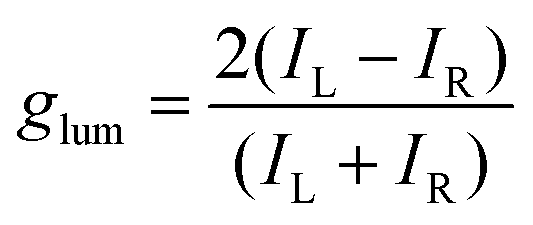DOI:
10.1039/D1MA00846C
(Review Article)
Mater. Adv., 2021,
2, 7620-7637
Imprinting chirality in inorganic nanomaterials for optoelectronic and bio-applications: strategies, challenges, and opportunities
Received
13th September 2021
, Accepted 18th October 2021
First published on 20th October 2021
Abstract
Chirality, ubiquitous in biological systems, is the property of the non-superimposable structures found in both macroscopic and nanoscopic objects. In the last ten years, the research on chiral inorganic nanomaterials has witnessed tremendous growth due to their shape, size, crystal facets-controlled unparalleled physical, chemical, optoelectronic, and bio-compatibility features. Due to the quantum confinement, the chiral nanomaterials exhibit high anisotropy factors and, therefore, there has been an upsurge in research activities around the chiroptical and (bio)recognition properties of optically active inorganic nanomaterials. Advancements in synthesis and characterization technologies have made it possible to prepare complex multi-functional materials. Inorganic nanoparticles are known to exhibit fascinating semiconducting or metallic behavior and when combined with chirality, their usability in various devices is enhanced drastically due to an additional degree of freedom over achiral systems. In this review, we discuss different chemical routes to transfer chirality from intrinsic chiral systems into achiral nanomaterials for optoelectronics and bio-applications, in the light of recent experimental findings. Despite the progress in the research field of nanomaterials, imprinting chirality, device fabrication, scalability, long-term stability, and reproducibility remain challenging tasks that need to be addressed. Since the synthesis and growth mechanisms of chiral inorganic systems can be found in literature, here, we shed light only on the complimentary metal-oxides semiconductors (CMOS), and bio-compatibility of either intrinsic or imprinted chiral nanomaterials and their potential applications in next-generation chiroptical devices. We also discuss feasible strategies to resolve existing issues related to the integration into devices and their operational aspects.

Prakash Chandra Mondal
| Prakash Chandra Mondal was born in a small village called Shyamnagar, part of the Sunderbans (means “beautiful forest”) island, West Bengal, India. He earned his PhD from the University of Delhi in 2013. He then moved to the Weizmann Institute of Science, Israel, and worked on molecular spintronics (2013–2016). Later, he worked on molecular electronics at the University of Alberta, Canada. Before joining the Department of Chemistry, at the Indian Institute of Technology (IIT) Kanpur as an Assistant Professor in 2019, he was a Marie-Curie Post-doctoral Fellow at the University of Valencia, Spain. Since 2021, he has been serving as the Analyst Board Member, RSC. |

Deepak Asthana
| Deepak Asthana obtained his PhD degree in Supramolecular and Materials chemistry in 2013 from Jawaharlal Nehru University, India. In 2014, he was awarded with JSPS postdoctoral fellow to work at Kyushu University, Japan, where he explored the field of triplet–triplet annihilation-based photon up-conversion. Later in 2016, he joined the ‘Molecular Magnets’ group at The University of Manchester, UK, where he worked on the synthesis of heterometallic multi-qubit systems. Currently, he is an Assistant Professor in the Department of Chemistry, Ashoka University, India. |

Ranjeev Kumar Parashar
| Ranjeev Kumar Parashar was born in Bihar, India. He received his BSc (Hons.) from Banaras Hindu University in 2017 and MSc in Chemistry from the Indian Institute of Technology (IIT), Kharagpur in 2019. He has qualified for the CSIR-NET examination to receive a junior research fellowship (JRF). Since July 2019, he has been working on nano-scale molecular electrochromic devices in the team of Dr Prakash Chandra Mondal at the Department of Chemistry, IIT Kanpur. |

Sakshi Jadhav
| Sakshi Jadhav is currently pursuing her MSc in Inorganic Chemistry from Ramnarain Ruia Autonomous College, University of Mumbai, India. She has completed her BSc in Chemistry from Bhavan's College, University of Mumbai. At present, she is working as an intern in a team led by Dr Prakash Chandra Mondal in the Department of Chemistry, Indian Institute of Technology (IIT) Kanpur. Her research interests include material science and nanotechnology. |
Key learning points
(1) Origin of homochirality and chirality transfer from intrinsic chiral systems to achiral inorganic nanomaterials.
(2) Various routes to functionalize chiral nanomaterials for governing size, shape, and optical properties.
(3) Recent trends in optoelectronic devices and bio-applications.
(4) Future scopes, challenges, and possible solutions.
|
1. Introduction
Chirality or handedness refers to materials, molecules, and architectures that lack, a plane, center, or axis of symmetry. In other words, an object that has non-superimposable mirror images is called chiral. The concept of chirality in molecules was first demonstrated by Louis Pasteur in 1848,1 while the phrase ‘chirality’ was first coined by Lord Kelvin in the year 1894.2 This finding has paved a new avenue for discovering many hidden phenomena at the macroscopic and also nanoscopic levels. Chirality appears as a ‘holy grail’ that can help us understand why nature prefers homochirality, which is a 100% enantiomeric excess (ee).3 In the recent past, chiral molecules have attracted the immense attention of researchers, particularly for their diverse applications in chiral optical switches, bio-sensors, and optoelectronic devices.4–7 Chiral systems show a high degree of chiral anisotropy that stems from strong resonance upon interacting with the electromagnetic waves. Thus, the chiral systems can rotate the plane-polarized electromagnetic waves (light in general) in either the left or right direction, depending on the optical configuration of the system. Of late, through the lenses of experiments and theoretical studies, Ron Naaman and others have found that organic chiral molecules such as DNA, oligopeptides, proteins, and polymers are capable of filtering the spin when a spin-polarized current passes through them.8–11 However, chiral inorganic nanomaterials are relatively less explored than (bio)organic molecules.12–16 Unlike the chiral organic materials, the exact understanding of the source of chirality in inorganic materials, including metallic or semiconductors, is still in the development stage.13,17,18 It has been observed that in many cases during the nanoparticle formation, the metal atoms in the cluster attained a lower symmetry and exhibited chiral effects.19–21 For instance, the Whetten group developed a method to prepare an optically active thiourea-based gold cluster compound Au28(SG)16 (where SG = N-γ-glutamyl-cysteinyl-glycine).22 The circular dichroism (CD) spectrum of this cluster exhibited a peak in the gold absorption band regions, which is an exciting feature observed with a chiral metal nanoparticle system. The Kelly group demonstrated that the microwave-assisted preparation of chiral luminescent CdS quantum dots (QDs) utilizing racemic and enantiomerically pure penicillamine.23 The CD spectra of semiconductor chiral CdS QDs display distinct signatures in the UV region (200–390 nm). Dynamic monitoring of the CD signals and quantum mechanical modeling by density functional theory (DFT) calculation suggest that the nanoparticle (NPs) core remains achiral whereas the surface acquires chirality and contributes to new signals in the CD spectrum.
A rather simpler approach to imprint chirality in an inorganic nanoparticle is to coat its surface with optically active moieties by performing surface engineering or ligand exchange. Surface engineering has found special applicability in the field of (chiral)nanoparticles (NPs).13,24–27 Two approaches are generally adopted for surface functionalization: one in which the nanoparticle core is coated with some inorganic shell such as ZnS or ZnSe to give core/shell or heterostructures, and the second in which the nanoparticle surface is protected with organic ligands.28,29 While core/shell type modification helps in removing surface defects, improves photo-stability and quantum efficiency, ligand exchange plays a vital function in controlling the overall usability of the induced chirality either in NPs or nanocrystals (NCs).30,31 Ligand exchange helps to tune the solubility or stability against aggregation, and further functionalization of nanoparticles.32,33 The nanoparticle surface can be modified with an intrinsic chiral ligand generally containing functional surface-active head groups such –NH2, –COOH, –SH, N3, etc., which would allow the further attachment of any desired moiety around the NP's surface.34–40 In general, the choice of functionalizing reagents depends very much on the nature of the nanoparticles; for example, thiol derivatives for gold and silver nanoparticles, carboxylic or phosphoric derivatives for metal oxides, and silane-based compounds for silica nanoparticles. This approach has been widely used in the field of biosensing and imaging and has been reviewed elsewhere.26,27,41,42 Here, we mainly focus on the three sections, the first section deals with the chirality transfer and surface engineering, the second part talks about optoelectronic device integration, and the third section discusses bio-applications of the chiral inorganic nanomaterials. A schematic overview encompassing the subject matter of this review is illustrated in Fig. 1.
 |
| | Fig. 1 A synopsis of the scope and applications of chiral inorganic nanomaterials, which is the key subject of the present review. | |
2. Origin of chirality and the surface engineering of chiral nanomaterials
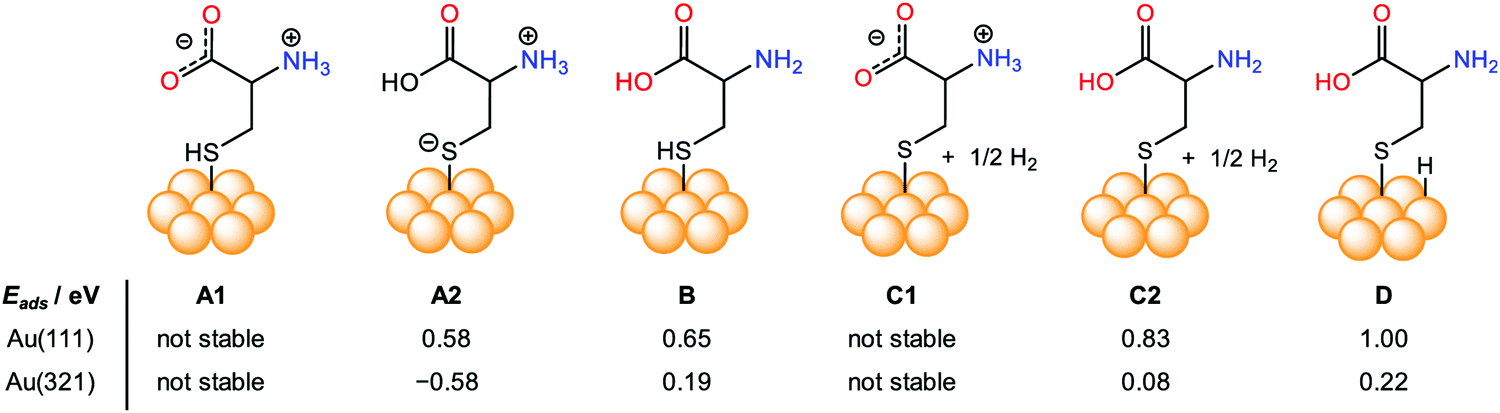
The chirality in any object appears due to the non-zero numerical function of the dot product between two components, electric dipole moment transition μ, and magnetic dipole moment transition m, according to the Rosenfeld equation.43R denotes the CD intensity at a given transition, and Im is the imaginary section of the scalar product. The concept of inducing chirality in the achiral inorganic NPs enriches the fundamental science domain and various applications. Inorganic chiral NPs originating from noble metals like gold and silver, or CdS/CdSe have emerged as appealing candidates for biocatalysts, sensing, optoelectronics, and spintronic applications, to name a few.44–48 Gold NPs have high Miller indices represented by ({hkl}, h ≠ k ≠ l ≠ 0), e.g., (321) and (17 11 9), which exhibit either intrinsic or induced chirality. However, chirality in an achiral nanoparticle can also be induced by capping its surface with intrinsic chiral moieties such as amino acids, proteins, peptides, and DNA, etc.49–52 To exploit chiral materials in practical applications, in-depth knowledge about chiral morphology at the atomic level is highly desired.53–56 In this regard, a systematic theoretical description to comprehend the interaction between chiral/achiral ligands and metal surfaces can help to attain a rational design, preparation, and better understanding of the chirality imprint phenomena.57,58 Aliphatic or aromatic thiols or dithiols are known to be strongly adsorbed on Au surfaces, forming stable Au–S interfaces and, therefore, are one of the most widely used NP surface modifier ligands.59–63 Though many theoretical studies were made to predict the chirality transfer mechanisms and dynamics, the studies discussing enantioselectivity remain sparse.64–69 This void was nearly filled by the work from the Ortuño group who carried out computational simulations using density functional theory (DFT), providing a profound understanding of the enantioselective interaction at an atomic level. Their study included different forms of chiral amino acids such as L-cysteine (neutral, ionic, zwitterion) adsorbed on both the chiral enantiomeric Au facets, Au(321), Au(32![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) and achiral Au(111), Au(110), Au(100) NPs surfaces.70 In their theoretical framework, 4 atomic layers were considered for (111) and (110), 8 for (100), and 16 for (321) and (32) facets. L-Cysteine growth adsorption energies on the multifacets Au NPs, Eads were computed according eqn (2):
) and achiral Au(111), Au(110), Au(100) NPs surfaces.70 In their theoretical framework, 4 atomic layers were considered for (111) and (110), 8 for (100), and 16 for (321) and (32) facets. L-Cysteine growth adsorption energies on the multifacets Au NPs, Eads were computed according eqn (2):| | | Eads = ES–L − ES − nEL | (2) |
where ES–L denotes the energy of L-cysteine deposited on Au, ES represents the energy of the Au NPs, nEL signifies the energy of n no. of L-cysteine units. While the different forms of L-cysteine grafted on Au(111) are endothermic, the zwitterionic form A2, in which a negative charge resides on the sulfur linked to the Au(321) surface through S− moieties, is the most stable form, having an adsorption energy of 0.58 eV (Fig. 2). From the quantum mechanical calculations, they concluded that the adsorption energy of the chiral amino acid, L-cysteine on the Au(321) facet increased as compared to the case when adsorbed on the Au(32) at a relatively lower surface coverage. This observation can help to rationalize the design of enantioselective experiments at the nanoscale. However, in their investigation, the computational studies on D-cysteine were not performed. The authors concluded that the computational tool might have immense potential to elucidate many complexities associated with the origin of chirality.
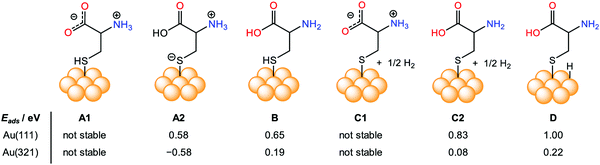 |
| | Fig. 2 Pictorial representation and comparison of the simulated surface adsorption energy, Eads (eV), of different forms of intrinsic L-cysteine amino acid (neutral, ionic, zwitterion) adsorbed on various crystal facets, including achiral Au(111), and chiral Au(321). Reprinted with permission.70 Copyright 2019, American Chemical Society. | |
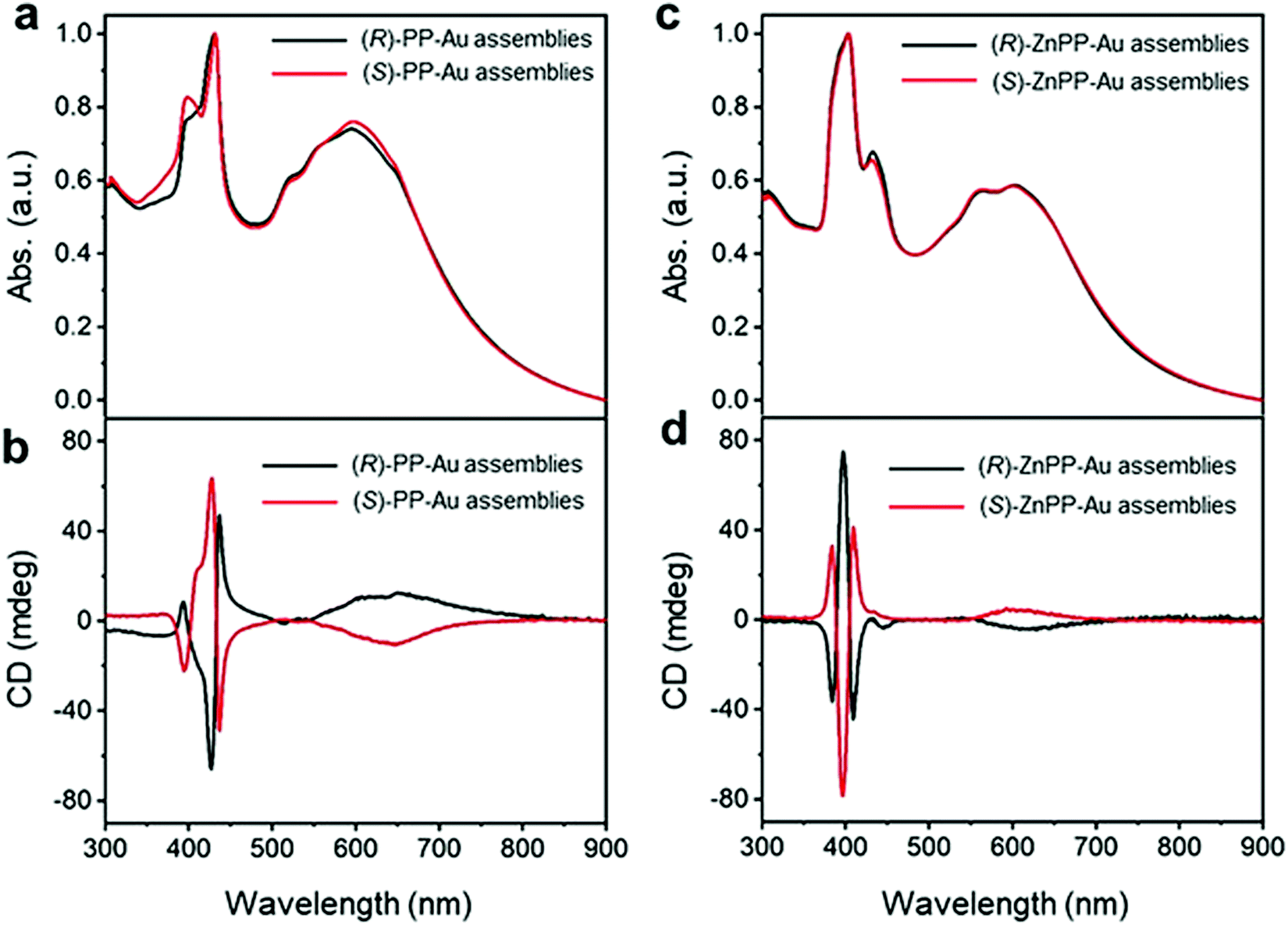
Recently, Yang and co-workers employed a new strategy for chirality transfer in achiral Au NPs superlattices.71 The group utilized the chiral porphyrin-based supramolecular assembly and exploited the van der Waals interactions to induce chirality. It was also claimed to be the first report of its kind on chirality transfer employing van der Waals interactions. Remarkably, it was observed that the chiral porphyrin supramolecular structure adopted J-type stacking and, upon metallization with zinc metal ion, changed to H-type stacking while maintaining its chirality. To induce the chirality in achiral Au NPs, first, the metallic NPs were functionalized with achiral n-octadecanethiol, resulting in core/shell heterostructures. Second, the chirality was introduced to the core/shell heterostructures by adding alkyl-terminated porphyrin or Zn–porphyrin solution. The presence of a new absorption band in the wavelength range of 500–800 nm in the UV-visible spectra (Fig. 3(a and c)) indicated the formation of two enantiomers such as (R)/(S)-PP-Au and (R)/(S)-ZnPP-Au assemblies. The additional spectral features appeared due to the extinction of metallic chiral Au NPs. More specifically, the CD spectra confirmed the induction of chirality, as positive and negative cotton effects revealed by (R)/(S)-PP-Au and (R)/(S)-ZnPP-Au assemblies could be seen (Fig. 3(b and d)). The TEM image (not shown here) revealed that the Au NPs formed spindle-like structures controlled by the chiral porphyrin. For a deeper understanding of the evolved chirality, the group performed additional experiments by changing the solvent, size, and functional group of the capping reagent. From this experiment, it was concluded that interactions between porphyrin moieties and alkyl chains play a crucial role in transferring chirality. Furthermore, the Zn porphyrin supramolecular structure was coordinated with that of oleylamine, which led to a stronger interaction between Au NPs due to the alkyl chain present in it, resulting in enrichment in the chirality. Key findings in the present work include that both the J-type chiral porphyrins and H-type chiral metalloporphyrins can imprint chirality into Au NPs.
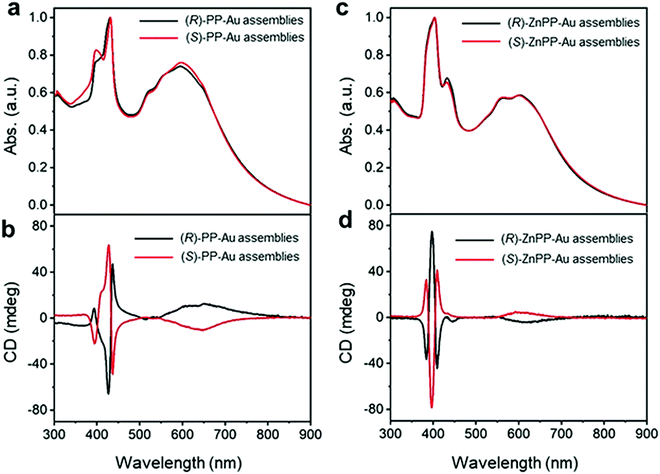 |
| | Fig. 3 (a) UV-Vis spectra recorded on (R)/(S)-PP-Au NPs, (c) (R)/(S)-ZnPP-Au NPs illustrating the appearance of new spectral features in the range of 500 to 800 nm. (b) CD spectra of (R)/(S)-PP-Au NPs, (d) (R)/(S)-ZnPP-Au NPs confirming the chirality transfer into metallic Au NPs. Reprinted with permission.71 Copyright 2021, Wiley-VCH. | |
Chirality can be transferred from chiral nanocluster to bound achiral ligand. Thus, the creation of intrinsically chiral metallic or semiconducting nanoparticle surfaces is crucial for many fundamental studies.72–74 For instance, the Salassa group recently demonstrated chirality transfer from an intrinsically chiral Au nanocluster to the achiral ligand.75 Chiral nanocluster Au38 was functionalized with the achiral 2-phenylethylthiolate (PET) ligand to form Au38(SR)24, and homochirality transfer took place making the achiral ligand VCD (vibrational circular dichroism) active. The induction of chirality was ascribed to asymmetric forms of gauche conformations. Moreover, it was observed that the chirality transfer relies on the site-specific binding on the surfaces of Au38. The dynamic behavior of chirality transfer phenomena was probed by the combined experimental (NMR techniques) and computational studies. Such chirality transfer can be harnessed in chirality-driven nanoscale molecular recognition, sensing, and catalysis. Self-assembly is a very simple yet extremely effective technique that has been extensively used to link the ligands and NPs.76–79 Chen and co-workers utilized the self-assembly method to attach achiral organic molecules rather than the traditional biomolecules to create Janus chiral Au NPs.80 They used organic linkers such as hexanethiolates and 3-mercapto-1,2-propanediol to produce vesicle-like, hollow nanostructures. As the surface coverage by the ligand 3-mercapto-1,2-propanediol and concentration of the NPs increased, the plasmonic circular dichroism (PCD) signals were enhanced. The PCD technique is advantageous over the CD, in particular, to detect the signals in the visible range.16,81 To support the experimental findings, first-principles calculations were performed to elucidate the genesis of chirality. The authors inferred that the nanoparticle dipoles that form a vertex on the hollow sphere surfaces are the prime reason for the initiation of chirality in Au NPs. Furthermore, the chiral NPs were able to differentiate between D-alanine and L-alanine.
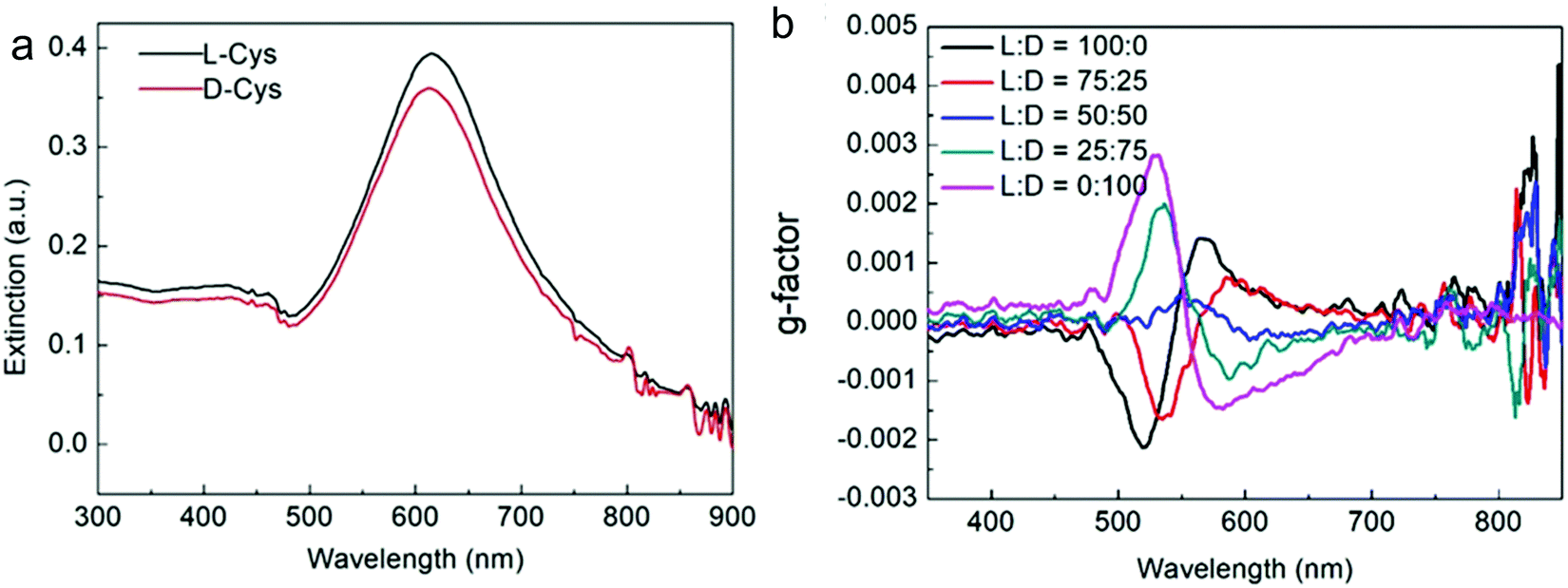
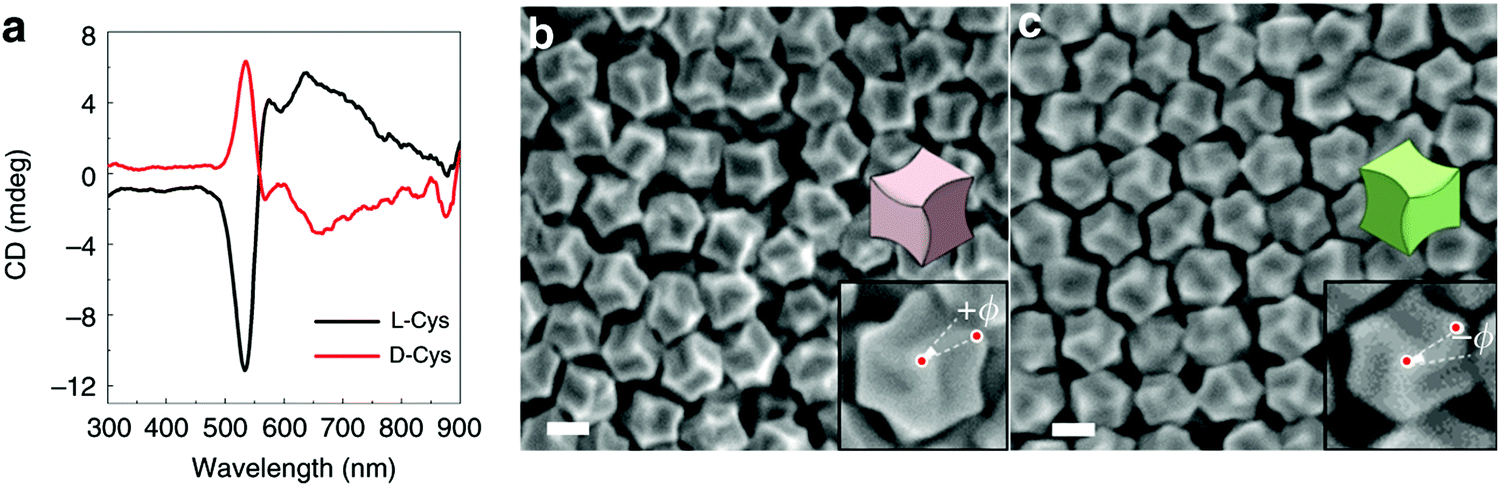
Nam and co-workers have made an outstanding contribution towards imprinting the chirality in achiral metallic nanoparticles by embedding biomolecules and exploring optoelectronics, catalysis, chiral sensing, and biomedical aspects of such systems.17,21,82–87 Recently, the Nam group unraveled the key roles played by the organic chiral inducer, cetyltrimethylammonium bromide (CTAB), and ascorbic acid (AA) in imprinting the chirality, controlling crystal facets, and growth mechanism.88 A two-step growth method that involved seed-mediation to prepare 3D chiral Au NPs was chosen. This stepwise growth process ensured the foundation of a chiral surface with a high miller-lattice index in the initial step and that maintained the interaction between molecules and highly enantioselective surface. Their method relies on the application of chiral amino acids and peptides as shape-controlling agents. In this stepwise growth, the Au NPs of around 150 nm size, which have high index facets exposed, were prepared. Amino acids or peptides were introduced in the system during the second step of the growth. Induced chirality leads to the direction-specific growth of the metallic NPs. They could prepare chiral NPs that display the highest g-factor in the visible region. The absorbance spectra of frehly prepared L-cys–Au NPs and D-cys–Au NPs are shown in Fig. 4a. The g-factor spectra recorded under different ratios indicate that L and D cysteine heavily affect the spectra when the concentration is varied from 100 to 0 (Fig. 4b). The formation of chiral NPs was ensured by CD and scanning electron microscopy (SEM) studies (Fig. 5a–c). Depending on the cysteine's handedness, L- or D-cysteine, the optical response of the NPs could be tuned. The NPs prepared using L-Cys exhibited an absorption peak at 534 nm for right-circularly polarized light, and an absorption peak at 638 nm for left-circularly polarized light. The CD spectra of Au NPs formed by the D-Cys-based displayed similar behavior with inverted peaks. Interestingly, the edge of the prepared rhombic dodecahedral nanoparticles showed twisting depending on the chiral nature of cysteine. The twisting angle of the edge(ø) changed its direction with a change in cysteine chirality. Using an equimolar mixture of L-Cys and D-Cys (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) formed achiral NPs and caused the disappearance of the CD signals. The present study is crucial for understanding small-molecule-induced chirality growth for future nanoscale optoelectronic applications.
:1) formed achiral NPs and caused the disappearance of the CD signals. The present study is crucial for understanding small-molecule-induced chirality growth for future nanoscale optoelectronic applications.
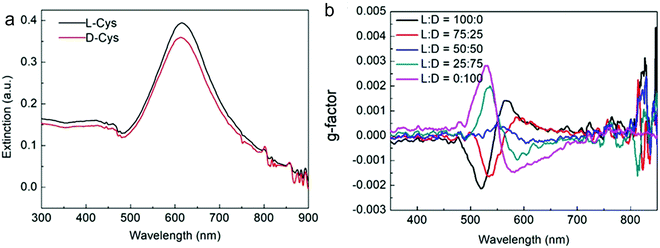 |
| | Fig. 4 (a) Absorbance spectra of L-Cys–Au NPs (solid black line) and D-Cys–Au NPs (solid red line). (b) The spectra affecting the g-factor of Au NPs recorded in different concentration ratios of L-Cys and D-Cys. Reprinted with permission.88 Copyright 2020, Springer Nature. | |
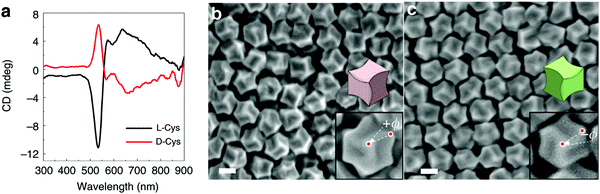 |
| | Fig. 5 (a) Visible-region CD spectra recorded on the L/D-cysteine-induced chiral metallic Au NPs. (b) SEM images of L-cysteine, and (c) D-cysteine induced chiral Au NPs. Reprinted with permission.88 Copyright 2020, Springer Nature. | |
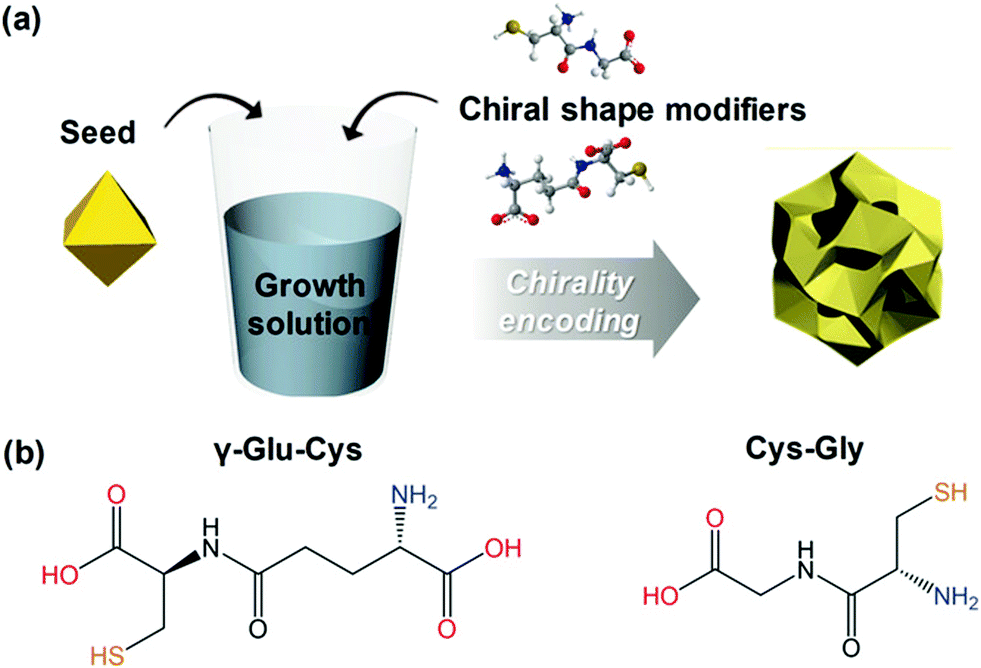
Among the biologically important systems, dipeptides are one of the most relevant systems having intrinsic chirality suitable for use in chirality transfer reactions. The Nam group has employed this impressive methodology and considered two different dipeptides, gamma-glutamyl–cysteine (γ-Glu–Cys) and cysteinyl–glycine (Cys–Gly), to induce chirality in 3-dimensional (3D) metallic Au NPs.87 The strategy is schematically illustrated in Fig. 6. As a result of chirality induction after the introduction of γ-Glu–Cys the Au NPs attained a cube-like morphology, while the Cys–Gly enforced a rhombic dodecahedron shape with elliptical cavities in each face. The SEM images show the reflection of surface-engineered Au NPs encoded with biomolecules and the resulting crystal facets (Fig. 7). To probe the origin of crystallographic chiral surfaces in Au NPs, 3D geometric models were employed, which entail the formation of chiral intermediates that drive the ultimate chirality. The present study demonstrates the dipeptide-mediated induction of the chirality in 3D Au NPs that can diversify their applications in several ways. The examples discussed so far fall in the category that discusses the induction of chirality in noble metallic NPs. However, such induction of chirality can also be used with inorganic semiconductor NPs. There are only limited examples of Chiral CdSe nanocrystals (NCs) that exhibit chiral-ligand-induced morphology and chiroptical behavior.89–94 He and co-workers could successfully modify the surface of CdSe-dot/CdS-rod NCs with the chiral amino acids such as L- and D-cysteine.95 The chirality in semiconductors was induced via a ligand-exchange method where L- and D-cysteine produced L-/D-Cys–CdSe/CdS chiral quantum nanocrystals that display exceptional CD and CPL characteristics. The study showed a remarkable anisotropic factor (glum) equal to 4.66 × 10−4 in CPL spectra with positive and negative line shapes obtained for D- and L-Cys–CdSe/CdS NCs, respectively. The anisotropic factor (glum) can be expressed as eqn 3:96
| | 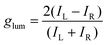 | (3) |
IL,
IR signify the corresponding intensities of the left- and right-handed CPL. Both the CD and CPL characteristics are significantly dependent on the concentration of chiral L/D cysteine, as at higher concentrations CD and CPL signals get suppressed. They further performed the DFT study and concluded that the orbital hybridization of either
L- or
D-cysteine with CdSe/CdS is the key factor of chirogenesis.
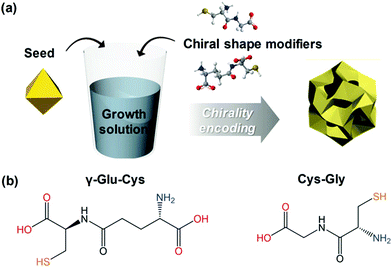 |
| | Fig. 6 (a) A schematic diagram indicating the preparation of chiral shape modifier-induced Au nanoparticles formation. (b) Chemical structures of the dipeptides are used as chirality inducers. Reprinted with permission.87 Copyright 2020, Wiley-VCH. | |
 |
| | Fig. 7 (a) SEM images reveal crystal facets of chiral Au NPs generated by γ-Glu–Cys, and (b) Cys–Gly, respectively. Reprinted with permission.87 Copyright 2020, Wiley-VCH. | |
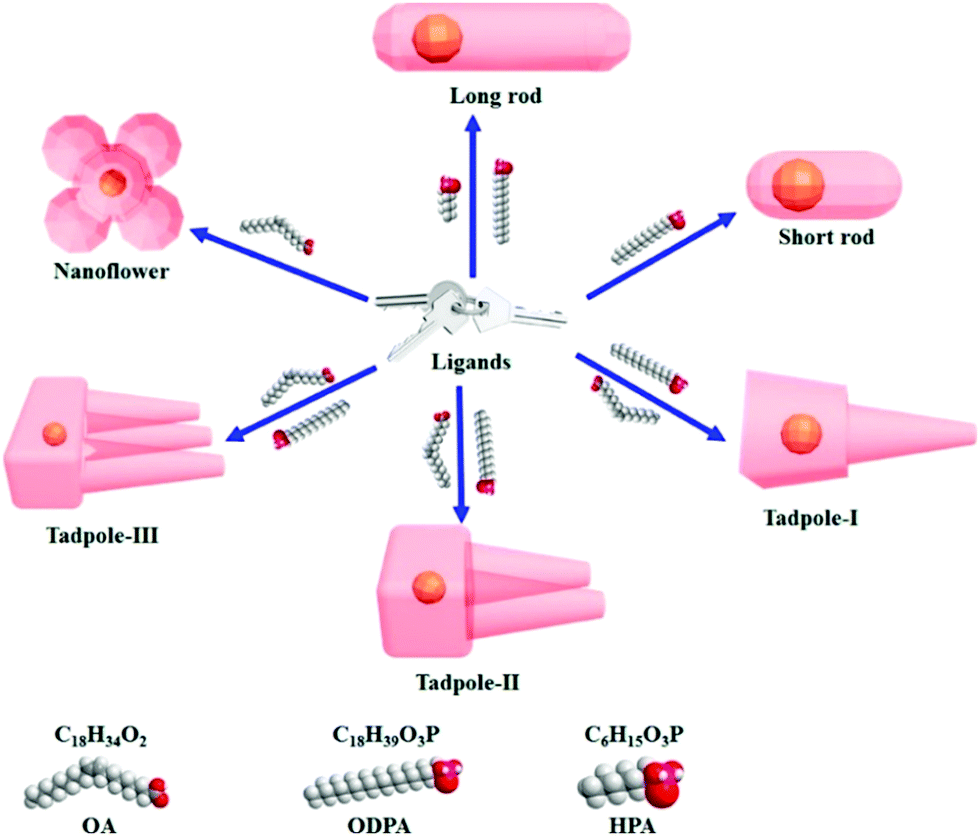
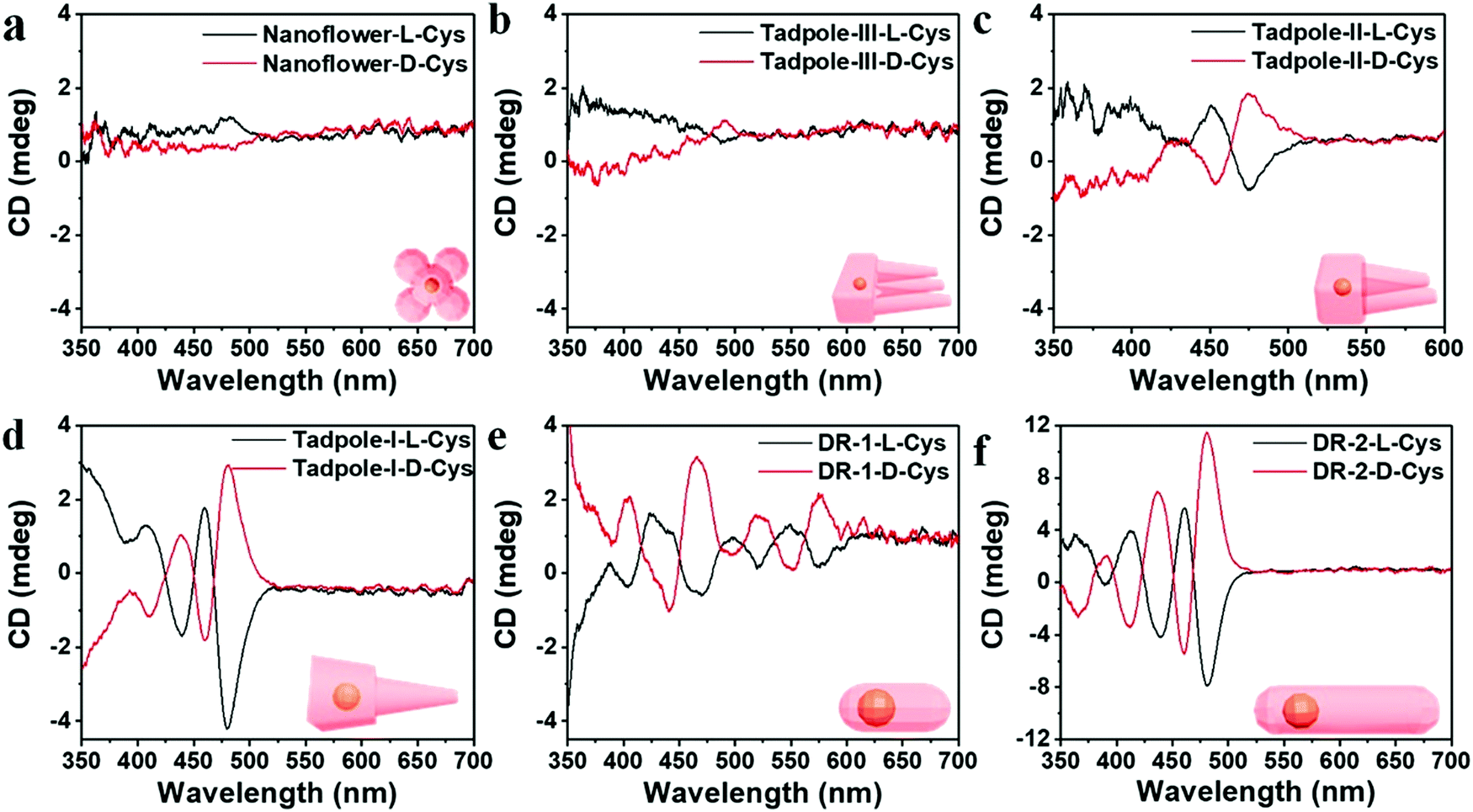
Cheng and co-workers utilized surface functionalization to prepare CdSe/CdS nanocrystals (NCs) of various morphologies including nanoflowers, tadpoles, etc. with precise shape control (Fig. 8).91 The detailed study provided a correlation between the shape and chirality induced by the ligand. In a typical procedure, CdSe/CdS NCs were first prepared followed by a post-synthesis ligand exchange reaction in aqueous media, NCs were modified with L-/D-cysteine. The UV-vis and photoluminescence (PL) spectral analysis of CdSe/CdS nanostructures were performed before and after ligand exchange. The formation of the CdS shell caused a red-shift in the absorption peak from 539 nm to 606 nm depending upon the morphology. Transmission electron microscopy (TEM) confirmed that the shape-directing agents successfully fine-tuned the morphology of the CdSe/CdS nanocrystals (Fig. 9). As can be seen from images (a–f), changing the concentration and nature of the ligand, nano-shapes such as nanoflowers, tadpoles, and dot/rods could be obtained. The obtained CdS shell phase was in agreement with the previous studies that suggest a wurtzite phase CdS growth when wurtzite CdSe seeds are used. It was observed that when the concentration was low or alkylphosphonic acid ligands were absent, the formation of spherical nanocrystals was favored (Fig. 9a–c). To explain the chiroptical changes in various morphologies, a quantitative comparison of CD responses was performed. The CdSe core used to prepare different morphologies of NCs was from the same batch and has been subjected to identical ligand exchange reactions. This implies that any change in CD spectra could be linked to the change in the nanostructure's geometry. As shown in Fig. 10, nanoflowers, tadpole and long dot/rod (DR) shaped structures, which have relatively thicker shells (>1 nm) and large CdS volumes, exhibited CD signals corresponding to CdS only. Conversely, the CD signal for the CdSe core was still present in the short DR sample. It was also observed that the anisotropy factor is enhanced as the morphology changes from the nanoflower shape to the tadpole shape.
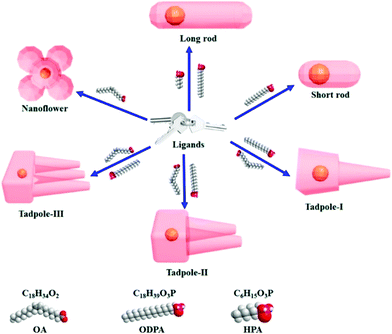 |
| | Fig. 8 A schematic diagram showing that chiral ligand-induced shape variations resulting in various anisotropic shapes can be achieved. Chemical structures of the chiral ligands are shown. Reprinted with permission.91 Copyright 2020, American Chemical Society. | |
 |
| | Fig. 9 (a–f) TEM images of CdSe/CdS NCs obtained in a variety of shapes including nanoflowers, Tadpole-III, Tadpole-II, Tadpole-I, short dot/rods, and long dot/rods. Reprinted with permission.91 Copyright 2020, American Chemical Society. | |
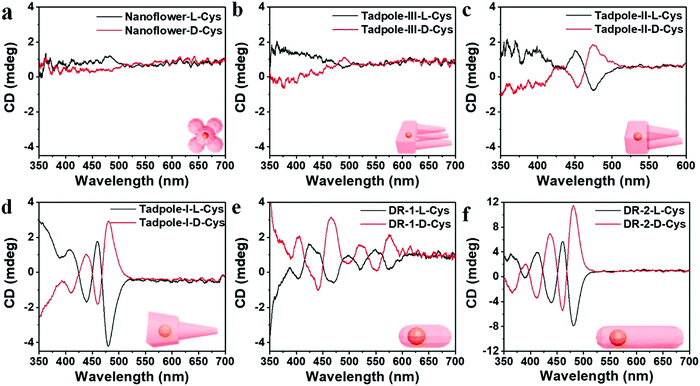 |
| | Fig. 10 (a–f) Mirror symmetry CD spectra of the L-/D-CdSe/CdS NCs having different shapes such as (a) nanoflowers, (b) Tadpole-III, (c) Tadpole-II, (d) Tadpole-I, (e) short dot/rods, and (f) long dot/rods. Reprinted with permission.91 Copyright 2020, American Chemical Society. | |
3. Optoelectronic applications
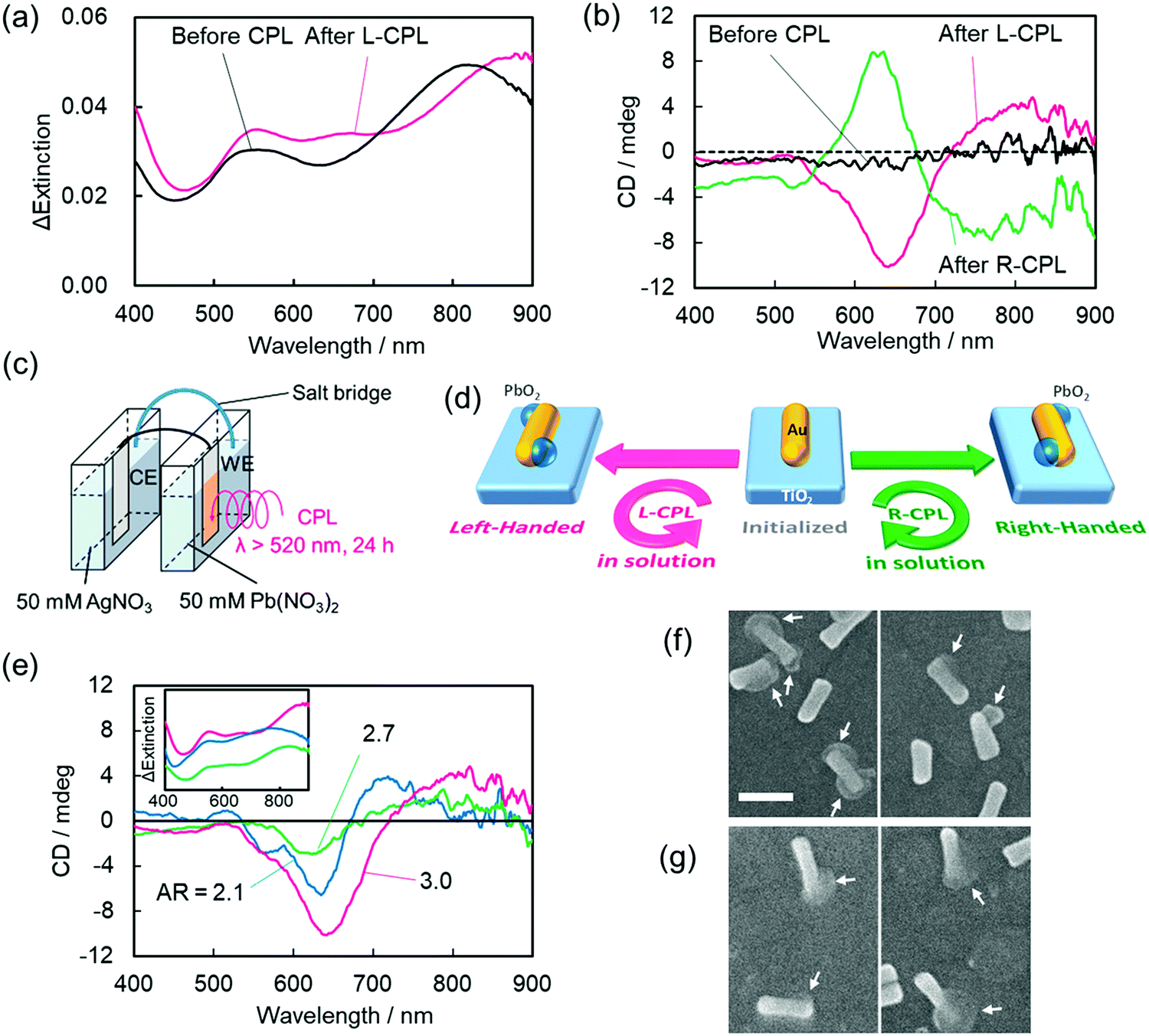
Optoelectronic devices are used to either modulate or detect light under an external electric field. Such devices enable photons to be used in the same fashion as electrons in an electronic device.97,98 Compared to conventional bulk systems, optical properties of nanomaterials benefit from a broad absorption window, higher quantum yields, narrow emission wavelengths, and fewer photo-bleaching risks, thanks to the strong light–matter interactions.99,100 Unlike chiral (bio)-organic materials, which usually contain a narrow absorption cross-section, display weak optical responses, and exhibit optical responses in the UV-region only, chiral plasmonic nanostructures are associated with stronger chiroptical responses and can interact with visible or near-infrared (NIR) light. These appealing features of chiral NPs significantly enhance their applicability in sensing, recognition, and optoelectronic devices. Avoiding any need for delicate (bio)molecular assembly, the Tatsuma research group successfully constructed a system based on gold nanorods deposited on a TiO2 surface in which handedness could be reversed via a photo-induced process.101 Interestingly, the chirality switch achieved by irradiating either with left or right-CPL could be maintained even in the absence of external stimuli. Sites with localized electric fields were then deposited with PbO2 making use of the plasmon-induced redox process to yield chiral nanostructures. Precursor gold nanorods (Au NRs) were first synthesized and were then adsorbed onto a TiO2 surface (film thickness ∼60 nm) present on a glass plate coated with indium-tin-oxide. As shown in Fig. 11, characteristic transverse and longitudinal mode extinction peaks (550 and 815 nm, respectively) and the absence of CD signals was observed. An electrochemical cell composed of two compartments (Fig. 11c) was used to illuminate the photo-responsive ITO/TiO2/AuNR-modified electrode in the presence of Pb(NO3)2 solution (50 mM) for 24 h. Illumination causes the formation of electron–hole (e–h) pairs at resonance sites on surface s of Au NRs. The holes formed around the resonance sites lead to the oxidation of Pb2+ and deposition of PbO2. Concurrently, excited electrons from the electron–hole pair systems are transferred from the lower energy valence band (VB) to the higher energy level conduction band (CB) of the TiO2 substrate. Irradiating the system with left-CPL, a red shift in the extinction bands could be seen. CD spectra showed peaks at around 500–600 nm, 600–700 nm, and 750–900 nm. The CD spectral profile for the irradiation (L-CPL light) of AuNRs prepared using different aspect ratio exhibited similar CD signals. Scanning electron microscopic images (Fig. 11f and g) show the existence of one or two PbO2 moieties on most AuNRs and the use of L-CPL and R-CPL produced mirror image geometries. The enantiomeric excess (ee) was found to be around ∼50%. The authors also successfully performed the chiral switching in air medium. The device is highly reproducible and thus allows the reversible switching of chirality.
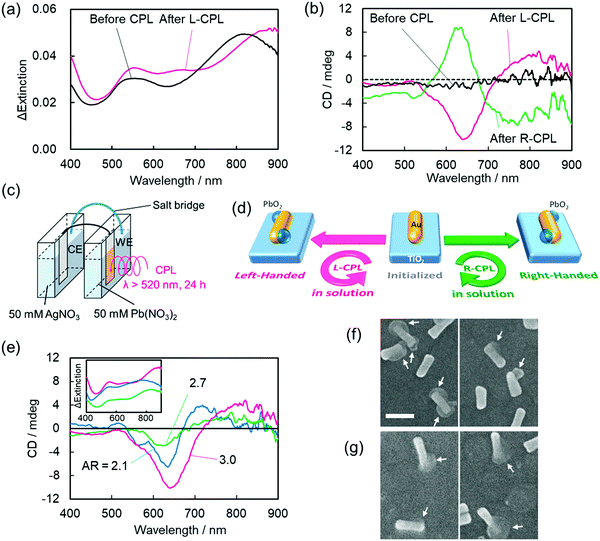 |
| | Fig. 11 (a) The change in extinction coefficient, (b) the visible region CD spectra of AuNRs deposited onto the TiO2 substrate before and after the deposition of lead oxide by either right- or left-CPL irradiation. (c) An electrochemical cell used for PbO2 deposition. (d) A schematic description of the device integration showing the formation of a chiral nanostructure. (e) CD signals of AuNRs after PbO2 deposition, (f and g) the corresponding SEM images of AuNRs. Reprinted with permission.101 Copyright 2020, American Chemical Society. | |
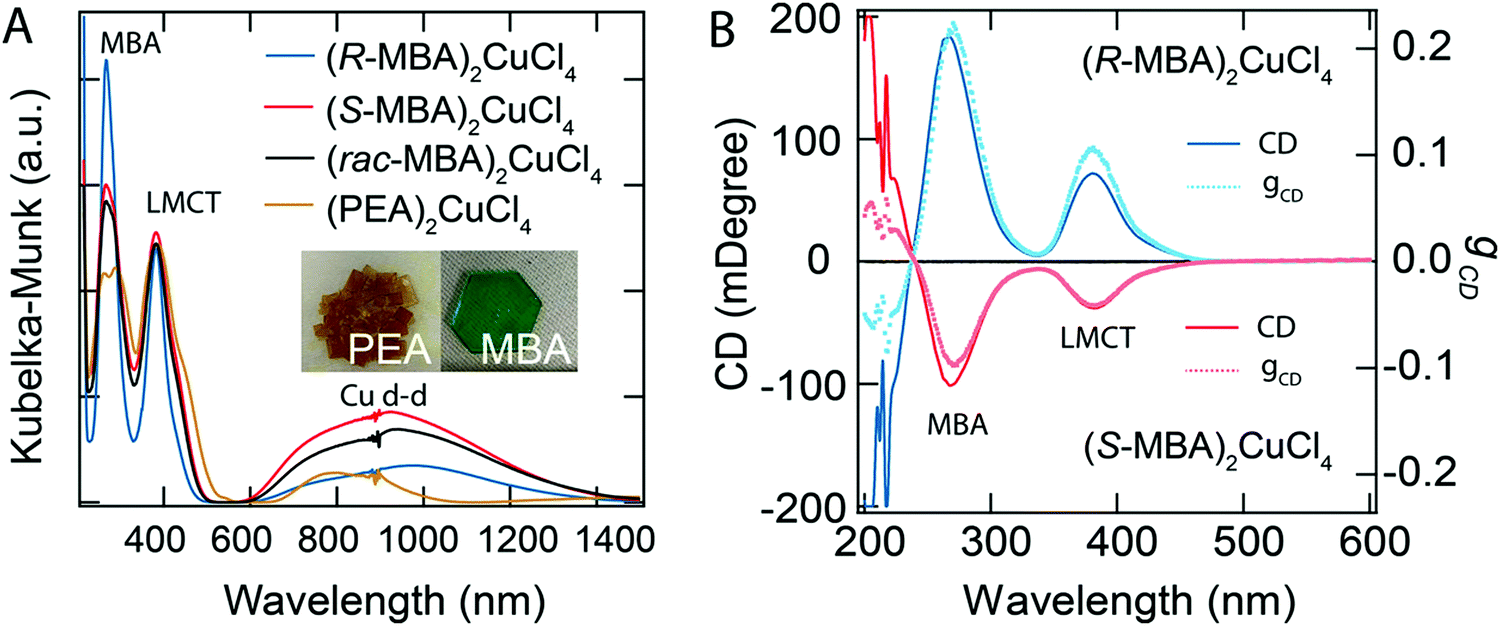
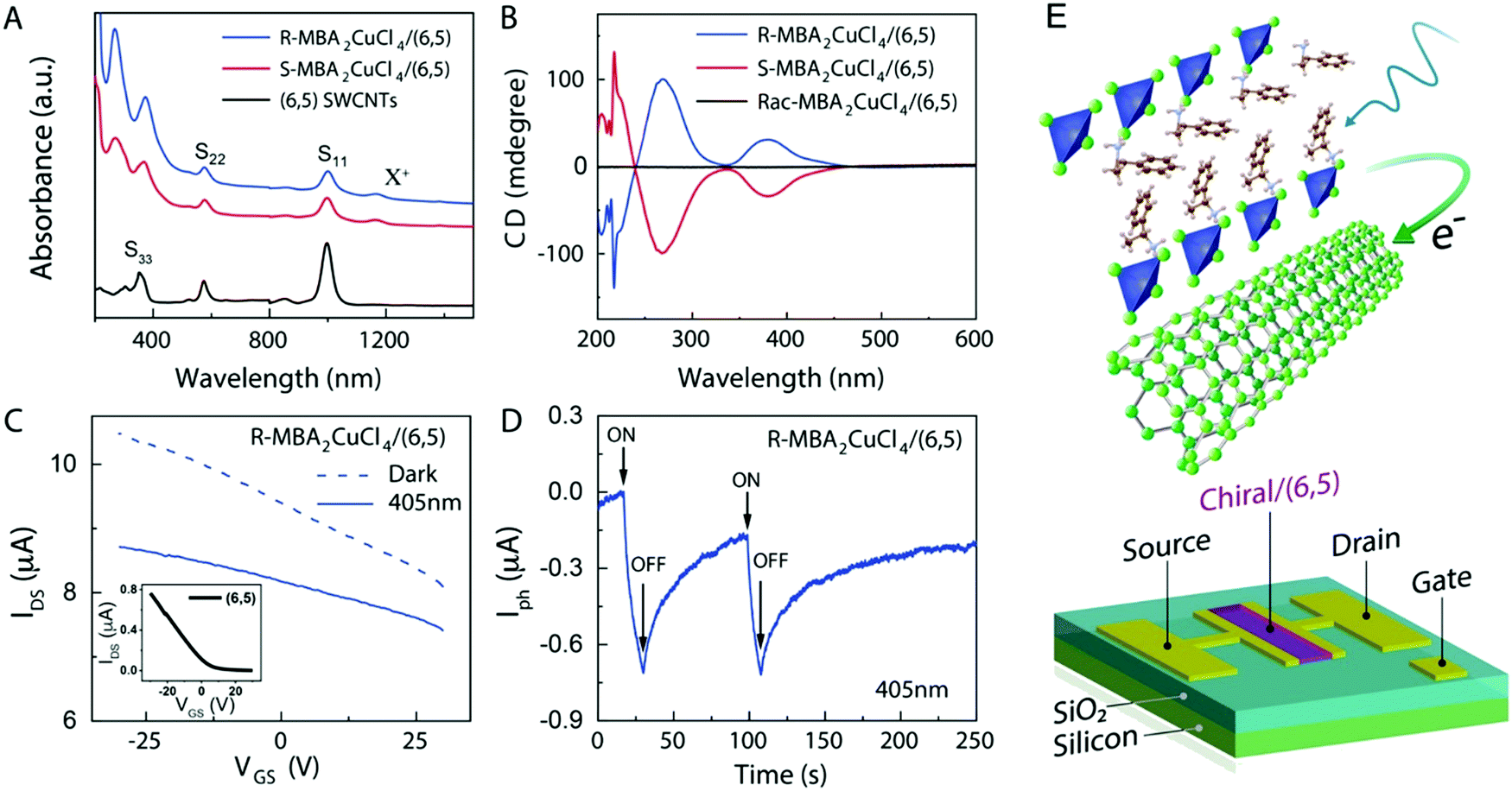
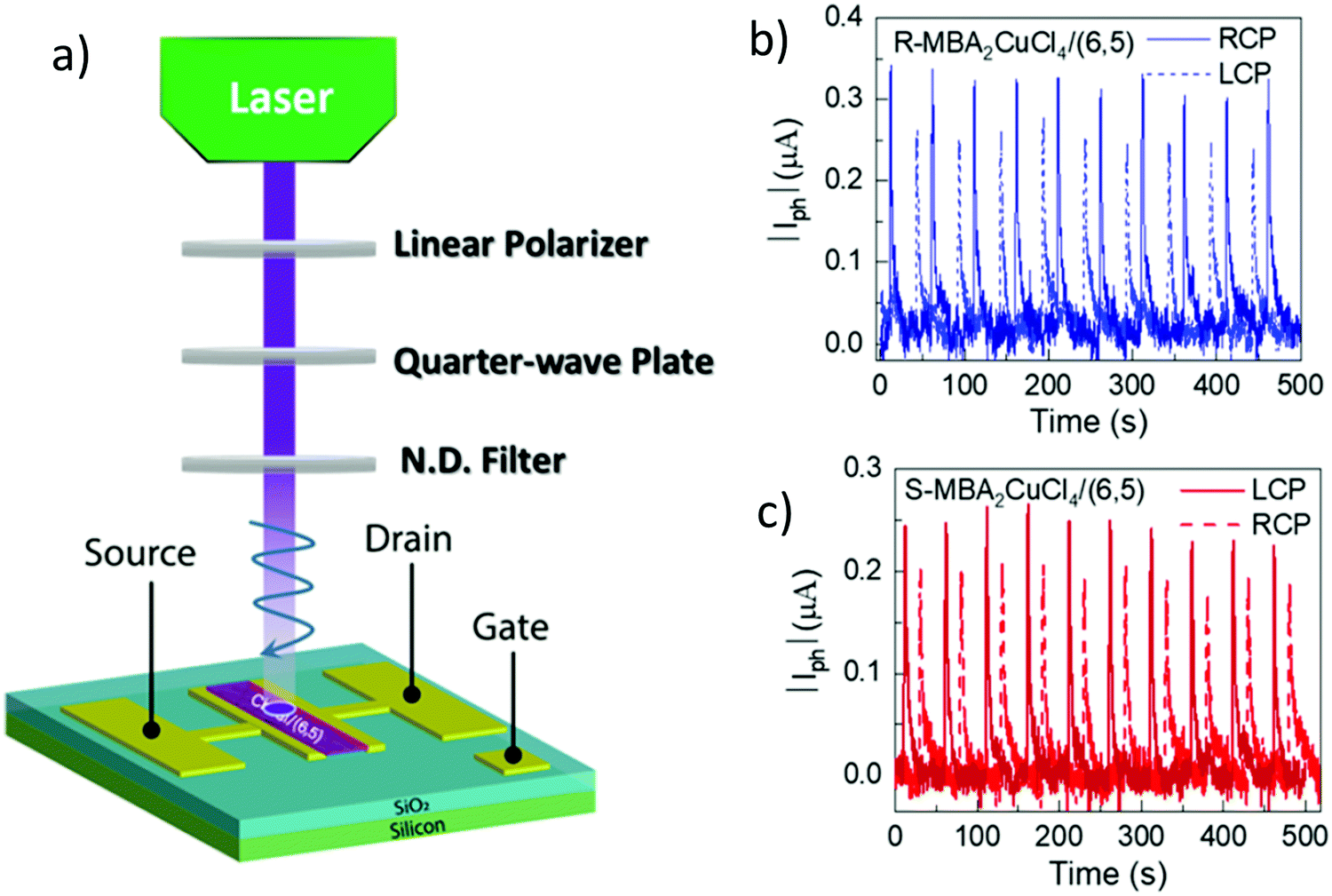
Circularly polarized luminescence and chirality are closely related properties. Materials that can detect CPL are of great importance for many applications at the nanoscale. Normally, the detector is coupled with the optical polarizer to achieve CPL detection. This requirement causes compromise with the sensitivity and resolution limits. Optically active systems interact differently with left or right-CPL and, therefore, display different absorption coefficients. Chiral metal halides are potentially important for the direct detection of CPL. Recently, a two-terminal device based on one-dimensional and quasi-two-dimensional perovskites has been demonstrated to be capable of direct photodetection of CPL.102,103 However these devices suffered from the need for high operational voltage and incapability of producing high output currents. One of the major issues associated with devices based on low-dimensional chiral metal halide semiconductors is low or sometimes even negligible conductivities/photoconductivity. Lu, Blackburn, and co-workers proposed a two-step strategy to circumvent this and prepared a device for the direct detection of CPL, which is comprised of chiral copper chloride and a carbon nanotube heterostructure.104 In the first step, zero-dimensional (0D) chiral copper chloride hybrids (R-/S-MBA)2CuCl4, where R/S-MBA stands for methylbenzylammonium, are prepared and characterized by CD spectroscopy. These hybrids are insulating in nature. Copper chloride hybrids thus formed are then coupled with single-walled carbon nanotubes (SWCNTs). The heterostructure MBA2CuCl4/SWCNT helps in transducing the CPL signal into electrical output while maintaining high anisotropy. The functioning of this junction based on spin-selective interfacial charge transfer is hugely facilitated by the excellent carrier transport properties of the SWCNT. The complete process involves the absorption of CPL photons by MBA2CuCl4, followed by electron transfer to the SWCNT. This system could work at voltages as low as 0.01 V with an anisotropy factor as high as 0.21. To ensure an effective electric current response, it is important to have a highly conductive material interface. A 10 nm thick highly enriched (6,5) SWCNT was spin-coated with a chiral MBA2CuCl4 layer making a 50 nm thick heterojunction. Linear absorption spectra and CD measurements for SWCNT and chiral heterojunctions are shown in Fig. 12. Field-effect transistor (FET) measurement was performed to investigate the direction of charge transfer. When illuminated with 405 nm laser light a significant decrease in source–drain current and a negative shift in threshold voltage is observed (Fig. 13). For optoelectronic detection of left (L) and right (R) CPL, a set-up consisting of a laser diode source generating 405 nm continuous-wave L/R-CPL, a polariser, a neutral density filter and a quarter-wave plate was prepared under an inert atmosphere (Fig. 14a). The detection system was calibrated to generate identical excitation L/R-CPL intensities. The CPL detection experiment for the heterojunctions of R-MBA2CuCl4/SWCNT exhibited a clear difference in photocurrent responses for right and left-handed CPL light. The photocurrent response (Iph) as a function of time (s) under applied bias for R-CPL was found to be larger than that for L-CPL (Fig. 14b and c). When S-MBA2CuCl4/SWCNT was tested, the photocurrent anisotropy was reversed.
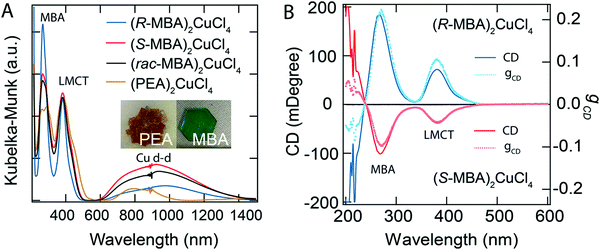 |
| | Fig. 12 (A) UV-vis-NIR spectra, and (B) CD spectra with the corresponding anisotropy factors of (R-/S-/rac-MBA)2CuCl4 and (PEA)2CuCl4. Optical images of (PEA)2CuCl4 and (R-MBA)2CuCl4 crystals are shown in the inset. Reprinted with permission.104 Copyright 2021, American Chemical Society. | |
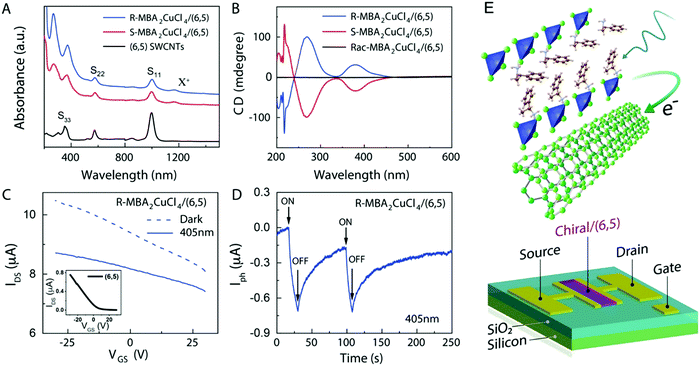 |
| | Fig. 13 (A) UV-Vis-NIR absorption spectra of SWNT, R- and S-MBA2CuCl4/SWCNT. (B) CD signals of R- and S-MBA2CuCl4/(6,5) SWCNT and (rac-MBA)2CuCl4/(6,5) SWCNT. (C) A comparison of the effects of dark and photoresponse conditions on the FET transfer curve. (D) The photocurrent response of (R-MBA)2CuCl4/SWCNT over time. (E) A schematic description of a three-terminal FET device. Reprinted with permission.104 Copyright 2021, American Chemical Society. | |
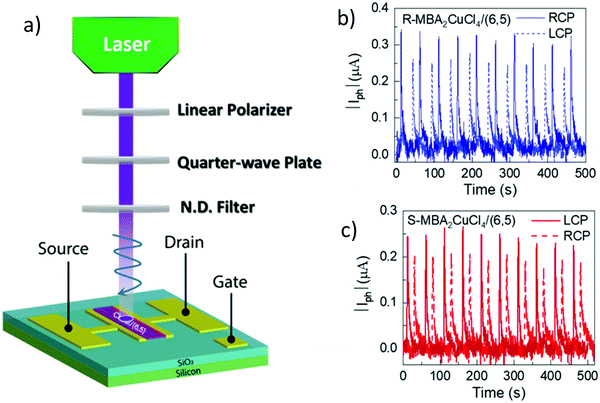 |
| | Fig. 14 (a) Schematic description of an experimental set-up used for creating left and right CPL. (b) Photocurrent (μA) variation as a function of time (s) using R-MBA2CuCl4/SWCNT, and (c) S-MBA2CuCl4/SWCNT when illuminated with a 405 nm pulsed laser light under VDS = 2 V. Reprinted with permission.104 Copyright 2021, American Chemical Society. | |
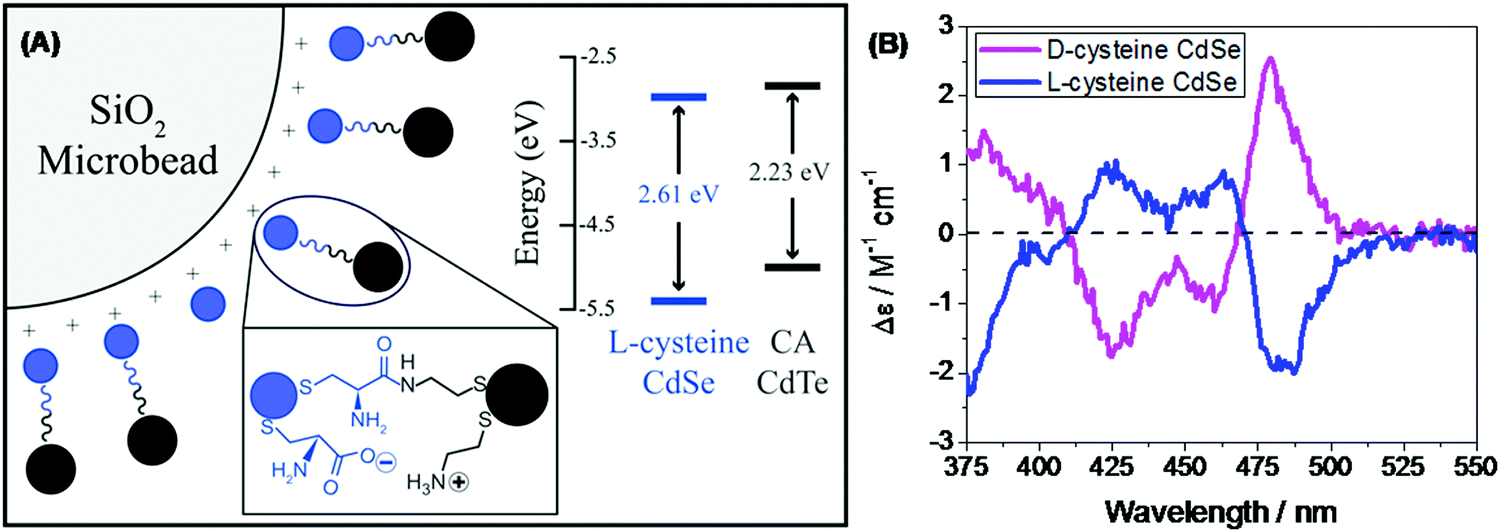
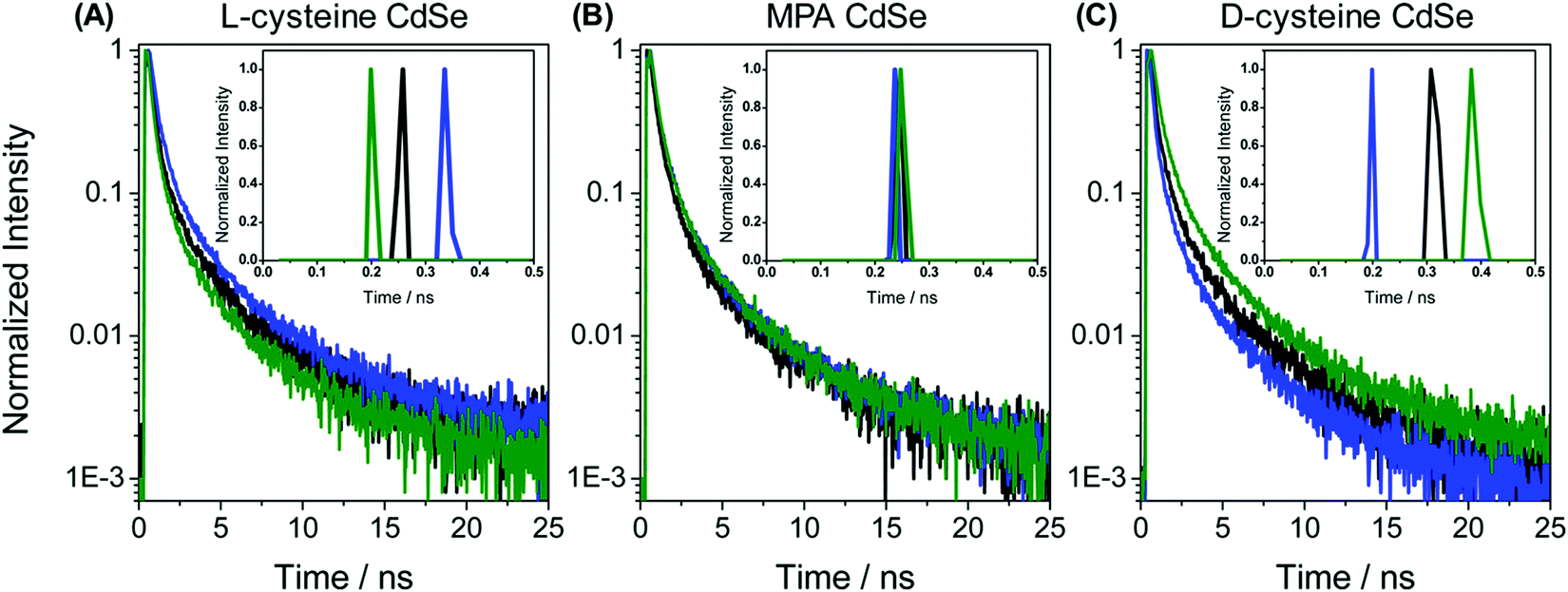
In the recent past, molecular spintronics has seen immense activity. A spintronic device utilizes both the spin and charge of the electron. Molecular chirality can be used in spintronic-based devices to control the spin-selective charge transport studies; however, the mechanism by which spin and chirality regulate the charge transport is still not fully understood.105,106 Findings from the Faraday rotation experiments suggested that spin-polarized excitons could be generated if circularized polarized light is used to photoexcite the quantum dots (QDs).107 Waldeck and co-workers investigated the profound effect that chiral QDs can exhibit spin-selective charge transport phenomena using molecular-scale spintronic devices.108 Chiral CdSe QDs were employed as electron acceptors templated on silica surfaces (SiO2 microbead, Fig. 15). Upon illumination with circularly polarized light, the QDs initiated the electron transfer. Corresponding electron-transfer rates for left and right CP light photoexcitation were measured and then used to calculate the electron-transfer rate asymmetry (Pet) and a 15-fold difference was observed. As can be seen in the scheme depicted above, the acceptor QD is ligand exchanged with cysteine making it chiral, whereas the donor QD is left achiral. The CD spectra, which show a very distinctive band around 475 nm, indicated that ligand chirality is imprinted on QD (Fig. 15B). The fluorescence lifetime for QD dyad assemblies containing achiral donor QD and acceptor QD (ligand exchanged with chiral capping ligand L-cysteine/D-cysteine or achiral ligand mercaptopropionic acid) were studied using excitation light as linear, clockwise (CW), and counterclockwise (CCW) circularly polarized forms (Fig. 16). It was found that when polarized light was used, the lifetime decay profile remained almost unchanged for the achiral acceptor and changed significantly for the chiral acceptor dyad assembly. For the D-cysteine functionalized acceptor QD, the decay was fastest when excitation light was CW-CPL and slowest when CCW-CPL was used. The L-cysteine capped acceptor QD dyad assembly showed the opposite behavior. The authors further demonstrated that asymmetry in electron transfer rates (Pet) is not only influenced by the chirality of the capping ligand but also depends on the QD exciton's CD intensity. The results confirmed that in quantum dyads comprised of chiral acceptor QD and achiral donor QD, electron transfer varies with the direction of circularly polarised excitation light and acceptor chirality. This work unequivocally demonstrates that not only bio-molecules but also chiral QDs can show spin-selective electron transfer processes supporting the chirality-induced spin selectivity (CISS) phenomena.33,109–111
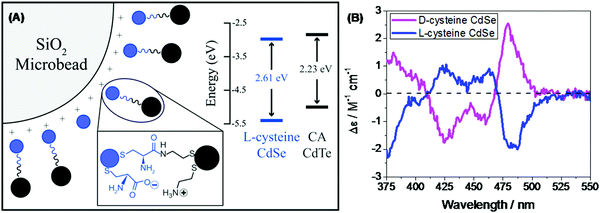 |
| | Fig. 15 (A) Schematic showing achiral donor CdTe QDs and the surface-functionalized chiral acceptor CdSe QD dyad assembled over a silica bead. The inset shows the energy levels of donor and acceptor QDs. (B) Mirror symmetry CD signals of the L-/D-Cys teine functionalized acceptor QD displaying opposite trends. Reprinted with permission.108 Copyright © 2017, American Chemical Society. | |
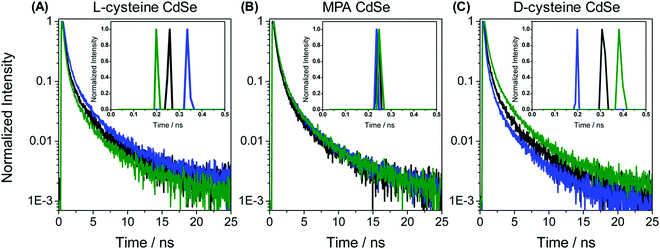 |
| | Fig. 16 (A–C) Lifetime decay profiles for acceptor CdSe QD functionalized with chiral L-cysteine, achiral mercaptopropionic acid, and chiral D-cysteine, respectively. Data represented in black, blue, and green correspond to the linear, clockwise, and counter-clockwise circularly polarized excitation light, respectively. Insets show the short parts of the lifetime distributions. Reprinted with permission.108 Copyright 2017, American Chemical Society. | |
Hybrid perovskites, where a cationic methylammonium is incorporated within the metal–halide structure, have attracted great attention for their applications in solar cells and other organic electronic devices.112 In this context, a lack of inversion symmetry and an additional feature of optical activity in chiral systems, particularly in hybrid perovskites obtained by using chiral organic cations, make them an apt system for spin-optoelectronic device fabrications. A recent perspective article by Wei and Ning discusses the design and application of chiral perovskites towards spin-optoelectronic and spintronic devices.113 Although chiral perovskites have been applied in spin-optoelectronic systems and there has been an increase in such devices, the theoretical aspects behind the work principle and factors affecting its efficiency have not been explored much.114–117 In this regard, Ning and co-workers performed theoretical calculations on a Pb–I-based structure and studied the impact on the spin filtering property of the system by changing halogen substitution on the chiral part, which is methylbenzylammonium (MBA), of the hybrid perovskite.118 In their study, they focused on the structure of chiral molecules responsible for inducing chirality in the hybrid perovskite. Following experimental results from (R-MBA)2PbBr4 that indicated an increase in CD intensity upon halogen substitution at the para-position, the Ning group first studied the hybrid perovskite systems obtained by substituting the para-position H-atom of S-MBA with F, Cl, and Br, and then substituting the H-atom that is near to the chiral carbon of S-MBA, respectively. Their investigation revealed the enhancement of the spin splitting energy from 13 meV to 73, 90, and 105 meV when the H-atom was substituted with F, Cl, and Br, respectively. This indicates the possibility of significant modulation of the output in spin-optoelectronic devices based on the chiral perovskites.
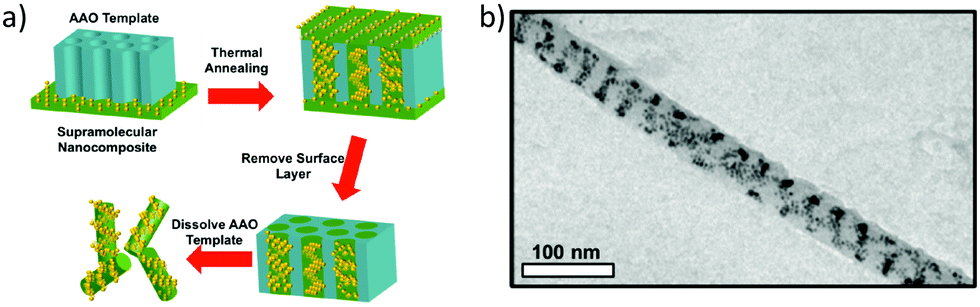
So far, we have discussed the induction of chirality mostly through the surface functionalization of the nanoparticles. We know that it is also possible to induce plasmonic chirality in the nanoparticles from the chiral molecules adsorbed on the surface through a dipole–dipole interaction so, in some cases, chiral induction could be achieved even in the absence of any chiral adsorbate.14 An interesting review by Liz-Marzán and co-workers discussed the recent progress in the templated assembly of achiral plasmonic nanoparticles exhibiting chiroptical activities.119 In an impressive example of utilizing supramolecular nanostructures to build a variety of nanoparticle assemblies in the cylindrical confinements, Xu and co-workers successfully prepared nanoparticle stacked-rings and helical nano-patterns.120 These assemblies displayed a chiral plasmonic response that was superior to the original material. A supramolecular assembly consisting of polystyrene-block-poly(4-vinylpyridine) with 3-n-pentadecylphenol was mixed with 9 vol% of dodecanethiol-capped gold nanoparticles of 4 nm. The mixture was drop-casted on the 35 nm Al2O3 membrane and annealed at 110 °C. The supramolecular part formed cylindrical hexagonal structures that were used to form one-dimensional nanowires of Au-NPs (Fig. 17). As can be seen in the in-plane TEM image (Fig. 17b), Au-NP forms a single helix having a pitch between 20 to 50 nm. By modifying the NP loading, the assembly pattern could be varied to yield stacked rings or single/double-helical structures. Nanoparticle helices generally display chiroptical activity in the 500–600 nm range. Using dark field-particle CD spectroscopy, the optical activity of a helical NP assembly was measured. A designated dip-peak was observed at around 800–1000 nm with a dissymmetry factor of 0.31 at λ = 895 nm, and 0.17 at λ = 960 nm. Near-infrared (NIR) chiral activity is highly promising towards the bimolecular application of nanoparticle-based devices.
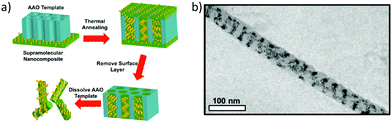 |
| | Fig. 17 (a) Schematic diagram showing the steps leading to the formation of Au-nanowires; (b) TEM image of a single-helical nanowire. Reprinted with permission.120 Copyright 2017, American Chemical Society. | |
Lin and co-workers investigated the formation of chiral assemblies of nanocomposites comprised of the achiral block copolymer and nanoparticles.121 They used theoretical calculations to get the predicted chirality and CD responses of the nanoparticle assembly in block copolymer nanochannels and then compared with experimental findings, which turned out to be in great agreement with the theoretical suggestions. A hybrid particle-field model was applied to study the obtained self-assembled nanostructures. In the bulk, block copolymers were assembled into hexagonal cylinders and when nanoparticles were added, these self-assembled nanostructures functioned as scaffolds and guide the nanoparticle's distribution. Nanoparticles followed the block copolymer self-assembled nanostructures, indicating that the block copolymer could be used to program the nanoparticles' spatial distributions. Simulations revealed that the lower loading of nanoparticles led to the formation of one-layer single and double helical structures. Increasing the loading caused the formation of double-layer helices, whereas further increments in loading destroyed the chiral nanostructure and did not form a triple-layer helical structure of the nanoparticles.
3.1 Bio-applications
Due to the exciting optical features and biocompatibility, chiral inorganic NPs have emerged as a tremendously efficient and productive framework to simplify biological complexities.122–126 This innovative domain contributes to improving the diagnostic strategies and exploring ways to promote a notable recovery. As such, it has effectively bridged the gap between the ever-growing challenges existing today and the solutions to overcome them. It could be a promising strategy for the evolution of diagnostic development. This section reveals some of the recent examples highlighting the capable execution of this principle.
The effective approach to uplifting the treatment of Alzheimer's disease (AD) is by precluding the aggression of amyloid-beta (Aβ). In this context, Tang and co-workers synthesized the achiral Au NPs, followed by the induction of chirality by functionalization with the chiral 3.3 nm L- and D-glutathione (GSH) (abbreviated as L3.3 and D3.3 respectively).127 Chiral Au NPs have been observed to enantioselectively curb the Aβ aggression and permeate the blood–brain-barrier (BBB) through several GSH channels that reside in the brain, resulting in the treatment of AD. The thioflavin T (ThT) fluorescence assay elucidated the free L- or D-GSH, indicating the sparse hindrance on Aβ42 fibrillization. Conversely, the inhibitory effect exhibited by the chiral GSH-stabilized Au NPs was huge and it greatly prevented Aβ42 fibrillization (Fig. 18a). The conformational transition of Aβ42 due to Au NPs was analyzed using circular dichroism (CD) spectroscopy showing L- and D-glutathione capped Aβ42 reverse optical features (Fig. 18b). It was further demonstrated that upon capping with the L- and D-glutathione AuNPs, the protein secondary structures, such as helix, beta, turn, and random, remained intact (Fig. 18c). The morphological changes of Aβ42 aggregates, which were the result of Au NPs and their inhibitory action, were investigated by atomic force microscopy (AFM) and transition electron microscopy (TEM) (Fig. 18d and e). Furthermore, in vivo mice experiments showed that the chiral NPs cross the BBB with ease due to their convenient size through simple intravenous injection. In particular, D3.3 improved the cellular viability by 80%, while L3.3 achieved the survival of 72%. The author inferred that the establishment of the chiral GSH ligand favored gold NPs and the extraordinary capacity towards the chiral recognition and enantioselective inhibition of Aβ fibrillization. Indeed, the observations and results proved that the employment of chiral nanomedicine will unlock the door to the treatment of AD. This arrangement of NPs with chiral activity makes it an apt candidate for the diagnosis of AD.
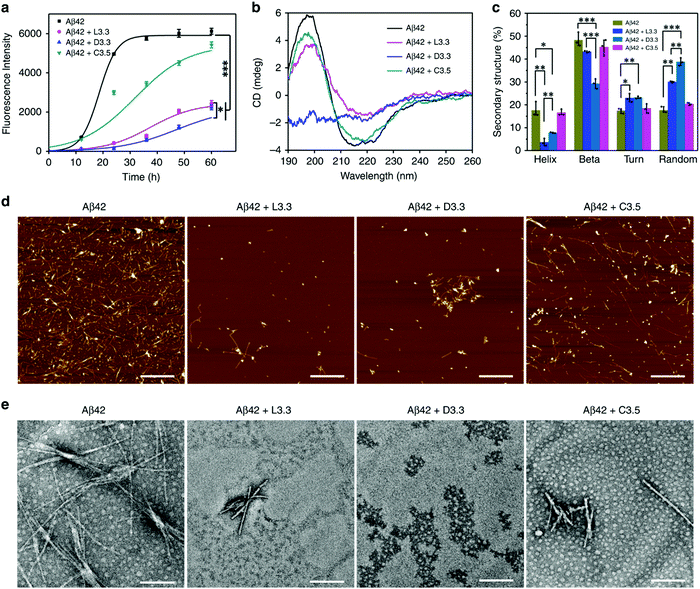 |
| | Fig. 18 (a) Fluorescence spectra of Aβ42 illustrate a considerable reduction of up to 63% in the maximum ThT intensity for (Aβ42 + D3.3) and 60% for (Aβ42 + L3.3). (b) CD spectra of Aβ42 show a positive peak at 195 nm and a negative peak at 215 nm, indicating the formation of the β-sheet structure, and a considerable reduction in both peaks was obtained on the addition of chiral Au NPs. (c) The intactness of protein secondary structure, (d) AFM images of free Aβ42 and with L3.3, D3.3, and (e) TEM images of free Aβ42 and with L3.3, D3.3. Reprinted with permission.127 Copyright 2020, Springer Nature. | |
Plasmonic metallic NPs are capable of amplifying the quality of chiral signals through surface plasmonic resonance (SPR), and this has captivated many researchers.128–130 A methodology has been formulated, based on the chiroptical activity of plasmonic gold NPs, for the identification of amyloids in the scrambled protein. The detection of amyloid fibrils, which are end products in several protein aggregation pathways, at low concentrations may reduce the risk of Parkinson's disease (PD). Liz-Marzán and co-workers employed gold nanorods (Au NRs) to probe the formed amyloid fibrils based on α-synuclein.131 There was no clear interaction shown by the Au NRs with the monomeric proteins but on the other end, the efficacious adsorption of Au NRs was observed on the helical protein fibrils by means of non-covalent interactions. The electrostatic interactions generated strong optical action near the SPR wavelengths. Concentration-dependent Au NR solutions (0.5 and 2.0 nM) revealed the different arrangements of α-synuclein fibrils confirmed by TEM images (Fig. 19). Moreover, when the concentration of NRs was increased, the TEM images displayed improved and extended coverage, which led to the infiltration of the α-synuclein fibrils surface, and thus extra NRs were most likely available in the solution (Fig. 19D).
 |
| | Fig. 19 TEM images of (A) α-synuclein fibrils, (B) α-synuclein fibrils mixed with 0.5 nM Au NRs, (C) 2.0 nM Au NRs, and (D) 3D chiral structure of Au NRs. Reprinted with permission.131 Copyright 2018, the National Academy of Sciences of the United States of America. | |
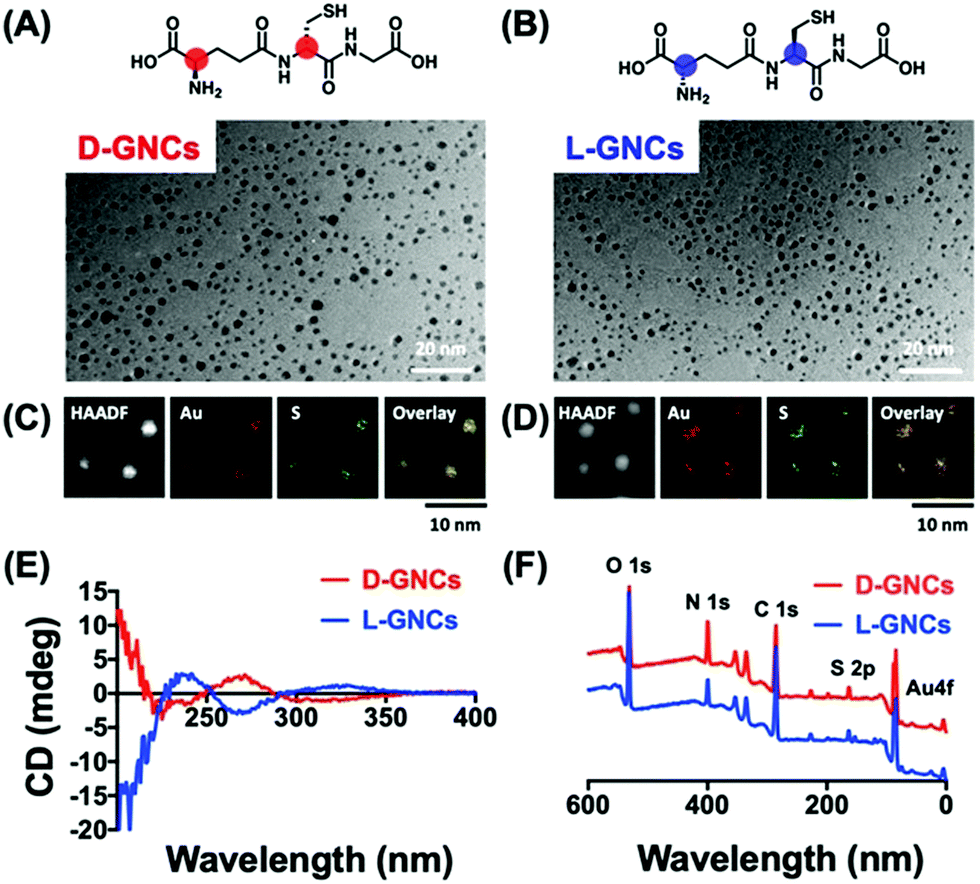
The concept of chiral inorganic NPs has made a considerable contribution in the field of microbiology.132,133 In this regard, Yan and co-workers engendered chirality into gold NPs by capping the surfaces with L-/D-cysteine for in vivo therapeutic applications.26 The cysteine (L/D)-capped chiral Au NPs manifested noteworthy inhibition of E. coli in vitro, as well as in vivo, as compared to the free L-/D-cysteine moieties. The lethal action of L-/D-cysteine-capped Au NPs towards the formation of E. coli was observed to be higher than that of the free L-/D-cysteine or the pristine Au NPs. Living organisms are known to show special selectivity towards specific chiral molecules. For example, amino acids and phospholipids are found in L-forms, whereas the nucleotides exist in their D-forms. Therefore, while designing new chiral biomaterials or drugs, it becomes important to have suitable chirality to minimize any opposite therapeutic effects that may result from the unfavoured chirality match. Recently, the Jiang group demonstrated the effects of positively charged/cationic gold nanocluster (GNC) surface functionalization with D- vs.L-forms of glutathione ligand on the toxicity and pharmacokinetics, which have been studied in great detail.134 Nanomaterials consisting of positive charges are expected to exhibit enhanced interactions with cell membranes that carry a negative charge. For this reason, cationic nanomaterials have found a range of biological applications. However, being cationic, such nanomaterials suffer from severe cytotoxicity and quick elimination from the blood plasma. To improve the clinical applications of cationic nanomaterials, it is desirable to reduce the cytotoxicity and decrease their clearance time. In this study, Jiang and co-workers used gold nanoclusters because they allow simple surface functionalization and can be easily cleared through renal pathways. The surface of GNCs was modified with D/L-glutathione and 4,6-diamino-2-pyrimidinethiol to make the surface positively charged (Fig. 20). To evaluate the biosafety of the prepared D-/L-GNCs, various cells were used. Experiments showed L-GNCs to be much more toxic than D-GNCs under similar conditions. Biocompatibility tests obtained by exposing D-/L-GNCs to the blood showed that D-GNCs perform much better than their L-counterparts. The difference in the biological behavior of D-/L-GNCs was studied by performing intravenous (i.v.) injection into mice. It was found that D-GNCs displayed a much longer retention time in blood plasma than the L-GNC or DL-GNC (achiral). The long retention time increases the circulation time in the blood and hence leads to a broader organ distribution of GNCs. The study demonstrated that by utilizing surface functionalization, the nanomaterials’ chirality could be reversed and their biosafety and in vivo performance could be drastically improved.
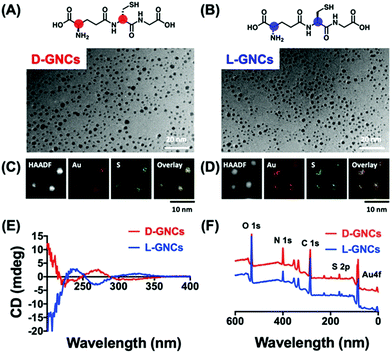 |
| | Fig. 20 (A and B) Chemical structure of chiral ligand D-/L-glutathione, (C and D) TEM images, and the corresponding elemental mapping analysis of the corresponding GNC's. (E) and (F) show the characterization of D-/L-GNCs by CD and X-ray photoelectron spectroscopy (XPS). Reprinted with permission.134 Copyright 2021, Wiley-VCH. | |
4. Conclusion and outlook
In summary, benefiting from controllable nanoscale size, large surface area, morphology, and quantum confinement, the metallic or semiconductor NPs have manifested excellent performance in various aspects of optics, physics, material science, and life science. The application of nanomaterials becomes more widespread when chirality is induced in achiral nanomaterials or intrinsic chiral nanomaterials are employed. Small chiral organic ligands are employed to induce chirality in achiral NPs, which act as symmetry breakers. Chiral nanomaterials possess intriguing optical features represented by circular dichromism, circular plasmonic luminescence, etc. The recent advances in this area have revealed that besides their optoelectronic device applications, chiral NPs are a boon towards the fields of disease diagnosis and therapies. The chiral NPs have been proved to be excellent candidates for the treatment of Alzheimer's Disease. In addition, the 3D chiral Au NRs-protein hybrid structures can act as a rich source for amyloid detection and expand the scope for the implication of template-driven plasmon chirality. Designing capable and influential antibacterial nano-scale agents that can cure bacterial infection is now possible due to the availability of such NPs. The future scope of this line of research is to study the applied mechanisms demonstrated by the chiral NPs in detail to comprehend the micro-steps and variations. The premise of this research is to evolve the application of this phenomenon and boost its activity for better results. Though the research progress made with the chiral NPs is laudable, this domain is still in the nascent phase, as the systematic understanding of chiral imprinting is crucial. Therefore, more experimental and theoretical studies are needed. During the process of nanoimprinting by chiral inducers, the formation of the cluster may occur instead of distinct NPs development. To prohibit such cluster formation, the concentration of the corresponding metal salt solution can be diluted. This induced chirality study has been experimentally performed on selected metals and semiconductors with mostly L and D cysteine amino acids. Thus, more experimental and theoretical studies are welcome with various other metals such as Cu, Ni, Fe, using chiral inducers, to understand their growth mechanisms. To make them fruitful bio-applications, more such studies are also necessary using such chirality-induced NPs. There is relatively little known about the magnetic field effect on chiral NPs, which is crucial for the rational design and construction of nanoscale spintronic devices. It seems that this domain is quite challenging because all the ferromagnetic metals (Fe, Co, Ni) are prone to oxidation during the formation of NPs. A feasible solution could be the electrochemical reduction of such metal salts followed by capping with such chiral ligands. Furthermore, this concept can be deployed to build heterostructures where at least two different NPs can be assembled. Since semiconducting NPs can conduct electricity and display good charge carrier concentration, NPs can also be applied in making electrode materials for devices. However, scalability, reproducibility, and operational aspects can be of major concern and would need to be resolved.
Author contributions
PCM: conceptualization; funding acquisition; investigation; supervision; writing – original draft. DA: writing – original draft. RKP: visualization; writing – review and editing. SJ: visualization; writing – review and editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
PCM acknowledges a start-up research grant from the Department of Science and Technology (SRG/2019/000391), New Delhi, and an initiation research grant for independent laboratory setup from IIT Kanpur (IITK/CHM/2019044), India. RKP acknowledges the Council of Scientific & Industrial Research (CSIR), New Delhi, for a junior research fellowship.
References
- L. Pasteur, Ann. Chim. Phys., 1848, 24, 442–459 Search PubMed.
- R. Bentley, Perspect. Biol. Med., 1995, 38, 188–229 CrossRef CAS PubMed.
- D. S. Sholl and A. J. Gellman, AIChE J., 2009, 55, 2484–2490 CrossRef CAS.
- B. L. Feringa, R. A. van Delden, N. Koumura and E. M. Geertsema, Chem. Rev., 2000, 100, 1789–1816 CrossRef CAS PubMed.
- J. W. Canary, Chem. Soc. Rev., 2009, 38, 747–756 RSC.
- I. Hodgkinson and Q. h. Wu, Adv. Mater., 2001, 13, 889–897 CrossRef CAS.
- H. Kim, S. W. Im, R. M. Kim, N. H. Cho, H.-E. Lee, H.-Y. Ahn and K. T. Nam, Mater. Adv., 2020, 1, 512–524 RSC.
- R. Naaman, Y. Paltiel and D. H. Waldeck, Nat. Rev. Chem., 2019, 3, 250–260 CrossRef CAS.
- P. C. Mondal, N. Kantor-Uriel, S. P. Mathew, F. Tassinari, C. Fontanesi and R. Naaman, Adv. Mater., 2015, 27, 1924–1927 CrossRef CAS PubMed.
- N. A. Kotov, L. M. Liz-Marzán and P. S. Weiss, ACS Nano, 2021, 15, 12457–12460 CrossRef CAS.
- R. Torres-Cavanillas, G. Escorcia-Ariza, I. Brotons-Alcázar, R. Sanchis-Gual, P. C. Mondal, L. E. Rosaleny, S. Giménez-Santamarina, M. Sessolo, M. Galbiati, S. Tatay, A. Gaita-Ariño, A. Forment-Aliaga and S. Cardona-Serra, J. Am. Chem. Soc., 2020, 142, 17572–17580 CrossRef CAS PubMed.
- G. Zheng, J. He, V. Kumar, S. Wang, I. Pastoriza-Santos, J. Pérez-Juste, L. M. Liz-Marzán and K. Y. Wong, Chem. Soc. Rev., 2021, 50, 3738–3754 RSC.
- C. Noguez and I. L. Garzón, Chem. Soc. Rev., 2009, 38, 757 RSC.
- Y. Xia, Y. Zhou and Z. Tang, Nanoscale, 2011, 3, 1374–1382 RSC.
-
J. Govan and Y. K. Gun’ko, SPR Nanoscience, 2016, vol. 3, pp. 1–30 Search PubMed.
- W. Ma, L. Xu, A. F. de Moura, X. Wu, H. Kuang, C. Xu and N. A. Kotov, Chem. Rev., 2017, 117, 8041–8093 CrossRef CAS PubMed.
- K. T. Nam and H. Kim, Science, 2021, 371, 1311 CrossRef CAS PubMed.
- X. Wang, M. Wang, R. Lei, S. F. Zhu, Y. Zhao and C. Chen, ACS Nano, 2017, 11, 4606–4616 CrossRef CAS PubMed.
- Q. Zhang, J. Gu, L. Zhang and J. Lin, Nanoscale, 2019, 11, 474–484 RSC.
- M. A. Ortunõ and N. López, Catal. Sci. Technol., 2019, 9, 5173–5185 RSC.
- H.-E. Lee, H.-Y. Ahn, J. Mun, Y. Y. Lee, M. Kim, N. H. Cho, K. Chang, W. S. Kim, J. Rho and K. T. Nam, Nature, 2018, 556, 360–365 CrossRef CAS PubMed.
- T. Gregory Schaaff, G. Knight, M. N. Shafigullin, R. F. Borkman and R. L. Whetten, J. Phys. Chem. B, 1998, 102, 10643–10646 CrossRef.
- M. P. Moloney, Y. K. Gun’ko and J. M. Kelly, Chem. Commun., 2007, 3900–3902 RSC.
- S. Jiang, K. Y. Win, S. Liu, C. P. Teng, Y. Zheng and M. Y. Han, Nanoscale, 2013, 5, 3127–3148 RSC.
- B. Zhou, X. Guo, N. Yang, Z. Huang, L. Huang, Z. Fang, C. Zhang, L. Li and C. Yu, J. Mater. Chem. B, 2021, 9, 5583–5598 RSC.
- Y. Xu, H. Wang, M. Zhang, J. Zhang and W. Yan, Nanomaterials, 2021, 11, 1621 CrossRef CAS PubMed.
- W. Wang, I. Erofeev, P. Nandi, H. Yan and U. Mirsaidov, Adv. Funct. Mater., 2021, 31, 2008639 CrossRef CAS.
- K. Wieszczycka, K. Staszak, M. J. Woźniak-Budych, J. Litowczenko, B. M. Maciejewska and S. Jurga, Coord. Chem. Rev., 2021, 436, 213846 CrossRef CAS.
- X. Shao, T. Zhang, B. Li, M. Zhou, X. Ma, J. Wang and S. Jiang, Inorg. Chem., 2019, 58, 6534–6543 CrossRef CAS PubMed.
- Y. Imanaka, T. Nakahodo and H. Fujihara, ChemistrySelect, 2017, 2, 5806–5809 CrossRef CAS.
- Y. Wang and T. Bürgi, Nanoscale Adv., 2021, 3, 2710–2727 RSC.
- B. P. Bloom, B. M. Graff, S. Ghosh, D. N. Beratan and D. H. Waldeck, J. Am. Chem. Soc., 2017, 139, 9038–9043 CrossRef CAS PubMed.
- B. P. Bloom, V. Kiran, V. Varade, R. Naaman and D. H. Waldeck, Nano Lett., 2016, 16, 4583–4589 CrossRef CAS PubMed.
- L. Bergquist and T. Hegmann, ChemNanoMat, 2017, 3, 863–868 CrossRef CAS.
- J. L. Y. Chen, C. Pezzato, P. Scrimin and L. J. Prins, Chem. – Eur. J., 2016, 22, 7028–7032 CrossRef CAS PubMed.
- S. Mokashi-Punekar and N. L. Rosi, Part. Part. Syst. Charact., 2019, 36, 5–9 CrossRef.
- K. Varga, S. Tannir, B. E. Haynie, B. M. Leonard, S. V. Dzyuba, J. Kubelka and M. Balaz, ACS Nano, 2017, 11, 9846–9853 CrossRef CAS PubMed.
- X. Shao, Y. Wu, S. Jiang, B. Li, T. Zhang and Y. Yan, Inorg. Chem., 2020, 59, 14382–14388 CrossRef CAS PubMed.
- N. Sunaga, T. Haraguchi and T. Akitsu, Symmetry, 2019, 11, 1094 CrossRef CAS.
- P. C. Mondal, P. Roy, D. Kim, E. E. Fullerton, H. Cohen and R. Naaman, Nano Lett., 2016, 16, 2806–2811 CrossRef CAS PubMed.
- L. Zhou, S. Jiang, X. Zhang, L. Fang and X. Guo, Electrophoresis, 2018, 39, 941–947 CrossRef CAS PubMed.
- P. Fu, M. Sun, L. Xu, X. Wu, L. Liu, H. Kuang, S. Song and C. Xu, Nanoscale, 2016, 8, 15008–15015 RSC.
- L. Rosenfeld, Z. Phys., 1929, 52, 161–174 CrossRef.
- L. Xiao, T. An, L. Wang, X. Xu and H. Sun, Nano Today, 2020, 30, 100824 CrossRef CAS.
- J. Liu, L. Yang, P. Qin, S. Zhang, K. K. L. Yung and Z. Huang, Adv. Mater., 2021, 133, 2005506 CrossRef PubMed.
- A. Guerrero-Martínez, J. L. Alonso-Gómez, B. Auguié, M. M. Cid and L. M. Liz-Marzán, Nano Today, 2011, 6, 381–400 CrossRef.
- W. Ma, L. Xu, L. Wang, C. Xu and H. Kuang, Adv. Funct. Mater., 2019, 29, 1805512 CrossRef.
- G. Koplovitz, G. Leitus, S. Ghosh, B. P. Bloom, S. Yochelis, D. Rotem, F. Vischio, M. Striccoli, E. Fanizza, R. Naaman, D. H. Waldeck, D. Porath and Y. Paltiel, Small, 2019, 15, 1804557 CrossRef PubMed.
- R. Fanelli, L. Milli, A. Cornia, A. Moretto, N. Castellucci, N. Zanna, G. Malachin, R. Tavano and C. Tomasini, Eur. J. Org. Chem., 2015, 6243–6248 CrossRef CAS.
- N. H. Cho, H. E. Lee, H. Y. Ahn, Y. Y. Lee, S. W. Im, H. Kim and K. T. Nam, Part. Part. Syst. Charact., 2019, 36, 1–5 Search PubMed.
- Z. Feng, Y. Yang, G. Xu, Y. Du and X. Sun, Electrophoresis, 2020, 41, 1060–1066 CrossRef CAS PubMed.
- H. Kim, S. W. Im, R. M. Kim, N. H. Cho, H.-E. Lee, H.-Y. Ahn and K. T. Nam, Mater. Adv., 2020, 1, 512–524 RSC.
- D. Biriukov and Z. Futera, J. Phys. Chem. C, 2021, 125, 7856–7867 CrossRef CAS.
- A. García-Etxarri, J. M. Ugalde, J. J. Sáenz and V. Mujica, J. Phys. Chem. C, 2020, 124, 1560–1565 CrossRef.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- S. E. Lohse and C. J. Murphy, Chem. Mater., 2013, 25, 1250–1261 CrossRef CAS.
- G. Carchini, N. Almora-Barrios, G. Revilla-López, L. Bellarosa, R. García-Muelas, M. García-Melchor, S. Pogodin, P. Błoński and N. López, Top. Catal., 2013, 56, 1262–1272 CrossRef CAS.
- Q. Tang and D. Jiang, Chem. Mater., 2016, 28, 5976–5988 CrossRef CAS.
- H. Häkkinen, Nat. Chem., 2012, 4, 443–455 CrossRef PubMed.
- J. Wang and A. Selloni, J. Phys. Chem. C, 2009, 113, 8895–8900 CrossRef CAS.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1170 CrossRef CAS PubMed.
- P. C. Mondal, C. Fontanesi, D. H. Waldeck and R. Naaman, ACS Nano, 2015, 9, 3377–3384 CrossRef CAS PubMed.
- P. C. Mondal and C. Fontanesi, ChemPhysChem, 2018, 19, 60–66 CrossRef CAS PubMed.
- G. Wulff, Z. Kristallogr. – Cryst. Mater., 1901, 34, 449–530 CrossRef CAS.
- R. Di Felice, A. Selloni and E. Molinari, J. Phys. Chem. B, 2003, 107, 1151–1156 CrossRef CAS.
- R. Di Felice and A. Selloni, J. Chem. Phys., 2004, 120, 4906–4914 CrossRef CAS PubMed.
- A. Kühnle, L. M. Molina, T. R. Linderoth, B. Hammer and F. Besenbacher, Phys. Rev. Lett., 2004, 93, 086101 CrossRef PubMed.
- B. Höffling, F. Ortmann, K. Hannewald and F. Bechstedt, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 045407 CrossRef.
- S. Monti, V. Carravetta and H. Ågren, J. Phys. Chem. Lett., 2016, 7, 272–276 CrossRef CAS PubMed.
- J. Morales-Vidal, N. López and M. A. Ortuño, J. Phys. Chem. C, 2019, 123, 13758–13764 CrossRef CAS.
- F. Yang, X. Liu and Z. Yang, Angew. Chem., Int. Ed., 2021, 60, 14671–14678 CrossRef CAS PubMed.
- M. Esposito, V. Tasco, F. Todisco, M. Cuscunà, A. Benedetti, M. Scuderi, G. Nicotra and A. Passaseo, Nano Lett., 2016, 16, 5823–5828 CrossRef CAS PubMed.
- M. Esposito, V. Tasco, F. Todisco, M. Cuscunà, A. Benedetti, D. Sanvitto and A. Passaseo, Nat. Commun., 2015, 6, 6484 CrossRef CAS PubMed.
- B. Frank, X. Yin, M. Schäferling, J. Zhao, S. M. Hein, P. V. Braun and H. Giessen, ACS Nano, 2013, 7, 6321–6329 CrossRef CAS PubMed.
- L. Riccardi, F. De Biasi, M. De Vivo, T. Bürgi, F. Rastrelli and G. Salassa, ACS Nano, 2019, 13, 7127–7134 CrossRef CAS PubMed.
- M. A. Boles, M. Engel and D. V. Talapin, Chem. Rev., 2016, 116, 11220–11289 CrossRef CAS PubMed.
- M. Gupta, S. S. Mohapatra, S. Dhara and S. K. Pal, J. Mater. Chem. C, 2018, 6, 2303–2310 RSC.
- H. Hinterwirth, S. Kappel, T. Waitz, T. Prohaska, W. Lindner and M. Lämmerhofer, ACS Nano, 2013, 7, 1129–1136 CrossRef CAS PubMed.
- S. Paul, B. B. Chakraborty, S. Anwar, S. B. Paul and S. Choudhury, Heliyon, 2020, 6, e03674 CrossRef PubMed.
- J. E. Lu, C. H. Yang, H. Wang, C. Yam, Z. G. Yu and S. Chen, Nanoscale, 2018, 10, 14586–14593 RSC.
- N. Berova, L. Di Bari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914 RSC.
- Y. Y. Lee, R. M. Kim, S. W. Im, M. Balamurugan and K. T. Nam, Nanoscale, 2020, 12, 58–66 RSC.
- J. Karst, N. H. Cho, H. Kim, H.-E. Lee, K. T. Nam, H. Giessen and M. Hentschel, ACS Nano, 2019, 13, 8659–8668 CrossRef CAS PubMed.
- H.-E. Lee, H.-Y. Ahn, J. Lee and K. T. Nam, ChemNanoMat, 2017, 3, 685–697 CrossRef CAS.
- N. H. Cho, G. H. Byun, Y.-C. Lim, S. W. Im, H. Kim, H.-E. Lee, H.-Y. Ahn and K. T. Nam, ACS Nano, 2020, 14, 3595–3602 CrossRef CAS PubMed.
- S. W. Im, H. Ahn, R. M. Kim, N. H. Cho, H. Kim, Y. Lim, H. Lee and K. T. Nam, Adv. Mater., 2020, 32, 1905758 CrossRef CAS PubMed.
- H. Kim, S. W. Im, N. H. Cho, D. H. Seo, R. M. Kim, Y.-C. Lim, H.-E. Lee, H.-Y. Ahn and K. T. Nam, Angew. Chem., Int. Ed., 2020, 59, 12976–12983 CrossRef CAS PubMed.
- H.-E. Lee, R. M. Kim, H.-Y. Ahn, Y. Y. Lee, G. H. Byun, S. W. Im, J. Mun, J. Rho and K. T. Nam, Nat. Commun., 2020, 11, 263 CrossRef CAS PubMed.
- U. Tohgha, K. K. Deol, A. G. Porter, S. G. Bartko, J. K. Choi, B. M. Leonard, K. Varga, J. Kubelka, G. Muller and M. Balaz, ACS Nano, 2013, 7, 11094–11102 CrossRef CAS PubMed.
- X. Gao, B. Han, X. Yang and Z. Tang, J. Am. Chem. Soc., 2019, 141, 13700–13707 CrossRef CAS PubMed.
- J. Hao, Y. Li, J. Miao, R. Liu, J. Li, H. Liu, Q. Wang, H. Liu, M.-H. Delville, T. He, K. Wang, X. Zhu and J. Cheng, ACS Nano, 2020, 14, 10346–10358 CrossRef CAS PubMed.
- U. Tohgha, K. Varga and M. Balaz, Chem. Commun., 2013, 49, 1844–1846 RSC.
- A. Ben Moshe, D. Szwarcman and G. Markovich, ACS Nano, 2011, 5, 9034–9043 CrossRef CAS PubMed.
- M. Sun, L. Xu, A. Qu, P. Zhao, T. Hao, W. Ma, C. Hao, X. Wen, F. M. Colombari, A. F. de Moura, N. A. Kotov, C. Xu and H. Kuang, Nat. Chem., 2018, 10, 821–830 CrossRef CAS PubMed.
- J. Cheng, J. Hao, H. Liu, J. Li, J. Li, X. Zhu, X. Lin, K. Wang and T. He, ACS Nano, 2018, 12, 5341–5350 CrossRef CAS PubMed.
- F. S. Richardson and J. P. Riehl, Chem. Rev., 1977, 77, 773–792 CrossRef CAS.
- J. Olesiak-Banska, M. Waszkielewicz and M. Samoc, Phys. Chem. Chem. Phys., 2018, 20, 24523–24526 RSC.
- Z. Guo, J. Li, C. Wang, R. Liu, J. Liang, Y. Gao, J. Cheng, W. Zhang, X. Zhu, R. Pan and T. He, Angew. Chem., Int. Ed., 2021, 60, 8441–8445 CrossRef CAS PubMed.
- G. Li, X. Fei, H. Liu, J. Gao, J. Nie, Y. Wang, Z. Tian, C. He, J. L. Wang, C. Ji, D. Oron and G. Yang, ACS Nano, 2020, 14, 4196–4205 CrossRef CAS PubMed.
- G. Yang, Y. Yu, B. Yang, T. Lu, Y. Cai, H. Yin, H. Zhang, N.-N. Zhang, L. Li, Y.-M. Zhang and S. X.-A. Zhang, Angew. Chem., Int. Ed., 2021, 60, 2018–2023 CrossRef CAS PubMed.
- K. Morisawa, T. Ishida and T. Tatsuma, ACS Nano, 2020, 14, 3603–3609 CrossRef CAS PubMed.
- C. Chen, L. Gao, W. Gao, C. Ge, X. Du, Z. Li, Y. Yang, G. Niu and J. Tang, Nat. Commun., 2019, 10, 1–7 CrossRef PubMed.
- L. Wang, Y. Xue, M. Cui, Y. Huang, H. Xu, C. Qin, J. Yang, H. Dai and M. Yuan, Angew. Chem., Int. Ed., 2020, 59, 6442–6450 CrossRef CAS PubMed.
- J. Hao, H. Lu, L. Mao, X. Chen, M. C. Beard and J. L. Blackburn, ACS Nano, 2021, 15, 7608–7617 CrossRef CAS PubMed.
- R. Naaman, Y. Paltiel and D. H. Waldeck, Nat. Rev. Chem., 2019, 3, 250–260 CrossRef CAS.
- B. Zhao, S. Yang, J. Deng and K. Pan, Adv. Sci., 2021, 2003681 CrossRef CAS PubMed.
- M. Ouyang, Science, 2003, 301, 1074–1078 CrossRef CAS PubMed.
- B. P. Bloom, B. M. Graff, S. Ghosh, D. N. Beratan and D. H. Waldeck, J. Am. Chem. Soc., 2017, 139, 9038–9043 CrossRef CAS PubMed.
- P. C. Mondal, C. Fontanesi, D. H. Waldeck and R. Naaman, Acc. Chem. Res., 2016, 49, 2560–2568 CrossRef CAS PubMed.
- R. Naaman, C. Fontanesi and D. H. Waldeck, Curr. Opin. Electrochem., 2019, 14, 138–142 CrossRef CAS.
- J. M. Abendroth, D. M. Stemer, B. P. Bloom, P. Roy, R. Naaman, D. H. Waldeck, P. S. Weiss and P. C. Mondal, ACS Nano, 2019, 13, 4928–4946 CrossRef CAS PubMed.
- G. Long, R. Sabatini, M. I. Saidaminov, G. Lakhwani, A. Rasmita, X. Liu, E. H. Sargent and W. Gao, Nat. Rev. Mater., 2020, 5, 423–439 CrossRef.
- Q. Wei and Z. Ning, ACS Mater. Lett., 2021, 3, 1266–1275 CrossRef CAS.
- Y. H. Kim, Y. Zhai, H. Lu, X. Pan, C. Xiao, E. A. Gaulding, S. P. Harvey, J. J. Berry, Z. V. Vardeny, J. M. Luther and M. C. Beard, Science, 2021, 371, 1129–1133 CrossRef CAS PubMed.
- L. Wang, Y. Xue, M. Cui, Y. Huang, H. Xu, C. Qin, J. Yang, H. Dai and M. Yuan, Angew. Chem., 2020, 132, 6504–6512 CrossRef.
- J. Ma, C. Fang, C. Chen, L. Jin, J. Wang, S. Wang, J. Tang and D. Li, ACS Nano, 2019, 13, 3659–3665 CrossRef CAS PubMed.
- G. Long, C. Jiang, R. Sabatini, Z. Yang, M. Wei, L. N. Quan, Q. Liang, A. Rasmita, M. Askerka, G. Walters, X. Gong, J. Xing, X. Wen, R. Quintero-Bermudez, H. Yuan, G. Xing, X. R. Wang, D. Song, O. Voznyy, M. Zhang, S. Hoogland, W. Gao, Q. Xiong and E. H. Sargent, Nat. Photonics, 2018, 12, 528–533 CrossRef CAS.
- Q. Wei, Q. Zhang, L. Xiang, S. Zhang, J. Liu, X. Yang, Y. Ke and Z. Ning, J. Phys. Chem. Lett., 2021, 12, 6492–6498 CrossRef CAS PubMed.
- D. Vila-Liarte, N. A. Kotov and L. M. Liz-Marzán, Chem. Sci., 2021 10.1039/D1SC03327A.
- P. Bai, S. Yang, W. Bao, J. Kao, K. Thorkelsson, M. Salmeron, X. Zhang and T. Xu, Nano Lett., 2017, 17, 6847–6854 CrossRef CAS PubMed.
- Q. Zhang, J. Gu, L. Zhang and J. Lin, Nanoscale, 2019, 11, 474–484 RSC.
- H. Zhang, S. Li, A. Qu, C. Hao, M. Sun, L. Xu, C. Xu and H. Kuang, Chem. Sci., 2020, 11, 12937–12954 RSC.
- W. Liu, H. S. Choi, J. P. Zimmer, E. Tanaka, J. V. Frangioni and M. Bawendi, J. Am. Chem. Soc., 2007, 129, 14530–14531 CrossRef CAS PubMed.
- F. Pinaud, D. King, H.-P. Moore and S. Weiss, J. Am. Chem. Soc., 2004, 126, 6115–6123 CrossRef CAS PubMed.
- Y. Li, Y. Zhou, H.-Y. Wang, S. Perrett, Y. Zhao, Z. Tang and G. Nie, Angew. Chem., Int. Ed., 2011, 50, 5860–5864 CrossRef CAS PubMed.
- Y. Zhou, Z. Zhu, W. Huang, W. Liu, S. Wu, X. Liu, Y. Gao, W. Zhang and Z. Tang, Angew. Chem., 2011, 123, 11658–11661 CrossRef.
- K. Hou, J. Zhao, H. Wang, B. Li, K. Li, X. Shi, K. Wan, J. Ai, J. Lv, D. Wang, Q. Huang, H. Wang, Q. Cao, S. Liu and Z. Tang, Nat. Commun., 2020, 11, 1–11 Search PubMed.
- M. E. Stewart, C. R. Anderton, L. B. Thompson, J. Maria, S. K. Gray, J. A. Rogers and R. G. Nuzzo, Chem. Rev., 2008, 108, 494–521 CrossRef CAS PubMed.
- L. Wang, M. Hasanzadeh Kafshgari and M. Meunier, Adv. Funct. Mater., 2020, 30, 2005400 CrossRef CAS.
- J. Mosquera, Y. Zhao, H. Jang, N. Xie, C. Xu, N. A. Kotov and L. M. Liz-Marzán, Adv. Funct. Mater., 2020, 30, 1902082 CrossRef CAS.
- J. Kumar, H. Eraña, E. López-Martínez, N. Claes, V. F. Martín, D. M. Solís, S. Bals, A. L. Cortajarena, J. Castilla and L. M. Liz-Marzán, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3225–3230 CrossRef CAS PubMed.
- C. Wang, P. Makvandi, E. N. Zare, F. R. Tay and L. Niu, Adv. Ther., 2020, 3, 2000024 CrossRef.
- U. T. Khatoon, G. V. S. N. Rao, M. K. Mohan, A. Ramanaviciene and A. Ramanavicius, J. Environ. Chem. Eng., 2018, 6, 5837–5844 CrossRef CAS.
- H. Tang, Q. Li, W. Yan and X. Jiang, Angew. Chem., Int. Ed., 2021, 60, 13829–13834 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Deepak
Asthana
*b,
Ranjeev Kumar
Parashar
a and
Sakshi
Jadhav
c
*a,
Deepak
Asthana
*b,
Ranjeev Kumar
Parashar
a and
Sakshi
Jadhav
c