
Medicinal chemistry updates on quinoline- and endoperoxide-based hybrids with potent antimalarial activity
Amad
Uddin
ab,
Meenal
Chawla
a,
Iram
Irfan
a,
Shubhra
Mahajan
a,
Shailja
Singh
b and
Mohammad
Abid
 *a
*a
aMedicinal Chemistry Laboratory, Department of Biosciences, Jamia Millia Islamia, Jamia Nagar, New Delhi-110025, India. E-mail: mabid@jmi.ac.in; Tel: +91 8750295095
bSpecial Centre for Molecular Medicine, Jawaharlal Nehru University, New Delhi-110067, India
First published on 7th November 2020
Abstract
The resistance of conventional antimalarial drugs against the malarial parasite continues to pose a challenge to control the disease. The indiscriminate exploitation of the available antimalarials has resulted in increasing treatment failures, which urges on the search for novel lead molecules. Artemisinin-based combination therapy (ACT) is the current WHO-recommended first-line treatment for the majority of malaria cases. Hybrid molecules offer a newer strategy for the development of next-generation antimalarial drugs. These comprise molecules, each with an individual pharmacological activity, linked together into a single hybrid molecule. This approach has been utilized by several research groups to develop molecules with potent antimalarial activity. In this review, we provide an overview of the pivotal roles of quinoline- and endoperoxide-based hybrids as inhibitors of the life-cycle progression of Plasmodium. Based on the exhaustive literature reports, we have collated the structural and functional analyses of quinoline- and endoperoxide-based hybrid molecules that show potency equal to or greater than those of the individual compounds, offering an effective therapeutics option for clinical use.
 Amad Uddin | Amad Uddin was born in 1991 and received his BSc (Hons) in Zoology in 2013 from the University of Delhi, New Delhi and MSc in Biosciences in 2015 from Jamia Millia Islamia, New Delhi. In December 2017, he registered on a PhD program under the joint supervision of Dr Mohammad Abid at the Department of Biosciences, Jamia Millia Islamia and Dr Shailja Singh at the Special Centre for Molecular Medicine, Jawaharlal Nehru University, New Delhi, India. Currently, he is a third year PhD student working on rational drug design and computational approaches for anti-malarial drug discovery. |
 Meenal Chawla | Meenal Chawla graduated in Zoology (Hons) from Kirori Mal College, Delhi University. She then completed her Master's degree in Biosciences from Jamia Millia Islamia. While a Master's student, she was the recipient of a Jawahar Bhawan Trust Scholarship for securing first position in the 1st year examinations. She passed the Junior Research Fellowship exam in June 2019. Her Master's thesis identified through in silico screening lead molecules that can target the FP-2 enzyme of the malaria parasite Plasmodium falciparum. She looks forward to carrying out biomedical research as it is at the heart of medical breakthroughs in healthcare. |
 Iram Irfan | Dr Iram Irfan is working as a Woman Scientist in the Department of Bioscience, Jamia Millia Islamia (JMI), New Delhi, India. She was awarded a PhD degree in Organic and Medicinal Chemistry from JMI in 2009 under the supervision of Professor Amir Azam. Currently she is working on ‘Structure-based design and synthesis of cysteine protease inhibitors as anti-malarial agents’, a project awarded by the Department of Science and Technology (DST), Government of India, under the supervision of Dr Mohammad Abid. Her area of research is antimalarial drug design and natural product modifications. |
 Shubhra Mahajan | Shubhra Mahajan graduated in Life Sciences from Kalindi College, Delhi University and is a post-graduate in Biosciences from Jamia Millia Islamia. Her Master's thesis was based on ‘Quinoline based derivatives of falcipain-2 inhibitors: anti-malarial and DNA binding studies’. She is currently working with the Ministry of Education, United Arab Emirates as a science teacher. |
 Shailja Singh | Dr Shailja Singh is working as an Associate Professor in the Special Centre for Molecular Medicine, Jawaharlal Nehru University (JNU), New Delhi. Dr Singh obtained her PhD in BioMedical Sciences from the University of Delhi, New Delhi. Dr Singh has received awards for her research work, i.e. the Young Biotechnologists Award 2010, Senior Innovative Young Biotechnologist Award 2013 and National Bio-Scientist Award 2018. A Gandhian Young Technological Innovation (GYTI) Award was made to her team in 2019. Shailja's extensive fundamental research on the biology of human parasites (malaria and Leishmania) has shown great potential in translational research towards drug discovery and development. |
 Mohammad Abid | Mohammad Abid was awarded a PhD degree in Organic & Medicinal chemistry from Jamia Millia Islamia (JMI), New Delhi, India. He undertook postdoctoral studies at UMR-8601, University Paris Descartes, France on antibacterial drug discovery targeting PDF and MetAP enzymes using a combined approach from September 2008 to January 2010. Dr Abid has held an academic position at JMI since December 2010. He also received a Young Scientist research grant from the Science & Engineering Research Board (SERB), Government of India. Recently he was awarded the prestigious Raman Postdoctoral Fellowship by the UGC to work as visiting Assistant Professor at Eppley Institute for Research in Cancer & Allied Diseases, University of Nebraska Medical Center (UNMC), Omaha, USA. Dr Abid's research group explores numerous interests within the discipline of structure-based drug design and medicinal chemistry. |
1. Introduction
Malaria is the most endemic, tropical parasitic, a vector-borne infectious disease transmitted by female Anopheles mosquitoes all around the globe.1 WHO in 2018 reported around 228 million cases and 405![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths from malaria worldwide, as compared with 231 million cases and 416000 estimated deaths in 2017.2 Six Plasmodium species that can cause zoonotic infection to humans are P. falciparum, P. vivax, P. ovale, P. knowlesi, P. simium and P. malariae.3,4P. falciparum is the most prevalent and most dangerous, responsible for over 90% of deaths and accounting for more than 95% of malaria cases globally.5,6 Widespread resistance limits the effectiveness of available antimalarial drugs, thus posing a significant challenge to public health. The situation is more precarious owing to the emergence of fast-spreading resistance in P. falciparum against chloroquine and artemisinin.6,7 This warrants the development of newer and safer drugs for the eradication of malaria. Amongst the oldest antimalarials, the alkaloid quinine, first isolated in 1820 from the bark of the cinchona tree, is on the WHO's list of essential medicines (Fig. 1). This is considered to be the safest and most effective antimalarial drug in the healthcare system.8,9 Based on the structure of quinine, chloroquine (CQ) was developed and used for malarial treatment because of its rapid efficacy and low toxicity.10 Many other quinolines such as amodiaquine, primaquine, and mefloquine were subsequently developed against chloroquine-resistant (CQR) strains of P. falciparum.11 Mechanistically, quinolines are recognized to inhibit the polymerization of heme, so preventing the disposal of polymers from the food vacuole to the cytoplasm, where hemozoin is formed. This leads to the intra-parasitic accumulation of free heme, which is lethal to the parasite. Some other vital targets that are involved in the antimalarial action of quinolines include tyrosine kinase, DNA, hemoglobin-degrading proteases, and phospholipases.12
000 deaths from malaria worldwide, as compared with 231 million cases and 416000 estimated deaths in 2017.2 Six Plasmodium species that can cause zoonotic infection to humans are P. falciparum, P. vivax, P. ovale, P. knowlesi, P. simium and P. malariae.3,4P. falciparum is the most prevalent and most dangerous, responsible for over 90% of deaths and accounting for more than 95% of malaria cases globally.5,6 Widespread resistance limits the effectiveness of available antimalarial drugs, thus posing a significant challenge to public health. The situation is more precarious owing to the emergence of fast-spreading resistance in P. falciparum against chloroquine and artemisinin.6,7 This warrants the development of newer and safer drugs for the eradication of malaria. Amongst the oldest antimalarials, the alkaloid quinine, first isolated in 1820 from the bark of the cinchona tree, is on the WHO's list of essential medicines (Fig. 1). This is considered to be the safest and most effective antimalarial drug in the healthcare system.8,9 Based on the structure of quinine, chloroquine (CQ) was developed and used for malarial treatment because of its rapid efficacy and low toxicity.10 Many other quinolines such as amodiaquine, primaquine, and mefloquine were subsequently developed against chloroquine-resistant (CQR) strains of P. falciparum.11 Mechanistically, quinolines are recognized to inhibit the polymerization of heme, so preventing the disposal of polymers from the food vacuole to the cytoplasm, where hemozoin is formed. This leads to the intra-parasitic accumulation of free heme, which is lethal to the parasite. Some other vital targets that are involved in the antimalarial action of quinolines include tyrosine kinase, DNA, hemoglobin-degrading proteases, and phospholipases.12
 | ||
| Fig. 1 WHO's list of essential antimalarial medicines. | ||
In 1972, artemisinin (ART), an enantiopure sesquiterpene lactone, was extracted from the Chinese medicinal plant Artemisia annua L. (sweet wormwood) by Youyou Tu (Nobel Prize 2015). It is used for the treatment of multi-drug-resistant (MDR) forms of P. falciparum.13 The most accepted theory for the mechanism of action of ART is its activation by heme to generate free radicals, which damage proteins required for parasite survival (Tse et al., 2019).14 The artemisinins are potent compounds against the pathogenic asexual blood stages of Plasmodium parasites and act on the transmissible sexual stages.15 The clinical use of ART is limited owing to its poor solubility and bioavailability. To overcome these issues, several ART derivatives (i.e., dihydroartemisinin, artemether, arteether, and artesunates) with better pharmacodynamic characteristics and pharmacokinetic properties were discovered.16 For an uncomplicated treatment of P. falciparum, WHO-approved artemisinin-based combination therapies (ACTs) are a first-line treatment. They have significantly controlled malaria incidence and mortality during the last two decades.17 ACTs are combinations of semi-synthetic artemisinin derivatives with a long-acting antimalarial drug (e.g., lumefantrine, mefloquine, amodiaquine, piperaquine, pyronaridine, sulfadoxine–pyrimethamine) which eliminates remaining parasites and limits the survival of resistant Plasmodium strains. However, there have been reports of decreased susceptibility of the parasite to artemisinins in the Greater Mekong Subregion (GMS), followed by ACTs' failure, worsening the situation to an extremely problematic level.18
This has compelled researchers to explore newer, multifaceted avenues to find antimalarial drugs. Early approaches include but are not limited to (a) chemical modification of known drugs, (b) drug repurposing (antifolates, tetracyclines, atovaquone), (c) use of natural products and their semi-synthetic analogs, (d) use of drug resistance reversers, and (e) identification of novel molecular targets for structure-based design.
A better understanding of the biology of the parasite provides many attractive targets that need to be investigated. The life-cycle of the parasite is complex as it involves the exoerythrocytic and erythrocytic cycle in humans (vertebrate) and sporogenic cycle in Anopheles mosquitoes. The parasitic cycle starts when sporozoites enter the human bloodstream during biting by an infected Anopheles mosquito and infect hepatocytes (liver cells), where they mature and produce exoerythrocytic merozoite forms that are set free to enter into the bloodstream.
Trophozoites are formed when erythrocytes are invaded by merozoites and finally mature into schizonts. Schizonts rupture and release the merozoites, which reinvade new erythrocytes. Asexual blood-stage-formed gametocytes enter into the gut of a blood-feeding mosquito, where they develop into male and female gametes. Zygotes are formed by the fusion of gametes that have matured into ookinetes and oocysts, and lastly, the sporozoites migrate into the salivary gland of the mosquito. The entry of the sporozoites into a new human host repeats the malaria life-cycle. Different drug targets studied in the malarial life-cycle include food vacuole, plasma membrane, apicoplast, mitochondrion, transporter, and cytosol. An overview of the different pathways, the enzymes that can be inhibited in these targets and the standard classes of antimalarial drugs with their possible inhibition point in the life-cycle of the parasite is shown in Fig. 2. A recent approach that has gained momentum worldwide is the development of antimalarial hybrids. Hybrid compounds are formed when two structural or pharmacophoric moieties combine into a single molecule. In the process, two molecules are covalently linked and developed into a hybrid molecule, each having its own activity. Hybrid molecules lessen the risk of drug resistance and can be designed to act on different stages of the life-cycle of the parasite.19 They were also found to be more efficient than the multi-component drugs because of the lower incidence of drug–drug adverse effects.20 The growing significance and advantages of hybrids and their potentials have been reviewed previously.19,21 Muregi et al. (2010) discussed the development of several endoperoxide- and CQ-based hybrids offering the potential to be developed for clinically use.21 Awasthi et al. (2017) elaborated on various physicochemical and pharmacokinetic properties as well as formulation for the development of clinically useful hybrid molecules.19 They also discussed the linking strategy and the selection of individual components in the design of potential antimalarial hybrid molecules. Here, we discuss the newer strategies used for linking two pharmacophoric units, followed by structure–activity relationship (SAR) optimization studies, to develop effective hybrid molecules. Overall, the review is an updated version of previous reviews and focuses on the recent developments in in vitro and in vivo studies on quinoline- and endoperoxide-based hybrid molecules as promising antimalarial drug candidates.22–24 This review identifies quinoline- and endoperoxide-based hybrids published till June 2020 in terms of their pre-clinical value.
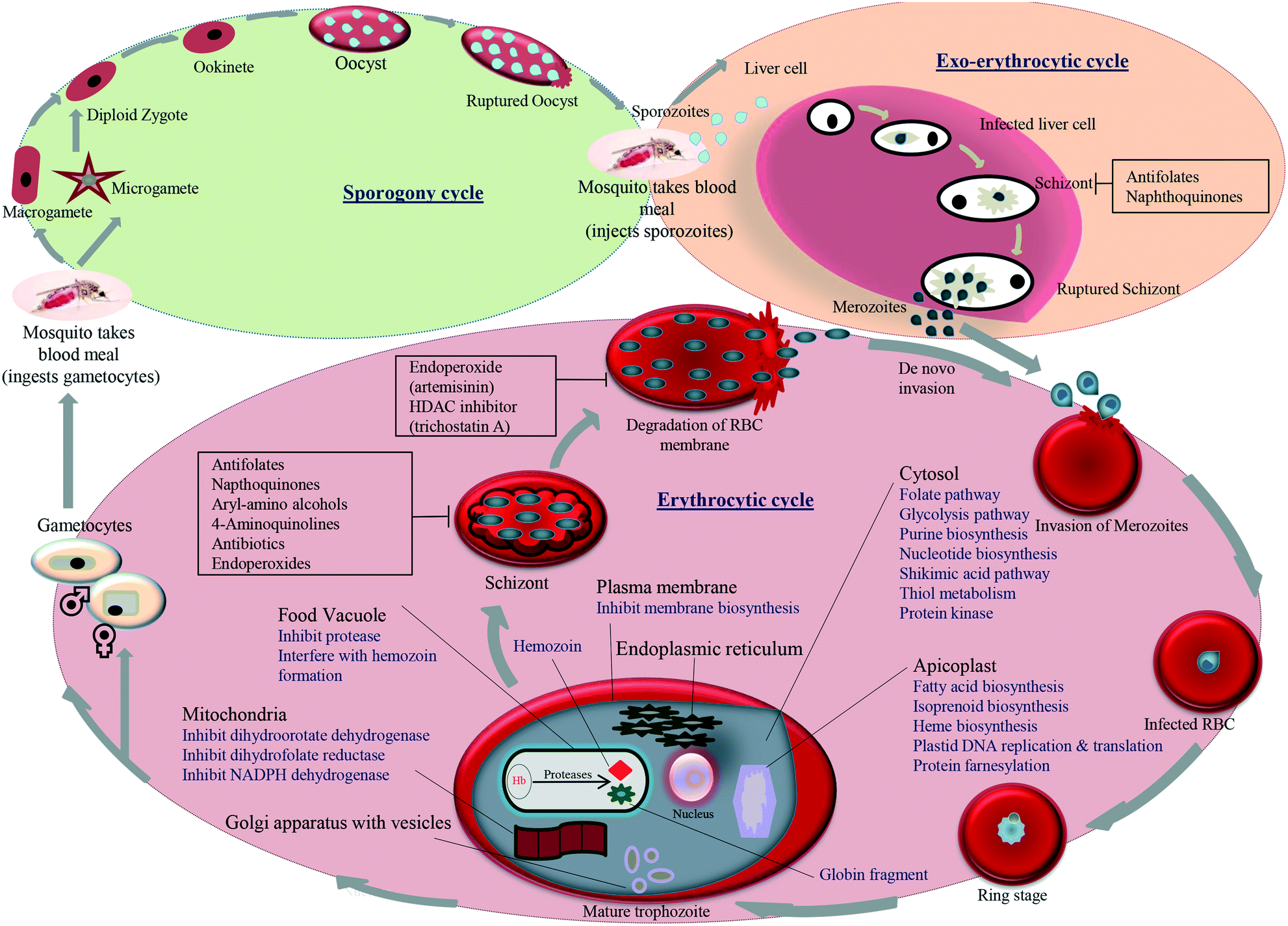 | ||
| Fig. 2 Different stages and potential drug targets in the life-cycle of P. falciparum. | ||
2. Quinoline-based hybrids
2.1 Chloroquine–astemizole hybrids
Whitlock et al. (2009)25 employed a hybridization strategy using two different moieties of CQ and astemizole linked by the core portion, each possessing significant antiplasmodial activities. The antiplasmodial activity was determined in a CQR strain (K1) of the P. falciparum parasite using CQ and astemizole as standard drugs. Hybrids 1–3 (Fig. 3) were found to be 3–10-fold more potent than CQ, where 1 (IC50 = 23 nM) showed maximum potency. Hybrids with conformationally constrained amino piperidine linkers (2 and 3) also showed potent activity against the K1 strain, with IC50 of 64 nM and 37 nM, respectively. In the cytotoxic studies, >100-fold selectivity for an anti-parasitic activity over cell-based cytotoxicity was observed for these hybrids. They were able to overcome resistance to CQ, much of which has been ascribed to mutations in the PfCRT gene. Hybrids 2 and 3, when tested in vivo in the P. berghei mouse malaria model, did not show activity that was better than CQ. A significant reduction in parasitemia at a dose of 4 × 50 mg kg−1 (2) and 4 × 20 mg kg−1 (3) was observed when administered intraperitoneally (i.p.). At this high dose, reduced parasitemia compared with CQ (99%) was observed in 13 animals, whereas 3 showed only 80% reduction at a dose of 4 × 20 mg kg−1 in 8 days.25 All the structures are given in Fig. 3, and their IC50 values are shown in Table 1.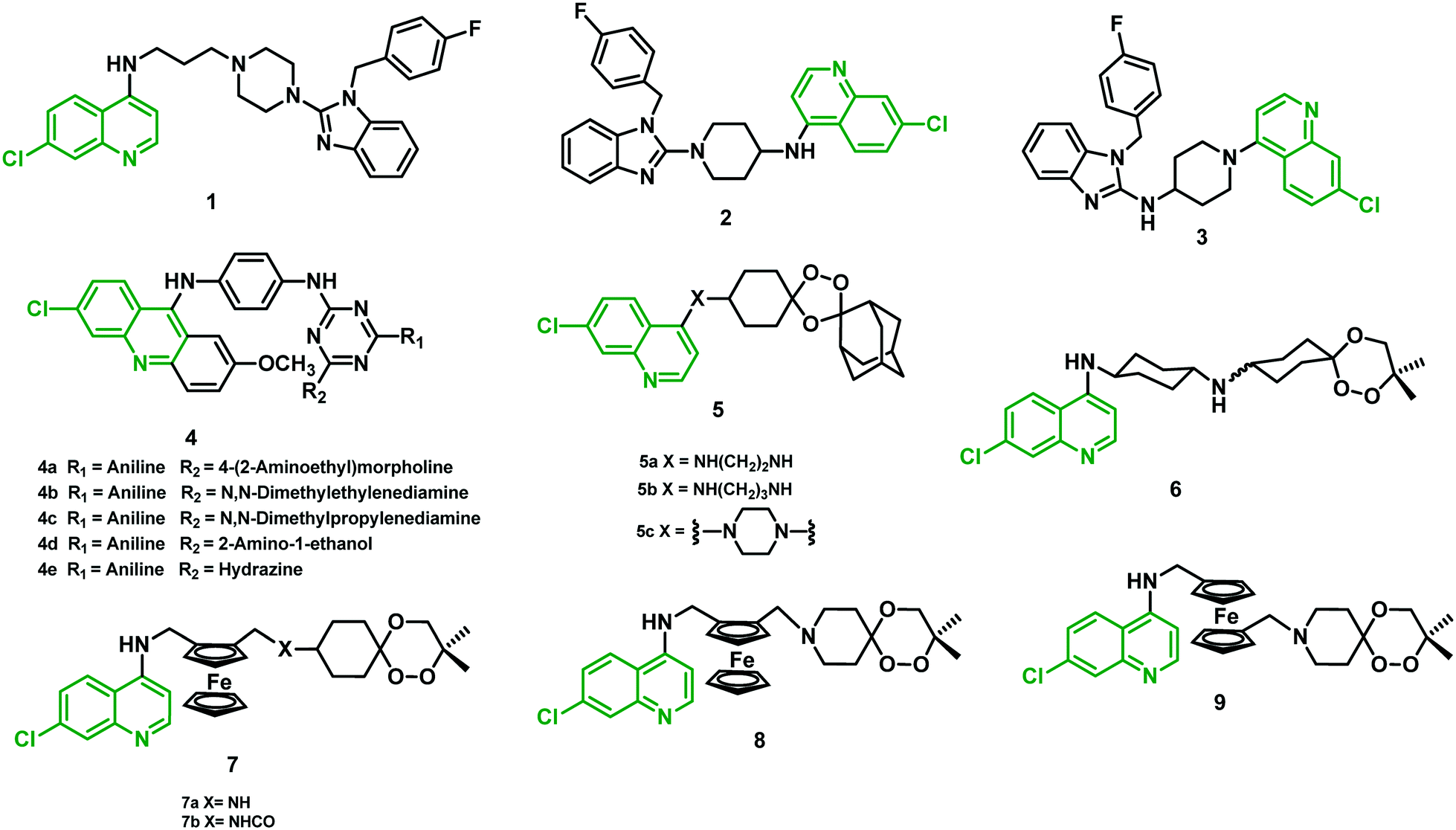 | ||
| Fig. 3 Chemical structure of chloroquine–astemizole (1–3), –acridines (4), –trioxolaquine (5) and –trioxaquine/trioxaferroquine (6–9) hybrids. | ||
| Hybrid | IC50 (nM) ± SD CQR | IC50 (nM) ± SD CQS |
|---|---|---|
| 1 | 23 | — |
| 2 | 64 | — |
| 3 | 37 | — |
| 4(a–e) | — | 4.21–9.46 |
| 5(a–c) | 3.60–26.21 | 6.13–12.61 |
| 6 | 7 ± 3 | — |
| 7(a, b) | 16 ± 4 | — |
| 8 | 71 ± 3 | — |
| 9 | 31 ± 2 | — |
2.2 9-Anilinoacridine-triazine hybrids
Acridines are known for their antimalarial effect due to their DNA intercalation, and binding to heme, and can also cause inhibition of the topoisomerase-II enzyme. Based on the potential effect of this core structure, Chauhan et al. (2009)26 synthesized a series of 9-anilinoacridine triazine hybrids and evaluated them against a chloroquine-sensitive (CQS) strain (3D7) of P. falciparum. Variation of diverse substituents on the triazine nucleus at the 4th and 6th positions was explored to determine the structure–activity relationship (SAR) among all the hybrids. Interestingly, hybrids 4c and 4e showed a two-fold increase in antimalarial potency compared with CQ, with IC50 of 4.21 nM and 4.27 nM, respectively. Moreover, hybrids 4a, 4b, and 4d displayed antimalarial activity comparable to CQ. All these hybrids exhibited a reasonably high selectivity index (SI) in the cytotoxic studies using Vero cells and MTT assay. Oral administration of lead hybrids (4a–e) in Swiss mice infected with a CQR strain (N-67) of P. yoelii at 100 mg kg−1 dose for four days was performed. Both 4a and 4c exhibited >95% suppression of parasitemia, with no significant protection to the treated mice in 28 days of survival assay. The study suggested that modification of substituent at positions 4 and 6 around the triazine nucleus might yield more effective antimalarial hybrids.262.3 1,2,4-Trioxolaquine hybrids
More recently, O'Neill et al. (2009)27 produced a 1,2,4-trioxolaquine hybrid (5a) that showed remarkable antiplasmodial activity in vitro against a CQS (3D7) (IC50 = 6.13 nM) and a CQR (K1) strain (IC50 = 3.60 nM) and thus was more effective than CQ (18 and 240 nM, respectively). They further established and optimized the structure by introducing a cyclic linker (5c) as well as an increased linker length (5b), which resulted in a reduction in antiplasmodial activity with increased IC50 values of CQS 12.61–CQR 26.21 and CQS 12.33–CQR 8.32 respectively.272.4 Trioxaquine and trioxaferroquine hybrids
Meunier's group carried forward the optimization of endoperoxide-AQ (Amino-quinoline) hybrids, and collaboration with Sanofi-Aventis resulted in the emergence of hybrid 6 (trioxaquine-PA1103/SAR116242). This was the first hybrid of an antimalarial drug to reach clinical trials. This hybrid was found equipotent to ART against several CQS and CQR strains of P. falciparum. It also cured 100% of mice in vivo at day 30, at low concentration (26–32 mg per kg per day), when administered either orally or i.p. In addition, no drug-related unfavorable effects were observed.28Robert et al. (2010)29 announced the synthesis of trioxaferroquine hybrids (7 and 8) containing a 1,2,4-trioxane moiety covalently linked to ferroquine (FQ), which is a synthetic ferrocenylquinoline. In vitro studies against two CQR strains (FcB1 and FcM29) of P. falciparum using hybrids 7, 8, and iso-trioxaferroquine (9) were performed. All the hybrids showed IC50 values in the range of 16–71 nM, compared with 145 and 735 nM against FcB1 and FcM29 for CQ. Hybrids were found equally potent to ART (10 and 18 nM, respectively), but slightly less active than FQ (6 and 7 nM). Further, in vivo studies with hybrid 7a orally administered at a very low dose (10 mg per kg per day) were able to clear parasitemia to below a detectable level in P. vinckei petteri-infected mice.29
2.5 Chloroquine–thiazolidinone hybrids
Kouznetsov et al. (2011)30 reported another series of CQ hybrids containing fragments of benzylamine at the C-4 position of the thiazolidinone moiety. None of the hybrids was found better than CQ against strain 3D7. Interestingly, hybrids 10a, 10b, 11 and 13 (IC50 values in the range 0.30–0.44 μM) were found to be more effective against the CQR Dd2 strain than CQ (IC50 = 0.50 μM). Hybrids 12 and 14 were found equipotent, with IC50 values 0.58 and 0.54 μM, respectively. These hybrids showed the ability to block β-hematin formation using FPIX (ferriprotoporphyrin IX) biocrystallization inhibition (FBIT) assay with low cytotoxicity in J774 murine macrophages and HepG2 cells (human hepatocellular carcinoma cells). In vivo studies (suppressive test for 4 days) on P. berghei ANKA-infected mice using hybrids 10a, 13, and 14 at a dose of 10 mg per kg per day were also performed. Hybrids 13 and 14 reduced parasitemia by 80% and 100%, respectively, whereas 10a inhibited parasite growth by merely 25%, taking CQ as a positive control (100% inhibition at the same dose). Moreover, 14 exhibited better lipophilicity, absorption, and distribution than CQ. Overall, benzyl amino fragments present in hybrids and the N-(aminoalkyl) thiazolidine-4-one moiety (13 and 14) displayed tremendous in vitro and in vivo antimalarial activity and a low level of cytotoxicity, suggesting that they can be further developed as possible antimalarial agents.302.6 Chloroquine–triazine hybrids
Chauhan et al. (2011)31 reported new hybrids made by linking 4-anilinoquinoline and triazine moieties and tested in vitro antimalarial activity against the CQS strain 3D7. Hybrids 15c (IC50 = 5.85 nM), 15d (IC50 = 5.92 nM), 15e (IC50 = 3.01 nM), 15f (IC50 = 7.03 nM) and 15g (IC50 = 6.41 nM) exhibited antimalarial potency better than that of CQ. These hybrids showed a selectivity index (SI) ranging from 92.06 to 610.98, indicating their selectivity towards parasite over Vero cells. In vivo studies suggested that hybrids 15a and 15b were orally active at a dose of 100 mg per kg per day for the next four days in Swiss mice infected by a CQR strain (N-67) of P. yoelii. Though the efficacy of lead hybrids 15a and 15b was modest, they were able to overcome CQ resistance. β-Hematin inhibition assay suggested that, apart from targeting heme, the possibility of another mechanism of action for the antimalarial activity of these hybrids cannot be ruled out.31 Further, the group reported another series of 4-anilinoquinoline–triazine hybrids using inexpensive starting materials, which they assessed in vitro against CQ-susceptible (3D7) and resistant (K1) strains of P. falciparum. Among all the hybrids, about sixteen exhibited potent antimalarial efficacy (IC50 values between 1.36 and 4.63 ng mL−1). Moreover, five hybrids (15h–15l) showed IC50 values in the range of 1.36–2.47 ng mL−1, more effective than CQ. These hybrids showed a high SI ranging between 117.37 and 3318.84, confirming their selectivity towards Plasmodium cells compared with Vero cells. All the hybrids showed potent β-hematin inhibitory activity (2.01–3.49 μg mL−1) compared with CQ (3.65 μg mL−1) when tested in vitro, possibly acting by inhibition of hemozoin formation. Based on docking studies, the selected hybrids 15h, 15i, 15j, and 15k were effective against the transketolase enzyme of P. falciparum, with IC50 values in the range of 72–85 μM compared with IC50 103 μM for the standard p-hydroxyphenylpyruvate. When tested in vivo in Swiss mice infected with CQR strain N-67 of P. yoelii, hybrids 15i and 15l at a dose 100 mg kg−1 for four days provided up to 99.99% protection to the treated mice. Moreover, 15l and 15m at 50 mg kg−1 dose administered by an oral route showed 77.8–99.8% reduction in parasitemia, confirming their pharmacological potential.32Rawat et al. (2013)33 synthesized 7-chloro-quinoline-triazine hybrids attached covalently via appropriate linkers and evaluated them against D6 (CQS) and W2 (CQR) strains of P. falciparum. Out of 28 hybrids, ten (16a–d, 16f–h, 16j, k, and 16m) showed better antimalarial activity (IC50 ranging from 0.11 to 0.42 μM) than CQ, while three, namely 16e, 16i, and 16l, were found to have IC50 ranging from 0.48 to 0.49 μM, which was comparable to CQ, against the W2 strain. None of them was associated with cytotoxic effects at 25 μM against three different cell lines (Vero, LLC-PK11, and HepG2). Overall, the activity profile of hybrids 16h and 16k could be beneficial for the next-generation drug candidates in malarial chemotherapy.33
Later, Sahu et al. (2016)34 also reported a series of 4-aminoquinoline-triazine hybrids consisting of 4-aminoquinoline, triazine nucleus and CQ, the nucleus of clociguanil, attached simultaneously with a suitable linker, to form a structure comprising a hydrophobic tail and hydrogen bond donor group head for inhibition of PfDHFR-TS (Plasmodium falciparum dihydrofolate reductase–thymidylate synthase). Percentages of dead rings, trophozoites, and schizonts were the parameters for the antimalarial activity of the hybrids. Compounds 17a and 17b showed potent activity against the 3D7 strain of P. falciparum at both 5 μg mL−1 and 50 μg mL−1 doses. In the case of 17a, the maximum number of dead rings, trophozoites, and schizonts was observed at 5 μg ml−1 (39.5%) and 50 μg ml−1 (48.5%), respectively, with an IC50 value of 46.33 μg mL−1. Hybrid 17b showed 35% dead rings, trophozoites, and schizonts at both 5 μg ml−1 and 50 μg ml−1 dose levels. It was concluded that the antimalarial activity of the hybrids increases many-fold as a result of the presence of primary, secondary, and tertiary amines in the same structure.34 The IC50 values of hybrids 10–17 are given in Table 2, and their structures are shown in Fig. 4.
| Hybrid | IC50 (nM) ± SD CQR | IC50 (nM) ± SD CQS |
|---|---|---|
| 10(a, b) | 300 ± 20–330 ± 50 | 300 ± 40–250 ± 30 |
| 11 | 350 ± 20 | 300 ± 30 |
| 12 | 580 ± 40 | 350 ± 50 |
| 13 | 440 ± 20 | 360 ± 20 |
| 14 | 540 ± 30 | 400 ± 30 |
| 15(a–m) | 3.01–7.03 | — |
| 16(a–m) | 110–420 | 100–430 |
| 17(a–b) | — | 155 |
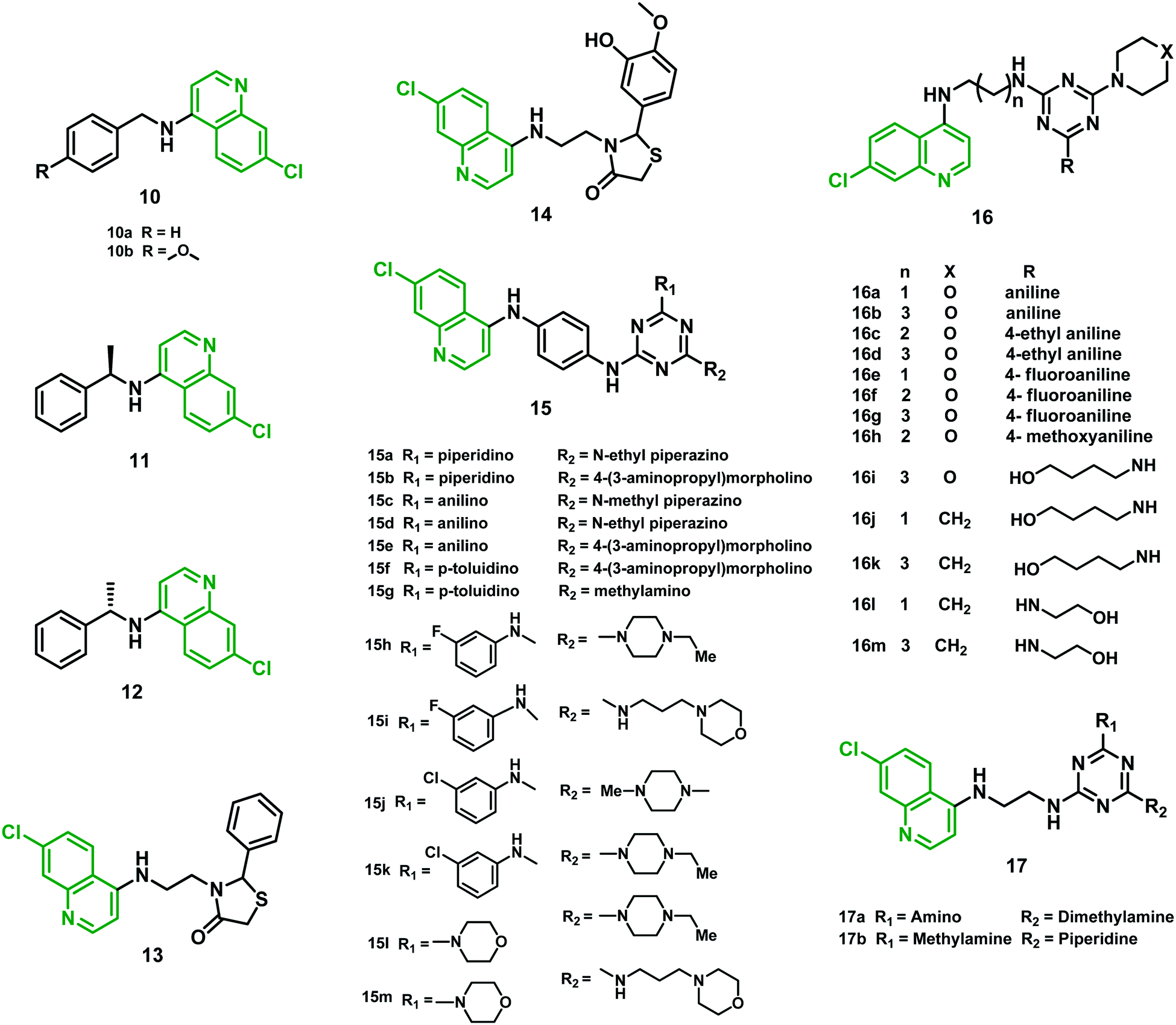 | ||
| Fig. 4 Chemical structures of chloroquine–benzylamine (10–12), –thiazolidinone (13–14), and –triazine (15–17) hybrids. | ||
2.7 4-Aminoquinoline–macrocyclic azalide hybrids
Azalide (AZ) was tested for in vitro and in vivo efficacy in prophylaxis of malaria and treatment in experimental models when used in combination with other malarial drugs. Poor efficacy was noticed when used alone. To improve its potential, Pesic et al. (2012)35 synthesized hybrid molecules consisting of 4-aminoquinoline covalently linked to a 15-membered azalide scaffold at position C-30 using linkers of different lengths.The in vitro antimalarial activity of the hybrids was compared with that of AZ and CQ. The antimalarial activity of cladinosyl analogs was found better than that of decladinosyl hybrids (18bvs.18a and 19bvs.19a) against CQR strain W2 and CQS strain 3D7A. Hybrids 19a and 19b were found to be the more potent pair relative to CQ (IC50 1.5–216-fold lower) and AZ (IC50 300–3838-fold lower) against the two tested P. falciparum strains. Hybrid 18b had the lower activity, but its cladinosyl analog 18a had activity improved by 56- to 150-fold over AZ. Meanwhile, when compared with CQ, only a 9-fold increase against the W2 strain was noticed. Further, 18a showed a satisfactory cytotoxicity and selectivity profile. Thus, the best candidate for further profiling in pharmacokinetic studies was found to be this hybrid molecule. The mechanism involved could be purely physicochemical since the tested hybrids have higher lipophilicity than their parent drugs and could be accumulated within the cells as a result to their penetration through the membranes. The efficacy could also be explained by changing the substrate specificity for the transport proteins involved in CQ efflux, or a combination of both. In vivo evaluation of compound 18a showed low oral efficacy, probably due to low absorption from the intestine. Overall, further development of these hybrids is unlikely owing to their poor in vivo efficacy.35
2.8 β-Amino-alcohol-tethered 4-aminoquinoline-isatin hybrids
Kumar et al. (2014)36 reported hybrids containing a 4-aminoquinoline tethered to an isatin core through a β-amino alcohol linker. Variation in the length of the alkyl chain at the C-4 position of the quinoline ring showed a great impact on the antimalarial efficacy of these hybrids. In vitro evaluation against CQR strain W2 of P. falciparum showed that efficacy was dependent on the nature of the substituent at the C-5 position of the isatin ring and the length of the alkyl chain. Hybrid 20a showed better efficacy than CQ, with IC50 of 24.9 nM. Moreover, hybrids 20b and 20c, with a chloro-substituent at the C-5 position of the isatin ring and shorter alkyl chain length (n = 2 and 3 respectively), were found most effective among them all, with IC50 of 11.7 and 13.5 nM, respectively. In the MTT assay against HCT116 (colon cancer cells) at 0.2, 2.0, and 20 μM they showed no sign of cytotoxicity.362.9 4-Aminoquinoline–imipramine hybrids
Imipramine, an anti-depressant drug, had shown mild intrinsic antimalarial activity. This drug also inhibited the P. falciparum chloroquine resistance transporter (PfCRT) linked with CQ efflux from the digestive vacuole in CQR parasites. Oliveira et al. (2015)37 developed a new hybrid by combining 4-aminoquinoline with imipramine as a strategy to reverse the CQR phenotype. The resulting hybrid 21 was effective at low nanomolar concentration against both CQS and CQR strains (IC50 = 3 nM and 5 nM, respectively). Hybrid 21 also demonstrated oral efficacy against the P. chabaudi model without any sign of toxicity. Additional alterations were performed in the linker as well as the aromatic head group of the imipramine moiety to eliminate any possible CNS side effects associated with it, which increased their lipophilicity and oral bioavailability. The resulting hybrid 22 demonstrated high in vitro activity against both CQS and CQR strains, with IC50 = 0.9 nM (D6) and 1.6 nM (Dd2), respectively. It also showed good cure rates at 4 × 46 mg kg−1 when tested in vivo.372.10 4-Aminoquinoline–purine hybrids
Rawat et al. (2017)38 synthesized novel hybrids containing 4-aminoquinoline and a purine moiety linked through an aliphatic diamine linker, common to the parent drug molecule. In vitro studies against both CQS (D6) and CQR (W2) strains of P. falciparum showed eight hybrids (23a–h), with IC50 values between 80 and 504 nM, exhibiting better efficacy against W2 strain when compared with chloroquine. Moreover, 23d showed six-fold more potency than chloroquine against the W2 strain. Hybrids 23c, 23d, 23f, and 23g (all having 1-methyl or 1-ethyl piperazine substituents) showed the best SI values. Compound 23d was also evaluated for heme-binding ability applying standard spectrophotometric methods, and the results observed were similar to those for CQ. Molecular docking analysis of these hybrids with HGPRT protein (hypoxanthine–guanine phosphoribosyltransferase) showed interactions with the interior part of the active site. The results were in agreement with the observed in vitro activities.38 All the hybrids (18–23) are shown in Fig. 5 and their IC50 values are shown in Table 3.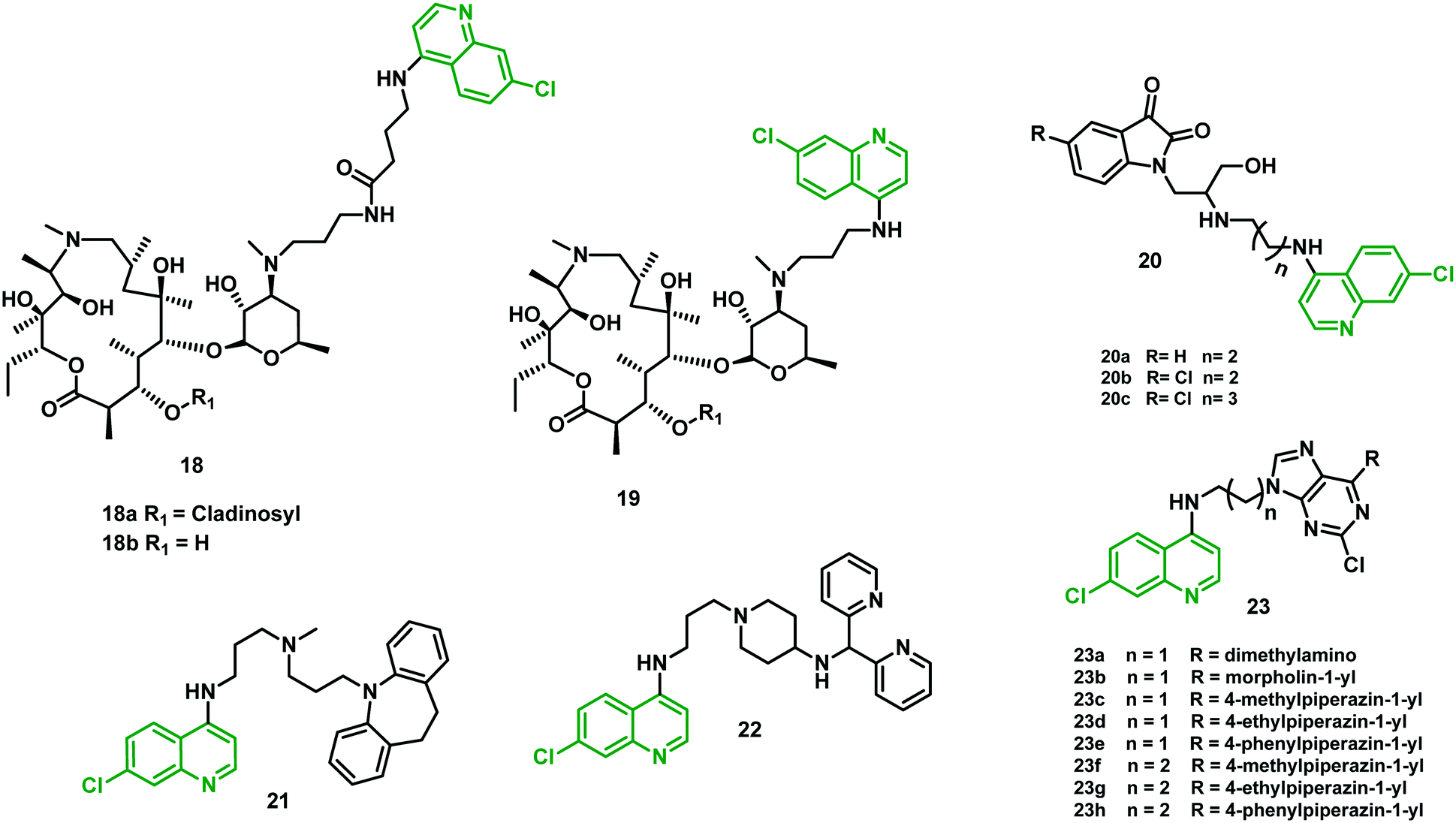 | ||
| Fig. 5 Chemical structures of chloroquine–cladinosyl (18 and 19), –aminoquinoline–isatin (20), –imipramine (21 and 22), and –purine (23) hybrids. | ||
| Hybrid | IC50 (nM) ± SD CQR | IC50 (nM) ± SD CQS |
|---|---|---|
| 18(a, b) | 48–295 | 77–636 |
| 19 | 2–9 | 3–11 |
| 20(a–c) | 11.7–24.9 | — |
| 21 | 5 | 3 |
| 22 | 1.6 | 9 |
| 23(a–h) | 80–504 | 61–187 |
2.11 Chloroquine–primaquine hybrids
Mueller et al. (2013)39 synthesized novel hybrids of primaquine and chloroquine with improved lipophilicity compared with the individual molecules that might promise adequate membrane permeability. The antiplasmodial activity of the hybrid 24 was assessed on the asexual blood stages of three strains of P. falciparum. It was found to be less potent than chloroquine for 3D7 (CQS) and Dd2 (CQR) strains (IC50 = 0.64 μM (3D7), 0.58 μM (Dd2)), ranging between the activities of primaquine and chloroquine. Interestingly, it had a profound activity against CQR strain K1 (IC50 = 0.08 μM), indicating an unexpected resistance-reversal effect. The inhibitory effect of 24 on the maturation of cultured P. falciparum NF54 (CQS) gametocytes from stage I to stage V was investigated after cultivating for seven days. The relative gametocyte numbers in cultures treated with 24 were lessened in comparison with the cultures treated with CQ and DMSO-treated controls, showing modest but noteworthy gametocytocidal effect.The hybrid 24 showed robust inhibitory activity against P. berghei liver-stage development, a prophylactic effect that was apparent in rodents in vitro. There was no complete suppression of intrahepatic parasites due to the prophylactic effect, but it showed a prophylactic delay in clinical malaria onset. It did not inhibit sporozoite motility in vitro and hepatocyte invasion. In vivo studies against P. berghei ANKA-infected mice by subcutaneous and intraperitoneal injection using 90 mg kg−1 dose were performed. The results showed that parasitemia did not develop in 66% of the mice. In contrast, the remaining ones developed parasitemia 12 days post-infection, implying the elimination of the parasites by an anti-liver-stage efficacy.39
2.12 Chloroquine–pyrimidine hybrids
Singh et al. (2013)40 reported chloroquine–pyrimidine hybrids using an expedient approach linking the 2,4-dichloropyrimidine unit to suitable derivatives of 4-amino-7-chloroquinoline or morpholine. The in vitro antiplasmodial activity against the D10 (CQS), as well as Dd2 (CQR), strains of P. falciparum was measured, applying CQ, artesunate (ASN), and MMV39004838 as positive controls. The study showed that hybrid 25a, bearing a morpholine group at the C-4 position of the pyrimidine, was active against the D10 strain, and 25b, bearing a piperidyl ring at the same position, was effective against both Dd2 (IC50 = 43 nM) and D10 (IC50 = 22.6 nM) strains with high SI value. The inhibition of hemozoin formation by hybrid 25bvia blocking of heme was observed by its binding capability with both heme and DNA.402.13 Chloroquine–sulfonamide hybrids
Krettli et al. (2015)41 prepared a few chloroquine–sulfonamide hybrids comprising aryl sulfonamide and amino quinoline units connected via an aliphatic linker of varied lengths. All the hybrids were evaluated against chloroquine- and sulfadoxine-resistant strain W2 of P. falciparum. All the hybrids displayed high potency in general, and ten of these had IC50 values ranging between 0.05 and 0.40 μM in the anti-HPR2 assay. This was better than CQ (IC50 = 0.46 μM) as well as sulfadoxine (IC50 > 15.5 μM). Moreover, hybrids 26a, 26b, and 26c were the most potent molecules in the series, with IC50 values of 0.10, 0.05, and 0.09 μM, respectively, and high SI values. In in vivo studies in P. berghei-infected mice, a dose of 10 mg kg−126a and 26c inhibited parasitemia by 47% and 49%, respectively, on day 5 after inoculation, whereas hybrids 26b and 26d were less potent as only 27% and 30% parasitemia inhibition was observed, respectively.41 Scopel et al. (2015)42 also reported quinoline–sulfonamide hybrids connected via hydrazine or hydrazide linkers. The most potent hybrid (27) was effective against CQS (3D7) as well as CQR (W2) strains, with IC50 of <0.19and <0.39, respectively. This hybrid was found to be non-toxic below the concentration of 100 μg mL−1 when tested in HepG2 and HeLa cells. It also showed in vivo efficacy against P. berghei, reducing the rate of blood-stage malaria parasites better than CQ at a dose of 10 mg mL−1.42 Prabhakar et al. (2016)43 prepared simpler N-(7-chloroquinolinyl-4-aminoalkyl) sulfonamide hybrids and found hybrid 28 with promising antimalarial activity against CQS (3D7) and CQR (K1) strains, with IC50 of 0.01 μM and 0.36 μM, similar to CQ. It also inhibited hemozoin formation (IC50 = 2.40 μM) significantly better than CQ (IC50 = 11.41 μM).432.14 Chloroquine–cinnamic acid hybrids
Zorc et al. (2016)44 designed N-cinnamoylated chloroquine hybrids linked by a hydrophobic and flexible butylamine/butyloxy linker. In vitro antimalarial potency was identified of between 15 and 141 nM against the sensitive strain 3D7 and between 11 and 111 nM against the resistant strain W2 of P. falciparum, compared with CQ (IC50 = 138 nM (W2)). Therefore, the cinnamoyl core appeared to be a valuable pharmacophore to increase the antiplasmodial potency of chloroquine. Most of the hybrids exhibited exceptional efficacy against erythrocytic and liver stages of the parasite, and they were non-toxic against human hepatoma cells (Huh7). The most potent hybrids (29c and 29e) were effective in vivo against the blood-stage infected P. berghei rodent malaria model. The animals showed survival extended by two to seven days when treated with 30 mg per kg per day for two days. However, 29c was extremely toxic at doses of 100 mg per kg per day, whereas the tested dosage of hybrid 29e did not show any toxicity.44 The structures of the hybrids (24–37) are given in Fig. 6, and their IC50 values are shown in Table 4.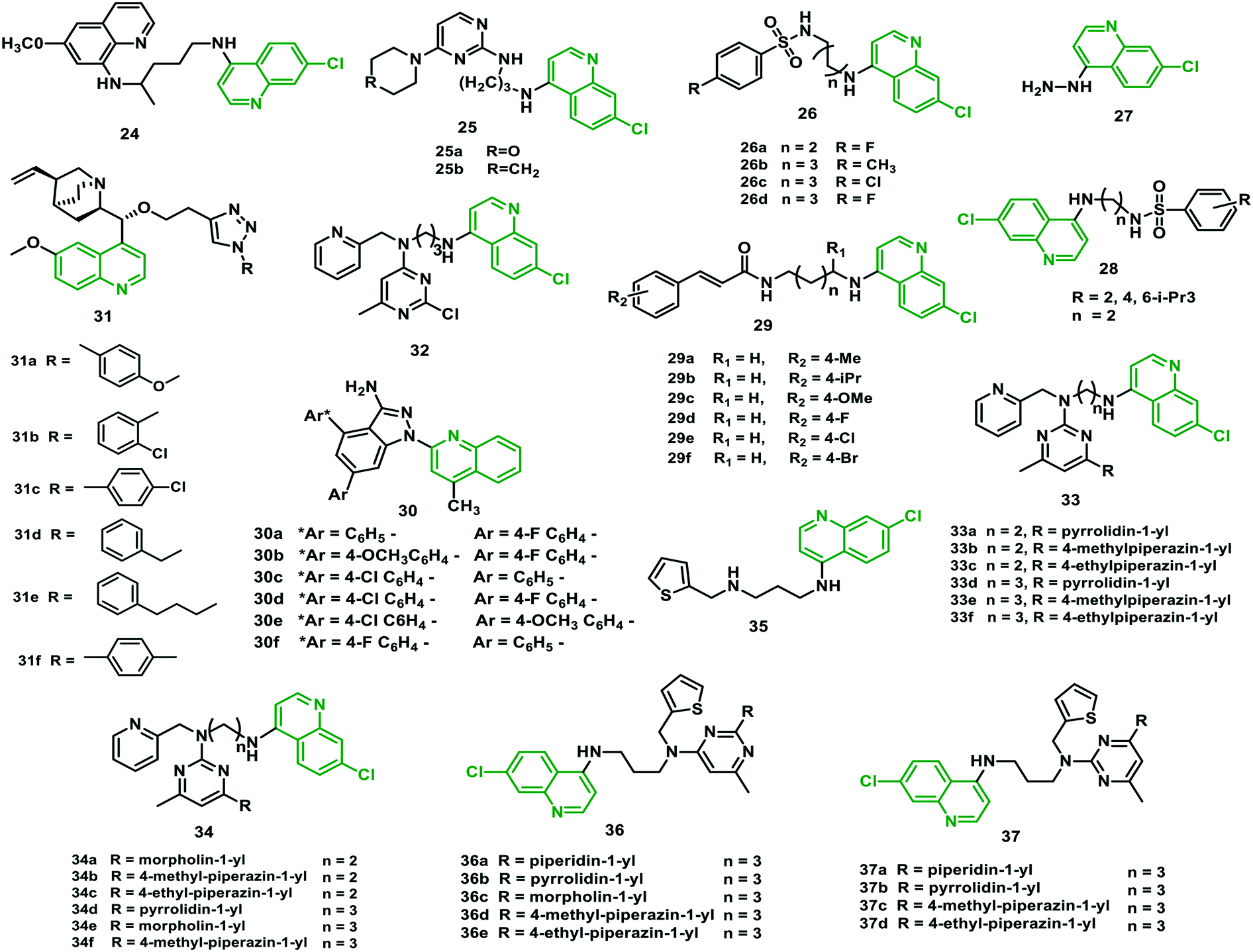 | ||
| Fig. 6 Chemical structures of chloroquine–primaquine (24), –pyrimidine (25), –sulfonamide (26–28), –cinnamic acid (29), –pyrazolopyridine (30), –triazole (31), and –pyrimidine (32–37) hybrids. | ||
| Hybrid | IC50 (nM) ± SD CQR | IC50 (nM) ± SD CQS |
|---|---|---|
| 24 | 580 ± 185 | 640 ± 46 |
| 25(a, b) | 43 ± 5.3 | 22.6 ± 2.2 |
| 26(a–d) | 50 ± 7–400 ± 70 | — |
| 27 | <0.19 μg mL−1 | <0.39 μg mL−1 |
| 28 | 360 | 10 |
| 29(a–f) | 11–111 | 15–141 |
| 30(a–f) | 1.92–2.92 μg ml−1 (EC50) | — |
| 31(a–f) | — | 27–43 |
| 32 | 319 | 1391 |
| 33(a–f) | 93–243 | 27–426 |
| 34(a–f) | 39–389 | 34–1144 |
| 35 | 75.1 | 423.9 |
| 36(a–e) | 31.2–409.1 | 37–83.7 |
| 37(a–d) | 18.9–40.1 | 32.8–44.6 |
2.15 Quinine–pyrazolopyridine hybrids
Saini et al. (2016)45 reported quinine–pyrazolopyridine hybrids and evaluated them in vitro for antimalarial potential using the schizont maturation inhibition assay. Hybrid 30d substituted with chalcone and fluorine at the para position of two phenyl rings was the most potent. Hybrids 30a–c and 30e, f, substituted with a halogen on at least one phenyl ring, also displayed antimalarial activity, with EC50 1.92–2.92 μg mL−1. When tested in vivo in P. berghei-infected Swiss albino mice at a dosage of 200 mg kg−1, 30d could inhibit parasitemia only 60.25%. In comparison, standard treatment cured all the five animals, with 100% suppression and a 100% survival rate. Overall, the study proved that halogen substitution on both the rings is responsible for the highest suppression of parasitemia among all these hybrids.452.16 Quinine–triazole hybrids
Panda et al. (2016)46 reported quinine–triazole hybrids and tested them in vitro against the blood stage of P. falciparum strain 3D7. Hybrids 31a–f, with IC50 = 43, 37, 41, 40, 30, and 27 nM respectively, were found better than the parent drug quinine (IC50 = 58 nM). The improved efficacy may be attributed to the lipophobic/hydrophilic properties of long alkyl chain linkers.462.17 Aminoquinoline–pyrimidine hybrids
Rawat et al. (2019)47 made a slight modification in the linker of previously reported aminoquinoline–pyrimidine hybrids to improve the efficacy. They introduced 2-formyl pyridyl and 2-formyl thiophenyl nuclei at the –NH group by reductive amination reaction to help accumulate these hybrids in the acidic food vacuole of the malaria parasite as well as to enhance the lipophilicity. In vitro antimalarial activity against D6 (CQS) and W2 (CQR) strains of P. falciparum resulted from the hybrid 33b, with IC50 = 27 nM (D6) and SI > 351. All the 23 hybrids (32, 33a–f, 34a–f, 35, 36a–e, 37a–d) showed better efficacy than CQ against the W2 strain. Moreover, 37d was the most effective (IC50 = 18.9 nM) against the W2 strain, with 22-fold better efficacy than CQ (IC50 = 421.5 nM). Hybrids 37c and 37d showed comparable efficacy to ART against the W2 strain. The heme-binding ability of 33b with hematin provided the clue for its primary mode of action. Molecular docking studies also demonstrated excellent binding interactions with PfDHFR.472.18 Chloroquine–1,2,3-triazole hybrids
Nath et al. (2019)48 reported a new series of 4-amino-7-chloroquinoline–1,2,3-triazole hybrids and screened them for antimalarial activity using Malaria SYBR Green-I Fluorescence (MSF) assay. All the hybrids were less potent than CQ against the CQS 3D7 strain, and hybrids 38a, 38b, and 38c demonstrated IC50 values of 39.9 nM, 40.00 nM, and 38.75 nM, respectively, against a CQR field isolate of P. falciparum (RKL-9). This was verified to be more efficient than CQ, suggesting the minimum possibility of cross-resistance with CQ. The results indicated that the efficacy diminishes with increment in the chain length of the aliphatic diamino linker connecting the triazole and 7-chloroquinoline scaffolds. No sign of significant cytotoxicity against Vero cells was observed, with the highest selectivity index shown by hybrid 38a. The mature gametocytes are targeted by these hybrids, which produce morphological damage in a dose-dependent manner. Hybrid 38b exhibited the highest potency among all these hybrids, with IC50 (nM (normal morphology)) of 8.50 μM. Hybrids 38a and 38c also targeted gametocytes, with IC50 (nM) of 10.71 μM and 12.03 μM, respectively. They showed promising schizonticidal action against asexual stages of the CQR strain of P. falciparum.48 The chemical structures of triazole, thiosemicarbazide, and piperidine hybrids (38–41) are shown in Fig. 7, and their IC50 values are given in Table 5.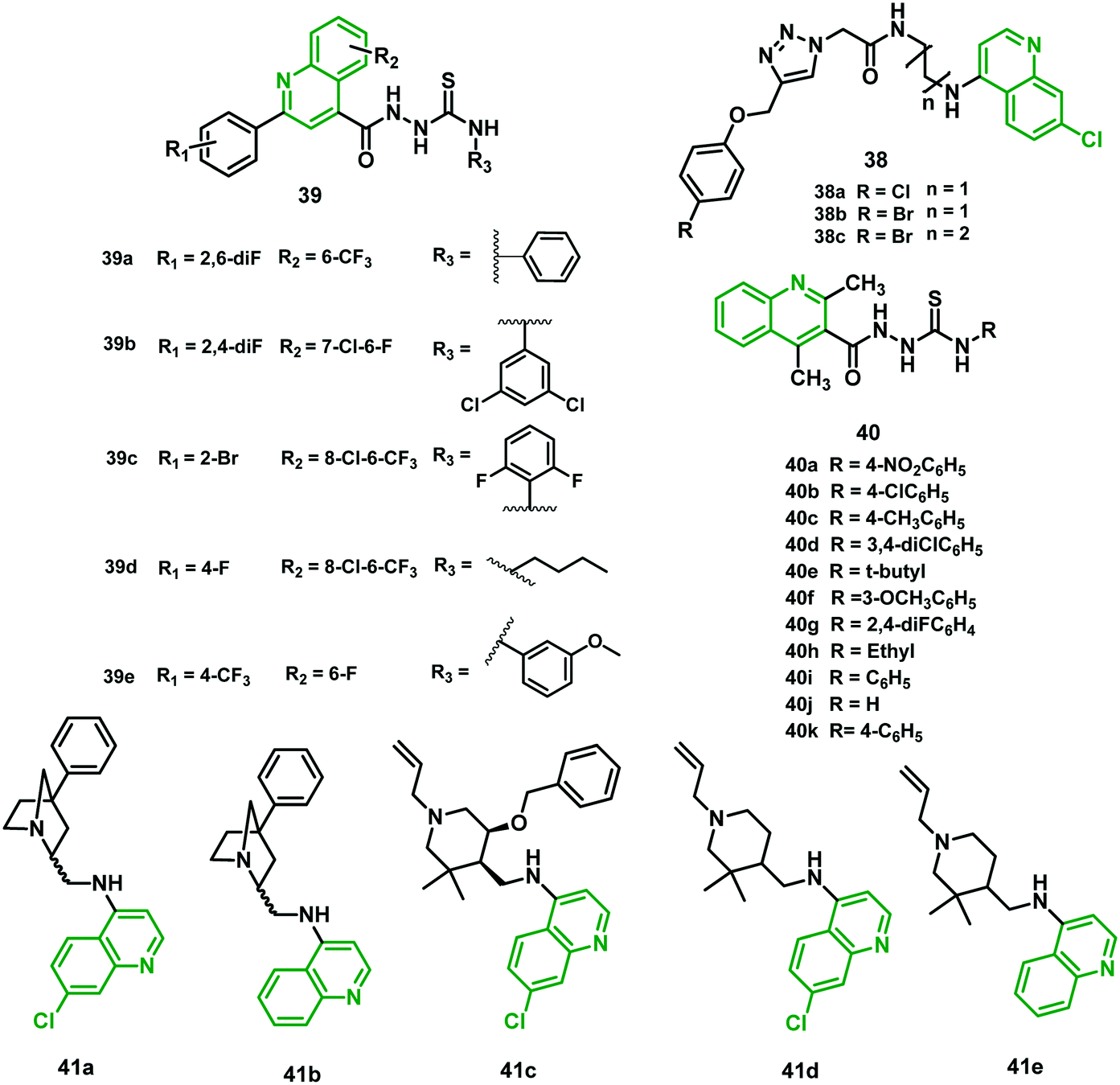 | ||
| Fig. 7 Chemical structure of chloroquine–triazole (38), quinine–thiosemicarbazide (39 and 40) and chloroquine–piperidine (41) hybrids. | ||
| Compound | IC50 (nM) ± SD CQR | IC50 (nM) ± SD CQS |
|---|---|---|
| 38(a–c) | 2.94–75.01 | 38.75–40.00 |
| 39(a–e) | 0.10–0.36 μg ml−1 | — |
| 40(a–k) | 0.02–0.26 μg mL−1 | — |
| 41(a–e) | 25 ± 8.9–69 ± 11.3 | 12 ± 1.0–236 ± 32 |
2.19 Quinoline–thiosemicarbazide hybrids
Patel et al. (2019)49 synthesized a novel series of fluorine-containing quinoline–thiosemicarbazide hybrids and evaluated them for antimalarial activity against a chloroquine-resistant P. falciparum strain. Hybrid 39a (IC50 = 0.19 μg mL−1) with –CF3 and –F groups, 39b (IC50 = 0.17 μg mL−1) with –F and –Cl groups, 39c (IC50 = 0.36 μg mL−1) with –CF3, –F, Cl and Br groups, 39e (IC50 = 0.10 μg mL−1) with –F and –NO2 groups, and 39d (IC50 = 0.28 μg mL−1) with –CF3, –F, and OCH3 groups showed remarkable activity compared with reference drug quinine (IC50 = 0.268 μg mL−1). Structure–activity relationship studies showed that analogs containing electron-withdrawing groups, such as trifluoro and chloro, as well as those containing electron-donating groups like methoxy and substituted methyl, showed significant antimalarial effectiveness. The substitution pattern on the quinoline ring and thiosemicarbazide linkage at distinct positions were discovered to have an immense impact on the biological activity. The hybrids containing –CF3 (39a, 39c) and –F (39b, 39e) substituted at the sixth position of the quinoline ring displayed remarkable antimalarial activity. In contrast, hybrids, bearing –Br at the sixth position of the quinoline ring and trifluoromethyl (–CF3) at the seventh and eighth positions of the quinoline ring showed modest antimalarial activity. In the cytotoxicity studies of 39a, 39b, 39c, and 39e in peripheral human blood lymphocyte cultures at three different concentrations (0.1, 1.0, and 10 μg mL−1) were not found toxic, and the results were similar to those for quinine. Overall, 39a, 39b, and 39e represented significant leads for the development of new antimalarial agents.49Later, Patel et al. also reported the microwave-assisted synthesis of quinoline–thiosemicarbazide hybrid derivatives from reaction of aryl thiosemicarbazides and N4-alkyl with ethyl 2,4-dimethyl quinoline-3-carboxylate. Compounds 40a (0.25 μg mL−1), 40b (0.15 μg mL−1), 40c (0.02 μg mL−1), 40d (0.19 μg mL−1), 40e (0.25 μg mL−1), 40f (0.10 μg mL−1), 40g (0.26 μg mL−1), 40h (0.15 μg mL−1), 40i (0.02 μg mL−1), and 40j (0.19 μg mL−1) showed higher inhibitory activity against a P. falciparum strain compared with quinine (0.26 μg mL−1). Additionally, a docking study of hybrids with PfDHFR-TS was done on three proteins: 4DP3, 3JSU, and 1J3K.
Compounds 40b (binding affinity = −8.32 kcal mol−1) and 40k (−8.22 kcal mol−1), compounds 40c (−7.23 kcal mol−1) and 40f (−5.82 kcal mol−1), and compounds 40b (−4.323 kcal mol−1) and 40f (−3.039 kcal mol−1) bound specifically to 3JSU, 4DP3, and 1J3K. Moreover, the interaction simulation of preferred compound 40d with protein 3JSU was considered through the study of molecular dynamics using the Schrodinger software.50
2.20 Chloroquine–piperidine hybrids
D'hooghe et al. (2020)51 recently synthesized four diverse series of quinoline–piperidine hybrids and assessed them against the NF54 (CQS) and K1 (CQR) strains of P. falciparum. Five 4-aminoquinoline–piperidines (41a–e) among all 18 hybrids synthesized exhibited exceptional antiplasmodial activities, with IC50 of 25–69 nM (K1), 2- to 7-fold more effective than chloroquine. The structural analysis signified that the occurrence of a chlorine atom at the 7-position of quinoline delivered the finest activity. However, the RI values (the ratios of the IC50 values of the resistant to sensitive strains) of the hybrids are roughly ten times less than for chloroquine. Furthermore, the results of the MTT assay showed no cytotoxicity, with SI values above 100. These five lead 4-aminoquinoline–piperidines might act by circumventing the chloroquine resistance mechanism and thus could exert their antimalarial activity by inhibition of crystallization of heme, though no biological studies confirm this theory.513. Artemisinin-based hybrids
3.1 Artemisinin–acridine hybrids
David D. N'Da et al. (2014)52 synthesized artemisinin–acridine hybrids. They screened for in vitro antiplasmodial activity against CQS (NF54) and CQR (Dd2) strains of P. falciparum using dihydroartemisinin (DHA), AS, and CQ for comparison. Hybrid 42 was most potent among the series, with IC50 of 2.6 nM (NF54) and 35.3 nM (Dd2). It was found to be seven-fold more effective than CQ (Dd2 strain) but had lower potency than DHA and AS against both the strains. Hybrid 42 also showed seven-fold superior gametocytocidal activity compared with chloroquine and high selectivity towards the parasitic cells, with lower cytotoxicity than emetine against Chinese hamster ovary (CHO) cells, with IC50 of 1.6 μM.523.2 Artemisinin–quinazoline hybrids
Tsogoeva et al. (2017)53 reported a few novel quinazoline–artemisinin hybrids and evaluated them against the 3D7 (CQS) strain of P. falciparum using their parent compounds quinazoline derivatives and artesunic acid as reference. Among all the hybrids, 43 (EC50 = 3.8 nM) and 44 (EC50 = 1.4 nM) showed antimalarial activity comparable to or even more efficient than DHA (EC50 = 2.4 nM).53 The chemical structures of the artemisinin hybrids (42–46) are given in Fig. 8 and their IC50 values are shown in Table 6.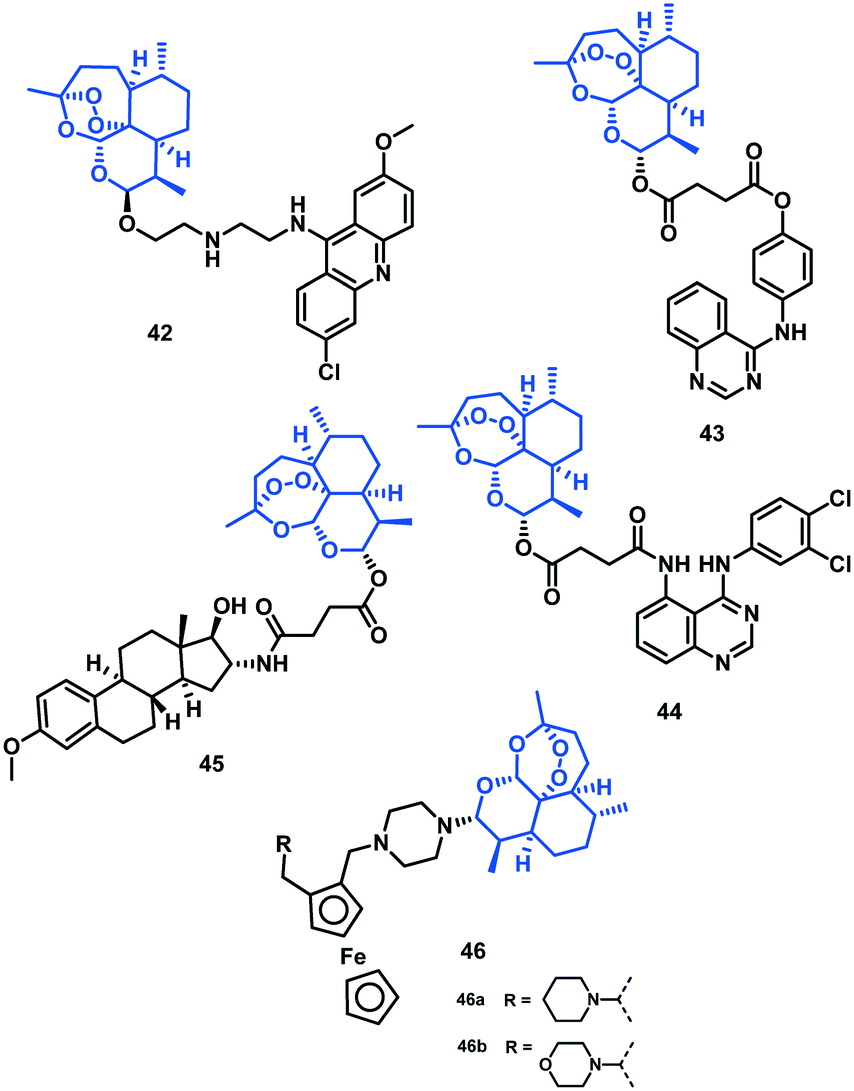 | ||
| Fig. 8 Chemical structures of artemisinin (42–46) hybrids. | ||
| Hybrid | IC50 (nM) ± SD CQR | IC50/EC50 (nM) ± SD CQS |
|---|---|---|
| 42 | 35.3 ± 3.6 | 2.6 ± 0.0 |
| 43 | — | 3.8 ± 1 |
| 44 | — | 1.4 ± 0.4 |
| 45 | — | 3.8 ± 0.8 |
| 46(a, b) | 0.8 ± 0.2–1.1 ± 1.1 | 3.3 ± 1.3–3.8 ± 1.4 |
3.3 Artemisinin–estrogen hybrids
Tsogoeva et al. (2018)54 reported the synthesis of artemisinin–estrogen hybrids and their antimalarial activity against the CQS 3D7 strain of P. falciparum. These hybrids exhibited excellent to moderate antimalarial efficacy, with EC50 values ranging from 3.8 to 128.8 nM, while their estrogen precursors showed no activity (EC50 > 16000 nM). Moreover, the best performing hybrid, 45, was twice as efficient as its parent artesunic acid (EC50 = 8.9 nM) and the standard drug chloroquine (9.8 nM). It also showed comparable efficacy with DHA (EC50 = 2.4 nM). It was concluded that a methoxy group instead of a benzyloxy subunit at C-3 of the estrogen moiety seems to be more beneficial for antimalarial activity of artemisinin–estrogen hybrids.54
3.4 Artemisinin–1,2-disubstituted ferrocene hybrids
David D. N'Da and Haynes et al. (2018)55 reported first artemisinin–ferrocene hybrids comprising 1,2-disubstituted ferrocene analogs added by a piperazine linker to an artemisinin core at the C-10 position. Ferrocene (in the Fe2+ state) undergoes facile oxidation to ferrocenium (Fe3+), and is finally reduced by NADH and glutathione (GSH) to produce ferrocene. The ensuing redox cycling involving ferrocene and ferrocenium increases the hydroxyl radical flux. Ferroquine is capable of producing micromolar amounts of hydroxyl radicals from H2O2. This ability can be exploited further through conjugation to an artemisinin core, whereas the produced hydroxyl radicals by ferroquine can activate oxidative stress by oxidizing reduced flavin cofactors that normally modulate levels of endogenous thiols required for expunging ROS (reactive oxygen species). In vitro antimalarial activity of artemisinin–ferrocene hybrids was investigated against three Plasmodium strains at asexual blood stages: the drug-resistant K1 and W2 and drug-sensitive NF54 strains. Hybrids 46a and 46b showed improved efficacy compared with artemether (AM), DHA, and AS towards the resistant K1 and W2 strains but was found to be less effective against the sensitive NF54 strain. The gametocytocidal action was resolved with PfNF54 early – (I–III) and late – (IV and V) stage gametocytes at 1.0 μM and 100 nM concentration. At 1.0 μM, these hybrids were equipotent with methylene blue and DHA against the early stage, but more effective against late-stage gametocytes as activity against late-stage gametocytes, in particular, reflects transmission-blocking capability. The hybrid 46b with morpholino ferrocenyl moiety was more specific towards the parasites than mammalian cells, but less so than DHA, possibly suggesting generalized toxicity. The RI value, the ratio of the IC50 values of the resistant to sensitive strains (IC50 K1/IC50 NF54 = 0.2, IC50 W2/IC50 NF54 = 0.4), was used as an indication of the potential for cross-resistance formation for each drug-resistant strain.554. Miscellaneous
4.1 Trifluoromethyl artemisinin–mefloquine hybrids
Bonnet-Delpon et al. (2005)56 designed and synthesized CF3-artemisinin–mefloquine hybrids as dual antimalarial molecules. Hybrid 47 was developed as an indivisible hybrid as piperidinyl amine of the mefloquine, which was covalently linked to the CF3-artemisinin moiety. In the divisible hybrid 48, the mefloquine was linked to the CF3-artemisinin via a diester linker, which can be easily hydrolyzed in vivo. Both the hybrids 47 and 48 were tested for in vitro antimalarial activity against four strains of P. falciparum that showed different degrees of resistance to mefloquine (F32 > Thai > FcB1 > K1, F32 being the most resistant) or chloroquine (K1 > FcB1 > F32 > Thai, K1 being the most resistant). Both were highly effective against all the tested strains in the low nanomolar range (IC50 = 2.4 to 17.2 nM). In vivo effect of both the hybrids was assessed with the murine malaria model infected with P. berghei strain NK173 by an i.p. administered dose of 35.5 μM kg−1. Hybrid 48 and to a lesser extent 47 were more efficient in controlling the parasitemia than the artemether at the administered dose.564.2 DHA–quinine hybrid
Walsh et al. (2007)57 prepared dihydroartemisinin–quinine hybrids via covalent linkage of the vinyl functionality of quinine with DHA. Hybrid 49 was screened against asynchronous, cultured blood-stage P. falciparum 3D7 and FcB1 by using a dehydrogenase assay (parasite lactate dehydrogenase assay). The study revealed that the hybrid displayed IC50 values of 8.95 nM (at 48 h) and 10.4 nM (at 72 h) against strain 3D7, and 9.59 nM (at 48 h) and 10.2 nM (at 72 h) against strain FcB1, better than quinine or artemisinin alone. The effectiveness of the hybrid was superior to that of a 1:1 mixture of quinine and artemisinin, which showed IC50 values of 31.8 nM (at 48 h) and 28.6 nM (at 72 h) against strain 3D7 and 27.9 nM (at 48 h) and 26.3 nM (at 72 h) against strain FcB1. The improved efficiency of the hybrid might be due to its increased cellular uptake compared with those of the individual drugs. Additionally, by chance it also acts as a ‘mutual prodrug’, where hydrolysis occurs in the ester group to give the individual components, and thus it provides an exceptional approach to delivering these antimalarial drugs to the protozoal site of action. In addition, the duration of the side effects generally associated with quinine regimens probably decreases as a result of the combination of the drugs in these hybrids.57
4.3 Artemisinin–primaquine hybrids
Primaquine (PQ) is the only accepted drug against the liver and hypnozoite stages in malaria infection. It also acts against the gametocytes, which disrupts the transmission of the infection to the mosquitoes. To further improve the efficacy, Lopes et al. (2011)58 linked PQ and ART drugs via covalent bonds to get primaquine–artemisinin hybrids. Inhibition of Plasmodium infection of liver cells by the hybrids was evaluated in an established in vitro infection system that employs a human hepatoma cell line (Huh7) and the rodent parasite P. berghei. The hybrids had compelling parasite-inhibitory effects, displaying IC50 values for 50 (12.5 nM) and 51 (9.1 nM) that are several-fold lower to the respective 1:1 PQ–ART mixture. There were no effects on Huh7 cell proliferation after treatment with the hybrids, measured by Alamar blue fluorescence, demonstrating that they selectively inhibit replication of the intracellular parasite. Hybrid 51 also acted by inhibiting cell invasion by the parasites, as revealed by the infection rate measured 4 hours after the addition of parasites to the cells. The chemical structures of these hybrids (47–59) are given in Fig. 9 and their IC50 values are shown in Table 7.
 | ||
| Fig. 9 Chemical structures of miscellaneous (47–59) hybrids. | ||
| Hybrid | IC50 (nM) ± SD CQR | IC50 (nM) ± SD CQS |
|---|---|---|
| 47 | 10.6 ± 0.2–17.2 ± 1.5 | — |
| 48 | 2.4 ± 0.4–6.6 ± 2.0 | — |
| 49 | 9.59 (95%CI) | 8.95 (95%CI) |
| 50 | 12.5 ± 1.1 | — |
| 51 | 9.1 ± 0.6 | — |
| 52(a–d) | 17.12 ± 0.44–29.24 ± 3.26 | 12.18 ± 1.21–28.99 ± 2.70 |
| 53 | 1.7 ± 0.3 | 4.5 ± 0.5 |
| 54 | 2.6 ± 1.0 | — |
| 55 | 8.4 ± 3.7–10.6 ± 2.3 | — |
| 56 | 63.17 ± 1.2 | 25.52 ± 4.45 |
| 57 | 146.2 ± 1.19 | 37.63 ± 1.18 |
| 58(a–b) | — | 9.3–27.1 |
| 59 | — | 24 ± 8 |
The therapeutic efficiency of the hybrids was then compared in vivo in C57BL/6 mice with P. berghei-infected red blood cells (iRBCs), and parasitemia was tracked every day until a level of 2–3% iRBCs was reached. Simultaneously, mice were treated daily for four days by subcutaneous injection of 31.8 μM kg−1 (equivalent to 9 mg kg−1 of ART) of 50, 51, or ART, an amount corresponding to the reported 90% effective dose (ED90) for ART administered by this route. Nine days post-infection, parasitemia reached zero for all mice, but it reoccurred at 12–13 days post-infection in mice that were treated with ART or hybrid 50. These results were probably due to hyperparasitemia since the mice did not show experimental cerebral malaria symptoms. In contrast, hybrid 51 did not show any parasitemia reoccurrence after the initial clearance of infections in the mice. Rather, throughout the experiment, they remained blood-stage-negative, with a 100% survival rate. Hence, 50 can be used to control ongoing blood-stage infection with greater effectiveness than the parent drug ART. Interestingly, 51 was less active in vitro but more effective in vivo when compared with 50, signifying improved pharmacokinetic properties and stability.58
4.4 DHA–aminoquinoline hybrids
David D. N'Da et al. (2013)59 synthesized hybrids of DHA and aminoquinoline entities linked by an ether bond to increase the half-life of DHA. Oxalate salts of 52a–52d were obtained by the reaction of these hybrids with oxalic acid to get more soluble and stable hybrids. In vitro antimalarial activity against CQS (D10) and CQR (Dd2) strains of P. falciparum using CQ and DHA as the reference drugs revealed oxalate salt hybrids 52a, 52b, 52c, and 52d had the most potent activity. They were found more effective than CQ (IC50 = 157.9 nM) against the Dd2 strain, with IC50 values between 17.2 and 38.9 nM. Hybrid 52c and its oxalate salt were 9- and 7-fold more efficient than CQ (17.12 nM; 20.76 nM, respectively). The study also revealed that the linker length of 2 or 3 Cs with or without an additional methylene substituent was suitable for their activity. Hybrids 52c and its oxalate salt displayed the best selectivity profiles based on both antiplasmodial activity and cytotoxicity. It was believed that these hybrids showed dual action as the artemisinin derivative acts on the early erythrocytic stages of P. falciparum and the chloroquine nucleus inhibits the polymerization of β-hematin.59Later, in 2013, the group studied the in vivo effect of the identified oxalate salts of hybrids 52c, 52a, 52b. The study revealed that the oxalate salts of 52c and 52a entirely cured mice at 50 mg kg−1 dose administered by the oral route and 15 mg kg−1via the i.p. route, while the artesunate provided a complete cure at 80 mg kg−1 for the oral route and 30 mg kg−1 i.p. A complete cure of malaria and clearance of parasitemia were not observed in the case of the oxalate salt of the hybrid 52b. The hybrids exhibited considerable antimalarial activity by the i.p. route, with ED50 values of 1.1, 1.4, and <0.8 mg kg−1 for the oxalate salts of hybrids 52c, 52a, and 52d, respectively. In this study, the optimum linker length was found to be a diamino alkyl chain comprising two carbon atoms, either methylated or unmethylated as in the oxalate salts of hybrids 52c and 52a. With the addition of another atom of carbon in the linker chain, as in the oxalate salt of 52b, survival rate was reduced by 100–66% at the same dosage.60
4.5 Artemisinin–(iso)quinoline hybrids
Çapcı et al. (2019)61 designed new ART–isoquinoline and ART–quinoline hybrids intended to treat MDR malaria. All the hybrids displayed effective antiplasmodial activity in the nanomolar to picomolar range against CQS (3D7) and CQR/MDR (Dd2 and K1) Plasmodium strains. The improved potency of the hybrids compared with their corresponding parent compounds was possibly due to the simultaneous uptake by the cells of both covalently linked pharmacophores, which are believed to have synergistic effects. The structures of hybrid 47–55 are given in Fig. 9.Hybrid 52, among other potent hybrids, showed EC50 values of 4.5 nM (3D7), 2.3 nM (Dd2) and 1.7 nM (K1). The suppressive activity of this hybrid was studied in P. berghei-infected mice for four consecutive days post-treatment. The mice treated subcutaneously with 105 μM kg−1 (40 mg kg−1) of ARE displayed a reduction of >99% in blood parasitemia, a dose that conferred 100% animal survival. Based on this, the same dose (105 μM kg−1), which was equivalent to 65 mg kg−1 of hybrid 52 was administered by a subcutaneous route as a starting point for testing these hybrids. At this dose, hybrid 53 decreased parasitemia by >99%, whereas at a dose of 35 μM kg−1, it decreased parasitemia by 95.2%. As a consequence of strong suppression of parasitemia, all mice treated with 105 μM kg−1 hybrid remained free of parasites up to 41 days post-infection and were considered cured, while at 35 μM kg−1, 80% of mice remained alive up to 41 days post-infection. This is considered to be much better than artesunate treatment at the same dosage in the experimental model.
Hybrid 53 inhibited the formation of β-hematin as effectively as chloroquine, assuming that only the 7-chloroquinoline pharmacophore of the hybrid accounts for its β-hematin inhibitory property.61
4.6 Novel G factor- or chloroquine–artemisinin hybrids
Dionissia A. Pepe et al. (2020)62 recently synthesized a series of novel hybrids of ART with either a 4-amino-7-chloroquinoline core of CQ or endoperoxide GmeP (phytormone endoperoxide G-factor analog) and derivatives of the same compounds with polyamines (PAs) spermidine (asymmetric PA) and homospermidine (symmetric longer chain PA), comprising either two molecules of GMeP endoperoxide or one or two of ART. Following this, their antimalarial activities were assessed against CQRP. falciparum FcB1/Colombia strain. Results showed that three (54, 55a, 55b) among the seven synthesized hybrids, containing one or two ART molecules per hybrid, were considerably more effective than the parent compound. The hybrid 54 of ART with GMeP was the most effective one, with an IC50 value of 2.6 nM, 21 times more potent than ART. Further, its SI (16372) was also significantly higher than that of ART, and it is the safest compound in the series.
The significantly lower IC50 value of the other two hybrids, 55a (IC50 = 8.4 nM, SI = 2108) and 55b (IC50 = 10.6 nM, SI = 983), comprising two ART molecules held via a PA chain, indicates that the second ART molecule does not present any benefit but lowers the SI values. The results also indicated that combinations between ACQ and endoperoxide GMeP were not effective. Further, the length and the geometry of the PA chain also did not appear to play any substantial role. This suggests that conjugation of two endoperoxide molecules can be employed for the development of safer and effective hybrids in the future.62
4.7 Ciprofloxacin-based hybrid
Mukhopadhyay et al. (2020)63 recently synthesized a new class of ciprofloxacin (fluoroquinolone-based antibiotic) hybrid molecules of acridine, quinolone, sulfonamide, and cinnamoyl pharmacophores. The hybrids were evaluated in vitro for their antimalarial activity against CQSPf 3D7, and CQRPf W2 strains using ciprofloxacin (CFX) and CQ as controls. All four hybrids exhibited significant antimalarial activity compared with CFX owing to their increased lipophilicity. The two most effective hybrids, 56 and 57, were tested against the Pf W2 strain, and showed 2 × 102- to 7 × 102-fold superior antimalarial activity compared with CFX. These were also more potent than CQ in the first life-cycle, with IC50 values 25.52 nM (3D7) and 63.17 nM (W2) and 37.63 nM (3D7) and 146.2 nM (W2), and also in the second life-cycle, with IC50 of 13.52 (3D7) and 30.64 (W2) and 14.83 nM (3D7) and 33.39 nM (W2), respectively. The hybrids displayed 3- and 7-fold higher activity, respectively, against the Pf W2 strain in the first cycle than did CQ. Also, these hybrids displayed 8- and 17-fold better resistance indices, respectively, than CQ. The cytotoxicity of hybrid 56 (CQ–CFX) was evaluated against mammalian fibroblast NIH3T3 cells by MTT assay using concentrations ranging from 0.3125 to 20 μM. The results revealed that the CQ–CFX hybrid did not produce any significant cytotoxicity up to 10 μM. Moreover, the hybrid did not display any hemolysis of RBCs at its highest tested concentration (test range: 10–45 μM). Further, the study revealed that CQ–CFX shows a stage-specific effect and is more effective during the ring and schizont stages. This study lacked in vivo validation of the efficacy of CQ–CFX, and there is a possibility that the ester prodrug may not survive in vivo.634.8 1,2,4-Trioxolane–carbonyl hybrids
O'Neill et al. (2010)64 targeted two subsections of carbonyl-based inhibitors: the carbonyl-containing peptides and the 1,2,4-trioxolane prodrugs. Among all of these, 58b showed potent antimalarial activity, with IC50 9.3 nM against the 3D7 strain of P. falciparum. Hybrids 58b and 58a acted as selective inhibitors of falcipain-2, with low-nanomolar activity (1.4 nM and 16.3 nM, respectively). These hybrids were thought to provide two independent mechanisms of action: release of carbonyl protease inhibitor from iron(II) decomposition and also release of cytotoxic C-radical species.644.9 Artemisinin–polypyrrole hybrids
Netropsin (NET), an antibiotic containing amide-linked N-methyl pyrrole units, showed a high affinity for adenine and thymine residues in the minor groove of DNA. Based on this, O'Neill et al. synthesized a series of ART–polypyrrole hybrids as minor groove binders and studied their antiplasmodial activity. They proposed that the ART hybrids have the possibility of increased and specific antimalarial activity as the malaria parasite P. falciparum has AT-rich DNA (>80%, compared with 60% in human). These hybrids were assessed in vitro against the CQS 3D7 strain of P. falciparum, and all hybrids showed IC50 values in the nanomolar range, but none of them was as effective as DHA. Moreover, hybrid 59 showed an IC50 value of 24 nM.655. Conclusions & future prospects
The development of hybrid molecules is a powerful method to generate new candidate antimalarial drugs, with some reaching clinical trials. It has gained momentum because of their promising biological effects. The remarkable developments made in the last few years in controlling the disease provide hope that malaria eradication is an achievable goal. The literature suggests that identification of linking fragments and the selection of the linker are the crucial steps in designing hybrid molecules. Here, we found, to the best of our knowledge, that most of the literature available on antimalarial hybrids has not shed light on their molecular mechanisms. Therefore, a detailed molecular understanding of the pathways involved in the mechanisms of action of these hybrid molecules is also required, so that more robust and effective hybrids can be designed and developed to achieve the goal. It is also evident that the molecular hybridization strategy can restore the efficacy against CQR strains of P. falciparum and could be beneficial for the next generation of drug candidates in malarial chemotherapy. To overcome the resistance to the available antimalarial remedies, new drug discovery efforts should also be integrated with combination therapy. The cross-talk mechanisms leading to additive and synergistic effects should be explored. Differential degrees of resistance of humans to this pathogen warrant in-depth analysis of the human genome concerning malarial infection. Study of the impact of the pathogen on the MHC (major histocompatibility complex) locus controlling the immune system could prove to be rewarding. With the current efforts, there is hope that more powerful drugs will be added to the arsenal in our fight against malaria in the near future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Mohammad Abid gratefully acknowledges the financial support of a Core Research Grant from the Science & Engineering Research Board (SERB), Government of India (File No. CRG/2018/003967). Amad Uddin is the recipient of a Senior Research Fellowship from ICMR (Fellowship/48/2019-ECD-II). Iram Irfan acknowledges the Department of Science and Technology (DST), Government of India for fellowship under its Women Scientists scheme (WOS-A, File no: SR/WOS-A/CS-116/2018).References
- B. Aneja, B. Kumar, M. A. Jairajpuri and M. Abid, RSC Adv., 2016, 6, 18364–18406 RSC.
- World health organization, World malaria report 2018, https://www.who.int/malaria/publications/world-malaria-report-2019/en, (accessed March 2020) Search PubMed.
- K. Plewes, S. J. Leopold, H. W. F. Kingston and A. M. Dondorp, Infect. Dis. Clin. North Am., 2019, 33, 39–60 CrossRef.
- P. Brasil, M. G. Zalis, A. D. Pina-Costa, A. M. Siqueira, C. B. Junior, S. Silva, A. L. L. Areas, M. P. Machado, D. A. M. Alvarenga, A. C. F. S. Santelli, H. G. Albuquerque, P. Cravo, F. V. S. Abreu, C. L. Peterka, G. M. Zanini, M. C. S. Mutis, A. Pissinatti, R. L. Oliveira, C. F. A. Brito, M. F. F. Cruz, R. Culleton and C. T. D. Ribeiro, Lancet Glob. Health, 2017, 5(10), e1038–e1046 CrossRef.
- P. Sondo, K. Derra, T. Lefevre, S. D. Nakanabo, Z. Tarnagda, O. Zampa, A. Kazienga, I. Valea, H. Sorgho, J. BoscoOuedraogo, T. R. Guiguemde and H. Tinto, Sci. Rep., 2019, 9, 1–9 CrossRef CAS.
- A. Uddin, V. Singh, I. Irfan, T. Mohammad, R. S. Hada, M. I. Hassan, M. Abid and S. Singh, Bioorg. Chem., 2020, 103, 104142 CrossRef CAS.
- V. Singh, R. S. Hada, A. Uddin, M. Abid, K. C. Pandey and S. Singh, Curr. Top. Med. Chem., 2020, 20, 377–389 CrossRef CAS.
- J. Achan, A. O. Talisuna, A. Erhart, A. Yeka, J. K. Tibenderana, F. N. Baliraine, P. J. Rosenthal and U. D'Alessandro, Malar. J., 2011, 10, 144 CrossRef CAS.
- World health organization, https://www.who.int/medicines/publications/essentialmedicines/en, (accessed March 2020), 2019.
- P. A. Fery, Compr. Nat. Prod. Chem., 2010, 501 Search PubMed.
- R. M. R. J. da Silva, M. O. Gandi, J. S. Mendonça, A. S. Carvalho, J. P. Coutinho, A. C. C. Aguiar, A. U. Krettli and N. Boechat, Bioorg. Med. Chem. Lett., 2019, 27, 1002–1008 CrossRef CAS.
- K. Kaur, M. Jain, R. P. Reddy and R. Jain, Eur. J. Med. Chem., 2010, 45, 3245–3264 CrossRef CAS.
- China Cooperative Research Group, Antimalarial studies on qinghaosu, Chin. Med. J., 1979, 92, 811–816 Search PubMed.
- E. G. Tse, M. Korsik and M. H. Todd, Malar. J., 2019, 18, 93 CrossRef.
- R. T. Eastman and D. A. Fidock, Nat. Rev. Microbiol., 2009, 7, 864 CrossRef CAS.
- E. Martino, M. Tarantino, M. Bergamini, V. Castelluccio, A. Coricello, M. Falcicchio, E. Lorusso and S. Collina, Future Med. Chem., 2019, 11, 1443–1459 CrossRef CAS.
- C. Giannangelo, F. J. I. Fowkes, J. A. Simpson, S. A. Charman and D. J. Creek, Trends Parasitol., 2019, 35, 529–543 CrossRef CAS.
- C. Nsanzabana, Trop. Med. Infect. Dis., 2019, 4, 26 CrossRef.
- D. Agarwal, R. D. Gupta and S. K. Awasthi, Antimicrob. Agents Chemother., 2017, 61(5), e00249-17 CrossRef.
- X. Nqoro, N. Tobeka and B. Aderibigbe, Molecules, 2017, 22, 2268 CrossRef.
- F. W. Muregi and A. Ishih, Drug Dev. Res., 2010, 71, 20–32 CAS.
- L. S. Feng, Z. Xu, L. Chang, C. Li, X. F. Yan, C. Gao, C. Ding, F. Zhao, F. Shi and X. Wu, Med. Res. Rev., 2020, 40, 931–971 CrossRef CAS.
- Y. Q. Hu, C. Gao, S. Zhang, L. Xu, Z. Xu, L. S. Feng, X. Wu and F. Zhao, Eur. J. Med. Chem., 2017, 139, 22–47 CrossRef CAS.
- A. Kumari, M. Karnatak, D. Singh, R. Shankar, J. L. Jat, S. Sharma, D. Yadav, R. Shrivastava and V. P. Verma, Eur. J. Med. Chem., 2019, 163, 804–829 CrossRef CAS.
- C. C. Musonda, G. A. Whitlock, M. J. Witty, R. Brun and M. Kaiser, Bioorg. Med. Chem. Lett., 2009, 19, 481–484 CrossRef CAS.
- A. Kumar, K. Srivastava, S. R. Kumar, S. K. Puri and P. M. Chauhan, Bioorg. Med. Chem. Lett., 2009, 19, 6996–6999 CrossRef CAS.
- N. C. Araújo, V. Barton, M. Jones, P. A. Stocks, S. A. Ward, J. Davies, P. G. Bray, A. E. Shone, M. L. Cristiano and P. M. O'Neill, Bioorg. Med. Chem. Lett., 2009, 19, 2038–2043 CrossRef.
- F. Cosledan, L. Fraisse, A. Pellet, F. Guillou, B. Mordmüller, P. G. Kremsner, A. Moreno, D. Mazier, J. P. Maffrand and B. Meunier, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17579–17584 CrossRef CAS.
- F. Bellot, F. Coslédan, L. Vendier, J. S. Brocard, B. Meunier and A. Robert, J. Med. Chem., 2010, 53, 4103–4109 CrossRef CAS.
- F. A. R. Ruiz, R. N. G. Sánchez, S. V. Estupinan, A. G. Barrio, D. F. T. Amado, B. M. P. Solórzano, J. N. Ruiz, A. R. M. Fernandez and V. Kouznetsov, Bioorg. Med. Chem., 2011, 19, 4562–4573 CrossRef.
- A. Kumar, K. Srivastava, S. R. Kumar, M. I. Siddiqi, S. K. Puri, J. K. Sexana and P. M. S. Chauhan, Eur. J. Med. Chem., 2011, 46, 676–690 CrossRef CAS.
- M. Sharma, K. Chauhan, S. S. Chauhan, A. Kumar, S. V. Singh, J. K. Saxena, P. Agarwal, K. Srivastava, S. R. Kumar, S. K. Puri, P. Shah, M. I. Siddiqid and P. M. S. Chauhan, MedChemComm, 2012, 3, 71–79 RSC.
- S. Manohar, S. I. Khan and D. S. Rawat, Chem. Biol. Drug Des., 2013, 81, 625–630 CrossRef CAS.
- S. Sahu, S. K. Ghosh, J. Kalita, M. Dutta and H. R. Bhat, Exp. Parasitol., 2016, 163, 38–45 CrossRef CAS.
- K. Starcevic, D. Pesic, A. Toplak, G. Landek, S. Alihodzic, E. Herreros, S. Ferrer, R. Spaventi and M. Peric, Eur. J. Med. Chem., 2012, 49, 365–378 CrossRef CAS.
- Nisha, J. Gut, P. J. Rosenthal and V. Kumar, Eur. J. Med. Chem., 2014, 84, 566–573 CrossRef CAS.
- R. Oliveira, D. Miranda, J. Magalhaes, R. Capela, M. J. Perry, P. M. O'Neill, R. Moreira and F. Lopes, Bioorg. Med. Chem., 2015, 23, 5120–5130 CrossRef CAS.
- P. L. Reddy, S. I. Khan, P. Ponnan, M. Tripathi and D. S. Rawat, Design, Eur. J. Med. Chem., 2017, 126, 675–686 CrossRef CAS.
- M. Lodige, M. D. Lewis, E. S. Paulsena, H. L. E. G. Pradel, L. Lehmann, R. Brune, G. Bringmann and A. K. Mueller, Int. J. Med. Microbiol., 2013, 303, 539–547 CrossRef.
- K. Singh, H. Kaur, P. Smith, C. D. Kock, K. Chibale and J. Balzarini, J. Med. Chem., 2013, 57, 435–448 CrossRef.
- L. C. S. Pinheiro, N. Boechat, M. D. L. G. Ferreira, C. C. S. Júnior, A. M. L. Jesus, M. M. M. Leite, N. B. Souza and A. U. Krettli, Bioorg. Med. Chem., 2015, 23, 5979–5984 CrossRef CAS.
- R. Reissoares, J. M. F. Silva, B. C. Carlos, C. C. Fonseca, L. S. A. Souza, F. V. Lopes, R. M. P. Dias, P. O. L. Moreira, C. Abramo, G. H. R. Viana, F. P. Varotti, A. D. Silva and K. K. G. Scopel, Bioorg. Med. Chem. Lett., 2015, 25, 2308–2313 CrossRef.
- S. Verma, S. Pandey, P. Agarwal, P. Verma, S. Deshpande, J. K. Saxena, K. Srivastava, P. M. S. Chauhana and Y. S. Prabhakar, RSC Adv., 2016, 6, 25584–25593 RSC.
- K. Pavic, I. Perkovic, P. Gilja, F. Kozlina, K. Ester, M. Kralj, D. Schols, D. H. Litina, E. Pontiki and B. Zorc, Molecules, 2016, 21, 1629 CrossRef.
- D. Saini, S. Jain, A. Kumar and N. Jain, EXCLI J., 2016, 15, 730–737 Search PubMed.
- H. M. Faidallah, S. S. Panda, J. C. Serrano, A. S. Girgis, K. A. Khan, K. A. Alamry, T. Therathanakorn, M. J. Meyers, F. M. Sverdrup, C. S. Eickhoff, S. G. Getchellc and A. R. Katritzky, Bioorg. Med. Chem., 2016, 24, 3527–3539 CrossRef CAS.
- S. S. Maurya, A. Bahuguna, S. I. Khan, D. Kumar, R. Kholiya and D. S. Rawat, Eur. J. Med. Chem., 2019, 162, 277–289 CrossRef CAS.
- I. Wadi, D. Prasad, N. Batra, K. Srivastava, A. R. Anvikar, N. Valecha and M. Nath, ChemMedChem, 2019, 14, 484–493 CrossRef CAS.
- D. B. Patel, K. D. Patel, N. P. Prajapati, K. R. Patel, D. P. Rajani, S. D. Rajani, N. S. Shah, D. D. Zala and H. D. Patela, J. Heterocycl. Chem., 2019, 56, 2235–2252 CrossRef CAS.
- D. B. Patel, D. G. Darji, K. R. Patel, D. P. Rajani, S. D. Rajani and H. D. Patel, J. Heterocycl. Chem., 2020, 57(3), 1183–1200 CrossRef CAS.
- T. V. de Walle, M. Boone, J. V. Puyvelde, J. Combrinck, P. J. Smith, K. Chibale and M. D’hooghe, Eur. J. Med. Chem., 2020, 198, 112330 CrossRef.
- J. P. Joubert, F. J. Smit, L. Plessis, P. J. Smith and D. D. N'Da, Eur. J. Pharm. Sci., 2014, 56, 16–27 CrossRef CAS.
- T. Frohlich, C. Reiter, M. M. Ibrahim, J. Beutel, C. Hutterer, I. Zeittrager, H. Bahsi, M. Leidenberger, O. Friedrich, B. Kappes, T. Efferth, M. Marschall and S. B. Tsogoeva, ACS Omega, 2017, 2, 2422–2431 CrossRef.
- T. Frohlich, A. Kiss, J. Wolfling, E. Mernyak, A. E. Kulmany, R. Minorics, I. Zupko, M. Leidenberger, O. Friedrich, B. Kappes, F. Hahn, M. Marschall, G. Schneider and S. B. Tsogoeva, ACS Med. Chem. Lett., 2018, 11, 1128–1133 CrossRef.
- C. D. Lange, D. Coertzen, F. J. Smit, J. F. Wentzel, H. N. Wong, L. M. Birkholtz, R. K. Haynes and D. D. N'Da, Bioorg. Med. Chem. Lett., 2018, 28, 3161–3163 CrossRef.
- F. Grellepois, P. Grellier, D. B. Delpon and J. P. Begue, ChemBioChem, 2005, 6, 648–652 CrossRef CAS.
- J. J. Walsh, D. Coughlan, N. Heneghan, C. Gaynor and A. Bell, Bioorg. Med. Chem. Lett., 2007, 17, 3599–3602 CrossRef CAS.
- R. Capela, G. G. Cabal, P. J. Rosenthal, J. Gut, M. M. Mota, R. Moreira, F. Lopes and M. Prudencio, Antimicrob. Agents Chemother., 2011, 55, 4698–4706 CrossRef CAS.
- M. C. Lombard, D. D. N'Da, J. C. Breytenbach, P. J. Smith and C. A. Lategan, Bioorg. Med. Chem. Lett., 2011, 21, 1683–1686 CrossRef CAS.
- M. C. Lombard, D. D. N'Da, C. T. V. Ba, S. Wein, J. Norman, L. Wiesner and H. Vial, Malar. J., 2013, 12, 71 CrossRef CAS.
- A. Capci, M. M. Lorion, H. Wang, N. Simon, M. Leidenberger, M. C. B. Silva, D. R. M. Moreira, Y. Zhu, Y. Meng, J. Y. Chen, Y. M. Lee, O. Friedrich, B. Kappes, J. Wang, L. Ackermann and S. B. Tsogoeva, Angew. Chem., Int. Ed., 2019, 58, 13066–13079 CrossRef CAS.
- D. A. Pepe, D. Toumpa, C. Andre-Barres, C. Menendez, E. Mouray, M. Baltas, P. Grellier, D. Papaioannou and C. M. Athanassopoulos, ACS Med. Chem. Lett., 2020, 11, 921–927 CrossRef CAS.
- S. Dana, P. Valissery, S. Kumar, S. K. Gurung, N. Mondal, S. K. Dhar and P. Mukhopadhyay, ACS Med. Chem. Lett., 2020, 11, 1450–1456 CrossRef CAS.
- P. Gibbons, E. Verissimo, N. C. Araujo, V. Barton, G. L. Nixon, R. K. Amewu, J. Chadwick, P. A. Stocks, G. A. Biagini, A. Srivastava, P. J. Rosenthal, J. Gut, R. C. Guedes, R. Moreira, R. Sharma, N. Berry, M. L. S. Cristiano, A. E. Shone, S. A. Ward and P. M. O'Neill, J. Med. Chem., 2010, 53, 8202–8206 CrossRef CAS.
- L. L. Pense, S. Sabbani, R. Sharma, I. Bhamra, E. Shore, A. E. Chadwick, N. G. Berry, J. Firman, N. C. Araujo, L. Cabral, M. L. S. Cristiano, C. Bateman, O. Janneh, A. Gavrila, Y. H. Wu, A. Hussain, S. A. Ward, P. A. Stocks, R. Cosstick and P. M. O'Neill, ChemMedChem, 2013, 8, 709–718 CrossRef.
| This journal is © The Royal Society of Chemistry 2021 |