
β-Lactam antibiotic targets and resistance mechanisms: from covalent inhibitors to substrates
Montserrat
Mora-Ochomogo
 and
Christopher T.
Lohans
*
and
Christopher T.
Lohans
*
Department of Biomedical and Molecular Sciences, Queen's University, Kingston, ON K7L 3N6, Canada. E-mail: christopher.lohans@queensu.ca
First published on 4th August 2021
Abstract
The β-lactams are the most widely used antibacterial agents worldwide. These antibiotics, a group that includes the penicillins and cephalosporins, are covalent inhibitors that target bacterial penicillin-binding proteins and disrupt peptidoglycan synthesis. Bacteria can achieve resistance to β-lactams in several ways, including the production of serine β-lactamase enzymes. While β-lactams also covalently interact with serine β-lactamases, these enzymes are capable of deacylating this complex, treating the antibiotic as a substrate. In this tutorial-style review, we provide an overview of the β-lactam antibiotics, focusing on their covalent interactions with their target proteins and resistance mechanisms. We begin by describing the structurally diverse range of β-lactam antibiotics and β-lactamase inhibitors that are currently used as therapeutics. Then, we introduce the penicillin-binding proteins, describing their functions and structures, and highlighting their interactions with β-lactam antibiotics. We next describe the classes of serine β-lactamases, exploring some of the mechanisms by which they achieve the ability to degrade β-lactams. Finally, we introduce the L,D-transpeptidases, a group of bacterial enzymes involved in peptidoglycan synthesis which are also targeted by β-lactam antibiotics. Although resistance mechanisms are now prevalent for all antibiotics in this class, past successes in antibiotic development have at least delayed this onset of resistance. The β-lactams continue to be an essential tool for the treatment of infectious disease, and recent advances (e.g., β-lactamase inhibitor development) will continue to support their future use.
Introduction
The β-lactams are the most widely used class of antibiotic.1,2 The antibiotics in this group are characterized by a reactive four-membered β-lactam ring, which is essential to their bactericidal activity.3 This β-lactam ring is generally fused with another heterocycle, and this bicyclic core is functionalized in several key positions, giving rise to several distinct subclasses including the penicillins, cephalosporins, and carbapenems. The biological activities of the β-lactam antibiotics are determined by their overall structures, as defined by their core scaffolds and substituents, which influence their interactions with target proteins and resistance mechanisms.β-Lactam antibiotics interfere with the synthesis of peptidoglycan, a core component of the bacterial cell wall that is composed of glycan strands reinforced with peptide bridges. Bacterial enzymes known as penicillin-binding proteins (PBPs)† catalyze the formation of peptide cross-links during the synthesis of peptidoglycan. It is this transpeptidation activity that is blocked by β-lactam antibiotics, weakening the cell wall and increasing susceptibility to osmotic stress, ultimately leading to autolysis.2,4 β-Lactam antibiotics are covalent inhibitors that target PBPs, acylating a catalytically essential serine residue with the reactive β-lactam core. This acylation step yields a stable acyl–enzyme complex (AEC), preventing the PBP from catalyzing transpeptidation.
The use of β-lactams as therapeutics is threatened by the emergence of β-lactam-resistant bacteria and the rapid dissemination of their resistance mechanisms. Enzymes that degrade β-lactam antibiotics, β-lactamases, are of particular clinical significance, and represent the most important resistance mechanism in Gram-negative bacteria.5–8 These enzymes efficiently catalyze the hydrolytic opening of the β-lactam ring, rendering the antibiotic unable to target PBPs. β-Lactamases are broadly categorized as serine β-lactamases (SBLs) and metallo-β-lactamases (MBLs) according to the mechanism by which they achieve this catalysis. While MBL catalysis is dependent on one or more zinc ions, SBLs interact with β-lactams in a similar fashion as PBPs, forming a covalent complex between the antibiotic and a serine residue. While this complex is relatively stable for PBPs, the SBL active site can catalyze its hydrolysis, releasing the degraded β-lactam.
While PBPs are the principal targets of β-lactam antibiotics, antibiotics in this class also covalently target other enzymes. Some β-lactam subclasses inhibit L,D-transpeptidases (Ldts), a family of enzymes that form alternative peptide cross-links in bacterial peptidoglycan. Although these enzymes are not essential in many bacteria,9 they play important roles in mycobacteria,10 and have attracted much recent interest as antibiotic targets.11 As with PBPs, the covalent complexes derived from β-lactams with Ldts tend to be stable, interfering with peptidoglycan transpeptidation. Some β-lactams are also observed to target other families of proteins, including some serine and cysteine proteases.12
β-Lactams are a structurally diverse group of antibiotics. The chemical structures of these β-lactams and the structures of the enzyme active sites with which they interact determines whether an antibiotic forms a stable complex that interferes with catalysis, or if the enzyme can deacylate this complex. In this review, we discuss the competing processes of β-lactam antibiotic development and bacterial resistance. We begin by providing an overview of the major subclasses of β-lactam antibiotics, and then describe their activities as covalent inhibitors of PBPs. We next introduce the SBLs and explore how they have achieved the ability to degrade β-lactam-derived covalent complexes, treating a covalent inhibitor as a substrate. While the four-membered β-lactam pharmacophore is the basis of inhibitors for other kinds of enzymes,13 this review specifically focuses on those β-lactams that are used as antibiotics. In addition, β-lactam resistance can be achieved through other mechanisms (e.g., MBL production, efflux), but we will focus on SBLs due to their covalent interactions with β-lactam antibiotics. Although resistance is always a threat to the clinical use of antibiotics, continuing medicinal chemistry efforts focusing on the β-lactam scaffold have preserved its central place in antibacterial chemotherapy. The ongoing studies characterizing these resistance mechanisms may help guide future antibiotic development, yielding new antibiotics that are less susceptible to inactivation by known enzymes.
β-Lactam antibiotics and β-lactamase inhibitors
Since the discovery and development of penicillin G as a therapeutic, β-lactams have played an essential role in the treatment of infectious disease. The β-lactam antibiotics are currently the most used antibacterial agents, representing more than half of antibiotic use worldwide.1 They are widely available, effective, and generally have low levels of toxicity. β-Lactams are divided into several distinct subclasses according to their structure, and the penicillin, cephalosporin, and carbapenem subclasses are commonly used for the treatment of a wide range of bacterial infections. A vast number of β-lactam antibiotics and variants have been generated by industrial and academic medicinal chemistry efforts, or have been identified as natural products.14 In this review, we focus on those that have reached the clinic.Chemically, β-lactam antibiotics are characterized by a core four-membered azetidinone (i.e., β-lactam) ring (Fig. 1). This group acylates serine and cysteine nucleophiles in the active sites of β-lactam targets, yielding a stable covalent acyl–enzyme complex (AEC) that interferes with catalysis. In most β-lactam subclasses, the β-lactam ring is fused to a five- or six-membered ring. Kinetic studies on the reactivity of β-lactams indicate that this fusion may not greatly enhance β-lactam ring reactivity,15 contrary to what might be expected based on the impact of ring fusion on the planarity of the β-lactam nitrogen. Specific binding interactions involving the active sites of β-lactam-targeted proteins also play a major role in determining the reactivity of these antibiotics, imparting a level of selectivity.15
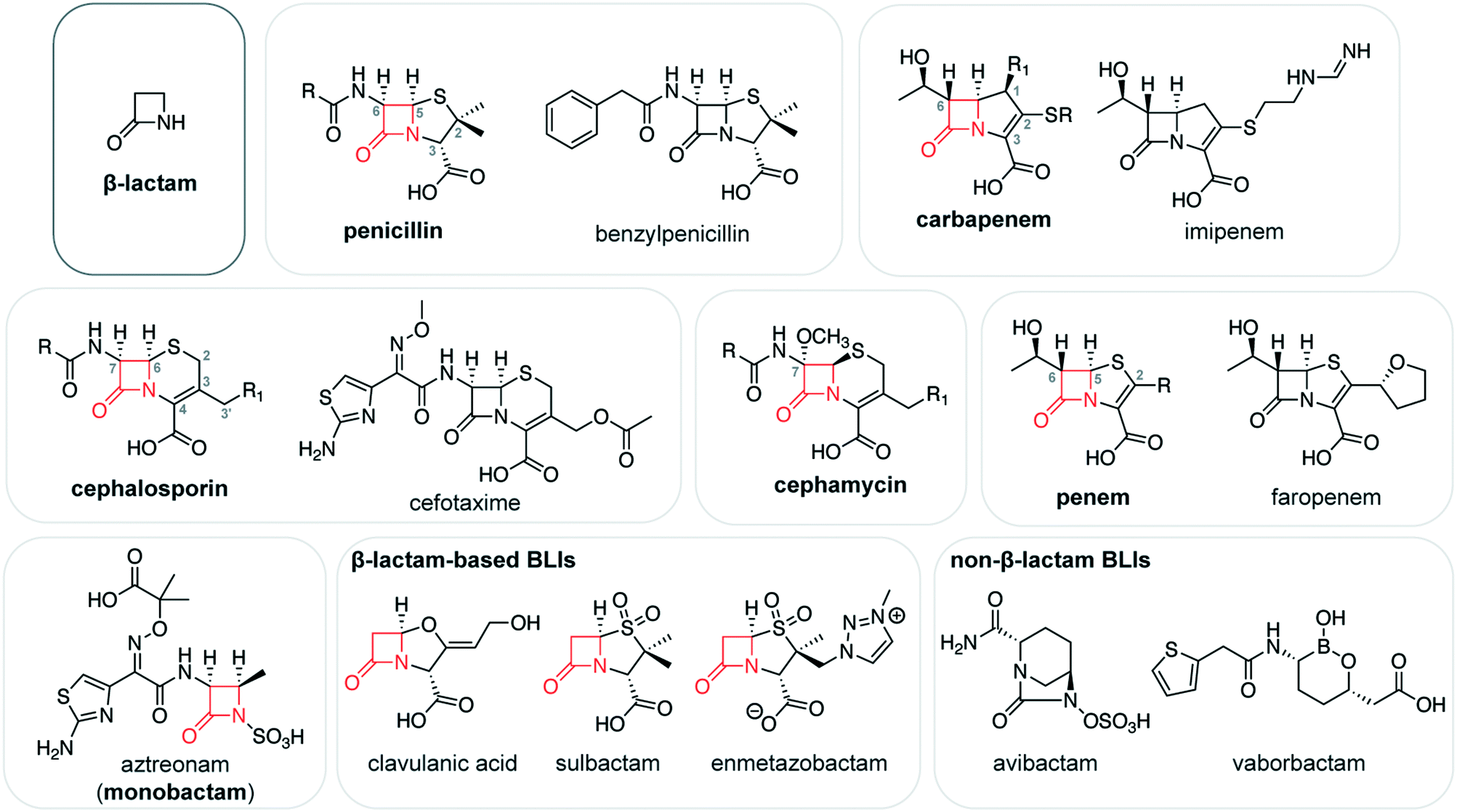 | ||
| Fig. 1 β-Lactam antibiotics and β-lactamase inhibitors. Chemical structure of the β-lactam ring, alongside generic structures of the major β-lactam antibiotic subclasses and representatives from these subclasses. The β-lactam ring in these structures is shown in red. Numbering typically begins from the atom in the top position of the ring fused to the β-lactam (e.g., sulfur in penicillins). The structures of common β-lactam-based and non-β-lactam-based β-lactamase inhibitors (BLIs) are also shown. | ||
Penicillins contain a core bicyclic ring system consisting of fused β-lactam and thiazolidine rings with a cis stereochemistry at C5 and C6 (Fig. 1). The thiazolidine ring bears two methyl substituents at C2 and a carboxylate group at C3. Penicillin antibiotics vary according to the substituent at C6. For example, penicillin G (or benzylpenicillin), the first penicillin to be discovered, bears a phenylacetamido substituent at this position. This C6 substituent has been the focus of extensive chemical modifications, enhancing the antibiotic target range and reducing susceptibility to β-lactamase-catalyzed degradation. These efforts have yielded penicillins with broad-spectrum activity against both Gram-positive and Gram-negative bacteria (e.g., ampicillin, amoxicillin), and that are relatively resistant to β-lactamase catalysis (e.g., oxacillin, methicillin). The penicillin temocillin also bears a methoxy group on C6, resulting in further β-lactamase resistance. The penicillin C3 carboxylate may also be esterified, as seen in prodrugs such as pivmecillinam.
Cephalosporins are structurally related to the penicillins, with a bicyclic core composed of a β-lactam ring fused to a six-membered dihydrothiazine ring with a double bond between C3 and C4 (Fig. 1). Like the penicillins, cephalosporins have a cis stereochemistry at C7 and C8. The dihydrothiazine ring bears a carboxylate group at C4 and a methyl or methylene group at C3, with the substituents on C3′ varying greatly. In many cephalosporins, this substituent is a leaving group which is eliminated following the opening of the β-lactam ring and migration of the C3–C4 double bond. The loss of the leaving group is not necessarily concerted with the opening of the β-lactam ring, and likely occurs at a later stage.16 Kinetic studies suggest that this elimination reaction interferes with the catalysis of some serine β-lactamases.17,18 The C3′ substituent can also be cleverly modified to enhance antibiotic uptake, as shown for the cephalosporin cefiderocol, which was recently approved for clinical use.19 Cefiderocol bears a siderophore substituent on C3′,20 promoting its transport into the bacterial periplasm in which its PBP targets are located.
Drug development efforts have focused extensively on the acylamido substituent at C7 of the cephalosporin core.21 Most first- and second-generation cephalosporins bear similar acylamido groups as found in earlier generations of penicillins. Third and fourth-generation cephalosporins (e.g., ceftazidime, cefotaxime) tend to have bulky C7 acylamido side chains containing a planar oxyimino group, which sterically interfere with β-lactamase catalysis.22 Structurally related to the cephalosporins, the cephamycins (e.g., cefoxitin) have a C7 methoxy substituent which also impacts β-lactamase-catalyzed degradation.18
The carbapenem subclass of β-lactams have a core consisting of a β-lactam ring fused to a five-membered pyrroline ring with a carbon at position 1 and a C2–C3 double bond (Fig. 1).23 The pyrroline ring bears a carboxylate group at C3 and a thioether substituent at C2, and many carbapenems have a 1β-methyl group for improved stability to human enzymes.24,25 All clinically used carbapenems bear a hydroxyethyl side chain at C6; the configuration at this position is inverted relative to penicillins and cephalosporins, and carbapenems have a trans stereochemistry at C5 and C6. Following the opening of the β-lactam ring, the pyrroline ring can undergo enamine–imine tautomerization, and the imine form inhibits the activity of some β-lactamases.26 Penems are structurally related to carbapenems, with a core composed of fused β-lactam and thiazoline rings (Fig. 1). Faropenem, an orally administered penem, bears a C6 hydroxyethyl side chain and a tetrahydrofuran substituent on C2.
Finally, the monobactam subclass of β-lactams consist of a core β-lactam ring that is not fused to any other rings. Currently, aztreonam is the main clinical representative of this group (Fig. 1), and new monobactams (e.g., LYS228) are currently in the antibiotic pipeline.27 The β-lactam ring of aztreonam has an acylamido substituent containing an oxyimino group, analogous to the side chains in many cephalosporins. The nitrogen of the monobactam β-lactam ring is sulfonylated, mimicking the C3/C4 carboxylate in other β-lactam antibiotics. Due in part to the inability of MBLs to degrade aztreonam at meaningful levels,28 combination therapies of aztreonam with β-lactams belonging to other classes are of great current interest.29,30
β-Lactam antibiotics are frequently administered with a β-lactamase inhibitor (BLI) that protects the antibiotic from SBL-catalyzed degradation.31–33 The first clinically used BLI, clavulanic acid, contains a β-lactam ring that acylates a nucleophilic serine in the SBL active site (Fig. 1). In the resulting AEC, the oxazolidine ring can undergo a fragmentation reaction, followed by tautomerization to yield a stable enamine form, or reaction with a second serine residue to irreversibly inhibit catalysis.34 Sulbactam and tazobactam, penicillin sulfone BLIs, are proposed to undergo a related fragmentation reaction, with the sulfone sulfur acting as a leaving group.35 The cross-linking and/or the tautomerization of the fragmented inhibitor species in the active site is thought to interfere with the SBL hydrolytic mechanism, thereby stabilizing the covalent complex and blocking SBL activity. The related cleavage of the bond between positions 1 and 5 has been exploited in other BLIs that have not reached the clinic such as the alkylidene penems and bromopenicillanic acid, which both undergo cyclization reactions in the AEC.36,37
SBL inhibitor development has largely shifted to scaffolds that do not contain β-lactam rings. The diazabicyclooctanes (DBOs; e.g., avibactam; Fig. 1) reversibly react with the SBL serine nucleophile, forming a carbamoyl–enzyme complex.38 Following the success of avibactam, a number of related inhibitors have been explored (e.g., relebactam, ETX2514, ETX1317), of which several have reached clinical studies and/or have been approved for use.39–41 Notably, there is great interest in developing DBOs which simultaneously target both SBLs and PBPs.42 SBL inhibitors based on a boronate scaffold have also encountered great success,43,44 with vaborbactam (Fig. 1) recently achieving approval for clinical use, and benzoxaboroles taniborbactam and QPX7728 progressing through clinical testing.45–47 These boronates are thought to act as transition state analogues, and some demonstrate the intriguing ability to inhibit both SBLs and MBLs.43,46
The development of β-lactam-based BLIs also continues. These efforts have yielded enmetazobactam, a penicillin sulfone that is currently in the clinical pipeline for use with the cephalosporin cefepime.48 Extensive efforts are also underway to develop inhibitors for MBLs;49,50 no such inhibitors are available in the clinic for the treatment of MBL-positive infections.
Penicillin-binding proteins
Peptidoglycan has a critical role in the bacterial cell wall, defining the structure of the cell and offering protection from osmotic pressure. Chemically, peptidoglycan is a mesh-like layer composed of glycan chains cross-linked by short peptide bridges. The final steps of peptidoglycan synthesis are (mostly)51 catalyzed by penicillin-binding proteins (PBPs), enzymes found in the bacterial periplasm.52 Bacteria produce several different PBPs which vary in terms of the catalytic domains present, the reactions catalyzed by those domains, and the overall functions of these enzymes in peptidoglycan synthesis and metabolism, cell division, and cell wall remodelling.52 PBPs with glycosyltransferase domains form glycosidic linkages between existing peptidoglycan and lipid II, a precursor composed of a lipid-modified disaccharide with a pentapeptide chain. Then, the transpeptidase domain found in all PBPs catalyzes the formation of covalent cross-links between the peptide chains (Fig. 2A).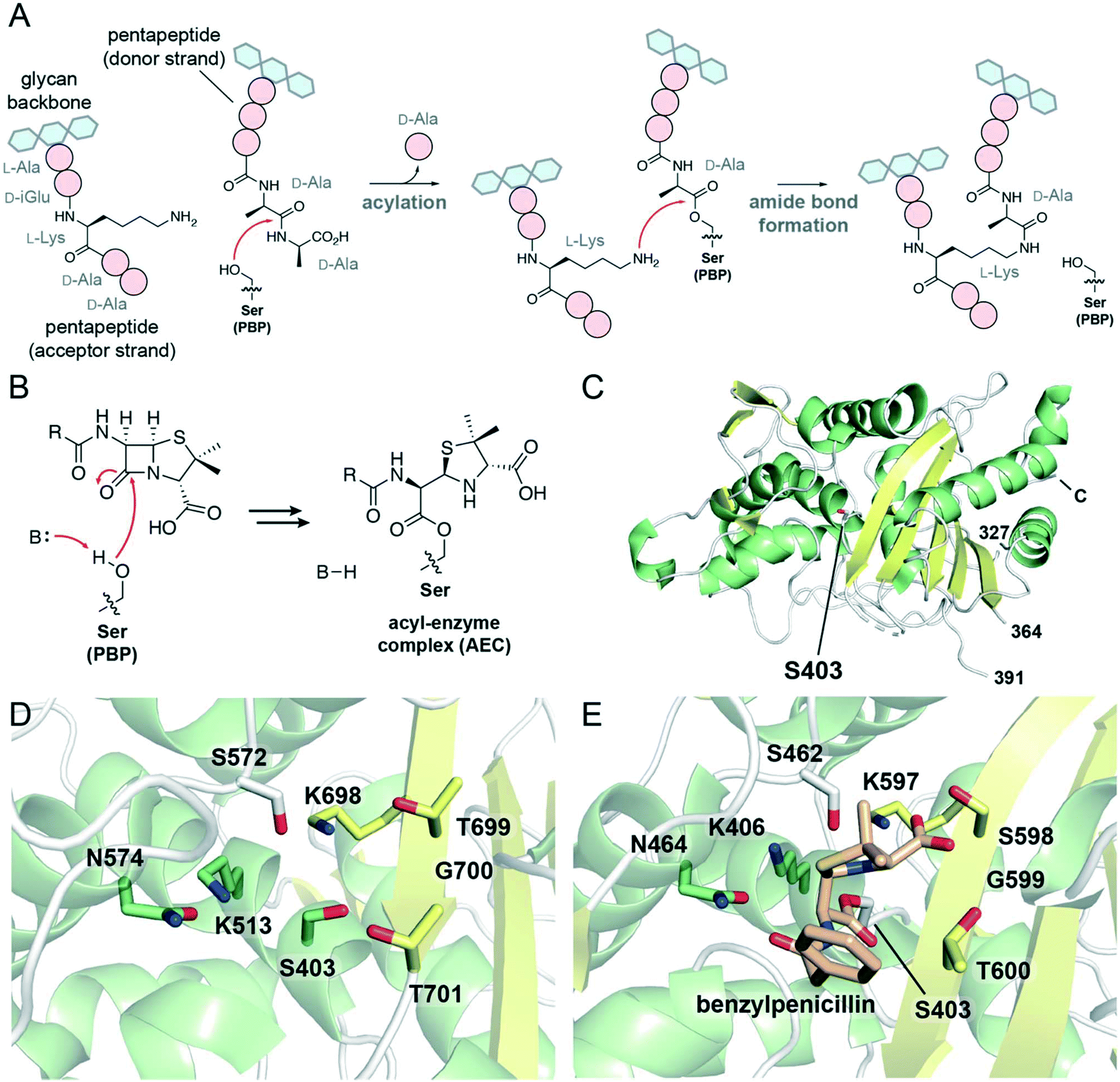 | ||
| Fig. 2 Mechanism, inhibition, and structures of PBP transpeptidases. (A) Scheme showing the role of PBP transpeptidases in the formation of 4 → 3 peptidoglycan cross-links. The PBP nucleophilic serine attacks a pentapeptide on the donor strand, forming a peptide–enzyme complex and releasing D-alanine. From the acceptor strand of peptidoglycan, a nucleophilic side chain (here, L-lysine) reacts with the peptide–enzyme complex, forming a cross-link and releasing the enzyme. (B) Scheme showing the acylation of the transpeptidase serine nucleophile with a penicillin antibiotic, yielding an acyl–enzyme complex. (C) View of the transpeptidase domain of PBP2a from Staphylococcus aureus (PBP 1MWT). The nucleophilic serine, Ser403, is represented as white sticks. α-Helices are shown in green, β-sheets in yellow, and loops in white. The N-terminal domains and the region between residues 364 and 391 are not displayed; the C-terminus is labeled. (D) View of the active site of PBP1b from Escherichia coli (PDB 3VMA). Conserved active site residues, including the nucleophilic serine Ser403, are shown as sticks, and are coloured according to local secondary structure. (E) View of the active site of the transpeptidase domain of PBP2a from S. aureus in complex with benzylpenicillin (PDB 1MWT). The nucleophilic serine, Ser403, is acylated with benzylpenicillin (shown as tan sticks). | ||
The mechanism of the transpeptidase domain begins with the nucleophilic attack of an active site serine residue onto a peptidoglycan pentapeptide chain at the amide bond connecting the fourth and fifth amino acid residues (Fig. 2A). This forms a covalent complex in which the transpeptidase serine is acylated with a tetrapeptide, accompanied by the loss of a D-alanine molecule. Then, a nucleophilic side chain on the third amino acid (e.g., L-lysine, meso-diaminopimelic acid) or bound to the third amino acid (e.g., pentaglycine) on the acceptor strand of peptidoglycan attacks the ester carbonyl of the peptide–PBP complex, forming an amide bond between the donor and acceptor peptide chains and releasing the PBP transpeptidase serine. This forms a 4 → 3 peptide cross-link in which the fourth residue of one peptide chain is covalently attached to the third residue of another peptide.
β-Lactam antibiotics covalently inhibit the transpeptidase domains of PBPs, disrupting peptidoglycan cross-linking which ultimately leads to cell death. The inhibitory complex formed between the β-lactam and the enzyme results from the nucleophilic attack of the transpeptidase serine onto the antibiotic β-lactam ring (Fig. 2B), resembling the first step of the transpeptidase mechanism. The covalent complexes derived from PBPs and β-lactams tend to be stable, resulting in long-lived inhibition of transpeptidase activity. Binding of the antibiotic to the enzyme active site has been attributed to structural similarities between the β-lactam antibiotic core and the terminal residues of the peptidoglycan pentapeptide chain (i.e., the D-Ala-D-Ala dipeptide; Fig. 2A).53
Crystallographic studies have characterized the structures of PBP transpeptidases from several bacterial species.52 These studies reveal that the structure of the transpeptidase domain is largely conserved, and is composed of two major subdomains: one subdomain consists of a β-sheet associated with α-helices, and the other is composed of α-helices (Fig. 2C).52 The active site serine nucleophile is found on one of these α-helices as part of a conserved SXXK motif, amidst other residues contributed by an adjacent β-strand and nearby active site loops (Fig. 2D). The lysine in this motif is proposed to act as a general base, activating the serine for nucleophilic attack onto the substrate pentapeptide chain. An active site loop presents an SXN motif, while a nearby β-strand contributes a KTG(T/S) motif to the active site. The lysine and serine/threonine residues in this latter motif likely activate the acceptor strand for nucleophilic attack.
Further crystallographic studies have demonstrated the interactions between β-lactam antibiotics and the transpeptidase active site. β-Lactams typically adopt a particular orientation in the AEC, interacting with many of the conserved elements mentioned above. As seen in the crystal structure of benzylpenicillin with PBP2a from Staphylococcus aureus (Fig. 2E), the C3 carboxylate hydrogen bonds to both hydroxyl-containing side chains in the KTG(T/S) motif.54 Similar interactions occur between this motif and the carboxylates of β-lactams belonging to other subclasses.55,56 The asparagine of the SXN motif hydrogen bonds to the carbonyl of the C6/C7 acylamido side chains of penicillins and cephalosporins, respectively,54,56 resembling interactions occurring between PBPs and their substrates.57 The C6 hydroxyethyl side chain of the carbapenems and penems also hydrogen bond to this asparagine residue.55
Although conserved elements in both the transpeptidase active site and the structures of β-lactam antibiotics govern their interactions, there is considerable variation in the efficacy of different β-lactams as PBP inhibitors both between and within subclasses. Studies profiling bacterial PBPs have revealed the preferential targeting of some enzymes by certain β-lactam subclasses, while other PBPs from the same species may show completely different trends.58–61 There is further heterogeneity within the different β-lactam subclasses, with the structures of the antibiotic substituents determining the spectrum of PBPs targeted and the potency of the resulting inhibition.60,61
Although the complexes derived from β-lactams and PBPs are typically stable, some PBPs catalyze the hydrolysis of these AECs.62,63 For many PBPs, the rate of hydrolysis is too slow to offer meaningful protection, while others catalyze this reaction at levels that provide a level of resistance.64 PBP5 from Pseudomonas aeruginosa demonstrates the ability to hydrolyze several β-lactams belonging to the penicillin, cephalosporin, and carbapenem subclasses.64 Structural studies revealed features resembling those found in the active sites of class A serine β-lactamases that may allow for this catalysis.64 Interestingly, a PBP from Streptomyces R61 catalyzes the C5–C6 fragmentation of penicillins.65,66 This fragmentation reaction yields an adduct resembling a peptide–enzyme complex, which could then be deacylated by hydrolysis or through reaction with particular amino acids and dipeptides.67
Some PBPs resist covalent inhibition by β-lactams by disfavouring the initial formation of the AEC.68 One such β-lactam-resistant PBP, PBP2a from S. aureus, contributes to the β-lactam resistant phenotype of methicillin-resistant S. aureus (MRSA). Access to the active site of PBP2a is allosterically controlled through interactions with peptidoglycan components, limiting the exposure of the serine nucleophile to most β-lactam antibiotics.69 Recombination and mutations in the genes of other PBPs has frequently lead to the production of modified PBPs with decreased affinity to β-lactam antibiotics, and such PBP variants are important resistance determinants in species such as Streptococcus pneumoniae and Neisseria spp.68
Serine β-lactamases
The production of β-lactamases is the most common resistance mechanism against β-lactam antibiotics in Gram-negative bacteria.5,31 These enzymes inactivate β-lactam antibiotics, preventing them from covalently targeting PBPs. Although other resistance mechanisms against β-lactams are widely distributed (e.g., efflux, target modification), β-lactamases are of particular clinical relevance as they are frequently encoded on plasmids and can be readily disseminated through horizontal gene transfer.70 Some β-lactamases are sufficient to confer resistance to β-lactam antibiotics alone, while enzymes with lower levels of activity offer sufficient protection when combined with other resistance mechanisms (e.g., decreased porin production, increased production of efflux pumps).71β-Lactamases are hydrolases that inactivate β-lactam antibiotics through a hydrolytic mechanism. Although thousands of different β-lactamase enzymes have been identified,72 they can be grouped into a few distinct classes according to structure and mechanism.5,73,74 The serine β-lactamases (SBLs), classes A, C, and D in the Ambler classification system, employ a two-step hydrolytic mechanism involving a nucleophilic serine residue. The metallo-β-lactamases (MBLs), which are assigned to class B, hydrolyze β-lactams using a water molecule activated by one or more zinc ions. As they do not interact covalently with β-lactam antibiotics, MBLs are not discussed further here, but the reader is directed to several excellent reviews.6,74–76
The mechanism by which β-lactam antibiotics interact with SBLs resembles the mechanism by which they target PBPs (Fig. 3A). Following initial substrate binding, SBL catalysis begins with the general base activated nucleophilic attack of an active site serine residue onto the antibiotic β-lactam ring, yielding a covalent AEC. While these AECs are relatively stable for PBPs, SBLs can often efficiently catalyze the deacylation of such complexes. To achieve this, a general base residue in the SBL active site promotes the attack of a hydrolytic water molecule onto the AEC ester carbonyl, hydrolyzing the covalent adduct and freeing the enzyme (Fig. 3A). The contributions of the general base residue(s) to SBL catalysis have been examined for all three classes of SBLs, with substitution of these residues typically yielding deacylation-deficient enzymes in which hydrolysis is impaired and the AEC is stabilized.77–79 There are notable differences regarding the precise residues involved and the overall mechanism of β-lactam degradation between the three Ambler classes of SBLs.
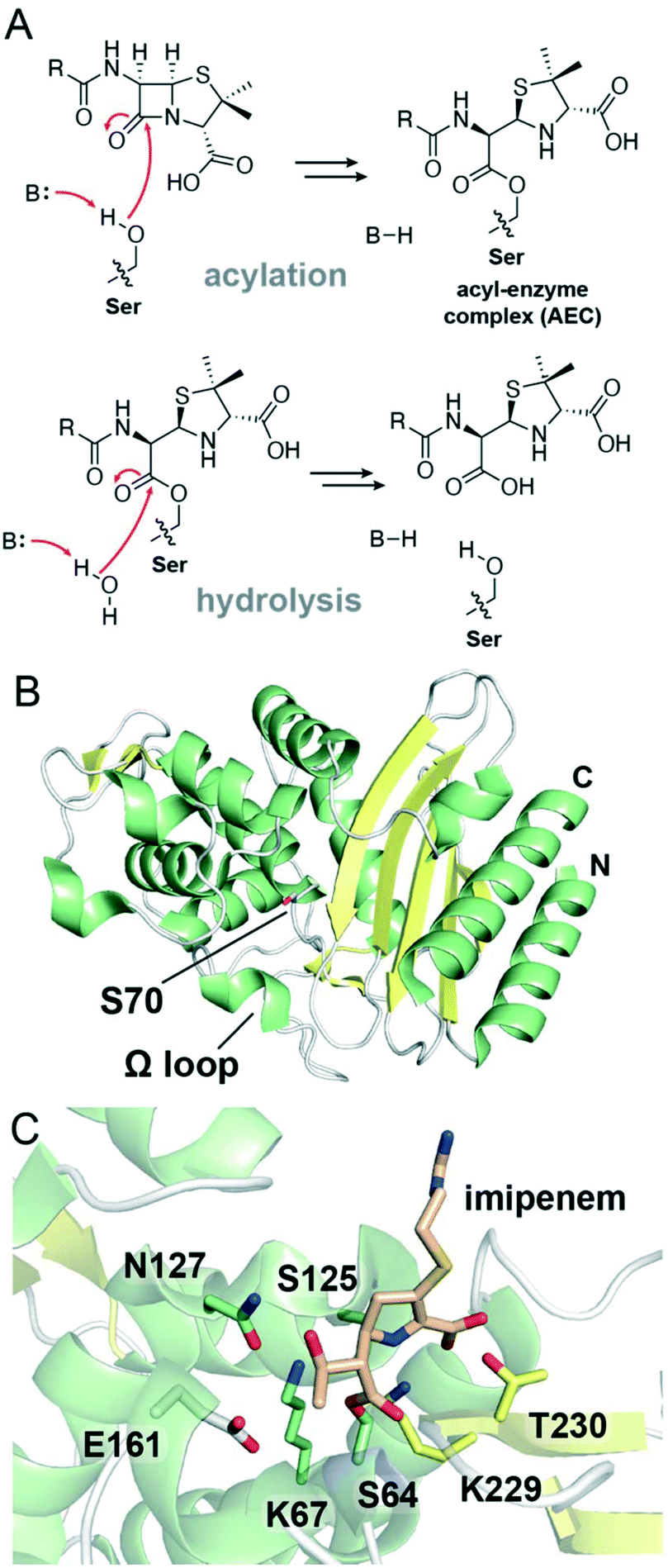 | ||
| Fig. 3 Overview of serine β-lactamase mechanism and structure of class A enzymes. (A) Reaction scheme showing the main steps in SBL catalysis. In the acylation step, the SBL nucleophilic serine residue attacks the substrate β-lactam ring, yielding an ester-linked acyl–enzyme complex. In the hydrolysis step, the nucleophilic attack of a water molecule onto the acyl–enzyme complex carbonyl occurs, hydrolyzing the antibiotic and releasing the serine. (B) View of a crystal structure of class A SBL TEM-1 (PDB 1M40). The nucleophilic serine, Ser70, is represented as white sticks. α-Helices are shown in green, β-sheets in yellow, and loops in white. The N- and C-termini are indicated. (C) View of the active site of class A SBL GES-1 in which the serine (Ser64; green sticks) is acylated with the carbapenem imipenem (tan sticks) (PDB 4GOG). Conserved active site residues are represented as sticks and are coloured according to local secondary structure elements. | ||
The active sites of SBLs and PBP transpeptidase domains are highly related, and modern SBLs and PBPs are thought to have a common ancestor.80,81 Some PBPs specialize as carboxypeptidases, hydrolytically cleaving D-alanine residues from the pentapeptide chains in peptidoglycan.82 SBLs have also specialized to favour hydrolysis, but target β-lactam antibiotics instead of peptidoglycan. The different classes of SBLs are thought to have evolved from PBPs in at least two distinct events, with the divergence of class C enzymes predating the divergence of the other two classes.81,83,84 Although there are similarities between PBPs and SBLs, the structural changes associated with SBL activity prevent these enzymes from efficiently hydrolyzing peptidoglycan.85
SBLs are commonly classified according to the β-lactam antibiotics that they can degrade. Enzymes such as TEM-1 and SHV-1 are penicillinases which confer resistance to penicillins and early generations of cephalosporins. The dissemination of these penicillinases among Gram-negative pathogens motivated the development of newer generations of cephalosporins (e.g., those with oxyimino-containing C7 side chains), which tend to be unsusceptible to their catalysis. However, this in turn led to the selection for bacteria that produce extended-spectrum β-lactamases (ESBLs) which hydrolyze many of the third and fourth generation cephalosporins.21 Although carbapenem antibiotics tend to be only slowly degraded by penicillinases and ESBLs, SBLs with carbapenemase activity currently threaten the clinical use of these antibiotics of last resort.23 New SBL (and MBL) variants with different substrate specificities continue to be identified, selected for in part due to the clinical use of β-lactam antibiotics.86,87
Class A serine β-lactamases
Following the introduction of the penicillins in the 1940s, class A SBLs soon emerged as a major resistance mechanism in hospital settings.21 SBLs belonging to this class are now very widespread, and are currently the most common variety of β-lactamase encountered.80 The class A SBL TEM-1 has been particularly extensively studied, and the evolution of TEM variants is well documented, providing invaluable insight into the relationship between the active site structure and substrate specificity of SBLs.88,89The first class A enzymes to be identified and characterized, such as TEM-1 and SHV-1, are typically penicillinases that degrade early penicillins (e.g., benzylpenicillin), while also offering lower levels of protection against first-generation cephalosporins.5 The clinical use of third-generation cephalosporins led to the selection of bacteria that produce class A extended-spectrum β-lactamases (ESBLs), including variants of the TEM and SHV enzymes as well as SBLs belonging to the CTX-M family.21,90 Similarly, the use of the carbapenem antibiotics is threatened by the dissemination of class A enzymes with carbapenemase activity, including members of the KPC and GES families.91 Class A SBLs are of great current clinical relevance, and all commonly used β-lactam antibiotics are degraded by at least some members of this class. Many class A enzymes are susceptible to classical SBL inhibitors such as clavulanic acid and sulbactam, but inhibitor-resistant variants are well-known and common.5 More recent inhibitor development efforts have yielded SBL inhibitors with better coverage against class A, such as avibactam and vaborbactam;31,38 however, SBLs with reduced susceptibility to these inhibitors are already known.92
The structural fold of the class A SBLs resembles that of the PBP transpeptidases, consisting of an α-helical subdomain and a subdomain consisting of an antiparallel β-sheet with associated α-helices (Fig. 3B).93 At the interface of these two subdomains, the nucleophilic serine residue is found at the terminus of an α-helix as part of an SXXK motif (Fig. 3C).94 A β-strand on the side of the active site bears a K(T/S) motif analogous to the PBP transpeptidase KTG(T/S) motif. The lysine and threonine/serine residues on this β-strand play a major role in β-lactam antibiotic binding, interacting with the C3/C4 carboxylate found in most β-lactams (Fig. 3C).79,94 Across from the β-sheet, a loop connecting two α-helices contributes serine and asparagine residues to the active site, resembling the transpeptidase SXN motif. This asparagine residue plays an important role in substrate binding, hydrogen bonding to the C6/C7 acylamido substituents of penicillins and cephalosporins. Other loops surrounding the active site play additional roles in substrate binding, determining the substrate specificity of these enzymes.
The Ω loop plays a critical role in the catalysis of class A SBLs.95 This loop bears a glutamate residue (Glu166; TEM-1 numbering) which is involved in both acylation and hydrolysis steps. Although there is some debate regarding the mechanism by which the nucleophilic serine is activated in the acylation step, both Glu166 and Lys73 (of the SXXK motif) are important for enzymatic activity, as demonstrated through kinetic studies.96 In the hydrolysis step, Glu166 acts as a general base, activating a water molecule for nucleophilic attack onto the ester carbonyl in the AEC.96 An oxyanion hole formed from the backbone amides of two active site residues (e.g., Ser70 and Ala237 in TEM-1) activates the carbonyl of the AEC during hydrolysis.97
Class A SBLs are typically efficient penicillinases that confer resistance to early penicillins such as benzylpenicillin (penicillin G) and ampicillin.5 However, their catalytic activity is often far lower against β-lactamase resistant penicillins such as methicillin and oxacillin.5 Crystallographic studies of the complex derived from a deacylation deficient variant of TEM-1 with benzylpenicillin revealed key binding interactions between the penicillin C3 carboxylate and C6 acylamido groups with conserved active site residues.98 The asparagine of the SXN motif hydrogen bonds to the C6 side chain, orienting the C6 side chain towards the Ω loop.98 Changes to the sequence and structure of the Ω loop play an important role in determining the penicillinase activity of class A SBLs, impacting their selectivity for different penicillins.95,99
While class A penicillinases can degrade early generation cephalosporins, the bulky C7 oxyimino-containing side chains found in most third and fourth generation cephalosporins (e.g., ceftazidime) are not easily accommodated in the active sites of these enzymes.22 Class A SBLs have achieved the ability to degrade such cephalosporins (i.e., ESBL activity) through a variety of different changes to the active site.88,89 Like with the penicillins, the Ω loop closely interacts with the C7 side chains of cephalosporins,100 and ESBL activity is often attributed to changes to this loop and the neighbouring β3 strand.88,101–103 These substitutions often decrease steric clash, allowing the cephalosporin side chain to be better accommodated in the enzyme active site.100 Substitutions to the Ω loop and the β3 strand can similarly enhance the activity of class A SBLs against aztreonam, an oxyimino-substituted monobactam.104,105 In contrast, the cephalosporinase activity of the CTX-M ESBLs may not require the broadening of the active site, and instead has been attributed to the mobility of an active site β-strand.106
Carbapenems are degraded very slowly by most class A SBLs, often forming stable AECs which resist hydrolysis. The stability of this AEC has been attributed to interactions between the carbapenem C6 hydroxyethyl side chain and the hydrolytic water molecule which decrease its nucleophilicity.107 Additional crystallographic studies indicate that the AEC derived from imipenem and TEM-1 adopts a conformation in which the ester carbonyl is not positioned in the oxyanion hole.107 Tautomerization of the carbapenem pyrroline ring in the AEC is also thought to disfavour hydrolysis.26,108
Some class A SBL families demonstrate the ability to efficiently degrade carbapenems, and crystallographic studies have yielded important insight into the mechanisms of catalysis. In the complex derived from SFC-1 and meropenem, the carbapenem hydroxyethyl side chain was oriented away from the position of the hydrolytic water molecule.109 Similarly, GES-5 controls the positioning of the carbapenem hydroxyethyl side chain, and favours an active site conformation in which the glutamate general base is appropriately positioned to activate the nucleophilic serine.94 Interactions involving the hydroxyethyl side chain are also important for the carbapenemase activity of KPC-2,110 and may involve a key tryptophan residue.111 Residues more distant from the active site (e.g., the Q241-R220 loop) also play critical roles in the deacylation of the carbapenem-derived AEC.112
Class C serine β-lactamases
Class C SBLs are a widely distributed group of serine β-lactamases most commonly associated with resistance to cephalosporins and cephamycins.71,113 Enzymes in this class are chromosomally encoded in bacteria belonging to many different clades, and plasmid-encoded class C SBLs have been widely disseminated. The name AmpC is often used to encompass the SBLs belonging to this class, although several distinct subgroups (e.g., CMY, ADC, FOX) have been described. Recent reports have aimed to standardize the discussion class C SBLs according to structural alignments.114,115 Regulation of class C SBL production has been extensively studied, and many enzymes in this class are induced in response to disruptions in peptidoglycan metabolism following exposure to β-lactam antibiotics.71 Mutations to genes encoding regulatory proteins and enzymes involved in peptidoglycan metabolism have often been observed in resistant bacteria, leading to hyperinducibility and/or hyperproduction.113Enzymes in this class tend to demonstrate high catalytic rates and affinities for early generation cephalosporins.116 In fact, the hydrolysis of some cephalosporins by class C enzyme P99 from Enterobacter aerogenes was observed to be diffusion limited.117 While the degradation of third and fourth generation cephalosporins and cephamycins tends to be slower, class C enzymes with extended spectrum cephalosporinase activity and enhanced cephamycinase activity are known.71 Class C SBLs also typically confer resistance to β-lactams belonging to the penicillin subclass, although some penicillins (e.g., oxacillin) act as inhibitors.5 However, most enzymes in this class are not able to degrade carbapenems.118 Nonetheless, the broad spectrum of activity associated with class C enzymes represents a therapeutic problem, exacerbated by the resistance of many enzymes of this class to SBL inhibitors such as clavulanic acid, sulbactam, and tazobactam.71
Similar to class A and D SBLs, the structural fold of class C SBLs consists of α-helical and combined α/β subdomains, although class C enzymes tend to be larger in size (Fig. 4A).119 The active site is found at the interface of the two subdomains, featuring the nucleophilic serine (as part of an SXXK motif) surrounded by active site loops and an adjacent β-strand (Fig. 4B).120 Instead of an SXN motif, the class C active site contains a YXN motif with a catalytically important tyrosine residue. The Ω loop also differs from the corresponding loop in class A enzymes, notably lacking a glutamate residue. Compared to the other classes of SBLs, the active site of class C SBLs is relatively exposed, which has been proposed to accommodate the bulky C7 side chains present in some cephalosporins.121
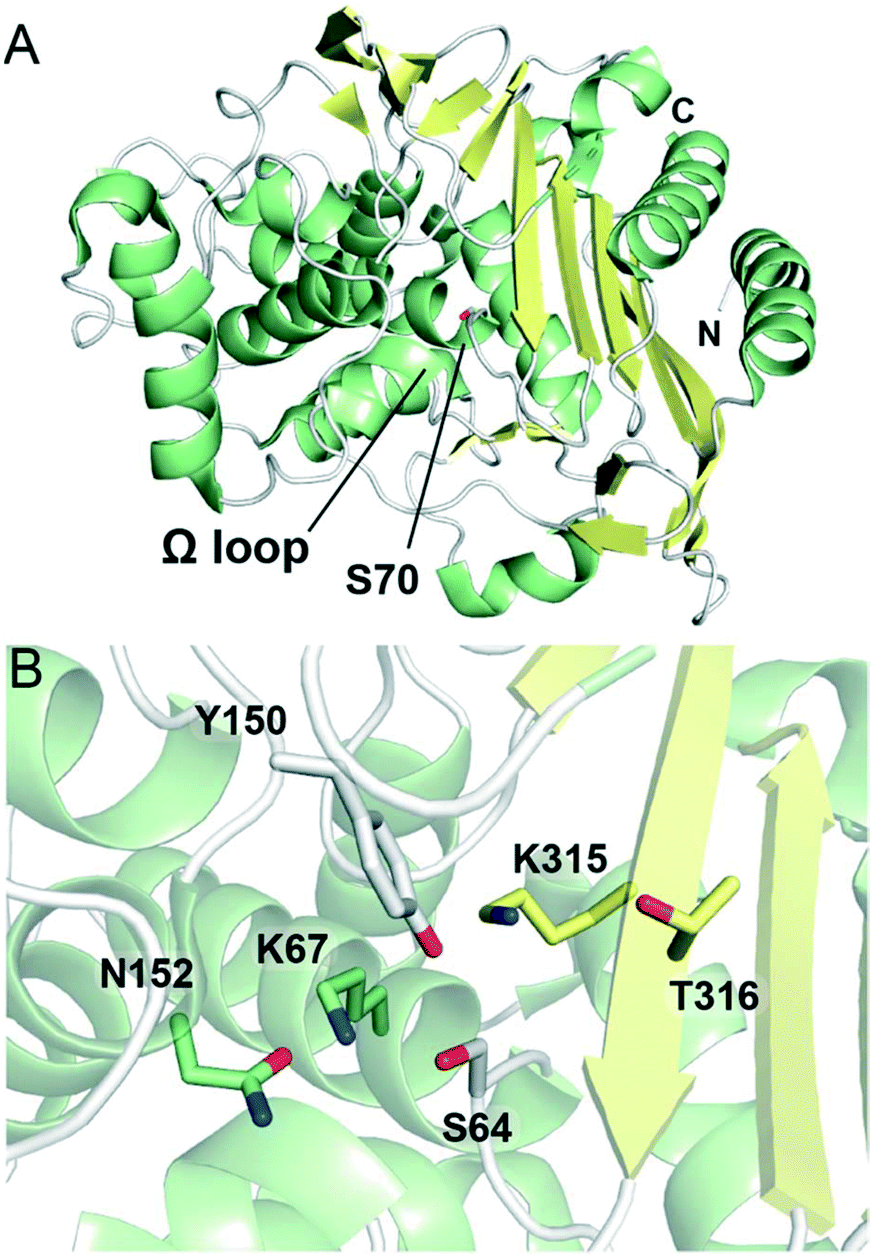 | ||
| Fig. 4 Structures of class C serine β-lactamases. (A) View of the crystal structure of class C SBL AmpC from Escherichia coli (PDB 1KE4). The nucleophilic serine, Ser70, is represented as white sticks. α-Helices are shown in green, β-sheets in yellow, and loops in white. The N- and C-termini are labeled. (B) View of the active site of AmpC (PDB 1IEL). Conserved active site residues are represented as sticks and are coloured according to local secondary structure elements. | ||
The precise mechanism of class C SBL catalysis has been the subject of debate.122 In the acylation step, Tyr150 (YXN motif; numbering for AmpC from Escherichia coli) and Lys67 (SXXK motif) are proposed to play important roles, activating the serine for attack onto the substrate β-lactam ring.123 Crystallographic and nuclear magnetic resonance spectroscopic studies suggest that the Tyr150 side chain does not occur as a phenolate in the active site,124,125 and it has been proposed that the neutral form of Lys67 abstracts a proton from Ser64 while Tyr150 activates the β-lactam ring nitrogen as a leaving group.126 Other studies have suggested that the substrate itself plays a role in catalysis, with the carboxylate group acting as a base.78 Lys67 and Tyr150 are also thought to be involved in the deacylation step, with Lys67 deprotonating the Tyr150 side chain such that the phenolate group can activate the catalytic water molecule required for hydrolysis.31,124
Class C SBLs are generally effective penicillinases, although there is significant variability in the rates at which different penicillins are hydrolyzed.5,71 Crystallographic studies indicate that the AECs derived from class C enzymes and penicillins are similar but not identical to those for other SBL classes, and there are notable conformational differences.22 The relatively open active site with a less prominent Ω loop would appear better capable of accommodating the C6 acylamido side chains of the penicillins.22 However, some class C enzymes are in fact inhibited by penicillins with bulkier C6 substituents (e.g., cloxacillin and oxacillin).5 Structural studies suggest that the C6 substituent of these penicillins disrupts the positioning of the thiazolidine nitrogen, disfavouring the transition state required for deacylation.78
In terms of clinical relevance, class C SBLs are most notable for their cephalosporinase activity, efficiently degrading early generation cephalosporins.5 In the AEC derived from cephalothin and AmpC, the cephalosporin C4 carboxylate group is shifted when compared to analogous complexes with class A enzymes, allowing the hydrolytic water molecule to enter the active site.127 Class C SBLs typically show lower levels of activity against later generation cephalosporins with oxyimino side chains, cephamycins, and the monobactam aztreonam.71 The conformation of the AEC derived from third generation cephalosporin ceftazidime with AmpC was proposed to interfere with the tetrahedral transition state required for hydrolysis, as with penicillins with bulky C6 substituents.120 However, minor changes to the active sites of class C SBLs can impart extended spectrum activity,121,128 often involving the Ω loop and other active site adjacent loops,129,130 resulting in improved substrate binding and turnover.131
Some class C SBLs have been observed to catalyze low levels of carbapenemase activity. CMY-10, a plasmid-encoded class C enzyme from Enterobacter aerogenes, has a broad substrate spectrum that includes the carbapenem imipenem.132 This activity was attributed to a three-residue deletion in a short loop (i.e., the R2 loop) across the active site from the Ω loop.132 This deletion opens up the active site, allowing for the accommodation of the carbapenem C2 thioether side chain.132 ADC-68, a chromosomally encoded class C enzymes from Acinetobacter baumannii, displays extended spectrum activity against third generation cephalosporins and carbapenems. This activity is thought to result from amino acid substitutions in the Ω loop and the nearby β8–β9 loop, opening up the active site for both cephalosporin C7 and carbapenem C2 side chains.133 Although these enzymes are relatively poor carbapenemases, they can offer protective effects when combined with other resistance mechanisms (e.g., decreased porin production).134
Class D serine β-lactamases
Historically, SBLs belonging to class D were rare and of little clinical relevance.135,136 Most enzymes in this class are grouped as OXAs, based on their ability to hydrolyze the penicillin oxacillin, and a number of other smaller groups have been distinguished. More recently, plasmid-encoded class D enzymes have become widely disseminated and many variants have been identified.72 Enzymes belonging to this class are now a major contributor of β-lactam resistance in Enterobacterales.135,137 Furthermore, OXA enzymes are commonly chromosomally encoded in A. baumannii, and have increased in clinical relevance alongside this antibiotic resistant pathogen.The first class D SBLs to be characterized were penicillinases, and were differentiated from other SBLs in part due to their catalytic activity against certain penicillins.5 However, as with the other SBL classes, the activity of known class D enzymes now extends to all of the other subclasses of β-lactam antibiotics. Subtle changes to the OXA enzyme active site have yielded enzymes with ESBL activity. Class D SBLs are also notable because of the carbapenemase activity demonstrated by some members. In addition, class D SBLs are often resistant to β-lactamase inhibitors such as clavulanic acid and sulbactam, and are poorly targeted by more recently developed SBL inhibitors such as avibactam.31 In spite of this, class D enzymes have not had the same clinical impact as some of their class A counterparts.
The overall fold and active site of the class D SBLs resembles those of other SBLs (Fig. 5A), although the class D active site is relatively hydrophobic (Fig. 5B).138–141 Instead of the asparagine residue found in the class A SXN motif, class D enzymes have a hydrophobic residue (typically valine) at this position. In addition, the active site β-strand and Ω loop contribute hydrophobic aromatic residues to the active site (Fig. 5B). Unusually, class D catalysis employs a carbamylated lysine residue (SXXK motif). This post-translationally modified residue, which results from the reversible reaction of the lysine ε-amino group with carbon dioxide, is thought to act as a general base in both acylation and the hydrolysis steps.136 The hydrophobicity of the class D active site is important for maintaining carbamylation, perturbing the basicity of the lysine side chain.136
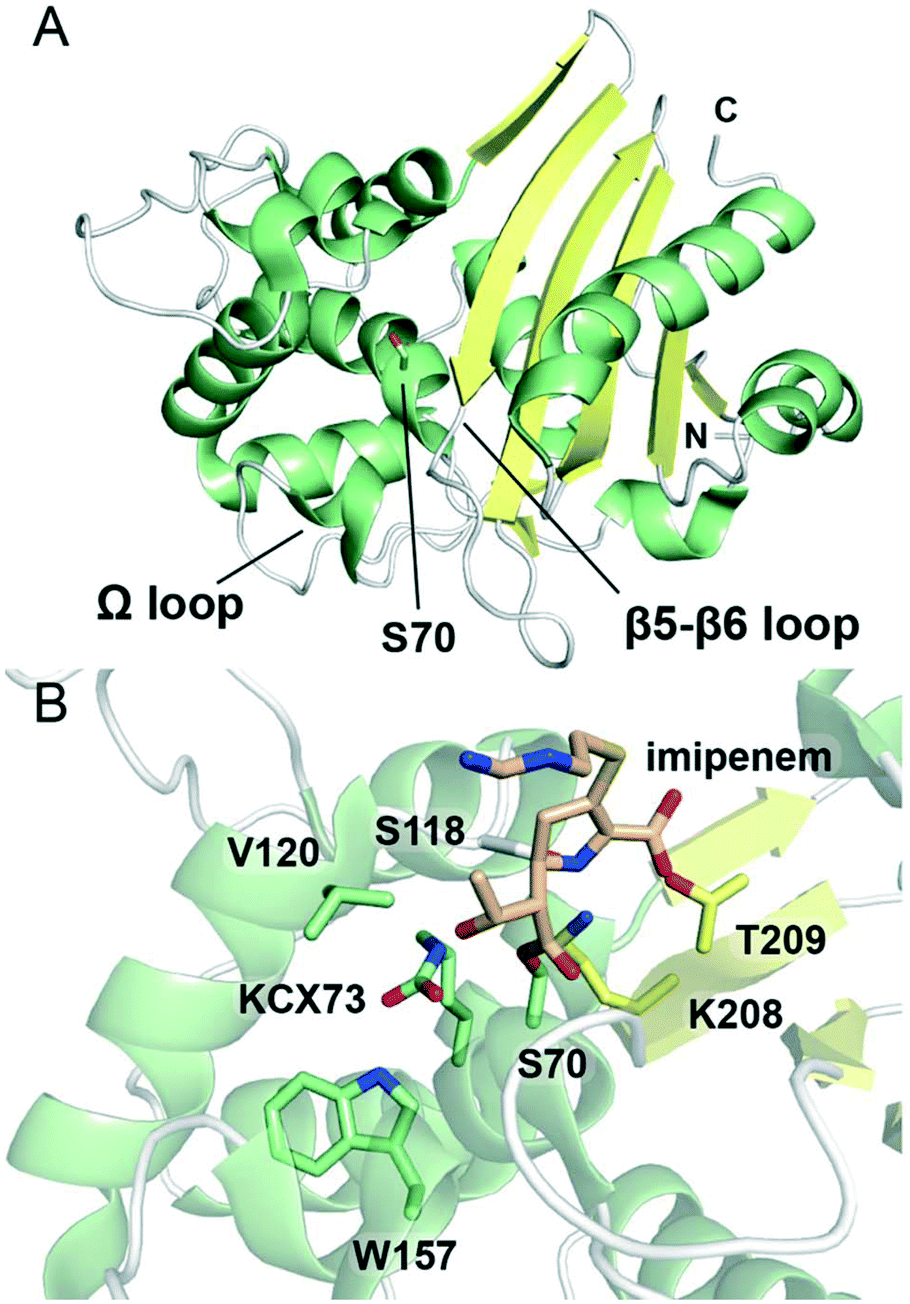 | ||
| Fig. 5 Structures of class D serine β-lactamases. (A) View of the crystal structure of class D SBL OXA-10 (PDB 1EWZ). The nucleophilic serine, Ser70, is represented as green sticks. α-Helices are shown in green, β-sheets in yellow, and loops in white. The N- and C-termini are indicated. (B) View of the active site of the complex derived from OXA-48 and the carbapenem imipenem (PDB 5QB4). The nucleophilic serine Ser70 is acylated by imipenem (tan sticks). The side chain amino group of Lys73 of the SXXK motif is in the form of a carbamate, and is indicated as KCX73. Conserved active site residues are represented as sticks, and are coloured according to local secondary structure. | ||
The profile of penicillins that are hydrolyzed by class D SBLs is determined largely by the fit of the penicillin C6 acylamido side chain in the active site. The valine (SXV motif) likely provides more room for penicillins with bulky side chains (e.g., the isoxazolyl group of oxacillin), while also altering the binding mode of the penicillin.138 The active sites of class D enzymes with oxacillinase activity such as OXA-1 have additional hydrophobic patches which form favourable binding interactions with the oxacillin C6 side chain.142
Some class D SBLs have achieved extended spectrum β-lactamase activity as the result of relatively minor active site changes. ESBL activity has been associated with amino acid substitutions on the Ω loop which enhance cephalosporin binding and hydrolysis.135,143 Other changes center on the β5–β6 loop (or the β6–β7 loop in some enzymes), which plays an important role in determining substrate specificity. A proline-to-serine substitution in the β5–β6 loop of OXA-24 and OXA-23 (yielding OXA-160 and OXA-225, respectively) resulted in increased hydrolytic activity against ceftazidime and cefotaxime.144 Crystallographic studies indicate that this single amino acid substitution relieves a steric clash with oxyimino-containing cephalosporin C7 side chains, enhancing binding. Interactions between the β5–β6 loop and the Ω loop also play important roles in determining ESBL activity, influencing substrate binding as well as the hydration state of the carbamylated lysine general base.145,146
The β5–β6 loop also contributes to the carbapenemase activity of class D SBLs.147 Class D enzymes often demonstrate very high affinities for carbapenem antibiotics, resulting in part from a hydrophobic bridge covering the active site; this bridge is typically composed of residues from the P loop and the β5–β6 loop (e.g., Tyr112 and Met223 in OXA-24),148 although such a bridge is not found in all class D carbapenemases (e.g., OXA-48). The hydrophobic bridge may also influence the tautomeric state of the carbapenem pyrroline ring,149 potentially favouring a form that is more susceptible to hydrolysis.26,150 While the carbapenem hydroxyethyl side chain is thought to interfere with the hydrolytic water molecule of many class A SBLs, structural and computational studies with class D SBLs indicate that hydrophobic active site residues may provide channels for water to access the active site.149,151 Recent studies have also revealed that class D SBLs can deacylate carbapenem-derived AECs through lactone formation, a mechanism which does not require a hydrolytic water molecule.152,153
L,D-Transpeptidases
The peptidoglycan of many bacteria consists predominantly of the 4 → 3 cross-links formed by PBP transpeptidase domains.154 However, alternative 3 → 3 cross-links are also observed in the peptidoglycan of some bacteria; in these cross-links, the third amino acid residue in one peptide chain is covalently attached to the third residue of another chain. These alternative cross-links are formed by L,D-transpeptidases (Ldts),9 enzymes which do not resemble PBPs in terms of sequence or structure.155,156 Genes encoding Ldts are widespread in bacteria, and some bacteria produce several different Ldt homologs.10,11 Although Ldts are not essential for all bacteria, they play a major role in peptidoglycan cross-linking in some groups (e.g., mycobacteria, Clostridioides difficile).157–159 In addition, Ldts have other roles in peptidoglycan modification, including the attachment of Braun's lipoprotein.160Initially, Ldts were considered to be a resistance mechanism against β-lactam antibiotic activity. A strain of Enterococcus faecium was observed to achieve decreased susceptibility to ampicillin by shifting the peptidoglycan cross-links from PBP-catalyzed 4 → 3 cross-links to Ldt-catalyzed 3 → 3 cross-links.161 However, later studies showed that some subclasses of β-lactam antibiotics do in fact inhibit Ldts, with the carbapenem and penem subclasses demonstrating particularly potent activity against some Ldts.11,162,163 As with PBPs, β-lactams target Ldts through a covalent mechanism, interfering with peptidoglycan transpeptidation. Whereas β-lactams acylate a serine residue in the active sites of PBPs and SBLs, the AECs derived from Ldts and β-lactams result from the acylation of a conserved cysteine residue.164
Crystallographic studies have revealed that the Ldt transpeptidase domain is composed of two β-sheets with associated α-helices, described as an ErfK/YbiS/YhnG fold (Fig. 6A).165,166 Notably, the Ldt transpeptidase domain differs significantly from the PBP transpeptidase domain (Fig. 2C), in spite of their related functions and inhibitor susceptibilities. The Ldt active site is centered around a conserved cysteine residue which is both required for transpeptidation and targeted by β-lactam antibiotics (Fig. 6B and C).167 This cysteine is found on an active site loop extending from a β-strand. Extending from two other strands of the same β-sheet, a long loop with secondary structure elements folds over the sheet, forming an active site “lid”. The Ldt active site is relatively hydrophobic (Fig. 6C), with the β-sheet and the lid contributing several aromatic residues.
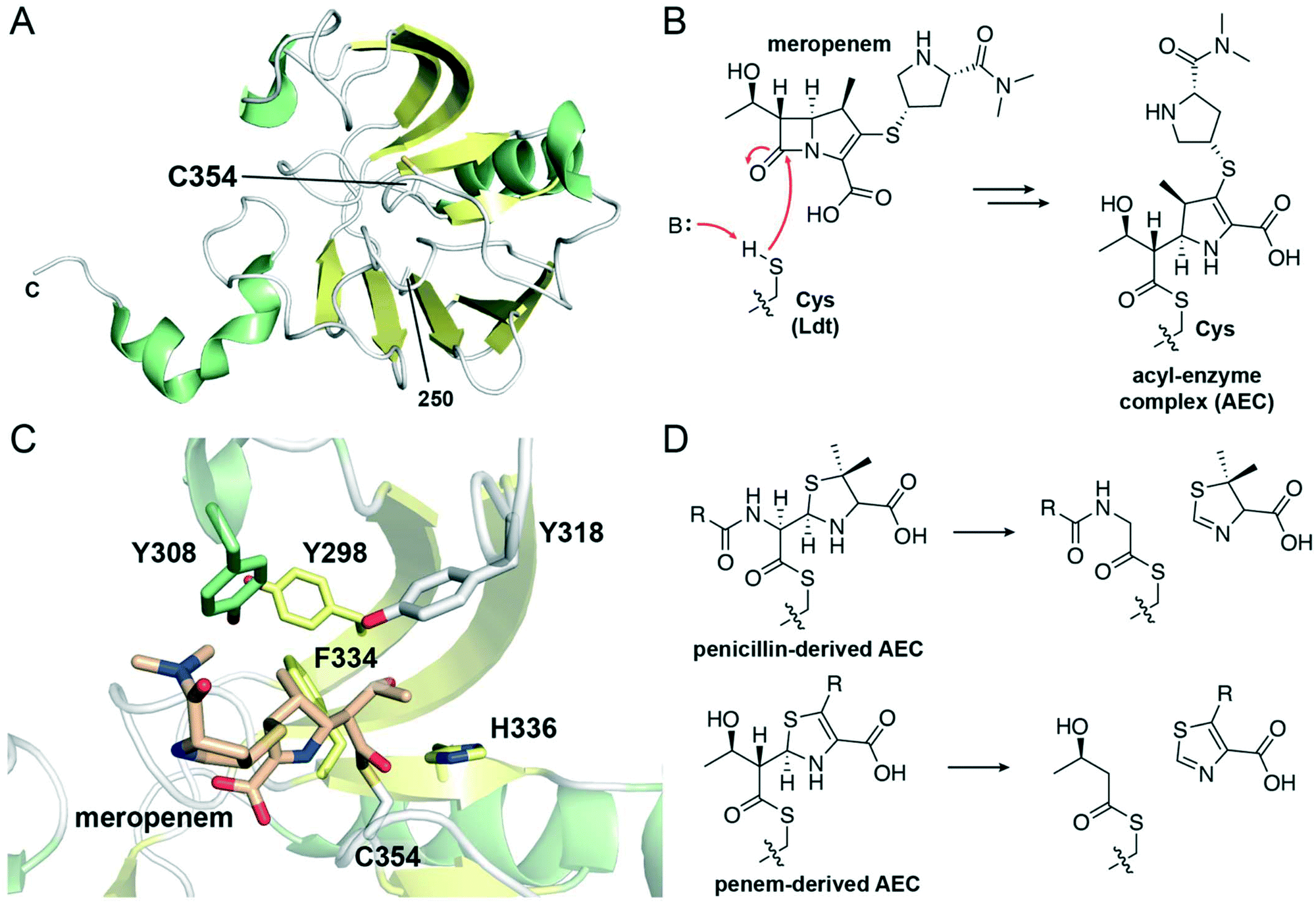 | ||
| Fig. 6 Structure and inhibition of L,D-transpeptidases. (A) View of the transpeptidase domain of LdtMt2 (PDB 6RLG), displaying the residues from 250 to the C-terminus. The nucleophilic cysteine, Cys354, is shown as white sticks. α-Helices are shown in green, β-sheets in yellow, and loops in white. The two N-terminal immunoglobulin fold-related domains are not shown. (B) Reaction scheme showing the acylation of the Ldt nucleophilic cysteine by the carbapenem meropenem, forming an acyl–enzyme complex. (C) View of the active site of LdtMt2 in which the nucleophilic cysteine, Cys354 (white sticks), is acylated with meropenem (tan sticks) (PDB 4GSU). General base His336 and selected hydrophobic active site residues are shown as sticks and are coloured according to local secondary structure. (D) Reaction scheme showing the C5–C6 fragmentation of penicillin-derived and penem-derived acyl-enzyme complexes by Ldts. | ||
The transpeptidase reaction catalyzed by the Ldts is mechanistically related to that catalyzed by PBPs.9,168 The active site cysteine nucleophilically attacks a peptidoglycan tetrapeptide (generated from a pentapeptide by a carboxypeptidase), releasing D-alanine and forming a covalent thioester-bound peptide–enzyme complex. Then, a nucleophilic amino acid from another peptidoglycan chain attacks the thioester carbonyl, forming a 3 → 3 cross-link and releasing the enzyme. The catalytic machinery responsible for this catalysis resembles that found in cysteine proteases. In the Ldt active site, the nucleophilic cysteine is part of a catalytic triad with a histidine residue and a third variable residue (e.g., glycine in the Ldt from Bacillus subtilis).165 This third residue orients the histidine such that it can activate the cysteine for catalysis.
The reaction of the Ldt cysteine nucleophile with a β-lactam antibiotic yields a thioester-based AEC (Fig. 6B). The structures of these complexes have been extensively studied, focusing especially on those derived from carbapenems.165,167 Unlike analogous complexes with PBPs and SBLs, fewer hydrogen bonding and ionic interactions are observed between β-lactams and the Ldt active site, and the conformation of the bound antibiotic varies.165,169–171 This lack of conserved interactions may in part explain the great range of potencies observed for different β-lactams as Ldt inhibitors.172,173
Although Ldts form covalent complexes with penicillins, the slow acylation rate limits their efficacy as inhibitors.172 Furthermore, Ldts can slowly catalyze the hydrolysis of the penicillin-derived complexes once they are formed. Studies with Ldtfm from Enterococcus faecium revealed that, prior to hydrolysis, C5–C6 fragmentation of the ampicillin-derived complex occurred (Fig. 6D).172 Related fragmentation reactions were observed with LdtMt2 from M. tuberculosis with several other penicillins.174 This reaction appears to resemble a fragmentation catalyzed by a PBP carboxypeptidase from a Streptomyces sp.65 However, this fragmentation reaction may occur more readily for Ldts, potentially due to stabilization of an enolate reaction intermediate by the cysteine sulfur.174
As with the penicillins, cephalosporins tend to be poor Ldt inhibitors.173,175 Mass spectrometric studies of Ldtfm with ceftriaxone showed relatively slow rates of acylation,172 similar to what was observed following kinetic studies of LdtMt1 from M. tuberculosis with representative cephalosporins.175 Following acylation, loss of the cephalosporin C3′ leaving group may occur, and the resulting AEC could be slowly hydrolyzed.172 Unusually, the AEC derived from Ldtfm and the cephalosporin nitrocefin was observed to undergo a recyclization reaction, re-forming the β-lactam ring of nitrocefin.176 This mechanism was proposed to decrease the antibacterial activity of nitrocefin against an E. faecium mutant.176
Of the different subclasses of β-lactams, the carbapenems and penems are typically the most potent Ldt inhibitors.11,173,175,177 However, while some Ldts are strongly targeted by these antibiotics, others are acylated slowly (e.g., Ldtfm) or not at all (e.g., LdtMt5).163,172 The complexes derived from Ldts and carbapenems are generally stable, with little-to-no hydrolysis occurring.11,172,174 Unusual degradation reactions have been crystallographically observed in the AECs derived from LdtMt2 and some carbapenems.162,178 A recent study reported that the complex derived from LdtMt2 and carbapenems is capable of undergoing recyclization,179 as was seen for Ldtfm with nitrocefin,176 while also noting low levels of formation of β-lactone products like those observed for class D SBLs.152
Analogous to the fragmentation reaction observed between Ldts and penicillins,172 the AECs derived from Ldts and penems undergo a rapid C5–C6 fragmentation reaction which releases a thiazole product and leaves the nucleophilic cysteine acylated with a 3-hydroxybutyryl group (Fig. 6D).174,177 Penems are very potent inhibitors of some Ldts, and are of interest as therapeutics for drug-resistant M. tuberculosis.180
Summary and future outlook
β-Lactams are a critical resource for the treatment of bacterial infections. The β-lactam scaffold has been extensively developed, yielding dozens of clinically used antibiotics with diverse core structures and functionalization. Considering the scale in which they are used, along with the range of resistance mechanisms observed in bacteria, it is remarkable how effective β-lactams continue to be. However, the number of isolates that are resistant to β-lactam antibiotics continues to rise, limiting the therapeutic options that are available.The clinical implementation of new β-lactam antibiotics is quickly followed by the identification of β-lactamases that can degrade them. This has played out repeatedly over the past decades, and will likely occur for any β-lactams developed in the future. The diversity of β-lactamases represents a critical problem, due to the substrate specificities of currently known enzymes and their ability to evolve to alter these specificities. Even if a β-lactam is not readily susceptible to β-lactamase-catalyzed degradation, other resistance mechanisms such as efflux pumps are available to bacteria.
While few new β-lactams have reached the clinic in recent years, there have been significant breakthroughs regarding β-lactamase inhibitors.47 New inhibitors based on diazabicyclooctane (e.g., avibactam) and boronate (e.g., vaborbactam) scaffolds have proven to be effective, and are approved for clinical use.31,43 Other inhibitors in the antibiotic pipeline display promising properties, such as taniborbactam which inhibits both SBLs and MBLs.181
Although β-lactams, PBPs, and SBLs have been the subject of extensive study, there is still much to learn. The discovery that β-lactam antibiotics target Ldts is relatively recent,182 and some β-lactams demonstrate activity against other groups of enzymes (e.g., bacterial signal peptidases,12,183,184 viral proteases).185 The sheer number of different β-lactam antibiotics, target proteins, and resistance mechanisms gives rise to a high level of complexity related to inhibition and catalysis. Modern experimental approaches (e.g., cryogenic electron microscopy) have opened new approaches for investigating the structures and functions of these proteins,186 and our understanding of peptidoglycan synthesis and cell wall remodeling continues to expand.187,188
Conflicts of interest
There are no conflicts to declare.Notes and references
- World Health Organization, WHO report on surveillance of antibiotic consumption: 2016–2018 early implementation, https://www.who.int/publications/i/item/who-report-on-surveillance-of-antibiotic-consumption.
- K. Bush and P. A. Bradford, Cold Spring Harbor Perspect. Med., 2016, 6, a025247 CrossRef.
- M. G. Page, in Antibiotic Discovery and Development, ed. T. J. Dougherty and M.J. Pucci, Springer, New York, 2012, pp. 79–117 Search PubMed.
- J. Flores-Kim, G. S. Dobihal, A. Fenton, D. Z. Rudner and T. G. Bernhardt, eLife, 2019, 8, e44912 CrossRef CAS PubMed.
- K. Bush, G. A. Jacoby and A. A. Medeiros, Antimicrob. Agents Chemother., 1995, 39, 1211–1233 CrossRef CAS PubMed.
- T. Palzkill, Ann. N. Y. Acad. Sci., 2013, 1277, 91–104 CrossRef CAS PubMed.
- K. Bush and P. A. Bradford, Clin. Microbiol. Rev., 2020, 33, e00047-19 CrossRef PubMed.
- P. D. Tamma, S. L. Aitken, R. A. Bonomo, A. J. Mathers, D. van Duin and C. J. Clancy, Clin. Infect. Dis., 2021, 72, e169–e183 CrossRef CAS.
- S. Magnet, L. Dubost, A. Marie, M. Arthur and L. Gutmann, J. Bacteriol., 2008, 190, 4782–4785 CrossRef CAS PubMed.
- R. Gupta, M. Lavollay, J.-L. Mainardi, M. Arthur, W. R. Bishai and G. Lamichhane, Nat. Med., 2010, 16, 466–469 CrossRef CAS PubMed.
- P. Kumar, A. Kaushik, E. P. Lloyd, S. G. Li, R. Mattoo, N. C. Ammerman, D. T. Bell, A. L. Perryman, T. A. Zandi, S. Ekins, S. L. Ginell, C. A. Townsend, J. S. Freundlich and G. Lamichhane, Nat. Chem. Biol., 2017, 13, 54–61 CrossRef CAS PubMed.
- M. Paetzel, R. E. Dalbey and N. C. Strynadka, Pharmacol. Ther., 2000, 87, 27–49 CrossRef CAS.
- M. I. Konaklieva, Curr. Med. Chem.: Anti-Infect. Agents, 2002, 1, 215–238 CrossRef CAS.
- R. B. Hamed, J. R. Gomez-Castellanos, L. Henry, C. Ducho, M. A. McDonough and C. J. Schofield, Nat. Prod. Rep., 2013, 30, 21–107 RSC.
- M. I. Page, Acc. Chem. Res., 1984, 17, 144–151 CrossRef CAS.
- R. F. Pratt and W. S. Faraci, J. Am. Chem. Soc., 1986, 108, 5328–5333 CrossRef CAS.
- W. S. Faraci and R. F. Pratt, Biochemistry, 1985, 24, 903–910 CrossRef CAS.
- W. S. Faraci and R. F. Pratt, Biochemistry, 1986, 25, 2934–2941 CrossRef CAS PubMed.
- S. Naseer, E. A. Weinstein, D. B. Rubin, K. Suvarna, X. Wei, K. Higgins, A. Goodwin, S. H. Jang, D. Iarikov and J. Farley, Clin. Infect. Dis., 2021, 72, e1103–e1111 CrossRef CAS PubMed.
- M. J. Miller and R. Liu, Acc. Chem. Res., 2021, 54, 1646–1661 CrossRef CAS PubMed.
- A. A. Medeiros, Clin. Infect. Dis., 1997, 24(Suppl 1), S19–45 CrossRef CAS PubMed.
- I. Trehan, F. Morandi, L. C. Blaszczak and B. K. Shoichet, Chem. Biol., 2002, 9, 971–980 CrossRef CAS PubMed.
- K. M. Papp-Wallace, A. Endimiani, M. A. Taracila and R. A. Bonomo, Antimicrob. Agents Chemother., 2011, 55, 4943–4960 CrossRef CAS PubMed.
- H. Kropp, J. G. Sundelof, R. Hajdu and F. M. Kahan, Antimicrob. Agents Chemother., 1982, 22, 62–70 CrossRef CAS PubMed.
- M. L. Hammond, J. Antimicrob. Chemother., 2004, 53(Suppl 2), ii7–9 CAS.
- C. J. Easton and J. R. Knowles, Biochemistry, 1982, 21, 2857–2862 CrossRef CAS PubMed.
- Q. Kong and Y. Yang, Bioorg. Med. Chem. Lett., 2021, 127799 CrossRef CAS PubMed.
- C. T. Lohans, J. Brem and C. J. Schofield, Antimicrob. Agents Chemother., 2017, 61, e01224-17 CrossRef PubMed.
- S. Maraki, V. E. Mavromanolaki, P. Moraitis, D. Stafylaki, A. Kasimati, E. Magkafouraki and E. Scoulica, Eur. J. Clin. Microbiol. Infect. Dis., 2021, 1–5 Search PubMed.
- M. F. Alghoribi, M. Alqurashi, L. Okdah, B. Alalwan, Y. S. AlHebaishi, A. Almalki, M. A. Alzayer, A. A. Alswaji, M. Doumith and M. Barry, Sci. Rep., 2021, 11, 9684 CrossRef CAS PubMed.
- C. L. Tooke, P. Hinchliffe, E. C. Bragginton, C. K. Colenso, V. H. A. Hirvonen, Y. Takebayashi and J. Spencer, J. Mol. Biol., 2019, 431, 3472–3500 CrossRef CAS PubMed.
- H. Douafer, V. Andrieu, O. Phanstiel and J. M. Brunel, J. Med. Chem., 2019, 62, 8665–8681 CrossRef CAS PubMed.
- F. van den Akker and R. A. Bonomo, Front. Microbiol., 2018, 9, 622 CrossRef PubMed.
- P. Stapleton, P.-J. Wu, A. King, K. Shannon, G. French and I. Phillips, Antimicrob. Agents Chemother., 1995, 39, 2478–2483 CrossRef CAS PubMed.
- D. Pagan-Rodriguez, X. Zhou, R. Simmons, C. R. Bethel, A. M. Hujer, M. S. Helfand, Z. Jin, B. Guo, V. E. Anderson and L. M. Ng, J. Biol. Chem., 2004, 279, 19494–19501 CrossRef CAS PubMed.
- W. Ke, P. Pattanaik, C. R. Bethel, A. Sheri, J. D. Buynak, R. A. Bonomo and F. van den Akker, PLoS One, 2012, 7, e49035 CrossRef CAS PubMed.
- S. A. Cohen and R. F. Pratt, Biochemistry, 1980, 19, 3996–4003 CrossRef CAS PubMed.
- D. E. Ehmann, H. Jahic, P. L. Ross, R. F. Gu, J. Hu, G. Kern, G. K. Walkup and S. L. Fisher, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11663–11668 CrossRef CAS.
- N. Gilbert, Nature, 2018, 555, S5–S7 CrossRef CAS PubMed.
- T. F. Durand-Réville, J. Comita-Prevoir, J. Zhang, X. Wu, T. L. May-Dracka, J. A. C. Romero, F. Wu, A. Chen, A. B. Shapiro and N. M. Carter, J. Med. Chem., 2020, 63, 12511–12525 CrossRef.
- T. F. Durand-Réville, S. Guler, J. Comita-Prevoir, B. Chen, N. Bifulco, H. Huynh, S. Lahiri, A. B. Shapiro, S. M. McLeod and N. M. Carter, Nat. Microbiol., 2017, 2, 1–10 Search PubMed.
- M. Rajavel, V. Kumar, H. Nguyen, J. Wyatt, S. H. Marshall, K. M. Papp-Wallace, P. Deshpande, S. Bhavsar, R. Yeole and S. Bhagwat, mBio, 2021, 12, e03058-20 CrossRef PubMed.
- A. Krajnc, P. A. Lang, T. D. Panduwawala, J. Brem and C. J. Schofield, Curr. Opin. Chem. Biol., 2019, 50, 101–110 CrossRef CAS.
- C. L. Tooke, P. Hinchliffe, A. Krajnc, A. J. Mulholland, J. Brem, C. J. Schofield and J. Spencer, RSC Med. Chem., 2020, 11, 491–496 RSC.
- A. Krajnc, J. r. Brem, P. Hinchliffe, K. Calvopiña, T. D. Panduwawala, P. A. Lang, J. J. Kamps, J. M. Tyrrell, E. Widlake and B. G. Saward, J. Med. Chem., 2019, 62, 8544–8556 CrossRef CAS PubMed.
- S. J. Hecker, K. R. Reddy, O. Lomovskaya, D. C. Griffith, D. Rubio-Aparicio, K. Nelson, R. Tsivkovski, D. Sun, M. Sabet and Z. Tarazi, J. Med. Chem., 2020, 63, 7491–7507 CrossRef PubMed.
- Pew Charitable Trusts, Antibiotics Currently in Global Clinical Development, https://www.pewtrusts.org/en/research-and-analysis/data-visualizations/2014/antibiotics-currently-in-clinical-development.
- I. Morrissey, S. Magnet, S. Hawser, S. Shapiro and P. Knechtle, Antimicrob. Agents Chemother., 2019, 63, e00514–e00519 CrossRef CAS PubMed.
- D. T. Davies and M. Everett, Acc. Chem. Res., 2021, 54, 2055–2064 CrossRef CAS PubMed.
- K. H. Tehrani, N. Wade, V. Mashayekhi, N. C. Brüchle, W. Jespers, K. Voskuil, D. Pesce, M. J. van Haren, G. J. van Westen and N. I. Martin, J. Med. Chem., 2021, 64, 9141–9151 CrossRef CAS PubMed.
- Q. M. Wang, R. B. Peery, R. B. Johnson, W. E. Alborn, W. K. Yeh and P. L. Skatrud, J. Bacteriol., 2001, 183, 4779–4785 CrossRef CAS PubMed.
- E. Sauvage, F. Kerff, M. Terrak, J. A. Ayala and P. Charlier, FEMS Microbiol. Rev., 2008, 32, 234–258 CrossRef CAS PubMed.
- D. J. Tipper and J. L. Strominger, Proc. Natl. Acad. Sci. U. S. A., 1965, 54, 1133–1141 CrossRef CAS PubMed.
- D. Lim and N. C. J. Strynadka, Nat. Struct. Biol., 2002, 9, 870–876 CAS.
- Z. Lu, H. Wang, A. Zhang, X. Liu, W. Zhou, C. Yang, L. Guddat, H. Yang, C. J. Schofield and Z. Rao, Mol. Pharmacol., 2020, 97, 287–294 CrossRef CAS PubMed.
- A. Singh, J. Tomberg, R. A. Nicholas and C. Davies, J. Biol. Chem., 2019, 294, 14020–14032 CrossRef.
- E. Sauvage, C. Duez, R. Herman, F. Kerff, S. Petrella, J. W. Anderson, S. A. Adediran, R. F. Pratt, J.-M. Frère and P. Charlier, J. Mol. Biol., 2007, 371, 528–539 CrossRef CAS PubMed.
- O. Kocaoglu, H.-C. T. Tsui, M. E. Winkler and E. E. Carlson, Antimicrob. Agents Chemother., 2015, 59, 3548–3555 CrossRef CAS PubMed.
- N. Curtis, D. Orr, G. W. Ross and M. G. Boulton, Antimicrob. Agents Chemother., 1979, 16, 533–539 CrossRef CAS PubMed.
- D. S. Sutaria, B. Moya, K. B. Green, T. H. Kim, X. Tao, Y. Jiao, A. Louie, G. L. Drusano and J. B. Bulitta, Antimicrob. Agents Chemother., 2018, 62, e00282-18 CrossRef PubMed.
- A. R. Sayed, N. R. Shah, K. B. Basso, M. Kamat, Y. Jiao, B. Moya, D. S. Sutaria, Y. Lang, X. Tao and W. Liu, Antimicrob. Agents Chemother., 2020, 65, e01956-20 CrossRef PubMed.
- C. Bebrone, C. Moali, F. Mahy, S. Rival, J. D. Docquier, G. M. Rossolini, J. Fastrez, R. F. Pratt, J.-M. Frère and M. Galleni, Antimicrob. Agents Chemother., 2001, 45, 1868–1871 CrossRef CAS.
- R. A. Nicholas, S. Krings, J. Tomberg, G. Nicola and C. Davies, J. Biol. Chem., 2003, 278, 52826–52833 CrossRef CAS PubMed.
- J. D. Smith, M. Kumarasiri, W. Zhang, D. Hesek, M. Lee, M. Toth, S. Vakulenko, J. F. Fisher, S. Mobashery and Y. Chen, Antimicrob. Agents Chemother., 2013, 57, 3137–3146 CrossRef CAS PubMed.
- J.-M. Frère, J.-M. Ghuysen, J. Degelaen, A. Loffet and H. R. Perkins, Nature, 1975, 258, 168–170 CrossRef.
- J.-M. Frère, J.-M. Ghuysen, H. Vanderhaeghe, P. Adriaens, J. Degelaen and J. De Graeve, Nature, 1976, 260, 451–454 CrossRef PubMed.
- A. Marquet, J.-M. Frère, J.-M. Ghuysen and A. Loffet, Biochem. J., 1979, 177, 909–916 CrossRef CAS.
- A. Zapun, C. Contreras-Martel and T. Vernet, FEMS Microbiol. Rev., 2008, 32, 361–385 CrossRef CAS PubMed.
- L. H. Otero, A. Rojas-Altuve, L. I. Llarrull, C. Carrasco-López, M. Kumarasiri, E. Lastochkin, J. Fishovitz, M. Dawley, D. Hesek and M. Lee, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16808–16813 CrossRef PubMed.
- M. Barlow, in Horizontal Gene Transfer, ed. M.B. Gogarten and J.P. Gogarten, L.C. Olendzenski Humana Press, Totowa, NJ, 2009, pp. 397–411 Search PubMed.
- G. A. Jacoby, Clin. Microbiol. Rev., 2009, 22, 161–182 CrossRef CAS.
- T. Naas, S. Oueslati, R. A. Bonnin, M. L. Dabos, A. Zavala, L. Dortet, P. Retailleau and B. I. Iorga, J. Enzyme Inhib. Med. Chem., 2017, 32, 917–919 CrossRef CAS PubMed.
- R. P. Ambler, Philos. Trans. R. Soc., B, 1980, 289, 321–331 CrossRef CAS PubMed.
- G. Bahr, L. J. González and A. J. Vila, Chem. Rev., 2021, 121, 7957–8094 CrossRef CAS PubMed.
- M. W. Crowder, J. Spencer and A. J. Vila, Acc. Chem. Res., 2006, 39, 721–728 CrossRef CAS.
- C. M. Rotondo and G. D. Wright, Curr. Opin. Microbiol., 2017, 39, 96–105 CrossRef CAS PubMed.
- K. D. Schneider, C. R. Bethel, A. M. Distler, A. M. Hujer, R. A. Bonomo and D. A. Leonard, Biochemistry, 2009, 48, 6136–6145 CrossRef CAS PubMed.
- A. Patera, L. C. Blaszczak and B. K. Shoichet, J. Am. Chem. Soc., 2000, 122, 10504–10512 CrossRef CAS.
- C. L. Tooke, P. Hinchliffe, R. A. Bonomo, C. J. Schofield, A. J. Mulholland and J. Spencer, J. Biol. Chem., 2020, 296, 100126 CrossRef.
- B. G. Hall and M. Barlow, Drug Resist. Updates, 2004, 7, 111–123 CrossRef CAS PubMed.
- I. Massova and S. Mobashery, Antimicrob. Agents Chemother., 1998, 42, 1–17 CrossRef CAS PubMed.
- A. S. Ghosh, C. Chowdhury and D. E. Nelson, Trends Microbiol., 2008, 16, 309–317 CrossRef CAS PubMed.
- A. Bulychev, I. Massova, K. Miyashita and S. Mobashery, J. Am. Chem. Soc., 1997, 119, 7619–7625 CrossRef CAS.
- J. R. Knox, P. C. Moews and J.-M. Frere, Chem. Biol., 1996, 3, 937–947 CrossRef CAS.
- R. F. Pratt, J. Med. Chem., 2016, 59, 8207–8220 CrossRef CAS.
- C. Fröhlich, J. A. Gama, K. Harms, V. H. Hirvonen, B. A. Lund, M. W. van der Kamp, P. J. Johnsen, Ø. Samuelsen and H.-K. S. Leiros, mSphere, 2021, 6, e00108-21 CrossRef PubMed.
- Z. Cheng, C. R. Bethel, P. W. Thomas, B. A. Shurina, J.-P. Alao, C. A. Thomas, K. Yang, S. H. Marshall, H. Zhang and A. M. Sturgill, Antimicrob. Agents Chemother., 2021, 65, e01714–e01720 CAS.
- T. Palzkill, Front. Mol. Biosci., 2018, 5, 16 CrossRef PubMed.
- M. G. Page, Clin. Microbiol. Infect., 2008, 14(Suppl 1), 63–74 CrossRef CAS.
- R. Cantón and T. M. Coque, Curr. Opin. Microbiol., 2006, 9, 466–475 CrossRef PubMed.
- J. Walther-Rasmussen and N. Høiby, J. Antimicrob. Chemother., 2007, 60, 470–482 CrossRef CAS.
- K. M. Papp-Wallace, M. L. Winkler, M. A. Taracila and R. A. Bonomo, Antimicrob. Agents Chemother., 2015, 59, 3710–3717 CrossRef CAS PubMed.
- G. Minasov, X. Wang and B. K. Shoichet, J. Am. Chem. Soc., 2002, 124, 5333–5340 CrossRef CAS PubMed.
- C. A. Smith, H. Frase, M. Toth, M. Kumarasiri, K. Wiafe, J. Munoz, S. Mobashery and S. B. Vakulenko, J. Am. Chem. Soc., 2012, 134, 19512–19515 CrossRef CAS PubMed.
- J. F. Petrosino and T. Palzkill, J. Bacteriol., 1996, 178, 1821–1828 CrossRef CAS PubMed.
- S. O. Meroueh, J. F. Fisher, H. B. Schlegel and S. Mobashery, J. Am. Chem. Soc., 2005, 127, 15397–15407 CrossRef CAS PubMed.
- N. C. Strynadka, R. Martin, S. E. Jensen, M. Gold and J. B. Jones, Nat. Struct. Biol., 1996, 3, 688–695 CrossRef CAS.
- N. C. Strynadka, H. Adachi, S. E. Jensen, K. Johns, A. Sielecki, C. Betzel, K. Sutoh and M. N. James, Nature, 1992, 359, 700–705 CrossRef CAS.
- I. Saves, O. Burlet-Schiltz, L. Maveyraud, J. P. Samama, J. C. Prome and J. M. Masson, Biochemistry, 1995, 34, 11660–11667 CrossRef CAS PubMed.
- M. P. Patel, L. Hu, V. Stojanoski, B. Sankaran, B. V. V. Prasad and T. Palzkill, Biochemistry, 2017, 56, 3443–3453 CrossRef CAS PubMed.
- Y. Pfeifer, A. Cullik and W. Witte, Int. J. Med. Microbiol. Suppl., 2010, 300, 371–379 CrossRef CAS.
- A. Matagne, A. M. Misselyn-Bauduin, B. Joris, T. Erpicum, B. Granier and J. M. Frère, Biochem. J., 1990, 265, 131–146 CrossRef CAS PubMed.
- X. Raquet, M. Vanhove, J. Lamotte-Brasseur, S. Goussard, P. Courvalin and J. M. Frère, Proteins, 1995, 23, 63–72 CrossRef CAS PubMed.
- C. Cantu III, W. Huang and T. Palzkill, J. Biol. Chem., 1996, 271, 22538–22545 CrossRef PubMed.
- E. Collatz, G. Tran Van Nhieu, D. Billot-Klein, R. Williamson and L. Gutmann, Gene, 1989, 78, 349–354 CrossRef CAS.
- Y. Chen, J. Delmas, J. Sirot, B. Shoichet and R. Bonnet, J. Mol. Biol., 2005, 348, 349–362 CrossRef CAS PubMed.
- L. Maveyraud, L. Mourey, L. P. Kotra, J.-D. Pedelacq, V. Guillet, S. Mobashery and J.-P. Samama, J. Am. Chem. Soc., 1998, 120, 9748–9752 CrossRef CAS.
- M. Kalp and P. R. Carey, Biochemistry, 2008, 47, 11830–11837 CrossRef CAS PubMed.
- F. Fonseca, E. I. Chudyk, M. W. van der Kamp, A. Correia, A. J. Mulholland and J. Spencer, J. Am. Chem. Soc., 2012, 134, 18275–18285 CrossRef CAS PubMed.
- W. Ke, C. R. Bethel, J. M. Thomson, R. A. Bonomo and F. van den Akker, Biochemistry, 2007, 46, 5732–5740 CrossRef CAS.
- K. M. Papp-Wallace, M. Taracila, C. J. Wallace, K. M. Hujer, C. R. Bethel, J. M. Hornick and R. A. Bonomo, Protein Sci., 2010, 19, 1714–1727 CrossRef CAS PubMed.
- I. M. Furey, S. C. Mehta, B. Sankaran, L. Hu, B. V. V. Prasad and T. Palzkill, J. Biol. Chem., 2021, 296, 100799 CrossRef CAS.
- P. D. Tamma, Y. Doi, R. A. Bonomo, J. K. Johnson and P. J. Simner, Clin. Infect. Dis., 2019, 69, 1446–1455 CrossRef CAS PubMed.
- M. G. P. Page, Antimicrob. Agents Chemother., 2020, 64, e02247-19 Search PubMed.
- A. R. Mack, M. D. Barnes, M. A. Taracila, A. M. Hujer, K. M. Hujer, G. Cabot, M. Feldgarden, D. H. Haft, W. Klimke, F. van den Akker, A. J. Vila, A. Smania, S. Haider, K. M. Papp-Wallace, P. A. Bradford, G. M. Rossolini, J.-D. Docquier, J.-M. Frère, M. Galleni, N. D. Hanson, A. Oliver, P. Plésiat, L. Poirel, P. Nordmann, T. G. Palzkill, G. A. Jacoby, K. Bush and R. A. Bonomo, Antimicrob. Agents Chemother., 2020, 64, e01841-19 CrossRef PubMed.
- R. Tripathi and N. N. Nair, J. Phys. Chem. B, 2016, 120, 2681–2690 CrossRef CAS PubMed.
- A. Bulychev and S. Mobashery, Antimicrob. Agents Chemother., 1999, 43, 1743–1746 CrossRef CAS PubMed.
- D.-W. Bae, Y.-E. Jung, B.-G. Jeong and S.-S. Cha, Arch. Biochem. Biophys., 2020, 693, 108570 CrossRef CAS PubMed.
- R. A. Powers and B. K. Shoichet, J. Med. Chem., 2002, 45, 3222–3234 CrossRef CAS PubMed.
- R. A. Powers, E. Caselli, P. J. Focia, F. Prati and B. K. Shoichet, Biochemistry, 2001, 40, 9207–9214 CrossRef CAS PubMed.
- P. Nordmann and H. Mammeri, Future Microbiol., 2007, 2, 297–307 CrossRef CAS PubMed.
- C. Fenollar-Ferrer, J. Frau, J. Donoso and F. Munoz, Theor. Chem. Acc., 2008, 121, 209–218 Search PubMed.
- A. Dubus, P. Ledent, J. Lamotte-Brasseur and J.-M. Frère, Proteins: Struct., Funct., Bioinf., 1996, 25, 473–485 CAS.
- Y. Chen, G. Minasov, T. A. Roth, F. Prati and B. K. Shoichet, J. Am. Chem. Soc., 2006, 128, 2970–2976 CrossRef CAS PubMed.
- Y. Kato-Toma, T. Iwashita, K. Masuda, Y. Oyama and M. Ishiguro, Biochem. J., 2003, 371, 175–181 CrossRef CAS PubMed.
- R. Tripathi and N. N. Nair, J. Am. Chem. Soc., 2013, 135, 14679–14690 CrossRef CAS PubMed.
- B. M. Beadle, I. Trehan, P. J. Focia and B. K. Shoichet, Structure, 2002, 10, 413–424 CrossRef CAS.
- R. A. Powers, Curr. Drug Targets, 2016, 17, 1051–1060 CrossRef CAS PubMed.
- J. M. Rodriguez-Martinez, P. Nordmann, E. Ronco and L. Poirel, Antimicrob. Agents Chemother., 2010, 54, 3484–3488 CrossRef CAS PubMed.
- H. Mammeri, L. Poirel, N. Fortineau and P. Nordmann, Antimicrob. Agents Chemother., 2006, 50, 2573–2576 CrossRef CAS PubMed.
- G. B. Tian, J. M. Adams-Haduch, M. Taracila, R. A. Bonomo, H. N. Wang and Y. Doi, Antimicrob. Agents Chemother., 2011, 55, 4922–4925 CrossRef CAS PubMed.
- J. Y. Kim, H. I. Jung, Y. J. An, J. H. Lee, S. J. Kim, S. H. Jeong, K. J. Lee, P. G. Suh, H. S. Lee, S. H. Lee and S. S. Cha, Mol. Microbiol., 2006, 60, 907–916 CrossRef CAS.
- J. H. Jeon, M. K. Hong, J. H. Lee, J. J. Lee, K. S. Park, A. M. Karim, J. Y. Jo, J. H. Kim, K. S. Ko, L. W. Kang and S. H. Lee, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 2924–2936 CrossRef CAS PubMed.
- H. Mammeri, H. Guillon, F. Eb and P. Nordmann, Antimicrob. Agents Chemother., 2010, 54, 4556–4560 CrossRef CAS PubMed.
- B. A. Evans and S. G. Amyes, Clin. Microbiol. Rev., 2014, 27, 241–263 CrossRef PubMed.
- D. A. Leonard, R. A. Bonomo and R. A. Powers, Acc. Chem. Res., 2013, 46, 2407–2415 CrossRef CAS.
- T. Naas and P. Nordmann, Curr. Pharm. Des., 1999, 5, 865–880 CAS.
- M. Paetzel, F. Danel, L. de Castro, S. C. Mosimann, M. G. Page and N. C. Strynadka, Nat. Struct. Biol., 2000, 7, 918–925 CrossRef CAS PubMed.
- L. Maveyraud, D. Golemi-Kotra, A. Ishiwata, O. Meroueh, S. Mobashery and J.-P. Samama, J. Am. Chem. Soc., 2002, 124, 2461–2465 CrossRef CAS PubMed.
- D. Golemi, L. Maveyraud, S. Vakulenko, S. Tranier, A. Ishiwata, L. P. Kotra, J.-P. Samama and S. Mobashery, J. Am. Chem. Soc., 2000, 122, 6132–6133 CrossRef CAS.
- S. Akhter, B. A. Lund, A. Ismael, M. Langer, J. Isaksson, T. Christopeit, H.-K. S. Leiros and A. Bayer, Eur. J. Med. Chem., 2018, 145, 634–648 CrossRef CAS PubMed.
- C. M. June, B. C. Vallier, R. A. Bonomo, D. A. Leonard and R. A. Powers, Antimicrob. Agents Chemother., 2014, 58, 333–341 CrossRef PubMed.
- L. M. Hall, D. M. Livermore, D. Gur, M. Akova and H. E. Akalin, Antimicrob. Agents Chemother., 1993, 37, 1637–1644 CrossRef CAS.
- J. M. Mitchell, J. R. Clasman, C. M. June, K.-C. J. Kaitany, J. R. LaFleur, M. A. Taracila, N. V. Klinger, R. A. Bonomo, T. Wymore, A. Szarecka, R. A. Powers and D. A. Leonard, Biochemistry, 2015, 54, 1976–1987 CrossRef CAS PubMed.
- V. H. A. Hirvonen, A. J. Mulholland, J. Spencer and M. W. van der Kamp, ACS Catal., 2020, 10, 6188–6196 CrossRef CAS.
- C. Fröhlich, V. Sørum, A. M. Thomassen, P. J. Johnsen, H.-K. S. Leiros and Ø. Samuelsen, mSphere, 2019, 4, e00024-19 CrossRef.
- F. De Luca, M. Benvenuti, F. Carboni, C. Pozzi, G. M. Rossolini, S. Mangani and J. D. Docquier, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 18424–18429 CrossRef CAS PubMed.
- E. Santillana, A. Beceiro, G. Bou and A. Romero, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 5354–5359 CrossRef CAS.
- C. A. Smith, N. K. Stewart, M. Toth and S. B. Vakulenko, Antimicrob. Agents Chemother., 2019, 63, e01202-19 Search PubMed.
- C. T. Lohans, E. I. Freeman, E. v. Groesen, C. L. Tooke, P. Hinchliffe, J. Spencer, J. Brem and C. J. Schofield, Sci. Rep., 2019, 9, 13608 CrossRef PubMed.
- C. A. Smith, N. T. Antunes, N. K. Stewart, M. Toth, M. Kumarasiri, M. Chang, S. Mobashery and S. B. Vakulenko, Chem. Biol., 2013, 20, 1107–1115 CrossRef CAS PubMed.
- C. T. Lohans, E. van Groesen, K. Kumar, C. L. Tooke, J. Spencer, R. S. Paton, J. Brem and C. J. Schofield, Angew. Chem., Int. Ed., 2018, 57, 1282–1285 CrossRef CAS PubMed.
- K. M. J. Aertker, H. T. H. Chan, C. T. Lohans and C. J. Schofield, J. Biol. Chem., 2020, 295, 16604–16613 CrossRef CAS PubMed.
- W. Vollmer, D. Blanot and M. A. De Pedro, FEMS Microbiol. Rev., 2008, 32, 149–167 CrossRef CAS PubMed.
- S. Biarrotte-Sorin, J.-E. Hugonnet, V. Delfosse, J.-L. Mainardi, L. Gutmann, M. Arthur and C. Mayer, J. Mol. Biol., 2006, 359, 533–538 CrossRef CAS PubMed.
- A. Aliashkevich and F. Cava, FEBS J., 2021 DOI:10.1111/febs.16066.
- M. Lavollay, M. Arthur, M. Fourgeaud, L. Dubost, A. Marie, N. Veziris, D. Blanot, L. Gutmann and J. L. Mainardi, J. Bacteriol., 2008, 190, 4360–4366 CrossRef CAS PubMed.
- L. Sütterlin, Z. Edoo, J.-E. Hugonnet, J.-L. Mainardi and M. Arthur, Antimicrob. Agents Chemother., 2017, 62, e01607-17 Search PubMed.
- M. Lavollay, M. Fourgeaud, J.-L. Herrmann, L. Dubost, A. Marie, L. Gutmann, M. Arthur and J.-L. Mainardi, J. Bacteriol., 2011, 193, 778–782 CrossRef CAS PubMed.
- S. Magnet, S. Bellais, L. Dubost, M. Fourgeaud, J. L. Mainardi, S. Petit-Frere, A. Marie, D. Mengin-Lecreulx, M. Arthur and L. Gutmann, J. Bacteriol., 2007, 189, 3927–3931 CrossRef CAS PubMed.
- J. L. Mainardi, R. Legrand, M. Arthur, B. Schoot, J. van Heijenoort and L. Gutmann, J. Biol. Chem., 2000, 275, 16490–16496 CrossRef CAS PubMed.
- M. A. Bianchet, Y. H. Pan, L. A. B. Basta, H. Saavedra, E. P. Lloyd, P. Kumar, R. Mattoo, C. A. Townsend and G. Lamichhane, BMC Biochem., 2017, 18, 8 CrossRef PubMed.
- M. Cordillot, V. Dubée, S. Triboulet, L. Dubost, A. Marie, J.-E. Hugonnet, M. Arthur and J.-L. Mainardi, Antimicrob. Agents Chemother., 2013, 57, 5940–5945 CrossRef CAS PubMed.
- J.-L. Mainardi, J.-E. Hugonnet, F. Rusconi, M. Fourgeaud, L. Dubost, A. N. Moumi, V. Delfosse, C. Mayer, L. Gutmann and L. B. Rice, J. Biol. Chem., 2007, 282, 30414–30422 CrossRef CAS.
- L. Lecoq, C. Bougault, J. E. Hugonnet, C. Veckerle, O. Pessey, M. Arthur and J. P. Simorre, Structure, 2012, 20, 850–861 CrossRef CAS PubMed.
- M. de Munnik, C. T. Lohans, P. A. Lang, G. W. Langley, T. R. Malla, A. Tumber, C. J. Schofield and J. Brem, Chem. Commun., 2019, 55, 10214–10217 RSC.
- H. S. Kim, J. Kim, H. N. Im, J. Y. Yoon, D. R. An, H. J. Yoon, J. Y. Kim, H. K. Min, S.-J. Kim, J. Y. Lee, B. W. Han and S. W. Suh, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 420–431 CrossRef CAS.
- S. A. Cochrane and C. T. Lohans, Eur. J. Med. Chem., 2020, 194, 112262 CrossRef CAS PubMed.
- F. Zhao, Y.-J. Hou, Y. Zhang, D.-C. Wang and D.-F. Li, Biochem. Biophys. Res. Commun., 2019, 510, 254–260 CrossRef CAS PubMed.
- S. Correale, A. Ruggiero, R. Capparelli, E. Pedone and R. Berisio, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1697–1706 CrossRef CAS.
- N. Caveney, A. Serapio-Palacios, S. Woodward, T. Bozorgmehr, G. Caballero, M. Vuckovic, W. Deng, B. Finlay and N. Strynadka, Antimicrob. Agents Chemother., 2021, 65, e01592-20 CrossRef PubMed.
- S. Triboulet, V. Dubée, L. Lecoq, C. Bougault, J.-L. Mainardi, L. B. Rice, M. Ethève-Quelquejeu, L. Gutmann, A. Marie, L. Dubost, J.-E. Hugonnet, J.-P. Simorre and M. Arthur, PLoS One, 2013, 8, e67831 CrossRef CAS.
- M. de Munnik, C. T. Lohans, G. W. Langley, C. Bon, J. Brem and C. J. Schofield, ChemBioChem, 2020, 21, 368–372 CrossRef CAS PubMed.
- C. T. Lohans, H. T. H. Chan, T. R. Malla, K. Kumar, J. J. A. G. Kamps, D. J. B. McArdle, E. van Groesen, M. de Munnik, C. L. Tooke, J. Spencer, R. S. Paton, J. Brem and C. J. Schofield, Angew. Chem., Int. Ed., 2019, 58, 1990–1994 CrossRef CAS PubMed.
- V. Dubée, S. Triboulet, J. L. Mainardi, M. Ethève-Quelquejeu, L. Gutmann, A. Marie, L. Dubost, J. E. Hugonnet and M. Arthur, Antimicrob. Agents Chemother., 2012, 56, 4189–4195 CrossRef PubMed.
- Z. Edoo, M. Arthur and J.-E. Hugonnet, Sci. Rep., 2017, 7, 9136 CrossRef PubMed.
- E. M. Steiner, G. Schneider and R. Schnell, FEBS J., 2017, 284, 725–741 CrossRef CAS PubMed.
- P. Kumar, V. Chauhan, J. R. A. Silva, J. Lameira, F. B. d'Andrea, S.-G. Li, S. L. Ginell, J. S. Freundlich, C. N. Alves, S. Bailey, K. A. Cohen and G. Lamichhane, Antimicrob. Agents Chemother., 2017, 61, e00866-17 Search PubMed.
- T. A. Zandi and C. A. Townsend, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2008610118 CrossRef CAS PubMed.
- N. Dhar, V. Dubee, L. Ballell, G. Cuinet, J. E. Hugonnet, F. Signorino-Gelo, D. Barros, M. Arthur and J. D. McKinney, Antimicrob. Agents Chemother., 2015, 59, 1308–1319 CrossRef PubMed.
- B. Liu, R. E. L. Trout, G.-H. Chu, D. McGarry, R. W. Jackson, J. C. Hamrick, D. M. Daigle, S. M. Cusick, C. Pozzi and F. De Luca, J. Med. Chem., 2019, 63, 2789–2801 CrossRef.
- J. L. Mainardi, J. E. Hugonnet, F. Rusconi, M. Fourgeaud, L. Dubost, A. N. Moumi, V. Delfosse, C. Mayer, L. Gutmann, L. B. Rice and M. Arthur, J. Biol. Chem., 2007, 282, 30414–30422 CrossRef CAS PubMed.
- M. Paetzel, R. E. Dalbey and N. C. Strynadka, Nature, 1998, 396, 186–190 CrossRef CAS PubMed.
- A. Allsop, G. Brooks, P. D. Edwards, A. C. Kaura and R. Southgate, J. Antibiot., 1996, 49, 921–928 CrossRef CAS PubMed.
- T. R. Malla, A. Tumber, T. John, L. Brewitz, C. Strain-Damerell, C. D. Owen, P. Lukacik, H. T. H. Chan, P. Maheswaran, E. Salah, F. Duarte, H. Yang, Z. Rao, M. A. Walsh and C. J. Schofield, Chem. Commun., 2021, 57, 1430–1433 RSC.
- N. A. Caveney, S. D. Workman, R. Yan, C. E. Atkinson, Z. Yu and N. C. J. Strynadka, Nat. Commun., 2021, 12, 2775 CrossRef CAS PubMed.
- E. J. Culp, N. Waglechner, W. Wang, A. A. Fiebig-Comyn, Y.-P. Hsu, K. Koteva, D. Sychantha, B. K. Coombes, M. S. Van Nieuwenhze and Y. V. Brun, Nature, 2020, 578, 582–587 CrossRef CAS PubMed.
- A. J. Egan, J. Errington and W. Vollmer, Nat. Rev. Microbiol., 2020, 18, 446–460 CrossRef CAS PubMed.
Footnote |
| † Abbreviations: ADC (Acinetobacter-derived cephalosporinases), AEC (acyl–enzyme complex), CMY (cephamycinase), CTX-M (cefotaximase), DBO (diazabicyclooctane), ESBL (extended spectrum β-lactamase), FOX (cefoxitinase), GES (Guiana extended spectrum), KPC (Klebsiella pneumoniae carbapenemase), Ldt (L,D-transpeptidase), MBL (metallo-β-lactamase), MRSA (methicillin-resistant Staphylococcus aureus), OXA (oxacillinase), PBP (penicillin-binding protein), SBL (serine β-lactamase), SHV (sulfhydryl variable), TEM (temoneira). |
| This journal is © The Royal Society of Chemistry 2021 |