
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTargeting the SARS-CoV-2-spike protein: from antibodies to miniproteins and peptides
Sebastian
Pomplun
 *
*
Massachusetts Institute of Technology, 77 Massachusetts Ave, Cambridge, MA 02139, USA. E-mail: pompluns@mit.edu
First published on 17th December 2020
Abstract
Coronavirus disease-19, caused by the novel β-coronavirus SARS-CoV-2, has created a global pandemic unseen in a century. Rapid worldwide efforts have enabled the characterization of the virus and its pathogenic mechanism. An early key finding is that SARS-CoV-2 uses spike proteins, the virus' most exposed structures, to bind to human ACE2 receptors and initiate cell invasion. Competitive targeting of the spike protein is a promising strategy to neutralize virus infectivity. This review article summarizes the discovery, binding modes and eventual applications of several classes of (bio)molecules targeting the spike protein: antibodies, nanobodies, soluble ACE2 variants, miniproteins, peptides and small molecules.
 Sebastian Pomplun | Sebastian Pomplun studied Chemistry and Pharmaceutical Technologies at the University ‘Sapienza’ in Rome. In 2015 he obtained his PhD in medicinal chemistry at the Max-Planck-Institute for Psychiatry in Munich. For a first postdoc, Sebastian joined the Chemistry R&D department of Roche Diagnostics in Penzberg, where he worked on bioconjugation and functional building blocks for peptide and oligonucleotide synthesis. In 2019 he joined the Pentelute Lab at MIT, developed combinatorial biohybrid nucleobase peptide libraries and worked on the discovery of SARS-CoV-2-spike targeting peptides. His research interest is focused on combining biological structures and synthetic molecules for the development of novel therapeutic modalities. |
Introduction
The novel β-coronavirus SARS-CoV-2 (SARS = severe acute respiratory syndrome) emerged in late 2019 from a zoonotic source and has since caused a global pandemic.1,2 SARS-CoV-2 infection can lead to coronavirus disease-19 (COVID-19) with symptoms ranging from mild disease to severe lung injury and multi-organ failure, eventually leading to death, especially in older patients with other co-morbidities.3 With 50 million confirmed cases and over 1.2 million deaths (as of November 2020), SARS-CoV-2 has spread further than the closely related SARS-CoV-1 and MERS-CoV.4,5Coronaviruses display multiple copies of the spike glycoprotein on their surface (Fig. 1A).6 The characteristic crown-like shape inspired this family of viruses being named corona (which in Latin means crown). Spike proteins7,8 are, therefore, the most exposed structures of the virus.9 The Spike protein is a trimeric fusion protein consisting of the subunits S1 and S2 (Fig. 1C). The S1 unit contains the receptor binding domain (RBD) responsible for recognition and engagement with host cells. The RBD can assume an ‘up’ or a ‘down’ conformation.10 As previously shown for SARS-Cov-1, the SARS-CoV-2 RBD binds with high affinity (Kd ∼ 10 nM) in ‘up’ conformation to human angiotensin converting enzyme 2 (ACE2) receptors for initiating eukaryotic cell invasion.11–13 Receptor binding destabilizes the prefusion trimer and is followed by proteolytic cleavage between units S1 and S2 (e.g., by the transmembrane serine protease TMPRSS2).14 This leads to structural rearrangements and cell invasion (Fig. 1B). Among several potential SARS-CoV-2 drug targets,15 the crucial function of the spike protein makes it a vulnerable and important target for virus neutralization.16 Neutralization mechanisms can rely on the direct competition for the ACE2 binding site, steric hindrance of ACE2 binding from adjacent binding sites or on the inhibition of structural rearrangements necessary for fusion.
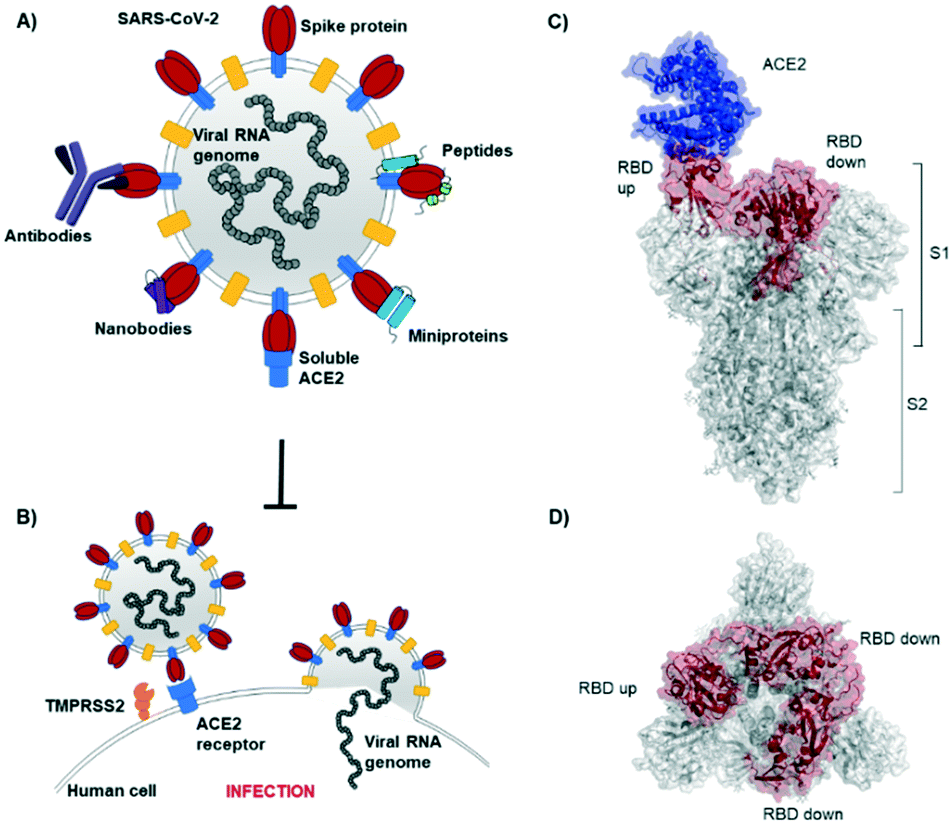 | ||
| Fig. 1 SARS-CoV-2 initiates cell entry upon spike protein binding to human ACE2 receptors: targeting the spike protein is a powerful strategy to neutralize virus infection. A) Schematic representation of a SARS-CoV-2 particle and several (bio) molecules binding to the spike protein. B) Simplified SARS-CoV-2 cell invasion mechanism scheme: the virus engages with human cells via SARS-CoV-2-spike protein binding to human ACE2 followed by proteolytic cleavage of the spike protein (by TMPRSS2) and fusion. C) Lateral representation of the trimeric SARS-CoV-2-spike protein. Human ACE2 (blue) is bound by one RBD (red) in ‘up’ conformation, the other two RBDs (red) are in ‘down’ conformation. The image was obtained by aligning PDB structures 6m0j and 6vsb with Pymol. D) Top view of the SARS-CoV-2-spike trimer (6vsb). | ||
In the past year, a considerable number of (bio)molecules targeting the spike protein has been identified. Many antibodies from the serum of SARS-CoV-2 convalescent patients have been isolated and characterized: spike protein binding is a primary mechanism of antibodies for virus neutralization. Also, minimized antibody mimetic scaffolds, such as nanobodies (mainly from cameloids), have been identified as neutralizing agents. Further, designer strategies for virus neutralization via spike protein binding include the use of variants of soluble human ACE2, rationally and computationally designed miniproteins and peptides. This review article outlines the discovery, binding, and applications of antibodies, nanobodies, miniproteins and peptides, targeting the SARS-CoV-2-spike protein (Table 1).
| Binding affinity to SARS-CoV-2-spike | Discovery strategy | Ref. | |
|---|---|---|---|
| Antibodies | Typically picomolar | Mainly isolation from patient serum | 17–21 |
| Nanobodies | Picomolar – nanomolar | Llama immunization, phage display | 23–26 |
| ACE2 variants | Nanomolar | Computational design, deep mutagenesis, affinity maturation | 27–30 |
| Miniproteins | Picomolar – nanomolar | Computational design, affinity maturation | 35 |
| Peptides | Nanomolar | Rational design, affinity selection – mass spectrometry | 36, 40 |
| Small molecules | Nanomolar | Serendipitous | 41 |
Antibodies
Humoral immune responses are important for protection against invading pathogens and include the production of specialized antibodies recognizing specific antigens of the invader. As the SARS-CoV-2-spike protein mediates entry into host cells and is prominently exposed at the viral surface, it is the main target of neutralizing antibodies.17 While in the future, the deployment of a vaccine might be a highly effective way to curtail the COVID-19 pandemic, neutralizing antibodies could offer more immediate solutions and be considered as treatment options for severe COVID-19 cases. Indeed, recent small-scale studies suggest that the treatment of COVID-19 patients with convalescent plasma improved clinical outcomes and decreased viral loads.17,18Antibodies from SARS-CoV-2 convalescent patients have been isolated and their activity and binding modes characterized. An impressive number of cryo-EM and crystal structures have been solved during this year, supporting the understanding of binding and neutralization mechanisms.
Studies describe antibodies with various binding modes to the SARS-CoV-2 spike protein. Ho et al. identified neutralizing antibodies from five COVID-19 patients. Some of the antibodies have potent virus neutralizing activity (<1 ng mL−1). Cryo-EM structures of three antibodies revealed binding to the RBD (down conformation, Fig. 2A), binding to the spike N-terminal domain (NTD) and binding to a quaternary epitope involving two copies of RBD. Also, Chi et al. solved the cryo-EM structure of an antibody binding to the NTD (Fig. 2B).19 The neutralizing activity of this antibody is likely due to steric hindrance interfering with the ACE2 engagement. Veesler et al. performed an extensive serological analysis study on ∼650 SARS-CoV-2 exposed individuals. After analyzing a large number of antibodies and their binding modes, they found the SARS-CoV-2 RBD being immunodominant and accounting for 90% of serum neutralizing activity.17 One of the antibodies, binding to two RBDs in the up conformation, is shown in Fig. 2C. Along this line of evidence, Bjorkman et al. found antibodies predominantly for the spike RBD, in addition to NTD binders. Further crystal structures of RBD binding neutralizing antibodies were solved by the groups of Wilson7 and Zhang.20
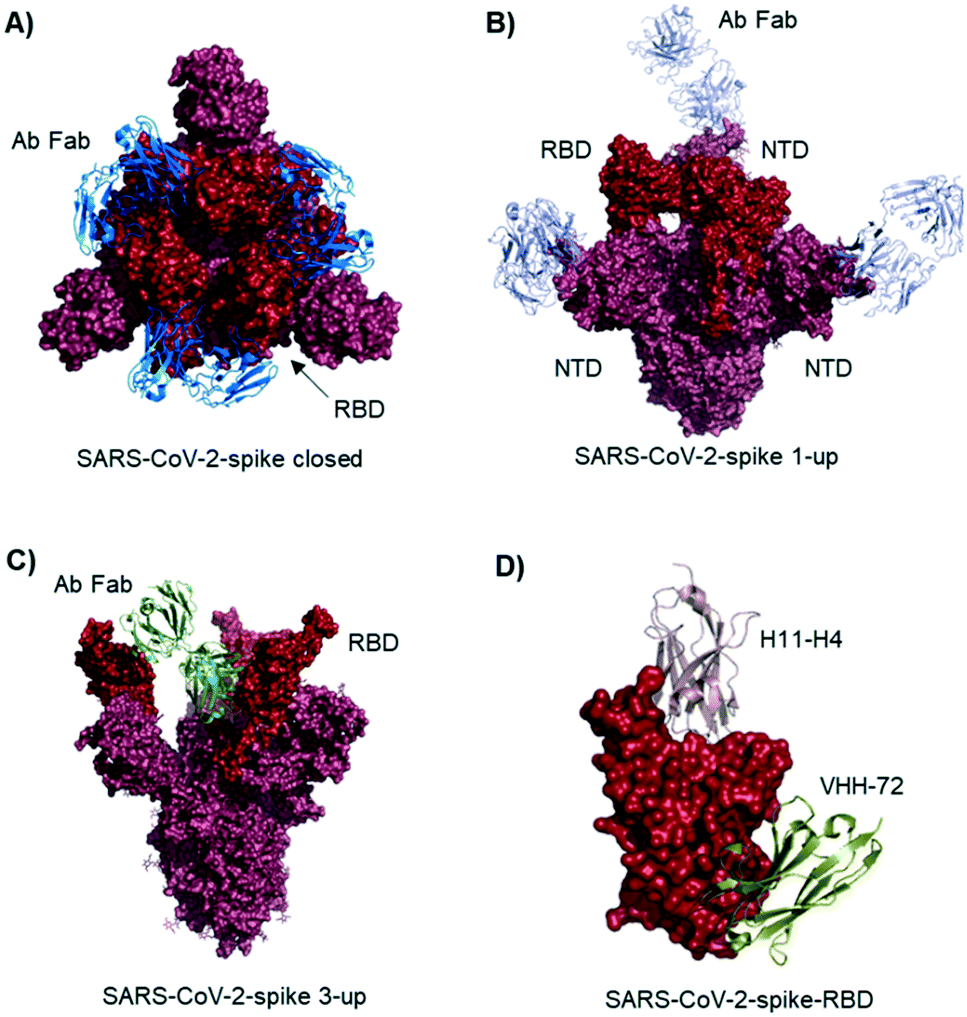 | ||
| Fig. 2 Antibodies and nanobodies can bind to the SARS-CoV-2-spike protein and neutralize virus infection. A) Top view of the spike trimer (surface shown in raspberry) with an antibody (from COVID-19 patient serum) Fab fragment (blue cartoons) bound to the RBD (red surface). All three RBDs are in ‘down’ conformation. PDB 6xey. B) Antibody Fab fragments (light blue cartoons) binding to the SARS-CoV-2-spike NTD (raspberry surface); PDB 7c21. C) SARS-CoV-2-spike protein with all three RBDs in ‘up’ conformation. One antibody Fab fragment (green cartoons) binds to two RBDs (red surface). PDB 7jw0 D) nanobodies H11-H4 (pink cartoon) and VHH-72 (gold cartoon) bound to SARS-CoV-2-spike RBD (red surface). Image obtained by combining PDB structures 6zh9 and 6waq. | ||
Many of the antibodies described above are putative candidates for the development of therapeutics. However, Kyratsous et al. described that novel spike mutants rapidly appeared after in vitro passaging in the presence of individual antibodies, resulting in loss of neutralization; escape mutants were not generated after treatment with a noncompeting antibody cocktail.21 Indeed, the serum of convalescent COVID-19 patients, containing such ‘antibody cocktails’ is an FDA approved emergency use therapy option.22 A retrospective study on the use of convalescent plasma by Bouvier et al. shows the therapeutic potential, but it pointed out that adequately powered, randomized controlled trials are still needed.18
Nanobodies
Nanobodies and other minimized antibody mimetics are interesting modalities for virus neutralization. These biomolecules maintain the ability of antibodies to bind with high potency and specificity to their antigens, while having a reduced grade of structural complexity.Using a phage display platform for fully human single domain antibodies Ying et al. selected binders that target several epitopes of SARS-CoV-2.23 They determined subnanomolar to low nanomolar affinities and identified variants with neutralizing activity in live virus infection assays.
McLellan et al. immunized llamas with prefusion-stabilized coronavirus spike protein (from SARS-CoV-1): one nanobody (VHH-72) shows cross-reactivity to SARS-CoV-2 and is neutralizing when used as an Fc fused dimer.24 Although targeting a site distal to the ACE2 binding surface (Fig. 2D), it causes enough steric hindrance to avoid RBD ACE2 engagement. Using a naive llama single-domain antibody library and PCR-based maturation, Naismith et al. found two potent RBD binding structures.25 Both were crystallized and analyzed (Fig. 1D, H11-H4). Also, these nanobodies exhibited virus-neutralizing activity in the form of Fc fusion complexes. Further synthetic llama nanobodies were described by Löw et al.:26 the authors describe variants with affinities for RBD as strong as 200 pM and neutralizing activity in live virus infection assays.
Soluble human ACE2 and derivatives
A promising strategy to prevent SARS-CoV-2 engagement with human cells is the use of soluble human ACE2 as a competitor: the natural binding partner of the spike protein in solution traps the RBD and inhibits binding to membrane-bound ACE2. Penninger et al. indeed showed a potent reduction of SARS-CoV-2 infection in cells and multiple human organoid models.27An advantage of using soluble ACE2 (and derivatives) to block spike proteins is the intrinsic resilience to viral mutational escape: several antibodies have been shown to lose activity after single mutations on the spike protein, with viruses eventually increasing their affinity for human ACE2.28 ACE2 traps increase their potency with growing virus affinity for membrane-bound ACE2.
Procko et al. used deep mutagenesis to generate ACE2 variants with increased affinity for the SARS-CoV-2-spike-RBD.29 They identified the best variants by displaying the ACE2 library on human cells, incubating with labelled RBD and sorting to select for the most potent binders. Wells et al. also engineered improved variants of ACE2:30 their strategy relied on the computational design of an improved binding interface and then a random mutagenesis/yeast display step to further optimize the sequence. Silva and collaborators at Neoleukin therapeutics used Rosetta “protein_mimic_designer” to engineer human ACE2 decoys. Their optimized structures include minimized variants that in a trimeric form show picomolar affinities for the SARS-CoV-2-spike protein, neutralize virus infection and display efficacious prophylaxis in hamster models when applied formulated as a nasal spray.28
Residues included in the α-helical N-terminus of ACE2 seem to provide the main interaction with the SARS-CoV-2-spike-RBD. Several studies early in 2020 suggested, after performing molecular dynamic simulations, that helix-1 alone or miniproteins consisting of helix-1 and helix-2 could bind to the RBD (Fig. 3A).31–33 Helix-1 mimetics had been shown to be successful for SARS-CoV-1 neutralization.34 In our laboratory, however, we tested helix-1 mimetics for SARS-CoV-2-spike-RBD binding, and did not observe association (data available as a preprint on BioRxiv).33 Experimental evidence for the two- helix miniproteins is still missing. A computational design strategy from the team around David Baker, however, resulted in ACE2 derived miniproteins with low nanomolar affinities and good thermostability.35
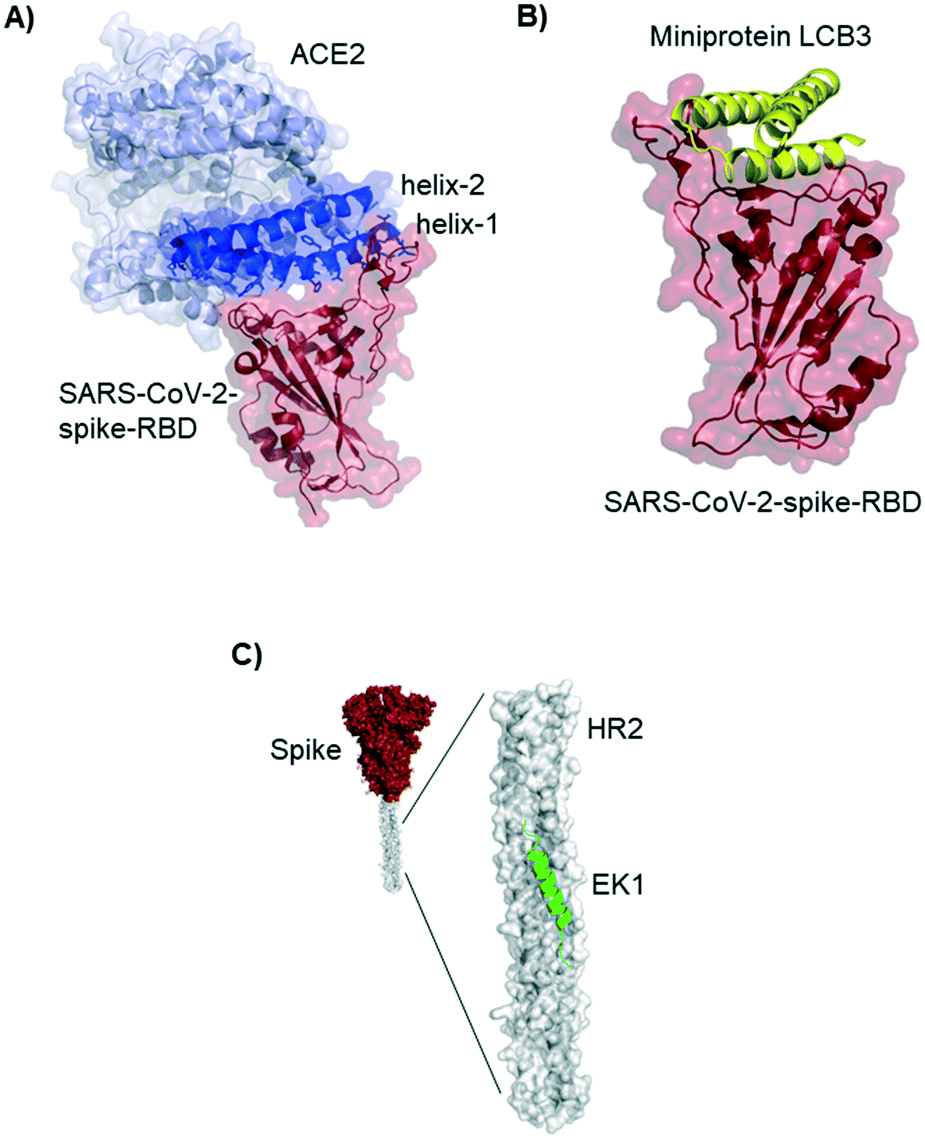 | ||
| Fig. 3 ACE2 derived and de novo designed miniproteins and peptides binding to the SARS-CoV-2-spike protein. A) Crystal structure 6m0j of ACE2 (blue) bound to SARS-CoV-2-spike-RBD (red). The ACE2 N-terminal helix-1 contains most of the residues interacting with the RBD and has been used as starting point for several design strategies for SARS-CoV-2 inhibitors. B) Cryo-EM structure (7jzm) of a computationally designed miniprotein (yellow cartoon) neutralizing SARS-CoV-2 in picomolar concentrations. C) Structural model (5zuv) of the fusion inhibitor EK1 (green cartoon) bound to the heptad repeat domain of the SARS-CoV-2 (grey surface). | ||
De novo discovery of miniproteins and peptides
Baker and colleagues also used computational de novo protein design strategies to engineer ACE2 competitive RBD binders from scratch.35 In SARS-CoV-2 infection assays of human cells the best miniprotein variant has a two digit picomolar inhibitory concentration. The structure of this 3-helix bundle bound to RBD is shown in Fig. 3B.Miniproteins are likely to have a fast clearance time (as most biomolecules with a molecular weight <60 kDa) in case of systemic applications. Their use in form of prophylactic nasal spray, however, is an interesting option for SARS-CoV-2 inhibition in the location of the first infection.
Lu et al. report a peptide targeting a completely different region of the spike protein: the peptide EK1 binds to the heptad repeat (HR2) region, thereby inhibiting structural rearrangement important for viral fusion with the cell membrane.36 The peptide was previously developed for SARS-CoV-1,37 but it is a pan-corona virus fusion inhibitor, also active in SARS-CoV-2 infection models.
In the Pentelute laboratory, we used a recently established high throughput affinity selection – mass spectrometry (AS – MS) platform38,39 to identify SARS-CoV-2-spike-RBD binders from synthetic peptide libraries (Fig. 4).40 Several peptides with a shared residue motif were selected and shown to associate to RBD with nanomolar affinities. While the peptides are not competing against ACE2 binding, their use as diagnostic tools is under investigation (information based on a BioRxiv preprint). The exposed position of the spike protein makes it, indeed, an intriguing structure for direct diagnostic detection.
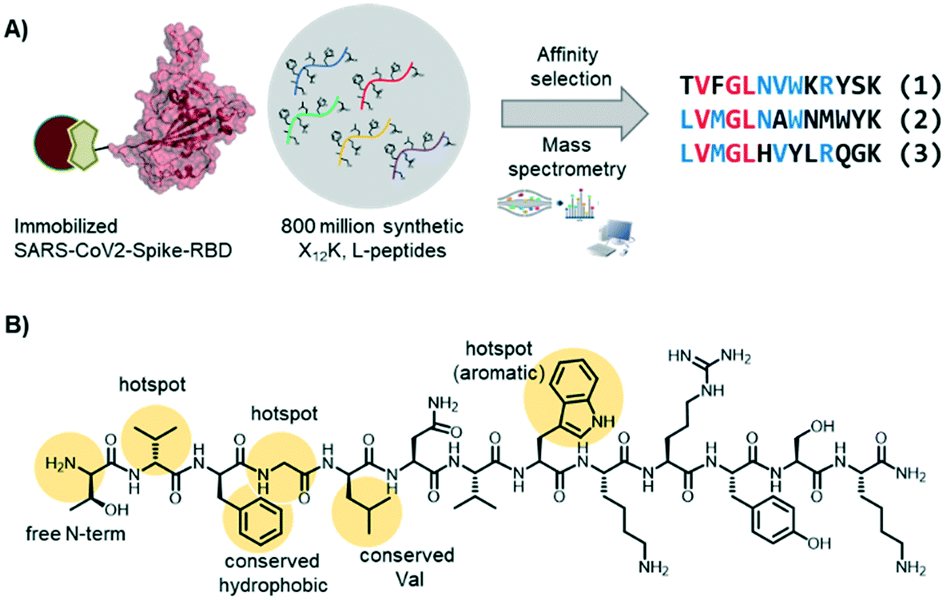 | ||
| Fig. 4 Short peptides with nanomolar affinity for the SARS-CoV-2-spike-RBD were identified by affinity selection – mass spectrometry. A) AS – MS scheme and sequences of peptide hits binding to SARS-CoV-2-spike-RBD. B) Structure of peptide hit 1 with important residue features evidence in yellow. | ||
Small molecules
To date, only one small molecule binding to the SARS-CoV-2-spike protein has been discovered. In a recent cryo-EM structure of the SARS-CoV-2 spike glycoprotein, an internal binding pocket was found to host the essential free fatty acid linoleic acid.41 Linoleic acid binding stabilizes a locked spike conformation giving rise to reduced ACE2 interaction in vitro. In human cells, linoleic acid supplementation synergizes with the COVID-19 drug remdesivir, with moderate inhibitory activity improvements.Conclusion and outlook
The emergence and spread of SARS-CoV-2 has caused a pandemic unmatched since the Spanish flu 1918. The SARS-CoV-2-spike protein is the spearhead of the virus: it binds to human cells with high avidity enabling viral cell invasion. However, it represents at the same time a vulnerable spot of the virus, which, among other potentially druggable virus proteins,42 makes it an intriguing target. Indeed, the human immune system can successfully neutralize virus infection with SARS-CoV-2-spike protein targeting antibodies. Also, many designer strategies to interfere with the SARS-CoV-2-spike ACE2 binding, based on affinity reagents of different nature, have been developed and shown high potential for COVID-19 treatment. Serum of convalescent COVID-19 patients containing neutralizing ‘antibody cocktails’ has been approved as emergency treatment; however, with the ongoing pandemic, solutions with broader applicability will be needed. Hopefully, several of the strategies and molecules described in this review might turn out as a robust starting point for developing efficacious COVID-19 prophylaxis or treatment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
I gratefully acknowledge Magdalena Vater and Matthias I. Gröschel for proofreading the manuscript and the Deutsche Forschungsgemeinschaft (DFG) for supporting me with a postdoctoral fellowship (PO 2413/1-1).Notes and references
- B. Hu, H. Guo, P. Zhou and Z. L. Shi, Nat. Rev. Microbiol., 2020 DOI:10.1038/s41579-020-00459-7.
- F. Wu, S. Zhao, B. Yu, Y. M. Chen, W. Wang, Z. G. Song, Y. Hu, Z. W. Tao, J. H. Tian, Y. Y. Pei, M. L. Yuan, Y. L. Zhang, F. H. Dai, Y. Liu, Q. M. Wang, J. J. Zheng, L. Xu, E. C. Holmes and Y. Z. Zhang, Nature, 2020, 579, 265–269 CrossRef CAS PubMed.
- W. J. Wiersinga, A. Rhodes, A. C. Cheng, S. J. Peacock and H. C. Prescott, JAMA, J. Am. Med. Assoc., 2020, 324, 782–793 CrossRef CAS.
- R. Lu, X. Zhao, J. Li, P. Niu, B. Yang, H. Wu, W. Wang, H. Song, B. Huang, N. Zhu, Y. Bi, X. Ma, F. Zhan, L. Wang, T. Hu, H. Zhou, Z. Hu, W. Zhou, L. Zhao, J. Chen, Y. Meng, J. Wang, Y. Lin, J. Yuan, Z. Xie, J. Ma, W. J. Liu, D. Wang, W. Xu, E. C. Holmes, G. F. Gao, G. Wu, W. Chen, W. Shi and W. Tan, Lancet, 2020, 395, 565–574 CrossRef CAS.
- J. Cui, F. Li and Z. L. Shi, Nat. Rev. Microbiol., 2019, 17, 181–192 CrossRef CAS PubMed.
- F. Li, Annu. Rev. Virol., 2016, 3, 237–261 CrossRef CAS PubMed.
- M. Yuan, N. C. Wu, X. Zhu, C. C. D. Lee, R. T. Y. So, H. Lv, C. K. P. Mok and I. A. Wilson, Science, 2020, 368, 630–633 CrossRef CAS PubMed.
- D. Wrapp, N. Wang, K. S. Corbett, J. A. Goldsmith, C. L. Hsieh, O. Abiona, B. S. Graham and J. S. McLellan, Science, 2020, 367, 1260–1263 CrossRef CAS PubMed.
- Z. Ke, J. Oton, K. Qu, M. Cortese, V. Zila, L. McKeane, T. Nakane, J. Zivanov, C. J. Neufeldt, B. Cerikan, J. M. Lu, J. Peukes, X. Xiong, H. G. Kräusslich, S. H. W. Scheres, R. Bartenschlager and J. A. G. Briggs, Nature, 2020 DOI:10.1038/s41586-020-2665-2.
- R. Henderson, R. J. Edwards, K. Mansouri, K. Janowska, V. Stalls, S. M. C. Gobeil, M. Kopp, D. Li, R. Parks, A. L. Hsu, M. J. Borgnia, B. F. Haynes and P. Acharya, Nat. Struct. Mol. Biol., 2020, 27, 925–933, DOI:10.1038/s41594-020-0479-4.
- Q. Wang, Y. Zhang, L. Wu, S. Niu, C. Song, Z. Zhang, G. Lu, C. Qiao, Y. Hu, K. Y. Yuen, Q. Wang, H. Zhou, J. Yan and J. Qi, Cell, 2020, 181, 894–904.e9 CrossRef CAS PubMed.
- J. Lan, J. Ge, J. Yu, S. Shan, H. Zhou, S. Fan, Q. Zhang, X. Shi, Q. Wang, L. Zhang and X. Wang, Nature, 2020, 581, 215–220 CrossRef CAS PubMed.
- J. Shang, G. Ye, K. Shi, Y. Wan, C. Luo, H. Aihara, Q. Geng, A. Auerbach and F. Li, Nature, 2020, 581, 221–224 CrossRef CAS.
- M. Hoffmann, H. Kleine-Weber, S. Schroeder, N. Krüger, T. Herrler, S. Erichsen, T. S. Schiergens, G. Herrler, N. H. Wu, A. Nitsche, M. A. Müller, C. Drosten and S. Pöhlmann, Cell, 2020, 181, 271–280.e8 CrossRef CAS PubMed.
- C. Liu, Q. Zhou, Y. Li, L. V. Garner, S. P. Watkins, L. J. Carter, J. Smoot, A. C. Gregg, A. D. Daniels, S. Jervey and D. Albaiu, ACS Cent. Sci., 2020, 6, 315–331 CrossRef CAS PubMed.
- T. Noy-Porat, E. Makdasi, R. Alcalay, A. Mechaly, Y. Levy, A. Bercovich-Kinori, A. Zauberman, H. Tamir, Y. Yahalom-Ronen, M. Israeli, E. Epstein, H. Achdout, S. Melamed, T. Chitlaru, S. Weiss, E. Peretz, O. Rosen, N. Paran, S. Yitzhaki, S. C. Shapira, T. Israely, O. Mazor and R. Rosenfeld, Nat. Commun., 2020, 11, 1–7 Search PubMed.
- L. Piccoli, Y. J. Park, M. A. Tortorici, N. Czudnochowski, A. C. Walls, M. Beltramello, C. Silacci-Fregni, D. Pinto, L. E. Rosen, J. E. Bowen, O. J. Acton, S. Jaconi, B. Guarino, A. Minola, F. Zatta, N. Sprugasci, J. Bassi, A. Peter, A. De Marco, J. C. Nix, F. Mele, S. Jovic, B. F. Rodriguez, S. V. Gupta, F. Jin, G. Piumatti, G. Lo Presti, A. F. Pellanda, M. Biggiogero, M. Tarkowski, M. S. Pizzuto, E. Cameroni, C. Havenar-Daughton, M. Smithey, D. Hong, V. Lepori, E. Albanese, A. Ceschi, E. Bernasconi, L. Elzi, P. Ferrari, C. Garzoni, A. Riva, G. Snell, F. Sallusto, K. Fink, H. W. Virgin, A. Lanzavecchia, D. Corti and D. Veesler, Cell, 2020, 1–19 Search PubMed.
- S. T. H. Liu, H. M. Lin, I. Baine, A. Wajnberg, J. P. Gumprecht, F. Rahman, D. Rodriguez, P. Tandon, A. Bassily-Marcus, J. Bander, C. Sanky, A. Dupper, A. Zheng, F. T. Nguyen, F. Amanat, D. Stadlbauer, D. R. Altman, B. K. Chen, F. Krammer, D. R. Mendu, A. Firpo-Betancourt, M. A. Levin, E. Bagiella, A. Casadevall, C. Cordon-Cardo, J. S. Jhang, S. A. Arinsburg, D. L. Reich, J. A. Aberg and N. M. Bouvier, Nat. Med., 2020, 26, 1708–1713, DOI:10.1038/s41591-020-1088-9.
- X. Chi, R. Yan, J. Zhang, G. Zhang, Y. Zhang, M. Hao, Z. Zhang, P. Fan, Y. Dong, Y. Yang, Z. Chen, Y. Guo, J. Zhang, Y. Li, X. Song, Y. Chen, L. Xia, L. Fu, L. Hou, J. Xu, C. Yu, J. Li, Q. Zhou and W. Chen, Science, 2020, 369, 650–655 CrossRef CAS PubMed.
- B. Ju, Q. Zhang, J. Ge, R. Wang, J. Sun, X. Ge, J. Yu, S. Shan, B. Zhou, S. Song, X. Tang, J. Yu, J. Lan, J. Yuan, H. Wang, J. Zhao, S. Zhang, Y. Wang, X. Shi, L. Liu, J. Zhao, X. Wang, Z. Zhang and L. Zhang, Nature, 2020, 584, 115–119 CrossRef CAS.
- A. Baum, B. O. Fulton, E. Wloga, R. Copin, K. E. Pascal, V. Russo, S. Giordano, K. Lanza, N. Negron, M. Ni, Y. Wei, G. S. Atwal, A. J. Murphy, N. Stahl, G. D. Yancopoulos and C. A. Kyratsous, Science, 2020, 1014–1018 CrossRef CAS.
- https//www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization#coviddrugs; Novemb. 2020.
- Y. Wu, C. Li, S. Xia, X. Tian, Y. Kong, Z. Wang, C. Gu, R. Zhang, C. Tu, Y. Xie, Z. Yang, L. Lu, S. Jiang and T. Ying, Cell Host Microbe, 2020, 891–898 CrossRef CAS.
- D. Wrapp, D. De Vlieger, K. S. Corbett, G. M. Torres, W. Van Breedam, K. Roose, L. van Schie, V.-C. C. -19 R. Team, M. Hoffmann, S. Pöhlmann, B. S. Graham, N. Callewaert, B. Schepens, X. Saelens and J. S. McLellan, Cell, 2020, 181, 1004–1015, DOI:10.1016/j.cell.2020.04.031.
- J. Huo, A. Le Bas, R. R. Ruza, H. M. E. Duyvesteyn, H. Mikolajek, T. Malinauskas, T. K. Tan, P. Rijal, M. Dumoux, P. N. Ward, J. Ren, D. Zhou, P. J. Harrison, M. Weckener, D. K. Clare, V. K. Vogirala, J. Radecke, L. Moynié, Y. Zhao, J. Gilbert-Jaramillo, M. L. Knight, J. A. Tree, K. R. Buttigieg, N. Coombes, M. J. Elmore, M. W. Carroll, L. Carrique, P. N. M. Shah, W. James, A. R. Townsend, D. I. Stuart, R. J. Owens and J. H. Naismith, Nat. Struct. Mol. Biol., 2020, 27, 846–854 CrossRef CAS.
- T. Custódio, H. Das, D. Sheward, L. Hanke, S. Pazicky, J. Pieprzyk, M. Sorgenfrei, M. Schroer, A. Gruzinov, C. Jeffries, M. Graewert, D. Svergun, N. Dobrev, K. Remans, M. Seeger, G. McInerney, B. Murrell, B. M. Hällberg and C. Löw, Nat. Commun., 2020, 11, 5588, DOI:10.1101/2020.06.23.165415.
- V. Monteil, H. Kwon, P. Prado, A. Hagelkrüys, R. A. Wimmer, M. Stahl, A. Leopoldi, E. Garreta, C. Hurtado del Pozo, F. Prosper, J. P. Romero, G. Wirnsberger, H. Zhang, A. S. Slutsky, R. Conder, N. Montserrat, A. Mirazimi and J. M. Penninger, Cell, 2020, 181, 905–913.e7 CrossRef CAS PubMed.
- T. W. Linsky, R. Vergara, N. Codina, J. W. Nelson, M. J. Walker, W. Su, C. O. Barnes, T.-Y. Hsiang, K. Esser-Nobis, K. Yu, Z. B. Reneer, Y. J. Hou, T. Priya, M. Mitsumoto, A. Pong, U. Y. Lau, M. L. Mason, J. Chen, A. Chen, T. Berrocal, H. Peng, N. S. Clairmont, J. Castellanos, Y.-R. Lin, A. Josephson-Day, R. S. Baric, D. H. Fuller, C. D. Walkey, T. M. Ross, R. Swanson, P. J. Bjorkman, M. Gale, L. M. Blancas-Mejia, H.-L. Yen and D.-A. Silva, Science, 2020, eabe0075 Search PubMed.
- K. K. Chan, D. Dorosky, P. Sharma, S. A. Abbasi, J. M. Dye, D. M. Kranz, A. S. Herbert and E. Procko, Science, 2020, 369, 1261–1265 CrossRef CAS PubMed.
- A. Glasgow, J. Glasgow, D. Limonta, P. Solomon, I. Lui, Y. Zhang, M. A. Nix, N. J. Rettko, S. Zha, R. Yamin, K. Kao, O. S. Rosenberg, J. V. Ravetch, A. P. Wiita, K. K. Leung, S. A. Lim, X. X. Zhou, T. C. Hobman, T. Kortemme and J. A. Wells, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 28046–28055 CrossRef CAS PubMed.
- Y. Han and P. Král, ACS Nano, 2020, 14, 5143–5147 CrossRef CAS PubMed.
- P. Chaturvedi, Y. Han, P. Král and L. Vuković, Adv. Theory Simul., 2020, 2000156, 1–6 Search PubMed.
- G. Zhang, S. Pomplun, A. R. Loftis, X. Tan, A. Loas and B. L. Pentelute, bioRxiv DOI:10.1101/2020.03.19.999318v2.
- D. P. Han, A. Penn-Nicholson and M. W. Cho, Virology, 2006, 350, 15–25 CrossRef CAS PubMed.
- L. Cao, I. Goreshnik, B. Coventry, J. B. Case, L. Miller, L. Kozodoy, R. E. Chen, L. Carter, A. C. Walls, Y.-J. Park, E.-M. Strauch, L. Stewart, M. S. Diamond, D. Veesler and D. Baker, Science, 2020, 431, eabd9909 Search PubMed.
- S. Xia, M. Liu, C. Wang, W. Xu, Q. Lan, S. Feng, F. Qi, L. Bao, L. Du, S. Liu, C. Qin, F. Sun, Z. Shi, Y. Zhu, S. Jiang and L. Lu, Cell Res., 2020, 30, 343–355 CrossRef CAS PubMed.
- S. Xia, L. Yan, W. Xu, A. S. Agrawal, A. Algaissi, C. T. K. Tseng, Q. Wang, L. Du, W. Tan, I. A. Wilson, S. Jiang, B. Yang and L. Lu, Sci. Adv., 2019, 5 DOI:10.1126/sciadv.aav4580.
- A. J. Quartararo, Z. P. Gates, B. A. Somsen, N. Hartrampf, X. Ye, A. Shimada, Y. Kajihara, C. Ottman and B. L. Pentelute, Nat. Commun., 2020, 11, 3183 CrossRef CAS PubMed.
- S. Pomplun, Z. P. Gates, G. Zhang, A. J. Quartararo and B. L. Pentelute, J. Am. Chem. Soc., 2020, 142(46), 19642–19651, DOI:10.1021/jacs.0c08964.
- S. Pomplun, M. Jbara, A. J. Quartararo, G. Zhang, J. S. Brown, Y.-C. Lee, X. Ye, S. Hanna and B. L. Pentelute, bioRxiv, 2020 DOI:10.1101/2020.09.29.317131.
- C. Toelzer, K. Gupta, S. K. N. Yadav, U. Borucu, A. D. Davidson, M. Kavanagh Williamson, D. K. Shoemark, F. Garzoni, O. Staufer, R. Milligan, J. Capin, A. J. Mulholland, J. Spatz, D. Fitzgerald, I. Berger and C. Schaffitzel, Science, 2020, 3255, eabd3255 Search PubMed.
- A. Saxena, J. Biosci., 2020, 45, 87 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |