
A niclosamide–tobramycin hybrid adjuvant potentiates cefiderocol against P. aeruginosa†
Liam
Berry
 a,
Marc
Brizuela
a,
Gregory
Jackson
a and
Frank
Schweizer
*ab
a,
Marc
Brizuela
a,
Gregory
Jackson
a and
Frank
Schweizer
*ab
aDepartment of Chemistry, University of Manitoba, Winnipeg, MB, Canada. E-mail: schweize@cc.umanitoba.ca
bDepartment of Medical Microbiology and Infectious Diseases, University of Manitoba, Winnipeg, MB, Canada
First published on 27th July 2021
Abstract
There is an urgent need for new therapies to overcome antimicrobial resistance (AMR) especially against Gram-negative bacilli (GNB). Multicomponent therapy combining antibiotics with enhancer molecules known as adjuvants is an emerging strategy to combat AMR. We have previously reported tobramycin-based adjuvants which are able to potentiate various antibiotics. In order to expand the repertoire of tobramycin hybrid adjuvants, a new hybrid containing niclosamide, an FDA approved anthelmintic which has recently demonstrated a variety of interesting biological effects, was synthesized. It was found that this conjugate can potentiate several antibiotics against multidrug-resistant GNB, including the recently approved siderophore cephalosporin cefiderocol. 8 μg ml−1 of the niclosamide–tobramycin hybrid in combination therapy against a pandrug-resistant strain of P. aeruginosa was able to lower the cefiderocol MIC 32-fold, from 8 μg ml−1 to 0.25 μg ml−1 in iron-rich media where siderophore uptake is reduced. These results indicate that a niclosamide–tobramycin hybrid adjuvant can serve to potentiate a newly approved antibiotic.
Introduction
Rising rates of multidrug-resistant (MDR) and extensively drug-resistant (XDR) bacterial infections represent one of the largest threats to global health.1 MDR Gram-negative bacteria (GNB) are particularly hard to treat due to their highly restrictive dual membrane system which limits the entry of many antibiotics into the cell.2 Even amongst GNB, some organisms such as Pseudomonas aeruginosa possess extremely restrictive membranes which severely limits effective treatment options. P. aeruginosa infections display troubling rates of antimicrobial resistance, are commonly hospital acquired and typically target immunocompromised patients.3 In light of the ongoing COVID-19 pandemic, ventilator associated pneumonia (VAP) caused by bacterial co-infection is a major concern.4P. aeruginosa is notable for causing VAP and MDR isolates of P. aeruginosa have been isolated from COVID-19 patients leading to increased levels of antibiotic usage and potentially increased rates of resistance.5As a result of the difficulty in treating MDR P. aeruginosa, new therapies against this pathogen are currently being developed. One such example is cefiderocol, a recently approved siderophore cephalosporin antibiotic with a novel method of cell entry.6 Cefiderocol is able to chelate iron and hijack the siderophore uptake mechanism that bacteria use to acquire iron from the environment, allowing it to bypass the outer membrane. Cefiderocol also displays enhanced stability towards β-lactamases and efficacy against a variety of MDR Gram-negative bacteria. Cefiderocol is used to treat complicated urinary tract infections and was approved by the FDA in September 2020 for the treatment of VAP and hospital-acquired pneumonia caused by GNB that are resistant to other antibiotics.7 Despite the promising efficacy of this antibiotic, antimicrobial resistance remains a concern. Clinical isolates have already been reported with cefiderocol resistance, presumably mediated through reduced expression of the pirA siderophore receptor gene.8 In order to maintain the clinical effectiveness of cefiderocol in light of reduced siderophore uptake, it may be advantageous to explore other methods of bypassing the outer membrane. Screening for compounds which enhance cefiderocol activity in iron-rich media where siderophore uptake is reduced may be one method of overcoming this problem.
Another approach to tackling antimicrobial resistance is the use of adjuvants. Adjuvants are molecules that possess little or no antimicrobial activity but are able to enhance the efficacy of partner antibiotics when used in combination therapy.9 We have recently reported several examples of tobramycin-based hybrid adjuvants that are able to potentiate a wide variety of antibiotics.10,11 These compounds are composed of a tobramycin antibiotic attached covalently to a partner antibiotic via an aliphatic hydrocarbon tether. We have previously demonstrated that these adjuvants are able to permeabilize the outer membrane of GNB as well as dissipate the proton motive force to de-energize efflux pumps.12 An interesting feature associated with these hybrids is that selection of the partner moiety modulates the potentiation effect and breadth of antibiotics synergized.13 In an effort to explore new combinations of antibiotics and tobramycin hybrid adjuvants, a niclosamide–tobramycin hybrid was synthesized. Niclosamide is an FDA approved non-antibiotic drug that is used to treat tapeworm infections but has recently been shown to display a wide variety of other biological effects.14 It is currently in several clinical trials for cancer therapy,15 and also demonstrates potent activity against Gram-positive bacteria.16 Although not active against GNB, niclosamide has been shown to disrupt quorum sensing and the secretion of virulence factors against P. aeruginosa,17 as well as reverse colistin-resistance in combination therapy.18,19 Despite the plethora of biological interactions, niclosamide suffers from poor bioavailability and water solubility which limits its use as a systemic agent.
A niclosamide–tobramycin hybrid molecule was synthesized, in part, to overcome the poor water solubility of niclosamide as well as to expand the repertoire of tobramycin-based antibiotic adjuvants due to niclosamide's diverse biological properties. Copper-assisted azide–alkyne cycloaddition (CuAAC)20 was chosen as the ligation method to join an azide derivative of niclosamide with a tobramycin derivative incorporating a 1-hexyne aliphatic tether. The resulting hybrid was found to potentiate a variety of antibiotics against wild-type Pseudomonas aeruginosa, Acinetobacter baumannii, and Escherichia coli. Notably, cephalosporins including cefiderocol were potentiated. The niclosamide–tobramycin hybrid was shown to synergize with cefiderocol against MDR/XDR clinical isolates of P. aeruginosa via checkerboard assay as well as with time-kill kinetics. Interestingly, it was also found that synergy with cefiderocol was lost under iron-depleted conditions. Overall, this report shows that a niclosamide–tobramycin adjuvant may be able to synergize with even the most potent antibiotics, increasing their effectiveness against resistant phenotypes.
Results and discussion
Synthesis of niclosamide–tobramycin hybrid
Niclosamide–tobramycin hybrid 7 was designed based on our previous studies with tobramycin hybrid adjuvants as outlined in Fig. 1.13 A 1,4-disubstituted 1,2,3-triazole linkage was recently demonstrated to retain the adjuvant activity of a tobramycin homodimer21 and thus CuAAC was chosen to link the two units. An azide containing analog of niclosamide (compound 3) was synthesized via 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) coupling between 5-chlorosalicylic acid 1 and 4-azidoaniline 2. Compound 3 was evaluated for microbiological activity to verify that the modification of the niclosamide scaffold would not result in a loss of activity (Tables S1 and S2†) and the results were comparable to our previously reported activity for niclosamide. Tobramycin 4 was N-Boc and O-TBDMS protected following previously established protocol,21 yielding 5 which contained a free OH at the C-5 position of the deoxystreptamine ring. Previous studies have shown that modifying this position results in tobramycin derivatives that lack the ribosomal binding activity characteristic of aminoglycosides but display enhanced membrane interaction.10 Alkylation with 6-iodohexyne followed by deprotection of the hydroxyl groups afforded amphiphilic tobramycin 6. A 6-carbon aliphatic tether was chosen as this alkyne fragment was shown to be effective for the above-mentioned tobramycin homodimer. Although previous tobramycin–fluoroquinolone hybrids utilized a 12-carbon tether, niclosamide is relatively more hydrophobic than ciprofloxacin and moxifloxacin, thus, a shorter alkyl chain was considered appropriate. Compounds 3 and 6 were coupled using a copper(I) catalyst followed by deprotection with trifluoroacetic acid (TFA) resulting in compound 7 which contained a niclosamide fragment attached to an amphiphilic tobramycin fragment via a triazole linkage.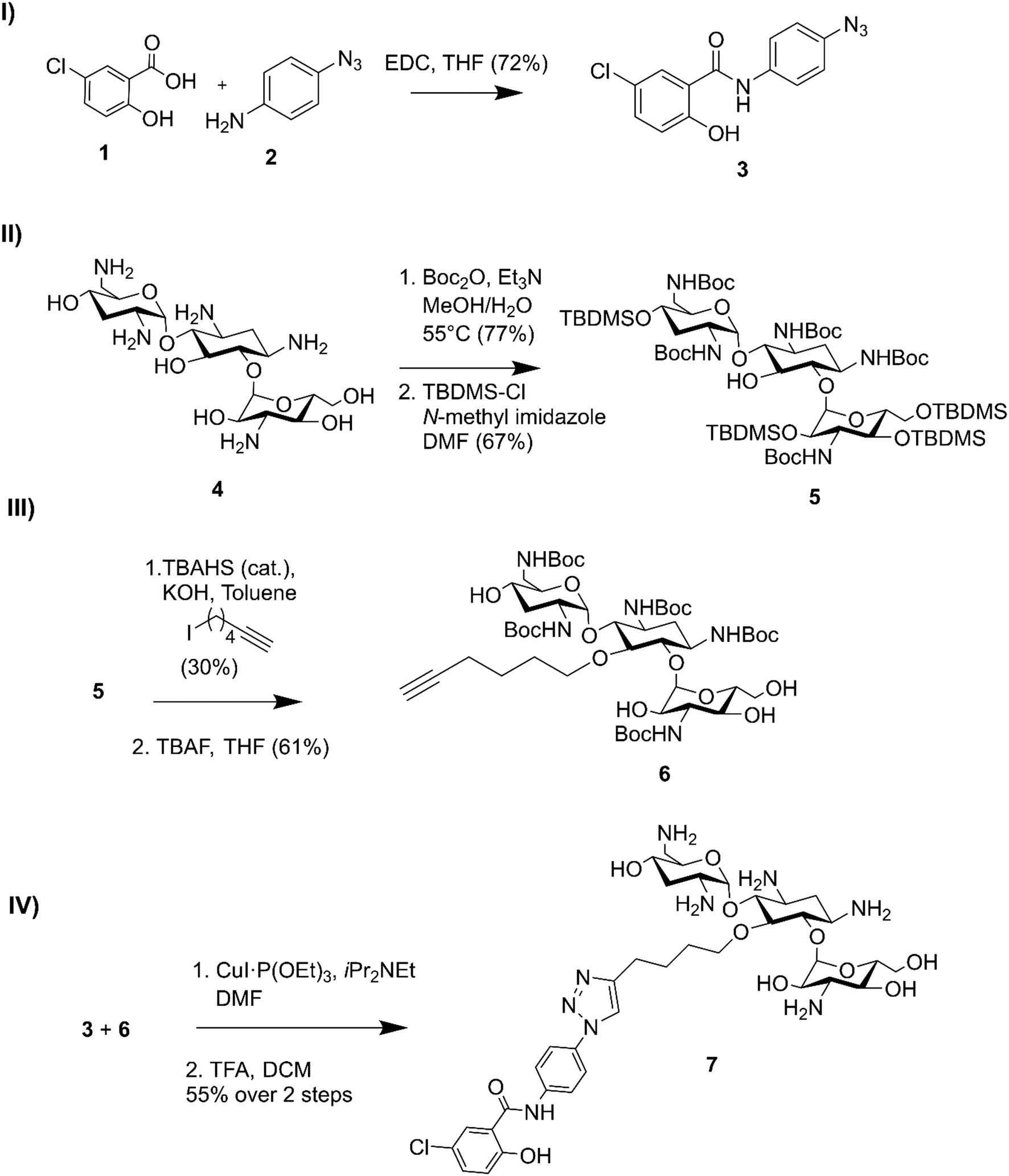 | ||
| Fig. 1 Synthesis of niclosamide tobramycin hybrid 7. I) Amide coupling of 3, II) tobramycin protection, III) alkylation followed by OTBDMS deprotection, IV) CuACC followed by N-Boc deprotection. | ||
Niclosamide–tobramycin hybrid 7 lacks intrinsic antibacterial activity
The antibacterial activity of 7 was initially evaluated against a panel of wild-type bacteria and compared to niclosamide and tobramycin (Table 1). As expected, niclosamide displayed potent activity against the Gram-positive organisms tested while tobramycin was active against Gram-negative organisms and Staphylococcus aureus. Compound 7 displayed very high MICs (≥64 μg ml−1) against all organisms tested confirming the expected loss of tobramycin ribosomal binding. The lack of intrinsic antibacterial activity of 7 also suggested that niclosamide's mechanism of action against Gram-positive bacteria was also lost. Many adjuvants lack stand-alone activity despite being able to synergize with other antibiotics.22 We therefore assessed the ability of 7 to potentiate various antibiotics in combination therapy.| Organism | Minimum inhibitory concentration (μg ml−1) | ||
|---|---|---|---|
| 7 | Niclosamide | Tobramycin | |
| P. aeruginosa PAO1 | 64 | >128 | 0.5 |
| A. baumannii ATCC 17978 | >128 | >128 | 0.5 |
| E. coli ATCC 25922 | >128 | >128 | 2 |
| S. aureus ATCC 29213 | >128 | 1 | 0.5 |
| MRSA ATCC 33592 | >128 | 0.25 | 2 |
| MRSE 61589 | 128 | 1 | 8 |
| E. faecalis ATCC 29212 | >128 | 2 | 8 |
| E. faecium ATCC 27270 | >128 | 2 | 16 |
Compound 7 potentiates different classes of antibiotics against Gram-negative bacteria
In order to assess the adjuvant capabilities of 7, we next studied the synergistic activity of hybrid 7 with 21 different antibiotics from various classes against wild-type P. aeruginosa, A. baumannii and E. coli. For the initial screening, a fixed concentration of 8 μg mL−1 of hybrid 7 in combination with each antibiotic was evaluated for the ability to inhibit bacterial growth prior to further analysis via checkerboard assay. The fold-potentiation was calculated by comparing the MIC of the antibiotic alone to the MIC in combination with the adjuvant. A decrease of the MIC in combination with 7 by at least 4-fold was interpreted as significant due to the inherent 2-fold margin of error in the microbroth dilution technique used to determine MIC.23 From the initial screen, compound 7 appeared to be more active against P. aeruginosa, and E. coli, with limited potentiation in wild-type A. baumannii (Fig. 2). Several classes of antibiotics were potentiated including efflux-susceptible antibiotics (fluoroquinolones, tetracyclines, and chloramphenicol) as well as antibiotics which have difficulty crossing the outer membrane (rifampicin, novobiocin). In addition, 7 was found to synergize with niclosamide, consistent with previous reports that show synergy between niclosamide and membrane-active agents.19 This also indicates the original mode of action of niclosamide is lost in hybrid 7.19 Based on the spectrum of antibiotics potentiated, it appears that compound 7 is most likely an adjuvant with a mechanism of action consistent with previously reported tobramycin hybrids (outer membrane destabilization and efflux pump inhibition).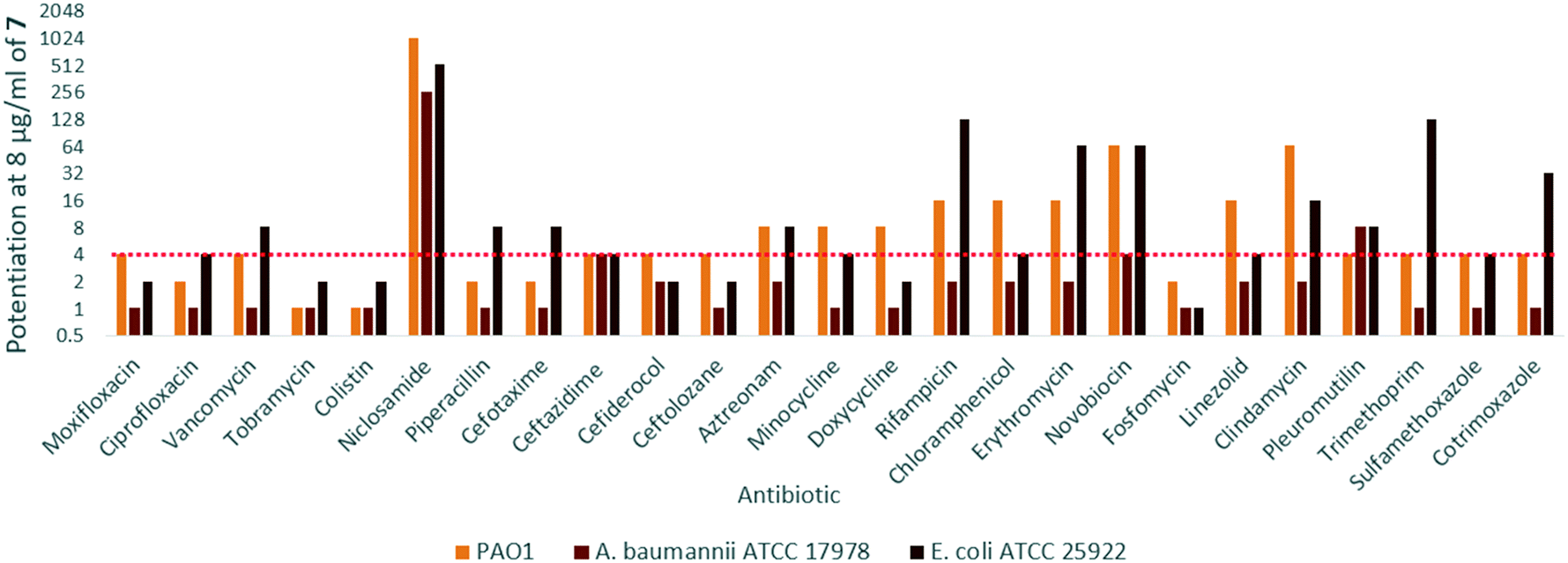 | ||
| Fig. 2 Fold potentiation of various antibiotics in combination with 8 μg ml−17 against wild-type Gram-negative bacteria, red dotted line denotes a 4-fold potentiation. Values at or above this line were determined to be significant due to the 2-fold margin of error inherent to MIC assays. | ||
Interestingly, 7 was also found to potentiate several generations of cephalosporins against PAO1. This effect was interesting as cephalosporins are one of the few classes of antibiotics effective against P. aeruginosa.24 We elected to further explore the synergy of hybrid 7 with various cephalosporins including cefotaxime, ceftazidime, cefiderocol and ceftolozane against MDR cephalosporin-resistant P. aeruginosa clinical isolates (Fig. 3). Significant potentiation was observed when 8 μg ml−17 was combined with cefiderocol against all isolates tested. Of particular note is the 32-fold potentiation seen in PA259, an XDR isolate resistant to all currently approved classes of antibiotics with the exception of colistin. As previously mentioned, this result was very promising since cefiderocol is approved to treat highly resistant P. aeruginosa infections. The combination of 7 and cefiderocol was chosen for a more detailed synergy evaluation against an expanded panel of clinical isolates.
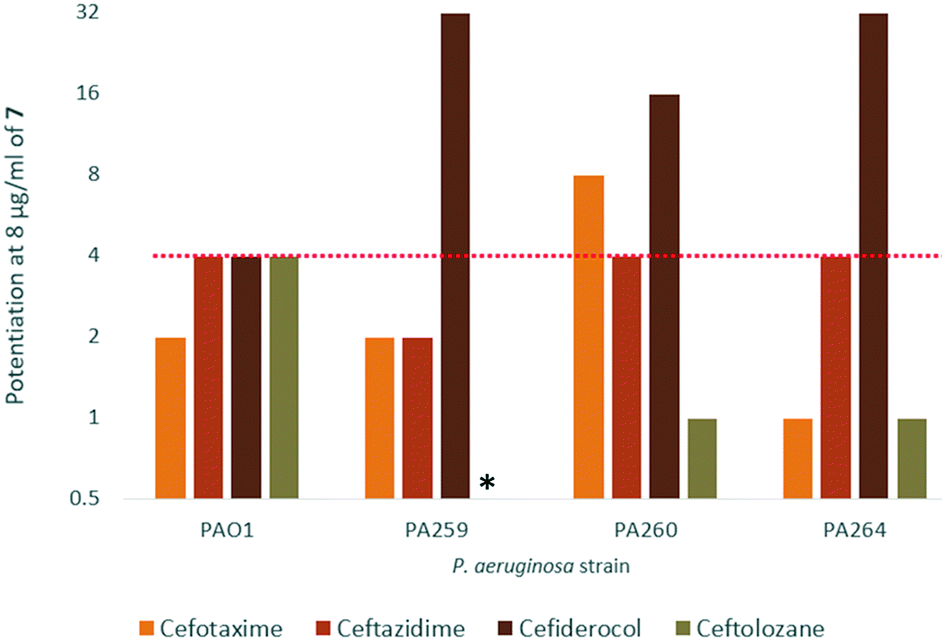 | ||
| Fig. 3 Fold potentiation of cephalosporins in combination with 8 μg ml−17 against wild-type and MDR clinical isolates of P. aeruginosa in MHB. Red dotted line denotes a 4-fold potentiation. *The combination of 7 and ceftolozane against PA259 was not evaluated. | ||
In order to demonstrate that neither niclosamide nor tobramycin alone was responsible for the observed synergy with cefiderocol, a checkerboard assay of each antibiotic was performed against PAO1. As expected, tobramycin or niclosamide alone were unable to change the MIC of cefiderocol in combination therapy, resulting in additive interactions for both agents (Table S4†). This shows that the adjuvant properties of 7 are unique to the hybrid structure which matches our previously established reports for tobramycin hybrid adjuvants.
It should also be noted that the media used for this assay was Mueller-Hinton broth (MHB). Due to cefiderocol's unique siderophore uptake mechanism, in vitro MIC values have been shown to differ depending on the level of iron present in the medium.6 Under iron-depleted conditions bacteria are believed to over-express siderophore secretion and uptake which enhances cefiderocol's outer membrane penetration. For this reason, current guidelines indicate that cefiderocol should be assessed using iron-depleted cation-adjusted Mueller-Hinton broth (ID-CAMHB) instead of the standard MHB used for microbroth dilution assays. However, to the best of our knowledge, the suitability of ID-CAMHB for assessing membrane-active adjuvants has yet to be determined. In addition, the interplay between siderophore uptake and membrane permeabilization would serve to further complicate the analysis. Thus, the initial synergy assay was performed in MHB in order to uncouple enhancing membrane penetration from siderophore uptake. This may limit the clinical relevance of the results obtained in MHB to situations where siderophore uptake is unavailable, such as in isolates with reduced expression of the pirA gene or in cases of hemochromatosis resulting in host iron overload conditions.25 The ability of hybrid 7 to potentiate cefiderocol in iron-replete media where cefiderocol faces reduced uptake was therefore assessed.
Hybrid 7 synergizes with cefiderocol against P. aeruginosa
To further confirm the observed synergy with cefiderocol against P. aeruginosa, a checkerboard assay was performed. The fractional inhibitory concentration (FIC) index for each isolate was calculated to assess the interactions between each agent. FIC for cefiderocol was calculated by dividing the MIC of cefiderocol in combination by the MIC of cefiderocol alone. Similar calculations were performed to obtain the FIC of 7. The FIC index was then calculated by adding the FIC indices of cefiderocol and the adjuvant. FIC index values of ≤0.5, 0.5 < x ≤ 4, and >4 were interpreted as synergistic, additive, and antagonistic interactions, respectively.23 As shown in Table 2, synergy was observed in most clinical isolates, but not in PA100036, PA86052, PA88949 and PA114228, which appeared extremely susceptible to cefiderocol (MIC ≤ 0.125 μg ml−1). Of particular note were strains PA259, PA101885 and PA109084 which had cefiderocol MIC values between 4–8 μg ml−1. In combination with 7 the MIC of cefiderocol in all of these strains was lowered significantly to ≤0.25 μg ml−1. In addition, synergy was observed in 2 out of 3 colistin-resistant clinical isolates tested. As the polybasic antibiotic colistin also exerts antimicrobial activity through membrane destabilization and cell lysis, colistin resistance is primarily mediated through modification of the negatively charged lipopolysaccharide (LPS) embedded in the outer membrane of GNB.26 Despite this, 7 retained adjuvant activity against these strains suggesting that LPS modification may not reduce its effectiveness as an adjuvant. Adjuvant activity was also compared to the well-known outer membrane permeabilizer polymyxin B nonapeptide (PMBN) (Table S6†). 8 μg ml−1 of PMBN was also able to potentiate cefiderocol in 3 out of 9 P. aeruginosa strains although 7 was superior overall.| Strain | MICFDC[MICCombination] | MIC7[MICCombination] | FIC index |
|---|---|---|---|
| (μg ml−1) | (μg ml−1) | ||
| a Denotes colistin-resistant isolates of P. aeruginosa. | |||
| PAO1 | 0.125[0.031] | 64[0.5] | 0.258 |
| PA259 | 8[0.25] | >128[4] | 0.031 < x <0.063 |
| PA260 | 1[0.063] | >128[0.125] | 0.063 < x < 0.063 |
| PA262 | 1[0.25] | >128[1] | 0.25 < x < 0.258 |
| PA264 | 0.5[0.031] | >128[0.125] | 0.125 < x < 0.126 |
| PA100036 | 0.063[0.063] | >128[0.25] | 1 < x < 1.002 |
| PA101885 | 4[0.063] | >128[2] | 0.017 < x < 0.031 |
| PA86052 | 0.031[0.031] | >128[0.25] | 1 < x < 1.001 |
| PA88949 | 0.016[0.016] | >128[0.25] | 1 < x < 1.002 |
| PA108590 | 2[0.031] | >128[4] | 0.016 < x < 0.047 |
| PA109084 | 4[0.016] | >128[0.5] | 0.004 < x < 0.008 |
| PA91433a | 0.25[0.016] | >128[1] | 0.016 < x < 0.023 |
| PA101243a | 0.5[0.063] | >128[2] | 0.125 < x < 0.141 |
| PA114228a | 0.125[0.063] | 32[16] | 1 |
To gain insight into the bacterial killing kinetics caused by the combination of niclosamide–tobramycin hybrid 7, a time-kill curve was generated for PA259 (Fig. 4). PA259 was chosen as it is XDR and showed the highest cefiderocol MIC. 4 μg ml−1 of cefiderocol was bacteriostatic after 24 hours while 8 μg ml−1 of 7 displayed little effect on bacteria growth. However, when 8 μg ml−1 of 7 was administered in combination with 4 μg ml−1 of cefiderocol, complete eradication was observed after 8 hours. This indicates that combinations of 7 and cefiderocol display bactericidal activity (defined as >3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log reduction in CFU mL−1) and further demonstrates how the activity of cefiderocol can be enhanced in combination therapy. Overall, these results indicate that even in iron-replete media where siderophore uptake is reduced, the activity of cefiderocol can be enhanced in combination therapy. This shows that hybrid 7 reverses the effect of reduced uptake caused by the presence of iron in the media and that membrane destabilization may serve to overcome reduced siderophore uptake.
log reduction in CFU mL−1) and further demonstrates how the activity of cefiderocol can be enhanced in combination therapy. Overall, these results indicate that even in iron-replete media where siderophore uptake is reduced, the activity of cefiderocol can be enhanced in combination therapy. This shows that hybrid 7 reverses the effect of reduced uptake caused by the presence of iron in the media and that membrane destabilization may serve to overcome reduced siderophore uptake.
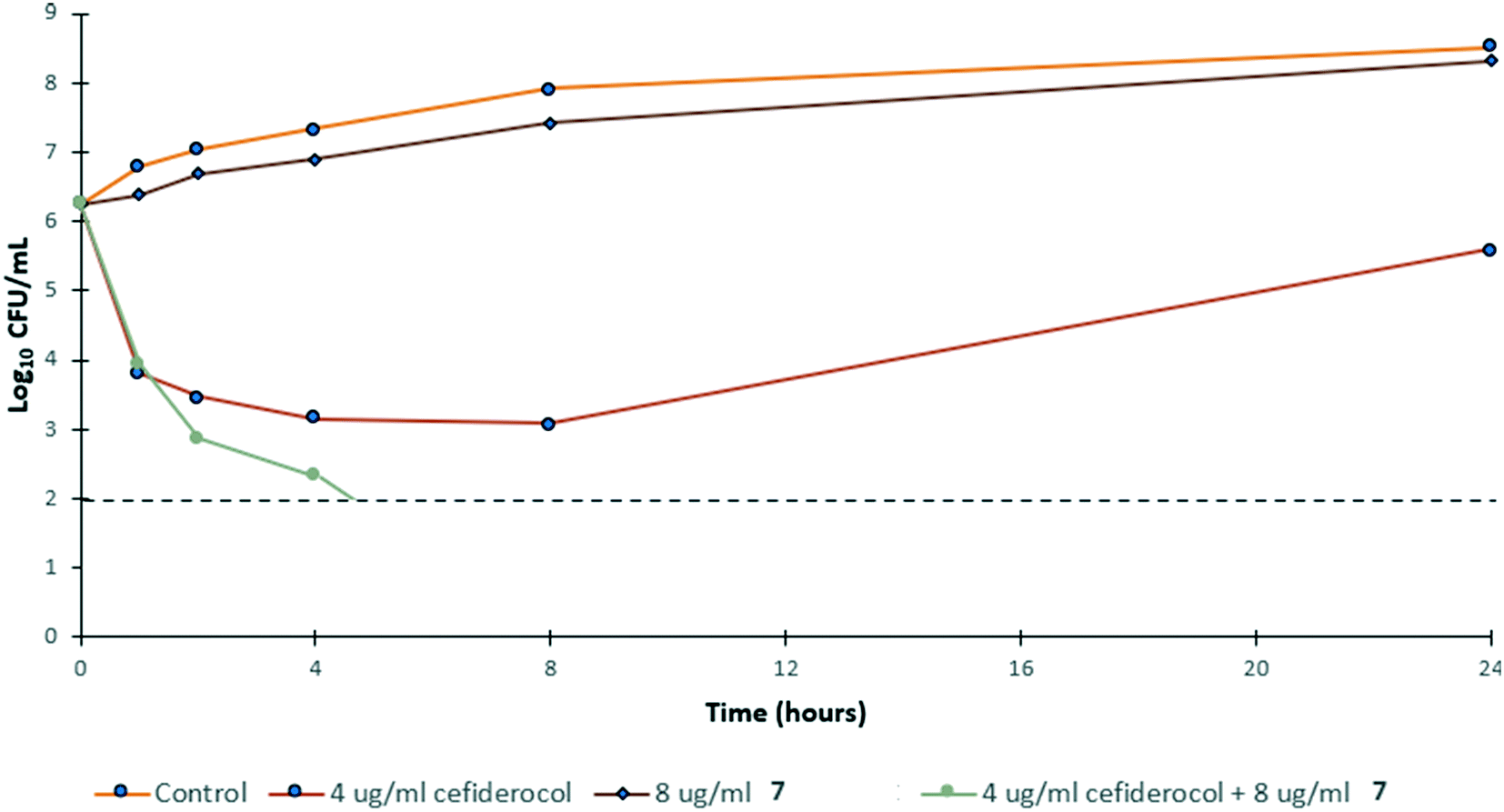 | ||
| Fig. 4 Time-kill kinetics of 7 in combination with ½ MIC of cefiderocol against PA259 in MHB. CFU mL−1 = colony forming units per mL. | ||
Synergy with cefiderocol was reduced against other gram-negative species
We next evaluated whether the potentiation of cefiderocol was unique to P. aeruginosa or could also be observed in other GNB. Checkerboard assays against A. baumannii (Table 3) and Enterobacteriaceae (E. coli, Klebsiella pneumoniae and Enterobacter cloacae, Table 4) revealed an overall reduction in synergy when compared to P. aeruginosa. Synergy was observed in wild-type A. baumannii ATCC 179781 but not in MDR clinical isolates. Synergy was also observed in 2 out of 4 strains of E. coli and 1 out of 3 strains of K. pneumoniae. Although some synergistic combinations were observed they were all limited to a 4-fold potentiation of cefiderocol, and no synergy was observed in E. cloacae. Overall, there appears to be a drastic reduction in synergy against Gram-negative organisms other than P. aeruginosa, indicating that the synergistic effects of 7 and cefiderocol appear to be P. aeruginosa specific. This may be a result of the inherently more restrictive outer membrane of P. aeruginosa compared to other Gram-negative organisms3 where cefiderocol faces fewer permeability challenges.| Strain | MICFDC[MICCombination] | MIC7[MICCombination] | FIC index |
|---|---|---|---|
| (μg ml−1) | (μg ml−1) | ||
| A. baumannii ATCC 17978 | 1[0.25] | >128[16] | 0.25 < x < 0.375 |
| AB031 | 1[0.5] | >128[0.25] | 0.5 < x < 0.502 |
| AB92247 | 0.25[0.125] | >128[8] | 0.5 < x < 0.563 |
| A. baumannii LAC-4 | 4[4] | >128[0.25] | 1 < x < 1.002 |
| Strain | MICFDC[MICCombination] | MIC7[MICCombination] | FIC index |
|---|---|---|---|
| (μg ml−1) | (μg ml−1) | ||
| E. coli ATCC 25922 | 0.125[0.063] | >128[0.25] | 0.5 < x < 0.502 |
| E. coli 107115 | 1[0.5] | >128[0.25] | 0.5 < x < 0.502 |
| E. coli 94393 | 1[0.25] | >128[1] | 0.25 < x < 0.258 |
| E. coli 94474 | 1[0.25] | >128[8] | 0.25 < x < 0.313 |
| KP113250 | 1[0.25] | >128[4] | 0.25 < x < 0.281 |
| KP113254 | 1[0.5] | >128[1] | 0.5 < x < 0.508 |
| KP116381 | 0.25[0.125] | >128[0.25] | 0.5 < x < 0.502 |
| E. cloacae 117029 | 1[0.5] | >128[4] | 0.5 < x < 0.531 |
| E. cloacae 118564 | 2[2] | >128[0.25] | 1 < x < 1.002 |
| E. cloacae 121187 | 2[2] | >128[0.25] | 1 < x < 1.002 |
Synergy was lost under iron-depleted conditions
As previously mentioned, ID-CAMHB is typically used to assess the activity of cefiderocol rather than MHB in order to maximize siderophore uptake. Unfortunately, when the synergy between 7 and cefiderocol again P. aeruginosa was assessed in ID-CAMHB a reduction in synergy was observed in all strains with the exception of wild-type PAO1 (Fig. 5). This may indicate that compound 7 would only be useful under conditions where siderophore uptake is unavailable (such as in resistant strains with reduced pirA expression). Increased membrane permeability may not confer any advantage to cefiderocol when siderophore uptake already serves to bypass the restrictive outer membrane of GNB. Alternatively, as this is the first reported instance of a membrane-active adjuvant being assessed in ID-CAMHB, this media may not be suitable for assessing the activity of similar compounds. Future work will explore the suitability of using ID-CAMHB to assess synergy of cefiderocol with membrane-active adjuvants. In addition, although a reduction in cefiderocol MIC against P. aeruginosa was seen in ID-CAMHB, PA259 still maintained a relatively high MIC of 4 μg ml−1 which according to the FDA and EUCAST guidelines indicates cefiderocol resistance.27 Overall, the 32-fold reduction in MIC observed in PA259 may still demonstrate the utility of adjuvant therapy in overcoming cefiderocol resistance despite the loss of synergy in ID-CAMHB, particularly in situations where siderophore uptake is limited.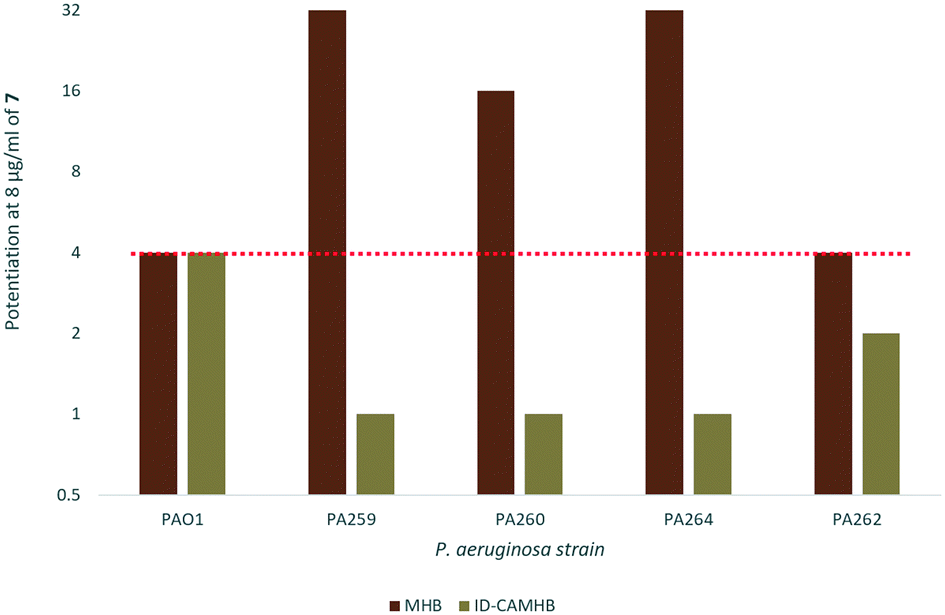 | ||
| Fig. 5 Fold-potentiation of 8 μg ml−17 in MHB compared to ID-CAMHB. Red dotted line denotes a 4-fold potentiation. | ||
Conclusion
A niclosamide–tobramycin hybrid 7 was successfully synthesized via CuAAC and assessed for synergy against several classes of antibiotics against GNB. It was found that 7 was able to potentiate a variety of antibiotics against wild-type P. aeruginosa and E. coli with limited activity against A. baumannii. Potent synergy was observed with cefiderocol against nearly all MDR clinical isolates of P. aeruginosa tested under iron-replete conditions and time-kill kinetics revealed complete bacterial killing at ½ MIC after 8 hours. Synergy observed appeared to be relatively specific to P. aeruginosa when compared to other GNB species. An overall loss of synergy was observed under ID-CAMHB conditions. Overall, this study indicates that a tobramycin–niclosamide hybrid can potentiate cefiderocol under iron-replete conditions. This is the first report on the potentiation of cefiderocol with an antibiotic adjuvant.Experimental
Synthesis
Tobramycin was purchased from AK Scientific Inc. (CA, USA) and all other reagents were purchased from Sigma-Aldrich (Oakville, ON, Canada) and were used without further purification. Air- and moisture-sensitive reactions were performed with dry solvents under inert nitrogen atmosphere. Reaction progress was monitored using thin-layer chromatography (TLC) plates visualized with UV light and staining with ninhydrin solution. Final compound 7 was fully characterized using 1-dimensional (1H and 13C) and 2-dimensional nuclear magnetic resonance (NMR) experiments as well as mass spectrometry. All NMR measurements were recorded on AMX-500 (500 MHz) Bruker instrument (Germany). High-resolution matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS) experiments were done on a Bruker ultraflextreme mass spectrometer (Germany) coupled to a time-of-flight mass analyzer.N-(4-Azidophenyl)-5-chloro-2-hydroxybenzamide (3)
5-Chlorosalicylic acid 1 (45 mg, 0.293 mmol) and EDC (112 mg, 0.586 mmol) were dissolved in tetrahydrofuran (2 mL) at room temperature and stirred for 15 minutes. 4-Azidoaniline 2 (51 mg, 0.299 mmol) was added and the reaction mixture was stirred at rt for 5 hours over which time the solution gradually turned from clear to yellow. The reaction was concentrated under vacuo and dissolved in 12 mL ethyl acetate, washed with 1 M HCl (3 × 5 mL), saturated sodium bicarbonate solution (3 × 5 mL) and brine (3 × 5 mL), dried over anhydrous Na2SO4 and concentrated in vacuo. The crude material was purified using flash chromatography using DCM as a solvent system to afford 3 as a brown solid (61 mg, 73%) 1H NMR (500 MHz, DMSO-d6) δ 11.81 (s, 1H), 10.47 (s, 1H), 7.93 (d, J = 2.6 Hz, 1H), 7.76 (dd, J = 9.0, 2.9 Hz, 2H), 7.48 (dd, J = 8.8, 2.6 Hz, 1H), 7.16–7.13 (m, 2H), 7.02 (d, J = 8.8 Hz, 1H).13C NMR (126 MHz, DMSO-d6) δ 165.4, 157.3, 136.7, 135.5, 135.3, 133.5, 128.7, 123.2, 122.8, 122.6, 122.5, 119.8, 119.5.
:1 mixture of methanol and water (150 mL) and treated with Boc2O (14.25 g, 65.29 mmol) in the presence of Et3N (8.0 mL, 57.4 mmol). The reaction mixture was stirred under reflux (at 55 °C) overnight (∼20 h), concentrated under vacuo, and thoroughly dried under high vacuum for 24 h to afford a white powdery solid (6.38 g, 77%). The dried crude penta-N-boc-protected tobramycin (1.13 g, 1.16 mmol) was dissolved in anhydrous DMF (6.0 mL) and treated with tert-butyldimethylsilyl chloride, TBDMSCl (1.13 g, 7.49 mmol), and N-methylimidazole (0.6 mL, 7.49 mmol). The reaction was stirred at rt for 4 days under nitrogen gas atmosphere, and the resulting mixture was poured into water (50.0 mL) and extracted with DCM (50 mL, ×3). The organic layer was dried over anhydrous Na2SO4, concentrated in vacuo, and purified by flash chromatography using gradient elution (hexanes/ethyl acetate, 15:1 to 8:1, v/v) to afford 5 (1.1 g, 67%) as a white solid. NMR data was consistent with a previous report.21
5-O-Hexyne-1,3,2′,6′,3′′-penta-N-Boc-tobramycin (6)
A solution of 5 (1.1 g, 0.78 mmol) in 4 mL toluene was treated with KOH (131 mg, 2.34 mmol) 2, 6-iodohexyne (0.2 mL, 1.55 mmol), and a catalytic amount of tetrabutylammonium hydrogen sulfate (TBAHS). The reaction was stirred at rt overnight, dispersed in 40 mL water and extracted with ethyl acetate (3 × 40 mL). The organic layers were combined, washed with brine (1 × 40 mL), dried over anhydrous Na2SO4, concentrated in vacuo, and purified by flash chromatography using gradient elution (hexanes/ethyl acetate, 15:1 to 8:1, v/v) to afford the alkylated product (331 mg, 30%) as a white solid. TBDMS deprotection of the hydroxyl groups was performed by dissolving 190 mg of the alkylated product in 2.5 mL anhydrous THF. The solution was treated with 0.1 mL tetrabutylammonium fluoride (TBAF) and stirred under nitrogen for 2 hours at rt. The reaction mixture was concentrated in vacuo, dissolved in 40 mL water and extracted with DCM (3 × 40 mL). The organic layers were combined, dried over anhydrous Na2SO4, concentrated in vacuo, and purified by flash chromatography using gradient elution (DCM/methanol, 25:1 to 20:1, v/v) to afford 6 (72 mg, 61%) NMR data was consistent with a previous report.21
Tobramycin–niclosamide hybrid 7
3 (8.2 mg, 0.027 mmol) and 6 (28.0 mg, 0.027 mmol) and CuI·P(OEt)3 (15 mg, 0.054 mmol) were dissolved in anhydrous DMF (3 mL) and treated with iPr2NEt (7.2 μL, 0.054 mmol). The reaction was stirred under nitrogen gas at rt for 2 h. The reaction mixture was concentrated under vacuum, dissolved in water (5 mL), and extracted with DCM (3 × 5 mL). The organic layers were combined, dried over anhydrous Na2SO4, concentrated in vacuo and the crude Boc-protected material was used in the next step without further purification. The crude residue was dissolved in 1 mL DCM and was treated with trifluoracetic acid (1 mL), stirred at rt for 2 hours and concentrated in vacuo. Purification by reverse-phase flash chromatography using gradient elution (methanol/water with 0.1% TFA, 20% methanol to 80% methanol, 7 eluted at 45% methanol) yielded off-white 7 (12.5 mg, 55%) 1H NMR (500 MHz, D2O) δ 7.91 (s, 1H, CH of triazole), 7.64–7.30 (m, 5H, niclosamide–Ph), 7.11–6.99 (m, 1H, niclosamide–Ph), 6.73–6.61 (m, 1 H, niclosamide–Ph), 5.18 (d, J = 2.6 Hz, 1H, anomeric H-1′), 4.99 (d, J = 3.5 Hz, 1H, anomeric H-1′′), 4.08 (dt, J = 8.5, 3.8 Hz, 1H, H-5′), 3.97 (t, J = 9.8 Hz, 1H, H-5′′), 3.88–3.51 (m, 10H, H-5, H-6, H-4, H-2′, H-4, H-2′′, H-4′′, O–CH2 of linker), 3.51–3.46 (m, 1H, H-6′′), 3.46–3.33 (m, 3H, H-1, H-3, H-3′′), 3.22–3.14 (m, 1H, H-6′), 3.10–3.01 (m, 1H, H-6′), 2.66–2.52 (m, 2H, triazole–CH2 of linker), 2.37 (dt, J = 12.6, 4.4 Hz, 1H, H-2), 2.07–1.92 (m, 2H, H-3′), 1.77 (q, J = 12.6 Hz, 1H, H-2), 1.68–1.45 (m, 4H, CH2 of linker). 13C NMR (126 MHz, D2O) δ 155.1 (niclosamide–CONH), 148.4, 137.5, 134.9, 134.9, 133.6, 128.8, 124.4 (CH of triazole), 122.0, 120.8, 120.3, 118.6, 117.6, 115.3, 101.3 (C-1′′), 92.7 (C-1′), 82.0 (C-4′′), 81.8 (C-5), 76.8 (C-5′′), 75.9 (C-5′), 73.2 (C-4), 73.1 (O–CH2 of linker), 68.5 (C-2′′), 64.8 (C-6), 63.1 (C-4′), 59.3 (C-6′′), 54.8 (C-3′′), 49.7 (C-1), 48.4 (C-3), 48.4, 47.2 (C-2′), 38.4 (C-6′′), 28.8 (CH2 of linker), 28.0 (C-3′), 27.7 (C-2), 25.1 (CH2 of linker), 24.4 (triazole–CH2 of linker). MALDI-TOF-MS m/z calcd for C37H54ClN9NaO11+ monoisotopic peak (M + Na)+: 858.352 found: 858.360.Antimicrobial susceptibility assay
Bacterial isolates used in this study were obtained from the American Type Culture Collection (ATCC) [reference strains], the Canadian National Intensive Care Unit (CAN-ICU) surveillance study28 and the Canadian Ward (CANWARD) surveillance study.29 Clinical isolates belonging to the CAN-ICU and CANWARD surveillance studies were recovered from patients suffering presumed infectious diseases entering or admitted in a participating medical center across Canada during the time of study. The in vitro antibacterial activities of the conjugates were assessed using the microbroth dilution method according to the Clinical and Laboratory Standards Institute (CLSI) guidelines.30 Bacterial cultures were grown overnight and diluted in saline to achieve a 0.5 McFarland turbidity, followed by 1:50 dilution in Mueller-Hinton broth (MHB) for inoculation to a final concentration of approximately 5 × 105 colony forming units per mL. Experiments were performed on 96-well plates. The tested agents were 2-fold serially diluted in MHB and incubated with equal volumes of bacterial inoculum at 37 °C for 18 h. The MIC was determined as the lowest concentration to inhibit visible bacterial growth in the form of visible turbidity, which was confirmed using an EMax Plus microplate reader (Molecular Devices, USA) at a wavelength of 590 nm. MHB with or without bacterial cells were used as positive or negative controls, respectively.
Checkerboard assay
The assay was performed on 96-well plates as previously described.31 The antibiotic was 2-fold serially diluted along the x axis, while the adjuvant was 2-fold serially diluted along the y axis to create a matrix in which each well contained a combination of both agents at different concentrations. Bacterial cultures grown overnight were diluted in saline to 0.5 McFarland turbidity, followed by 1:50 dilution in MHB and inoculation of each well to a final concentration of approximately 5 × 105 colony forming units per mL. Wells consisting of only MHB with or without bacterial cells were used as positive or negative controls, respectively. Plates were incubated for 18 h at 37 °C and inspected for visible turbidity, which was confirmed using EMax Plus microplate reader (Molecular Devices, USA) at a wavelength of 590 nm. Fractional inhibitory concentration (FIC) of antibiotic was calculated by dividing the MIC of antibiotic in the presence of conjugates by the MIC of antibiotic alone. Likewise, the FIC of conjugates was calculated via dividing the MIC of conjugates in the presence of antibiotic by the MIC of conjugates alone. The FIC index was obtained by the summation of both FIC values. The FIC indices were interpreted as synergistic, indifferent or antagonistic for values of ≤0.5, 0.5 < x ≤ 4 or >4, respectively. ID-CAMHB was prepared from MHB as previously described.32
Author contributions
LB: conceptualization, investigation, writing – original draft, MB: investigation (microbiology assays), GJ: investigation (synthesis of 6), FS: conceptualization, project administration, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Canadian Institutes of Health Research (CIHR) in the form of a pilot project (162159) and the Natural Sciences and Engineering Research Council of Canada (NSERC) in the form of a discovery grant (DG 2018-06047). LB acknowledges support from the University of Manitoba in the form of a University of Manitoba Graduate Fellowship.References
- E. D. Brown and G. D. Wright, Nature, 2016, 529, 336–343 CrossRef CAS PubMed.
- H. I. Zgurskaya, C. A. López and S. Gnanakaran, ACS Infect. Dis., 2016, 1, 512–522 CrossRef PubMed.
- Z. Pang, R. Raudonis, B. R. Glick, T. J. Lin and Z. Cheng, Biotechnol. Adv., 2019, 37, 177–192 CrossRef CAS PubMed.
- H. C. C. Póvoa, G. C. Chianca and N. L. P. P. Iorio, Infect. Dis. Ther., 2020, 9, 417–420 CrossRef PubMed.
- M. Gregorova, D. Morse, T. Brignoli, J. Steventon, F. Hamilton, M. Albur, D. Arnold, M. Thomas, A. Halliday, H. Baum, C. Rice, M. B. Avison, A. D. Davidson, M. Santopaolo, E. Oliver, A. Goenka, A. Finn, L. Wooldridge, B. Amulic, R. J. Boyton, D. M. Altmann, D. K. Butler, C. McMurray, J. Stockton, S. Nicholls, C. Cooper, N. Loman, M. J. Cox, L. Rivino and R. C. Massey, eLife, 2020, 9, 1–13 CrossRef PubMed.
- G. G. Zhanel, A. R. Golden, S. Zelenitsky, K. Wiebe, C. K. Lawrence, H. J. Adam, T. Idowu, R. Domalaon, F. Schweizer, M. A. Zhanel, P. R. S. Lagacé-Wiens, A. J. Walkty, A. Noreddin, J. P. Lynch III and J. A. Karlowsky, Drugs, 2019, 79, 271–289 CrossRef CAS PubMed.
- E. Yusuf, H. I. Bax, N. J. Verkaik and M. van Westreenen, J. Clin. Med., 2021, 10, 1068 CrossRef CAS PubMed.
- S. Malik, M. Kaminski, D. Landman and J. Quale, Antimicrob. Agents Chemother., 2020, 64, e01221-20 CrossRef PubMed.
- G. D. Wright, Trends Microbiol., 2016, 24, 862–871 CrossRef CAS PubMed.
- B. K. Gorityala, G. Guchhait, S. Goswami, D. M. Fernando, A. Kumar, G. G. Zhanel and F. Schweizer, J. Med. Chem., 2016, 59, 8441–8455 CrossRef CAS PubMed.
- R. Domalaon, T. Idowu, G. G. Zhanel and F. Schweizer, Clin. Microbiol. Rev., 2018, 31, e00077-17 CrossRef PubMed.
- X. Yang, S. Goswami, B. K. Gorityala, R. Domalaon, Y. Lyu, A. Kumar, G. G. Zhanel and F. Schweizer, J. Med. Chem., 2017, 60, 3913–3932 CrossRef CAS PubMed.
- T. Idowu, D. Ammeter, M. Brizuela, G. Jackson, S. Alam and F. Schweizer, Bioorg. Med. Chem. Lett., 2020, 30, 127575 CrossRef CAS PubMed.
- W. Chen, R. A. Mook, R. T. Premont and J. Wang, Cell. Signalling, 2018, 41, 89–96 CrossRef CAS PubMed.
- S. Burock, S. Daum, U. Keilholz, K. Neumann, W. Walther and U. Stein, BMC Cancer, 2018, 18, 297 CrossRef.
- R. Rajamuthiah, B. B. Fuchs, A. L. Conery, W. Kim, E. Jayamani, B. Kwon, F. M. Ausubel and E. Mylonakis, PLoS One, 2015, 10, e0124595 CrossRef PubMed.
- F. Imperi, F. Massai, C. R. Pillai, F. Longo, E. Zennaro, G. Rampioni, P. Visc and L. Leoni, Antimicrob. Agents Chemother., 2013, 57, 996–1005 CrossRef CAS PubMed.
- R. Ayerbe-Algaba, M. L. Gil-Marqués, M. E. Jiménez-Mejías, V. Sánchez-Encinales, R. Parra-Millán, M. E. Pachón-Ibáñez, J. Pachón and Y. Smani, Front. Cell. Infect. Microbiol., 2018, 8, 348 CrossRef CAS.
- R. Domalaon, P. Malaka De Silva, A. Kumar, G. G. Zhanel and F. Schweizer, Antimicrob. Agents Chemother., 2019, 63, e02574-18 CrossRef PubMed.
- R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 565–598 CrossRef.
- T. Idowu, D. Ammeter, H. Rossong, G. G. Zhanel and F. Schweizer, J. Med. Chem., 2019, 62, 9103–9115 CrossRef CAS PubMed.
- H. Douafer, V. Andrieu, O. Phanstiel and J. M. Brunel, J. Med. Chem., 2019, 62, 8665–8681 CrossRef CAS PubMed.
- F. C. Odds, J. Antimicrob. Chemother., 2003, 52, 1 CrossRef CAS PubMed.
- N. Yaneja and H. Kaur, Microbiol. Insights, 2016, 9, 9–19 Search PubMed.
- J. M. Kidd, K. Abdelraouf and D. P. Nicolau, Antimicrob. Agents Chemother., 2019, 64, e01767-19 Search PubMed.
- K. Jeannot, A. Bolard and P. Plésiat, Int. J. Antimicrob. Agents, 2017, 49, 526–535 CrossRef CAS PubMed.
- P. J. Simner and R. Patel, J. Clin. Microbiol., 2020, 59, e00951-20 CrossRef PubMed.
- G. G. Zhanel, M. DeCorby, N. Laing, B. Weshnoweski, R. Vashisht, F. Tailor, K. A. Nichol, A. Wierzbowski, P. J. Baudry, J. A. Karlowsky, P. Lagacé-Wiens, A. Walkty, M. McCracken, M. R. Mulvey, J. Johnson, Canadian Antimicrobial Resistance Alliance (CARA) and D. J. Hoban, Antimicrob. Agents Chemother., 2008, 52, 1430–1437 CrossRef CAS PubMed.
- D. J. Hoban and G. G. Zhanel, J. Antimicrob. Chemother., 2013, 68 Suppl 1, i3–i5 CrossRef PubMed.
- Clinical and Laboratory Standards Institute, Performance standards for antimicrobial susceptibility testing, Clin. Lab. Stand Institute, Wayne, PA, 28th edn, 2018 Search PubMed.
- L. Berry, R. Doma laon, M. Brizuela, G. G. Zhanel and F. Schweizer, MedChemComm, 2019, 10, 517–527 RSC.
- M. A. Hackel, M. Tsuji, Y. Yamano, R. Echols, J. A. Karlowsky and D. F. Sahm, Antimicrob. Agents Chemother., 2018, 62, e01968-17 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1md00206f |
| This journal is © The Royal Society of Chemistry 2021 |