Ruane, G. E. Crofoot, C. K. McDonald, D. M. Brainard, L. Lad, S. Swaminathan, W. I. Sundquist, R. Sakowicz, A. E. Chester, W. E. Lee, E. S. Daar, S. R. Yant and T. Cihlar, Nature, 2020, 584, 614–618 CrossRef CAS PubMed.
- R. L. Sahani, R. Diana-Rivero, S. K. V. Vernekar, L. Wang, H. Du, H. Zhang, A. E. Castaner, M. C. Casey, K. A. Kirby, P. R. Tedbury, J. Xie, S. G. Sarafianos and Z. Wang, Viruses, 2021, 13, 479 CrossRef CAS PubMed.
- L. Sun, A. Dick, M. E. Meuser, T. Huang, W. A. Zalloum, C.-H. Chen, S. Cherukupalli, S. Xu, X. Ding, P. Gao, D. Kang, E. De Clercq, C. Pannecouque, S. Cocklin, K.-H. Lee, X. Liu and P. Zhan, J. Med. Chem., 2020, 63, 4790–4810 CrossRef CAS PubMed.
- L. Sun, T. Huang, A. Dick, M. E. Meuser, W. A. Zalloum, C. H. Chen, X. Ding, P. Gao, S. Cocklin, K. H. Lee, P. Zhan and X. Liu, Eur. J. Med. Chem., 2020, 190, 112085 CrossRef CAS PubMed.
- S. K. V. Vernekar, R. L. Sahani, M. C. Casey, J. Kankanala, L. Wang, K. A. Kirby, H. Du, H. Zhang, P. R. Tedbury, J. Xie, S. G. Sarafianos and Z. Wang, Viruses, 2020, 12, 452 CrossRef CAS PubMed.
- L. Wang, M. C. Casey, S. K. V. Vernekar, R. L. Sahani, J. Kankanala, K. A. Kirby, H. Du, A. Hachiya, H. Zhang, P. R. Tedbury, J. Xie, S. G. Sarafianos and Z. Wang, Eur. J. Med. Chem., 2020, 204, 112626 CrossRef CAS PubMed.
- L. Wang, M. C. Casey, S. K. V. Vernekar, R. L. Sahani, K. A. Kirby, H. J. Du, H. C. Zhang, P. R. Tedbury, J. S. Xie, S. G. Sarafianos and Z. Q. Wang, Acta Pharm. Sin. B, 2021, 11, 810–822 CrossRef CAS PubMed.
- G. Wu, W. A. Zalloum, M. E. Meuser, L. Jing, D. Kang, C. H. Chen, Y. Tian, F. Zhang, S. Cocklin, K. H. Lee, X. Liu and P. Zhan, Eur. J. Med. Chem., 2018, 158, 478–492 CrossRef CAS PubMed.
- M. Trunzer, B. Faller and A. Zimmerlin, J. Med. Chem., 2009, 52, 329–335 CrossRef CAS PubMed.
- S. Chandrappa, K. Vinaya, T. Ramakrishnappa and K. S. Rangappa, Synlett, 2010, 2010, 3019–3022 CrossRef.
- Z. Wang, Z. Yu, D. Kang, J. Zhang, Y. Tian, D. Daelemans, E. De Clercq, C. Pannecouque, P. Zhan and X. Liu, Bioorg. Med. Chem., 2019, 27, 447–456 CrossRef CAS PubMed.
- V. J. Wacher, J. A. Silverman, Y. Zhang and L. Z. Benet, J. Pharm. Sci., 1998, 87, 1322–1330 CrossRef CAS PubMed.
- O. Pornillos, B. K. Ganser-Pornillos, B. N. Kelly, Y. Hua, F. G. Whitby, C. D. Stout, W. I. Sundquist, C. P. Hill and M. Yeager, Cell, 2009, 137, 1282–1292 CrossRef PubMed.
- A. Adachi, H. E. Gendelman, S. Koenig, T. Folks, R. Willey, A. Rabson and M. A. Martin, J. Virol., 1986, 59, 284–291 CrossRef CAS PubMed.
- Schrödinger Small-Molecule Drug Discovery Suite 2019-1, Schrödinger, LLC, New York, NY, 2019 Search PubMed.
- Schrödinger Release 2019-1: Maestro, Schrödinger, LLC, New York, NY, 2019 Search PubMed.
- G. M. Sastry, M. Adzhigirey, T. Day, R. Annabhimoju and W. Sherman, J. Comput.-Aided Mol. Des., 2013, 27, 221–234 CrossRef PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. TiradoRives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Docking of PF74, synthesis and chemical data for all intermediates. NMR spectra for all final compounds. See DOI: 10.1039/d1md00292a |
| ‡ Equal contributions. |
| This journal is © The Royal Society of Chemistry 2021 |