
Deglycase-activity oriented screening to identify DJ-1 inhibitors†
Igor
Maksimovic
 ab,
Efrat
Finkin-Groner
c,
Yoshiyuki
Fukase
c,
Qingfei
Zheng
b,
Shan
Sun
c,
Mayako
Michino
c,
David J.
Huggins
cd,
Robert W.
Myers
c and
Yael
David
*abde
ab,
Efrat
Finkin-Groner
c,
Yoshiyuki
Fukase
c,
Qingfei
Zheng
b,
Shan
Sun
c,
Mayako
Michino
c,
David J.
Huggins
cd,
Robert W.
Myers
c and
Yael
David
*abde
aTri-Institutional PhD Program in Chemical Biology, New York, New York 10065, USA
bChemical Biology Program, Memorial Sloan Kettering Cancer Center, New York, New York 10065, USA. E-mail: davidshy@mskcc.org
cTri-Institutional Therapeutics Discovery Institute, 413 East 69th Street, New York, NY 10021, USA
dDepartment of Physiology, Biophysics and Systems Biology, Weill Cornell Medicine, New York, New York 10065, USA
eDepartment of Pharmacology, Weill Cornell Medicine, New York, New York 10065, USA
First published on 2nd June 2021
Abstract
The oncoprotein and Parkinson's disease-associated enzyme DJ-1/PARK7 has emerged as a promiscuous deglycase that can remove methylglyoxal-induced glycation adducts from both proteins and nucleotides. However, dissecting its structural and enzymatic functions remains a challenge due to the lack of potent, specific, and pharmacokinetically stable inhibitors targeting its catalytic site (including Cys106). To evaluate potential drug-like leads against DJ-1, we leveraged its deglycase activity in an enzyme-coupled, fluorescence lactate-detection assay based on the recent understanding of its deglycation mechanism. In addition, we developed assays to directly evaluate DJ-1's esterase activity using both colorimetric and fluorescent substrates. The resulting optimized assay was used to evaluate a library of potential reversible and irreversible DJ-1 inhibitors. The deglycase activity-oriented screening strategy described herein establishes a new platform for the discovery of potential anti-cancer drugs.
Introduction
The deposition of post-translational modifications (PTMs) on proteins is carefully regulated and crucial to cellular function.1 Enzymatically introduced and removed marks such as phosphorylation, ubiquitination, acetylation and methylation, are prevalent, dynamic, tightly controlled and well-studied.2 By contrast, their non-enzymatic counterparts, which are the result of reactive metabolites directly modifying amino acid side chains to form non-enzymatic covalent modifications (NECMs), are spontaneous, often irreversible, and understudied.3–5One such NECM, generally termed glycation, occurs through the chemical reaction between an amine-containing side chain and the carbonyl of a reducing sugar or a metabolic intermediate via the Maillard reaction.4–7 Thereafter, through multiple rearrangements, the marks form an array of adducts called advanced glycation end products (AGEs).8 These include cross links between proteins as well as with other cellular macromolecules. While some of the initial adducts can spontaneously hydrolyze or be susceptible to enzymatic regulation, many of the later AGE products, including the cross links, are irreversible and only expunged through turnover.3,4,9
Like other NECMs, glycation adducts can affect protein function by (1) occupying sites of other regulatory PTMs, (2) introducing alternative 3-dimensional structure that can alter its function and stability, or (3) changing the electrostatic topologies of protein surfaces affecting its capacity to interact with other cellular components.5,8,10
Due to their long half-lives and unstructured lysine and arginine rich tails, histone proteins are highly susceptible to accumulation of non-enzymatic modifications, with a resulting pathophysiological impact.11 This is a crucial property as due to their proximity to the genetic material, histone PTM patterns are key determinants of chromatin structure, function and subsequently cellular physiology.12,13 Indeed, histone glycation via ribose or the glycolytic by-product methylglyoxal (MGO), has been shown to affect PTM patterning and chromatin compaction. Specifically, an acute MGO exposure attenuates the electrostatic interactions between cationic histones and anionic DNA, causing chromatin decompaction and hyper-transcription, whereas a chronic or higher dose of MGO induces cross links between histones and DNA resulting in compacted chromatin and hypo-transcription.10,14
As methylglyoxal is an extremely reactive dicarbonyl, its cellular levels are heavily regulated enzymatically via the glyoxalase/glutathione scavenging cascade.15 Furthermore, early arginine MGO-glycation intermediates can be converted via PAD4 into citrulline.16 More generally, unmodified DNA and protein side chains can be regenerated by the enzyme DJ-1 (PARK7).10,14,17 DJ-1 is an oncoprotein with roles in oxidative stress sensing and cellular protection against redox damage.18,19 DJ-1 contains a highly conserved cysteine residue (Cys106, Fig. 1A), which in conjunction with a nearby histidine (His126), forms a catalytic dyad (Fig. 1B) enabling DJ-1 to function as a weak amidase (Fig. 2A).20,21 This amidase activity facilitates the removal of early DNA or protein MGO adducts. With increased metabolic rates and a reliance on anaerobic respiration through glycolysis (Warburg effect), cancer cells produce greater amounts of deleterious species such as MGO.22 To avoid the associated accumulated ROS damage, the cells depend on scavenging (e.g. glutathione/glyoxalase) or repair (e.g. DJ-1) systems.23 Cancer cells having reduced DJ-1 (via RNAi knock-down) are more susceptible to MGO-induced cell death, suggesting that DJ-1's deglycase activity plays a role in maintaining cancer cell survival.10,14,17
 | ||
| Fig. 1 DJ-1 is a multifunctional protein with deglycase activity. (A) DJ-1 has roles in ROS sensing and scavenging, as a transcription regulator, and was found to be an oncoprotein. (B) Crystallographic data shows reversible covalent binding of isatin inhibitor 1 to the DJ-1 active site Cys106, adjacent to catalytic dyad His126 and stabilized by nearby Glu18. | ||
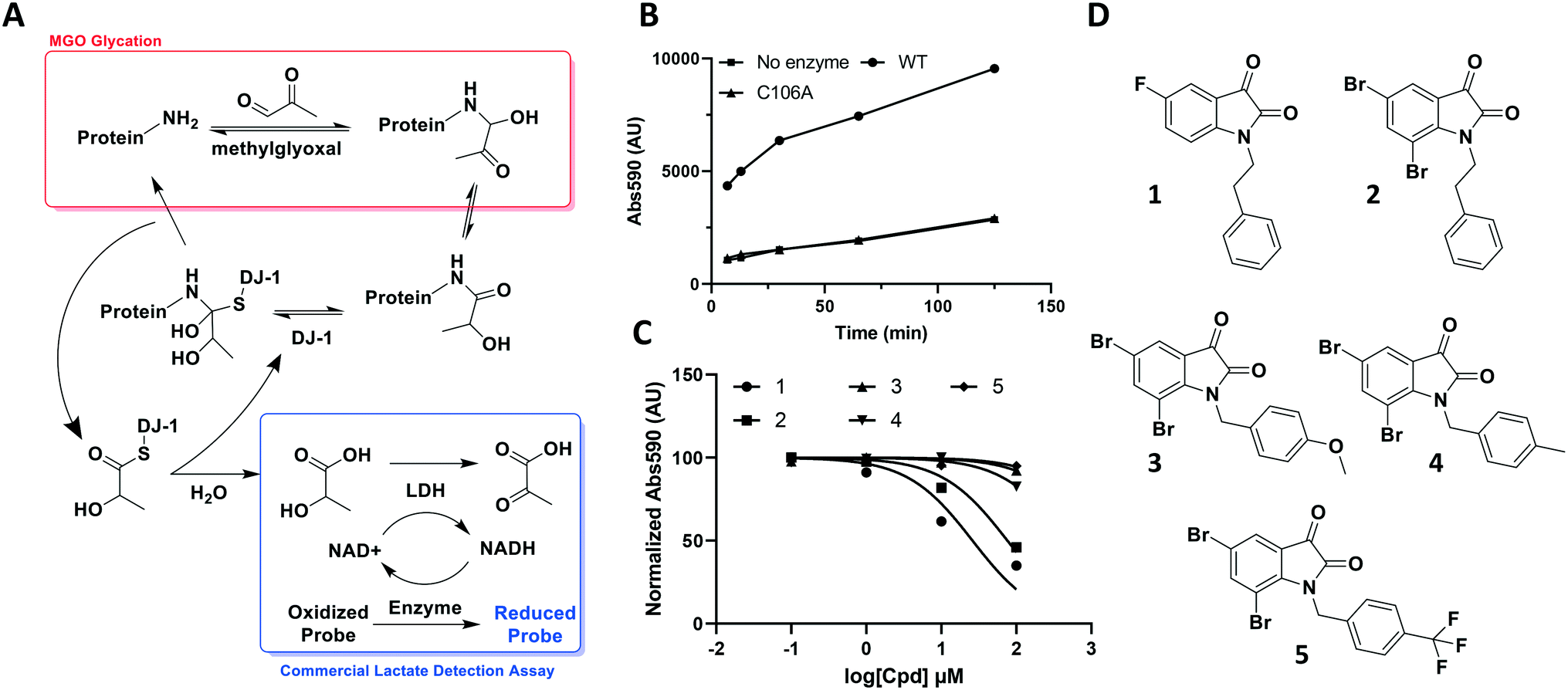 | ||
| Fig. 2 DJ-1's deglycase function can be leveraged to generate a reproducible readout to evaluate potential modulators. (A) Scheme showing the role of DJ-1 in protein deglycation, as well as detection of the lactate product by a commercial lactate detection assay. (B) Lactate assay signal/noise determination of detected absorbance against time after commercial assay mix addition. (C) Characterization of various isatin analogs as DJ-1 inhibitors using lactate assay. Normalized endpoint absorbance vs. log[cpd] (μM) (D) structures of compounds tested in (C). | ||
Following its implication as a potential target for anti-cancer therapy, the reactivity of Cys106 has recently been exploited in the identification of three covalent DJ-1 inhibitors. The first consists of an epoxide warhead (in addition to an alkyne group which enables click chemistry) appended to an optimized amino-epoxycyclohexenone-scaffold. This covalent, irreversible inhibitor was shown to selectively label reduced Cys106 in DJ-1 and enrich it in several cell lines based proteomic analyses.24 In addition, a fragment-based approach in conjunction with crystallographic structural studies was used to identify a covalent, reversible isatin-based DJ-1 inhibitor (Fig. 1B). The isatin compound was demonstrated to inhibit DJ-1's ability to reduce the formation of glyoxal glycation in live cells.25 Following these discoveries, a bis-isatin series of inhibitors were designed to target the DJ-1 homodimer. This bivalent scaffold was demonstrated to inhibit cancer cell growth both in vitro and via intratumor injection in mouse xenografts.26 Together, these compounds demonstrate the potential for targeting this system and the need for a powerful and reliable assay to evaluate new candidate scaffolds against DJ-1.
Here, we report the development of a simple, robust, and continuous fluorescence-based assay that monitors the esterase activity of DJ-1. We further demonstrate that the active site Cys106 is required for this activity. We utilize this assay, in conjunction with structure–activity relationship and in silico modeling, to successfully evaluate a small library of potential DJ-1 modulators and report several novel isatin-based reversible and irreversible inhibitors.
Results and discussion
The aldehyde of methylglyoxal is susceptible to nucleophilic attack by protein side chains such as the ε-amine of lysine or the guanidino arginine tails, and nucleic acid bases such as deoxyguanosine. Once the initial amino carbinol is formed, the reaction cascades via a Schiff base intermediate into a stabilized Amadori product. Numerous rearrangement and crosslinking reactions then occur, introducing complexity through a plethora of modifications and decreasing the likelihood of the reaction being reversed.27 However, in its nascent stage, the modification is still labile and vulnerable to reversal by housekeeping enzymes such as DJ-1.Through its catalytically active Cys106, which is nestled in a nucleophilic elbow and activated by nearby His126, DJ-1 is capable of forming a hemithioacetal with unstable early MGO-modifications.21 The unmodified macromolecule is liberated upon collapse of the tetrahedral intermediate and the resulting DJ-1-MGO thioester is hydrolyzed to yield L/D lactate and regenerated DJ-1.17,23 Initially, we sought to take advantage of this biochemical mechanism and quantitate DJ-1 activity using a commercial fluorescence-based NADH-coupled L-lactate assay kit (Fig. 2A). To this end, we incubated 50 μM DJ-1 directly with 5 mM MGO for 20 min at 37 °C to effect enzyme glycation and subsequent intermolecular deglycation before adding the assay mix. We validated that DJ-1 was capable of generating lactate at a materially higher rate than its catalytically inactive C106A mutant (Fig. 2B). These data show that there is a signal “burst” up to 5 minutes after adding the assay mix that corresponds to lactate release from the initial incubation of DJ-1 with the MGO that had built up. Thereafter, a new steady state of lactate production by DJ-1 is achieved. As expected, both of these kinetic states are higher with the wild type enzyme than with the catalytically dead mutant or in the absence of enzyme. Using this assay, we evaluated a small series of isatin analogs, including one of the published compounds (compound 1, Fig. S1,†2C, and 2D) by incubating the compounds with DJ-1 for 20 min before adding MGO for additional 20 min and then, the lactate detection reagents as described above. Two compounds (1 and 2) showed significant reductions in DJ-1 activity upon addition. These data, along with the inactivity of C106A mutant, confirm that the L-lactate production was catalysed by DJ-1.
While these results affirmed that quantitating lactate production is a viable method for assaying DJ-1 activity, this approach suffered from several drawbacks. First, the high concentrations of enzyme (50 μM) required in the assay render the minimum potential IC50 too high, making it impossible to measure Ki values below half of the [E] (25 μM). Secondly, DJ-1 is both the enzyme and the substrate which makes varying their concentrations independently as well as determining the Km for the substrate impossible. Additionally, despite utilizing a fluorescent readout, which is expected to increase assay sensitivity, even under optimized conditions the signal to noise ratio for active vs. catalytically inactive DJ-1 is modest at 4-fold. Indeed, the new compounds that were screened did not show similar potency to the reported compounds, which suggests limited quantitative reliability of the assay and rendering it inappropriate for a rigorous medicinal chemistry campaign.25
To address these limitations, we turned toward an ester-based substrate, para-nitrophenyl acetate (6, PNPAc), which hydrolysis releases a conjugated phenolate that absorbs light at 405 nm (Fig. 3A).28 DJ-1 was mixed with assay buffer containing the PNPAc substrate and the reaction was monitored continuously at 405 nm using a plate-based spectro-photometer for 30 minutes to quantitate PNP production. A titration of the hydrolysed substrate against its absorbance was measured to determine the absolute value of each absorbance unit recorded (Fig. S2†). We subsequently optimized the assay by iteratively titrating the enzyme and then the substrate to calculate the enzyme's KM and kcat with PNPAc (Fig. 3B, and Table 1). Thereafter, we evaluated the same collection of isatin analogs and established that these compounds are indeed effective at inhibiting DJ-1 activity, in line with previous reports (Fig. S3† and 3C). While this continuous assay provided a fast, inexpensive, coupling enzyme-free approach, it still required too high enzyme concentrations, was limited by a modest dynamic range (due to non-enzymatic PNPAc hydrolysis and low absolute absorbance), and by the fact that certain compounds may absorb within the assay range.
 | ||
| Fig. 3 Continuous absorbance assay to quantitate the DJ-1 esterase activity. (A) Para-nitrophenyl acetate substrate and visible phenolate. (B) Michaelis–Menten analysis to determine DJ-1 enzymatic parameters. Data reported N = 2. (C) Use of assay to characterize isatin-based inhibitors. | ||
| Substrate | [DJ-1] (nM) | K M (μM) | V max (pM s−1) | k cat/KM (s−1 M−1) |
|---|---|---|---|---|
| PNPAc | 550 | 2850 ± 250 | 47.2 ± 1.6 | 0.296 ± 0.028 |
| DiFMUAc | 550 | 151 ± 17 | 713 ± 32 | 84.2 ± 10.0 |
In an effort to further improve assay sensitivity, we explored a use of a fluorescent substrate, namely 4-methylumbelliferyl acetate (MUAc).29 DJ-1 was incubated with the substrate and product release was monitored by fluorescence (excitation: 358 nm, emission: 455 nm). Although we observed product formation using wild type DJ-1 but not the C106A mutant, we recognized that the near neutral pH of our assay was too low for maximal assay sensitivity under continuous monitoring conditions (data not shown). While MU-phenolate is highly fluorescent, the phenol pKa (7.8) is higher than the assay pH such resulting in a significant portion of the product being present in a protonated and less-fluorescent form under the reaction conditions. Increasing the assay pH not only poorly reflects physiological conditions but also increases the rate of non-enzymatic substrate hydrolysis, decreasing the assay signal to noise ratio.
To overcome these issues, we utilized a modification of the MUAc substrate in which the phenolic carbon is flanked on each side by an electronegative fluoride and designate this compound as 6,8-difluoro-4-methylumbellliferyl acetate (7, DiFMUAc, Fig. 4A).30 The inductive effect of the fluorides stabilizes the enolate tautomer, reducing the pKa of the DiFMU phenol to 4.7. Thus, at physiological pH, the reaction product is fully ionized, resulting in the expected increase in absolute fluorescence and signal to noise sensitivity (Fig. S4†). We performed a titration experiment of the hydrolysed analog of DiFMUAc (DiFMU) to quantify the fluorescent signal recorded (Fig. S5†). Using this novel substrate in continuous assay format, we optimized the individual kinetic assay parameters through iterative enzyme and substrate titrations to calculate the KM and kcat of the enzyme with this substrate (Fig. 4B). Notably, these values correlate well with previously reported kinetic parameters of DJ-1 with other substrates as well.31,32 The DiFMUAc substrate was found to have a 19-fold lower KM for DJ-1 and a 15-fold higher Vmaxvs. PNPAc, resulting in a 280-fold higher kcat/KM (Table 1).
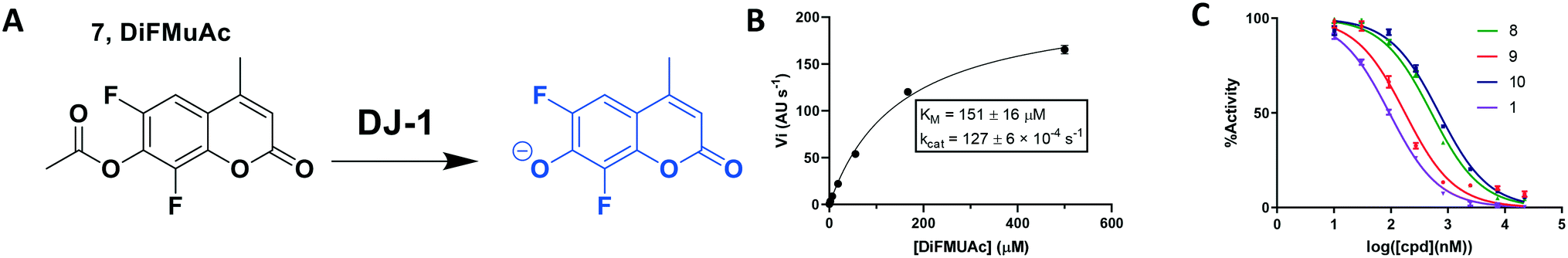 | ||
| Fig. 4 Kinetic fluorescent assay to quantitate DJ-1 esterase activity. (A) 6,8-Difluoro-4-methylumbelliferyl acetate (7, DiFMUAc) substrate and fluorescent phenolate. (B) Michaelis–Menten analysis to determine substrate/enzyme parameters for DIFMUAc. N = 2. (C) Inhibition of DJ-1 following a 60 min pre-incubation with the compounds noted. | ||
Using this optimized in vitro assay, we initiated expanded SAR studies on isatin-analogs and other reported DJ-1 inhibitors (Fig. S6† and 4D). Our starting point was the isatin molecule (compound 1) that had been shown to have between 100 and 300 nM DJ-1 inhibitory and binding activities, respectively.25 We carefully analyzed previously reported SAR and crystallographic data of the free and isatin-bound active site. Given the apparent underutilization of the active site (inhibitor binding pocket) by the published isatin analogs, we reasoned that there was room for expansion in several positions around the isatin core. We focused on two key goals, namely occupying the entire active site pocket and probing the surface of the protein. To this end we synthesized and tested a series of isatin derivatives for DJ-1 inhibition, probing the effect of varying each position on the indole ring as well as determining the tolerance to larger changes (Table 2).
|
|
|||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | IC50 (μM) |
| 1 | CH2CH2Ph | F | H | H | 0.118 |
| 2 | CH2CH2Ph | Br | H | Br | 0.0626 |
| 3 | CH2Ph–p-OMe | Br | H | Br | 0.267 |
| 4 | CH2Ph–p-Me | Br | H | Br | 0.676 |
| 5 | CH2Ph–p-CF3 | Br | H | Br | 2.41 |
| 11 | CH2Me | F | H | H | 0.118 |
| 12 | CH2C(C2H4)Ph | F | H | H | 0.356 |
| 13 | CH2CH2C(Me)3 | F | H | H | 0.851 |
| 14 | CH2CH2BzImzMe | F | H | H | NA |
| 15 | CH2CH2Ph | OMe | H | H | 2.93 |
| 16 | CH2CH2Ph | COOH | H | H | 9.79 |
| 17 | CH2CH2Ph | CH2Ph | H | H | NA |
| 18 | CH2Me | CH2N4 | H | H | NA |
| 19 | CH2CH2Ph | H | CH2COOH | H | 2.35 |
| 20 | CH2CH2Ph | H | H | H | 0.701 |
| 21 | CH2CH2Ph | F | H | OMe | 0.0546 |
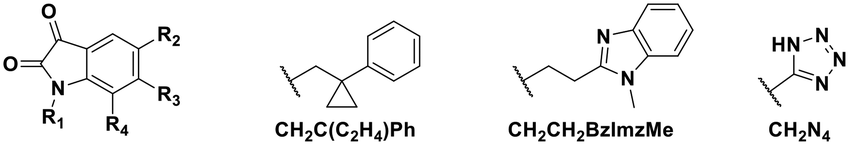
The crystal structure of known inhibitor compound 1 shows that the phenylethyl substituent on the isatin N1 protrudes through a Leu128- and Asn76-flanked gate into a narrow polar pocket formed between two alpha helices (Fig. 5A). Decreasing the size of this hydrophobic moiety to an ethyl group (compound 11) resulted in no change in inhibition, suggesting that the phenyl group contributes minimally to the binding. Similarly, constraining the N1 alkyl benzyl linker (compound 1) via the introduction of a bulky cyclopropyl substituent on the benzylic carbon of the phenylethyl group (compound 12) also had minimal effect on the inhibitory potency relative to compound 1 (3-fold change in binding). The docking model of 12 reveals that the cyclopropyl occupies the hydrophobic area but the narrow pocket cannot accommodate the phenyl ring which is subsequently ejected toward the solvent area (Fig. 5B). By contrast, replacing the phenyl group of 1 with a bulky t-butyl moiety (compound 13) resulted in a 7-fold drop in potency. Further replacement of the phenyl group of 1 with a bulky N-methyl benzimidazole substituent (compound 14) yielded an inactive compound. This effect was also observed when comparing compounds 2 and 4, where shortening of the phenyl ring linker while maintaining the overall length through a para-methyl group resulted in a 10-fold decrease in potency. Interestingly, the restrictions imposed by this channel may have been partially overcome through the inclusion of a para-methoxy (compound 3) instead of a para-methyl (compound 4) in modified linker class resulting in a 2.5-fold increase in potency over its more hydrophobic counterpart. However, replacing the methyl with a larger electron withdrawing trifluoromethyl (compound 5) caused a further 3.5-fold decrease in potency, which implies that the electronic state of the terminal phenyl group is important.
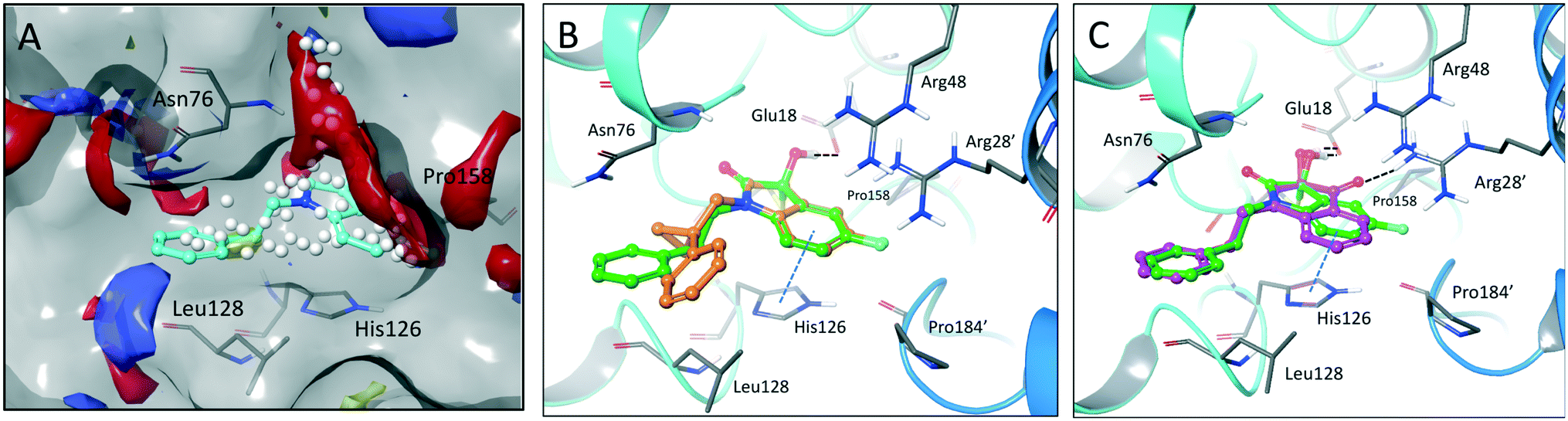 | ||
| Fig. 5 Binding site mapping and covalent docking models. (A) Overlay of the DJ-1 binding site map with the crystal structure of DJ-1 in complex with 1 (PDB: 6AFL). Yellow indicates hydrophobic area. Red and blue indicates areas favourable for hydrogen bond acceptors and donors respectively. White spheres delineate the size of the site. Superimposed docking poses of 12 (B, orange) and 9 (C, magenta) with the DJ-1 crystal structure in complex with 1. | ||
Examination of the crystallographic data for 1 demonstrates the existence of a small additional pocket between fluorine at C-5 of the isatin, and the nearby residues His126 and Pro158 (Fig. 5A). To leverage this cavity, we substituted the larger halide bromine at the C5 position. Addition of bromides at this C5 as well as the previously unexplored C7 (compound 2, Fig. S7†) resulted in a slight increase in potency over the published compound 1. The desfluoro analog of 1 (compound 20) was less active by ∼6-fold. Similarly, replacing the fluorine of 1 with a methoxy (compound 15) resulted in an almost 25-fold reduction in affinity whereas replacement with carboxylate (compound 16), benzyl (compound 17) and tetrazole (compound 18) functionalities led to >100-fold reductions in activity. Appending a carboxymethyl group to C6 (compound 19), with the aim of introducing an electrostatic interaction with positively charged Arg48, reduced the inhibitory potency by 3-fold (vs. compound 20). We were able to capitalize on the space between the isatin and the active site pocket by introducing a C7-methoxy group (compound 21, Fig. S7†), which resulted in a slight increase in inhibitory activity vs.1. Compounds 21 and 2 represent our most potent reversible DJ-1 inhibitors with IC50 values of approximately 50 nM.
The reversible covalent alpha-ketoamide functionality of the isatin scaffold is an inherent advantage in terms of selectivity and affinity for DJ-1 inhibition, however, it poses a potential issue for molecular longevity in vivo. To circumvent such potential PK issues as well as find more “potent” inhibitors, we sought to migrate away from the dicarbonyl motif and identify irreversible, covalent inhibitors of DJ-1. This strategy was supported by the previous identification of the amino-epoxycyclohexenone DJ-1 inhibitor (compound 8).24 We synthesized this inhibitor as well as several additional non-isatin analogs in which the C3 carbonyl was modified. Replacing the C3 carbonyl with cyanide (compound 22 010624), epoxide (compound 23 010710), dimethyl (compound 24 010904), or with geminal hydroxyl and methyl groups (compound 25 010708) yielded inactive compounds against DJ-1 (Fig. S8†).
While this might suggest that deviation from the isatin scaffold was not tolerated by DJ-1's active site, we were successful in synthesizing and evaluating another series of non-isatin inhibitors of DJ-1 (Table 3). Using our standard kinetic fluorescent assay, we initially observed only mild inhibition by all the compounds, including the published epoxycyclohexenone. However, modification of the assay format to include a 60–90 min preincubation of DJ-1 with the inhibitors at room temperature, resulted in a reduction of the IC50 values of all the compounds in Table 3 except for the isatin-derived molecules like 1 (Fig. S9†). The observed time-dependent inhibition suggested the possibility of covalent chemistry occurring between DJ-1 (Cys106) and the electrophilic warheads incorporated into these inhibitors. Decreases in the activity of DJ-1 in the absence of inhibitors was also observed as a result of the preincubation, likely due to oxidation of active site nucleophile Cys106 (no additional thiol reductants present in the assay buffer). The most potent covalent inhibitor in our set (compound 26) had a chlorine in place of the electrophilic C3 carbonyl of 1, to produce an electrophilic alpha-chloroamide and yielded a binding pose well accommodated to the pocket (Fig. S10†). Notably, shifting the reactive center one carbon away from the indole ring via a chlorovinyl (compound 27) reduced the compound activity 74-fold indicating that the compound is firmly wedged in the pocket prior to covalent modification. Adding a second chlorine atom at this position (compound 28) reduced the IC50 by 8-fold, possibly due to the increased steric bulk. Replacing the chlorine of 26 with a fluorine (compound 10) further reduced the inhibitory activity, possibly because it is a poorer leaving group than chlorine.
| Compound | IC50 (μM) | Structure |
|---|---|---|
| 8 | 0.51 |
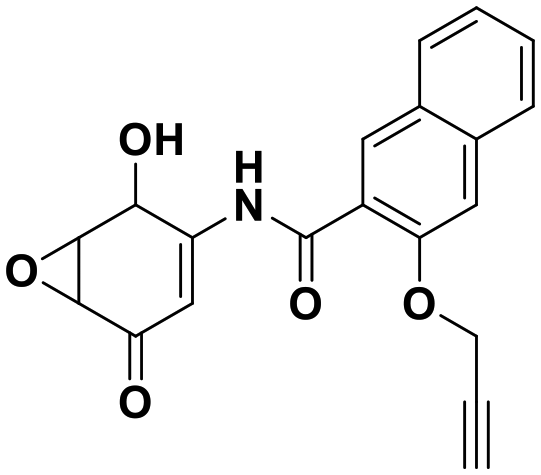
|
| 9 | 0.18 |
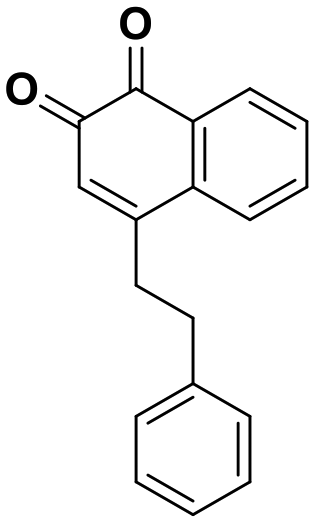
|
| 10 | 0.70 |

|
| 26 | 0.085 |
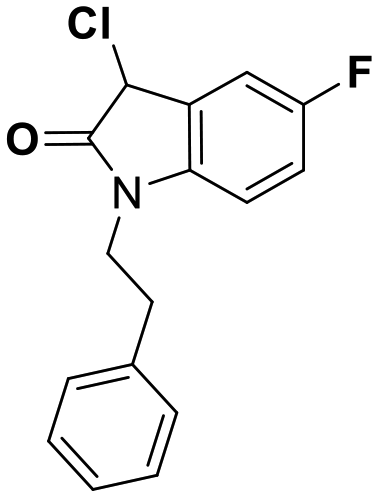
|
| 27 | 15 |
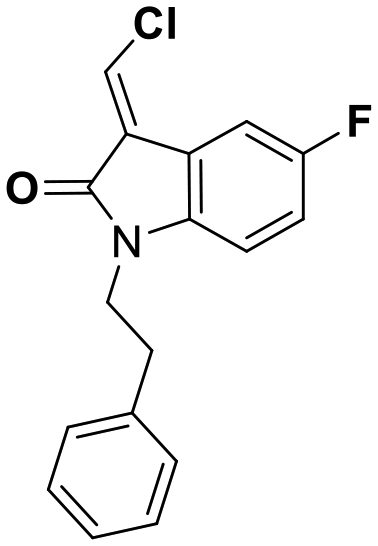
|
| 28 | 1.7 |
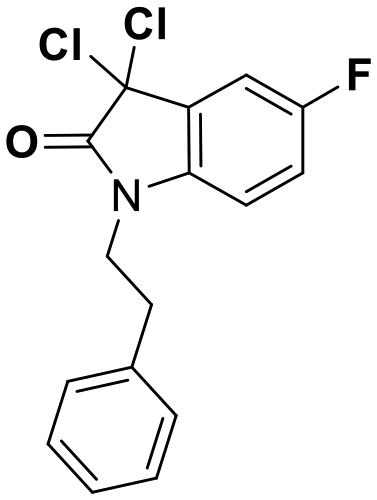
|
To evaluate whether these compounds formed reversible or irreversible covalent bonds with the Cys106, we incubated either negative control isatin 1, or positive control epoxycyclohexenone 8, as well as compounds 10, 26, 27, and 28 with DJ-1 for 2 hours, then removed the small molecules with size exclusion spin columns before analysing the protein's mass change by mass spectrometry. As expected, compounds 1 and 8 showed no change or a mass shift corresponding to an addition, respectively (Fig. S11A and B†). Compounds 10 and 26 exhibited the chemically expected mass shift which supports the claim that they are irreversible covalent molecules (Fig. S11C and D†). Conversely, compound 28 showed no change indicating that neither of the geminal chlorides can be displaced and the compound binds reversibly only (Fig. S11E†). However, compound 27 showed an idiosyncratic mass shift of +259.32 rather than the expected +267.30 which suggests that while the reaction is likely irreversible, a more complex conversion is occurring (Fig. S11F†).
Interestingly, expanding the indole fused ring into a naphthyl and removing the N1 nitrogen yielded a novel potent aromatic scaffold, compound 9. The covalent docking model of compound 9 in the DJ-1 active site revealed a similar binding pose to compound 1 with the C4 ethyl benzyl substituents of both compounds aligning almost perfectly (Fig. 5C). The nucleophilic Cys106 is predicted to form a covalent bond with the electrophilic C2 of compound 9 and π-interactions between the naphthoquinone ring and His126, in addition to hydrogen bonds between the ring and Glu18 and the C1 carbonyl and Arg28 that are predicted to stabilize the compound. The IC50 of compound 9 (0.18 μM) was similar (∼2-fold) to that of our most potent irreversible covalent inhibitor compound 26. This scaffold opens new chemical space with the potential for optimization into a more potent and pharmacokinetically stable DJ-1 inhibitor.
In summary, we identified deficiencies in existing DJ-1 assays and showed that the esterase activity of DJ-1 can be further exploited using DiFMUAc substrate to develop a simple, fluorescence-based continuous enzyme assay with increased sensitivity. We utilized this assay to define the kinetic parameters of DiFMUAc with DJ-1 and demonstrated its use as a versatile and potentially high-throughput tool for screening DJ-1 inhibitors. Additionally, we have expanded the SAR for isatin-analog based DJ-1 inhibitors, including the identification of a novel irreversible covalent inhibitor. These molecules, as well as the screening platform described herein, should greatly enhance our efforts to establish the potential of DJ-1 inhibitors as anti-cancer therapeutics.
Conclusions
The enzymatic activity of DJ-1 can be quantitated continuously and with increased sensitivity using DiFMUAc as an alternative esterase substrate yielding a fluorescent product. Using this assay, we discovered novel, potent isatin-based reversible and irreversible covalent inhibitors. Further work in developing these scaffolds, as well as determining and optimizing their selectivity will be crucial before these compounds may be used to leverage cancer cells' DJ-1 dependence, as a therapeutic strategy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We especially thank the Tri-Institutional Therapeutics Discovery Institute (TDI), a 501(c)(3) organization, synthetic and analytical chemistry group for their assistance in synthesizing and characterizing the compounds described herein. TDI receives financial support from Takeda Pharmaceutical Company, TDI's parent institutes (Memorial Sloan Kettering Cancer Center (MSKCC), the Rockefeller University, and Weill Cornell Medicine) and from a generous contribution from Mr. Lewis Sanders and other philanthropic sources. Work in the David Lab is supported by the Josie Robertson Foundation, the Pershing Square Sohn Cancer Research Alliance, the NIH (CCSG core grant P30 CA008748, MSK SPORE P50 CA192937, R21 DA044767 and R35 GM138386), the Parker Institute for Cancer Immunotherapy (PICI), and the Anna Fuller Trust. In addition, the David Lab is supported by the Mr. William H. Goodwin and Mrs. Alice Goodwin Commonwealth Foundation for Cancer Research, and the Center for Experimental Therapeutics at MSKCC.References
- T. Jenuwein and C. D. Allis, Science, 2001, 293, 1074–1080 Search PubMed.
- A. J. Bannister and T. Kouzarides, Cell Res., 2011, 21, 381–395 Search PubMed.
- Q. Zheng, I. Maksimovic, A. Upad and Y. David, Protein Cell, 2020, 11, 401–416 Search PubMed.
- H. Talasz, S. Wasserer and B. Puschendorf, J. Cell. Biochem., 2002, 85, 24–34 Search PubMed.
- I. Maksimovic and Y. David, Trends Biochem. Sci., 2021 DOI:10.1016/j.tibs.2021.04.004.
- F. J. Tessier, Pathol. Biol., 2010, 58, 214–219 Search PubMed.
- M. Hellwig and T. Henle, Angew. Chem., Int. Ed., 2014, 53, 10316–10329 Search PubMed.
- A. Perrone, A. Giovino, J. Benny and F. Martinelli, Oxid. Med. Cell. Longevity, 2020, 2020, 3818196 Search PubMed.
- B. S. Szwergold, S. Howell and P. J. Beisswenger, Diabetes, 2001, 50, 2139–2147 Search PubMed.
- Q. Zheng, N. D. Omans, R. Leicher, A. Osunsade, A. S. Agustinus, E. Finkin-Groner, H. D'Ambrosio, B. Liu, S. Chandarlapaty, S. Liu and Y. David, Nat. Commun., 2019, 10, 1289 Search PubMed.
- S. L. Commerford, A. L. Carsten and E. P. Cronkite, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 1163–1165 Search PubMed.
- B. Bartholomew, Annu. Rev. Biochem., 2014, 83, 671–696 Search PubMed.
- K. Luger, A. W. Mader, R. K. Richmond, D. F. Sargent and T. J. Richmond, Nature, 1997, 389, 251–260 Search PubMed.
- J. J. Galligan, J. A. Wepy, M. D. Streeter, P. J. Kingsley, M. M. Mitchener, O. R. Wauchope, W. N. Beavers, K. L. Rose, T. Wang, D. A. Spiegel and L. J. Marnett, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9228–9233 Search PubMed.
- N. Rabbani, M. Xue and P. J. Thornalley, Biochem. Soc. Trans., 2014, 42, 419–424 Search PubMed.
- Q. Zheng, A. Osunsade and Y. David, Nat. Commun., 2020, 11, 3241 Search PubMed.
- G. Richarme, M. Mihoub, J. Dairou, L. C. Bui, T. Leger and A. Lamouri, J. Biol. Chem., 2015, 290, 1885–1897 Search PubMed.
- M. A. Wilson, Antioxid. Redox Signaling, 2011, 15, 111–122 Search PubMed.
- M. J. Devine, H. Plun-Favreau and N. W. Wood, Nat. Rev. Cancer, 2011, 11, 812–823 Search PubMed.
- X. Tao and L. Tong, J. Biol. Chem., 2003, 278, 31372–31379 Search PubMed.
- K. Honbou, N. N. Suzuki, M. Horiuchi, T. Niki, T. Taira, H. Ariga and F. Inagaki, J. Biol. Chem., 2003, 278, 31380–31384 Search PubMed.
- M. V. Liberti and J. W. Locasale, Trends Biochem. Sci., 2016, 41, 211–218 Search PubMed.
- N. Matsuda, M. Kimura, B. B. Queliconi, W. Kojima, M. Mishima, K. Takagi, F. Koyano, K. Yamano, T. Mizushima, Y. Ito and K. Tanaka, Sci. Rep., 2017, 7, 12816 Search PubMed.
- J. Drechsel, F. A. Mandl and S. A. Sieber, ACS Chem. Biol., 2018, 13, 2016–2019 Search PubMed.
- S. Tashiro, J. M. M. Caaveiro, M. Nakakido, A. Tanabe, S. Nagatoishi, Y. Tamura, N. Matsuda, D. Liu, Q. Q. Hoang and K. Tsumoto, ACS Chem. Biol., 2018, 13, 2783–2793 Search PubMed.
- X. B. Chen, H. Y. Zhu, K. Bao, L. Jiang, H. Zhu, M. D. Ying, Q. J. He, B. Yang, R. Sheng and J. Cao, Acta Pharmacol. Sin., 2021 DOI:10.1038/s41401-020-00600-5.
- T. Wang, M. D. Streeter and D. A. Spiegel, Bioorg. Med. Chem. Lett., 2015, 25, 4881–4886 Search PubMed.
- B. S. Hartley and B. A. Kilby, Biochem. J., 1952, 50, 672–678 Search PubMed.
- W. Shao and J. Wiegel, Appl. Environ. Microbiol., 1995, 61, 729–733 Search PubMed.
- M. Burger, T. J. Zimmermann, Y. Kondoh, P. Stege, N. Watanabe, H. Osada, H. Waldmann and I. R. Vetter, J. Lipid Res., 2012, 53, 43–50 Search PubMed.
- J. Y. Lee, J. Song, K. Kwon, S. Jang, C. Kim, K. Baek, J. Kim and C. Park, Hum. Mol. Genet., 2012, 21, 3215–3225 Search PubMed.
- J. Chen, L. Li and L. S. Chin, Hum. Mol. Genet., 2010, 19, 2395–2408 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ESI figures, materials and methods, and characterization of chemical compounds. See DOI: 10.1039/d1md00062d |
| This journal is © The Royal Society of Chemistry 2021 |