
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew prodrugs and analogs of the phenazine 5,10-dioxide natural products iodinin and myxin promote selective cytotoxicity towards human acute myeloid leukemia cells†
Elvar Örn
Viktorsson
 ab,
Reidun
Aesoy
c,
Sindre
Støa
a,
Viola
Lekve
c,
Stein Ove
Døskeland
d,
Lars
Herfindal
c and
Pål
Rongved
*a
ab,
Reidun
Aesoy
c,
Sindre
Støa
a,
Viola
Lekve
c,
Stein Ove
Døskeland
d,
Lars
Herfindal
c and
Pål
Rongved
*a
aSchool of Pharmacy, Department of Pharmaceutical Chemistry, University of Oslo, PO Box 1068 Blindern, N0316 Oslo, Norway. E-mail: pal.rongved@farmasi.uio.no
bSchool of Health Sciences, Faculty of Pharmaceutical Sciences, University of Iceland, Hofsvallagata 53, IS-107 Reykjavik, Iceland
cCentre for Pharmacy, Department of Clinical Science, University of Bergen, Jonas Lies vei 87, N-5021 Bergen, Norway
dDepartment of Biomedicine, University of Bergen, Jonas Lies vei 91, N-5021 Bergen, Norway
First published on 19th April 2021
Abstract
Novel chemotherapeutic strategies for acute myeloid leukemia (AML) treatment are called for. We have recently demonstrated that the phenazine 5,10-dioxide natural products iodinin (3) and myxin (4) exhibit potent and hypoxia-selective cell death on MOLM-13 human AML cells, and that the N-oxide functionalities are pivotal for the cytotoxic activity. Very few structure–activity relationship studies dedicated to phenazine 5,10-dioxides exist on mammalian cell lines and the present work describes our efforts regarding in vitro lead optimizations of the natural compounds iodinin (3) and myxin (4). Prodrug strategies reveal carbamate side chains to be the optimal phenol-attached group. Derivatives with no oxygen-based substituent (–OH or –OCH3) in the 6th position of the phenazine skeleton upheld potency if alkyl or carbamate side chains were attached to the phenol in position 1. 7,8-Dihalogenated- and 7,8-dimethylated analogs of 1-hydroxyphenazine 5,10-dioxide (21) displayed increased cytotoxic potency in MOLM-13 cells compared to all the other compounds studied. On the other hand, dihalogenated compounds displayed high toxicity towards the cardiomyoblast H9c2 cell line, while MOLM-13 selectivity of the 7,8-dimethylated analogs were less affected. Further, a parallel artificial membrane permeability assay (PAMPA) demonstrated the majority of the synthesized compounds to penetrate cell membranes efficiently, which corresponded to their cytotoxic potency. This work enhances the understanding of the structural characteristics essential for the activity of phenazine 5,10-dioxides, rendering them promising chemotherapeutic agents.
Introduction
Phenazine 5,10-dioxides (1, Fig. 1) constitute an oxidized subclass of phenazines (2, Fig. 1), often endowed with anti-infective1–4 and tumor growth-inhibiting properties.5–9 Iodinin (1,6-dihydroxyphenazine 5,10-dioxide, 3) and myxin (1-hydroxy-6-methoxyphenazine 5,10-dioxide, 4) are well-known examples of natural origin, both containing N-oxide functionalities rarely observed in natural products.10 Herfindal et al. demonstrated that the purple pigment iodinin (3) possesses potent and selective cytotoxic properties towards cell lines derived from patients with acute myeloid leukemia (AML).11 Moreover, iodinin (3) showed lower toxicity than the standard anti-AML drug daunorubicin for non-cancerous cells like hepatocytes, cardiomyoblasts (H9c2) and normal rat kidney (NRK) epithelial cells.11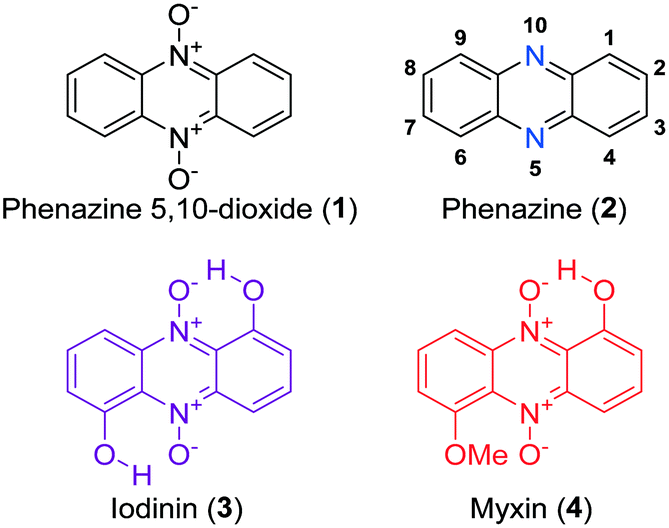 | ||
| Fig. 1 The structure of phenazine 5,10-dioxide (1), phenazine (2) with numbered positions and the natural products iodinin (3) and myxin (4). | ||
Iodinin (3) was one of the first phenazines to be discovered, first isolated in 1939 by Davis et al., from cultures of Brevibacterium iodinum.12,13 Its weak to moderate antimicrobial activities have been known for decades,3,14,15 and early studies by Hano et al. also showed iodinin (3) to expand the lifetime of mice with Ehrlich sarcoma relative to placebo control.16 Although evidence from in vitro experiments strongly suggest iodinin (3) to be a promising candidate to pursue in new options for AML treatment,11,17 a major drawback is the very poor solubility in aqueous solvents. This issue needed to be resolved in order to obtain a preclinical evaluation of iodinin's potential as an anticancer drug. The poor aqueous solubility stems from the two intramolecular ion–dipole bonds resulting in low solute–solvent interactions (see 3, Fig. 1). Myxin (4) was isolated from Sorangium sp. in 1966 (ref. 18) and has received attention from the medicinal chemistry community mainly because of its antimicrobial profile.3,4,19 In fact, myxin (4) was earlier marketed as a cupric complex (Unitrop®) in veterinary medicine for treatment of topical infections.20
Two modes of action have been proposed for the biological activity of myxin (4) and iodinin (3). Hollstein et al. demonstrated that both of them are capable of DNA intercalation, especially in guanine–cytosine rich regions of the DNA-double strand at concentrations similar to those required for other known intercalators like ethidium bromide and actinomycin.21,22 They also showed myxin (4) to affect the incorporation rate of CTP and GTP into RNA, but not of ATP and UTP.21 The other proposed mechanism is by generation of reactive oxygen species (ROS). Gates et al.23 showed phenazine 5,10-dioxides like myxin (4) being able to undertake a bio reductive activation to form a radical intermediate of the mother compound (Scheme 1), which further collapses to unleash the cytotoxic hydroxyl radical (˙OH).23 They found also that myxin (4) can induce cell death of human colorectal HCT-116 cells under both aerobic and anaerobic conditions.23 It is widely accepted that hydroxyl radicals cause DNA damage and are able to abstract hydrogens from the deoxyribose sugars in the DNA-double strand helix.24 This is a typical result for drugs that unleash ˙OH radicals such as tirapazamine (10, Fig. 3), as earlier demonstrated by Gates and co-workers.25
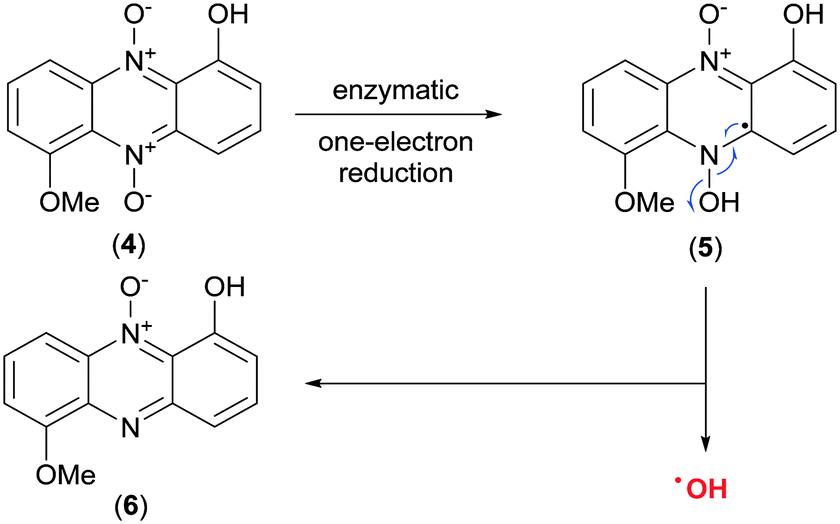 | ||
| Scheme 1 Discharge of the hydroxyl radical (˙OH) upon bio-reductive activation of myxin (4).23 | ||
We have recently published an efficient synthesis of iodinin (3), myxin (4) and several alkylated analogs, and studied their cytotoxic effects on human AML-cells (MOLM-13).26 The 1-hydroxy-6-(2-ethoxy-2-oxoethoxy) phenazine 5,10-dioxide (7) was found to be the most potent analog with ability to induce cell death, showing EC50 at sub-micromolar concentrations (see Fig. 2).
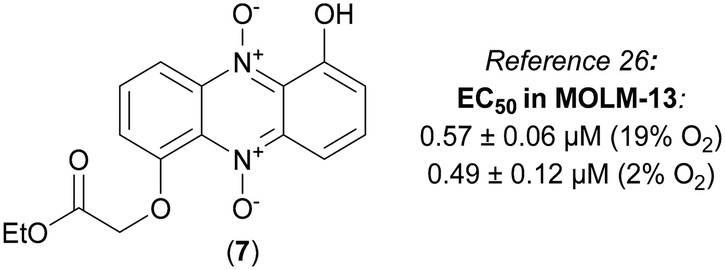 | ||
| Fig. 2 The structure of compound 7, a highly cytotoxic analog of iodinin (3) and myxin (4). | ||
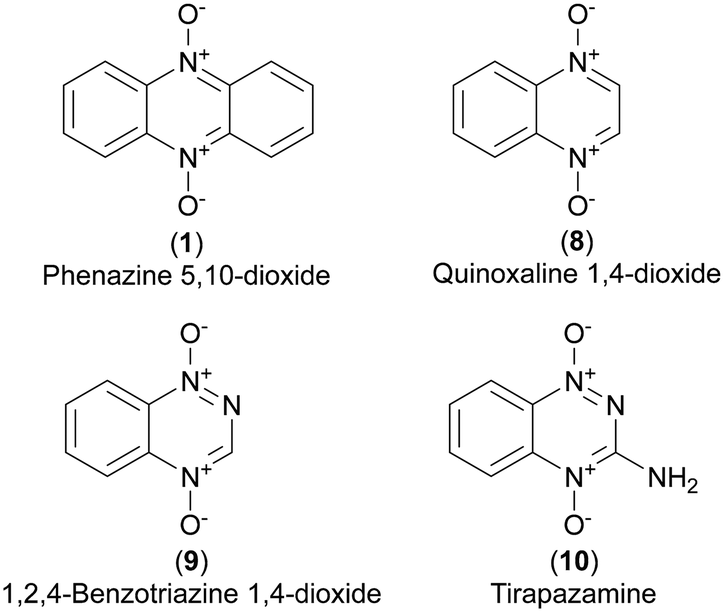 | ||
| Fig. 3 The core structures of known hypoxia selective scaffolds and tirapazamine. | ||
Several N-oxidized heterocyclic aromatic scaffolds have been reported to act more potently in hypoxic than normoxic environment. Examples are phenazine 5,10-dioxides (1),8,27 quinoxaline 1,4-dioxides28 (8) and 1,2,4-benzotriazine 1,4-dioxides (9).29 The latter class includes tirapazamine (3-amino-1,2,4-benzotriazine 1,4-dioxide, 10), a lead prototype of hypoxia selective prodrugs and a subject of several phase I, II and III clinical trials for treatment of various cancers.30–32 The rapid cell growth and incomplete vasculature in tumors generate a hypoxic environment33 where hypoxia selective drugs could play a role in antineoplastic treatment.34 Cimmino et al. reviewed phenazines and cancer, concluding that phenazine 5,10-dioxides have the highest potential among phenazines to be exploited for cancer therapy.5
We wanted to further exploit the anti-cancer potential of the phenazines by (i) synthesis of bio-activatable prodrugs of O-functionalized phenazine 5,10-dioxides with increased aqueous solubility, (ii) increase their potency against cancer cells by rationally optimize the ring substitution patterns, or by utilizing other related scaffolds. Further we wanted to (iii) assess cytotoxicity of the analogs on cancer cells and non-cancerous cell lines to develop a SAR study and to be able to understand why some analogs are superior leads compared to others. Taken together, in the present study we show that improvement of bioactivity as well as physico-chemical properties of simple molecules like the phenazines gives new insight in how this scaffold can be optimized for future drug development. The phenazine 5,10-dioxides require a bioreductive process to discharge an ˙OH radical and can therefore be viewed as prodrugs (Scheme 1). In this paper however, the term prodrug is specifically used for analogs with side chain functionalities attached to their phenols and are generally regarded as labile towards aqueous or enzymatic hydrolysis (esters, carbonates, carbamates). In theory, such compounds could have better physicochemical properties than the lead iodinin (3), improving factors such as solubility and membrane crossover. Once entered the cell, such prodrugs could either release their cytotoxic hydroxyl radical before or after hydrolysis of the side chain. We also aim to investigate if other non-cleavable substituents than those published earlier26 can alter the anti-AML activity of the phenazine 5,10-dioxide scaffold.
Results and discussion
Synthesis of compounds
Iodinin (3) and myxin (4), synthesized according to our previously described procedure,26 served as scaffold for the synthesis of a small catalogue of iodinin and myxin prodrugs. The ester analogs 11 and 12 were prepared by reacting iodinin (3) with pivaloyl chloride at −40 °C and valeric anhydride at 0 °C respectively, under basic conditions catalyzed by DMAP (Scheme 2). By similar means, mono- and bis-substituted carbonates (13 and 14) were produced by reacting iodinin (3) with ethyl chloroformate. The reactions forming 11–14 occurred rapidly since the reaction mixture quickly shifted color from deep purple towards brown yellow.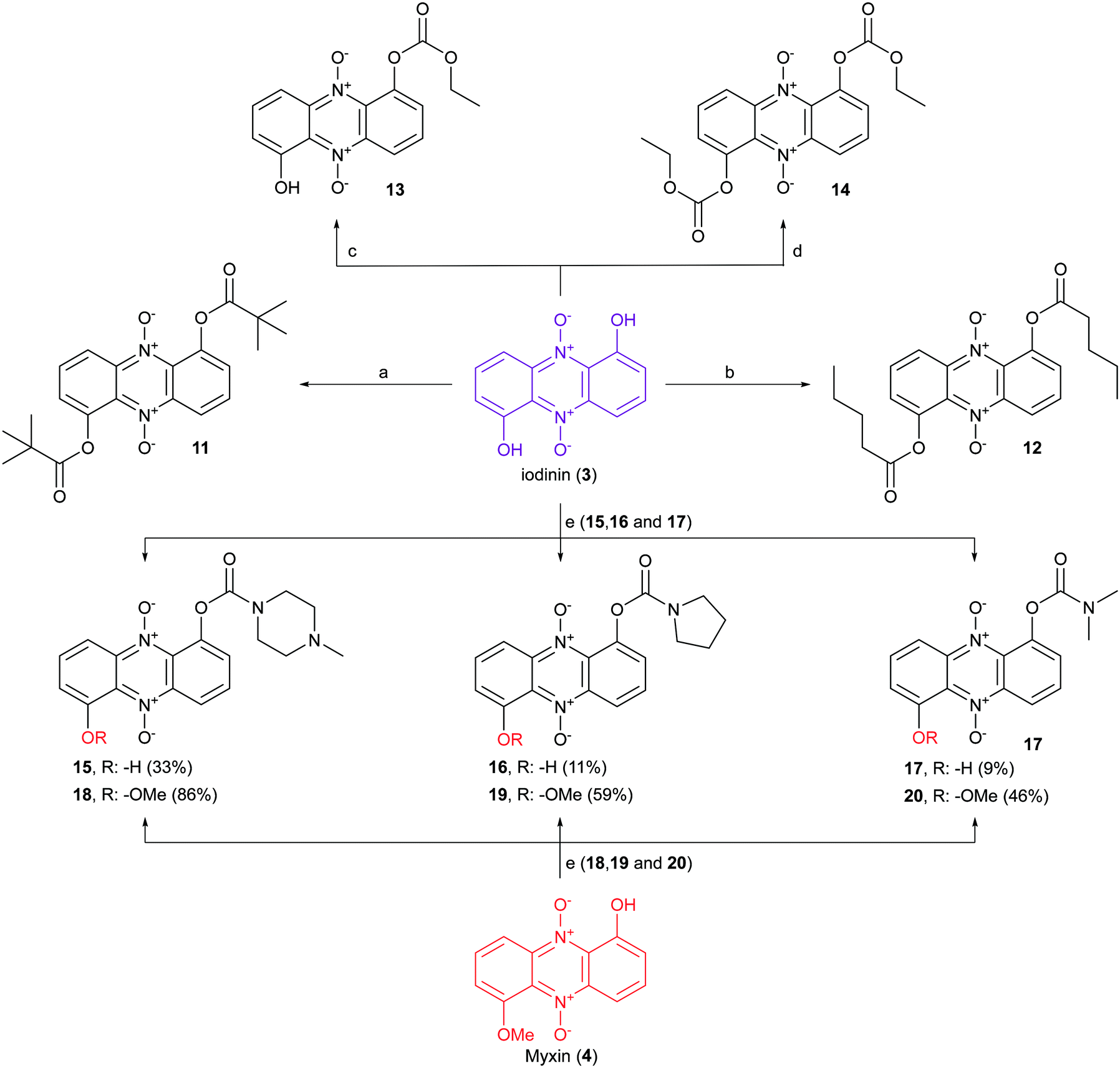 | ||
| Scheme 2 Synthesis of iodinin (3) and myxin (4) prodrugs. Reagents and conditions: a) pivaloyl chloride, Et3N, DMAP, PhMe −40 °C, 30 min, 39%. b) Valeric anhydride, Et3N, DMAP, PhMe, 0 °C, 16 h, 39%. c) Ethyl chloroformate, Et3N, DMAP, PhMe −40 °C, 30 min, 25%. d) Ethyl chloroformate, Et3N, DMAP, PhMe, 0 °C, 90 min, 67%. e) A corresponding carbamoyl chloride (or HCl salt), DABCO, THF, rt. | ||
Next in line were carbamate functionalities. Attempts to react iodinin (3) with a carbamoyl chloride in PhMe with DMAP and Et3N only resulted in trace conversions (TLC analysis). Iodinin (3) showed no nucleophilicity towards any isocyanates in attempts to form primary carbamates. However, when DABCO was exposed to iodinin (3) and a neat carbamoyl chloride, the reaction mixture started to produce fume. The cherry red carbamate analogs 15–17 were thus synthesized as depicted in Scheme 2. The yields from these reactions aimed to mono-functionalize iodinin (3) were modest (9–33%). Myxin (4) was rapidly transformed into the orange-colored carbamate-analogs 18–20 using the same method in good yields (see Scheme 2). The higher yields for myxin (4) can be explained by its far superior solubility in organic media compared to iodinin (3) and the functionalization of only a single phenol group instead of two. Interestingly DABCO does not deprotonate phenols, because if such a deprotonation did occur, the reaction mixture would instantly switch color from red to dark green, like for the K2CO3/18-crown-6 system used for alkylations of iodinin (3).26 This suggests that DABCO activates the carbamoyl chloride allowing the neutral phenol moiety of the phenazine to attack the DABCO–carbamoyl chloride complex as depicted in Scheme 3.
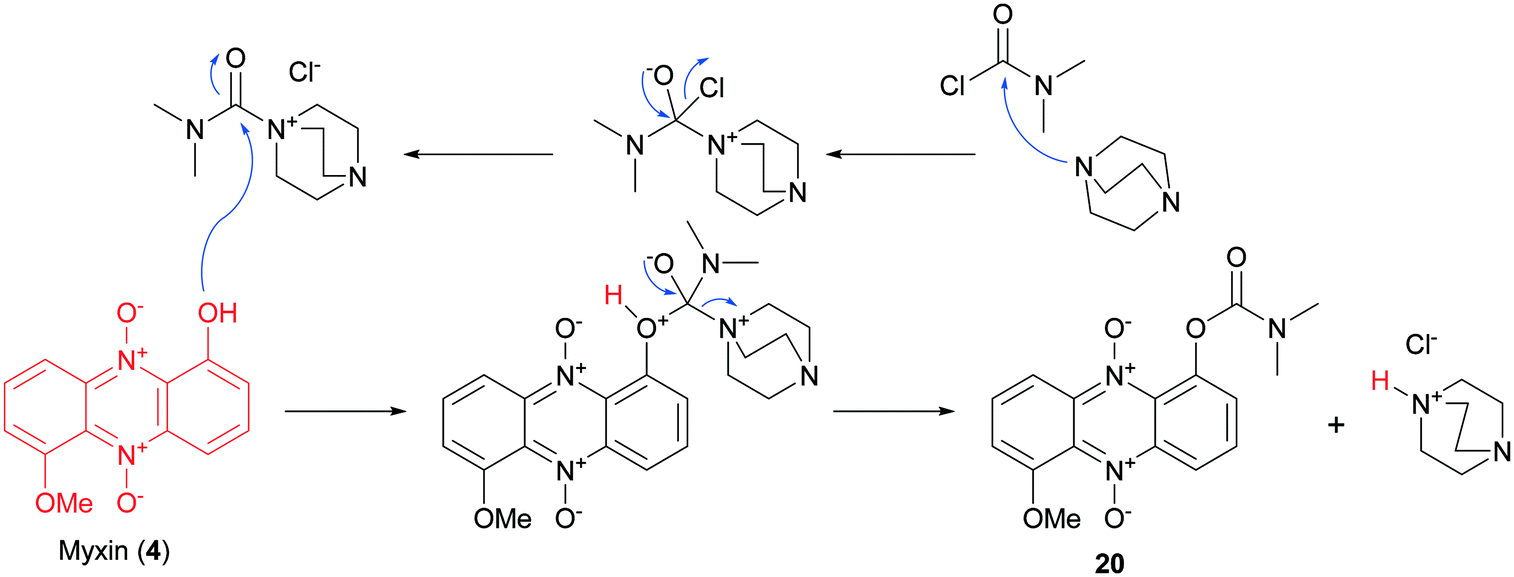 | ||
| Scheme 3 A proposed reaction mechanism for DABCO enforced carbamoylation of myxin (4). | ||
The next step was to construct phenazine 5,10-dioxides lacking an oxygen substituent in position 6. 1-Hydroxyphenazine 5,10-dioxide (21) was synthesized by two separate strategies (see Scheme 4). First, commercially obtained 1-hydroxyphenazine (22) was oxidized to 21 using pulse-wise addition of mCPBA in toluene over a period of 5–6 hours, as reported previously for the synthesis of iodinin (3).26 This method yielded the dioxide (21) in 49% yield at a single gram scale. In an alternative approach, a Beirut reaction35 was undertaken as cyclohexane-1,2-dione and benzofuroxan (23) were condensed in neat diethylamine. This gave a filtered bright red crude that was oxidized using mCPBA. This 2-step procedure gave 2 g of isolated 1-hydroxyphenazine 5,10-dioxide (21) in 40% yield.
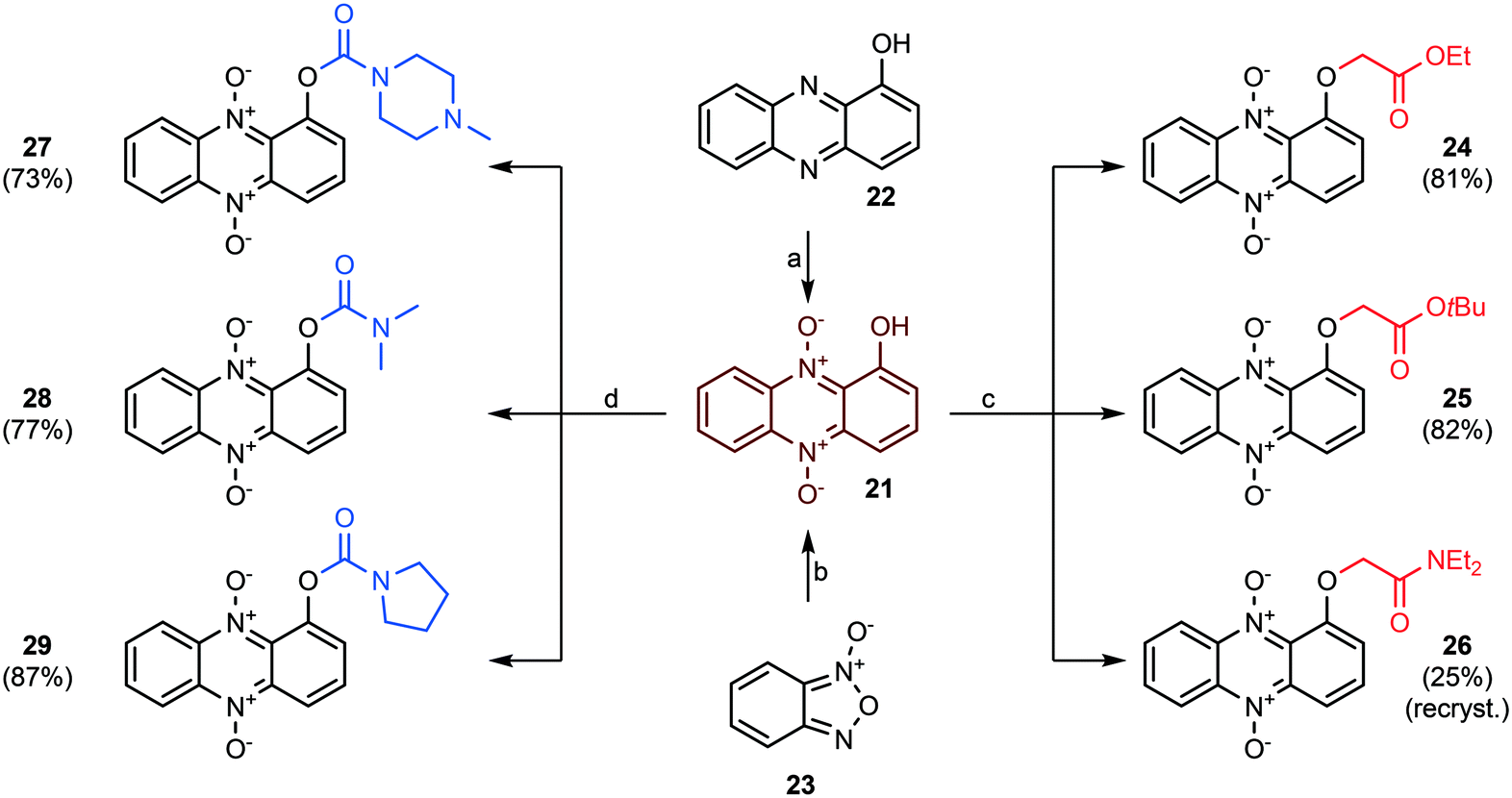 | ||
| Scheme 4 The synthesis and functionalization of 1-hydroxyphenazine 5,10-dioxide (21). Reagents and conditions: a) mCPBA, PhMe, 80 °C, 5 h, 49% b) 1,2-cyclohexanedione, diethylamine, 0 °C → rt, 90 min, then filtration and mCPBA oxidation as in a), 40% over 2 steps c) ethyl-/or tertbutyl bromoacetate (or 2-chloro-N,N-diethylacetamide and KI), K2CO3, 18-crown-6, DMF. rt, 2–5 hours. d) A carbamoyl chloride (or the corresponding hydrochloride salt), DABCO, THF, rt, 1–3 hours. | ||
The reaction of 21 with ethyl bromoacetate in the presence of K2CO3 and 18-crown-6 gave compound 24 in 84% yield. This molecule was synthesized to serve as an analog to 7 (see Fig. 2), a highly cytotoxic compound in MOLM-13 cells.26 The same step, when repeated with only tert-butyl bromoacetate, produced 25, and again with 2-chloro-N,N-diethylacetamide (in addition to KI) gave 26 in 25% yield after recrystallization from hot EtOH. The main building block 21 was also exposed to carbamoyl chlorides and DABCO forming carbamate analogs 27–29 in 73–87% yields (see Scheme 4).
7,8-Disubstituted 1-methoxyphenazines 36–39 were synthesized via the classic condensation procedure using 1,2-benzoquinones and O-phenylenediamines as starting materials. Oxidation of 3-methoxycatechol (30) using ortho-chloranil in cold ether afforded the corresponding 1,2-benzoquinone 31 after filtration of the reaction mixture which was immediately transferred to an acidic toluene solution containing a corresponding ortho-phenylenediamine (32–35) (see Scheme 5). These efforts afforded 7,8-disubstituted-1-methoxyphenazines 36–39 isolated in moderate to good yields.
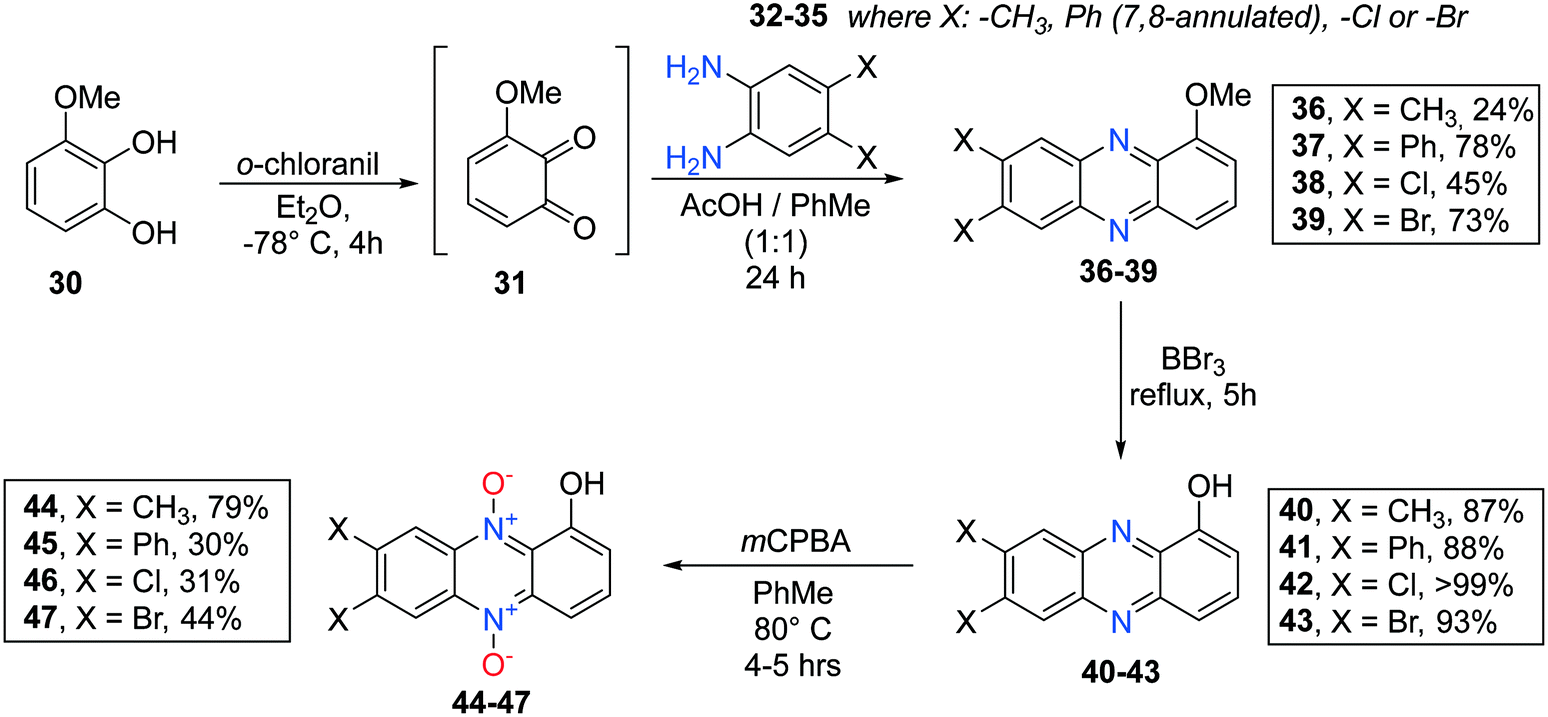 | ||
| Scheme 5 Synthesis of 7,8-substituted phenazine 5,10-dioxides 44–47. | ||
Similar work had been performed earlier by Cushman et al.36 and Huigens III and co-workers.1 Next, each 1-methoxyphenazine (36–39) was individually reacted in BBr3 neat under reflux to afford the corresponding 1-hydroxyphenazines 40–43 in yields 87% and higher.
The 1-hydroxyphenazines 40–43 were then exposed to a pulse-wise addition of mCPBA in toluene at 80 °C to give 7,8-disubstituted-1-hydroxyphenazine 5,10-dioxides 44–47 in non-optimized, yet acceptable yields for further use (see Scheme 5). Especially, 7,8-dibromo and 7,8-dichloro substituted analogs 45 and 46 were difficult to purify due to poor solubility and high lipophilicity resulting in tailing on the silica gel. They were thus not subjected to biological testing but the crudes after flash column chromatography could be used for further synthesis.
7,8-Disubstituted-1-hydroxyphenazine 5,10-dioxides 44–47 were subsequently functionalized with ethyl-bromoacetate (Scheme 6). These compounds were also synthesized to serve as mimics for the cytotoxic compound 7 (see Fig. 2). In addition, compounds 44–47 were also reacted with the hydrochloride salt of 4-methyl-1-piperazinecarbonyl chloride in order to give piperazine analogs 52–55 in good yields (Scheme 6). 7,8-Dimethyl-1-hydroxyphenazine 5,10-dioxide (44) was also functionalized affording the diethylamide analog 56 and the pyrrolidine carbamate 57 (Scheme 7).
 | ||
| Scheme 6 Functionalization of 7,8-substituted phenazine 5,10-dioxides 48–55. Reagents and conditions: a) ethyl bromoacetate, K2CO3, 18-crown-6, DMF. Rt, 2–5 hours. b) 4-Methyl-1-piperazinecarbonyl chloride hydrochloride, DABCO, THF, rt, 3–4 hours. *Rapid decomposure of the compound observed in NMR analysis (CDCl3 solution). | ||
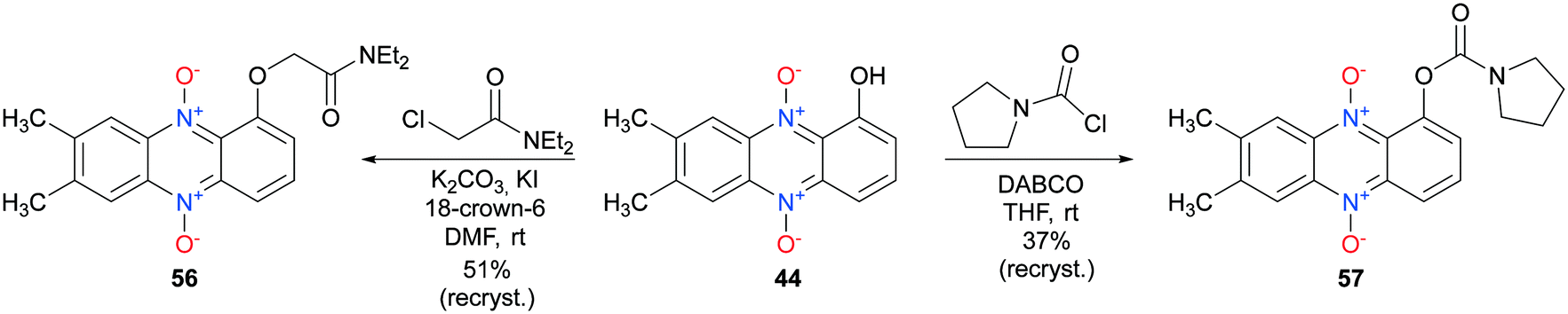 | ||
| Scheme 7 Synthesis of 7,8-dimethyl-1-hydroxyphenazine 5,10 dioxide analogs 56 and 57. | ||
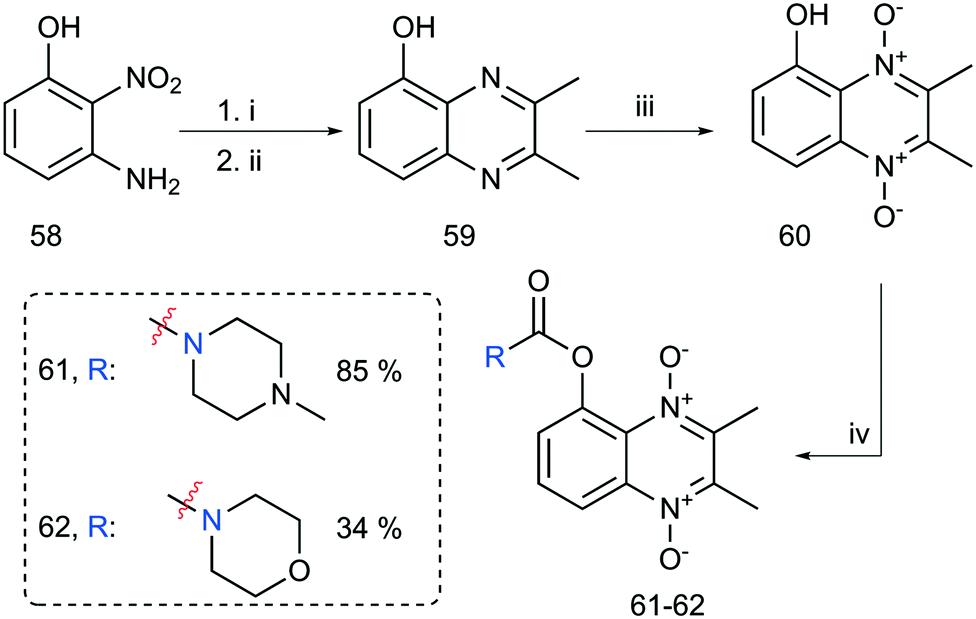
The last step was the synthesis of 2-ring analogs. 2,3-Dimethylquinoxalin-5-ol (59) was prepared by reducing 3-amino-2-nitrophenol (58) with sodium dithionite refluxing in a basic mixture of MeOH/H2O (see Scheme 8). The crude mixture was concentrated to a slurry in vacuo, diluted with AcOH–PhMe (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) and subsequently exposed to 2,3-diketo-butane yielding 59 in 79% yield over 2 steps. A subsequent mCPBA oxidation gave the corresponding dioxide 60 in 80% yield after flash column chromatography. Further functionalization with carbamoyl chlorides gave carbamate analogs 61 and 62 as well. Interestingly, 60 did not tolerate the presence of K2CO3/18-crown-6 system in alkylation attempts. TLC analysis revealed multiple inseparable products formed in combination with unreacted starting material. An offered explanation is that DABCO does not deprotonate the phenol functionality of 60 like the K2CO3/18-crown-6 reaction conditions.
:3) and subsequently exposed to 2,3-diketo-butane yielding 59 in 79% yield over 2 steps. A subsequent mCPBA oxidation gave the corresponding dioxide 60 in 80% yield after flash column chromatography. Further functionalization with carbamoyl chlorides gave carbamate analogs 61 and 62 as well. Interestingly, 60 did not tolerate the presence of K2CO3/18-crown-6 system in alkylation attempts. TLC analysis revealed multiple inseparable products formed in combination with unreacted starting material. An offered explanation is that DABCO does not deprotonate the phenol functionality of 60 like the K2CO3/18-crown-6 reaction conditions.
 | ||
| Scheme 8 Synthesis of functionalized bicyclic aromatic N-oxides. Reaction conditions: i) Na2S2O4, Na2CO3, H2O/MeOH, reflux, 2 h. ii) 2,3-Diketo-butane (diacetyl), AcOH/PhMe, 20 h, 79% over two steps. iii) mCPBA. PhMe, 80 °C, 4.5 h, 80%. iv) Corresponding carbamoyl chloride, DABCO, THF. | ||
Cytotoxicity of the synthesized compounds
Our previous findings show iodinin (3), myxin (4) and compound 7 to be cytotoxic towards the MOLM-13 human AML cell line.11,26 Myhren et al. also found that iodinin (3) favored apoptosis induction in AML cell lines over normal cell lines like cardiomyoblasts, in fact higher selectivity compared to the anthracycline daunorubicin (DNR).11 We wanted to investigate the cytotoxic activity of iodinin (3), myxin (4) and compound 7 in cells derived from non-cancerous tissues, in addition to the cytotoxicity towards AML cells. The two nonmalignant cell lines, NRK (derived from rat kidney epithelium) and H9c2 (rat cardiomyoblasts) were incubated with these compounds as well as tirapazamine (10). The results confirmed the leukemic cell selectivity of the lead compound iodinin (3) (Table 1).11 Myxin (4) killed the MOLM-13 AML cells at concentrations around 100-fold lower compared to those needed to kill NRK cells under hypoxic conditions (Table 1). To the authors' best knowledge, such high selectivity of myxin (4) towards cancer cells has not been described previously. Compound 7, priorly shown to be more potent than both iodinin (3) and myxin (4),26 was also less toxic to NRK and H9c2 cells than AML cells (Table 1).| Cpd # | EC50 (μM) MOLM-13 | EC50 (μM) NRK | EC50 (μM) H9c2 | Permeability (PAMPA) | |||
|---|---|---|---|---|---|---|---|
| 19% O2 | 2% O2 | 19% O2 | 2% O2 | 19% O2 | P eff | Classification | |
| Cell viability was assessed by the WST-1 proliferation assay and Hoechst staining 24 h after treatment of cells with various concentrations of iodinin (3) or its analogues. Non-linear regression analyses were performed using SigmaPlot in order to calculate the given EC50 values. The EC50 values of tirapazamine (10; TPZ) are based on microscopic evaluation (marked with *), whereas the EC50 values of iodinin and its analogues are based on the WST-1 results, and all are adjusted relative to untreated control cells in each experiment. The data are average of 4–6 experiments for MOLM-13 and 2–3 for NRK and H9c2 cells, ± SEM. a Poor solubility in DMSO. n.d.: not determined b Ref: Viktorsson et al.26Peff: permeability calculated as described in methods section. c Denotes that n ≥ 3. For all other samples, n = 2. Classification: ip: impermeable, pp: poor permeability, ip: intermediate permeability, p: permeable.48 | |||||||
| 3 (iodinin) | 2.0 ± 0.07b | 0.79 ± 0.10b | >50 | >50 | >50 | −5.64c | Intermediate |
| 4 (myxin) | 1.4 ± 0.30b | 0.77 ± 0.13b | 77 ± 11 | 76 ± 14 | 46 ± 4.1 | −4.94 | High |
| 7 | 0.57 ± 0.06b | 0.49 ± 0.12b | 12 ± 1.7 | 18 ± 1.5 | 15 ± 0.9 | −4.84c | High |
| 10 (TPZ) | 95 ± 8*b | 22 ± 2*b | >100* | 35 ± 6.3* | >100* | — | — |
| 11 | 11.0 ± 0.7 | 8.9 ± 1.71 | n.d. | n.d. | >25 | −6.06 | Low |
| 12 | 36 ± 0.1 | 22 ± 2.8 | n.d. | n.d. | >25 | −7.74 | Impermeable |
| 13 | 50 ± 0.4 | 66 ± 2.2 | n.d. | n.d. | >100 | −7.32c | Impermeable |
| 14 | 6.1 ± 0.05 | 4.0 ± 0.33 | >25 | >25 | >25 | −4.75 | High |
| 15 | 1.5 ± 0.16 | 1.4 ± 0.06 | 53 ± 2.0 | 59 ± 1.1 | 100 ± 13 | −5.96c | Low |
| 16 | 0.86 ± 0.05 | 0.79 ± 0.089 | 9.6 ± 0.38 | 15 ± 1.0 | 12 ± 0.7 | −4.74 | High |
| 17 | 0.89 ± 0.03 | 0.70 ± 0.04 | 49 ± 1.8 | 62 ± 1.3 | 64 ± 9.2 | −5.06 | High |
| 18 | 1.0 ± 0.06 | 0.98 ± 0.08 | 26 ± 2.1 | 39 ± 4.2 | 26 ± 0.9 | −5.74 | Low |
| 19 | 2.0 ± 0.11 | 1.8 ± 0.17 | 32 ± 2.5 | 35 ± 1.5 | 35 ± 3.3 | −5.1 | High |
| 20 | 1.9 ± 0.07 | 1.7 ± 0.09 | 22 ± 1.7 | 29 ± 0.7 | 35 ± 1.8 | −5.3 | High |
| 21 | 11 ± 1.8 | 12 ± 0.04 | 79 ± 4.2 | >100 | >100 | −4.74 | High |
| 24 | 1.5 ± 0.14 | 1.7 ± 0.29 | 26 ± 0.8 | 25 ± 0.2 | 20 ± 2.9 | −4.93 | High |
| 25 | 2.2 ± 0.09 | 1.9 ± 0.11 | 26 ± 0.7 | 25 ± 1.9 | 20 ± 2.1 | −4.76 | High |
| 26 | 1.2 ± 0.08 | 1.4 ± 0.15 | 13 ± 0.01 | 16 ± 1.0 | 23 ± 0.1 | −5.2 | High |
| 27 | 1.5 ± 0.18 | 1.9 ± 0.05 | 25 ± 0.2 | 37 ± 1.9 | 36 ± 11 | −4.85 | High |
| 28 | 1.3 ± 0.15 | 2.1 ± 0.16 | 19 ± 1.3 | 25 ± 0.6 | 16 ± 2.6 | −4.95 | High |
| 29 | 1.5 ± 0.18 | 1.2 ± 0.15 | 18 ± 0.6 | 21 ± 2.4 | 13 ± 0.5 | −4.91 | High |
| 44 | 2.8 ± 0.25 | 2.3 ± 0.10 | >50 | >50 | >50 | −5.45 | Intermediate |
| 45 | —a | >10a | n.d. | n.d. | n.d. | −7.23 | Impermeable |
| 48 | 8.3 ± 0.78 | 6.3 ± 0.25 | 140 ± 2 | >100 | >100 | −4.93 | High |
| 50 | 0.41 ± 0.05 | 0.38 ± 0.04 | 3.1 ± 0.02 | 4.7 ± 0.09 | 1.3 ± 0.2 | −5.16 | High |
| 51 | 2.4 ± 0.03 | 1.6 ± 0.09 | 10 ± 0.7 | 9.7 ± 2.40 | 3.3 ± 0.50 | −5.24 | High |
| 52 | 0.63 ± 0.05 | 0.54 ± 0.06 | 12 ± 1.0 | 13 ± 0.7 | 9.4 ± 0.55 | −4.97 | High |
| 53 | 2.8 ± 0.18 | 1.8 ± 0.10 | 2.0 ± 0.06 | 7.5 ± 0.38 | 2.3 ± 0.05 | −5.26c | High |
| 54 | 0.34 ± 0.04 | 0.32 ± 0.06 | 11 ± 1.0 | 15 ± 1.8 | 3.2 ± 0.10 | −4.97c | High |
| 55 | 0.41 ± 0.04 | 0.23 ± 0.03 | 6.2 ± 0.02 | 7.6 ± 0.60 | 2.2 ± 0.53 | −4.88 | High |
| 56 | 1.8 ± 0.24 | 3.4 ± 0.8 | 21 ± 0.3 | 52 ± 1.2 | 41 ± 5.7 | −5.18 | High |
| 57 | 1.5 ± 0.02 | 0.78 ± 0.11 | 6.6 ± 0.04 | 8.9 ± 0.06 | 8.7 ± 1.15 | −4.85 | High |
| 60 | >200 | >200 | n.d. | n.d. | n.d. | −4.35 | High |
| 61 | >200 | >200 | n.d. | n.d. | n.d. | −4.48 | High |
| 62 | >200 | >200 | n.d. | n.d. | n.d. | −4.65 | High |
Ester-, carbonate-, and carbamate prodrugs of iodinin or myxin
The iodinin ester and carbonate analogs 11–14 exhibited low potency towards MOLM-13 cells compared to iodinin (3) and myxin (4). Based on the high activity of mono-alkylated iodinin analogs reported earlier (such as 7),26 the mono-functionalized carbonate compound 13 was anticipated to be highly active and therefore more potent than the di-functionalized compound 14. Surprisingly, 13 had low potency (EC50 ∼ 50 μM, in normoxia, MOLM-13, Table 1). To verify that this discrepancy in activity between analog 13 and 14 was not due to degradation of analog 13 during storage, the compound was resynthesized, purified by recrystallization, and immediately retested yielding similar results. In order to understand the reason behind the lack of cytotoxicity of some of the analogs, all compounds were tested for membrane permeability in a parallel artificial membrane permeability assay (PAMPA). Although some cytostatics, like the anthracyclines are impermeable, and rely on active transport for cellular internalization37,38 we suspected that lack of ability to diffuse over membranes could explain the lack of activity towards the AML cell line. Both 12 and 13 were found to be impermeable in the PAMPA assay, and their low toxicity towards MOLM-13 cells may, at least in part, be explained by their lack of diffusion across the cell membrane (Table 1). In comparison, iodinin (3), myxin (4) and compound 7, were all permeable in the PAMPA assay (Table 1). The bis-O-substituted analog 14 proved to be more potent than compound 13 with an EC50 of 6 μM for MOLM-13 cells in normoxia (Table 1) which correlates to a higher permeability of compound 14 (Table 1). However, since the ester- and carbonate analogs (11–14) show lower EC50 values towards the AML cells compared to their parent compounds, they appear unsuited as iodinin (3) prodrugs for anti-AML therapy.Mono-substituted carbamate analogs of iodinin (15–17) or myxin (18–20) displayed high potency towards the MOLM-13 cells and were all equally or more potent than their respective parent compounds iodinin (3) and myxin (4) towards MOLM-13 cells (Table 1). Of iodinin carbamates 15–17, compound 15 had lowest potency, which corresponds with it having lower permeability compared to compounds 16 and 17. This is likely due to the tertiary amine of N-methyl piperazine becoming positively charged at physiological pH (compounds 15 and 18) and therefore reducing the ability for membrane crossover. Myxin carbamate analogs 19 and 20 resided within the high permeability group. Although the piperazine analogs 15 and 18 displayed low permeability according to PAMPA results, they were both highly cytotoxic towards the MOLM-13 cell line. None of the iodinin or myxin carbamates (15–20) appeared to be more cytotoxic under hypoxic conditions (2% O2, Table 1). The lack of hypoxia selectivity for 15–20 might be caused by an electron withdrawing effect of the carbamate group making the aromatic phenazine system less electron rich. In comparison, a phenazine like myxin (4) shows greater hypoxia selectivity where the phenol- and the methoxy-functionalities contribute to a higher electron density in the aromatic ring system. To reflect on that note, SAR-studies conducted on tirapazamine (10) showed weakly electron donating groups to be more likely affording a higher hypoxic cytotoxicity ratio.29 When tested against the NRK- and H9c2 cells, the carbamates (15–20) showed EC50 values about 15–80 times higher than for the AML cells (Table 1). In short conclusion, carbamates of iodinin and myxin represent an attractive new class of anti-AML therapeutic agents with an attractive therapeutic index like the lead compound myxin (3).
1-Hydroxyphenazine 5,10-dioxide and analogs
Construction of a phenazine 5,10-dioxide scaffold lacking an –OH group in position 6 resulted in 1-hydroxyphenazine 5,10-dioxide (21). This transformation alone resulted in a significant loss of cytotoxic activity (EC50: 11 ± 1.8) against MOLM-13 cells compared to the leads iodinin and myxin (Table 1). Interestingly, the introduction of either alkyl- or carbamate sidechains to the phenol of 21 re-established the cytotoxic activity (compounds 24–29, Table 1). This finding indicates that the N-oxide in position 10 adjacent to the functionalized phenol in 21 has become less stable after the phenol functionalization as an intramolecular ion–dipole stabilization is no longer possible. The effect of this destabilization is therefore likely to render compounds 24–29 more prone to unleash the hydroxyl radical for DNA-damage than 21. All the analogs (24–29) were highly permeable in the PAMPA assay (Table 1), even the ionizable piperazine analog 27 (opposite to piperazine compounds 15 and 18). On average, these analogs (24–29) only had around 14 times lower EC50 values in AML cells than the non-malignant NRK cells, therefore not outperforming iodinin- or myxin carbamates 15–20 in terms of cytotoxic selectivity towards the MOLM-13 cell line. These findings therefore suggest that the oxygen-based substituent in position 6 of the aromatic ring system plays an important role for the observed AML-selectivity of the compounds. However, it is important to point out that we have only synthesized and tested compounds with –H, –OH or O-derivatized analogs in positions 1 and 6. More comprehensive SAR studies on these two positions involving different combinations of functional groups such as –OH, –OR, −alkyl, −halogens, –NH2 or N-derivatized analogs could shed light on even more active and selective analogs towards MOLM-13 cells. However, the synthesis of some of these compounds could prove to be challenging, especially in terms of the oxidation step required to form the N-oxides in positions 5 and 10. To reflect on that note, 1,6-dihydroxyphenazine was oxidized efficiently with mCPBA in order to yield iodinin (3) whereas mCPBA oxidation of 1-hydroxy-6-methoxyphenazine could not yield myxin (4).26In conclusion, compounds 24–29 exhibited poorer selectivity between AML-cells and non-cancerous cell lines compared to monosubstituted carbamates of both iodinin and myxin (compounds 15–20).
7,8-Disubstituted 1-hydroxyphenazine 5,10-dioxide analogs and bicyclic derivatives
The addition of various substituents to positions 7 and 8 of 1-hydroxyphenazine 5,10-dioxide (21) increased the cytotoxic potential. 7,8-Dimethyl-1-hydroxyphenazine 5,10-dioxide (44) had higher potency against AML cells (EC50 = 2.8 μM, MOLM-13, normoxia) than 1-hydroxyphenazine 5,10-dioxide (21) (EC50 = 11 μM, MOLM-13 cells, normoxia). Addition of an aromatic ring (7,8-annulated) to 21 resulted in a practically insoluble compound (45) which did not give any response in any of the cells tested, most likely because of very poor solubility in the cell media (1H-NMR spectra in saturated DMSO-d6 solution confirmed the very low solubility, supplementary information, Fig. S37†), or insufficient cell membrane penetration (Table 1). However, when this compound was functionalized with a piperazine side chain (compound 53), the bioactivity was re-established (EC50 of 2.8 μM, MOLM-13, normoxia, see Table 1). This effect of the piperazine side chain was also reflected in the increase in permeability from impermeable for compound 45 to highly permeable for compound 53 (Table 1). The transformation of 45 to 53 gave a cytotoxic compound which did not distinguish between AML-cells and normal cells (Table 1), suggesting that addition of a ring to the phenazine core structure alters the mode of action for cytotoxicity to one that is ubiquitous for all cells and not only for AML cells. In conclusion, four-ring analogs of 1-hydroxyphenazine 5,10-dioxide do not appear suited for further research towards potential anti-cancer drugs.Four new analogs turned out to be particularly potent in MOLM-13 cells; the 7,8-dimethyl and 7,8-dihalogen-piperazine analogs 52, 54 and 55 and the 7,8-dichloro analog 50 bearing the 2-ethoxy-2-oxoethoxy side chain (see Table 1). The 7,8-dichloropiperazine analog 54 was found to be more selective towards leukemic cell lines compared with NRK-cells by a factor of more than 30 at 19% O2 and a factor of 45 in a hypoxic setting. The 7,8-dichloro-analog 54 and the 7,8-dibromo-analog 55 were the most potent of all the compounds tested against MOLM-13 cells, both at high and low oxygen levels. In agreement with their toxicity, these compounds were all highly permeable in the PAMPA assay (Table 1). However, in addition to the increased efficacy towards AML cells, the H9c2 cardiomyoblast cell line was also more responsive to the dihalogenated compounds 50, 51, 54 and 55, particularly compared to the NRK epithelial cell line. This indicates that the dihalogenated compounds 50, 51, 54 and 55 may not be suitable as lead compounds in AML therapy due to potential adverse effects on heart tissue.
The 7,8-dimethyl piperazine analog 52 had high potency towards AML cells, and was also found to be less toxic towards the non-cancerous cell lines compared to the halogenated compounds 50, 54 and 55 (Table 1). The 7,8-dimethyl pyrrolidine carbamate 57 showed comparable activity in MOLM-13 to that of myxin (4) and relatively good hypoxia selectivity was observed, but it was more toxic to NRK cells than iodinin (3) and myxin (4). As mentioned earlier, the iodinin-pyrrolidine analog 16 was also found to be more toxic than the iodinin carbamates 15 and 17. This could indicate that a pyrrolidine-carbamate based side-chain is unsuited for further development of a phenazine 5,10-dioxide anti-AML agents. The 7,8-dimethyl analogs 56 and 48 differ in their side chain (ethyl ester vs. diethyl amide) and the diethylamide 56 was more potent than the ethyl ester 48 towards the AML cell line MOLM-13 (Table 1). Both these analogs were less toxic in NRK and H9c2 compared to their 1-hydroxyphenazine 5,10-dioxide analogs 26 and 24.
Finally, a series of bicyclic aromatic N-oxides with carbamate side chains were evaluated (60–62). Despite their two N-oxides and their high membrane permeability (Table 1), we did not detect any cytotoxic activity towards the AML cell line MOLM-13 (EC50 > 200 μM, Table 1), indicating that three-cyclic aromatic rings are important for the cytotoxic activity.
Modes of action
Three main parameters which are anticipated to affect the activity of the 1-hydroxyphenazine 5,10-dioxide analogs are: 1) ability to cross biological membranes, 2) ability to unleash the cytotoxic hydroxyl radical (˙OH) via enzymatic bio-reductive activation, and 3) ability to intercalate into DNA which results in inhibited DNA replication, transcription or generation of strand breaks. In addition, metal chelation39 and redox cycling40 cannot be ruled out as factors contributing to the cytotoxicity. For instance, if the carbamate bonds of analogs 27–29 are enzymatically hydrolyzed and their N-oxides consequently reduced intracellularly as well, their common metabolic product is 1-hydroxyphenazine (22, 1-HP). 1-HP (22) is a well-known virulence factor of Pseudomonads aeruginosa, known to promote ROS production (the superoxide radical; O2˙−) via redox cycling.41,42 Further, 1-HP (22) can chelate iron and has been reported to cause iron starvation to the cells of A. fumigatus.42 Natural phenazines like 1-HP (22) can also reduce Fe(III) to Fe(II)42 which serves as the predominant catalyst of intracellular Fenton type reactions, that further can induce oxidative stress via production of ROS,43 leading to depletion of intracellular anti-oxidants.44 For each individual compound, the mode of action could therefore be a result of multiple factors acting simultaneously as is common for antineoplastic antibiotics, such as the anthracyclines, actinomycin, mitomycin and others. The anthracycline class of cancer chemotherapeutic agents have been used extensively for decades to treat various forms of cancer. Being potent inhibitors of nucleic acid synthesis, their mode of action is considered to stem from several factors acting simultaneously: 1) DNA-intercalation of their planar aromatic aglycone moiety, 2) ROS formation (O2˙− production leading to subsequent formation of H2O2 and ˙OH in the presence of Fe(II)) and 3) inhibition of topoisomerase II.45 Since myxin and iodinin are known intercalators,13,21,22 it requires further investigations to see if their derivatives presented herein have similar intercalating capabilities as their parent compounds. However, it is noteworthy that the bicyclic structures 60–62 showed no cytotoxicity in MOLM-13 indicating that intercalation might be a very important contributor to the phenazine 5,10-dioxides mode of action in addition to their ˙OH discharge.In correlation to carbamate metabolism in vivo, the anticancer drug irinotecan is an FDA approved carbamate prodrug which is biodegraded to the active camptothecin phenol-analog SN-38 by human liver microsomal carboxylesterases.46,47 Thus, whether the carbamates presented in this work (such as 15–20etc.) are hydrolyzed and cleaved from its parent 1-hydroxyphenazine 5,10-dioxide analog by intracellular enzymes prior to cell death is not yet established. We can therefore not exclude the possibility that the analogs themselves can generate a higher toxicity by e.g. generate ROS or facilitate DNA-intercalation prior to hydrolysis of the carbamate functional group.
To identify molecular structures which could be determinant for the desired anti-AML activity, we performed a Kernel-based partial least square regression (see ESI† document 2 for details on methods and results). The KPLS model showed that –OH substituents in position 1 and 6 of the analogs were associated with high activity towards AML cells, but not NRK cells. This suggests that analogs should be mono-substituted with an –OR group in order to retain AML selectivity, but this is probably not the case if the analogs are prodrugs where cleavage of the linker from the phenazine parent compound will result in an –OH group. The nature of the linker was also important for activity. In general, carbamate-linkers were associated with high activity, whereas carbonate-linkers related to low activity. Taken together, these data could be used to further optimize analogs of the phenazine 5,10-dioxide scaffold to develop highly active and selective compounds against AML.
Conclusions
In order to improve the drug properties of the phenazine 5,10-dioxide analogs, we added substituents to increase solubility in aqueous media, improve cellular drug internalization, and at the same time maintain the parts of the molecule which are crucial for their cytotoxic potential. Our previous report identified the N-oxides as imperative for the cytotoxic potential.26 The results presented herein further strengthen our hypothesis that the phenazine 5,10-dioxide is a promising scaffold to develop new chemotherapeutic candidates against acute myeloid leukemia. Analogs with only a single oxygen substituent (–OH or –OCH3) in the 6th position gives 1-hydroxyphenazine 5,10-dioxide (21), which is more convenient to synthesize than iodinin (3) and myxin (4). Although 1-hydroxyphenazine 5,10-dioxide (21) is not as potent as the lead compounds iodinin (3) and myxin (4), the cytotoxic potency of 21 could be restored by introduction of various side chains. Furthermore, adding substituents to the 7- and 8-positions of 1-hydroxyphenazine 5,10-dioxide scaffold afforded 7,8-dimethyl and 7,8-dihalogen analogs of superior potency against MOLM-13 cells compared with iodinin (3) and myxin (4). However, these were also found to be more toxic towards the non-malignant cell lines NRK and H9c2.We conclude that both iodinin and myxin carbamate prodrugs (15–20) and the 7,8-dimethyl analog 52 meet criteria to be investigated further as leads in terms of antileukemic activities, displaying both high potency and cell selectivity towards the MOLM-13 cell line. Published SAR studies on phenazine 5,10-dioxides are scarce in literature, but utilization of in silico KPLS models showed that the oxygen substituent in position 1 and 6 was involved in the AML selectivity observed for the analogs. Moreover, it also supported our findings that introduction of a fourth ring to create a tetracyclic structure shifted the activity to general cytotoxic, without preferences towards AML cells. We believe the synthetic methods and biological trends derived from the present work could pave the way, not only for enhanced research on phenazine 5,10-dioxides, but also to afford novel drug candidates for the treatment of AML with less side-effects than current standards in AML induction chemotherapy can offer.
Experimental
Synthesis of compounds
All information for synthetic experiments including general information, synthetic procedures, characterization data and figures of NMR spectra are provided in the ESI.†Cell maintenance and experimental conditions
All cell culturing media and serum were from Sigma-Aldrich (St. Louis, MO, USA). Product no. are given in parentheses. The human acute myeloid leukemia cell line MOLM-13 (ACC, 554)48 was cultured in RPMI medium (R5886) enriched with 10% fetal bovine serum (F7524) and 0.2 mM L-glutamine (G7513). The normal rat kidney epithelial (NRK, ATCC, CRL-6509) and the rat cardiomyoblast (H9c2. ATCC, CRL-1446) cells were cultured in DMEM medium (D6429) enriched with 10% fetal bovine serum (F7524). All cell lines were additionally supplemented with 100 IU mL−1 penicillin and 100 μg mL−1 streptomycin (P0781). The cells were cultured in a humidified atmosphere either under 2% or 20% O2 at 37 °C.All analogs were dissolved in DMSO (D5879, Sigma-Aldrich) at concentrations between 2.5 and 20 mM before testing. For cytotoxic testing, the MOLM-13 cells were seeded as 20000 cells/well, while NRK and H9c2 cells were seeded as 5000 cells/well in 96-well Microplates (#167008, Thermo Scientific™ Nunc™ MicroWell™) with 100 μL medium/well. The MOLM-13 cells were seeded at the day of the experiment, while the NRK and H9c2 cells were seeded the day before the experiment, to allow them to attach to the surface of the wells before adding compounds. The cells were exposed to various concentrations of iodinin or its analogs for 24 h before the cell proliferation reagent WST-1 was used to assess the viability, in accordance with the manufacturer's instructions (11644807001, Roche Diagnostics, Sigma-Aldrich). The cells were next fixed in 2% buffered formaldehyde (pH 7.4) with the DNA-specific dye Hoechst 33342 (Polysciences Inc.) and assessed for cell death by UV-microscopy based on nuclear morphology.49 The highest concentration of analog corresponded to 1% of DMSO, which alone gave less than 7% cell death as judged by nuclear morphology.
EC50 values were determined by analyses of WST-1 signal results as well as microscopic evaluation of cell death by four-parameter regression analyses using the SigmaPlot software (Systat Software Inc. San Jose, CA):
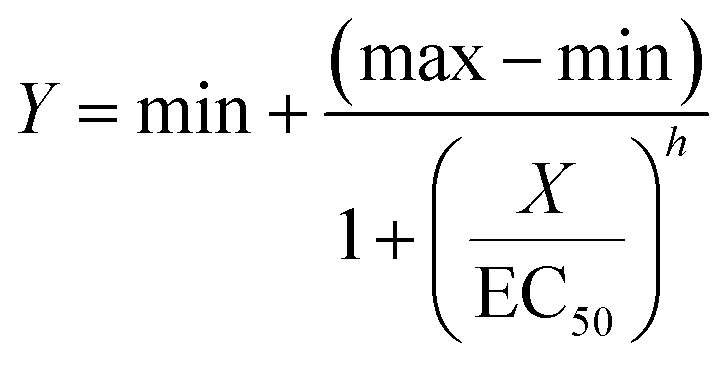
PAMPA (parallel artificial membrane permeability assay)
The membrane permeability of the analogs was investigated using the Corning Gentest™ pre-coated parallel artificial membrane permeability assay (PAMPA) Plate System (Cording Discovery Labware, Bedford, USA). The analog stock solution was diluted in PBS to 50 μM, and 300 μL was added to the donor plate. 200 μL PBS was added to the receiver plate, the plate system assembled, and incubated in the dark at room temperature. After 5 hours of incubation, 50 μl of the donor or acceptor well were injected into a reversed phase HPLC column (Kromasil 100–5 C18 150–4.6 mm, Akzo Nobel, Sweden) connected to a Merck-Hitachi LaChrome HPLC-system (VWR, WestChester, USA) with a L-7455 diode array detector. Mobile phase A was 0.05% aqueous TFA, and mobile phase B was acetonitrile. The flow rate was 1.6 ml min−1 and compounds eluted between 1 and 4.5 minutes was eluted during a 6.5 min gradient from 90:10% mobile phase A:B to 0:100% mobile phase A:B. The ratio of the compound concentrations in the donor and acceptor compartment was calculated after integration of the peaks at 285 nm. The value of the effective permeability, logPeff, was calculated as described in Bennion et al.50
Membrane permeability were classified by the range defined by Bennion et al., allowing us to divide the compounds into four groups, compounds with high permeability (logPeff > −5.33), intermediate permeability (logPeff > −5.66 and <−5.33), low permeability (logPeff > −6.14 and <−5.66) impermeable (logPeff < −6.14).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Oslo, School of Pharmacy, The Research Council of Norway, The Norwegian Cancer Society and Novo Pre Seed for funding this research. We also acknowledge professor Frode Rise and senior engineer Dirk Petersen for their maintenance of excellent NMR-facility at the University of Oslo. We would like to express gratitude as well to Dr. Alexander H. Åstrand for proofreading the experimental part of this work.References
- A. T. Garrison, Y. Abouelhassan, D. Kallifidas, F. Bai, M. Ukhanova, V. Mai, S. Jin, H. Luesch and R. W. Huigens, Angew. Chem., Int. Ed., 2015, 54, 14819–14823 CrossRef CAS PubMed.
- A. T. Garrison, Y. Abouelhassan, V. M. Norwood, D. Kallifidas, F. Bai, M. T. Nguyen, M. Rolfe, G. M. Burch, S. Jin, H. Luesch and R. W. Huigens, J. Med. Chem., 2016, 59, 3808–3825 CrossRef CAS PubMed.
- M. Weigele, G. Maestrone, M. Mitrovic and W. Leimgruber, Antimicrob. Agents Chemother., 1970, 10, 46–49 CAS.
- S. M. Lesley and R. M. Behki, J. Bacteriol., 1967, 94, 1837–1845 CrossRef CAS PubMed.
- A. Cimmino, A. Evidente, V. Mathieu, A. Andolfi, F. Lefranc, A. Kornienko and R. Kiss, Nat. Prod. Rep., 2012, 29, 487–501 RSC.
- H. Cerecetto, M. González, M. L. Lavaggi, A. Azqueta, A. López de Cerain and A. Monge, J. Med. Chem., 2005, 48, 21–23 CrossRef CAS PubMed.
- H. Cerecetto, M. Gonzalez, M. L. Lavaggi, M. A. Aravena, C. Rigol, C. Olea-Azar, A. Azqueta, A. L. d. Cerain, A. Monge and A. M. Bruno, Med. Chem., 2006, 2, 511–521 CrossRef CAS PubMed.
- M. Lavaggi, M. Cabrera, C. Pintos, C. Arredondo, G. Pachón, J. Rodríguez, S. Reymondo, J. Pacheco, M. Cascante, C. Olea-Azar, A. L. de Ceráin, A. Monge, H. Cerecetto and M. González, ISRN Pharmacol., 2011, 11 Search PubMed.
- N. Guttenberger, W. Blankenfeldt and R. Breinbauer, Bioorg. Med. Chem., 2017, 25(22), 6149–6166 CrossRef CAS PubMed.
- Y. Zhao, G. Qian, Y. Ye, S. Wright, H. Chen, Y. Shen, F. Liu and L. Du, Org. Lett., 2016, 18, 2495–2498 CrossRef CAS PubMed.
- L. Myhren, G. Nygaard, G. Gausdal, H. Sletta, K. Teigen, K. Degnes, K. Zahlsen, A. Brunsvik, Ø. Bruserud, S. Døskeland, F. Selheim and L. Herfindal, Mar. Drugs, 2013, 11, 332 CrossRef CAS PubMed.
- G. R. Clemo and A. F. Daglish, J. Chem. Soc., 1950, 1481–1485 RSC.
- J. G. Davis, Zentralblatt für Bakteriologie, Parasitenkunde, Infektionskrankheiten und Hygiene, 1939, 100, 273–276 CAS.
- H. McIlwain, Biochem. J., 1943, 37, 265–271 CrossRef CAS PubMed.
- I. Tanabe, N. Iio, R. Imanura, T. Matsumoto and A. Obayashi, Memories of the Faculty of Agriculture, Kagoshima Univeristy, 1976, vol. 10, pp. 127–125 Search PubMed.
- K. Hano, H. Iwata and K. Nakajima, Chem. Pharm. Bull., 1965, 13, 107–113 CrossRef CAS PubMed.
- H. Sletta, K. F. Degnes, L. Herfindal, G. Klinkenberg, E. Fjaervik, K. Zahlsen, A. Brunsvik, G. Nygaard, F. L. Aachmann, T. E. Ellingsen, S. O. Doskeland and S. B. Zotchev, Appl. Microbiol. Biotechnol., 2014, 98, 603–610 CrossRef CAS PubMed.
- E. A. Peterson, D. C. Gillespie and F. D. Cook, Can. J. Microbiol., 1966, 12, 221–230 CrossRef CAS PubMed.
- R. M. Behki and S. M. Lesley, J. Bacteriol., 1972, 109, 250–261 CrossRef CAS PubMed.
- G. Manius, R. Tscherne, R. Venteicher and A. Secker, J. Pharm. Sci., 1981, 70, 1024–1026 CrossRef CAS PubMed.
- U. Hollstein and P. L. Butler, Biochemistry, 1972, 11, 1345–1350 CrossRef CAS PubMed.
- U. Hollstein and R. J. Van Gemert, Biochemistry, 1971, 10, 497–504 CrossRef CAS PubMed.
- G. Chowdhury, U. Sarkar, S. Pullen, W. R. Wilson, A. Rajapakse, T. Fuchs-Knotts and K. S. Gates, Chem. Res. Toxicol., 2012, 25, 197–206 Search PubMed.
- B. Balasubramanian, W. K. Pogozelski and T. D. Tullius, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 9738–9743 CrossRef CAS PubMed.
- V. Junnotula, U. Sarkar, S. Sinha and K. S. Gates, J. Am. Chem. Soc., 2009, 131, 1015–1024 CrossRef CAS PubMed.
- E. Ö. Viktorsson, B. Melling Grøthe, R. Aesoy, M. Sabir, S. Snellingen, A. Prandina, O. A. Høgmoen Åstrand, T. Bonge-Hansen, S. O. Døskeland, L. Herfindal and P. Rongved, Bioorg. Med. Chem., 2017, 25, 2285–2293 CrossRef CAS PubMed.
- M. L. Lavaggi, M. Nieves, M. Cabrera, C. Olea-Azar, A. López de Ceráin, A. Monge, H. Cerecetto and M. González, Eur. J. Med. Chem., 2010, 45, 5362–5369 CrossRef CAS PubMed.
- V. Junnotula, A. Rajapakse, L. Arbillaga, A. López de Cerain, B. Solano, R. Villar, A. Monge and K. S. Gates, Bioorg. Med. Chem., 2010, 18, 3125–3132 CrossRef CAS PubMed.
- M. P. Hay, S. A. Gamage, M. S. Kovacs, F. B. Pruijn, R. F. Anderson, A. V. Patterson, W. R. Wilson, J. M. Brown and W. A. Denny, J. Med. Chem., 2003, 46, 169–182 CrossRef CAS PubMed.
- M. Loredana and O. Ian, Curr. Clin. Pharmacol., 2006, 1, 71–79 CrossRef PubMed.
- J. von Pawel, R. von Roemeling, U. Gatzemeier, M. Boyer, L. O. Elisson, P. Clark, D. Talbot, A. Rey, T. W. Butler, V. Hirsh, I. Olver, B. Bergman, J. Ayoub, G. Richardson, D. Dunlop, A. Arcenas, R. Vescio, J. Viallet and J. Treat, J. Clin. Oncol., 2000, 18, 1351–1359 CrossRef CAS PubMed.
- B. G. Wouters, L. H. Wang and J. M. Brown, Ann. Oncol., 1999, 10(Suppl 5), S29–S33 CrossRef PubMed.
- P. Vaupel and L. Harrison, Oncologist, 2004, 9, 4–9 CrossRef PubMed.
- J. M. Brown and W. R. Wilson, Nat. Rev. Cancer, 2004, 4, 437–447 CrossRef CAS PubMed.
- C. H. Issidorides, M. A. Atfah, J. J. Sabounji, A. R. Sidani and M. J. Haddadin, Tetrahedron, 1978, 34, 217–221 CrossRef CAS.
- M. Conda-Sheridan, L. Marler, E.-J. Park, T. P. Kondratyuk, K. Jermihov, A. D. Mesecar, J. M. Pezzuto, R. N. Asolkar, W. Fenical and M. Cushman, J. Med. Chem., 2010, 53, 8688–8699 CrossRef CAS PubMed.
- H. H. Lee, B. F. Leake, R. B. Kim and R. H. Ho, Mol. Pharmacol., 2017, 91, 14–24 CrossRef CAS PubMed.
- M. Okabe, M. Unno, H. Harigae, M. Kaku, Y. Okitsu, T. Sasaki, T. Mizoi, K. Shiiba, H. Takanaga, T. Terasaki, S. Matsuno, I. Sasaki, S. Ito and T. Abe, Biochem. Biophys. Res. Commun., 2005, 333, 754–762 CrossRef CAS PubMed.
- R. S. Fraser and J. Creanor, Biochem. J., 1975, 147, 401 CrossRef CAS PubMed.
- H. M. Hassan and I. Fridovich, J. Bacteriol., 1980, 141, 156–163 CrossRef CAS PubMed.
- S. Sinha, X. Shen, F. Gallazzi, Q. Li, J. W. Zmijewski, J. R. Lancaster and K. S. Gates, Chem. Res. Toxicol., 2015, 28, 175–181 Search PubMed.
- B. Briard, P. Bomme, B. E. Lechner, G. L. A. Mislin, V. Lair, M.-C. Prévost, J.-P. Latgé, H. Haas and A. Beauvais, Sci. Rep., 2015, 5, 8220 Search PubMed.
- J. A. Lemire, J. J. Harrison and R. J. Turner, Nat. Rev. Microbiol., 2013, 11, 371–384 Search PubMed.
- M. Muller, Free Radical Biol. Med., 2002, 33, 1527–1533 CrossRef CAS PubMed.
- M. B. Martins-Teixeira and I. Carvalho, ChemMedChem, 2020, 15, 933–948 CrossRef CAS PubMed.
- A. K. Ghosh and M. Brindisi, J. Med. Chem., 2015, 58, 2895–2940 CrossRef CAS PubMed.
- S. P. Sanghani, S. K. Quinney, T. B. Fredenburg, W. I. Davis, D. J. Murry and W. F. Bosron, Drug Metab. Dispos., 2004, 32, 505–511 CrossRef CAS PubMed.
- Y. Matsuo, R. A. F. MacLeod, C. C. Uphoff, H. G. Drexler, C. Nishizaki, Y. Katayama, G. Kimura, N. Fujii, E. Omoto, M. Harada and K. Orita, Leukemia, 1997, 11, 1469–1477 CrossRef CAS PubMed.
- L. Myhren, I. M. Nilssen, V. Nicolas, S. O. Doeskeland, G. Barratt and L. Herfindal, Eur. J. Pharm. Biopharm., 2014, 88, 186–193 CrossRef CAS PubMed.
- B. J. Bennion, N. A. Be, M. W. McNerney, V. Lao, E. M. Carlson, C. A. Valdez, M. A. Malfatti, H. A. Enright, T. H. Nguyen, F. C. Lightstone and T. S. Carpenter, J. Phys. Chem. B, 2017, 121, 5228–5237 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, characterization data, NMR spectra, detailed results from PAMPA assay, KPLS-data. See DOI: 10.1039/d1md00020a |
| This journal is © The Royal Society of Chemistry 2021 |