
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLimitations of conjugated polymers as emitters in triplet–triplet annihilation upconversion†
Riley
O'shea
ab,
Can
Gao
 c,
Siobhan
Bradley
a,
Tze Cin
Owyong
ab,
Na
Wu
ab,
Jonathan M.
White
b,
Kenneth P.
Ghiggino
a and
Wallace W. H.
Wong
*ab
c,
Siobhan
Bradley
a,
Tze Cin
Owyong
ab,
Na
Wu
ab,
Jonathan M.
White
b,
Kenneth P.
Ghiggino
a and
Wallace W. H.
Wong
*ab
aARC Centre of Excellence in Exciton Science, School of Chemistry, University of Melbourne, Parkville, VIC 3010, Australia. E-mail: wwhwong@unimelb.edu.au
bSchool of Chemistry, Bio21 Institute, University of Melbourne, Parkville, VIC 3010, Australia
cBeijing National Laboratory for Molecular Sciences, Key Laboratory of Organic Solids, Institute of Chemistry, Chinese Academy of Sciences, Beijing, China
First published on 11th October 2021
Abstract
A series of poly(phenylene-vinylene)s (PPV) was synthesized with two monomer building blocks containing methoxy and 2-ethylhexyloxy (MEH) sidechains and sterically bulky fluorenyloxy sidechains. Variations in monomer incorporation were achieved by adjusting the ratio of the monomer precursors in the synthesis of the PPVs using the Gilch polymerization method. The increased incorporation of the bulky monomer led to expected variation in photophysical properties, in particular, the enhancement of photoluminescence quantum yield. Using palladium(II) tetraphenyltetrabenzoporphyrin (PdTPTBP) as the triplet sensitizer, all polymers displayed upconverted emission in solution with the comonomer ratio for the fluorenyloxy and MEH units at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 showing the best performance with upconversion efficiency of 0.135%. This efficiency was up to three times higher than that of two commercially available PPVs, MEH-PPV and Super yellow PPV, under the same experimental conditions. Further analysis of the efficiency of the photochemical processes involved revealed several limitations to the performance of these conjugated polymer emitters. While increasing steric bulk of the sidechains improved the photoluminescence quantum yield, it also caused variation in excited state energies of the polymers through the series. This led to poor alignment with the triplet energy of our chosen sensitizer for some of the polymers. Even when the triplet–triplet energy transfer efficiency was high, the contact triplet pair formation efficiency was found to be very low. We reasoned that the structural conformation of the polymers hindered the chromophore orbital overlap during triplet collisions required for triplet–triplet annihilation.
:10 showing the best performance with upconversion efficiency of 0.135%. This efficiency was up to three times higher than that of two commercially available PPVs, MEH-PPV and Super yellow PPV, under the same experimental conditions. Further analysis of the efficiency of the photochemical processes involved revealed several limitations to the performance of these conjugated polymer emitters. While increasing steric bulk of the sidechains improved the photoluminescence quantum yield, it also caused variation in excited state energies of the polymers through the series. This led to poor alignment with the triplet energy of our chosen sensitizer for some of the polymers. Even when the triplet–triplet energy transfer efficiency was high, the contact triplet pair formation efficiency was found to be very low. We reasoned that the structural conformation of the polymers hindered the chromophore orbital overlap during triplet collisions required for triplet–triplet annihilation.
Introduction
Triplet–triplet annihilation (TTA) upconversion is a photochemical process by which absorption of two lower energy photons can produce one photon of higher energy.1 TTA upconversion is possible under relatively low excitation power compared to other photon upconversion methods such as two-photon absorption. As such, one of its most attractive applications is in harvesting solar photons that are below the bandgap of photovoltaic cells, thereby raising the photovoltaic efficiency above the Shockley–Queisser limit.2,3 For TTA upconversion to occur, five photochemical processes must proceed effectively in sequence (Fig. 1). Two types of chromophores are usually required, a triplet sensitizer and an emitter. The triplet sensitizer absorbs incoming lower energy photons. The singlet excited state generated on the triplet sensitizer is quickly converted to the triplet excited state via intersystem crossing (ISC). Through triplet–triplet energy transfer (TET), the triplet excited state of the emitter species is populated. When triplet excited states on two emitter species encounter each other, they can undergo TTA to generate one emitter species in its singlet excited state while returning the other emitter to its ground state. The singlet excited emitter can then decay radiatively producing a photon at a higher energy than the photon initially absorbed by the sensitizer. The upconversion quantum yield (ΦUC) is a product of all the quantum yields for the photochemical processes involved, given by the equation:| ΦUC = ΦISCΦTETΦTTAΦPL | (1) |
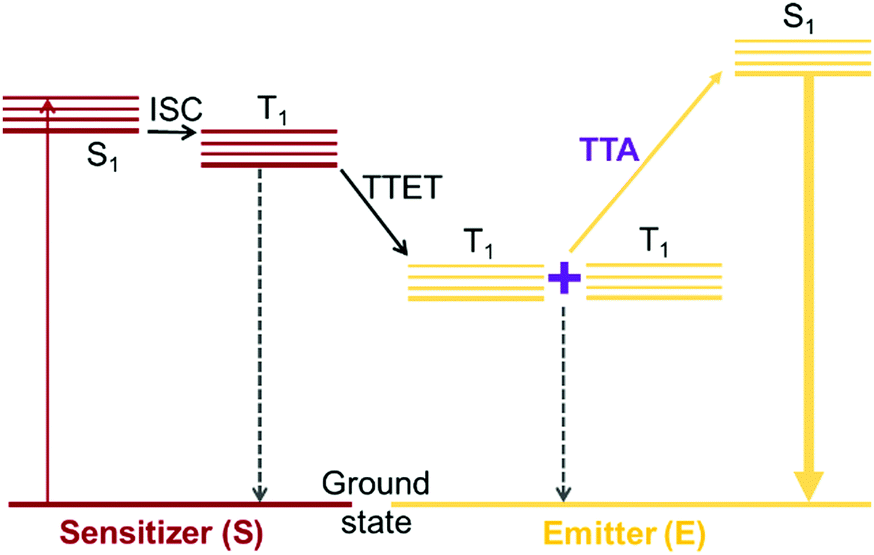 | ||
| Fig. 1 Jablonski diagram depicting the processes involved in TTA upconversion with a sensitizer-emitter pair. | ||
The most studied and efficient TTA upconversion systems consists of transition metal complex triplet sensitizers and small molecule emitters. For example, platinum(II) or palladium(II) octaethylporphyrin (PtOEP or PdOEP) as the triplet sensitizers and 9,10-diphenylanthracene (DPA) and derivatives thereof as the emitters have been reported by many researchers.5–7 While the best reported TTA upconversion performance is in solution,5 there have been many investigations into solvent-free systems that are more amenable for integration with photovoltaic cells.8–11 The main challenge in solvent-free systems is ensuring efficient TET and TTA processes without molecular diffusion. In this context, conjugated polymers as emitters offer the possibility of enhanced triplet excited state diffusion along the polymer backbone and rapid intramolecular TTA.12
Very few examples exist of polymer-based emitters, and even fewer of conjugated polymers.13–17 A promising conjugated polymer emitter is a commercially available poly(phenylenevinylene) (PPV) copolymer with the trade name “super yellow PPV”.16 Upconverted emission was reported using super yellow PPV with palladium(II) tetraphenyl-tetrabenzoporphyrin (PdTPTBP) as the triplet sensitizer in thin films. The use of PPV emitters over other conjugated polymers may be particularly advantageous owing to their large singlet–triplet excited state energy gap.18 This may mitigate losses due to reabsorption of upconverted light by having a sufficiently large anti-Stokes shift, thereby reducing the spectral overlap with the absorbance of the triplet sensitizer. Additionally, Schmidt and co-workers have shown that the triplet exciton transport may proceed via a fast intramolecular diffusion along the PPV backbone,12 which cannot be realized in the case of small molecule emitters that may only proceed via intermolecular diffusion. However, the use of PPV or other conjugated polymer emitters is still underdeveloped, and only one set measurements of upconversion quantum yields exist to date, using poly(phenylene-ethynylene) (PPE) copolymers as emitters.19
Given the work on super yellow PPV,16 we devised a series of polymer emitters with a similar structural motif (Fig. 2). The proposed PPV copolymer P1–6 bears similarity to super yellow PPV in that it contains both alkoxy and aryloxy sidechains on the comonomer units. The proposed copolymer P1–6 uses the bulky fluorenol sidechains (comonomer denoted “n”) to suppress aggregation caused quenching of the polymer emission. The alkoxy sidechains (comonomer denoted “m”), which in this case are from the commonly known MEH-PPV, are used to redshift the polymer emission. It is noteworthy that the copolymer ratio in super yellow PPV would have been optimised for electroluminescence performance rather than upconversion. Therefore, a range of comonomer ratios can be explored to find the optimal value for upconversion emission using the PPV copolymers P1–6. This can be achieved via a Gilch polymerization strategy,20 such that a statistically regio-random polymer can be generated but with fixed ratios (n:m) of the two comonomers. While our PPV series showed improved TTA upconversion performance over MEH-PPV and Super yellow PPV, the maximum ΦUC obtained was 0.135% under high excitation intensity. Apart from discussions on performance across the polymer series, the efficiency of each photochemical step in the TTA upconversion process was analysed alongside sensitizer phosphorescence lifetime and polymer triplet excited state decay data. Limitations to the TTA upconversion performance for conjugated polymers as emitters were revealed.
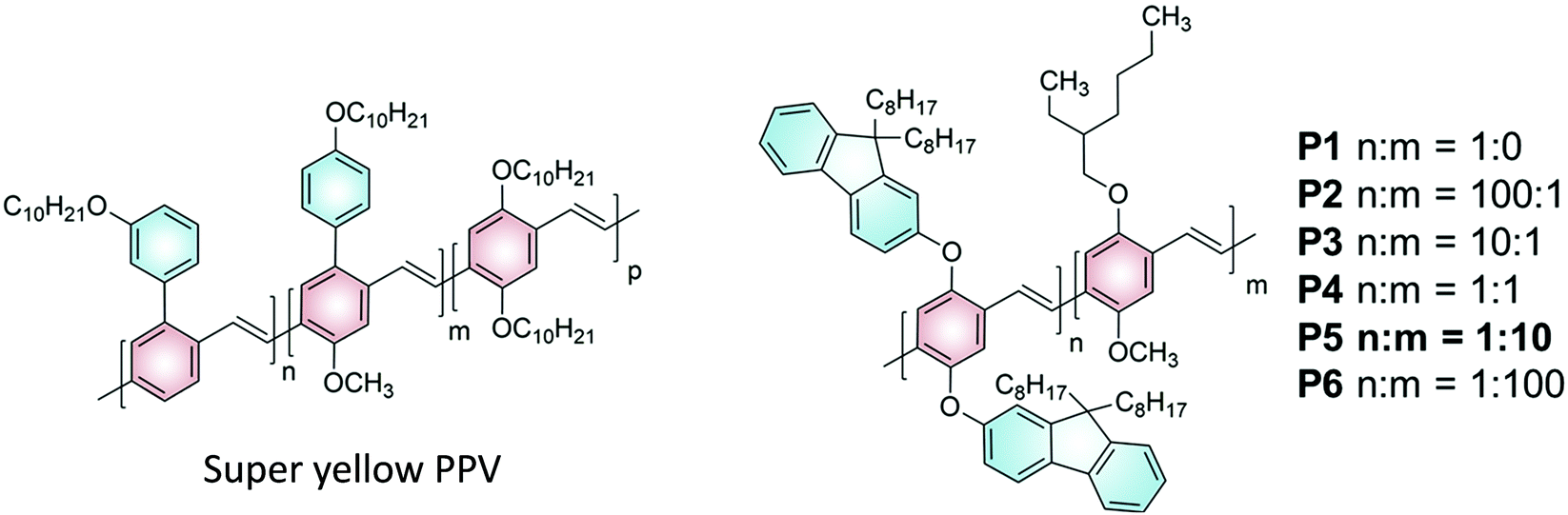 | ||
| Fig. 2 Structures of Super yellow PPV (left) and a proposed structure of a novel PPV copolymer P1–6 (right). | ||
Results and discussion
Synthesis
The synthesis of the PPV emitter first begins with an Ullmann coupling between diethyl terephthalate 1 and dioctyl fluorenol 2 to furnish compound 3 in a 67% yield (Fig. 3). Compound 3 is then reduced with lithium aluminium hydride to give the corresponding diol 4 in a 62% yield (Fig. 3). The diol 4 can then be converted to the Gilch monomer 5via thionyl chloride in a 57% yield. The single crystal structure of the monomer 5 was then obtained by slow evaporation from a chloroform solution (Fig. S10, ESI†). For more detail see Tables S1 and S2 (ESI†). PPV copolymers P1–6 were then generated by Gilch polymerization with both monomer 5 and the MEH monomer unit 6 (Fig. 3) with yields varying between 50 to 76% for differing comonomer ratios (Table S3, ESI†).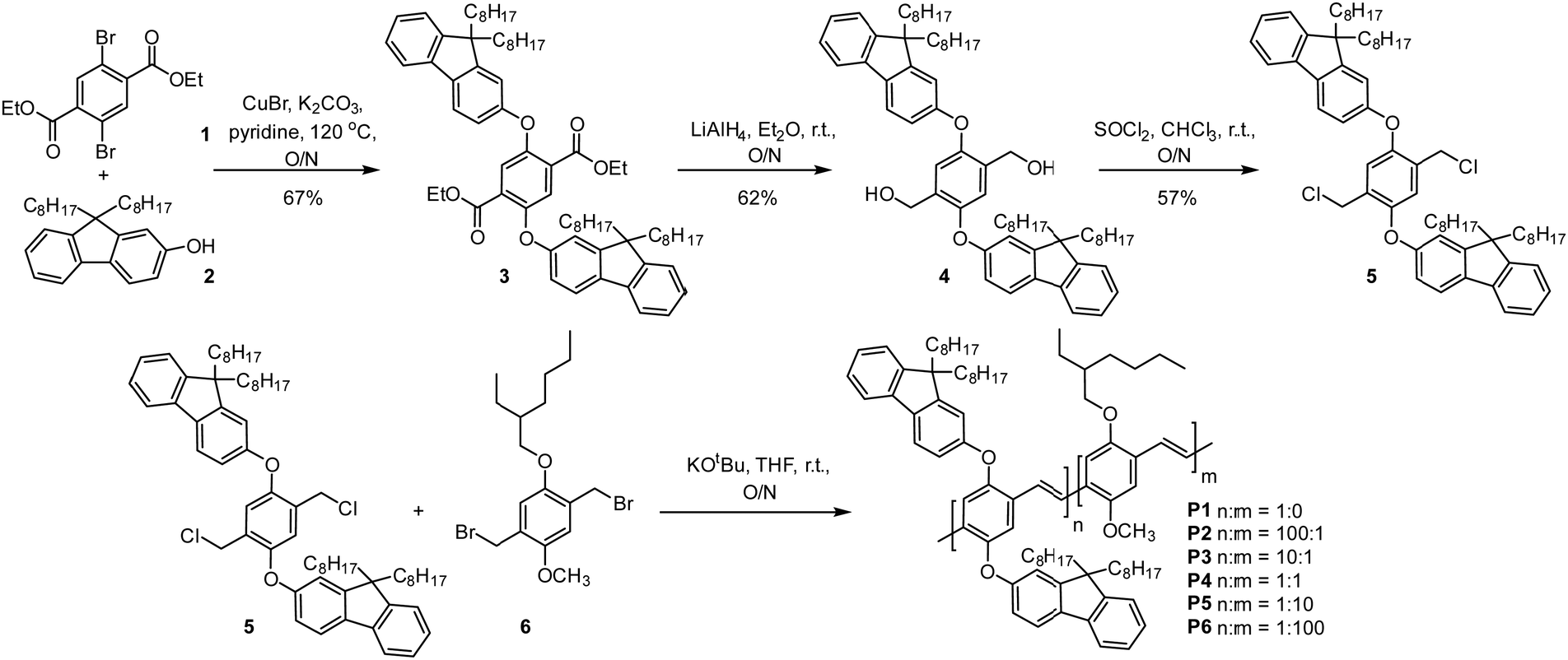 | ||
| Fig. 3 Synthesis of PPV copolymers P1–6 starting from dibromodiethylterephthalate 1 and dioctylfluorenol 2. | ||
The average molecular weight of the samples varies with the comonomer ratio, with a general decreasing trend in molecular weight for an increasing proportion of MEH monomer 6 (Table 1). Most of the PPVs apart from P2 and P3 have a similar chain length of 44 to 55 units (4–5 chromophores).21 Both the solution and solid state ΦPL increased with increasing proportion of the fluorenyloxy monomer 5 going from P6 to P1. As the aggregation of the polymer backbone was the commonly accepted cause for photoluminescence quenching in PPV materials, the sterically bulky monomer 5 reduced aggregation resulting in enhanced ΦPL.
| Polymer | n:m | M w (g mol−1) | M n (g mol−1) | Đ | Degree of polymerisationa | Average # of chromophoresb | Solution ΦPLc (%) | Solid state ΦPL (%) |
|---|---|---|---|---|---|---|---|---|
| a Degree of polymerisation determined from the ratio of Mn and the average monomer molecular weight based on the initial stoichiometry used in the polymerization. b Calculated based on the degree of polymerisation assuming 11 repeat units.21 c Solution PLQY measured at 0.5 mg mL−1via integrating sphere method with a reabsorption correction.22 d GPC data from Sigma Aldrich product specification sheet. e Photoluminescence quantum yield values for films and solution of super yellow PPV taken from Monkman and coworkers.23 | ||||||||
| P1 | 1:0 |
113000 |
54000 |
2.1 | 59 | 5 | 87.0 ± 0.8 | 55.4 ± 0.6 |
| P2 | 100:1 |
368000 |
147000 |
2.5 | 162 | 15 | 84.7 ± 0.9 | 38.3 ± 0.9 |
| P3 | 10:1 |
23000 |
100000 |
2.4 | 117 | 11 | 74.4 ± 0.8 | 35.7 ± 0.6 |
| P4 | 1:1 |
110000 |
28000 |
3.9 | 48 | 4 | 47.8 ± 0.9 | 12.2 ± 0.4 |
| P5 | 1:10 |
52000 |
18000 |
2.9 | 56 | 5 | 34.3 ± 0.3 | 4.9 ± 0.1 |
| P6 | 1:100 |
33000 |
13000 |
2.6 | 49 | 4 | 32.0 ± 0.3 | 4.8 ± 0.1 |
| MEH-PPV | — | 62500 |
20100 |
3.1 | 77 | 7 | 15.5 ± 0.1 | — |
| Super yellowd | — | 1300000 |
200000 |
6.5 | 458 | 42 | 69 ± 3e | 17 ± 1e |
The maximum absorbance and emission wavelengths were blue shifted with increasing portion of bulky monomer 5 (Fig. 4). This is likely due to the increase in twisting of conjugated backbone from increased incorporation of bulky monomer 5 leading to a decrease in average conjugation length along the polymer backbone. In addition, the decrease in the portion of more electron-rich MEH units would reduce the HOMO energy leading to increased HOMO–LUMO energy gap. It is expected that these spectral shifts will have significant impact on upconversion performance. Additionally, the two peaks in the UV-Vis spectra at ∼300 nm and ∼500 nm vary inversely, this can be attributed to the absorption from the fluorenyl units. The decrease in the peak intensity at ∼300 nm corresponds to a decrease in the presence of the fluorenyl side chains throughout the copolymer series going from P1 to P6.
 | ||
| Fig. 4 Solution UV-Vis absorbance (a) and photoluminescence spectra (b) from the PPV copolymers P1–6 and MEH-PPV. | ||
Upconversion photophysics
All the PPV copolymers P1–P6 display red-to-yellow upconversion when paired with PdTPTBP as a sensitizer in chloroform solution (Fig. 5a and b). Upconversion was then compared between each polymer using 1.0 mg mL−1 and with 5.0 μM PdTPTBP. The optimal comonomer ratio for the PPV copolymers, was found to be n:m = 1:10 (P5) in terms of upconverted emission intensity. The upconverted emission is lower for n:m = 1:100 (P6) despite an increase in quenching of the sensitizer's phosphorescence. This may be in part due to the slight decrease in ΦPL between P5 and P6 (Table 1).
 | ||
| Fig. 5 Upconverted emission spectra from PPV copolymers P1–6 with PdTPTBP as the sensitizer under 632 nm excitation, without (a) and with (b) a 600 nm low bandpass filter. Inset in Fig. 5b is the image of TTA upconversion from PPV copolymer P5/PdTPTBP dilute solution with 632 nm excitation. (c) Energy diagram depicting the calculated singlet and triplet excited state energies obtained by DFT of the model oligomers compared to the triplet excited state energy of PdTPTBP. (d) Upconversion quantum yield for PPV copolymers P1–6, MEH-PPV and Super yellow PPV. (e) Dependence of upconversion quantum yield on excitation intensity for polymer P5. (f) Upconversion power threshold for polymer P5 (determined to be 370 mW cm−2). | ||
The upconversion emission intensity and ΦUC for copolymers P1–P4 were very low (<0.002%) with very poor sensitizer phosphorescence quenching (Fig. 5a and Table 2). This contrasts with the ΦPL enhancement with increasing portion of bulky monomer 5. One explanation for the low performance of P1–P4 could be energy mismatch between the triplet excited state of the sensitizer and that of these polymers.
| Polymer | Triplet lifetime (μs) | Phosphorescence lifetime (μs) | Φ UC (%) | Φ PL (%) | Φ TET (%) | Φ TTA (%) |
|---|---|---|---|---|---|---|
| a Calculated from the ratio of the phosphorescence integrated emission between the polymer upconversion sample and sensitizer only sample (see eqn (5)). b Calculated using eqn (1). | ||||||
| Sensitizer only | — | 123 | — | — | — | — |
| P1 | 84.3 | 56.1 | 0.0003 | 87.0 | 6.8 | 0.005 |
| P2 | 144 | 53.1 | 0.001 | 84.7 | 16.2 | 0.007 |
| P3 | 145 | 56.3 | 0.001 | 74.4 | 22.5 | 0.006 |
| P4 | 82.9 | 29.6 | 0.002 | 47.8 | 76.7 | 0.006 |
| P5 | 131 | 29.9 | 0.135 | 34.3 | 86.3 | 0.460 |
| P6 | 113 | 43.5 | 0.032 | 32.0 | 90.7 | 0.111 |
| MEH-PPV | 73.7 | 13.5 | 0.039 | 15.5 | 92.7 | 0.274 |
| Super yellow | 47 | 36.6 | 0.029 | 69.0 | 93.5 | 0.045 |
To determine the extent of this electronic effect, time-dependent density functional theory (TD-DFT) calculations were performed on PPV oligomers. Geometry optimization of the oligomers were achieved using PM6 level of theory and energies of the first singlet excited state (S1) and the first triplet excited state (T1) were calculated using B3LYP/6-31G(d) (Fig. 5c). The alkyl sidechains were truncated in the model oligomers to reduce computational demand (see Supporting Information for details). These oligomers bear either methoxy sidechains (OMe), or fluorenyloxy side chains (OAr), or a simple 1:1 ratio of the two (OMeOAr). For all cases, both the HOMO and LUMO distributed along the PPV backbone (Fig. S12, ESI†). The calculated energies for S1 ranged from 2.11 eV for the OMe oligomer to 2.34 eV for the OAr oligomer. These calculated values were comparable to the peak emission energies from the PL data which were ∼0.1 eV smaller through the series (Fig. 4b). The calculated energies for T1 followed the same trend as the energies for S1. The energy of T1 for OMe oligomer was the smallest at 1.51 eV with OMeOAr at 1.70 eV and OAr at 1.79 eV. When these values are compared to the phosphorescence energy of the sensitizer at 1.56 eV, only the OMe oligomer was expected to undergo triplet energy transfer via an exoergic process. With an increased portion of fluorenyloxy units a significant energy barrier arises. This agrees with the poor sensitizer phosphorescence quenching by copolymers P1–P4 with bulky monomer to MEH monomer ratio n ≥ m.
The best copolymer P5 was chosen for further optimization of upconversion performance which were found to be 0.5 mg mL−1 of polymer with 7.5 μM of PdTPTBP (Fig. S15–S18, ESI†). The performance of P5 was then compared to two commercially available PPVs, MEH-PPV and Super yellow PPV, under the same experimental conditions (Fig. 5d and Fig. S19, ESI†). Both commercial polymers showed lower upconversion emission intensity than P5. The ΦUC was then measured for all of the PPV copolymers P1–6 including the two commercially available PPVs via an integrating sphere method. The highest performing emitter was copolymer P5 with ΦUC at 0.135% significantly outperforming MEH-PPV with ΦUC at 0.039%. Super yellow PPV showed even lower ΦUC of 0.029%.
Given the significant variation in ΦUC between polymer samples, transient absorbance (TA) decay data were obtained to determine the polymer triplet excited state dynamics of the polymers (Table 2). Additionally, the phosphorescence decay of the sensitizer in the presence of the polymer emitter was measured to determine the difference in triplet transfer rates between samples (Table 2). For the transient absorbance lifetimes measurements, a second order decay is expected following the expression:
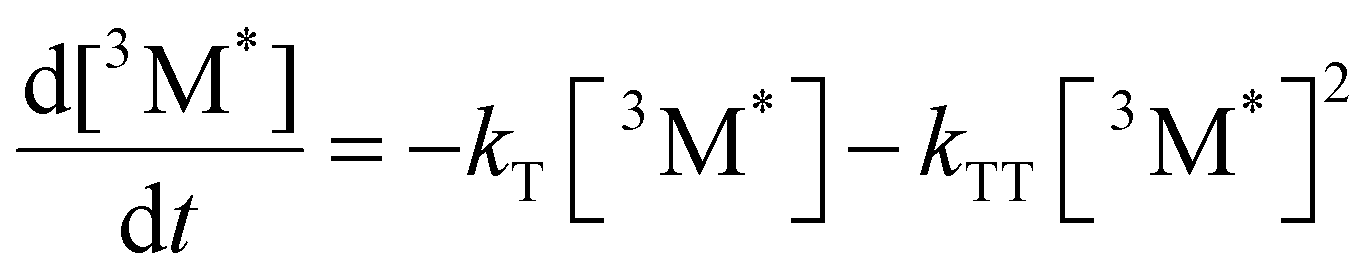 | (2) |
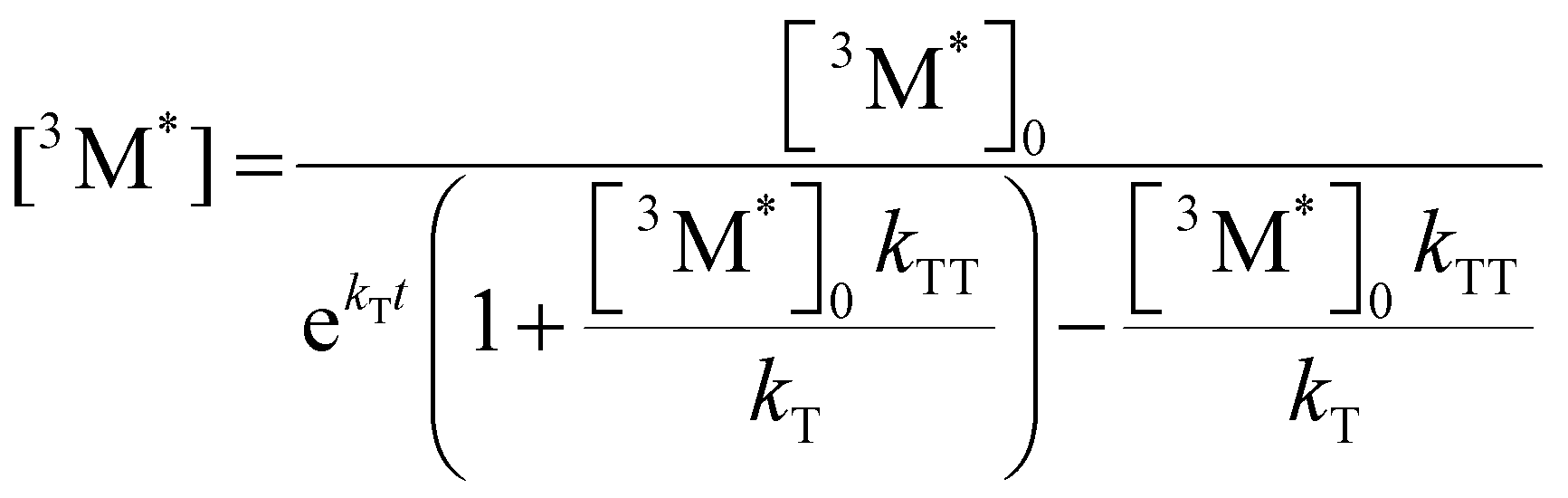 | (3) |
 | (4) |
From eqn (4), the triplet lifetimes of the polymers can be extracted (see Table 2). However, there is no obvious trend seen in relation to ΦUC and all values are roughly within the same order of magnitude, with an average value of 117 μs across the polymer series. If an intramolecular TTA mechanism were to be significant, a fast second order contribution to the decays might be expected. For MEH-PPV, triplet lifetimes during intramolecular TTA events have been calculated to be in the order of 10 fs,12 due to a rapid exciton diffusion process with values for kTT = 1014 M−1 s−1. In the case observed here, an intermolecular TTA mechanism is most likely to occur, but the low ΦTTA values suggest polymer collisions are ineffective (see later discussion). The sensitizer phosphorescence lifetime for each sample appear to correlate with ΦTET, except for P6 and Super yellow which have a slightly increased lifetime than expected. It should also be noted that the phosphorescence lifetime can be affected by differences in molar concentration of the polymer chains due to the different molecular weights. However, Super yellow PPV has the largest molecular weight but does not have the longest phosphorescence lifetime, while P6 has the lowest molecular weight but does not have the shortest phosphorescence lifetime. This suggests that other factors influence the effectiveness of the triplet energy transfer, such as the energetic offset identified by TD-DFT calculations.
The ΦTET were calculated by comparing the degree of phosphorescence quenching of the sensitizer by the polymers to the phosphorescence of a sensitizer-only sample. The ΦTET for P5, P6, MEH-PPV and Super yellow PPV were all close to 90% (Table 2). The ΦTET for the copolymers decreased with increasing content of the bulky fluorenyloxy monomer going from P6 to P1. This is again in agreement with the poor triplet excited state energy alignment between the polymers and the sensitizer apparent in TD-DFT calculations. The triplet state lifetimes of the polymers are ∼100 μs (Table 2) and, assuming diffusional interactions (the second order diffusion rate constant in chloroform is 5.8 × 109 M−1 s−1), there should be multiple collisions between triplet excited polymer chains for those polymers where sensitization was efficient (e.g.P5, P6). Even allowing for spin statistical factors, the observed very low quantum yields of upconversion suggest that steric restrictions and other factors must make most collisions ineffective in leading to annihilation and formation of emitting singlet states. Nevertheless, for the later part of the polymer series, a significant change can be seen in the ΦTTA, with P5 clearly above all the other polymers with a value of 0.46%, nearly double that of the second highest; MEH-PPV with a value of 0.27%. It is unclear why P5 has a significantly higher ΦTTA. As a general trend, the ΦTTA increases throughout the polymer series.
Since the ΦUC value was low for all samples, the upconversion power threshold was only determined for P5 (Fig. 5e). The value determined was 370 mW cm−2, which is particularly high compared to most upconversion systems. This is likely from the difficulty in having effective collisions between triplet polymer chains that lead to annihilation, as discussed above. The upconversion quantum yield was also determined as a function of excitation intensity above and below the power threshold for P5 (Fig. 5f). The quantum yield appears to plateau as the excitation intensity approaches 1000 mW cm−2. At high excitation intensity, the sample should be saturated with triplet excited state species and the ΦUC would reach a limit.
Conclusions
A series of PPV copolymers P1–P6 was synthesized all of which display weak TTA upconversion emission with PdTPTBP as the sensitizer. The upconversion quantum yields were measured for all copolymers and compared to two commercially available PPVs. The best ΦUC obtained was 0.135% for the PPV copolymer P5 with n:m = 1:10. The PPV copolymers show a general trend whereby ΦUC increases with an increased proportion of the MEH comonomer unit. However, this is contrary to the ΦPL, which decreases with an increased proportion of the MEH comonomer unit. The quenching of phosphorescence of the sensitizer and the rate of decay of the triplet exciton on the polymer emitter were also measured. The variation in the efficiency of sensitizer quenching, can be related to the reduction in steric bulk between the PPV copolymers P1–P6. An increased proportion of the MEH comonomer unit reduces the steric bulk allowing more facile approach of the sensitizer to the polymer backbone facilitating triplet energy transfer. The power intensity threshold for PPV copolymer P5 was found to be 370 mW cm−2.
Our study revealed several limitations in using conjugated emitters for TTA upconversion. Firstly, while the PL concentration quenching problem can be mitigated by introduction of sterically bulky sidechains, these sidechains have significant negative impact on the TET process between the sensitizer and the polymer. In addition, theoretical calculations show the bulky sidechains can also affect the polymer energetics, leading to poor triplet energy alignment between the sensitizer and the polymer. This also has an impact on the TET process. Secondly, the triplet excited state lifetime of the PPV materials in this study is at least an order of magnitude shorter than that for molecular emitters such as DPA.7 Finally, the ΦTTA values for all samples were very low at <1%. ΦTTA is determined by the contact triplet pair formation efficiency and the factor f,4 which is a measure of the probability of generating the singlet excited state after TTA. Even assuming a minimum f of 1/9, there appears to be a low contact triplet pair formation efficiency for PPV materials, and this is a major hurdle in achieving better ΦUC. A possible reason for low contact triplet pair formation efficiency is the steric interference imposed by the structural conformation of the polymers that inhibits the chromophore orbital overlap during the triplet collisions required for TTA. These issues might be addressed by moving to an intramolecular TTA mechanism with triplet sensitizers tethered to the conjugated polymer emitter to produce a high intrachain concentration of polymer chromophore triplets.
Experimental section
General experimental information
Commercial reagents were purchased from Univar, Sigma-Aldrich, AK Scientific, Matrix Scientific, Ajax Finechem, and Labchem, and were used as received. The reference MEH-PPV polymer was purchased commercially from Lumtec (https://lumtec.com.tw/). Anhydrous toluene, diethyl ether, dichloromethane, and tetrahydrofuran were obtained from alumina drying columns. For reactions carried out under inert conditions, standard Schlenk techniques were used. Solvents were sparged with nitrogen gas for several hours prior to use, and the reaction vessels were sealed with a rubber septum under a nitrogen atmosphere. Thin layer chromatography (TLC) was done using Merck-Millipore Silica gel glass plates (60G F254), with a 254 nm and 365 nm light mercury lamp used for identifying spots. 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were obtained in CDCl3, on a 400 MHz Varian spectrometer. NMR peaks were referenced to the CHCl3 solvent peak. UV-Vis spectroscopy was performed on Agilent Technologies Cary 50 UV-Vis. Photoluminescent spectroscopy was performed on Varian Cary Eclipse fluorimeter. Absolute photoluminescence quantum yield of the samples was determined via an integrating sphere method using an integrating sphere accessory (F3018, Horiba Jobin Yvon) on a Fluorolog-3 fluorimeter.Synthesis procedures
1H NMR (400 MHz, CHCl3, δ): 7.64 (d, J = 6.0Hz, 4H), 7.58 (s, 2H), 7.32 (m, 4H), 7.24 (m, 2H), 7.02 (s, 2H), 6.89 (d, J = 7.7Hz, 2H), 4.21 (q, J = 7.1Hz, 4H), 1.94 (m, 8H), 1.15 (m, 49H), 0.81 (t, J = 6.8Hz, 12H), 0.66 (m, 8H); 13C NMR (100 MHz, CDCl3, δ): 164.5, 157.2, 153.0, 151.3, 150.5, 140.5, 136.6, 127.9, 126.8, 126.5, 123.7, 122.8, 120.6, 119.2, 116.2, 113.0, 61.5, 55.2, 40.4, 31.8, 30.1, 29.3, 23.8, 22.6, 14.1, 14.0; HRMS (ESI) m/z: [M]+ calcd for C70H94O6, 1030.7050; found, 1030.7050.
1H NMR (400 MHz, CHCl3, δ): 7.66 (d, J = 8.2Hz, 4H), 7.32 (m, 6H), 7.05 (d, J = 1.8Hz, 2H), 7.01 (s, 2H), 6.93 (dd, J = 8.2, 1.9Hz, 2H), 4.68 (d, J = 6.3Hz, 4H), 1.94 (m, 8H), 1.11 (m, 42H), 0.82 (t, J = 7.1Hz, 12H), 0.66 (m, 8H); 13C NMR (100 MHz, CDCl3, δ):156.6, 153.1, 150.8, 150.5, 140.5, 136.9, 132.1, 126.8, 126.6, 122.8, 120.7, 119.3, 118.5, 116.9, 113.7, 61.0, 55.2, 40.4, 31.8, 30.0, 29.3, 29.2, 23.8, 22.6, 14.1; HRMS (ESI) m/z: [M]+ calcd for C66H90O4, 946.6839; found, 946.6837.
1H NMR (400 MHz, CHCl3, δ): 7.67 (t, J = 7.3Hz, 4H), 7.31 (m, 6H), 7.02 (m, 6H), 4.61 (s, 4H), 1.94 (m, 8H), 1.12 (m, 44H), 0.84 (m, 13H), 0.66 (m, 8H); 13C NMR (100 MHz, CDCl3, δ):156.4, 153.1, 150.9, 150.5, 140.5, 137.1, 129.8, 126.8, 126.6, 122.8, 120.7, 120.0, 119.3, 117.5, 113.8, 55.3, 40.4, 40.4, 31.8, 30.0, 29.3, 29.2, 23.8, 22.6, 22.6, 14.1; HRMS (ESI) m/z: [M]+ calcd for C66H88O2Cl2, 982.6161; found, 982.6159.
General polymerization method
Potassium tert-butoxide (20 eq.) was dissolved in dry degassed THF (made to 0.4 M) and stirred at room temperature under nitrogen atmosphere. Then the monomer(s) (1 eq.) were added dropwise as a solution of dry degassed THF (made to 0.035 M) to the mixture. The mixture was then allowed to stir overnight at room temperature. The mixture was then diluted with THF and poured into 10 times the reaction volume of methanol with vigorous stirring. The suspension was then centrifuged to collect the polymer, which was then washed 3 times with methanol and then dried in a vacuum oven overnight. Example procedure for P4 provided below. See Table S2 in the ESI,† for synthesis details for P1 to P6. GPC traces is shown in Fig. S11 (ESI†).Example procedure: synthesis of P4
Potassium tert-butoxide (0.22) was dissolved in 4 mL of dry degassed THF and stirred at room temperature under nitrogen atmosphere. Then 2,2′-((2,5-bis(chloromethyl)-1,4-phenylene)bis(oxy))bis(9,9-dioctyl-9H-fluorene) (0.05 g) and 1,4-bis(bromomethyl)-2-((2-ethylhexyl)oxy)-5-methoxybenzene (0.021 g) were added dropwise as a solution in 1 mL of dry degassed THF to the mixture. The mixture was then allowed to stir overnight at room temperature. The mixture was then diluted with THF and poured into 50 mL of methanol with vigorous stirring. The suspension was then centrifuged to collect the polymer, the polymer was then washed 3 times with methanol and then dried in a vacuum oven overnight. Mass = 0.035g (59%).Upconversion experiments
Upconversion samples were degassed three times via freeze–pump–thaw cycles prior to analysis. Upconversion spectra were taken using an Ocean Optics USB spectrometer with 300 μm fiber optic cable. The ΦUC of the polymer samples was determined with 632 nm excitation (band pass filtered HeNe laser) with an intensity of 985 mW cm-2via an integrating sphere method with a LABSPHERE (model number: 4P-GPS-053-SL) and detected with a liquid nitrogen cooled CCD camera from Princeton Instruments (series number: SP2500). Band pass filters for both short and long pass filters were obtained from Thorlabs (Edgepass filter range).The values for the triplet energy transfer quantum yields were determined by comparing the integrated emission from the sensitizer with/without the polymer present, using the following equation:
 | (5) |
Transient absorbance and phosphorescence decay measurements were performed with identical sample conditions as per upconversion emission samples. Phosphorescence decay measurements used 632 nm laser pulse, with detection set to observe emission at 790 nm. Transient absorbance measurements used 632 nm laser pulse, with 830 nm probe. Triplet excited state concentrations (y-axis) were obtained from transient absorbance values using the Beer–Lambert law and an extinction coefficient of 1.19 × 105 M−1 cm−1.25
Author contributions
R. O. and W. W. H. W. conceived the experiments. R. O., C. G., S. B., T. C. O., N. W. and J. M. W. conducted the experiments. R. O., K. P. G. and W. W. H. W. performed the analysis. R. O., C. G. and T. C. O. generated and formatted the figures. All authors reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the ARC Centre of Excellence in Exciton Science (CE170100026).Notes and references
- T. F. Schulze and T. W. Schmidt, Energy Environ. Sci., 2015, 8, 103–125 RSC.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- Y. Y. Cheng, B. Fückel, R. W. MacQueen, T. Khoury, R. G. C. R. Clady, T. F. Schulze, N. J. Ekins-Daukes, M. J. Crossley, B. Stannowski, K. Lips and T. W. Schmidt, Energy Environ. Sci., 2012, 5, 6953–6959 RSC.
- Y. Zhou, F. N. Castellano, T. W. Schmidt and K. Hanson, ACS Energy Lett., 2020, 5, 2322–2326 CrossRef CAS.
- T. Ogawa, N. Yanai, A. Monguzzi and N. Kimizuka, Sci. Rep., 2015, 5, 10882 CrossRef CAS PubMed.
- S. H. Lee, M. A. Ayer, R. Vadrucci, C. Weder and Y. C. Simon, Polym. Chem., 2014, 5, 6898–6904 RSC.
- C. Gao, S. K. K. Prasad, B. Zhang, M. Dvořák, M. J. Y. Tayebjee, D. R. McCamey, T. W. Schmidt, T. A. Smith and W. W. H. Wong, J. Phys. Chem. C, 2019, 123, 20181–20187 CrossRef CAS.
- E. M. Gholizadeh, S. K. K. Prasad, Z. L. Teh, T. Ishwara, S. Norman, A. J. Petty, J. H. Cole, S. Cheong, R. D. Tilley, J. E. Anthony, S. Huang and T. W. Schmidt, Nat. Photonics, 2020, 14, 585–590 CrossRef CAS.
- S. Fischer, J. C. Goldschmidt, P. Löper, G. H. Bauer, R. Brüggemann, K. Krämer, D. Biner, M. Hermle and S. W. Glunz, J. Appl. Phys., 2010, 108, 044912 CrossRef.
- N. Nishimura, J. R. Allardice, J. Xiao, Q. Gu, V. Gray and A. Rao, Chem. Sci., 2019, 10, 4750–4760 RSC.
- P. Xia, E. K. Raulerson, D. Coleman, C. S. Gerke, L. Mangolini, M. L. Tang and S. T. Roberts, Nat. Chem., 2020, 12, 137–144 CrossRef CAS PubMed.
- I. Lyskov, E. Trushin, B. Q. Baragiola, T. W. Schmidt, J. H. Cole and S. P. Russo, J. Phys. Chem. C, 2019, 123, 26831–26841 CrossRef CAS.
- D. Hertel, H. Bässler, R. Guentner and U. Scherf, J. Chem. Phys., 2001, 115, 10007–10013 CrossRef CAS.
- F. Laquai, G. Wegner, C. Im, A. Büsing and S. Heun, J. Chem. Phys., 2005, 123, 074902 CrossRef PubMed.
- S. Baluschev, P. E. Keivanidis, G. Wegner, J. Jacob, A. C. Grimsdale, K. Müllen, T. Miteva, A. Yasuda and G. Nelles, Appl. Phys. Lett., 2005, 86, 061904 CrossRef.
- V. Jankus, E. W. Snedden, D. W. Bright, V. L. Whittle, J. A. G. Williams and A. Monkman, Adv. Funct. Mater., 2013, 23, 384–393 CrossRef CAS.
- T. Mori, S. Hamzawy, P. Wagner, A. J. Mozer and A. Nattestad, J. Phys. Chem. C, 2021, 125, 14538–14548 CrossRef CAS.
- A. Köhler and D. Beljonne, Adv. Funct. Mater., 2004, 14, 11–18 CrossRef.
- R. O’Shea, C. Gao, T. C. Owyong, J. M. White and W. W. H. Wong, Mater. Adv., 2021, 2, 2031–2035 RSC.
- H. G. Gilch and W. L. Wheelwright, J. Polym. Sci., Part A-1: Polym. Chem., 1966, 4, 1337–1349 CrossRef CAS.
- H. Meier, U. Stalmach and H. Kolshorn, Acta Polym., 1997, 48, 379–384 CrossRef CAS.
- C. Würth, M. Grabolle, J. Pauli, M. Spieles and U. Resch-Genger, Nat. Protoc., 2013, 8, 1535–1550 CrossRef PubMed.
- E. W. Snedden, L. A. Cury, K. N. Bourdakos and A. P. Monkman, Chem. Phys. Lett., 2010, 490, 76–79 CrossRef CAS.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS.
- A. P. Monkman, H. D. Burrows, M. da, G. Miguel, I. Hamblett and S. Navaratnam, Chem. Phys. Lett., 1999, 307, 303–309 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and mass spectra, polymerisation data, X-ray crystallography data summary and supplementary photophysical data. CCDC 1992328. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ma00684c |
| This journal is © The Royal Society of Chemistry 2021 |