
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Variation in solvato-, AIE- and mechano-fluorochromic behavior for furanyl and thiophenyl-substituted anthranyl π-conjugates: the role of tiny flanking donor groups†
Madhuparna
Chakraborty
and
Manab
Chakravarty
 *
*
Department of Chemistry, BITS-Pilani Hyderabad Campus, Jawahar Nagar, Shameerpet, Hyderabad-500078, Telangana, India. E-mail: manab@hyderabad.bits-pilani.ac.in
First published on 30th August 2021
Abstract
Only a few reports exist on thiophene or furan-linked typical tetraphenylethene or difluoroboron systems known as aggregation-induced emission-active fluorogens (AIEgens) with a substantial difference in solvato and mechano-fluorochromic (MFC) features. With a pressing demand for finding a smart strategy to achieve MFC-active materials, we herein report unsymmetrically substituted anthracene-vinyl-phenothiazine π-conjugates (TAPTZ and FAPTZ) linked with furan or thiophene as a tiny flanking donor group. Both the compounds are easily synthesized using an economic path by avoiding metal catalysts and harsh reaction conditions. In solvatofluorochromic studies, by varying polarity of solvents from hexane to acetonitrile, the bathochromic shifts of 118 nm (3082 cm−1 Stokes shift) for TAPTZ and 112 nm (2840 cm−1 Stokes shift) for FAPTZ are noticed. Such electron-rich but conformationally twisted molecules create a twisted intramolecular charge transfer (TICT) state responsible for the solvatofluorochromic feature. A relatively more significant change in the excited state dipole moment for TAPTZ causes the difference. Both these molecules exhibit blue-shifted AIE features (faint orange to intense yellow), where TAPTZ has appeared to be a relatively better AIEgen with a 65 nm average particle size. Interestingly, FAPTZ has emerged as a stronger emitter than TAPTZ in the solid state. By grinding in a mortar and pestle or by quick pressing (in an infrared pellet maker: 20 MPa), TAPTZ and FAPTZ display reversible MFC features with a 15 nm and 22 nm redshift, respectively. Single crystal study discloses the difference in the molecular twisting and packing in the solid-state for these analogs. A relatively large number of intramolecular interactions (dominated by S⋯S and C⋯S) make the crystal packing stronger for TAPTZ and make such a difference in the sensitivity. Thus, we found a decent impact of the flanking donor groups thiophene/furan on the emission behavior under different environments. The observed fact is further supported by powder X-ray diffraction and lifetime measurement studies. Also, the intermolecular interactions are quantified by Hirshfeld-surface analyses to validate the empirical facts further. Finally, FAPTZ is documented as a favorable platform for rewritable optical-recording/security-based applications.
Introduction
The urge for the design and synthesis of stimuli-responsive small molecules offers ample demand in academics and research on security displays, sensors, and memory-based applications.1 Amongst them, mechano-fluorochromic properties (MFC) have gained substantial attention due to the generation of admirable multiple colorations just by grinding or rubbing the compound without performing cumbersome synthetic modifications.2 There are multiple small organic molecular systems discovered with stimuli-responsive properties.2b,3a,b One of the essential needs for MFC-active solid-state fluorophores is to attain twisted conformation offering numerous intermolecular interactions.4 In such a context, small heterocycles such as furan and thiophene exhibited a wide difference in MFC and electronic properties.5–8 Nevertheless, thiophene and furan are generally considered weak electron-donors and are explored commonly in the donor–π–acceptor like systems.6 Mostly, thiophene has been established as a promising donor in semiconductor/transistor-based applications.7,8 In contrast, the related investigations with furan are inadequate. However, recent studies revealed that the furan is also a favorable building block for generating semiconductors9a,b and has better charge carrier mobility.10 The greater extent of biodegradation, smaller size, and high planarity make the choice of furan more favorable despite having relatively inferior aromaticity and polarizability features compared to thiophene. In particular, a household AIE-framework tetraphenylethene (TPE)-linked furan TPE-F (Fig. 1) was found to be superior in exhibiting chromic behavior to that of analogous TPE-T under external stimuli.11 Additionally, the furan/thiophene-linked β-diketo building blocks were incorporated with the TPE system (TDK/FDK) and a variation in solvatofluorochromism, AIEE-behaviour, and (non)reversible MFC properties was identified.12 Apart from the TPE system, the other difluoroboron β-diketonate was attached to thiophene or furan (TBD and FBD) and recognized as a reversible stimuli-responsive material with subtle differences between thiophene and furan analogs. The extent of molecular planarity, intermolecular interactions, and the crystal packing density were found to play a role behind the variation in the emission color.11–13 | ||
| Fig. 1 Known furan/thiophene substituted MFC-active compounds. | ||
Thiophene or furan resides almost in the molecular plane with a slight twisting angle in all the reports above. However, the impact on emission color variation from the furan/thiophene analogs upon solvent or external pressure stimuli looks highly promising. Nevertheless, few molecules are only reported on the comparative solid-state and solvatofluorochromic features between furan and thiophene-conjugated analogs. In our current research on unsymmetrically substituted anthracene-based π-conjugated fluorophores, we noticed that a highly planar anthracene system could avoid disc-like molecular packing in the presence of flanking aryl/heteroaryl mainly due to the conformational variation (twisted at different torsion angles), resulting in the formation of AIEgens.14 The van der Waals interaction between the anthracenyl peripheral H's and these groups creates such twisted stable conformation. In such a system, thiophene analogs appeared to be better AIEgens than furan.15 However, solvato- and mechano-fluorochromic features were not promising for those molecules. Based on our interest in creating multi-stimuli-responsive materials,16 we herein focus on introducing anthracene–π–phenothiazine conjugates linked with furan or thiophene as flanking donor groups (Fig. 2).
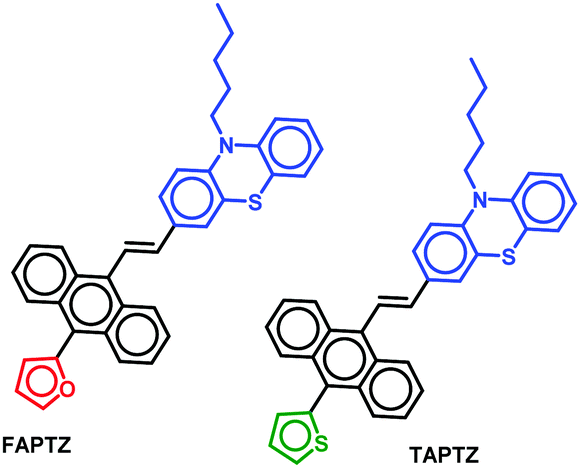 | ||
| Fig. 2 Thiophene/furan-flanked anthracene–π–phenothiazine molecules. | ||
The electron-rich and bulky phenothiazine unit is chosen due to its capability for forming weak C–H⋯S/S⋯π/C–H⋯π intermolecular interactions and the nonplanar structure of such a core would favor exhibiting AIE-features by avoiding the cofacial π⋯π stacking. Moreover, highly electron-rich phenothiazine can create a variation in the electronic distribution within the system where the anthracenyl part would be relatively electron-poor. Thus, the system would display the solvatofluorochromic properties. The impact of flanking small heterocycles into this system would draw special attention. AIEgens can emit in the solid-state, and the possible conformational changes under mechanical force can offer MFC-feature. We found dissimilarity in terms of the emission behavior in the solution and aggregated and solid-state due to such a tiny difference in molecular structure (thiophene/furan). Thus, the variation in electronic feature, size factor and aromaticity between thiophene and furan can govern such a subtle disparity in stimuli-responsive emission feature for these unsymmetrically substituted π-conjugates. Besides, the difference in intermolecular interactions has also created a contrast in the solid-state emission features. All the observed properties are analyzed with the help of molecular structure analyses, powder X-ray diffraction (PXRD), and fluorescence lifetime studies.
Results and discussion
Synthesis and characterization
The established synthetic procedure finds a broad scope to tune the substitution effect; the planned molecules FAPTZ (furan-flanked) and TAPTZ (thiophene-flanked) are conveniently synthesized in a decent yield by simple, inexpensive, and efficient Horner–Wadsworth–Emmons (HWE) reactions at room temperature (i.e., energetically economic) from easily accessible phosphonates (Scheme S1, ESI†) and phenothiazine-carbaldehyde (Scheme 1).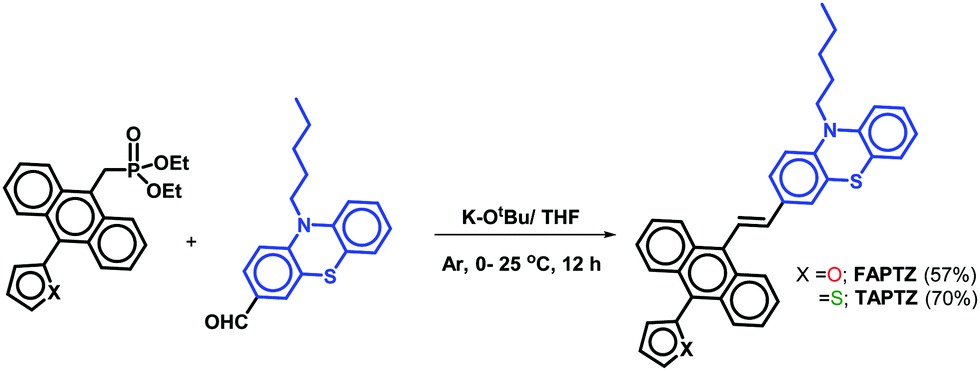 | ||
| Scheme 1 Synthesis of FAPTZ and TAPTZ. | ||
An expensive metal/ligand combination with high-temperature requirement is escaped to access such molecules where thiophene/furan units are introduced via Friedel–Crafts arylation reaction.17 The reaction can be scaled up on a g scale, indicating easy access to these materials for real-world applications. These compounds are soluble in most organic polar/nonpolar solvents and well-characterized by IR, NMR (1H/13C), and mass-spectroscopic studies. Finally, the molecules are crystallized by the room-temperature crystallization method, and the molecular structure with (E)-configuration is explicitly established by single-crystal X-ray diffraction studies (SCXRD; the details are discussed later). The phenothiazine unit's presence raises the thermal stability of the π-conjugates that show stability up to 400 °C and beyond as confirmed by thermogravimetric analysis (TGA, Fig. S1, ESI†).
Solvatofluorochromic studies
Primarily, the photophysical behaviors in the solution state were examined for these π-conjugates by changing the nature of the solvent with different polarities. A sharp maximum absorption at λabs ∼ 405–410 nm was detected for both the analogs in absorption spectroscopy (see Fig. S2, ESI† for all the absorption and emission spectra) due to π–π* transition, and there was no impressive shift observed by changing the polarity of the solvent, indicating an unaffected ground state. A slightly higher λabs for the furan analog dictates a relatively better conjugation than that of thiophene. The emission spectra reveal a cleaner discernible coloration for TAPTZ than FAPTZ in most of the solvents of different polarities (Fig. 3). The emission at relatively longer λmax for furan (FAPTZ) has further supported a more planar molecular structure for the furan analog. By varying ET(30)18a [a measure of microscopic solvent polarity] of different solvents, a promising positive solvatochromic effect (redshift) is noticed with both the compounds. The solvatochromic range between hexane (nonpolar) and MeCN (polar) is marginally better with a 118 nm redshift (3082 cm−1 Stokes shift) for TAPTZ than FAPTZ displaying a 112 nm redshift (2840 cm−1 Stokes shift). All the photophysical parameters, including the solvatochromic shifts for both the compounds, are listed in Table 1.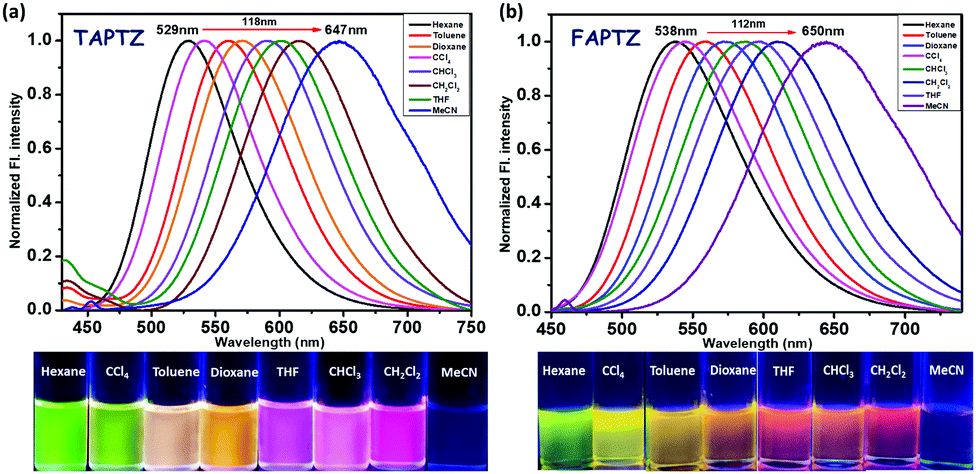 | ||
| Fig. 3 Normalized emission spectra of (a) TAPTZ and (b) FAPTZ, in different solvents. Concentration of the probe: 10 μM, λex = 405 nm. The image is taken under 365 nm UV light. | ||
| Solvent | Abs λmax (nm) TAPTZ/FAPTZ | Emi λmax (nm) TAPTZ/FAPTZ | Stokes shift ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) A − F cm−1TAPTZ/FAPTZ A − F cm−1TAPTZ/FAPTZ |
Quantum yield ϕf (%) TAPTZ/FAPTZ | Dipole moment | E T (30) |
|---|---|---|---|---|---|---|
|
A − F (cm−1): the wavenumber difference between absorbance and fluorescence. Relative quantum yields are measured using quinine sulfate as the standard (see the Experimental section). |
||||||
| Hexane | 402/404 | 529/538 | 5972/6165 | 14/13 | 0.08 | 31.0 |
| CCl4 | 409/413 | 541/553 | 5966/6130 | 8.8/13 | 0 | 32.5 |
| Toluene | 406/412 | 560/566 | 6774/6604 | 10/10 | 0.43 | 33.9 |
| 1,4-Dioxane | 407/412 | 571/579 | 7057/7001 | 8.9/7.2 | 1.3 | 36.0 |
| THF | 407/413 | 602/605 | 7959/7684 | 5.7/19 | 1.75 | 37.4 |
| CHCl3 | 408/412 | 590/595 | 7561/7465 | 5.7/22 | 1.15 | 39.1 |
| CH2Cl2 | 409/412 | 615/618 | 8190/8091 | 3.2/7.5 | 1.60 | 40.7 |
| Acetonitrile | 408/410 | 647/650 | 9054/9005 | 0.32/6.4 | 3.45 | 45.6 |
The overall difference of 242 cm−1 between TAPTZ and FAPTZ in the Stokes shift (between hexane and MeCN) and the considerable color variation are noteworthy (Fig. 3 and Table 1). The redshift may be attributed to the presence of the TICT (twisted intramolecular charge transfer) state, where the phenothiazine unit can be noted as an electron donor and anthracene as an electron acceptor. The role of significant twisting arises from the flanking thiophene and furan groups. The electronic structure is confirmed by performing a DFT study (Density Functional Theory) on these molecules. Ground-state optimization using DFT at the CAMB3LYP-6-31G(d,p) level identifies that the electron clouds in the highest occupied molecular orbital (HOMO) level are majorly located on the phenothiazine core (owing to the presence of two heteroatoms), while the lowest unoccupied molecular orbital (LUMO) level is predominantly placed over the anthracene ring with a slight contribution by flanking and phenothiazine groups (Fig. 4). Such electronic distribution can also support the ICT feature in the molecules. This is also consistent with the observed solvatofluorochromic properties.
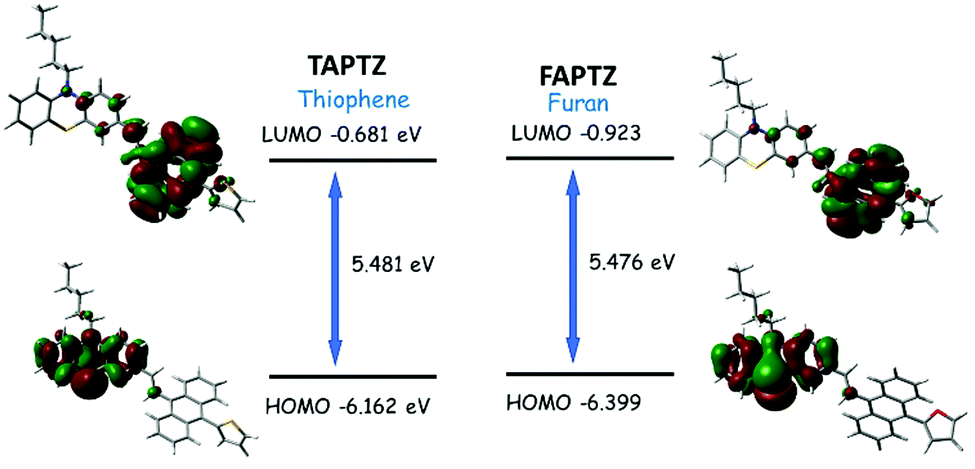 | ||
| Fig. 4 Optimized [using the CAMB3LYP-6-31G(d,p) level] molecular structure of TAPTZ and FAPTZ with the electronic distribution in the HOMO and LUMO. | ||
The LUMO and HOMO are relatively more stabilized for furan analogs, and the lower energy gap can cause longer wavelength absorption and emission for FAPTZ. The relatively lower quantum yields can be attributed to the presence of heavier sulfur atoms for TAPTZ. The DFT-optimized molecular structures demonstrate a more important π-conjugation for FAPTZ with a −67° furan-anthracene torsion angle than TAPTZ where thiophene is oriented in almost orthogonal (−88°) geometry with anthracene (Fig. S3, ESI†). The interplanar angles (58–66°) between anthracene and phenothiazine units are practically equal for FAPTZ and TAPTZ (Fig. S3, ESI†), indicating nearly identical coplanarity.
Moreover, a largely twisted conformational structure can create a more polar and energetically stabilized TICT state in the polar solvent, resulting in poor quantum yield with a high redshift.18b The plot of ET(30) vs. Stokes shift and emission λmax (Fig. S4 and S5, ESI†) also supports the possible formation of the TICT state. The TICT state is typically more convincing in the typical D–A system, but the generation of the TICT state from such electron-rich and conformationally twisted systems is possible where phenothiazine acts as the donor and anthracene as an acceptor.19 Moreover, anthracene and phenothiazine cores are coupled together through a trans-vinylidene spacer. Twisted conformation arises from the interactions between peripheral H-atoms of vinylidene and anthracenyl systems. However, the fluorescence (FL) intensity enhancement upon increasing the viscosity of the solvent medium (glycerol in methanol, Fig. S6, ESI†) indicates a substantial twisting in the molecular structures with such bulky rotors and creates the formation of a TICT state.18b The TICT state formation can also be favored due to the reversible first oxidation potential of phenothiazine upon photoexcitation and subsequent intramolecular rotation.20 Although the conventional Lippert–Mataga equation might be inappropriate to apply for such a molecular shape, we have examined the Lippert–Mataga plot [Fig. S7 and Table S1, ESI†]. The Lippert–Mataga plot [Stokes shift against orientation polarizability (Δf), reveals a quick rise in the Stokes shift with a polar solvent. It also confirms the enhanced dipole moment in the excited state,21 showing 14.37 D for TAPTZ and 13.91 for FAPTZ [Table S1, ESI†]. This marginal change in dipole moment for FAPTZ validates a slightly better redshift for the thiophene analog.
AIE-properties
The AIE properties are the most exciting photophysical behavior that has widened the application scope of the fluorophores in the solid/aggregated state and gained attention in the last two decades.22 Acetonitrile was preferred as a good and water-miscible solvent for these studies due to the weakest emission. The AIE properties were examined by measuring absorption (Fig. S8, ESI†) and emission spectra (Fig. 5) for each compound in 10 μM acetonitrile solution upon slow addition of water fraction [a bad solvent fw(v/v%)]. The reduction of FL intensity was observed initially due to the polarity effect until fw = 40–50% (Fig. 5b and d) for both cases. There was a sudden upturn in the FL intensity for TAPTZ from fw = 40%, and the intensity was maximum at fw = 90% with a 73 nm blue-shift (yellow emission at λmax 574 nm). For FAPTZ, the FL enhancement started at fw = 50% and reached maximum until fw = 70% with an 81 nm blue-shift (yellow emission at λmax 569 nm). Thus, both these compounds exhibit a similar AEE-effect (aggregation-enhanced emission) in terms of the emission color but more pronounced for TAPTZ (14 times enhancement) in comparison to FAPTZ (only five times enhancement). The ups and downs in the FL intensity at higher fw for FAPTZ were observed earlier due to the formation of a nanoparticle suspension with both crystalline and amorphous characters that control the emission intensity in the solution.14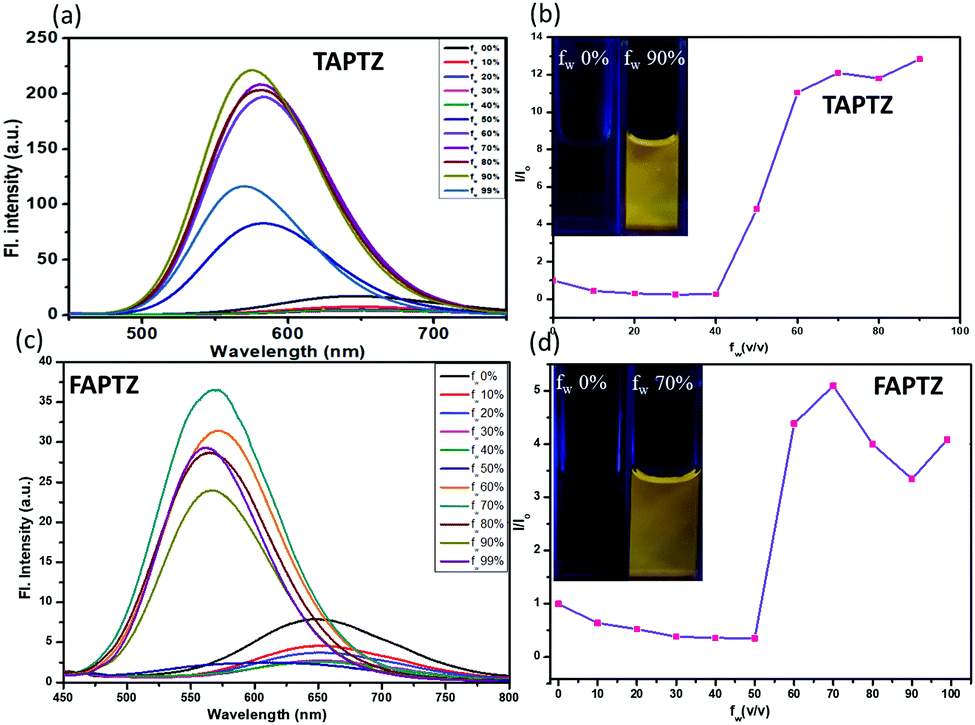 | ||
| Fig. 5 Emission spectra of (a) TAPTZ and (c) FAPTZ. (b and d) The I/I0 plot (I0: FL intensity before addition of water; I: FL intensity after addition of water) [concentration of the probe: 10 μM, λex = 405 nm]. The image is taken at fw = 0% and fw = 90% (TAPTZ) and 70% (FAPTZ) under 365 nm UV light at different fw in acetonitrile. | ||
The aggregation at the highest FL intensity is recognized by the average size of the particles calculated from dynamic light scattering (DLS) studies that display 65 nm for TAPTZ and 85 nm for FAPTZ (Fig. S9, ESI†). The restricted intramolecular motion during the aggregation was earlier established as the primary cause for AIE behavior.22 Such a highly twisted conformation with large rotors in an olefinic stator would mostly avoid the co-facial intermolecular aggregation. Thus, the intramolecular motion will be limited to fluoresce through a radiative channel. The torsion angle of 67° for thiophene would favor showing better AIE-properties than the furan-analog. The blue shift indicates the reduction in the molecular coplanarity upon aggregation. Blue-shifted AIE properties were also noticed earlier in a similar system.23,24 A cross-aggregation (J-type) is found from the crystal packing (vide infra), and that causes a lack of conjugation to have a blue-shifted emission. The smaller particle size is possibly supporting to have more AIE-effect for TAPTZ.25 The observed AIE properties are also competent enough to emit in the solid-state, and such a variously twisted conformation of the molecules will tend to exhibit MFC characteristics and thus, is explored.
MFC-Studies
Initially, the solid-state photophysical properties are investigated before and after grinding both the pristine samples to examine the change in emission color. The solid-state UV-Vis spectrum displays a broad and substantial absorption in the region of 440 nm, mainly due to π–π* transition for both the pristine samples (Fig. S10, ESI†). Upon grinding in a mortar and pestle or applying pressure (20 MPa) using an IR-pellet maker, the change in absorption spectrum is slightly blue-shifted (∼17 nm) for TAPTZ, but almost no difference for FAPTZ (Fig. 6), indicating almost similar behaviour in the ground state. However, the solid-state FL spectrum for TAPTZ is noticeably red-shifted from 550 nm to 565 nm (15 nm) after grinding. The absolute quantum yield (ϕf, %) for the pristine sample is 9.1, and that was weakened to 6.02 after grinding. Thus, one can visualize the sharp FL switching from light green to yellow emitting solid (15 nm contrast) through the naked eye under 365 nm UV-light after grinding the solid. On fumigation of the ground sample with hexane, the original green color returns. Notably, thermal energy could not bring back the actual green color. On the other hand, the furan analog FAPTZ emitted a relatively brighter green light (emission λmax = 537 nm) with ϕf (%) 24 and switched to 559 nm upon grinding.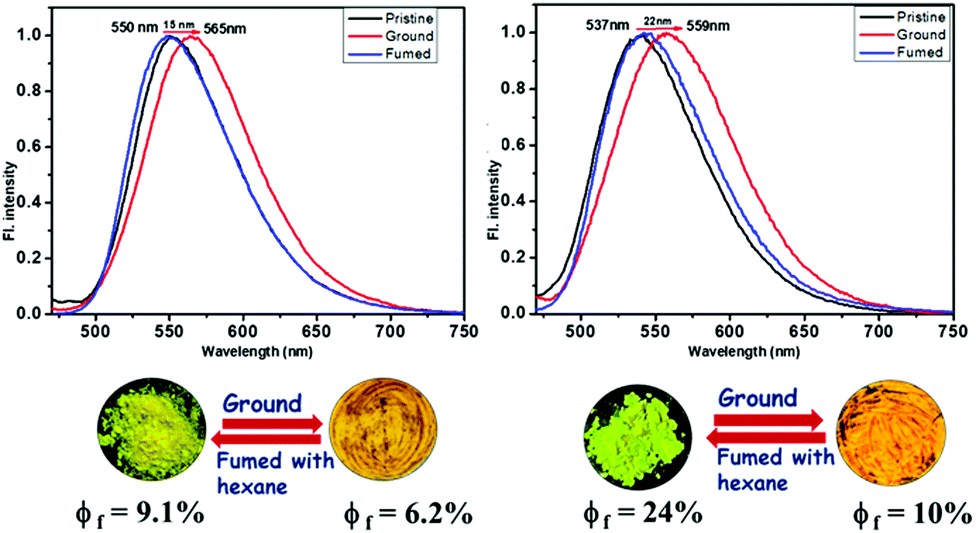 | ||
| Fig. 6 Pristine and ground samples of TAPTZ and FAPTZ along with the corresponding (reversible) emission spectra [λex = 420 nm]. The images are taken under 365 nm UV-light. | ||
Additionally, the excited state's fluorescence lifetime (ns) was determined for solid TAPTZ and FAPTZ in both the pristine and ground states (Fig. S12 and Table S2, ESI†). All the related parameters are stated in Table 2. More Φf (%) for FAPTZ can be elucidated with a relatively better ratio Kr/Knr [radiative rate constant (Kr) and non-radiative rate constant (Knr)]. Grinding the compounds resulted in a decrease in the quantum yield and could be deciphered with lower Kr/Knr values (Table 2).
| Samples | States | λ maxabs (nm) | λ maxem (nm) | Φ f (%) | τ (ns) | K r/Knr |
|---|---|---|---|---|---|---|
| TAPTZ | Pristine | 440 | 550 | 9.1 | 0.65 | 0.100 |
| Ground | 423 | 565 | 6.2 | 1.16 | 0.060 | |
| FAPTZ | Pristine | 436 | 537 | 24.0 | 0.57 | 0.323 |
| Ground | 438 | 559 | 10.0 | 0.54 | 0.121 |
SCXRD studies
It is essential to examine the single-crystal X-ray structures for these π-conjugates to obtain a molecular-level understanding and investigate the cause for the FL-switching upon grinding. Typically, such FL switching is regulated by the change in the supramolecular assembly. The crystal structures were easily determined with suitably grown single crystals, obtained from 30% EtOAc in hexane through a room-temperature slow evaporation technique. Both the compounds TAPTZ and FAPTZ crystallized in a triclinic system with a P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group [see Table S3 (ESI†) for all the crystallographic parameters]. Predominantly, the molecular structures determined from SCXRD have presented the fundamental difference between these two molecules in the solid-state (Fig. 7).
space group [see Table S3 (ESI†) for all the crystallographic parameters]. Predominantly, the molecular structures determined from SCXRD have presented the fundamental difference between these two molecules in the solid-state (Fig. 7).
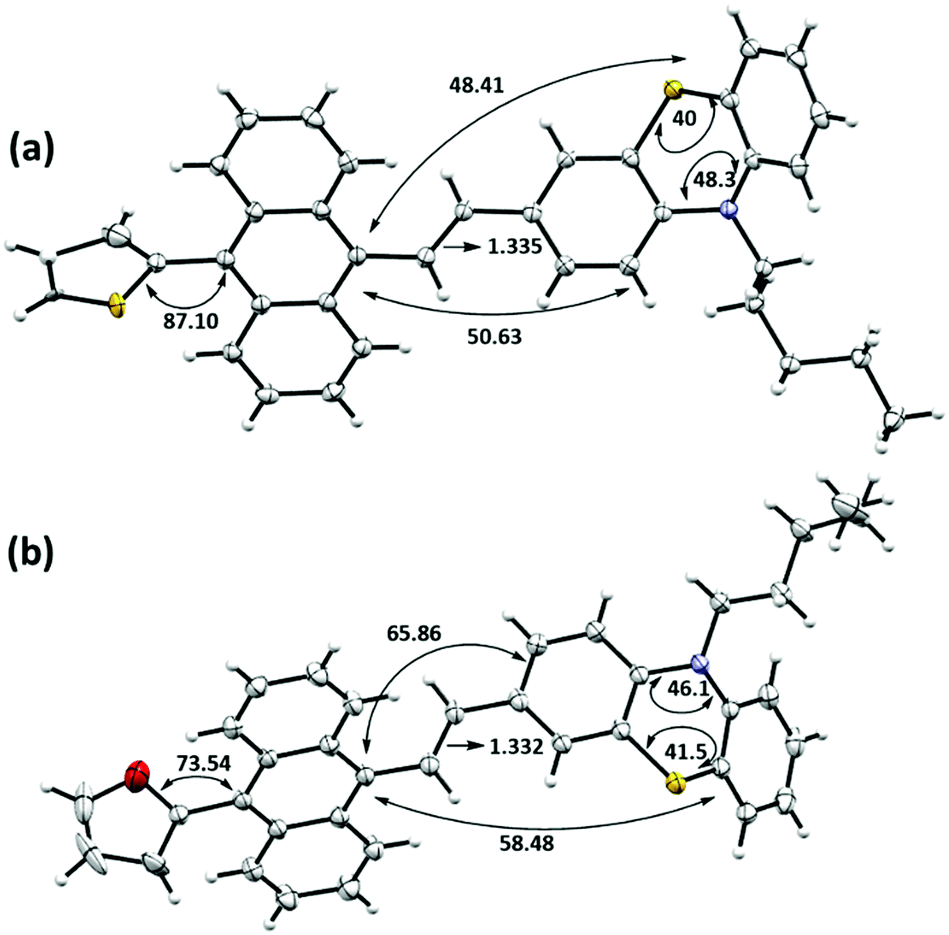 | ||
| Fig. 7 Single-crystal X-ray structures (ORTEP diagram with 50% probability level) and few selected bond angles (°) and distances (Å): (a) TAPTZ (the disordered part is removed for clarity) and (b) FAPTZ. TAPTZ was solved with a slight disorder in the thiophene ring but refined successfully. | ||
Both the structures are significantly twisted in the solid-state and emit through a radiative channel to exhibit solid-state emission properties. Direct attachment of the thiophene or furan unit as a flanking donor causes the torsion angle of 87.1° and 73.5° with anthracene for TAPTZ and FAPTZ, respectively. A relatively smaller size of the furan ring enables a smaller torsion angle but does not exhibit the emission at a longer wavelength in the solid state. A further detailed inspection indicates much smaller angles between anthracene and phenothiazine (∼50°) plane for TAPTZ than FAPTZ that attains an interplanar angle of ∼66°. Such a difference would facilitate more π-electron conjugation in TAPTZ, resulting in emission at a higher wavelength. A slightly longer (0.003 Å) olefinic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond also supports better conjugation in TAPTZ than FAPTZ. It is worth noting that the DFT optimized molecular structures of TAPTZ and FAPTZ had almost equal coplanarity between anthracene and phenothiazine in the molecular state, and the differences are only observed in the solid-state due to the non-covalent interactions.
C bond also supports better conjugation in TAPTZ than FAPTZ. It is worth noting that the DFT optimized molecular structures of TAPTZ and FAPTZ had almost equal coplanarity between anthracene and phenothiazine in the molecular state, and the differences are only observed in the solid-state due to the non-covalent interactions.
Next, we emphasized supramolecular interactions and mode of molecular crystal packing for TAPTZ and FAPTZ, which offer profound comparisons to recognize the switching of solid-state fluorescence behavior upon grinding.26,27 The intermolecular interactions for these crystals are designated in Table 3 and Fig. 8. The differences in crystal packing from three different axes are presented in Fig. S13 (ESI†). The almost equal number of C⋯H interactions do not bring much variance in the MFC behavior. However, no π⋯π interactions are observed because of such twisted conformations of these molecules, and cross J-type aggregation is perceived. The distances (Å) between two anthracenyl centroids are 9.743 and 5.010 for TAPTZ and FAPTZ, respectively. However, for TAPTZ, the flanking thiophene causes two additional interactions (C⋯S and S⋯S) that help to make the crystal packing more compact and can be further supported with a higher melting point for TAPTZ (165 °C) in comparison to FAPTZ (153 °C).
| Compounds | C⋯H | O⋯H | S⋯S | S⋯H | C⋯S | H⋯H |
|---|---|---|---|---|---|---|
| TAPTZ (10 interactions) | 2.887, 2.885 | 3.524 | 2.789 | 3.335 | 2.399 | |
| 2.861, 2.829, 2.803, 2.816 | ||||||
| FAPTZ (8 interactions) | 2.833, 2.818 | 2.680 | 2.306 | |||
| 2.898, 2.747 | ||||||
| 2.865, 2.816 |
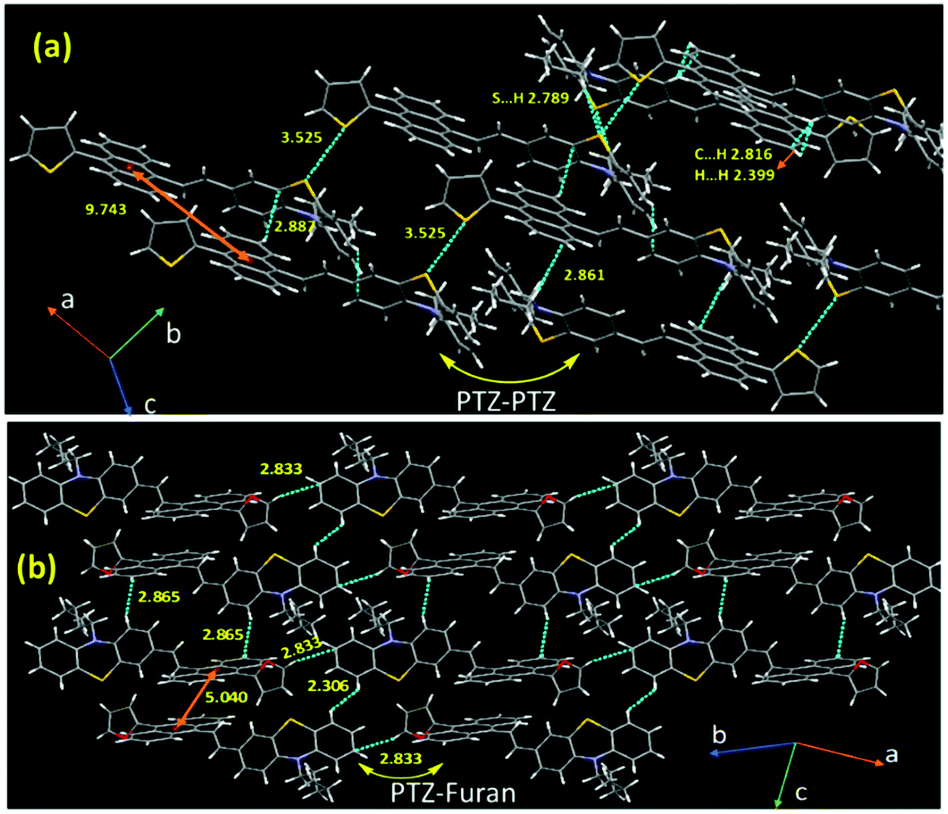 | ||
| Fig. 8 Crystal packing for (a) TAPTZ and (b) FAPTZ showing a few intermolecular interactions (d/Å). | ||
The TAPTZ molecules are oriented so that PTZ–PTZ units are closer, whereas PTZ–furan has appeared to be joined for FAPTZ in the crystal lattice (Fig. 8). The C⋯H interactions for both the compounds enforce more flexibility and are more helpful to exhibit MFC behavior.28a,b Nevertheless, a relatively more number of interactions for TAPTZ achieve little higher rigidity and higher crystal lattice energy29 which thus becomes less sensitive. Notably, both these compounds display MFC-feature under grinding/pressure by improving the degree of planarization in molecular conformation that would elucidate the observed redshift.30 The penetration of hexane vapor into the distorted crystal packing will permit molecules to develop flexibility and rearrange the molecular conformation to return to the pristine crystal packing to induce the authentic emission.
This repeatable fluorescence switching using FAPTZ is almost similar (change of ±10 nm wavelength) even after multiple grinding/fuming (Fig. S11, ESI†) processes. Thus, the material is supportive of numerous cycles of reversible color change upon solvent exposure.
PXRD analysis
To gain a deeper insight into the above slight variation in MFC-feature, powder X-ray diffraction (PXRD) studies are performed. Both these analogs demonstrate many sharp and intense diffraction signals in the pristine state (Fig. 9), specifying a well-ordered crystalline structure. However, TAPTZ has appeared to be relatively more crystalline than FAPTZ as deciphered before. Some of the diffraction planes are designated based on the simulated diffraction patterns for both the compounds, and the diffractions are consistent with the experimentally determined signals. The reversible MFC behavior for both the compounds is supported by the diffractogram for pristine, ground, and fumed samples. The crystal to amorphous state transformation is noticed after grinding because almost no diffraction peaks at the same 2θ are observed (Fig. 9). The original intense diffractions recur upon fuming with hexane. It depicts a considerable decrease in the crystallinity (transform towards the amorphous phase) after grinding the solid and the crystallinity is restored after fuming and fuming with hexane assists in rearranging the molecules to reach the original crystalline from the amorphous state. Thus, changes in the PXRD pattern support the difference in displaying MFC features for such a phase transition in these molecules.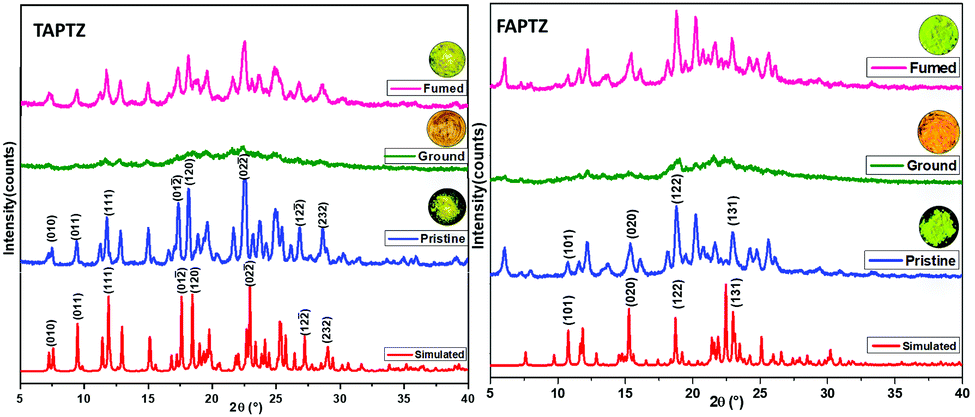 | ||
| Fig. 9 The PXRD patterns of TAPTZ and FAPTZ in pristine, ground, and fumed states. | ||
Hirshfeld surface analysis
The crystal packing showed the contribution of C⋯H interactions inside the crystal lattice. The quantitative analyses of intermolecular interactions are performed through the Hirshfeld surface generations (Fig. S14, ESI†). A significant amount of C⋯H type interactions (33.3%) is acting on the FAPTZ crystal compared to TAPTZ (30.5%). Notably, there are 4.5% C⋯C interactions for TAPTZ, whereas FAPTZ shows 3% of such interactions. The higher ratios of C⋯H/C⋯C interactions for FAPTZ indicate good MFC behavior in comparison to TAPTZ.31 This Hirshfeld analysis is very well aligned with our experimentally observed features and the crystal packing analysis. Thus, the difference in the intermolecular interactions between these two molecules can elucidate the slight difference in MFC-feature for these two analogs.Application as an optical rewritable recorder
Further, such MFC-active materials are also recognised as a platform for rewritable optical recording. Such FL-switching is very much appropriate for real-world applications. An economical filter paper-based rewritable optical recorder is created by dispersing TAPTZ on the filter paper. Thus, the yellow-emitting surface becomes ready for the writing. The letters are suitably printed on this yellow-emitting surface by pressing with a metal spatula. The letters emit orange and are visible with bare eyes under 365 nm UV-torch (Fig. 10). The orange emitting part can further return to a yellow-emitting surface by fuming with hexane. Thus, this TAPTZ-coated filter paper is documented as an encouraging MFC material for repetitive usage in optical recording.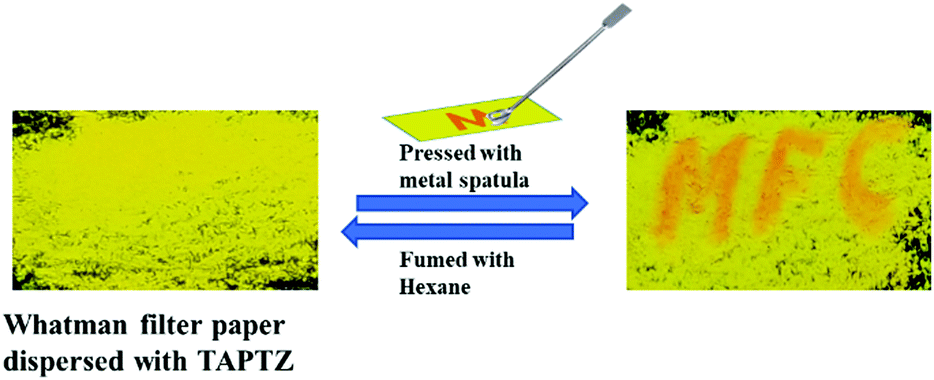 | ||
| Fig. 10 Application of TAPTZ as a rewritable optical recorder. | ||
Experimental
Materials
All of the chemicals were purchased from Merck, and the solvents for column chromatography were purchased from Finar. Spectroscopic grade solvents used for UV-vis and fluorescence spectroscopy were obtained from Sisco Research Laboratories (SRL). All the experiments were performed at room temperature, 298 ± 2 K. 1H, and 13C NMR spectra were recorded on Bruker 400 MHz spectrometers with an operating frequency of 101 MHz for 13C. Chemical shifts (δ) are reported in ppm relative to the residual solvent signal (δ 7.26 for 1H NMR and δ 77.0 for 13C NMR).Methods and measurements
where Φf = quantum yield of the sample, Φref = quantum yield of quinine sulfate (0.54), aR = area under the curve of emission spectra of quinine sulfate in 0.1 M H2SO4, AS = absorbance of the probe in different solvents, AR = absorbance of quinine sulfate in 0.1 M H2SO4, ηS = refractive index of the corresponding solvent, and ηR = refractive index of water. The error: ±10%.
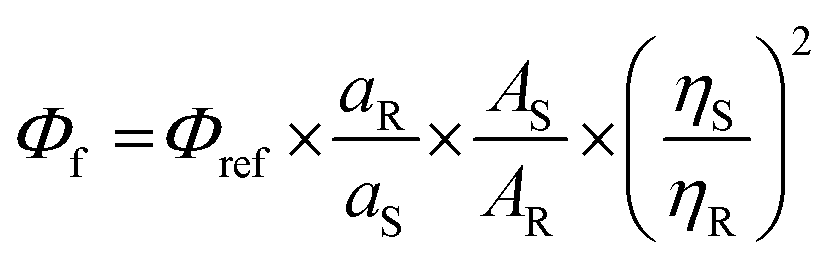
Viscosity effect
The viscochromic properties were studied by measuring absorption and emission spectra (Fig. S6, ESI†) for both TAPTZ and FAPTZ in 10 μM MeOH solution upon incremental addition of glycerol fraction [highly viscous liquid fg (v/v%): fraction of glycerol in methanol]. The FL intensities for TAPTZ and FAPTZ increased linearly with the gradual addition of glycerol (fg, v/v%) in MeOH until fg = 60% and there was a sudden enhancement in the FL intensity at fg > 60%.Dynamic light scattering (DLS) measurement
The average particle size in the aggregated state was found using a Malvern particle size analyzer (zeta sizer nano-ZS), keeping a concentration of 10 μM.Thermogravimetric analysis
The TGA thermograms were recorded using Themys One+, Setaram instrument keeping the temperature range 30–650 °C with 10 °C min−1 under a N2 atmosphere.Single crystal X-ray measurement
All measurements were performed on a Rigaku XtaLAB P200 diffractometer using multi-layer mirror monochromated Cu-Kα radiation (λ = 1.54184 Å). The data were collected at a temperature of −173 ± 1 °C to a maximum 2θ value of 149.8°. Data were collected and processed using CrysAlisPro (Rigaku Oxford Diffraction). The linear absorption coefficient, μ, for Cu-Kα radiation is 18.391 cm−1. An empirical absorption correction was applied, which resulted in transmission factors ranging from 0.227 to 0.593. The data were corrected for Lorentz and polarization effects. The structure was solved by direct methods (SIR2011)32 and expanded using Fourier techniques. The non-hydrogen atoms were refined anisotropically. Hydrogen atoms were refined using the riding model. All calculations were performed using the Olex 2 crystallographic software package except for refinement, which was performed using SHELXL Version 2014/7.33Density functional theory
The DFT studies were performed on these molecules using a CAM-B3LYP 631-G(d,p) basis set. The HOMO and LUMO energies were calculated, and corresponding cubes were generated using the Gaussian 09 package.Hirshfeld surface analyses34
Hirshfeld surface analysis and void space calculation is a quantitative tool to understand non-covalent interactions. We have generated Hirshfeld surfaces for TAPTZ and FAPTZ with an iso-value of 0.5 a.u. The surface is generated, and the interactions are shown in terms of de and di, where de and di are distances of an atom external or internal to the generated Hirshfeld surfaces; together, this pair (de and di) generates a 2D fingerprint plot. The different colors on the fingerprint plot represent the frequency of occurrence of interaction. A red spot represents the direct interaction between two atoms. Ultimately it gives a normalized contact distance (dnorm). The dnorm values are mapped onto the Hirshfeld surface using a red, white, and blue color scheme; red, white, and blue regions correspond to the strong, medium, and weak interactions, respectively. All the Hirshfeld surfaces were generated using Crystal Explorer 3.1 software.Synthesis and characterization
Conclusions
In conclusion, two new anthranyl π-conjugates TAPTZ and FAPTZ linked with thiophene or furan as a flanking donor are easily synthesized. Thiophene analogs are found to be superior in displaying solvatofluorochromic features to the furan analog, with a slightly better variation in the excited state dipole moment. The emission wavelength in solution dictates a better conjugation in FAPTZ than TAPTZ, and that is interpreted by the DFT-optimized molecular structures, presenting better π-electronic conjugations in FAPTZ from a comparatively lesser torsion angle between furan and anthracene by keeping the rest almost similar. Contrarily, the solid-state emission appears at a lower wavelength for FAPTZ because of the higher torsion angle between anthracene (acceptor) and PTZ (donor). TAPTZ has appeared to be a better blue-shifted AIEgen with a relatively smaller particle size. The tilted conformation and a cross-packing enable these molecules to be emissive in the solid-state and exhibit reversible red-shifted MFC-feature. A relatively lower redshift and sensitivity for TAPTZ are described by more substantial crystal packing with two extra C⋯S and S⋯S interactions than FAPTZ. Such dissimilarity is deduced with SCXRD, PXRD, and fluorescence lifetime studies. Hirshfeld surface analyses indicate quantitatively more C⋯H interactions in FAPTZ than TAPTZ. Thus, this work has provided a thought on the effect of a tiny change in a flanking donor that can impact the solvato-, AIE- and MFC-features, which would develop new MFC-active materials. The practical application of reversible security writing demonstrates the efficacy of these π-conjugates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank DST-SERB (CRG/2018/000456) for financial support. MC thanks BITS-Pilani Hyderabad for NMR/X-ray facilities. We thank Prof Durba Roy for the DFT calculations.References
-
(a) R. Gao, X. Fang and D. Yan, J. Mater. Chem. C, 2019, 7, 3399–3412 RSC
; (b) Q. Li and Z. Li, Acc. Chem. Res., 2020, 53, 962–973 CrossRef CAS PubMed
-
(a) Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073–1095 CrossRef CAS PubMed
-
(a) G. Zhang, J. Sun, P. Xue, Z. Zhang, P. Gong, J. Peng and R. Lu, J. Mater. Chem. C, 2015, 3, 2925–2932 RSC
-
(a) X. Huang, L. Qian, Y. Zhou, M. Liu, Y. Cheng and H. Wu, J. Mater. Chem. C, 2018, 6, 5075–5096 RSC
- Y. Yin, Z. Chen, Y. Yang, G. Liu, C. Fan and S. Pu, RSC Adv., 2019, 9, 24338–24343 RSC
-
(a) W. Sharmoukh, J. Cong, B. A. Ali, N. K. Allam and L. Kloo, ACS Omega, 2020, 5(27), 16856–16864 CrossRef CAS PubMed
-
(a) G. Barbarella, M. Melucci and G. Sotgiu, Adv. Mater., 2005, 17, 1581 CrossRef CAS
- M. El-Sayed, H. Müller, G. Rheinwald, H. Lang and S. Spange, Chem. Mater., 2003, 15, 746–754 CrossRef CAS
-
(a) A. Pachariyangkun, M. Suda, S. Hadsadee, S. Jungsuttiwong, P. Nalaoh, P. Pattanasattayavong, T. Sudyoadsuk, H. M. Yamamoto and V. Promarak, J. Mater. Chem. C, 2020, 8, 17297–17306 RSC
-
(a) O. Gidron and M. Bendikov, Angew. Chem., Int. Ed., 2014, 53, 2546 CrossRef CAS PubMed
- Z. Zhao, H. Nie, C. Ge, Y. Cai, Y. Xiong, J. Qi, W. Wu, R. T. K. Kwok, X. Gao, A. Qin, J. W. Y. Lam and B. Z. Tang, Adv. Sci., 2017, 4, 1700005 CrossRef PubMed
-
(a) W. Liu, Y. Wang, J. Yang, X. Li, X. Wang and L. Ma, Dyes Pigm., 2020, 175, 108149 CrossRef CAS
- W. A. Morris, T. Butler, M. Kolpaczynska and C. L. Fraser, Mater. Chem. Front., 2017, 1, 158–166 RSC
- M. Z. K. Baig, D. Majhi, R. N. P. Tulichala, M. Sarkar and M. Chakravarty, J. Mater. Chem. C, 2017, 5, 2080–2387 Search PubMed
- W. Xu, M. M. S. Lee, Z. Zhang, H. H. Y. Sung, I. D. Williams, R. T. K. Kwok, J. W. Y. Lam, D. Wang and B. Z. Tang, Chem. Sci., 2019, 10, 3494–3501 RSC
- T. Jadhav, B. Dhokale, Y. Patil, S. M. Mobin and R. Misra, J. Phys. Chem. C, 2016, 120(42), 24030–24040 CrossRef CAS
- M. Z. K. Baig, G. Pallikonda, P. Trivedi, R. N. P. Tulichala, B. Ghosh and M. Chakravarty, ChemistrySelect, 2016, 1, 4332 CrossRef CAS
-
(a) C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS
- S. Sasaki, G. P. C. Drummen and G. Konishi, J. Mater. Chem. C, 2016, 4, 2731–2743 RSC
- J. Zhang, B. Xu, J. Chen, L. Wang and W. Tian, J. Phys. Chem. C, 2013, 117(44), 23117–23125 CrossRef CAS
- U. Subuddhi, S. Haldar, S. Sankararaman and A. K. Mishra, Photochem. Photobiol. Sci., 2006, 5, 459–466 CrossRef CAS PubMed
-
(a) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC
- Z. Yang, W. Qin, J. W. Y. Lam, S. Chen, H. H. Y. Sung, I. D. Williams and B. Z. Tang, Chem. Sci., 2013, 4, 3725–3730 RSC
- B. Prusti, P. Sarkar, S. K. Pati and M. Chakravarty, J. Mater. Chem. C, 2021, 9, 9555–9570 RSC
- N. Zhang, H. Chen, Y. Fan, L. Zhou, S. Trepout, J. Guo and M. H. Li, ACS Nano, 2018, 12, 4025–4035 CrossRef CAS PubMed
- B. Fang, M. Chu, Z. Wu, Y. Shi, Y. S. Zhao and M. Yin, J. Mater. Chem. C, 2019, 7, 4434–4440 RSC
- B. Roy, M. C. Reddy and P. Hazra, Chem. Sci., 2018, 9, 3592–3606 RSC
-
(a) A. Ekbote, S. M. Mobin and R. Misra, J. Mater. Chem. C, 2018, 6, 10888–10901 RSC
-
Mechanochromic fluorescent materials phenomena, materials and applications, ed. J. Xu and Z. Chi, Royal Society of Chemistry, 2014, p. 167 Search PubMed
- Y. Ren and T. Baumgartner, Inorg. Chem., 2012, 51, 2669–2678 CrossRef CAS PubMed
- B. Roy, M. C. Reddy and P. Hazra, Chem. Sci., 2018, 9, 3592–3606 RSC
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, C. Giacovazzo, M. Mallamo, A. Mazzone, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2012, 45, 357 CrossRef CAS
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2100035 and 2100036. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ma00664a |
| This journal is © The Royal Society of Chemistry 2021 |