
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient energy storage in mustard husk derived porous spherical carbon nanostructures†
Atin
Pramanik‡
 *a,
Shreyasi
Chattopadhyay‡
a,
Goutam
De
*ab and
Sourindra
Mahanty
*a
*a,
Shreyasi
Chattopadhyay‡
a,
Goutam
De
*ab and
Sourindra
Mahanty
*a
aCSIR-Central Glass & Ceramic Research Institute, 196 Raja S. C. Mullick Road, Kolkata 700032, India. E-mail: mahanty@cgcri.res.in; ap294@st-andrews.ac.uk; g.de@bose.res.in
bS. N. Bose National Centre for Basic Sciences, Kolkata, India
First published on 13th September 2021
Abstract
An environment-friendly synthesis of highly porous spherical carbon nanostructures (PSCNs), in situ doped with N and S, from mustard seed waste has been accomplished. The synthesised PSCNs have an interconnected network, abundant active interfaces, heteroatom rich content, and notably high porosity/surface area which are favourable for fast ion transport and efficient charge storage. This active material (PSCN), when employed as a lithium-ion battery (LIB) half-cell anode, shows a specific charge capacity of 714 mA h g−1 at a current density of 100 mA g−1 even after 550 cycles with 112% capacity retention and high restoration capability. Furthermore, PSCN//LiFePO4 full cell LIBs show an excellent performance with a highly reversible capacity of ∼195 mA h g−1 at a current density of 50 mA g−1 for 400 cycles. The PSCN electrode also exhibited a specific capacitance of 257.8 F g−1 at a current density of 0.1 A g−1 with ∼93% capacity retention after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles, when used as an electrochemical supercapacitor in aqueous 3 M KOH electrolyte. This work shows the preparation of high value and advanced carbon nanostructured material from renewable bio-mass waste for high-performance electrochemical energy storage applications.
000 cycles, when used as an electrochemical supercapacitor in aqueous 3 M KOH electrolyte. This work shows the preparation of high value and advanced carbon nanostructured material from renewable bio-mass waste for high-performance electrochemical energy storage applications.
Introduction
Presently, Li-ion batteries and electrochemical supercapacitors are the two front-running technologies for energy storage solutions. However, increasing energy demand and rising environmental concerns have created a strong research impetus for developing eco-friendly, cost-effective, lightweight, and sustainable electrode materials.1 Carbon, in different forms, continues to be the dominant electrode material for both of these applications.2 Particularly, recent research has shown that morphology tailored hierarchically porous nanostructures can yield considerably enhanced electrochemical properties.1,3,4 This is so because of the existence of abundant macropores that facilitate mass transport by improving the wettability of the electrode/electrolyte interfaces thereby shortening the diffusion pathways for the charged species and the presence of micro-and/or mesopores that provides the high surface area desired for increased reaction sites.5,6 In this vein, porous carbon derived from biomass appears to be an attractive prospect for achieving desirable molecular structures and architectures.7–14 Not only will it provide a green route with a low carbon footprint, but it will also provide energy sustainability due to the abundance, low cost, and easy processing of the naturally occurring biomass. Realizing this, a number of research studies have been devoted to extracting porous carbon from various biomasses and studying their electrochemical properties as LIB anodes e.g., wheat straw,15 rice husk,16 coconut oil,9 avocado seed waste,17 starch,7 green tea leaves,18 coffee oil,19 sweet potato,8 banana peel20etc., and also as supercapacitor electrodes e.g., apple waste,21 tannin,22 petals,23 sugar cane bagasse,24,25 sewage sludge,26Moringa oleifera leaves,27 sunflower stalks,28 puffed rice,29 popcorn,30 coconut shells,31Moringa oleifera stems,32 Camellia petals,33etc. Some excellent recent review articles on biomass derived carbon for various energy storage applications validate the importance of this approach.34–37 From the morphological point of view, synthesis of spherical porous carbon has always proven to be difficult without using hard templates or structure directing agents. Kim et al. prepared solid spheroidal carbon by a pyrolysis technique with a large average diameter of ∼3 μm and a BET surface area of 5.05 m2 g−1.19 Pol et al. synthesized carbon microspheres (average diameter ∼2 μm) through decomposition of mesitylene (C9H12).38 Chen et al. used polyacrylonitrile nanospheres as a template to synthesise carbon nanoparticles.39 To date, there is no report available on the synthesis of spherical porous carbon hydrothermally using a template free technique, to the best of our knowledge. In the present work, we have demonstrated a facile hydrothermal synthesis, followed by an activation process for the preparation of porous spherical carbon nanostructures in situ doped with N and S from black mustard seed husks and investigated their applicability as Li-ion battery anodes and also, as supercapacitor electrodes.Experimental
Chemicals
Mustard seed husks were purchased from a local oil mill, in India. Sulfuric acid, H2SO4 (98%, Merck, Germany), isopropanol (99%, Merck, Germany), hydrochloric acid, HCl (38%, Merck, Germany), and potassium hydroxide, KOH (Merck, Germany) were used as received. For all the experiments, water purified by a 3-stage Millipore Mill-Q system (Merck, Germany) was used.Material synthesis
Before use, the mustard seed husks were pulverized to small particles by hand grinding and washed several times with a dilute H2SO4 solution. Then, after further washing with water and isopropanol solution, they were dried fully. 4.0 g of dried mustard seed husk was put into a sulfuric acid and water solution (4:96 by volume) and loaded into a stainless-steel autoclave (Teflon-lined). The autoclave with the reaction mixture was heated at 180 °C for 24 h in an oven. Thereafter, the reaction vessel was cooled naturally and the resulting black product was washed repeatedly with deionized water and isopropanol for removal of the soluble impurities. Finally, it was dried by keeping in a vacuum oven at 120 °C for 12 h.
Preparation of activated spherical porous carbon nanostructures (PSCNs)
The carbon precursor was properly mixed with KOH (1:4 weight ratio) by a hand grinding method for 1 h. The resulting KOH-impregnated carbon precursor was firstly dried at 90 °C for 12 h in an oven and then annealed under an argon atmosphere for 2 h at 800 °C in a tubular furnace at a ramp rate of 5 °C min−1 for activation. After the activation process, the sample was thoroughly rinsed with 5% HCl solution to remove the unreacted KOH completely along with other inorganic impurities. Finally, water and isopropanol were used to clean the product thoroughly. After drying in an oven at 110 °C for 12 h, the desired activated carbon nanostructure (PSCN) was obtained. A schematic representation of the synthesis steps is shown in Scheme 1.
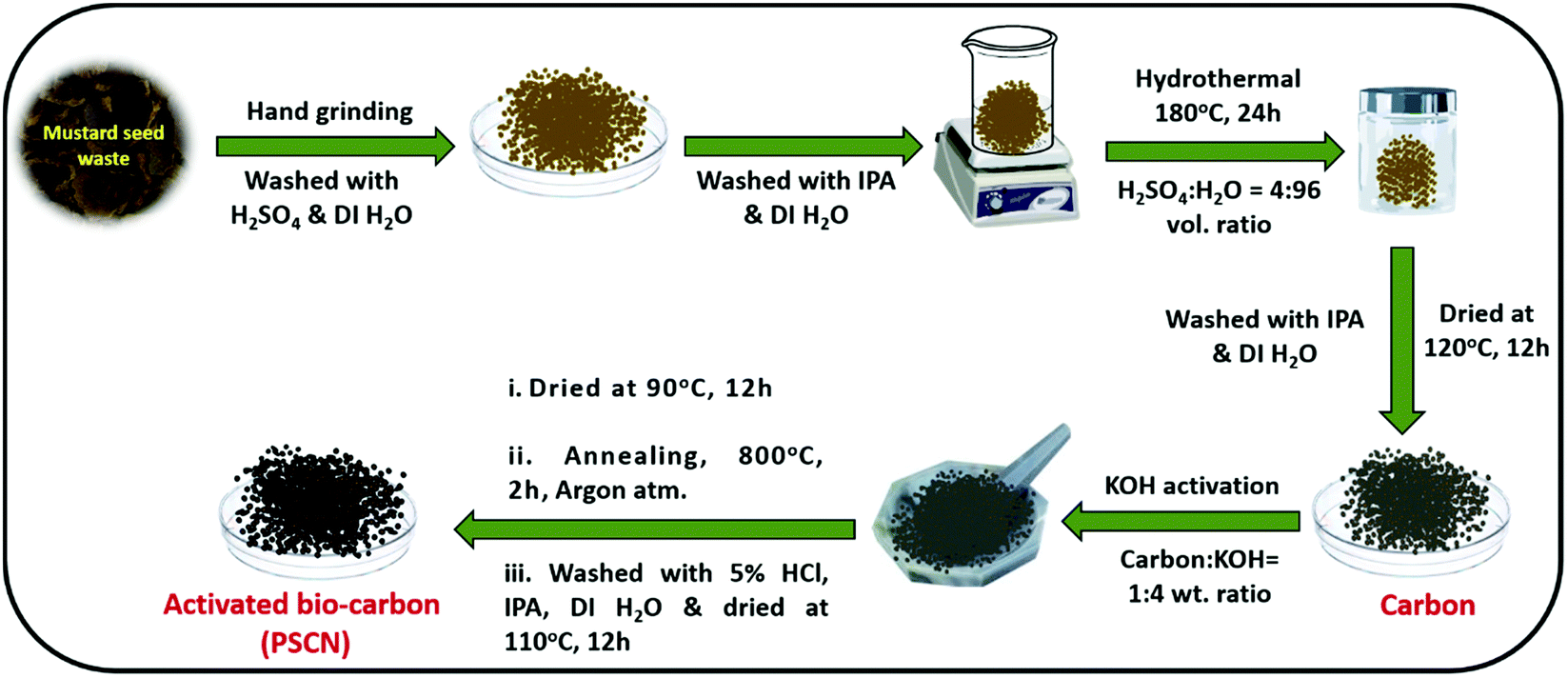 | ||
| Scheme 1 Schematic representation for the synthesis of activated N,S-doped spherical porous carbon nanostructures (PSCN). | ||
Material characterisation
X-Ray diffractograms were recorded by an X-ray diffractometer (Philips X’Pert, the Netherlands) in the 2θ range of 10–80° at a scan rate of 1° min−1. Cu-Kα radiation (λ = 1.5406 Å) was used at 40 kV and 40 mA. Fourier transformed infrared (FTIR) studies were carried out in the wavenumber region of 4000–500 cm−1 by a BOMEN infrared spectrophotometer in transmission mode. A Renishaw In Via Reflex micro Raman spectrometer, with argon ion laser excitation (514 nm), was used to obtain the Raman spectra. The morphological features were investigated by a ZEISS Supra 35 (Germany) field emission scanning electron microscope (FESEM) and a 300 kV Tecnai G2 30ST (FEI) transmission electron microscope (TEM). A Netzsch Thermal Analyzer (STA449F, Germany) was used for thermogravimetric analysis (TGA) in air at a heating rate of 10 °C min−1. X-ray photoelectron spectroscopic (XPS) studies were performed using a PHI 5000 VersaProbe II spectrophotometer (Physical Electronics Inc., USA) using a monochromatized Al Kα (∼1486.6 eV) X-ray beam of size ∼100 μm. In order to neutralize the static charges generated on the sample surface during XPS measurements, a dual-beam charge neutralization system was operated. Also, prior to data acquisition, Ar+ ion sputtering was done on the sample surface (∼2 mm × 2 mm) at 2 kV for removing any contaminants (estimated etching depth ∼ 30 ± 5 nm).Electrochemical characterisation
:10:10 weight ratio in n-methyl pyrrolidinone solvent. The prepared slurry was coated onto pre-cleaned copper foil and dried at 110 °C for 5 h in a vacuum oven. The dried coated foils were then pressed at 4.0 tons per sq. inch, and electrodes were cut into circular disks (dia = 15 mm). The active mass loading in each electrode was ∼2.0 mg. The coin cells were fabricated in an Ar atmosphere inside a glove box (M'BRAUN, Germany) where the oxygen and moisture levels were controlled at <0.5 ppm each. LIB half cells were assembled using lithium metal circular discs (dia = 15 mm), which act as the counter electrode and also, as the reference electrode. LIB full cells were made using a PSCN electrode as the anode and a LiFePO4 (coated on aluminum foil) electrode as a cathode. In all cases, 1.0 M LiPF6 in EC:DMC (1:2 vol%) was used as the electrolyte and Celgard 2300 as the separator. The LiFePO4 cathode electrodes were fabricated by the same standard slurry casting procedure in which the weight ratio of LiFePO4 powder (MTI corporation, USA), Super-P carbon, and polyvinylidene fluoride (PVDF) binder was 80:10:10. The prepared slurry was coated on aluminium foil (thickness 20 μm) and dried at 110 °C for 5 h in a vacuum oven. After pressing at 4.0 ton per sq. inch, electrodes were cut into a circular disk (dia. = 15 mm). A typical active mass loading was ∼6.3 mg based on the mass balance calculation according to our previous work.6
:10:10 weight ratio and using N-methyl-2-pyrrolidone (NMP) as the solvent. The slurry was coated onto nickel foam (dimension 1.5 cm × 1.5 cm) to form a working electrode. The electrodes were dried at 110 °C for ∼5 h in a vacuum oven for evaporation of the residual solvents. The active mass loading in the electrode was ∼2 mg cm−2. The electrodes were kept dipped in 3 M KOH solution for proper wetting before undertaking the electrochemical tests. In three-electrode cell configurations, Pt mesh (2 cm × 2 cm) and Ag/AgCl (3 M KCl) were used as the counter and the reference electrode respectively. A 3 M aqueous solution of KOH was used as an electrolyte. A galvanostat-potentiostat (PGSTAT 300N, Autolab, the Netherlands) was employed to conduct cyclic voltammetry (CV) and galvanostatic charge–discharge (GCD) measurements. For three-electrode cells, CVs were recorded in the potential window of −1.0 to 0.0 V at various scan rates of 2, 5, 10, 20, 50, and 100 mV s−1. GCD measurements were performed in the same potential window at various current densities of 0.1, 0.2, 0.5, 1, 5, and 10 A g−1. Electrochemical impedance spectra (EIS) were recorded by a galvanostat-potentiostat (PGSTAT302N, Autolab, The Netherlands) within the frequency range of 100 kHz to 0.1 Hz using an AC amplitude of 10 mV.
Results and discussion
The morphology of the mustard seed husk-derived carbon was examined by FESEM and TEM. FESEM image of the as-prepared carbonized material shows agglomerated spherical particles of ∼100–200 nm in diameter (Fig. 1a). On the other hand, the PSCNs upon treatment with KOH activation followed by annealing show more distinct spheres with considerably less agglomeration (Fig. 1b). It is plausible that during the activation process KOH would etch out the ionic impurities which are likely to be accumulated in higher concentrations in the agglomerated interfaces. Simultaneous reaction with the ionic and other organic impurities, and the presence of negative OH− groups on the carbon nanostructures during the activation process at elevated temperature result in dissolution of the interfaces, and interparticle repulsion. After the removal of ionic and decomposed organic matters through washing the final activated material, the PSCNs appear to be less agglomerated with clear boundaries. The porous structure of the PSCNs is not clearly visible in the FESEM micrograph (Fig. 1b). The low resolution TEM image of the PSCNs shows the presence of interconnected spheres (Fig. 1c). The high magnification TEM image (Fig. 1d) reveals a nanoporous structure and presence of thinner spherical multi-layer constituents. This kind of nanostructure is beneficial for achieving a high surface area, as well as easy electrolyte accessibility, and would shorten the diffusion pathway to facilitate ion-transport. The selected area diffraction (SAD) pattern (Fig. 1d, inset) reveals the predominantly amorphous nature of the PSCN carbon nanospheres. | ||
| Fig. 1 FESEM micrographs of PSCN before (a) and after (b) activation. TEM images of PSCN (c and d) at different magnifications (inset of 1d shows the SAED pattern). | ||
FTIR and TGA of the final activated sample (PSCN) were performed and the results are presented in Fig. S1 and S2 (ESI†), respectively. TGA in air shows an initial mass loss (∼2.5%) due to the elimination of adsorbed water, and then the carbonaceous material was decomposed in two steps with a total loss of 90.5% up to the temperature, 800 °C. The corresponding FTIR shows (Fig. S2, ESI†) peaks corresponding to –OH, CH–, and C–C, and weak peaks for other functional groups and peaks originating from residual silica. A thorough XPS analysis of the PSCN was undertaken to understand the functionality present in the PSCN. The surface survey scan (Fig. S3, ESI†) shows the presence of binding energy peaks related to S2p, C1s, N1s and O1s at 164, 284.45, 401 and 532 eV, respectively. Peaks observed at the binding energy values of 103.2 and 154.3 eV are due to Si (Fig. S3, ESI†). It is noteworthy to mention that the presence of S2p and N1s in XPS can be attributed to the in situ incorporation of S and N in the graphitic carbon skeleton of PSCN. It may be noted that the pungent odour of mustard is due to the existence of propenyl isothiocyanate (H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2NCS),40 which is believed to be the source of doping of S and N during its decomposition in an inert atmosphere.
CHCH2NCS),40 which is believed to be the source of doping of S and N during its decomposition in an inert atmosphere.
For a detailed understanding, deconvolutions of high-resolution spectra have further been undertaken and shown in Fig. 2. The deconvoluted C1s spectrum shows (Fig. 2a) sp2 CC and sp3 C–C related peaks at binding energy values of 284.07 and 284.47 eV, respectively.41 Other peaks at 285.05, 285.82, 287.03, 288.58, and 290.07 eV can be assigned to C–N, C–O/C–S, CO, O–CO, and π–π* transitions in sp2 carbons.41,42Fig. 2b shows the fitted N1s spectrum with four peaks at 398.77, 400.30, 401.58, and 402.76 eV corresponding to pyridnic-N, pyrrolic-N, graphitic-N, and oxide forms of nitrogen, respectively.41,43 Notably, the presence of pyridinic-N, pyrrolic-N, and graphitic-N indicates doping of nitrogen in the graphitic carbon skeleton of the PSCN. The S2p spectrum of the PSCN has been resolved in three different binding energy peaks (Fig. 2c) corresponding to thiophene-S at 163.96 (S2p3/2) and 165.27eV (S2p1/2), and oxidized sulfur at 169 eV.43,44 Interestingly, a comparison of the fitted N1s and S2p suggests that nitrogen can accommodate itself in a graphitic framework via different configurations like pyridinic, pyrrolic, and graphitic-N, but sulfur can only exist in a thiophene-like structure after calcining the sample at 800 °C.44 A semiquantitative estimation from XPS shows that the PSCN contain about 1.4 and 1 at% of N and S, respectively. Therefore, thorough XPS analyses confirm successful in situ doping of heteroatoms N and S as well as associated functional groups which might help in achieving enhanced electrochemical properties of PSCNs. It is noteworthy that the heat-treated mustard husk usually contains a significant amount of silica and other cationic species.45 However, besides in situ N and S doping we observed the presence of only a low amount of silica with a maximum value of ∼7 wt% as confirmed by TGA (Fig. S2, ESI†). It is expected that most of the cationic species including a significant amount of silica were dissolved out during the alkali (KOH) activation process and washing with aqueous HCl.
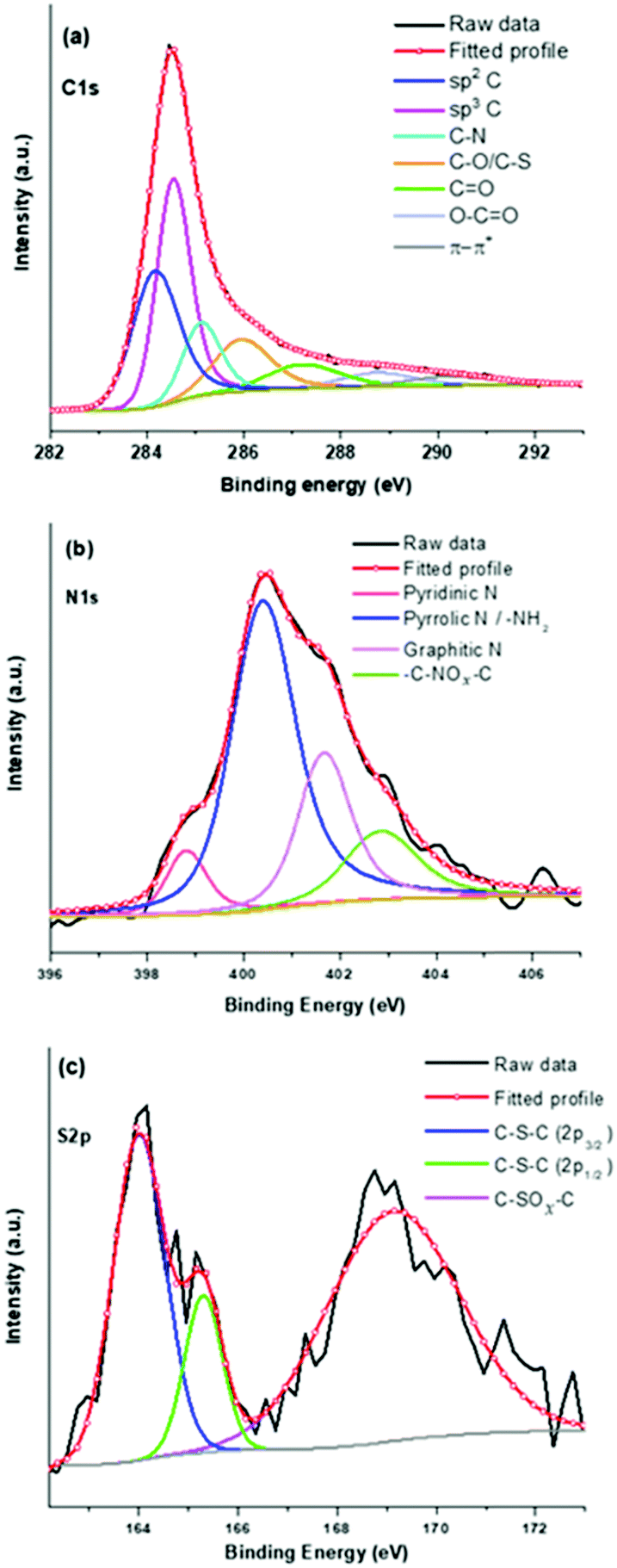 | ||
| Fig. 2 High-resolution deconvoluted XPS spectra of PSCNs: (a) C1s, (b) N1s, and (c) S2p. | ||
In order to verify the phase of the synthesized PSCN derived from mustard seed husks, powder X-ray diffraction (XRD) analyses were carried out. The X-ray diffractogram, shown in Fig. 3a, shows a prominent broad peak at around 2θ = ∼22.8° and a small hump at ∼43.3° which could be identified with the reflections from (002) and (100) lattice planes of carbon (JCPDS file: 00-001-0646).35 No other peaks due to any possible second phase could be observed. The nature of the diffractogram suggests an amorphous nature of PSCN, but with a high degree of graphitization, which is beneficial for the improvement of the electrical conductivity. Being a powerful tool, Raman spectroscopy was employed to further analyze the nature of mustard seed husk-derived activated spherical carbon nanostructures and the presence of structural defects. The Raman spectrum of PSCN (Fig. 3b) shows two strong peaks at ∼1348 and 1589 cm−1 corresponding to the D and G bands of graphitic carbon along with the conventional 2D (G′) band at 2819 cm−1, indicating the graphitic nature of PSCN.46 The intensity ratio of the D and G band (ID/IG), calculated from Fig. 3b, shows a value of ∼0.94, which, along with the broadening of D and G bands indicates the existence of structural defects within the graphitic framework.47,48 For further structural analysis, the D and G bands were deconvoluted using Lorentzian fit which resulted in five peaks (Fig. 3c). In Fig. 3b, two peaks centered at 1348 and 1586 cm−1 can be attributed to the conventional D and G bands originating due to sp3 and sp2 hybridized carbon respectively.47,48 Other peaks at 1204, 1518 and 1615 cm−1 correspond to D*, D** and D′, respectively, which have been commonly found in disordered carbon materials.46,49 The D* peak could originate from the sp3 rich phase of the disordered structure present in amorphous PSCN, and the phonon density of states in small graphitic clusters or C–H vibrations in hydrogenated carbons resulting in the generation of a D** peak.46,49,50 Therefore, the Raman spectral analyses suggest the existence of structural defects which can be attributed to the in situ incorporation of N and S in the graphitic framework, as observed in XPS analysis, as well as to the probable edge defects related to the morphology. The presence of such defects could have a significant influence on the electrochemical performance of PSCN.
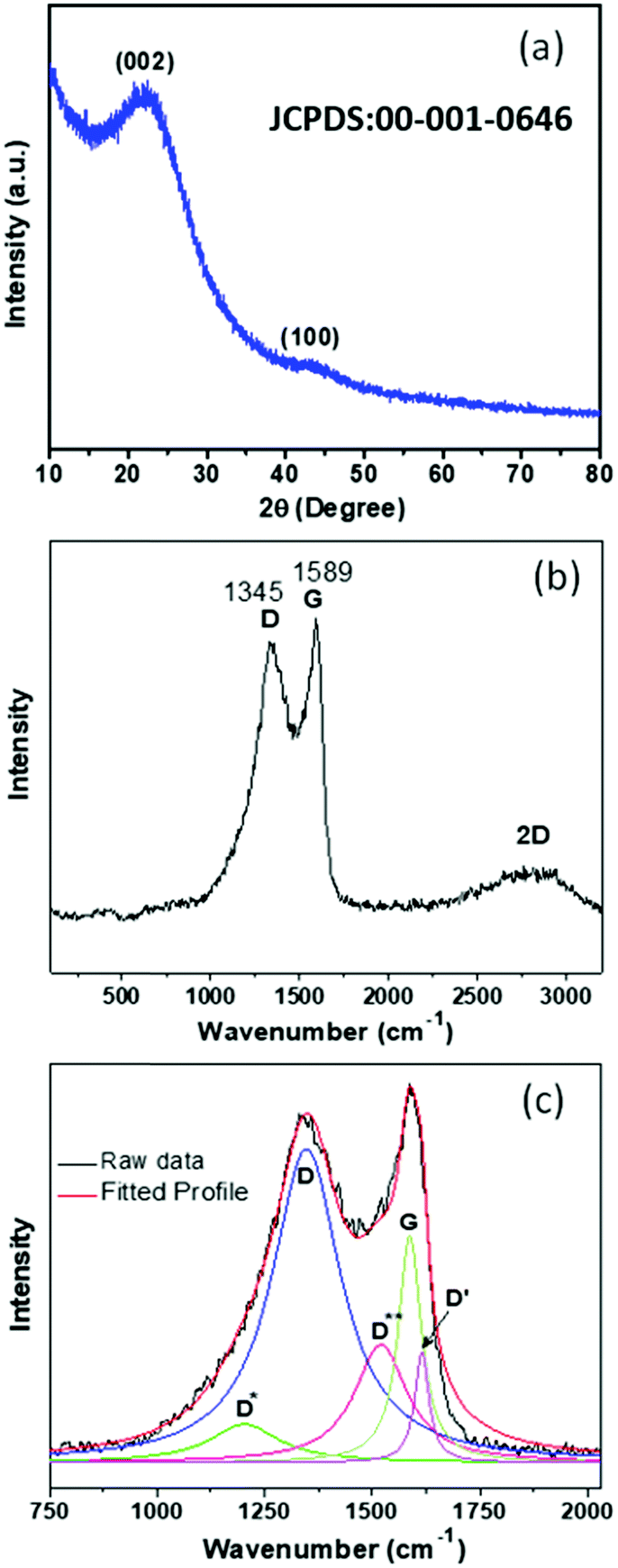 | ||
| Fig. 3 (a) X-ray diffractogram and (b) Raman spectrum of PSCNs. (c) Deconvoluted multiple peaks fit for D and G bands. | ||
Nitrogen adsorption–desorption isotherms, measured at 77 K, are shown in Fig. 4. The PSCN material exhibits a type-I isotherm with a sharp adsorption knee at high relative pressure. The specific surface area is calculated to be 618 m2 g−1, based on the standard BET method. The BJH pore size distribution (inset in Fig. 4) was obtained from the adsorption branch of the isotherm and the average pore size was calculated to be ∼3 nm with a pore volume of 0.98 cc g−1 which implies existence of nanopores on the carbon spheres. As a large specific surface area with nanopores is known to be favorable for high-rate electrochemical processes, we believe that the PSCNs might offer a high energy storage property.
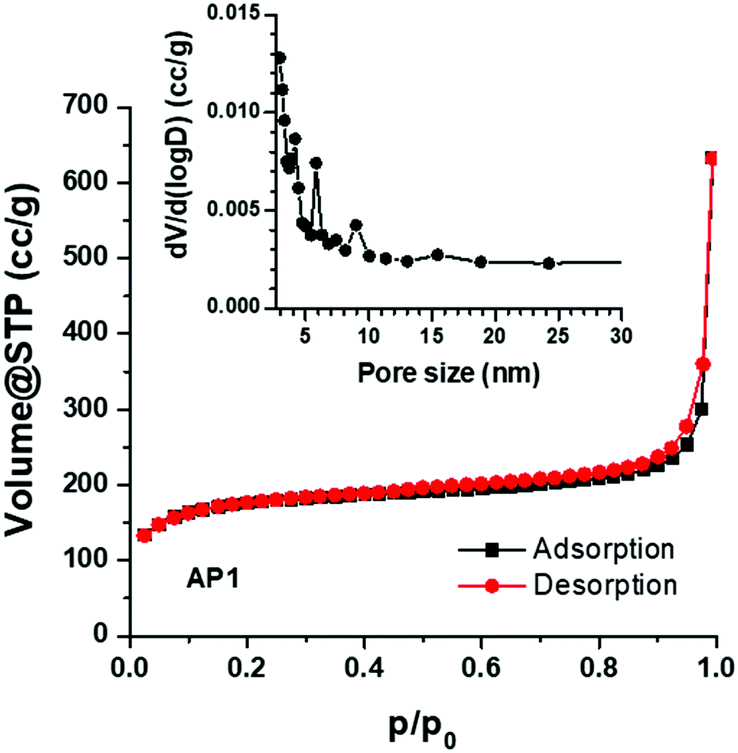 | ||
| Fig. 4 Absorption-desorption isotherms of the synthesized PSCNs. The inset shows the pore size distribution. | ||
Electrochemical performance as a half-cell lithium-ion battery (LIB) anode
The electrochemical performance of PSCN has been examined firstly against Li/Li+ in a half-cell configuration. Fig. 5a shows the CV profiles for the initial four consecutive cycles at a scan rate of 0.1 mV s−1 within a potential window of 0.005–3.0 V. In the 1st scan, the intense cathodic peak was observed at 0.68 V, the irreversible nature of which indicates lithium intercalation into carbon and formation of a solid electrolyte interface (SEI) layer.51 Another peak was observed at 1.30 V, which could be due to the partial decomposition of electrolyte. Another redox couple, observed at 1.64/2.35 V, is possibly due to the presence of N and S in the PSCN and can be assigned to the following electrochemical reactions: CNx + yLi+ + ye− ⇌ LiyCNx and CSx + yLi+ + ye− ⇌ LiyCSx.52,53 However, from the second cycle onward, the cathodic peak at 2.35 V does not appear, and the corresponding anodic peak at 1.64 V also becomes smaller, indicating it to be an irreversible process. No further changes are apparent in the CV profiles after the 2nd cycle. A pair of peaks at 1.90/1.65 V was observed in the CV and is due to the in-situ doping of N and S atoms in the PSCN material. However, the CV curves exhibit a somewhat quasi-rectangular shape, which is indicative of reversible adsorption of non-faradaic nature.53 Thus, faradaic adsorption/desorption of Li+ ions onto the disorder sites on the surface of carbon is indicated by the observed distortion of rectangular shape.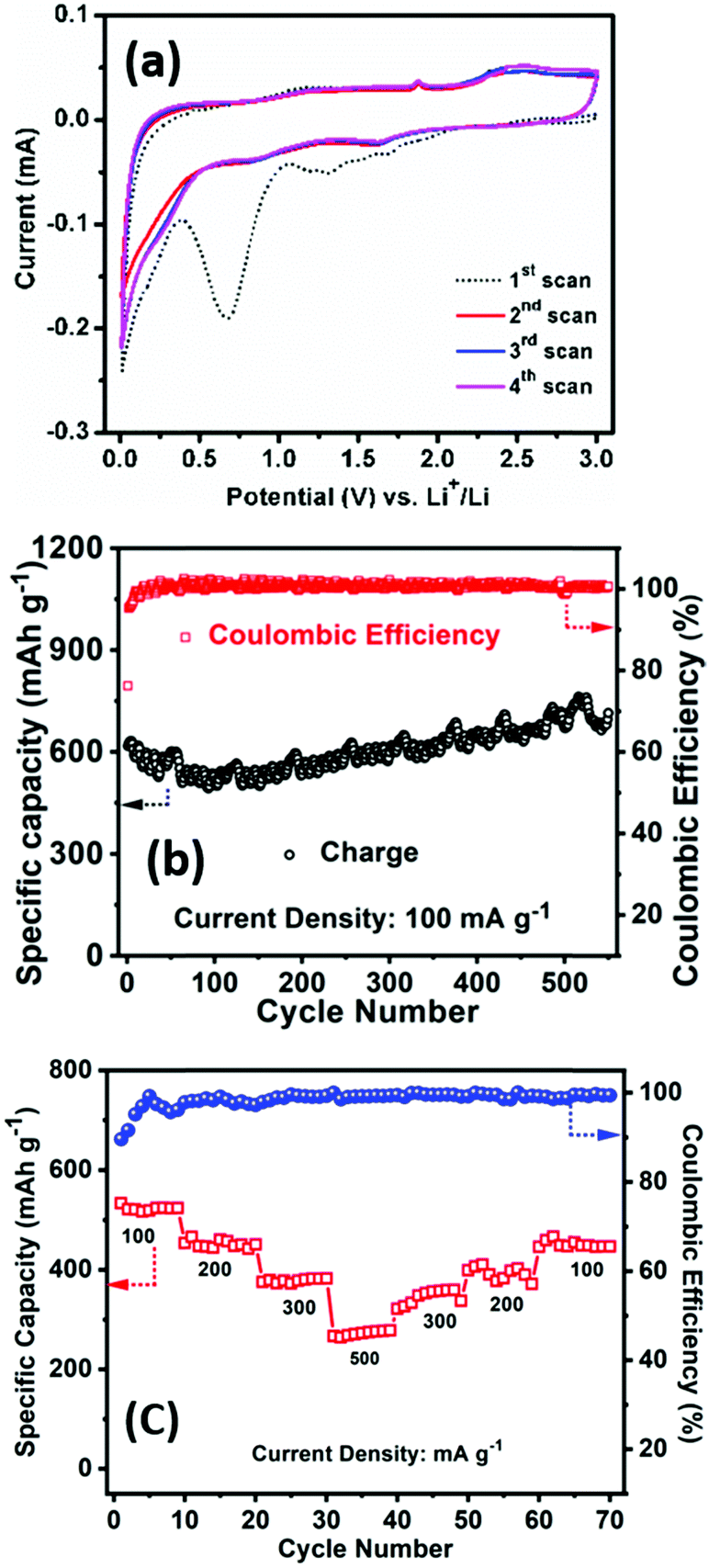 | ||
| Fig. 5 Electrochemical half-cell LIB anode performance of the PSCNs. (a) Cyclic voltammetry of the PSCN electrode for the first four cycles at 0.1 mV s−1 at 0.01–3.0 V, (b) cycling performance at 100 mA g−1 current density, and (c) rate performance at different current densities (100–500 mA g−1). | ||
The galvanostatic charge/discharge experiments have been conducted at different current densities to measure the specific capacity of the electrodes. The first cycle charge/discharge has been done at 100 mA g−1 current density and a high specific capacity of ∼617 mA h g−1 is obtained with ∼75% of coulombic efficiency. It might be the extent of irreversible trapping of lithium inside the bulk of the carbon that would adversely affect the first cycle coulombic efficiency through formation of SEI on the surface of the electrode, which is quite common for anode materials. Occurrence of capacity fading for the initial few cycles is also quite common for carbon-based materials. The cyclability test has been carried out at a higher current density of 100 mA g−1 for 550 cycles. The charge capacity was found to be ∼714 mA h g−1 after 550 continuous cycles with ∼100% coulombic efficiency, signifying ∼112% capacity retention with respect to the value at the first cycle (Fig. 5b). These results are superior to the values previously reported for other bio-material derived carbons (Table 1).7–9,15–20,54–56 The high surface area of the PSCNs and the presence of sulfur and nitrogen in the PSCNs could be factors for their excellent performance as LIB anodes. However, it is observed from Fig. 5b that initially there is a slightly decreasing nature up to 50 cycles but thereafter there is a gradual increase in capacity. The phenomenon of capacity increase during the continuous cycling process is well known and frequently reported in the literature for various anode materials,4,57 and explained to be due to the kinetically activated reversible electrolyte degradation57 leading to extra lithium ion insertion/extraction during the cycling processes.
| Material source and synthesis | Morphology | BET surface area (m2 g−1) | Potential region (V) | Reversible capacity (mA h g−1)/current density (mA g−1) | Cycle number/retention % | Ref. |
|---|---|---|---|---|---|---|
| Wheat straw | Fibrous structure | 24.9 | 0.0–3.0 | 344/18500 | 300/∼86 | 15 |
| Rice husk | Porous block | 52 | 0.0–3.0 | 396/74.4 | 100/∼100 | 16 |
| — | 1200 | 0.0–3.0 | 1055/— | —/— | 54 | |
| Carbon nanoparticles from coconut oil/incineration, piranha treatment | Quasi-spherical morphology | 133 | 0.0–3.0 | ∼250/100 | 90/∼32 | 9 |
| Avocado seed waste derived carbon/pyrolysis | Agglomerated particle | — | 0.0–2.0 | 315/100 | 100/∼99.9 | 17 |
| Starch derived carbon/pyrolysis | Ultrathin carbon sheets | 970 | 0.001–3.0 | ∼220/5C | 400/∼95 | 7 |
| Green tea leave/pyrolysis | Agglomerated particle | 146.3 | 0.005–3.0 | 471/37.2 | 50/∼89 | 18 |
| Coffee oil derived carbon/dry autoclaving | Spherical and dumbbell-like carbon | 5.05 | 0.01–2.0 | 274/100 | 250/97 | 19 |
| Sweet potato-derived carbon/pyrolysis | Nanoparticle | 79.1 | 0.01–3.0 | 170/100 | 50/— | 8 |
| Banana peel derived carbon/pyrolysis | Agglomerated structure | 217 | 0.0–3.0 | 600/500 | 600/— | 20 |
| Hair/pyrolysis | Carbon Microtubes | 306.9 | 0.005–3.0 | ∼658/∼200 | 1000/∼65 | 55 |
| Soybean/pyrolysis | Porous network | 1089.8 | 0.01–2.8 | 360/50 | 500/— | 56 |
| Mustard seed husk derived carbon (PSCN) | Spherical porous carbon | 618 | 0.01–3.0 | 714/100 | 550/112 | This work |
The rate capability at different current density and the capacity restoration ability of PSCN electrodes is shown in Fig. 5c. The cell has been tested with 100 mA g−1 to 500 mA g−1 of current density with ten-cycle intervals for each rate. It is noted that at 100 mA g−1 of current density the specific charge capacity is 545 mA h g−1, but when the current density is doubled to 200 mA g−1, still the capacity is quite high with a value of 466 mA h g−1i.e., 85.5% of its initial capacity. Furthermore, when the current density is increased to five times its initial rate (500 mA g−1), 51.3% of its initial capacity is still obtained (280 mA h g−1). After that, the cell has been subjected back to its initial current rate (100 mA g−1) and a capacity of 474 mA h g−1 is observed indicating restoration of ∼87% of its initial capacity.
Electrochemical performance as a full-cell lithium-ion battery (LIB)
Inspired by the half-cell LIB performance and to demonstrate the potential practical applications, we further evaluated the full-cell performance of the PSCN anode by coupling it with a commercial LiFePO4 cathode. Fig. 6a shows the cyclic voltammograms of the LiFePO4//PSCN full cell for the first four consecutive scans recorded at 2 mV s−1 in the potential window of 1.0–3.6 V. It is observed that from the 2nd cycle onward, the voltammograms nearly trace each other indicating that the charge/discharge processes occurring in the LiFePO4//PSCN full cell are of excellent reversibility. The average voltage profiles are at ∼3 V vs. Li/Li+, with high reversibility. Fig. 6b shows the cycling performances of the LiFePO4//PSCN full cell at 50 mA g−1 current density for 400 cycles. The first cycle-specific charge capacity is ∼195 mA h g−1 with ∼87% coulombic efficiency. The specific charge capacity has been calculated based on the PSCN electrode. From the second cycle itself and onwards, the coulombic efficiency increases to ∼91%. The cell shows a capacity decay during the initial cycles but after ∼100 cycles, the capacity starts increasing again. Observation of such initial decay in capacity suggests that further optimization, including mass balance for the full cell assembly, might be beneficial. It could also be due to the fact that initially the SPCN electrode was not fully activated. But, when the electrode gets activated gradually during repeated cycling, the capacity increases and shows a stable extraordinary cycling performance for continuous 400 cycles with the same current rate. It shows a charge capacity of ∼246 mA h g−1 after 400 cycles, indicating excellent cycling stability.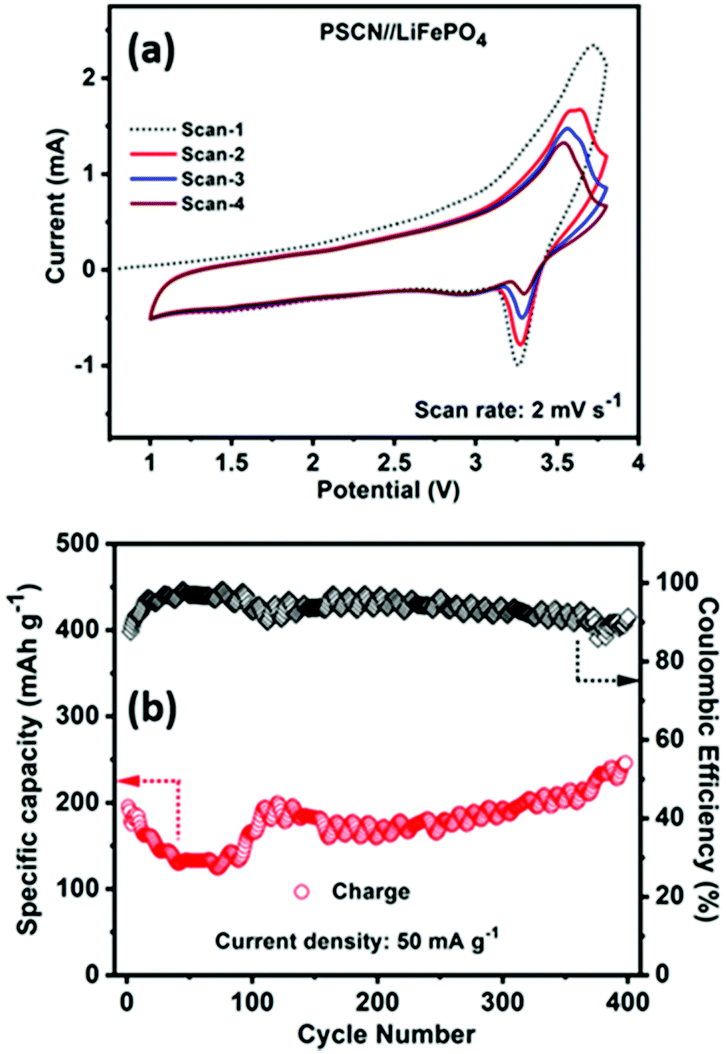 | ||
| Fig. 6 Full cell LIB performance of a PSCN anode coupled with a LiFePO4 cathode. (a) Cyclic voltammetry profiles at a scan rate of 2 mV s−1 at 1.0–3.6 V, and (b) cycling performance and the corresponding coulombic efficiencies at 50 mA g−1 of current density. | ||
Electrochemical supercapacitor performance in a three-electrode configuration
For evaluating the electrochemical capacitive performances of PSCN, conducting cyclic voltammetry (CV), galvanostatic charge/discharge (GCD), and electrochemical impedance spectroscopy (ESI†) for a three-electrode system in 3 M KOH aqueous solution were carried out (Fig. 7). The CV curves were measured within a potential window of 0.0 to −1.0 V at various scanning rates of 2–100 mV s−1. The CV curves of the PSCN electrode obtained at 2 mV s−1 (Fig. 7a) unveiled a nearly perfect rectangular shape with the combination of a very small pseudocapacitive nature, due to the N-doping in the sample,58 which is typically observed for ideal electric double layer capacitors. However, a small hump suggests minor co-occurrence of the faradaic reactions. Interestingly, the quasi-rectangular shape of the CV curves was maintained for all scan rates from 5 to 100 mV s−1 without showing any appreciable distortion even at high scan rates. These results demonstrate the excellent rate capability of the PSCNs, and imply it to be a perfectly reversible double layer capacitor. The GCD profiles of the PSCN electrode was measured at 0.1–10 A g−1 current densities within the potential window of 0.0 to −1.0 V (Fig. 7b). The symmetric triangular-shaped GCD curves show the reversible charge/discharge performance and excellent EDLC behaviour, concurring with the CV features.58 Furthermore, there has been a very low voltage drop (IR drop) between charge and discharge even at high current densities implying outstanding reversibility and rate performance. The specific capacitance of the electrode can be evaluated from GCD curves using the following equation:32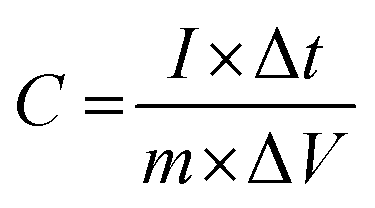
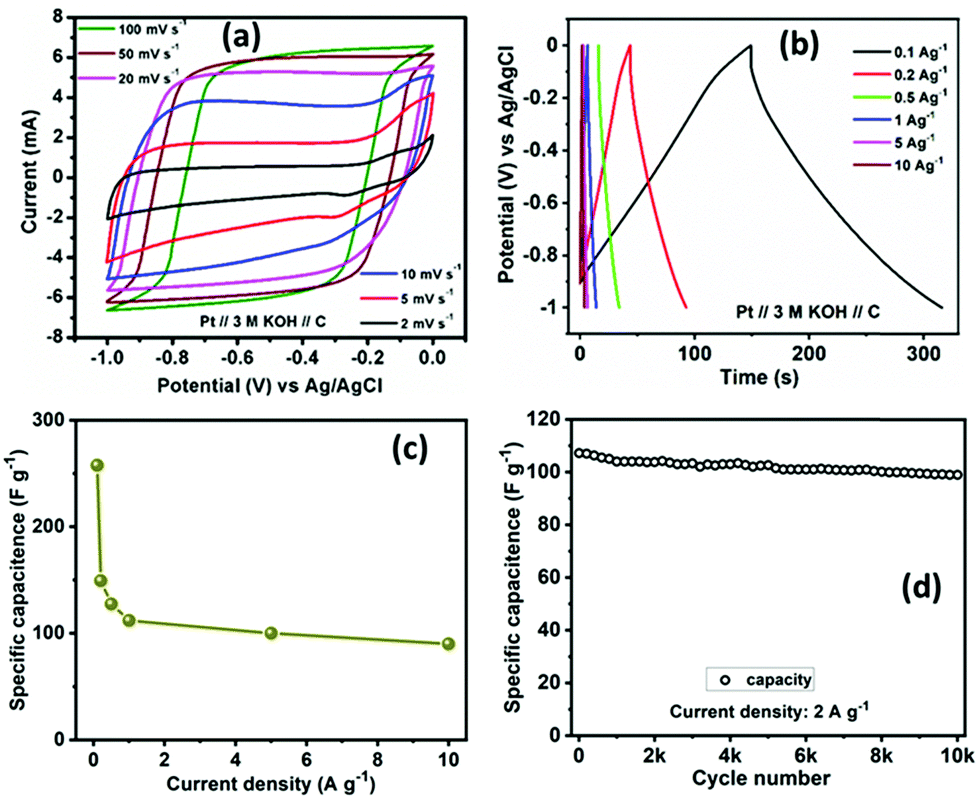 | ||
| Fig. 7 Electrochemical supercapacitor performances of the PSCN electrode in a three-electrode configuration, (a) cyclic voltammetry at various scan rates at 0.0 to −1.0 V, (b) GCD profile at 0.1 to 10 A g−1 current density, (c) variation of specific capacitance vs. current density, and (d) cycling stability test at 2 A g−1 current density for 10000 cycles. | ||
The specific capacitance of the PSCN electrode has been calculated to be 257.8 F g−1 at 0.1 A g−1 current density (Fig. 7c). As the current density is increased, the specific capacitance decreases and values of 149.3, 127.5, 112, 100, and 90 F g−1 at 0.2, 0.5, 1, 5, and 10 A g−1 respectively have been obtained (Fig. 7c). In other words, ∼35% of capacitance retention is observed when the current density is increased to 100 times its initial rate, implying an extraordinary rate capability of the PSCN. Long-term cycling stability is one of the important criteria for a supercapacitor electrode. The cycle stability test of the SPC electrode was done by continuous 10000 GCD measurements at 2 A g−1 current density (Fig. 7d). The electrode reveals ∼93% retention of its initial capacitance after 10000 cycles exhibiting a remarkable cycling stability. A comparison of electrochemical performance of PSCN with other carbon materials derived from various bio-sources is given in Table 2.21–27,29–33,59–63
| Material source and synthesis | Morphology | BET surface area (m2 g−1) | Electrolyte | Capacity (F g−1)/current density (A g−1) | Cycle number/retention % | Ref. |
|---|---|---|---|---|---|---|
| Apple waste/hydrothermal | — | 2000 | 2 M H2SO4 | 264/∼0.001 | — | 21 |
| Biomass-derived/pyrolysis | Agglomerated structure | 3120 | 6 M KOH | 315/0.1 | 10000/95.8 |
59 |
| Tannin-derived carbons/pyrolysis | — | 2000 | 1 M H2SO4 | 37/0.2 | 10000/∼55 |
22 |
| Carbon membranes derived from petals/pyrolysis | Wrinkled flakes | 509 | KCl | 154/10 mV s−1 | 10000/∼90 |
23 |
| Sugar cane bagasse with urea/pyrolysis | 3D Agglomerated flakes | 623.54 | 6 M KOH | 323/1 | 5000/∼100 | 24 |
| Sewage sludge derived carbon (N-doped)/hydrothermal | Agglomerated particle | 1043 | KOH | 286.68/1 | 10000/97.55 |
26 |
| Moringa oleifera leaves/pyrolysis | Crumpled porous structure | 817 | 6 M KOH | ∼49/1 | 20000/∼100 |
27 |
| Waste bagasse/hydrothermal | 3D-hierarchical honeycomb-like texture | 3151 | 6 M KOH | 413/1 | 10000/93.4 |
25 |
| Sunflower stalk derived carbon/pyrolysis | Spherical or ellipsoidal particle agglomerates | 1505 | 6 M KOH | 365/0.1 | 15000/95 |
60 |
| Cotton stalk/pyrolysis | Pore structure | 1960 | 1 M H2SO4 | 254/0.2 | 10000/∼100 |
61 |
| Wood/reflux | Carbon fibers | 3220 | 1 M H2SO4 | 280/0.5 | 2000/99.3 | 62 |
| Sunflower stalk derived carbon/pyrolysis | Spherical or ellipsoidal particle agglomerates | 1505 | 6 M KOH | 365/0.1 | 15000/95 |
63 |
| Carbon nanosheets derived from puffed rice/pyrolysis | Nanosheets | 3326 | 6 M KOH | 334/0.5 | 11000/97 |
29 |
| Popcorn-derived porous carbon/pyrolysis | Porous flakes | 3301 | 6 M KOH | 348/0.2 | 10000/95 |
30 |
| Coconut shell derived/pyrolysis | Nanosheets | 1874 | 6 M KOH | 268/1 | 5000/— | 31 |
| Moringa oleifera stem carbon/pyrolysis | Porous nanosheets | 2250 | 6 M KOH | 283/0.5 | 20000/∼100 |
32 |
| Camellia petals and ammonium persulfate derived carbon/pyrolysis | Porous nanosheets | 1122 | 6 M KOH | 227/0.5 | 10000/∼100 |
33 |
| Mustard seed husk derived carbon (PSCNs) | Spherical porous carbon | 618 | 3 M KOH | 257.8/0.1 | 10000/∼93 |
This work |
To understand the underlying reasons for such excellent cycling stability, electrochemical impedance spectroscopy was carried out in the as-assembled state and after 10000 cycles (Fig. S4a and b; ESI†). Fig. S4b (ESI†) reveals the appearance of a semicircle in the Nyquist plot in the high frequency region for the as-assembled state indicating the co-occurrence of a faradaic process with the solution resistance (Rs) and charge transfer resistance (Rct) being 0.73 Ω and 26 Ω respectively. But, after 10000 cycles (Fig. S4b, ESI†), the semicircle disappears and the Nyquist plot inclines to become parallel to the Y-axis signifying highly capacitive behaviour. The solution resistance remains low (0.59 Ω) even after 10000 cycles indicating excellent stability of the PSCN electrode material.
Thus, the above experimental results demonstrate that our PSCN material has excellent electrochemical reversibility when employed as an electrode in LIB half-cell, LIB full cell, and supercapacitor applications. The ultrahigh electrochemical activity and remarkable cycling stability with high specific capacity could be attributed to the following probable reasons: (i) interconnected 3D spherical micro/meso/microporous carbon nanoparticles with ordered arrangement ensures sufficient active site accessibility, and faster ion transmission; (ii) high surface area with large volume of macropore network can act as an ion reservoir to store electrolyte ions and advance the accessibility of the inner surfaces of PSCN materials; (iii) the 3D spherical frameworks impart structural stability; (iv) the presence of multidirectional porosity provides efficient pathways for fast ion transport; and (v) in situ N and S doping in the PSCNs enhances the electronic conductivity facilitating charge transport.
Conclusions
In summary, we present here a simple synthesis process to prepare a carbon material (PSCN) from mustard husk waste which is cost-effective, environment-friendly and sustainable. The derived carbon material is in situ N- and S-doped and has a high surface area (618 m2 g−1) with a spherical nanostructure, and is found to be suitable for electrochemical energy storage applications. When used as LIB half-cell anodes, the PSCN shows a reversible capacity of 714 mA h g−1 at 100 mA g−1 after 550 cycles. Furthermore, the full-cell LIB (PSCN//LiFePO4) performance shows a very stable reversible capacity of ∼195 mA h g−1 at 50 mA g−1 current density after 400 cycles. As an electrochemical supercapacitor electrode, the PSCN exhibits enhanced performances of 257.8 F g−1 specific capacitance at 0.1 A g−1 current density with retention of 93% after 10000 cycles. Therefore, the PSCN, derived from abundant mustard husk biomass could find a range of real-world applications in sustainable energy related areas.
Author contributions
Atin Pramanik: Conceptualization, methodology, writing – original draft. Shreyasi Chattopadhyay: methodology. Goutam De: methodology, review & editing. Sourindra Mahanty: conceptualization, writing – review & editing.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
A. P. thanks CSIR India for a senior research fellowship (Award No. 31/15(136)/2017-EMR-I). S. C. thanks UGC, India for a research fellowship (Award No. F.2-44/2011(SA-I)).Notes and references
- N. S. Choi, Z. Chen, S. A. Freunberger, X. Ji, Y. K. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 9994–10024 CrossRef CAS PubMed.
- J. Jiang, G. Nie, P. Nie, Z. Li, Z. Pan, Z. Kou, H. Dou, X. Zhang and J. Wang, Nano-Micro Lett., 2020, 12, 1–30 CrossRef PubMed.
- A. Pramanik, S. Maiti and S. Mahanty, J. Mater. Chem. A, 2014, 2, 18515–18522 RSC.
- A. Pramanik, S. Maiti, M. Sreemany and S. Mahanty, J. Nanoparticle Res., 2016, 18, 93 CrossRef.
- J. Huang, J. Wang, C. Wang, H. Zhang, C. Lu and J. Wang, Chem. Mater., 2015, 27, 2107–2113 CrossRef CAS.
- A. Pramanik, S. Maiti, M. Sreemany and S. Mahanty, ChemistrySelect, 2017, 2, 7854–7864 CrossRef CAS.
- M. Bakierska, M. Lis, J. Pacek, M. Świętosławski, M. Gajewska, A. Tąta, E. Proniewicz and M. Molenda, Carbon Nanotube Fibers Yarns, 2019, 145, 426–432 CAS.
- P. Zheng, T. Liu, J. Zhang, L. Zhang, Y. Liu, J. Huang and S. Guo, RSC Adv., 2015, 5, 40737–40741 RSC.
- R. R. Gaddam, D. Yang, R. Narayan, K. V. S. N. Raju, N. A. Kumar and X. S. Zhao, Nano Energy, 2016, 26, 346–352 CrossRef CAS.
- Y. Yao and F. Wu, Nano Energy, 2015, 17, 91–103 CrossRef CAS.
- M. R. P. E. Irisarri and A. Ponrouch, J. Electrochem., 2015, 162, A2476–A2482 Search PubMed.
- X. Hao, J. Wang, B. Ding, Y. Wang, Z. Chang, H. Dou and X. Zhang, J. Power Sources, 2017, 352, 34–41 CrossRef CAS.
- Y. Xu, K. Wang, J. Han, C. Liu, Y. An, Q. Meng, C. Li, X. Zhang, X. Sun, Y. Zhang, L. Mao, Z. Wei and Y. Ma, Adv. Mater., 2020, 32, 1–10 Search PubMed.
- Y. Yang, Q. Lin, B. Ding, J. Wang, V. Malgras, J. Jiang, Z. Li, S. Chen, H. Dou, S. M. Alshehri, T. Ahamad, J. Na, X. Zhang and Y. Yamauchi, Carbon Nanotube Fibers Yarns, 2020, 167, 627–633 CAS.
- L. Chen, Y. Zhang, C. Lin, W. Yang, Y. Meng, Y. Guo, M. Li and D. Xiao, J. Mater. Chem. A, 2014, 2, 9684–9690 RSC.
- L. Wang, Z. Schnepp and M. M. Titirici, J. Mater. Chem. A, 2013, 1, 5269–5273 RSC.
- T. J. Yokokura, J. R. Rodriguez and V. G. Pol, ACS Omega, 2020, 5, 19715–19720 CrossRef CAS PubMed.
- S. W. Han, D. W. Jung, J. H. Jeong and E. S. Oh, Chem. Eng. J., 2014, 254, 597–604 CrossRef CAS.
- K. Kim, R. A. Adams, P. J. Kim, A. Arora, E. Martinez, J. P. Youngblood and V. G. Pol, Carbon Nanotube Fibers Yarns, 2018, 133, 62–68 CAS.
- E. M. Lotfabad, J. Ding, K. Cui, A. Kohandehghan, W. P. Kalisvaart, M. Hazelton and D. Mitlin, ACS Nano, 2014, 8, 7115–7129 CrossRef CAS PubMed.
- L. Guardia, L. Suárez, N. Querejeta, R. Rodríguez Madrera, B. Suárez and T. A. Centeno, ACS Sustainable Chem. Eng., 2019, 7, 17335–17343 CrossRef CAS.
- J. Castro-Gutiérrez, N. Díez, M. Sevilla, M. T. Izquierdo, J. Ghanbaja, A. Celzard and V. Fierro, ACS Sustainable Chem. Eng., 2019, 7, 17627–17635 CrossRef.
- X. Yu, Y. Wang, L. Li, H. Li and Y. Shang, Sci. Rep., 2017, 7, 45378 CrossRef CAS PubMed.
- J. Liu, Y. Deng, X. Li and L. Wang, ACS Sustainable Chem. Eng., 2016, 4, 177–187 CrossRef CAS.
- P. Yu, Y. Liang, H. Dong, H. Hu, S. Liu, L. Peng, M. Zheng, Y. Xiao and Y. Liu, ACS Sustainable Chem. Eng., 2018, 6, 15325–15332 CrossRef CAS.
- Z. X. Xu, X. Q. Deng, S. Zhang, Y. F. Shen, Y. Q. Shan, Z. M. Zhang, R. Luque, R. Luque, P. G. Duan and X. Hu, Green Chem., 2020, 22, 3885–3895 RSC.
- L. Peng, Y. Cai, Y. Luo, G. Yuan, J. Huang, C. Hu, H. Dong, Y. Xiao, Y. Liang, Y. Liu and M. Zheng, ACS Sustainable Chem. Eng., 2018, 6, 12716–12726 CrossRef CAS.
- M. Chaudhary, A. K. Nayak, R. Muhammad, D. Pradhan and P. Mohanty, ACS Sustainable Chem. Eng., 2018, 6, 5895–5902 CrossRef CAS.
- J. Hou, K. Jiang, M. Tahir, X. Wu, F. Idrees, M. Shen and C. Cao, J. Power Sources, 2017, 371, 148–155 CrossRef CAS.
- J. Hou, K. Jiang, R. Wei, M. Tahir, X. Wu, M. Shen, X. Wang and C. Cao, ACS Appl. Mater. Interfaces, 2017, 9, 30626–30634 CrossRef CAS PubMed.
- L. Sun, C. Tian, M. Li, X. Meng, L. Wang, R. Wang, J. Yin and H. Fu, J. Mater. Chem. A, 2013, 1, 6462–6470 RSC.
- Y. Cai, Y. Luo, H. Dong, X. Zhao, Y. Xiao, Y. Liang, H. Hu, Y. Liu and M. Zheng, J. Power Sources, 2017, 353, 260–269 CrossRef CAS.
- T. Wei, X. Wei, L. Yang, H. Xiao, Y. Gao and H. Li, J. Power Sources, 2016, 331, 373–381 CrossRef CAS.
- B. M. Matsagar, R.-X. Yang, S. Dutta, Y. Sik Ok and K. C.-W. Wu, J. Mater. Chem. A, 2021, 9, 3703–3728 RSC.
- B. Szczęśniak, J. Phuriragpitikhon, J. Choma and M. Jaroniec, J. Mater. Chem. A, 2020, 8, 18464–18491 RSC.
- L. Miao, Z. Song, D. Zhu, L. Li, L. Gan and M. Liu, Mater. Adv., 2020, 1, 945–966 RSC.
- Q. Chen, X. Tan, Y. Liu, S. Liu, M. Li, Y. Gu, P. Zhang, S. Ye, Z. Yang and Y. Yang, J. Mater. Chem. A, 2020, 8, 5773–5811 RSC.
- V. G. Pol, E. Lee, D. Zhou, F. Dogan, J. M. Calderon-Moreno and C. S. Johnson, Electrochim. Acta, 2014, 127, 61–67 CrossRef CAS.
- A. Chen, K. Xia, L. Zhang, Y. Yu, Y. Li, H. Sun, Y. Wang, Y. Li and S. Li, Langmuir, 2016, 32, 8934–8941 CrossRef CAS PubMed.
- S. V. Eaton, Bot. Gaz., 1942, 104, 82–89 CrossRef CAS.
- J. Xu, M. Wang, N. P. Wickramaratne, M. Jaroniec, S. Dou and L. Dai, Adv. Mater., 2015, 27, 2042–2048 CrossRef CAS PubMed.
- Z. Tian, J. Li, G. Zhu, J. Lu, Y. Wang, Z. Shi and C. Xu, Phys. Chem. Chem. Phys., 2015, 18, 1125–1130 RSC.
- S. N. Bhange, S. M. Unni and S. Kurungot, J. Mater. Chem. A, 2016, 4, 6014–6020 RSC.
- S. Yang, L. Zhi, K. Tang, X. Feng, J. Maier and K. Müllen, Adv. Funct. Mater., 2012, 22, 3634–3640 CrossRef CAS.
- T. Kamal and M. A. Siddiqui, Mater. Res. Express, 2018, 5, 86509 CrossRef.
- A. Kaniyoor and S. Ramaprabhu, AIP Adv., 2012, 2, 32183 CrossRef.
- C. He, S. Wu, N. Zhao, C. Shi, E. Liu and J. Li, ACS Nano, 2013, 7, 4459–4469 CrossRef CAS PubMed.
- E. Kang, Y. S. Jung, A. S. Cavanagh, G. H. Kim, S. M. George, A. C. Dillon, J. K. Kim and J. Lee, Adv. Funct. Mater., 2011, 21, 2430–2438 CrossRef CAS.
- J. Schwan, S. Ulrich, V. Batori, H. Ehrhardt and S. R. P. Silva, J. Appl. Phys., 1996, 80, 440–447 CrossRef CAS.
- C. W. Lee, S. B. Yoon, H. K. Kim, H. C. Youn, J. Han, K. C. Roh and K. B. Kim, J. Mater. Chem. A, 2015, 3, 2314–2322 RSC.
- Z. Li, Z. Xu, X. Tan, H. Wang, C. M. B. Holt, T. Stephenson, B. C. Olsen and D. Mitlin, Energy Environ. Sci., 2013, 6, 871–878 RSC.
- Z. W. Seh, Y. Sun, Q. Zhang and Y. Cui, Chem. Soc. Rev., 2016, 45, 5605–5634 RSC.
- B. Shan, Y. Cui, W. Liu, Y. Zhang, S. Liu, H. Wang, L. Sun, Z. Wang and R. Wu, ACS Sustainable Chem. Eng., 2018, 6, 14989–15000 CrossRef CAS.
- G. T. K. Fey and C. L. Chen, J. Power Sources, 2001, 97–98, 47–51 CrossRef CAS.
- J. Zhu, S. Liu, Y. Liu, T. Meng, L. Ma, H. Zhang, M. Kuang and J. Jiang, ACS Sustainable Chem. Eng., 2018, 6, 13662–13669 CrossRef CAS.
- D. Zhang, G. Wang, L. Xu, J. Lian, J. Bao, Y. Zhao, J. Qiu and H. Li, Appl. Surf. Sci., 2018, 451, 298–305 CrossRef CAS.
- G. Zhou, D. Wang, F. Li, L. Zhang, N. Li, Z. Wu, L. Wen, G. Q. Lu and H. Cheng, Chem. Mater., 2010, 22, 5306–5313 CrossRef CAS.
- S. Feng, Z. Liu, Q. Yu, Z. Zhuang, Q. Chen, S. Fu, L. Zhou and L. Mai, ACS Appl. Mater. Interfaces, 2019, 11, 4011–4016 CrossRef CAS PubMed.
- S. Dong, X. He, H. Zhang, X. Xie, M. Yu, C. Yu, N. Xiao and J. Qiu, J. Mater. Chem. A, 2018, 6, 15954–15960 RSC.
- J. He, D. Zhang, Y. Wang, J. Zhang, B. Yang, H. Shi, K. Wang and Y. Wang, Appl. Surf. Sci., 2020, 515, 146020 CrossRef CAS.
- X. Tian, H. Ma, Z. Li, S. Yan, L. Ma, F. Yu, G. Wang, X. Guo, Y. Ma and C. Wong, J. Power Sources, 2017, 359, 88–96 CrossRef CAS.
- Z. Jin, X. Yan, Y. Yu and G. Zhao, J. Mater. Chem. A, 2014, 2, 11706–11715 RSC.
- X. Wang, S. Yun, W. Fang, C. Zhang, X. Liang, Z. Lei and Z. Liu, ACS Sustainable Chem. Eng., 2018, 6, 11397–11407 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: FTIR spectrum, TGA, XPS survey scan and Nyquist plots of PSCN in a three-electrode configuration. See DOI: 10.1039/d1ma00679g |
| ‡ Present address: School of Chemistry, University of St Andrews, KY16 9ST, Scotland, UK. |
| This journal is © The Royal Society of Chemistry 2021 |