
A primer on harnessing non-enzymatic post-translational modifications for drug design
Marcus J. C.
Long
a,
Phillippe
Ly
b and
Yimon
Aye
 *b
*b
aUniversity of Lausanne, 1015 Lausanne, Switzerland
bSwiss Federal Institute of Technology in Lausanne (EPFL), 1015 Lausanne, Switzerland. E-mail: yimon.aye@epfl.ch
First published on 26th October 2021
Abstract
Of the manifold concepts in drug discovery and design, covalent drugs have re-emerged as one of the most promising over the past 20-or so years. All such drugs harness the ability of a covalent bond to drive an interaction between a target biomolecule, typically a protein, and a small molecule. Formation of a covalent bond necessarily prolongs target engagement, opening avenues to targeting shallower binding sites, protein complexes, and other difficult to drug manifolds, amongst other virtues. This opinion piece discusses frameworks around which to develop covalent drugs. Our argument, based on results from our research program on natural electrophile signaling, is that targeting specific residues innately involved in native signaling programs are ideally poised to be targeted by covalent drugs. We outline ways to identify electrophile-sensing residues, and discuss how studying ramifications of innate signaling by endogenous molecules can provide a means to predict drug mechanism and function and assess on- versus off-target behaviors.
Covalent interactions between proteins and small molecules have found diverse uses in biology. Although there are different models for how covalent ligand–protein interactions occur, critical aspects include that these are time-dependent, typically irreversible, and inherently promiscuous. These considerations to some extent can render analysis of covalent binding more complex than non-covalent binding, and further highlight that off-target effects (as a function of time) must also be considered carefully when dealing with covalent binders. Nonetheless, by tuning reactivity, localization, longevity and other key parameters, covalent binders have emerged to be particularly useful for numerous aspects in biology.
One of the principle uses of covalent molecules in biology is chemical probes for investigating protein–protein interactions or enzyme activity; generally, such molecules leverage the indelible mark left by covalent binders to tag proteins for further analysis. Numerous covalent probes display broad reactivity and short half-lives in cells, allowing proximity profiling to be enacted through rapid covalent labeling of proteins resident in specific locales in which the reactive species is liberated.1 Other covalent probes show selective reactivity, albeit for a specific functional group, such as cysteine, methionine, and lysine, or even a modified functional group, such as dimedone that labels sulfenic acids and likely some similar oxidation states of sulfur.2,3 These probes allow irreversible labeling of specific residues (and functionally coupled residues) to be assessed on a relatively large scale (hundreds to thousands of residues). Conversely, chemical probes can enact specific protein functionalization and neofunctionalization, or labeling for visualization, for instance of expression, activity, or subcellular locale. This is achieved most commonly through reactive domains (SNAP, CLIP, HALO4) that can be fused to a protein of interest, or through modification of a specific inhibitor.5
Another key use of covalent interactions in biological systems is in drugs.6–9 There are some drugs that appear to function through a relatively non-specific mechanism, such as the covalent multiple sclerosis drug DMF and its later generation analogs, or the non-covalent inhibitor midostaurin, an approved acute myeloid leukemia therapy. However, the majority of approved drugs have a major associated target. In terms of covalent binding, this means that drugs react kinetically much faster with their target than other potential targets. Kinetic selectivity can be obtained in numerous ways, including through increasing overall reactivity (although this will also increase promiscuity), through creating the electrophile as a consequence of enzymatic activity (mechanism-based inhibition, which we will not discuss) and through ligandable interactions, that we discuss below.
Before moving forward, it is important to note that the goals of drugs and probes are different. One exists to cure a disease: the other is a detection agent. However, at the molecular level both usually aim to label a specific target or targets. Clearly, the selectivity preferences of probes depend on the nature of the intended goal and can span for low selectivity to particularly high. On the other hand, for a drug, off-target labeling is acceptable, so long as it is not overall detrimental to the effect of the drug, or the patient. In this way, to some extent, the main difference between a chemical probe and a drug is ranked on how well the former can identify protein targets, whereas the latter focuses on a holistic effect on an organism. Nonetheless, it is the interaction between the drug and its target(s) that is responsible for the positive effect of the drug, and it is this aspect we will focus on here.
Analysis of many modern ligandable covalent drugs shows that they consist of two distinct parts. One section, which is typically considerably larger than the other (upwards of ∼250 Daltons), is a “ligandable” fragment. This piece binds to protein(s) non-covalently, inhibiting the activity of the target. The second portion is typically lower in molecular weight (∼60 Daltons) but contains a weakly-reactive Michael acceptor. The combination of the twain forges a molecule that, ideally, binds the target in a rapid-reversible manner, ushering formation of a covalent bond selectively to the target protein, yielding an irreversibly-bound complex. This choreography imparts enzyme-like rate acceleration to covalent-bond formation, which in the absence of ligand binding, is (ideally) negligible: many acrylamide-containing covalent inhibitors, for instance, have second-order rates of inhibition >105 M−1 s−1 [t1/2(labeling) <10 s, with protein and inhibitor both at 1 μM]; the rate constant for the second-order reaction (k2) between acrylamide and cysteine <0.1 M−1 s−1 [t1/2(labeling) >107 s, with protein and inhibitor both at 1 μM]. Although older modus operandi to achieve covalent inhibition, e.g., direct alkylation and mechanism-based inhibition exist, ligandable covalent binding has become a go-to strategy.10,11
It is clear that for ligandable designs, we need both a non-covalent binding site on the target protein matching the non-covalent moiety of the inhibitor, and a nearby nucleophilic residue to trap out the enzyme-inhibitor complex. Should there be no reactive residue, but the reversible portion can still bind the protein, the inhibitor will function reversibly. For such interactions, in patients and model organisms, upon inhibitor clearance (or removal of the compound from media in cultured cells), inhibition will diminish. However, for proteins that form a covalent bond to the bound inhibitor, inhibition is sustained until new protein is synthesized, regardless of clearance (Fig. 1). Thus, covalent appendages can help home in on specific targets of more promiscuous non-covalent binders, and raise the burden on targeted cells, while minimizing the impact of clearance. Several other benefits of covalent binders have also been proposed, including overcoming competitive ligands, and targeting “shallow binding sites”. Work has justifiably focused on matching the ligandable interaction with the protein of interest (POI), and picking cysteines of convenience or genetic aberrations (e.g., oncogenic rasG12C)12–14 as targets. This drive has led to a good deal of drugs approved in this millennium, that target cysteines including: afatinib15 and zanubrutinib.16,17 Many similar molecules are in clinical trials and drives to identify novel ligandable interactions are common. Recently this strategy was extended to targeting N-terminal amines,18 through approval of voxelotor to treat sickle cell disease.19,20 Herein we will discuss an alternative design strategy harnessing a ligandable interaction to orchestrate a selective privileged thiol–electrophile interaction. Privileged thiol–electrophile interactions are referred to as such because reactions between such matched thiol/electrophile pairs are intrinsically rapid. However, equally, or perhaps more importantly, interactions between matched thiol/electrophile pairs lead to phenotypically dominant outputs. This means that even when occupancy of the ligand on the target is low, changes in signaling can occur by virtue of a new property of the ligand–protein complex. These properties, in nature, allow specific protein thiols to sense and respond to specific (sets of) signaling electrophiles, which are themselves often toxic, or are products of toxic processes or cellular damage. We will discuss why such an approach is conceptually different from the traditional viewpoint, and we will describe ways to predict mode of action, validate on target phenotypes, and data derived from such studies.
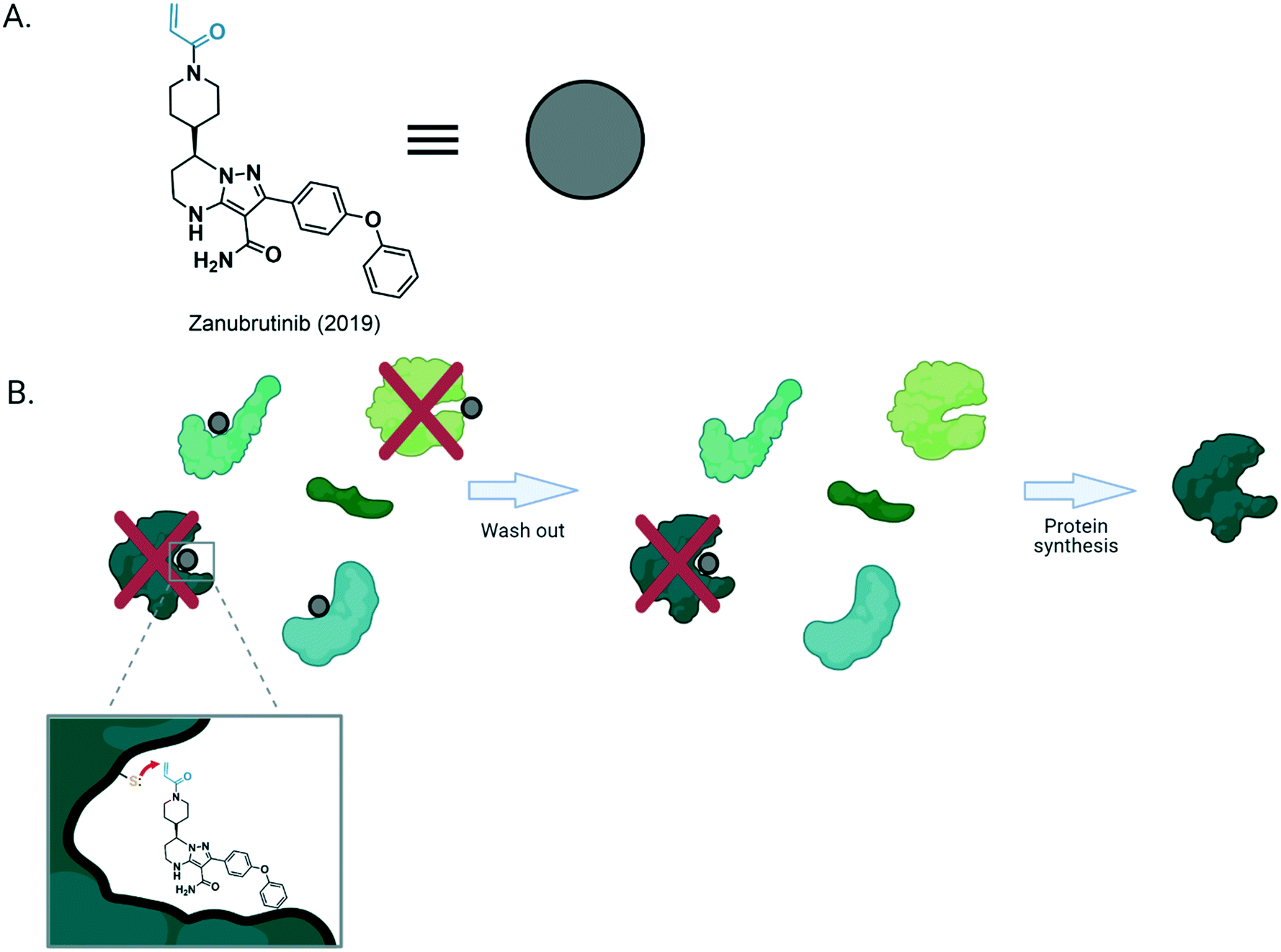 | ||
| Fig. 1 (A) Structure of zanubrutinib, a covalent drug approved in 2019 for treatment of mantle cell lymphoma. The black ligand motif of the molecule allows binding to the adenosine-triphosphate binding pocket of its target, while the blue (Michael acceptor-based electrophilic motif) is covalently modified by nucleophilic residue from the protein. (B) General structure of a covalent inhibitor bound to its target. After binding, the Michael acceptor situated proximal to the nucleophilic residue, favors the formation of a covalent bond between the target protein and the drug molecule. This forms the irreversibly-bound ligand–protein complex that remains in vivo until [degradation and] new protein synthesis. However, if the reactive residue (Cys in this example) is not present, the inhibition will be reversible. | ||
Privileged thiol–electrophile interactions
In the 1950's Betty and Jim Millar21 established a strong link between exposure to reactive electrophiles and cancer. This link was extended to other disease etiologies, like drug-induced liver injury22 and neurodegeneration.23 So, when electrophiles were detected in cells and dietary samples, it was first thought that these molecules were unwanted and dangerous. Contrary to such expectations, the preponderance of data shows that life has harnessed naturally-produced electrophiles to change decision making and meaningfully modulate proteins controlling important signaling pathways: low concentrations of native electrophile externally added to cell culture media promote growth and aid recovery and survival under stress. However, at higher concentrations, these electrophiles behave as billed and are toxic. This hormesis (or both stimulatory and inhibitory properties) indicates that electrophiles have multiple biological behaviors, and that they are uniquely context-dependent.2Electrophiles created in an unstressed cell are degraded rapidly,24 constricting their diffusion distance likely to much shorter than the length of a cell, perhaps even shorter than the diameters of some organelles.2 However, to signal, an endogenous electrophile must covalently engage a target, a time-dependent process. For our purposes, we will consider only proteins, although such arguments could apply to other macromolecules. Thus, a sufficient amount of labeled protein must arise prior to clearance of the electrophile for phenotypes to be observed. Assuming that one binding event leads to inhibition of one target protein molecule, then typically ∼50% occupancy is minimally necessary to trigger a phenotype. Such a scenario mimics a genetic state in which one allele of a protein is functional and another is not. For numerous, although likely not a majority of genes, such a state can elicit a phenotypic change relative to wild-type. However, for numerous proteins, including enzymes in important pathways, ≫50% activity reduction is required to elicit a phenotypic change relative to wild-type.25 Such criteria set a high burden in terms of necessary occupancy, especially for electrophiles, to achieve, given that electrophiles are short-lived and their concentrations in healthy cells must be limited as they are toxic.
High occupancy of an electrophile on a specific protein may be engendered by coordinating localized electrophile release with localization of the target protein (Fig. 2A). This choreography stimulates the intended bimolecular reaction over others that may occur if the electrophile were in the bulk of the cell. To analyze this process, we must consider concentration of electrophile and protein, and the second-order rate of reaction (k2). Owing to limitations/concerns underlying probes, and an inability to measure concentrations locally, we do not know particularly well local cellular concentration of even the most well-studied endogenous electrophile, 4-hydroxynonenal (HNE), or how long elevated HNE levels are sustainable.2,24 However, HNE concentrations can unlikely exceed low millimolar levels, a concentration that when administered externally, is toxic; such levels are likely not sustainable. Although there are exceptions, most protein concentrations are not significantly more than micromolar in cells,26,27 with many being lower. However, subcellular or localized protein concentrations are less established. The k2 for HNE reacting with cysteine is ∼1 M−1 s−1. A similar k2 has been reported for numerous protein cysteines engaging with HNE in vitro. At 1 mM HNE, the half-life of labeling of a cysteine of typical reactivity under pseudo first-order kinetics is ∼12 minutes; at 0.1 mM HNE, t1/2 = 120 minutes. Thus, the localized model results in relatively slow labeling even with optimistic HNE concentrations. This model also does not also necessarily lead to selective labeling of the target protein. This is because there are almost certainly many cysteines present in any given cellular region, many of which have k2(HNE) = 1 M−1 s−1. This concern may be assuaged since the vast majority of electrophile-labeling events may not affect protein-activity/function (although few have been adequately studied). However, as electrophile labeling is ostensibly irreversible,28 and it can introduce reactive groups, e.g., aldehydes into the protein, the reasoning above seems an unsatisfying defense (Fig. 2B).
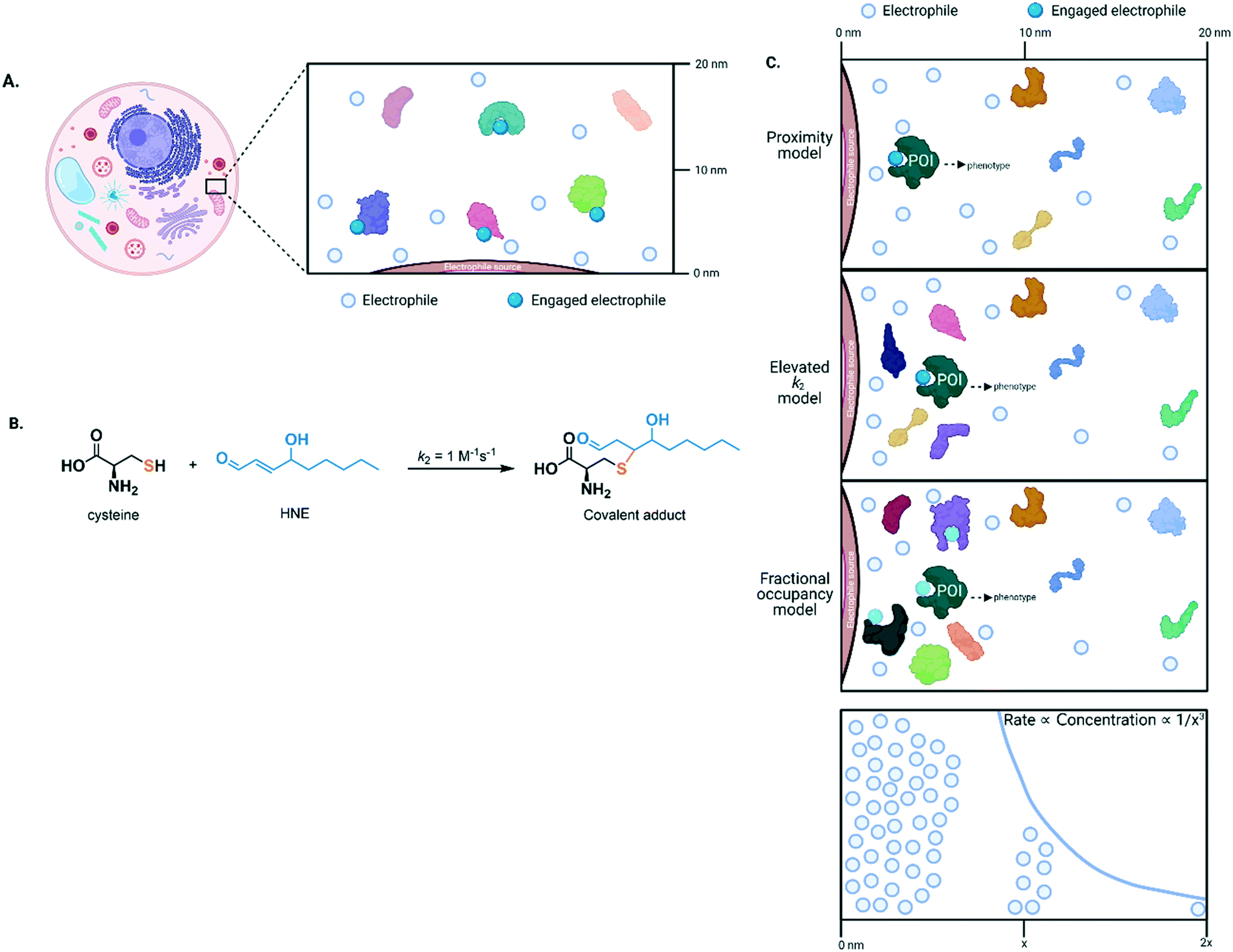 | ||
| Fig. 2 (A) Because of their high reactivity, electrophiles covalently engage with nearby targets, and covalent bond formation occurs in a time-dependent manner. The scheme shown exemplifies a case where more than 50% of protein labeling (by the electrophile) is required to elicit a phenotypic change. Should the protein have enzymatic properties, this functional output may be reflected in modulation of enzymatic activity. (B) Native lipid-derived electrophilic metabolite, HNE, reacts with free/non-protein-bound cysteine to form an aldehyde-containing product that can undergo subsequent nucleophilic substitution. (C) Top three panels: there are three possible models to explain how low concentration of HNE (generated from a point source, e.g., from an organellar lipid membrane during lipid peroxidation) can selectively modify a protein target and thus promote signaling. X-axis designates distance from the point source. The proximity model (top panel) is relatively slow and unselective at labeling, given that many proteins have at least one cysteine residue. On the other hand, the elevated k2 model (middle) allows high occupancy to be achieved in a short time and/or greatly modulates bulk activity in a more impactful manner than the fractional occupancy model (bottom). Lowest panel: the rate of labeling (axis in Y direction as drawn) is proportional to the concentration of the electrophile that decreases as a function of the spherical volume of the cell (axis in X direction as drawn). POI, protein of interest. | ||
Thus, other mechanisms likely help promote electrophile-labeling selectivity and allow signaling to occur at low HNE concentrations. In our opinion the most likely mechanisms involve elevating k2 (for HNE, this could be up to ∼108 fold, the ratio of the diffusion-limited k2 for protein labeling: k2(cysteine)), and/or mechanism of activity modulation.2,29 For the former, by elevating k2, a high occupancy can be achieved in a short time, prior to off-target labeling, regardless of the presence of other proteins. For the latter, the more impact a single electrophile labeling event has on the bulk contingent of the target (or other) protein(s), the less percentage labeling is needed. Note that lowering required threshold occupancy is particularly important for second-order reactions, where driving high occupancy becomes increasingly longer duration relative to a pseudo-first order process. To give optimal outputs, the mechanisms above could function in tandem. Electrophile-sensor proteins functioning through dominant pathways and displaying kinetically-favorable modification rates are termed privileged first responders (PFRs).24
The privileged, few?
From the discussion above, we can predict that since cysteine is present at least once in most proteins, native reactive electrophiles like HNE, or related small-molecule covalent drugs, will label multiple proteins under prolonged bolus-dosing conditions. Such promiscuity is most easily shown by fluorescence imaging of SDS-PAGE analysis of lysates of cells which had been treated with a clickable variant of native electrophiles such as HNE,30,31 fumarate,32 or itaconate,33 or related Michael-acceptor-based drug, dimethyl fumarate34 prior to lysis, and were then treated with a clickable dye. The number of bands appearing in the soluble proteome is impressive. However, such gel-based analysis does not show occupancy of the targets, so many of the detected labeling events may not be significant. Conversely, for specific proteins, labeling may only be able to occur in specific locales that may not be exposed to the electrophile (the distribution of bolus dosed electrophile across different organelles/locales within the cell is rarely known). As touched on above, for many targets, covalent labeling may also not well lead to functional output. Similar multi-target- and time-dependent-labeling events are observed in aged tissues using HNE-antibodies,35 although HNE-antibodies should be treated with caution36 as they are likely not particularly specific and correct control experiments cannot easily be used to validate them.More quantitative data from several laboratories brought to light that electrophiles are not as promiscuous as may have been expected. Iso-TOP-ABPP, a method that uses cysteine-reactive chemical probes to assay labeling of hundreds to thousands of cysteines, typically within lysates, demonstrated that significant (>75%) labeling of a handful out of ∼1000 cysteines occurs upon treatment of lysates with relatively high HNE concentrations.37 Extrapolating this ratio over the whole cysteome predicts hundreds to thousands of HNE-sensitive cysteines. This ballpark analysis agrees with more proteome-wide data from dosing/mass-spectrometry regimens that indicated there are hundreds38 of HNE-sensitive proteins.39–41 Experiments using bolus dosing of cells with alkyne- or azide-modified electrophiles, biotin-click, and streptavidin enrichment reach similar numbers. Labeling of some of these sensors, for related electrophiles at least, appears to be stimulated by oxidative stress.42In situ derivation of a chemically-diverse mixture of lipid-derived electrophilic species specifically in the mitochondria, showed enrichment principally of mitochondrial proteins, giving weight to location being critical to determine lipid-modification processes.
In the few cases where different chemotypes have been compared, it has transpired that labeling is unexpectedly chemoselective. For instance, iso-TOP-ABPP measurements showed little overlap of targets between HNE and a prostaglandin-derived electrophile. Other examples of this phenomenon exist, although PFRs are not always chemoselective, especially for proteins with multiple sensor cysteines.
Limitations thus far
Many of the above methods were conducted in lysates and/or deploy procedures where dose and timing of electrophile is exaggerated relative to what likely occurs in cells. Such conditions likely artificially elevate chances of observing a labeling event because mass spectrometry is particularly sensitive, and HNE labels targets not only in a dose but also time-dependent manner.24 Furthermore, as the majority of methods leverage probes that detect unmodified cysteines they cannot absolutely and/or directly quantitate occupancy, sensors with moderate reactivity, or cysteines sensitive to the environment established under experimental conditions, may have been identified. Conversely, as reactive cysteines are likely sensitive to their environment, the procedures used could readily mask reactive cysteines.Identifying phenotypically-relevant sensors
It is also clear that the above experiments are blind to dominant signaling behavior, and ultimately give limited insight into links between ligand binding and biological output in general. Once targets of specific signaling molecules are assigned, typically their role in biological signaling can be addressed, separately, to fill in some of the gaps outlined above. For instance, should labeling of a POI be necessary for a specific phenotype, its knockout/knockdown/loss of function should diminish the associated phenotype observed on electrophile treatment. This approach has been applied to numerous traditional biological problems, including a host of screens. A plethora of published data demonstrate that this stratagem has not worked well with reactive electrophiles. There are numerous potential reasons for lack of success, including: detection methods not identifying the correct sensors; the manifold pathways and target proteins modulated by these reactive small molecules synergize to a “compound” phenotype with no single target necessary or sufficient for cellular response; there are multiple sensors, of which the majority are sufficient, but not necessary to cause signaling (Fig. 3). The first reason likely contributes to the issues in this area, as discussed above. The second reason is essentially untestable in the form it is presented. The third reason is mutually exclusive to the second and is further testable provided we can form precisely defined electrophile-modified states and interrogate them in an unperturbed cell. Should labeling of an individual POI be sufficient to trigger labeling, the state so formed, will necessarily change signaling.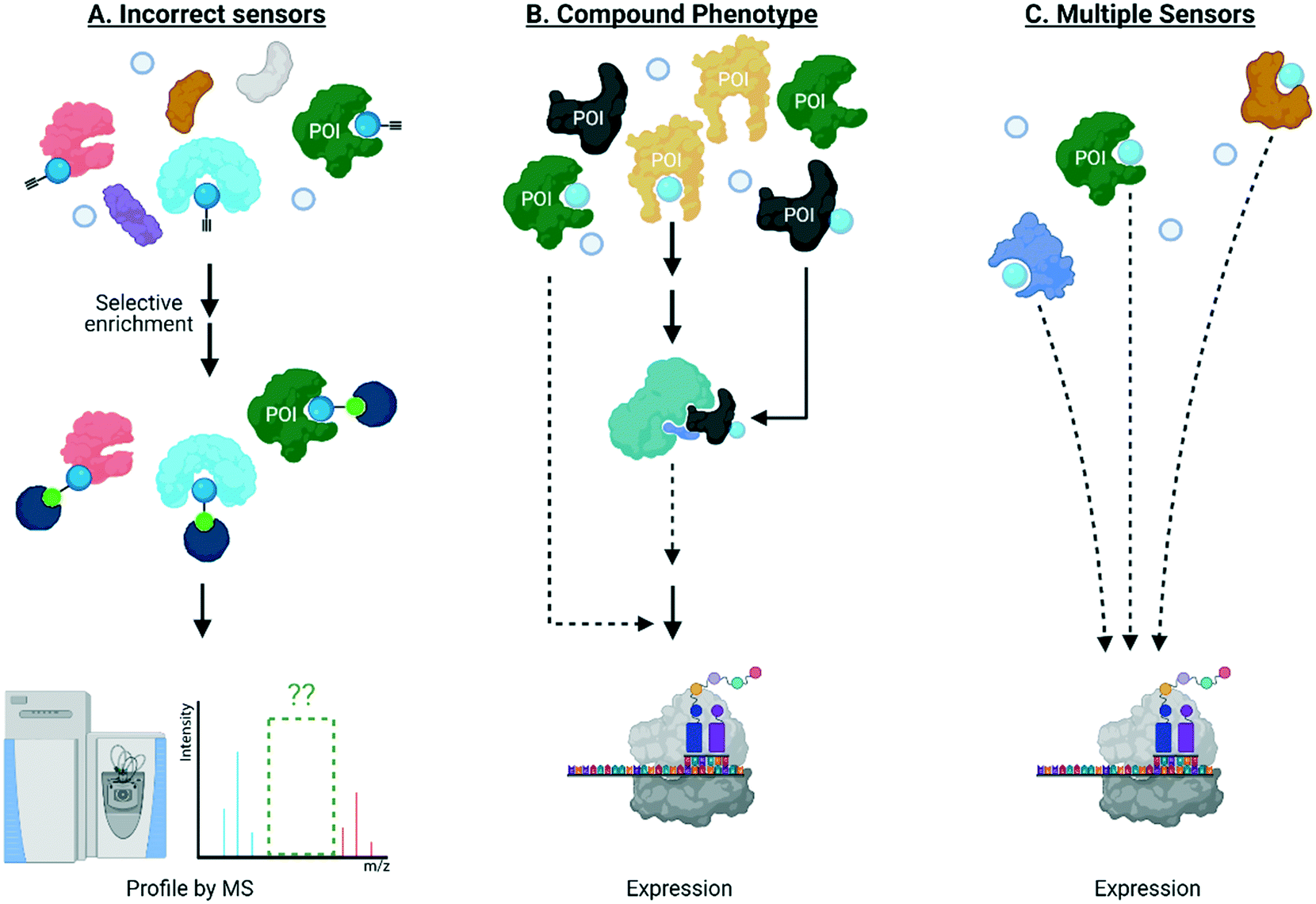 | ||
| Fig. 3 Traditional knockdown/knockout/loss-of-function screens of a protein of interest (POI) labeled by electrophiles have not worked well for the following reasons: (A) the kinetically-privileged electrophile-sensor protein has not been correctly identified by state-of-the-art methods which remain limited in terms of, for instance, reliance on indirect readout using proxy electrophiles, limited ability to identify targets with low ligand occupancy, and lack of spatiotemporal resolution in intact living systems; (B) multiple pathways and targets must be modified to achieve the observed overall phenotype, such as protein translation for instance; and (C) multiple sensors are present and their labeling/site occupancy is sufficient, but modification is not necessary to cause signaling. Effect on translation is shown just as an example but many biological processes have been implicated to be affected by electrophilic protein modificaitons. | ||
REX technologies a new way to study electrophile–protein engagement
To investigate if electrophile engagement by specific sensors be sufficient to trigger signaling, we developed T-REX.43 This method uses a chemical probe bearing a defined photocaged ligand (probe) to label a specific, preordained protein with a specific electrophile of choice, providing that protein be a kinetically privileged sensor of the specific electrophile in question. This constraint is applied because the native electrophile is offered from the probe to the POI pseudo-intramolecularly. Then, the POI can engage with the electrophile, or the POI can leave the electrophile to diffuse away, irreversibly; under T-REX conditions, second order labeling of the POI by the released electrophile is minimal. The amount of electrophile released to the POI from the probe, and the amount of electrophile on the POI, are quantifiable because the (proto)electrophile (i.e. both in the probe and that released) contains a biorthogonal handle allowing fluorophore tagging. Thus, T-REX can quantitatively assess POI electrophile occupancy. The majority of proteins are not labelled under T-REX. Furthermore, proteins that are labelled by T-REX in cells show rapid in vitro k2(HNE).2,43,44 These observations are consistent with T-REX labeling only the best electrophile sensors. Finally, HNE released to the bulk proteome under T-REX does not affect numerous HNE-sensitive signaling processes. Thus, phenotypes measured under T-REX are due to labeling of the POI.For several HNE-sensitive proteins, T-REX delivery efficiency (DE) between 15 and 50% triggers signaling phenotypes.30,31,44–50 In several instances the phenotypes stimulated are similar in magnitude to those observed under bolus dosing, or when an inhibitor is used to fully shut down POI activity. Thus, specific electrophile labeling of single proteins is sufficient to trigger signaling. Signaling changes measured are significantly greater than predicted based on measured occupancy. This result demonstrates that dominant signaling modes occur in redox sensing, and as this has now been shown for several different proteins/pathways (NF-κB,43,51 PI3K/Akt,43,49,52 DNA-damage,51 and antioxidant response30,31,43,45–47,50 signaling), we cautiously postulate that such behaviors are not uncommon. Such results reconcile some indirect data that had shown that the occupancy on postulated electrophile sensors necessary to trigger signaling was relatively small.2,44,45,50
T-REX-mediated electrophile release is chemospecific. ∼10 Different electrophiles so far, and likely many more, are compatible with this method. T-REX thus can perform unbiased (i.e. independent of permeation, metabolism, distribution, etc.) structure activity relationships (SAR) to identify an optimal match between electrophile and target protein in terms of both kinetics and phenotype/dominance in cells and organisms. These considerations all together led us to propose that PFRs53 are ideal for covalent drug design and that T-REX is an ideal means to predict drug phenotypes and inform on covalent drug design (Fig. 4).
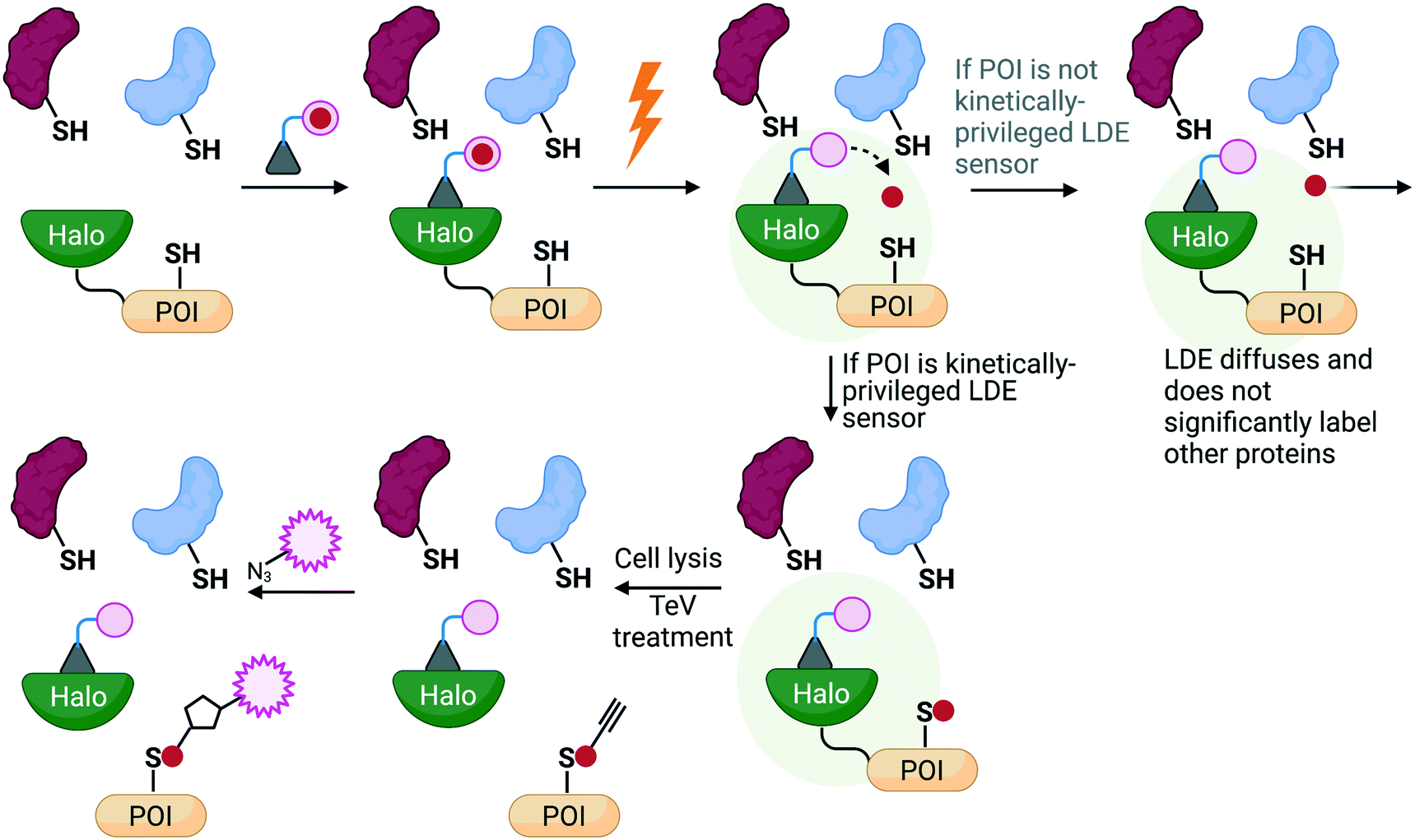 | ||
| Fig. 4 An overview of the principles behind REX technologies. A photocaged probe bearing a haloalkane binds to a Halo protein tethered to the POI that is expressed in living systems. Upon irradiation (0.5–5 mW cm−2, 365 nm, 3–10 min), the released electrophile is offered to the POI pseudo-intramolecularly, where two possibilities can occur: 1) the liberated ligand does not label the POI and diffuses away (whereby sub nM-μM amount of electrophile released has been shown to be insufficient to significantly label the background proteome to cause an effect; or 2) the released electrophile does label the POI and results in fractional labeling that can be detected after click-coupling with a fluorophore. Importantly, this target-specific labeling can be directly linked to a biological response of interest that can be measured using relevant downstream assays. | ||
Translating T-REX data
In our translational studies, we investigated the isoform-specific inhibition of Akt3 kinase by HNE-derived electrophiles. Akt is upregulated in numerous cancers, and is a downstream effector of PIK3, for which an inhibitor, alpelisib, was recently approved to treat breast cancer.54 However, Akt has 3 different isoforms, encoded at differing genetic loci in humans and linked to different cancers. Thus, there is a growing need for Akt-isoform specific inhibitors. Some success has been reached with partly Akt1-selective inhibitors, with drugs such as MK-2206 entering phase II clinical trials.55–57 However, there is generally a pressing need for drug strategies targeting specific isoforms or specific alleles that is only partially met. Some success has been achieved in this area, such as the approval of Ayvakit to treat patients with a PDGFRA exon 18 mutation, although much work is left to be done.58 In the case of Akt isoforms, Akt3 is particularly difficult to target, despite being implicated in triple negative breast cancer etiology. Intriguingly, when comparing electrophile sensitivity of Akt isoforms by T-REX, Akt3 emerged as the most HNE-reactive isoform. Electrophile labeling occurs on a cysteine unique to Akt3, C119.49 C119 lies in an isoform-divergent linker region separating the two principal domains of Akt, the pleckstrin homology domain and the kinase domain. Critically, Akt3(C119) electrophile engagement leads to dominant negative (DN) inhibition of Akt activity. Akt2 is labelled by HNE, although this occurs approximately four-fold less efficiently than for Akt3 and is not DN. Akt1 is not targeted by electrophiles.We chose to derive an inhibitor from the Akt1-semi-selective inhibitor, MK-2206, that targets Akt3 ∼100-fold less effectively. This inhibitor binds to an allosteric site common to all Akts. This choice was made because C119 is removed from the active site, where many Akt-targeted molecules bind: the MK-2206-binding site is closer to the flexible linker region. We also wanted to pit the two segments of the chimeric inhibitor together, so it was good that the reversible interaction favors Akt1, and the covalent portion favors Akt3. The resulting molecule, MK-HNE and its second-generation inhibitor, MK-FNE, were selective inhibitors of Akt3, showing no labeling of Akt1, and weaker labeling of Akt2 (∼3.5-fold <Akt3).52 MK-FNE enacted irreversible inhibition of Akt3 and Akt2. For Akt3, inhibition was DN: when a mutant Akt3 that cannot bind MK-HNE [Akt3(C119S)] was expressed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with wild type Akt3, 100% inhibition was retained, even post drug washout. Repeating this experiment with Akt2, significant regain of activity was observed. MK-FNE emerged to be more selectively cell line toxic than MK-2206, and it further showed positive synergy with Akt3 (and not Akt1/2) knockdown in sensitive lines. MK-2206 showed positive synergy with Akt1 (and not Akt2/3) knockdown. MK-FNE emerged to be more potent and showed less side effects than MK-2206 in mouse xenograft models of breast cancer (Fig. 5).
:1 with wild type Akt3, 100% inhibition was retained, even post drug washout. Repeating this experiment with Akt2, significant regain of activity was observed. MK-FNE emerged to be more selectively cell line toxic than MK-2206, and it further showed positive synergy with Akt3 (and not Akt1/2) knockdown in sensitive lines. MK-2206 showed positive synergy with Akt1 (and not Akt2/3) knockdown. MK-FNE emerged to be more potent and showed less side effects than MK-2206 in mouse xenograft models of breast cancer (Fig. 5).
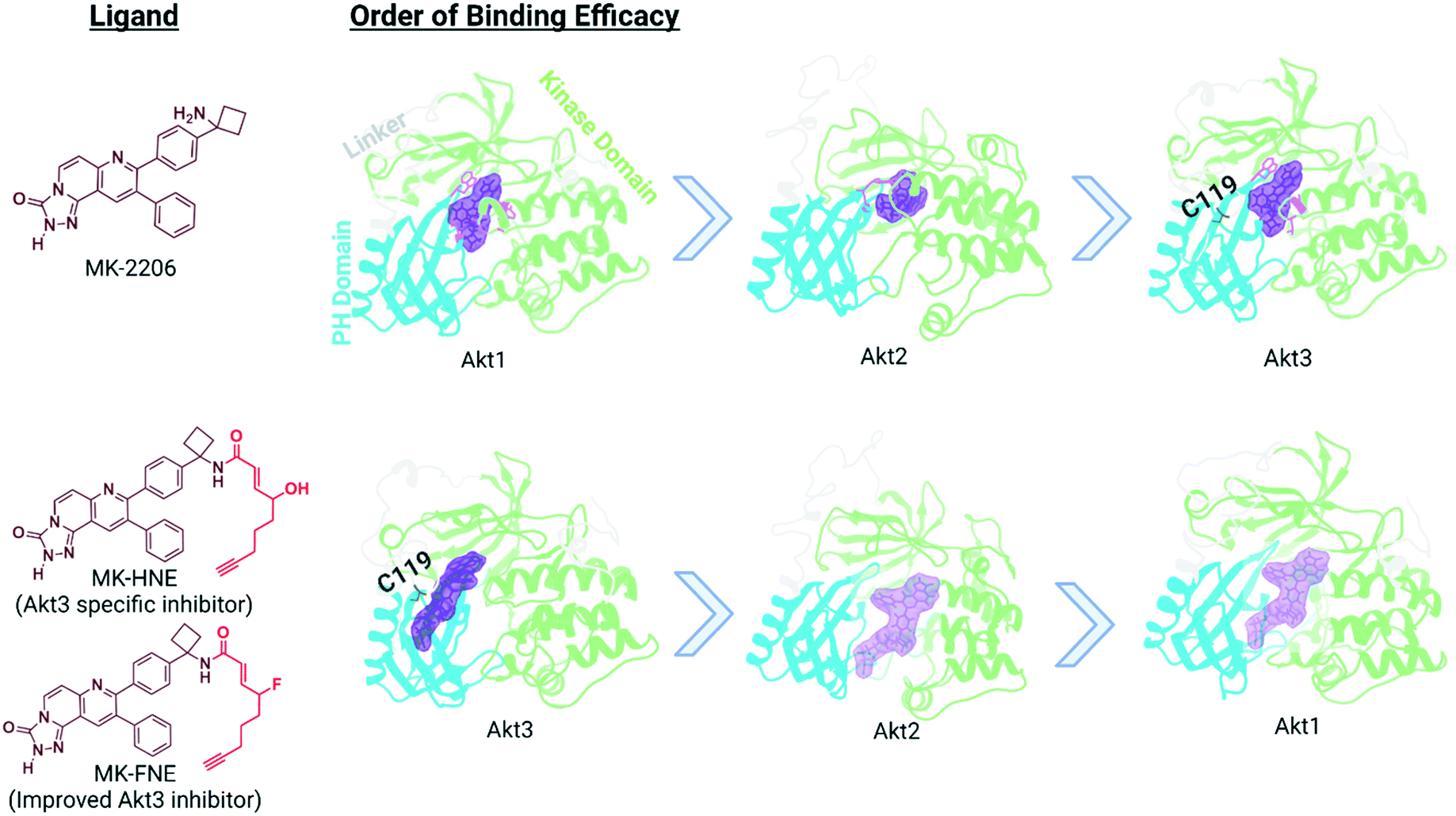 | ||
| Fig. 5 Binding efficacy of MK-2206 and MK-H(F)NE to three different isoforms of Akt: Akt1 (PDB 3O96), Akt2 homology model based on AKT1 crystal structure (PDB 6HHG), and Akt3 homology model based on Akt1 crystal structure (PDB 3O96). The domains for each isoform of Akt is color-coded: pleckstrin homology domain in blue, linker region in light gray, kinase domain in light green, and proposed allosteric binding site in purple.59 Based upon our studies, the binding of MK-HNE and second generation MK-FNE has a reversal in binding efficacy compared to MK-2206, due to the covalent interaction between the electrophilic Michael acceptor motif (red in structure) and C119. | ||
T-REX studies of Akt3 labeling had shown that Akt(T305) phosphorylation was unaffected by HNEylation. Conversely, MK-2206 treatment of cells reduced Akt3(T305) phosphorylation. When we examined Akt3(T305) phosphorylation after Mk-FNE treatment, we saw no change, despite Akt3 inhibition being significant. Thus, the mode of action of MK-FNE is dominated by electrophile–protein engagement rather than MK-2206 engagement.
Going forward
From the above experiments we learned that HNE labeling of Akt3 is rapid, DN, and inhibits Akt3 activity: this profile is not common to other Akt isoforms. From these observations, we elected to develop an Akt3-selective candidate molecule, that emerged to be quite selective for Akt3. T-REX, therefore, can inform on covalent drug design, and further can predict bespoke traits of the electrophile–protein interaction, while also informing on target profiles. These points are encouraging for Akt3-selective inhibitor design. It is important to consider what aspects of T-REX assisted in making such data possible and further what aspects of this strategy could be improved or could be better leveraged in the future.Akt3's HNE sensitivity was discovered using a medium-throughput T-REX screen. This is an arduous process that is likely not possible on the whole proteome and further compounds inherent limitations of T-REX. We have now established G-REX,60,61 a method for unbiased profiling of electrophile-sensitive proteins. Hits from G-REX will be fed into our pipeline later. However, generally, any electrophile panning program can be used to generate candidate PFRs for study by T-REX, and then precision drug design as we outline. We indeed encourage laboratories claiming to have discovered HNE-sensitive proteins to interrogate HNE-labeling and phenotypes induced by unambiguous labeling prior to making such claims.
The ramifications of Akt3-HNE labeling were evaluated rigorously in cell culture, relative to a defunct mutant and relative to an isoform that did not sense electrophiles. These experiments were also repeated in zebrafish, where similar labeling and signaling were observed, in an isoform-specific manner. Showing robustness of phenotypes and transmission is certainly important prior to going forward to drug discovery. However, such experiments would ideally be carried out in the model organism of choice for drug testing, which in our case, as for many preclinical studies, was mice. Unfortunately, mice are not applicable to T-REX as our photouncaging methodology is not compatible with whole mice. T-REX may, be applicable to specific tissues, or organs, and circulating cells, like blood cells. Time will tell how important advanced T-REX testing be for triaging hybrid molecules.
The effects of T-REX and MK-FNE were studied at the level of Akt activity, Akt3 phosphorylation, various immediate downstream signaling processes, and occupancy. These agreed well and together provide a very convincing picture that the HNE-like moiety is the determinant of MK-FNE activity, in terms of how MK-FNE impacts Akt3 (isoform selectivity, DN, and phosphosite modification). However, we did not develop a detailed picture of the changes in transcriptome, chromatin, etc., that occur upon Akt3 T-REX. These could have been rigorously compared with MK-2206 treatment, and the hybrid molecule. Leveraging deep sequencing methods would allow establishment of more quantitative signatures arising due to target engagement. Comparison of signatures caused upon T-REX to those from drug candidates could also allow testing of on-target behavior in a more detailed and holistic manner that could be expanded to other tissues and models. Our early experiments in cells and fish,68 indicate the T-REX protocol is applicable to RNA-seq experiments, although these are best performed against a sensor cysteine mutant control, or relative to release of a molecule that does not trigger expected phenotypes. These studies have been used to uncover hitherto unknown modes of action of approved drugs, effectively the inverse of the strategy we propose for drug design.68 Of course, given T-REX can only achieve low occupancy on the target, comparisons with saturating concentrations of a drug, may prove to be difficult to draw in some instances. This concern dwindles the more dominant the signaling phenotypes and the higher the occupancy achievable by T-REX. Clearly, such sensors are the most likely to be of use for drug mining.
Typically, the root cause of inhibition in classic inhibitor designs is derived from the non-covalent interaction; in most cases it is unknown how covalent electrophile labeling of the cysteine affects activity (although in some cases oxidation of the targeted residue stimulates activity62). What is clear is that targeting PFRs is accessible to scenarios in which non-covalent interaction with the target is not inhibitory. This is important because there has been an increase in assays screening for protein binders, yielding a wealth of binders reported in the literature. It will be important to test how effective such covalent inhibitors can be in the future.
The DE Akt3 was approximately 20%, and inhibition was DN.49 This DE is not the highest we have reported. However, DE(Akt3) is higher than several proteins we have studied, such as enzyme ribonucleotide reductase (RNR) and mRNA-binding protein HuR. For HuR, there was no change in activity upon electrophile labeling, even though similar DE stimulated Ube2V1 to promote Ube2N activity in cells. Such an outcome for HuR could arise due to: DE being too low; lack of/insufficiency of dominance of signaling; because the HuR activity measured was not affected by electrophile labeling; or possibly because of experiment-specific variations such as protein expression/expression of associated factors; etc. Whatever it be, as the phenotype engendered by the electrophilic moiety becomes less impactful, T-REX experiments may be less able to predict phenotypes. In such scenarios, it would be logical to change the electrophile released in a bid to improve signaling properties, which T-REX offers a means to achieve; we have already shown that even small changes in ligand can lead to large changes in phenotypic output even when labeling of the target protein is similar.68 Regardless of the effects of such SAR, there remain benefits in terms of targeting kinetically privileged cysteines, as they are inherently reactive, a property that will transpire to good inhibitor kinetics, although it is unknown how necessary such properties are to derive a ligandable covalent drug. Many covalent drugs have been developed without ostensibly targeting PFRs. Investigating the electrophile sensitivity and privileged signaling properties of cysteines targeted by approved drugs using a method like T-REX would also therefore be insightful.
The discussion above, nonetheless, shows it is important that threshold parameters be established for translating data from T-REX to a ligandable inhibitor. Although precise thresholds may not be realizable (due to mechanistic changes, locale-specific sensing effects, etc.), having a series of experimentally-validated criteria derived from numerous electrophile scaffolds and different PFRs could certainly be useful to triage hits from screening efforts. We now possess enough proteins to perform such experiments, although not all of these have accommodating reversible ligands. Akt, fortunately is the target of numerous drugs, and HNE(FNE)-derivatization of these molecules could readily inform on how Akt isoform affinity/selectivity affects drug efficacy and isoform selectivity. Similar chemical structural changes carried out across a series of ligandable interactions could inform other variables, such as linker preferences, although these are more likely to be idiosyncratic, as has been found for many chimeric drugs.
Another clear issue going forward is how well is the molecular design able to suppress HNE's pleotropic tendencies? We know that kinetically we have reduced the inherent reactivity of HNE by several orders of magnitude by converting the aldehyde to an amide. The kinact/ki for Akt3/MK-FNE is ∼104 M−1 s−1, which is ∼6 orders of magnitude faster than the standard rate of a β-substituted enamide with a cysteine.2,49,52 This is close to the maximum rate enhancement possible for HNE (108-fold) with a protein of interest. The kinact/ki is also faster than any second order rate reported for reaction between HNE and a target protein, although these rates are poorly studied in general, and we may never know all the best sensors. For MK-FNE we evaluated off-target effects both in mice (these were less than MK-2206,) and positive synergy of MK-FNE was observed only with Akt3 knockdown, whereas positive synergy was only observed with Akt1 for MK2206 in cell culture. As our T-REX studies had shown that C119 within Akt3 was the cysteine attacked by MK-HNE/FNE we were also able to perform some experiments with Akt3(C119S), which is resistant to covalent targeting by MK-FNE/HNE. Prior to advanced testing, performing experiments on xenografts of cells expressing Akt3(C119S) at endogenous levels would be an important step.
A related issue is, is HNE a good handle for drug design? It is true that the hydrophobic portion of HNE is not present in many drugs, although similar functions are present in approved drugs such as fulvestrant,63 Symmetrel64 and Flumadine,65 and tamoxifen.66 Click staining of xenografts treated with MK-FNE also showed significant permeation into the tumor post oral gavage. However, how hydrophobic tails affect tissue and subcellular distribution is another issue we have yet to address.
Conclusion
We therefore believe that we have provided good evidence that biological activity can be modulated in a DN fashion through specific covalent engagement with a specific electrophile.67 If this engagement be ushered through a ligandable interaction, even if that ligandable interaction should lead to measurable effects on the target protein, the covalent handle can exert its effects in a manner predictable based on data derived from studying protein-specific electrophile labeling. Using T-REX or other strategies that may arise to model protein specific electrophile engagement we can hone our resources to focus on the most responsive target proteins, and interrogate off-target effects in ways that were not previously possible. Such insights are also important, as assessing targets and on-target effects of drugs continues to be a complex process.Author contributions
Concept and outline: Y. A.; writing: M. J. C. L., Y. A.; figures: P. L.; figure legends: P. L., Y. A.; final proof-editing: all authors.Conflicts of interest
REX-technologies and inhibitors derived from their applications have been filed for US-patent applications by the authors' former institution, Cornell University.Acknowledgements
This research was supported by Swiss National Science Foundation (SNSF) Project funding (184729) and EPFL. Figures were made with http://BioRender.com. All procedures at Cornell University were approved by the Institutional Animal Care and Use Committee (IACUC) and performed in accordance with the guidelines of the NIH. All procedures at EPFL were performed in accordance with the Swiss regulations on animal experimentation (Animal Welfare Act SR 455 and Animal Welfare Ordinance SR 455.1), in the EPFL zebrafish unit, cantonal veterinary authorization VD-H23.References
- M. J. C. Long, Y. Zhao and Y. Aye, RSC Chem. Biol., 2020, 1, 42–55 RSC.
- Key Reference: S. Parvez, M. J. C. Long, J. R. Poganik and Y. Aye, Chem. Rev., 2018, 118, 8798–8888 CrossRef CAS PubMed.
- Redox Biochemistry, ed. R. Banerjee, D. F. Becker, M. B. Dickman, V. N. Gladyshev and S. W. Ragsdale, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2007 Search PubMed.
- J. Wilhelm, S. Kühn, M. Tarnawski, G. Gotthard, J. Tünnermann, T. Tänzer, J. Karpenko, N. Mertes, L. Xue, U. Uhrig, J. Reinstein, J. Hiblot and K. Johnsson, Biochemistry, 2021, 60, 2560–2575 CrossRef CAS PubMed.
- Key Reference: M. J. C. Long, J. R. Poganik and Y. Aye, J. Am. Chem. Soc., 2016, 138, 3610–3622 CrossRef CAS PubMed.
- Key Reference: J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, Nat. Rev. Drug Discovery, 2011, 10, 307–317 CrossRef CAS PubMed.
- A. K. Ghosh, I. Samanta, A. Mondal and W. R. Liu, ChemMedChem, 2019, 14, 889–906 CrossRef CAS PubMed.
- S. De Cesco, J. Kurian, C. Dufresne, A. K. Mittermaier and N. Moitessier, Eur. J. Med. Chem., 2017, 138, 96–114 CrossRef CAS PubMed.
- R. A. Bauer, Drug Discovery Today, 2015, 20, 1061–1073 CrossRef CAS PubMed.
- P. A. Schwartz, P. Kuzmic, J. Solowiej, S. Bergqvist, B. Bolanos, C. Almaden, A. Nagata, K. Ryan, J. Feng, D. Dalvie, J. C. Kath, M. Xu, R. Wani and B. W. Murray, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 173–178 CrossRef CAS PubMed.
- M. J. C. Long, A. Kulkarni and Y. Aye, ChemBioChem, 2021, 22 DOI:10.1002/cbic.202100051.
- J. M. Ostrem, U. Peters, M. L. Sos, J. A. Wells and K. M. Shokat, Nature, 2013, 503, 548–551 CrossRef CAS PubMed.
- R. S. Goody, M. P. Müller and D. Rauh, Cell Chem. Biol., 2019, 26, 1338–1348 CrossRef CAS PubMed.
- D. Y. Yoo, A. D. Hauser, S. T. Joy, D. Bar-Sagi and P. S. Arora, ACS Chem. Biol., 2020, 15, 1604–1612 CrossRef CAS PubMed.
- R. T. Dungo and G. M. Keating, Drugs, 2013, 73, 1503–1515 CrossRef CAS PubMed.
- Y. Guo, Y. Liu, N. Hu, D. Yu, C. Zhou, G. Shi, B. Zhang, M. Wei, J. Liu, L. Luo, Z. Tang, H. Song, Y. Guo, X. Liu, D. Su, S. Zhang, X. Song, X. Zhou, Y. Hong, S. Chen, Z. Cheng, S. Young, Q. Wei, H. Wang, Q. Wang, L. Lv, F. Wang, H. Xu, H. Sun, H. Xing, N. Li, W. Zhang, Z. Wang, G. Liu, Z. Sun, D. Zhou, W. Li, L. Liu, L. Wang and Z. Wang, J. Med. Chem., 2019, 62, 7923–7940 CrossRef CAS PubMed.
- Y. Y. Syed, Drugs, 2020, 80, 91–97 CrossRef CAS PubMed.
- B. Metcalf, C. Chuang, K. Dufu, M. P. Patel, A. Silva-Garcia, C. Johnson, Q. Lu, J. R. Partridge, L. Patskovska, Y. Patskovsky, S. C. Almo, M. P. Jacobson, L. Hua, Q. Xu, S. L. Gwaltney, C. Yee, J. Harris, B. P. Morgan, J. James, D. Xu, A. Hutchaleelaha, K. Paulvannan, D. Oksenberg and Z. Li, ACS Med. Chem. Lett., 2017, 8, 321–326 CrossRef CAS PubMed.
- J. Han, S. L. Saraf and V. R. Gordeuk, Pharmacotherapy, 2020, 40, 525–534 CrossRef CAS PubMed.
- A. Hutchaleelaha, M. Patel, C. Washington, V. Siu, E. Allen, D. Oksenberg, D. D. Gretler, T. Mant and J. Lehrer-Graiwer, Br. J. Clin. Pharmacol., 2019, 85, 1290–1302 CrossRef CAS PubMed.
- A. A. Taylor, J. Biol. Chem., 2017, 292, 12008–12009 CrossRef CAS PubMed.
- Key Reference: K. Mitra, W. Kang, K. Tanis, S. Pacchione, M. Su, Z. Wang, K. Bleicher, T. Griffiths, G. Laws, M. Kuhls, A. Podtelezhnikov, J. Monroe, T. Johnson, D. Marsh, I. Knemeyer, Q. Chen, J. Lebron and F. Sistare, Drug Metab. Pharmacokinet., 2019, 34, S42 CrossRef.
- J. R. Poganik and Y. Aye, Front. Aging Neurosci., 2020, 12, 1 CrossRef CAS PubMed.
- X. Liu, M. J. C. Long and Y. Aye, Trends Biochem. Sci., 2019, 44, 75–89 CrossRef CAS PubMed.
- W. F. Eanes, T. J. S. Merritt, J. M. Flowers, S. Kumagai, E. Sezgin and C.-T. Zhu, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19413–19418 CrossRef CAS PubMed.
- J. R. Wiśniewski, M. Y. Hein, J. Cox and M. Mann, Mol. Cell. Proteomics, 2014, 13, 3497–3506 CrossRef PubMed.
- R. Milo, P. Jorgensen, U. Moran, G. Weber and M. Springer, Nucleic Acids Res., 2010, 38, D750–D753 CrossRef CAS PubMed.
- M. J. C. Long, J. R. Poganik, S. Ghosh and Y. Aye, ACS Chem. Biol., 2017, 12, 586–600 CrossRef CAS PubMed.
- M. J. C. Long, L. Wang and Y. Aye, Antioxid. Redox Signaling, 2020, 33, 1077–1091 CrossRef CAS PubMed.
- X. Fang, Y. Fu, M. J. C. Long, J. A. Haegele, E. J. Ge, S. Parvez and Y. Aye, J. Am. Chem. Soc., 2013, 135, 14496–14499 CrossRef CAS PubMed.
- S. Parvez, Y. Fu, J. Li, M. J. C. Long, H.-Y. Lin, D. K. Lee, G. S. Hu and Y. Aye, J. Am. Chem. Soc., 2015, 137, 10–13 CrossRef CAS PubMed.
- R. A. Kulkarni, D. W. Bak, D. Wei, S. E. Bergholtz, C. A. Briney, J. H. Shrimp, A. Alpsoy, A. L. Thorpe, A. E. Bavari, D. R. Crooks, M. Levy, L. Florens, M. P. Washburn, N. Frizzell, E. C. Dykhuizen, E. Weerapana, W. M. Linehan and J. L. Meier, Nat. Chem. Biol., 2019, 15, 391–400 CrossRef CAS PubMed.
- W. Qin, Y. Zhang, H. Tang, D. Liu, Y. Chen, Y. Liu and C. Wang, J. Am. Chem. Soc., 2020, 142, 10894–10898 CrossRef CAS PubMed.
- I. Kastrati, M. I. Siklos, E. L. Calderon-Gierszal, L. El-Shennawy, G. Georgieva, E. N. Thayer, G. R. J. Thatcher and J. Frasor, J. Biol. Chem., 2016, 291, 3639–3647 CrossRef CAS PubMed.
- B. P. Mihalas, G. N. De Iuliis, K. A. Redgrove, E. A. McLaughlin and B. Nixon, Sci. Rep., 2017, 7, 6247 CrossRef PubMed.
- H. Zhang, N. Lyn, A. Haghani and H. J. Forman, in Aging, ed. S. P. Curran, Springer US, New York, NY, 2020, vol. 2144, pp. 237–244 Search PubMed.
- Key Reference: C. Wang, E. Weerapana, M. M. Blewett and B. F. Cravatt, Nat. Methods, 2014, 11, 79–85 CrossRef CAS PubMed.
- M. Golizeh, T. Geib and L. Sleno, Rapid Commun. Mass Spectrom., 2016, 30, 1488–1494 CrossRef CAS PubMed.
- S. G. Codreanu, B. Zhang, S. M. Sobecki, D. D. Billheimer and D. C. Liebler, Mol. Cell. Proteomics, 2009, 8, 670–680 CrossRef CAS PubMed.
- Y. Chen, Y. Cong, B. Quan, T. Lan, X. Chu, Z. Ye, X. Hou and C. Wang, Redox Biol., 2017, 12, 712–718 CrossRef CAS PubMed.
- B. K. Chacko, S. B. Wall, P. A. Kramer, S. Ravi, T. Mitchell, M. S. Johnson, L. Wilson, S. Barnes, A. Landar and V. M. Darley-Usmar, Redox Biol., 2016, 9, 57–66 CrossRef CAS PubMed.
- T. D. Cummins, A. N. Higdon, P. A. Kramer, B. K. Chacko, D. W. Riggs, J. K. Salabei, L. J. Dell'Italia, J. Zhang, V. M. Darley-Usmar and B. G. Hill, Free Radical Biol. Med., 2013, 59, 56–68 CrossRef CAS PubMed.
- S. Parvez, M. J. C. Long, H.-Y. Lin, Y. Zhao, J. A. Haegele, V. N. Pham, D. K. Lee and Y. Aye, Nat. Protoc., 2016, 11, 2328–2356 CrossRef CAS PubMed.
- S. L. Surya, M. J. C. Long, D. A. Urul, Y. Zhao, E. J. Mercer, I. M. EIsaid, T. Evans and Y. Aye, ACS Chem. Biol., 2018, 13, 1824–1831 CrossRef CAS PubMed.
- J. R. Poganik, A. K. Van Hall-Beauvais, M. J. C. Long, M. T. Disare, Y. Zhao and Y. Aye, HCA, 2020, 103 DOI:10.1002/hlca.202000041.
- H.-Y. Lin, J. A. Haegele, M. T. Disare, Q. Lin and Y. Aye, J. Am. Chem. Soc., 2015, 137, 6232–6244 CrossRef CAS PubMed.
- M. J. Long, H.-Y. Lin, S. Parvez, Y. Zhao, J. R. Poganik, P. Huang and Y. Aye, Cell Chem. Biol., 2017, 24, 944–957.e7 CrossRef CAS PubMed.
- M. J. C. Long, D. A. Urul, S. Chawla, H.-Y. Lin, Y. Zhao, J. A. Haegele, Y. Wang and Y. Aye, Biochemistry, 2018, 57, 216–220 CrossRef CAS PubMed.
- Key Reference: M. J. C. Long, S. Parvez, Y. Zhao, S. L. Surya, Y. Wang, S. Zhang and Y. Aye, Nat. Chem. Biol., 2017, 13, 333–338 CrossRef CAS PubMed.
- J. R. Poganik, M. J. C. Long, M. T. Disare, X. Liu, S.-H. Chang, T. Hla and Y. Aye, FASEB J., 2019, 33, 14636–14652 CrossRef CAS PubMed.
- Y. Zhao, M. J. C. Long, Y. Wang, S. Zhang and Y. Aye, ACS Cent. Sci., 2018, 4, 246–259 CrossRef CAS PubMed.
- Key Reference: X. Liu, M. J. C. Long, B. D. Hopkins, C. Luo, L. Wang and Y. Aye, ACS Cent. Sci., 2020, 6, 892–902 CrossRef CAS PubMed.
- M. J. C. Long and Y. Aye, Cell Chem. Biol., 2017, 24, 787–800 CrossRef CAS PubMed.
- F. André, E. Ciruelos, G. Rubovszky, M. Campone, S. Loibl, H. S. Rugo, H. Iwata, P. Conte, I. A. Mayer, B. Kaufman, T. Yamashita, Y.-S. Lu, K. Inoue, M. Takahashi, Z. Pápai, A.-S. Longin, D. Mills, C. Wilke, S. Hirawat and D. Juric, N. Engl. J. Med., 2019, 380, 1929–1940 CrossRef PubMed.
- C. X. Ma, V. Suman, M. P. Goetz, D. Northfelt, M. E. Burkard, F. Ademuyiwa, M. Naughton, J. Margenthaler, R. Aft, R. Gray, A. Tevaarwerk, L. Wilke, T. Haddad, T. Moynihan, C. Loprinzi, T. Hieken, E. K. Barnell, Z. L. Skidmore, Y.-Y. Feng, K. Krysiak, J. Hoog, Z. Guo, L. Nehring, K. B. Wisinski, E. Mardis, I. S. Hagemann, K. Vij, S. Sanati, H. Al-Kateb, O. L. Griffith, M. Griffith, L. Doyle, C. Erlichman and M. J. Ellis, Clin. Cancer Res., 2017, 23, 6823–6832 CrossRef CAS PubMed.
- A. P. Myers, P. A. Konstantinopoulos, W. T. Barry, W. Luo, R. R. Broaddus, V. Makker, R. Drapkin, J. Liu, A. Doyle, N. S. Horowitz, F. Meric-Bernstam, M. Birrer, C. Aghajanian, R. L. Coleman, G. B. Mills, L. C. Cantley, U. A. Matulonis and S. N. Westin, Int. J. Cancer, 2020, 147, 413–422 CrossRef CAS PubMed.
- Y. Oki, M. Fanale, J. Romaguera, L. Fayad, N. Fowler, A. Copeland, F. Samaniego, L. W. Kwak, S. Neelapu, M. Wang, L. Feng and A. Younes, Br. J. Haematol., 2015, 171, 463–470 CrossRef CAS PubMed.
- U.S. Food and Drug Administration, 2020.
- M. Rehan, M. A. Beg, S. Parveen, G. A. Damanhouri and G. F. Zaher, PLoS One, 2014, 9, e109705 CrossRef PubMed.
- Y. Zhao, M. J. C. Long, Y. Wang, S. Zhang and Y. Aye, ACS Cent. Sci., 2018, 4, 246–259 CrossRef CAS PubMed.
- Key Reference: M. J. C. Long, C. Rogg and Y. Aye, Acc. Chem. Res., 2021, 54, 618–631 CrossRef CAS PubMed.
- C. E. Paulsen, T. H. Truong, F. J. Garcia, A. Homann, V. Gupta, S. E. Leonard and K. S. Carroll, Nat. Chem. Biol., 2012, 8, 57–64 CrossRef CAS PubMed.
- J. F. R. Robertson and M. Harrison, Br. J. Cancer, 2004, 90, S7–S10 CrossRef CAS PubMed.
- V. Vernier, Toxicol. Appl. Pharmacol., 1969, 15, 642–665 CrossRef CAS PubMed.
- A. M. Wallbank, Exp. Biol. Med., 1969, 131, 1025–1027 CrossRef CAS PubMed.
- M. Clemons, S. Danson and A. Howell, Cancer Treat. Rev., 2002, 28, 165–180 CrossRef CAS PubMed.
- M. J. C. Long, ChemBioChem, 2021, 22, 814–817 CrossRef CAS PubMed.
- J. R. Poganik, K. T. Huang, S. Parvez, Y. Zhao, S. Raja, M. J. C. Long and Y. Aye, Nat. Commun., 2021, 12, 5736 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |