
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComprehensive determination of the high-pressure structural behaviour of BaTiO3†
Craig L.
Bull
 *ab,
Christopher J.
Ridley
a,
Kevin S.
Knight
cd,
Nicholas P.
Funnell
a and
Alexandra S.
Gibbs
ae
*ab,
Christopher J.
Ridley
a,
Kevin S.
Knight
cd,
Nicholas P.
Funnell
a and
Alexandra S.
Gibbs
ae
aISIS Neutron and Muon Facility, Rutherford Appleton Laboratory, Chilton, OX11 OQX, UK. E-mail: craig.bull@stfc.ac.uk; Tel: +44 1235 445706
bSchool of Chemistry, University of Edinburgh, David Brewster Road, Edinburgh EH9 3FJ, UK
cDepartment of Materials Science and Engineering, University of Sheffield, Sheffield S1 3JD, UK
dDepartment of Earth Sciences, The Natural History Museum, Cromwell Road, London, SW15 5BD, UK
eSchool of Chemistry, University of St Andrews, Fife, KY16 9SA, UK
First published on 17th August 2021
Abstract
We have mapped the phase diagram of BaTiO3 more extensively than previous attempts using high-pressure neutron-powder diffraction. The mapping of the phase diagram has been performed using isothermal compression at fixed temperatures (175, 225, 290, 480 K) within each of the known crystallographic phases, up to ∼6 GPa using a large volume press. The crystallographic structure of each phase has been measured, and the determined absolute atomic displacements of all atoms within the cell have provided detailed information on the order of the phase transitions observed, and the behaviour of the ferroelectric dipole moment.
1 Introduction
The physical and crystallographic properties of BaTiO3 have been extensively studied since its discovery in the 1940s. Interest in BaTiO3 stems from its favourable ferroelectric properties and high dielectric constant, proving an extremely valuable commodity in the production of capacitors and transducers; the material is also a model candidate for understanding the thermodynamics of proper ferroelectric phase transitions with Devonshire theory, where the polarisation is the order parameter.1 Consequently many reviews and studies can be found summarising its properties, from many different characterisation techniques.2–11Above 393 K BaTiO3 has the aristotype perovskite structure (cubic, Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m), where the centre of symmetry forbids the formation of a spontaneous dipole moment and hence is paraelectric (Fig. 1).12 Upon cooling below the Curie temperature (Tc = 393 K) the symmetry reduces (tetragonal, P4mm), accompanied by the displacement of the Ti atoms within the TiO6 octahedra giving rise to a dipole moment, becoming ferroelectric. As the two space-groups are related, symmetry and the Landau and Liftschitz conditions (as outlined in13) are satisfied, and the transition is permitted to be second order in nature (Fig. 1).14 With decreasing temperature further reductions in crystallographic symmetry are observed at 268 K (orthorhombic, Bmm2) and 183 K (rhombohedral, R3m).15 With decreasing temperature the spontaneous polarisation is enhanced as the Ti atom moves further away from the idealised position of the cubic structure (Fig. 1).15,16 This series of phase transitions can be understood using symmetry adapted basis vectors,17 as the progressive condensation of the triply degenerate Γ4− mode resulting in distortions away from the cubic parent structure. Condensation of the cubic axis [100] component results in the tetragonal phase; the [110] component results in the orthorhombic phase, with the [111] component finally condensing to form the rhombohedral phase.
m), where the centre of symmetry forbids the formation of a spontaneous dipole moment and hence is paraelectric (Fig. 1).12 Upon cooling below the Curie temperature (Tc = 393 K) the symmetry reduces (tetragonal, P4mm), accompanied by the displacement of the Ti atoms within the TiO6 octahedra giving rise to a dipole moment, becoming ferroelectric. As the two space-groups are related, symmetry and the Landau and Liftschitz conditions (as outlined in13) are satisfied, and the transition is permitted to be second order in nature (Fig. 1).14 With decreasing temperature further reductions in crystallographic symmetry are observed at 268 K (orthorhombic, Bmm2) and 183 K (rhombohedral, R3m).15 With decreasing temperature the spontaneous polarisation is enhanced as the Ti atom moves further away from the idealised position of the cubic structure (Fig. 1).15,16 This series of phase transitions can be understood using symmetry adapted basis vectors,17 as the progressive condensation of the triply degenerate Γ4− mode resulting in distortions away from the cubic parent structure. Condensation of the cubic axis [100] component results in the tetragonal phase; the [110] component results in the orthorhombic phase, with the [111] component finally condensing to form the rhombohedral phase.
 | ||
| Fig. 1 Structure of BaTiO3 in its four known crystallographic phases at ambient pressure. Top left: Cubic (Pmm). The regular undistorted octahedra is shown (central light blue titanium atom, surround by 6 red oxygen atoms). The cubic unit cell is shown with the barium (brown sphere) atom at its centre. Top right: Tetragonal (P4mm). The distorted octahedra are shown in which there are 1 long, 4 medium and 1 short Ti–O bonds, which arise from displacement in the z-direction of the oxygen atoms. Bottom left: Orthorhombic (Bmm2). The further distorted TiO6 is shown which is a result of displacement in both the x and z direction of the oxygen atoms giving rise to two short, medium and long Ti–O bonds in the octahedra. Bottom right: Rhombohedral (R3m). The octahedra is at its most distorted with displacement of the oxygen atoms in all three crystallographic directions giving rise to 3 short and three long Ti–O bonds in the octahedra. The opaque green arrow indicates the approximate direction of the dipole in each ferroelectric phase. | ||
As the symmetry changes between the tetragonal-orthorhombic-rhombohedral structures are not directly related by subgroup relations, they cannot be second-order in nature. While these transitions are often described using Landau theory,18 there are a number of experimental observations which cannot easily be explained by the displacive model, such as the mixture of observed Ti displacements observed in all phases from X-ray fine structure measurements, and strong diffuse X-ray scattering.19 An order–disorder model has been used to provide a mechanism to explain these discrepancies, which allows for a distribution of the ferroelectric displacements of the Ti atom toward each of the eight possible 〈111〉 TiO6 octahedral faces, giving rise to disorder between chains and long range correlations in chains in the 〈100〉 direction. Overall, this model gives rise to the average rhombohedral crystallographic symmetry, strongly supported experimentally by observations made in X-ray single crystal, PDF and XAS measurements.20–22 However, these results are in contradiction to Raman and inelastic neutron measurements which support a displacive nature to the transitions.23 One recent study has showed that it is possible to combine both the order–disorder and displacive mechanism to describe the cubic-tetragonal phase transition.24 A recent quantum mechanical DFT study suggested that all phases of BaTiO3 have local distortion towards the [111] direction (an octahedral face) and this can be used to understand the presence of unexpected 1st order Raman modes.19 Recent work by Shao and Zuo25 show that within a local volume of 35 nm3 which is tetragonal in symmetry there are ∼15 nm sized domains which are higher symmetry.
The high-pressure behaviour of BaTiO3 has been studied using many different characterisation techniques.26–31 The Curie temperature, dielectric constant and spontaneous polarisation of BaTiO3 have been shown to decrease at high pressure.30 The tetragonal to cubic phase transition is observed at 2 GPa at 290 K,31–34 and isothermal compression of BaTiO3 results in the vanishing of ferroelectricity through the same series of phase transitions observed with increasing temperature.31,35,36 However, the nature of the transitions have been reported to gradually change with the first-order character weakening at higher pressure.29,37 The work of Ishidate et al determined the low-temperature and high-pressure phase diagram of BaTiO3 by dielectric measurements up to ∼6.5 GPa,35 and to a large extent confirmed by Hayward and Salje using Landau theory.38
Darlington et al studied the structure of BaTiO3 using high-resolution neutron-powder diffraction, in 5 K intervals from 150 to 425 K. They were able to obtain absolute oxygen displacements, and determine the spontaneous polarisation in reasonable agreement with the literature.18 The work of Hayward et al reports a high pressure study close to 3.2 GPa at four temperatures (between 298–130 K) going from the cubic, to tetragonal, to the orthorhombic phase. They show that the transition temperatures are reduced, and that the Ti atom displacement is smaller in the orthorhombic phase (and comparable in the tetragonal phase) compared to that at ambient pressure.36
In the present study we have used neutron-powder diffraction to follow the high-pressure structural behaviour of BaTiO3 at four temperatures (up to ∼6 GPa, in the range 175–480 K). The compressive behaviour of each of the four phases has been determined from ambient pressure, and all phase transitions in the sequence have been observed. Through advances in high pressure apparatus for neutron diffraction39 we have been able to determine more crystallographic data points with increased crystallographic precision compared to previous studies.
2 Experimental
Polycrystalline BaTiO3, obtained from Johnson Matthey Materials Technology, has been used throughout this study. The sample was further annealed to reduce microstrain in the bulk material, as has been done previously by Darlington et al.18 The reduction in micro strain is observed in the splitting of the resolved reflections in the powder diffraction pattern. The ideal unstressed tetragonal phase of BaTiO3 has a c/a ratio of 1.01, which was verified through characterisation on the high resolution powder diffractometer (HRPD) at the ISIS neutron and muon source, UK. The diffraction pattern confirmed the single phase nature of the sample with a c/a ratio of 1.01.2.1 High pressure neutron diffraction
High-pressure neutron diffraction experiments were performed on the PEARL diffractometer at the ISIS Neutron and Muon Source, UK.39 The sample was loaded into an encapsulated gasket machined from null-scattering TiZr alloy.40 In the sample chamber, a small piece of lead was included for which the pressure and temperature equation of state has been well characterised41 and perdeuterated methanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethanol (4:1) was included as a hydrostatic pressure transmitting medium.42 The gasket assembly was clamped between a pair of single toroidal profiled anvils43 machined from toughened zirconia alumina.39 The gasket and anvil assembly was then mounted in a modified V3 Paris-Edinburgh press44 which permits variation in temperature from 120–480 K. Diffraction patterns were obtained in the 90° scattering geometry, permitting access to a d-spacing range of 0.5–4.1 Å. Data of sufficient quality were collected at each pressure point to determine crystal symmetry and atomic coordinates. The few instances where the crystal symmetry could not be identified unambiguously were limited by instrument resolution; no further improvement could be gained by additional measurement time. The data were processed and corrected using Mantid,45 and the diffraction patterns were analysed by Rietveld refinement using the GSAS suite of programmes.46
:ethanol (4:1) was included as a hydrostatic pressure transmitting medium.42 The gasket assembly was clamped between a pair of single toroidal profiled anvils43 machined from toughened zirconia alumina.39 The gasket and anvil assembly was then mounted in a modified V3 Paris-Edinburgh press44 which permits variation in temperature from 120–480 K. Diffraction patterns were obtained in the 90° scattering geometry, permitting access to a d-spacing range of 0.5–4.1 Å. Data of sufficient quality were collected at each pressure point to determine crystal symmetry and atomic coordinates. The few instances where the crystal symmetry could not be identified unambiguously were limited by instrument resolution; no further improvement could be gained by additional measurement time. The data were processed and corrected using Mantid,45 and the diffraction patterns were analysed by Rietveld refinement using the GSAS suite of programmes.46
3 Results
Fig. 2 shows the regions of the BaTiO3 phase diagram for which neutron-powder diffraction data have been measured in the current study. Also shown, are the previous determined phase boundaries determined by Ishidate et al.35 The figure also shows the powder diffraction profile of characteristic peaks in the ∼2 Å region of the diffraction pattern which give can be used to fingerprint the phase present. We now present the results of the four isothermal compressions which each start in a distinct crystallographic structure of BaTiO3.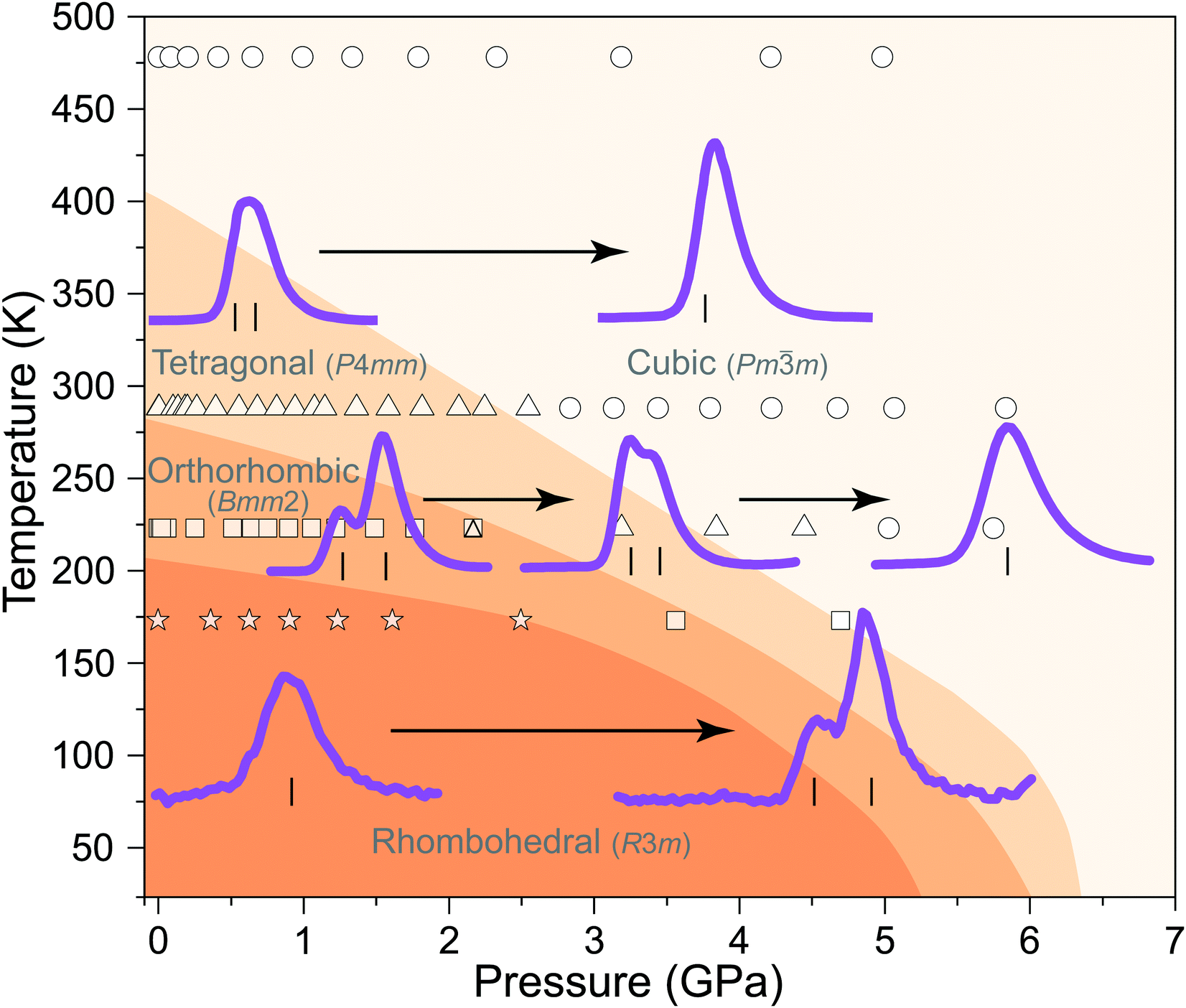 | ||
| Fig. 2 Phase diagram, of BaTiO3 as a function of pressure and temperature. The shaded regions show the phase boundaries as defined by the work of Ishidate et al.35 The open symbols show the temperatures and pressures in which the phases have been observed in the current study by neutron-powder diffraction, open circles – cubic phase, open triangles – tetragonal phase, open squares – orthorhombic phase and the open stars the rhombohedral phase. The overlapped triangle/square symbol is the region of observed phase co-existence. Also shown are the profile of the neutron diffraction pattern of each phase in the region of 2 Å, the relative intensity and shape of the diffraction peaks observed in this region are diagnostic of the phase of BaTiO3 present, the vertical tick marks show the positions of the reflections giving rise to the profile in each symmetry. There is inherent peak asymmetry to higher d-spacing in the peak profiles as a results of means by which the neutrons are produced at a t–o–f source and is clearly see in the peak profiles shown here. | ||
3.1 Compression of cubic phase at 480 K
At ambient pressure above 393 K, BaTiO3 is found in the cubic phase (space group Pmm). Throughout the present study, to aid with distortion comparisons at lower symmetry, the Ti atoms will be sited at 0,0,0 in the unit cell for all crystallographic phase. In the cubic phase the Ba is sited at ½,½,½ and the O atom at the 0,0,½ position (on Wyckoff sites 1a, 1b and 3d respectively). The structure is depicted in Fig. 1. At ambient pressure (480 K) this gives rise to an undistorted TiO6 octahedra with a Ti–O distance of 2.006 Å.
As expected the unit-cell volume reduces upon compression as shown in Fig. 3. The individual bond lengths of Ti–O and Ba–O show the same trend as the unit cell volume. Also shown in Fig. 3 is a fit to the volume by a Birch–Murnaghan equation of state (EoS). Both second- and third-order descriptions are shown for comparison and determined using the PASCal programme.47 The determined bulk modulus (B0) its pressure derivative (B′) and ambient pressure volume (V0) are detailed in Table 1. A representative diffraction pattern and associated Rietveld fit to the data are shown in Fig. 3.
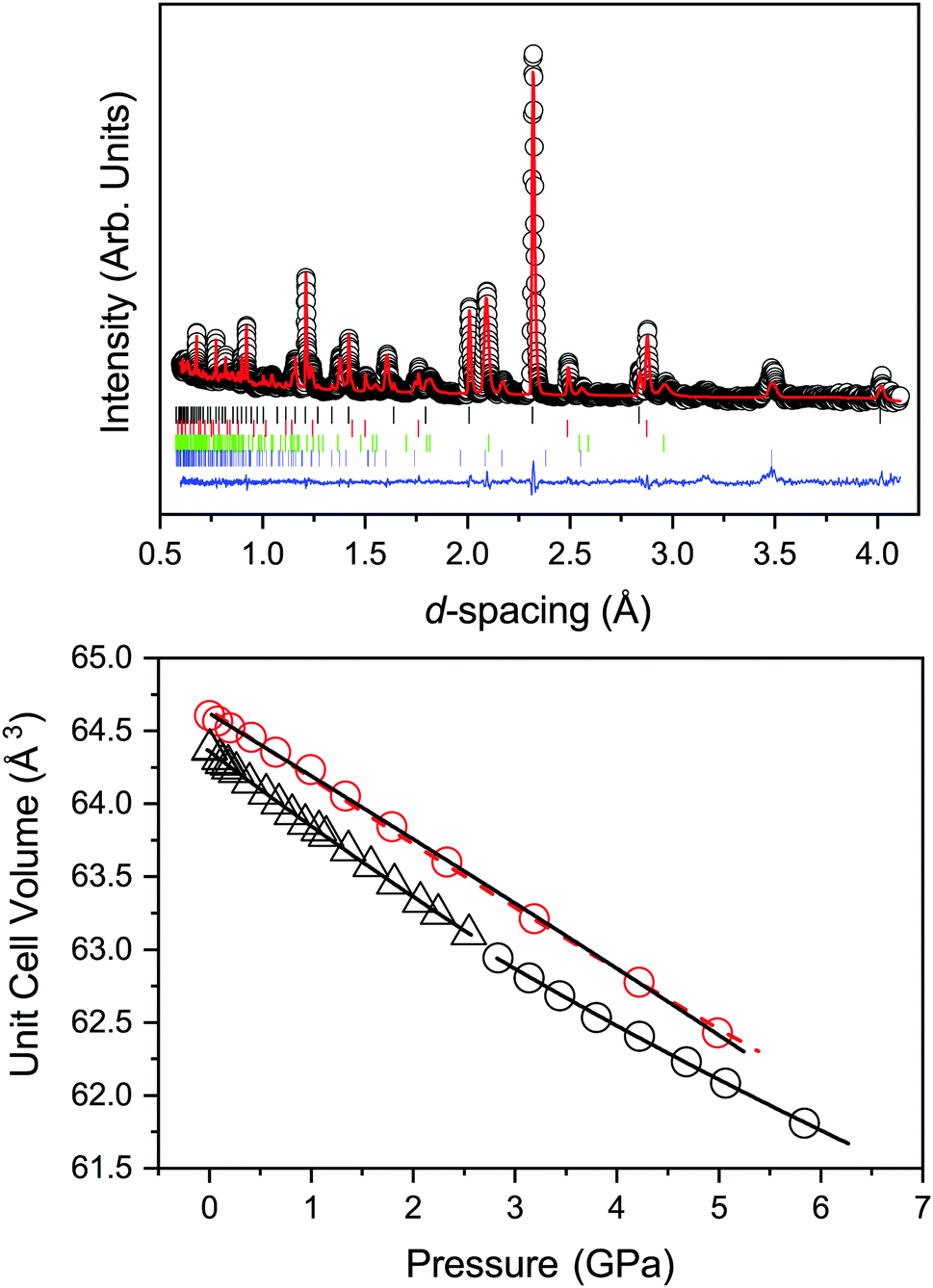 | ||
| Fig. 3 Top: Neutron powder diffraction pattern with Rietveld refined profile for cubic BaTiO3 at 0.1 GPa and 480 K. Data are shown as open black circles, the calculated profile in red, and the residual in blue. The tick marks show the reflection positions for (from top to bottom) cubic BaTiO3 (black), Pb (red), Al2O3 (green), and ZrO2 (blue). Bottom: Variation in unit-cell volume of BaTiO3 with increasing pressure at 480 and 290 K. The open red circles show the experimental data for the cubic phase at 480 K, the open black squares and black circles are the tetragonal and cubic phases at 290 K respectively. The solid lines show the fit to the data by a 3rd order Birch–Murnaghan equation of state and for the 480 K data set the dashed red line is the fit of a 2nd order Birch–Murnaghan equation of state to the data. Values for the equation of states are given in Table 1. | ||
| Temp. (K) | 2nd Order | 3rd Order | B′ | ||
|---|---|---|---|---|---|
| V 0 (Å) | B 0 (GPa) | V 0 (Å) | B 0 (GPa) | ||
| 480 (c) | 64.64(1) | 136(2) | 64.62(1) | 152(4) | −3(1) |
| 290 (t) | 64.36(1) | 124.6(7) | 64.36(1) | 122(2) | 7(2) |
| 290 (c) | 64.08(2) | 150(2) | 64.22(15) | 127(23) | 10(6) |
| 225 (o) | 128.63(1) | 116(2) | 128.14(1) | 109(4) | 14(6) |
| 225 (t) | 64.09(9) | 142(8) | — | — | — |
| 175 (r) | 64.26(1) | 124(2) | — | — | — |
| Temp. (K) | X 1 K (TPa−1) | X 2 K (TPa−1) | X 3 K (TPa−1) |
|---|---|---|---|
| 480 (c) | 2.29(5) | 2.29(5) | 2.29(5) |
| 290 (t) | 3.95(4) | 1.85(1) | 1.85(1) |
| 290 (c) | 1.91(4) | 1.91(4) | 1.91(4) |
| 225 (o) | 4.01(9) | 2.57(9) | 1.60(7) |
| 225 (t) | 2.45(26) | 1.82(3) | 1.20(41) |
| 175 (r) | 2.74(21) | 2.37(18) | 2.71(20) |
3.2 Compression of tetragonal phase at 290 K
At 290 K, the ambient pressure phase of BaTiO3 is tetragonal (P4mm). The origin of the polar space group was set by fixing Ti at the 0,0,0 position, Ba sited at ½,½,½ + δza and the O atoms described by 0,0,½ + δz1 and 0,½,δz2. The Ba and O atoms are constrained by symmetry to displace along the z-direction only (relative to the cubic structure by δza, δz1,2 respectively). The oxygen atoms move around the fixed central Ti atom forming the TiO6 octahedra. In this phase the TiO6 octahedra are distorted so that there are three distinct bond lengths within the octahedra with one long (2.20 Å), four medium (2.00 Å), and one short (1.84 Å) bond lengths at ambient pressure (see Fig. 1).The variation of the tetragonal unit-cell volume on compression is shown in Fig. 3. As reported previously, at ∼2.1 GPa there is a transition from the tetragonal to cubic phase (see Fig. 2).35 The difference in the diffraction pattern of the two phases is evident in the region around the h00 and 00h reflections in the tetragonal phase in comparison to the 00h reflection of the cubic phase, in the d-spacing range of the PEARL instrument this is most evident in 200 and 002 reflections centred around ∼1.98 Å (Fig. 2). At 2.6 GPa it is clear that there is a splitting of the reflection indicative of the tetragonal phase, however, at the next pressure point (2.8 GPa) no such splitting is observed and hence the data are fit to the cubic structure and the peak is indexed as the cubic 200 reflection (see Fig. 4). To look at the nature of this transition the ratio of the axis lengths a and c of the unit cell (a value of 1 indicating cubic symmetry) can be plotted as function of pressure (see Fig. 4). There is a smooth reduction in the c/a ratio, and above ∼2.6 GPa the value suddenly decreases to 1 over a pressure step of 0.2 GPa. The variation in unit-cell volume with pressure has also been fitted with a Birch–Murnaghan equation of state in both the tetragonal and cubic phase at 290 K the determined values are detailed in Table 1 and the fit shown in Fig. 3.
 | ||
Fig. 4 Top left panel: Variation in c/a ratio of BaTiO3 in the tetragonal phase (open triangle) at 290 K with increasing pressure. The c/a ratio goes to unity upon transformation to cubic symmetry (open circle). The insets show the diffraction pattern and Rietveld fits in the region around 2 Å – (A) fit to the diffraction pattern at 2.5 GPa with tetragonal structure, (B) fit to the diffraction pattern at 2.7 GPa with tetragonal structure and (C) same diffraction pattern fitted with the cubic structure. Top right panel: Variation in the Ti–O distances within the TiO6 octahedra with increasing pressure at 290 K. Up to 2.6 GPa three distinct Ti–O distances are present in the tetragonal phase (Ti–O1 (black squares), Ti–O1 (red squares) and Ti–O2 (blue triangles)) and for the cubic phase only one Ti–O bond distance (open triangles) is found in the undistorted octahedra. In both panels the vertical dotted line shows the phase boundary between the tetragonal and cubic phase. Bottom right panel: Variation in axial length ratios of BaTiO3 upon compression at 225 K. The orthorhombic ratio 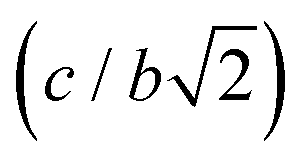 as open squares, the c/a ratio for the tetragonal phase as open triangles. The c/a ratio goes to unity upon transformation to cubic symmetry (open circle). Dashed vertical lines indicate the phase boundaries. Also shown are the diffraction patterns (and associated Rietveld fits) from the region around 2 Å for the orthorhombic phase labelled (D and E) unfitted mixed phase of orthorhombic and tetragonal, (F) tetragonal phase and (G) cubic phase. Bottom right panel: Variation in the Ti–O bond distances in the TiO6 octahedra in the orthorhombic phase of BaTiO3 with increasing pressure at 225 K. as open squares, the c/a ratio for the tetragonal phase as open triangles. The c/a ratio goes to unity upon transformation to cubic symmetry (open circle). Dashed vertical lines indicate the phase boundaries. Also shown are the diffraction patterns (and associated Rietveld fits) from the region around 2 Å for the orthorhombic phase labelled (D and E) unfitted mixed phase of orthorhombic and tetragonal, (F) tetragonal phase and (G) cubic phase. Bottom right panel: Variation in the Ti–O bond distances in the TiO6 octahedra in the orthorhombic phase of BaTiO3 with increasing pressure at 225 K. | ||
Fig. 4 shows the variation in the three Ti–O bond distances with increasing pressure of both the tetragonal and cubic phase at 290 K. It can be seen that with increasing pressure the three different Ti–O bonds in the tetragonal phase converge towards a value similar to that of the singular value of the cubic phase value of ∼2 Å. A table of all refined structural parameters derived from diffraction data described in this work are given in the ESI.†
3.3 Compression of orthorhombic phase at 225 K
At 225 K and ambient pressure, BaTiO3 is orthorhombic, Bmm2 (a non standard setting used to maintain consistency with the earlier literature) and contains two formula units. The origin of the polar space group was set by fixing Ti at the 0,0,0 position, with Ba at ½,½,δza and O1 at 0,½,δz1 and O2 at ¼ + δx2,0, ¼ + δz2. The displacement of the O and Ba atoms relative to the cubic phase are described by the displacement za for the Ba and by δz1, δx2 and δz2 for the O atoms. Refinement of the atomic positions permits the Ba atom to move in the z-direction only, and the O atoms to move around the fixed central Ti atom. In this phase the TiO6 octahedra possess three distinct bond lengths with two long (2.14 Å), two short (1.88 Å) and two medium (1.99 Å) length bonds at ambient pressure (see Fig. 1). The degrees of freedom of the O atoms also permits the O–Ti–O bond angles with the octahedra to no longer be 180° and at ambient pressure are ∼176°. In terms of the diffraction pattern of the structure it is visually differentiable from the other phases by the relative intensities of the reflections for example the 020/202 reflections centred around 1.95 Å show a distinctly different intensity ratio to that of the equivalent (200/002) reflections in the tetragonal phase at ambient pressure (Fig. 2).As expected, the volume (per formula unit) of the orthorhombic phase of BaTiO3 at 225 K decreases with pressure (see ESI†). The variation in unit-cell volume can be described by a Birch–Murnaghan EoS as detailed in Table 1. The change in axial-length ratio of the orthorhombic phase is shown in Fig. 4 and decreases linearly with increasing pressure. At 2.2 GPa there is a change in the diffraction pattern that cannot be described by only the orthorhombic phase but as a mixed phase with coexistence of the tetragonal and orthorhombic phase (see inset Fig. 4). Beyond this pressure the pattern is described by the tetragonal phase and the c/a ratio decreases indicating decrease of unit cell distortion and shown in Fig. 4. At 5 GPa however, it is possible to fit the data set with either the cubic or tetragonal symmetry with the same overall quality of fit indicators. Hence in Fig. 4 a value for the c/a ratio of 1.002 is shown but also 1. As the data can be fitted equally well with a higher symmetry and visually the fit around the region of the 200 reflection is better. We suggest that the 5 GPa data set is actually in the cubic phase. However, it should be noted that this result is governed by the resolution limit of the current measurement using the PEARL instrument. With increasing pressure the Ti–O bonds of the TiO6 octahedra in the octahedral phase tend towards a reduced variance of values (Fig. 4).
3.4 Compression of rhombohedral phase
Below 215 K at ambient pressure BaTiO3 is in the rhombohedral phase (R3m). The polar direction is along [111] and hence the Ti is located at 0,0,0, the displacement of the Ba atom described by ½ + δxa,½ + δxa,½ + δxa and that of the oxygen described as 0 + δx1,½ + δy1, ½ + δx1, where δxa, δx1 and δy1 are the displacements which give rise to the dipole (Fig. 1). The TiO6 octahedron contain three short, 1.92(2) Å, and three longer, 2.09(2) Å, Ti–O bonds.In contrast to the work of Hayward et al, the study of the rhombohedral phase has been approached by cooling at ambient pressure. In terms of diffraction, this phase is significantly different to that of the orthorhombic phase at the same pressure see Fig. 2.36 The sharp 200 reflection is observed at ∼2 Å in contrast to the split 020/202 reflections of the orthorhombic phase (see Fig. 2).
With increasing pressure the unit-cell volume reduces with increasing pressure at 175 K (see ESI†). There are limited data points and hence only a second order Birch–Murngahan equation of state can be fitted reliably, results detailed in Table 1. Above 2.5 GPa there is a change in the diffraction pattern and it cannot be described by the rhombohedral phase. Fig. 5 shows how the diffraction pattern around the region of the 200 reflection of the rhombohedral phase evolves with increasing pressure, showing the transformation to the orthorhombic phase and the corresponding fit. Although the diffraction patterns up to ∼2.5 GPa are fitted with the rhombohedral phase of BaTiO3, with increasing pressure the results of the refinements show no observable trend in bond length behaviour.
 | ||
| Fig. 5 Variation in diffraction pattern and associated Rietveld fit of BaTiO3 with increasing pressure at 175 K in region of 2 Å. From top to bottom data collected at 2.5, 3.6 and 4.7 GPa. In each row there is a fit to the data with the rhombohedral phase (left panel) and the orthorhombic phase (right panel). In each plot the data is shown as the open circles, the Rietveld fit as the solid red line and the blue trace the residual of the fit. The vertical tick marks show the reflection positions for the 200 reflection for the rhombohedral phase and 020/202 reflections for the orthorhombic phase. At 2.5 GPa, the similarity in fit at 1.98 Å leads us to rely on the fitting metrics for the overall pattern instead. At the other pressures, the difference in visual fit quality is clear. | ||
The compressive behaviour of BaTiO3 was also performed at 120 and 155 K however, in both experiments crystalline methanol was also formed upon compression. The presence of the crystalline methanol in the diffraction profile hindered refinement of the data as a result of peak overlap, however, it was possible to determine that no phase transition to the orthorhombic phase was observed up to a pressure of 4.8 GPa at 120 K.
4 Discussion
4.1 Compression behaviour of BaTiO3
The bulk modulus of each of the phases of BaTiO3 has been determined and are detailed in Table 1. There are some interesting anomalies upon compression of some of the phases. The negative B' of the cubic (Pmm) phase at 480 K suggests abnormal softening of the elastic modulus with compression. Similar behaviour has been seen in other materials for example ZrO2.48 We suggest that this softening could described by one of three possible mechanisms. As described above and in detail elsewhere19,21 BaTiO3 has a local structure which is disordered, even in the cubic phase, though our crystal structure parameterisation assumes perfect ordering with no local distortions. The appropriateness of this approximation depends on the length scales being considered, which in the present study may be insufficient to ignore the effects of local distortion. A gradual change in the local structure in the cubic phase with increasing pressure may be the cause for the observed softening of the material. A second explanation is that softening of the material can be a prelude to a phase transition, however, this softening is seen throughout the pressure range studied at 480 K. A third explanation may be that that there are anomalies in the higher elastic constants in cubic BaTiO3 which may result in a negative B′. Anomalies have already been described for the tetragonal phase of BaTiO3 which is also locally disordered. Brillouin scattering from the cubic phase49 suggests large pressure gradients in shear wave velocities with negative curvature, such anomalous behaviour may well be a result of “soft mode” behaviour, often observed in high-symmetry phases near the critical pressure of a second order phase transition.50
At 290 K we note that 2nd order B0 is higher in the cubic phase, 150(2) GPa, compared to that of the tetragonal phase, 124.6(7) GPa (see Table 1). This observation is corroborated by sensitive ultrasonic measurements,51 with values at 290 K of 139.2(3.1) GPa and 195.0(5.0) GPa for the tetragonal and cubic phase respectively.51 Previous Brillouin scattering measurements at as function of pressure gave a B0 of 139.4 GPa.49 We have also determined the derivative of the bulk modulus to be ∼7 for the tetragonal phase and the previous study by Fischer et al derive a value of 10.51 We note that the number of data points determined in each phase at 290 K in particular the cubic phase may not be sufficient to support the fit by a 3rd order EoS and the determined values are only shown for completeness and comparison. However, the value of B0 derived from the third order fit is closer in value to that derived previously by a X-ray diffraction study of 135 GPa with a derivative of 6.4 (errors not provided for either value).31
The orthorhombic phase appears to be more compressible than the tetragonal and cubic phases with a bulk modulus of 116(2) GPa compared to the tetragonal phase at 225 K with B0 of 142(8) GPa. It should be noted that there are fewer data points for each phase with decreasing temperature with which to determine the compressibility of each phase and this may well affect the fitting of the data to an equation of state. Two separate theoretical determinations of B0 give values of 135 GPa and 97.5 GPa.52,53
The bulk modulus of the rhombohedral phase of BaTiO3 at 175 K can only be reliably fitted with 2nd order Birch–Murnaghan EoS. Fitting with a 3rd order EoS gives a negative value of B′. This result is likely to be a result of the number of data points measured and the small pressure range over which the phase exists. The determined values are in close agreement with those determined by computational methods who predict values 126 GPa and 103.5 GPa.52,53 By visual inspection of the data it may be that some softening of the bulk modulus may be occurring with increasing pressure, although not as pronounced as the numerical fitting may suggest, the individual median compressibilities are given in Table 1 and can be compared for all phases at each temperature measured.
4.2 Phase transitions of BaTiO3
As previously discussed the ideal value for the tetragonal c/a ratio at 290 K is 1.01. The c/a ratio is a means to describe the strain in the system (and in the ESI,† the variation in spontaneous strain for the transition observed is detailed). The abrupt change in the c/a ratio from 1.004 to 1 is consistent with a first order improper ferroelastic transition, caused by a discontinuity in the order parameter (the polarisation). We also note that there is a disagreement in the transition pressure relative to that observed by other methods with the value ranging from 1.6 GPa,51 1.9 GPa,54 2.35 GPa49 and ∼2.6 GPa (this study). These discrepancies may be a result of the different measurement techniques sensitivities to the transition, rates of compression (which are relatively slow in the current study), sample volume effects and potentially temperature effects as the Clapeyron slope of the tetragonal to cubic phase transition is quite steep with a value of −46 K GPa−1.54 However, our value is in agreement with that determined from Raman spectroscopy by Meja-Uriarte et al, 2–2.8 GPa.32At 225 K BaTiO3 is orthorhombic, but at 2.2 GPa the diffraction pattern can be indexed as mixed phase containing both orthorhombic and tetragonal phases, consistent with a first order transition. The pressure of this transition is in close agreement with that reported previously of 2.2 GPa (Fig. 2).35 The subsequent transition from tetragonal to cubic symmetry is also first-order like in nature. The exact pressure at which it occurs is harder to determine as the differentiation between the cubic and tetragonal phase becomes limited by the instrument ability to resolve the 200/002 reflections in the tetragonal phase from the 200 reflection in the cubic phase (Fig. 4). The evidence suggests that the tetragonal-cubic phase transition at 225 K occurs just before 5 GPa and is again first order in nature as the behaviour of the c/a ratio is similar (Fig. 4) within the pressure step size used in the current study and if the transition was second order a more gradual change in c/a ratio would be observed (over a wide pressure range). It has been discussed in the literature that the transitions of BaTiO3 at pressure become more second-order like in nature however, we suggest the transition remains very much first-order like in nature.29 However, this transition appears to occur at a higher pressure than that reported previously by Ishidate et al but again may be for similar reasons to that given above such as errors in reported pressure, sample volume effects and measurement specific sensitivities (see Fig. 2).35
At ambient pressure and 175 K the material is observed to be in the rhombohedral phase, at 3.6 GPa the diffraction pattern shows a change in profile, in particular around the reflection centred around 2.00 Å. Fig. 5 shows the diffraction pattern at 175 K at a series of pressures as it transforms from the rhombohedral phase to the orthorhombic phase. Shown at 2.5, 3.6 and 4.7 GPa are the Rietveld profile fit to the measured diffraction patterns with both the rhombohedral and orthorhombic structures in the diagntostic region around 2 Å. At 2.5 GPa there is very little to differentiate the visually fits between the two phases shown in Fig. 5 however, fitting with the orthorhombic phase gives a fit with a significant higher R-factor for the fit of the whole pattern compared to that for the rhombohedral phase fit. At 3.6 GPa there is no discernible difference in the overall quality of fit metrics of the Rietveld refinement of diffraction pattern to either the orthorhombic or rhombohedral phase and both show similar misfit across the whole diffraction pattern and the more complex profile suggests a mixed phase region is being observed at this pressure, with a small improvement in the fit of the orthorhombic phase across the whole diffraction pattern. At 4.7 GPa it becomes clear across the whole diffraction pattern that it is best fitted with the orthorhombic structure as shown in Fig. 5.
The phase diagram of BaTiO3 from previous studies is shown in Fig. 2 with the addition of the data described in this manuscript. The phase transition borders are in reasonable agreement with those determined experimentally previously by Ishidate et al.35 and to a lesser extent that described by Landau theory work of Hayward and Salje.38 In the current study some of the phases are found to exist at higher pressures than suggested by other techniques, however, this is not uncommon as described above. The fact that we do not observe some of the phase transitions upon compression at lower temperature does not indicate they do not exist it is simply we were unable to compress the sample to the required pressure to induce the structural transformation at the given temperature.
4.3 Absolute displacements and spontaneous polarisation
The spontaneous polarisation (P) can be determined using the expression below | (1) |
 is the apparent charge of atom j and have the value of +3.6 for Ba, +5.7 for Ti and −3.1 for O.55 These values differ from the formal valencies (+2, +4 and −2) as the ions are not rigid and are polarised in the field as result of their displacements.56Δj is the absolute displacement from the high symmetry position in the cubic phase. e is the coulombic charge (1.9 × 10−19 C) and Vc the volume occupied by one formula unit.
is the apparent charge of atom j and have the value of +3.6 for Ba, +5.7 for Ti and −3.1 for O.55 These values differ from the formal valencies (+2, +4 and −2) as the ions are not rigid and are polarised in the field as result of their displacements.56Δj is the absolute displacement from the high symmetry position in the cubic phase. e is the coulombic charge (1.9 × 10−19 C) and Vc the volume occupied by one formula unit.
To calculate the absolute displacements (Δj) we make the assumption that the phase transitions are driven by the softening of a transverse optic mode at the zone centre (as discussed in detail by Senn et al.24) and hence, it is possible to determine, relative to the high symmetry form, the absolute displacement in the direction of the polarisation (Δ). We assume that the centre of mass of the unit cell is invariant55 for a soft mode transitions and hence for the sum of atoms (j) displaced with mass (mj) where mBa = 137.33, mTi = 47.867 and mO = 15.999,
 | (2) |
The refinement of the crystal structure only yields relative atomic displacements:
 | (3) |
 | (4) |

These calculations require the space groups of the three distinct ferroelectric phases to be polar and for the tetragonal and orthorhombic phase the polar direction to be along the c-axis and for the rhombohedral phase to be polar along the [111] axis. Absolute co-ordinates for each phase are defined in Table 2.
| Atom | x | y | z |
|---|---|---|---|
| Tetragonal | |||
| Ba | a/2 | a/2 | c/2 + ΔBa |
| Ti | 0 | 0 | Δ Ti |
| O1 | 0 | 0 | c/2 + ΔO1 |
| O2 | 0 | a/2 | Δ O2 |
| Orthorhombic | |||
| Ba | a/2 | b/2 | Δ Ba |
| Ti | 0 | 0 | Δ Ti |
| O1 | 0 | b/2 | Δ O1 |
| O2 | a/4 + ΔO2x | 0 | c/4 + ΔO2z |
| Rhombohedral | |||
| Ba | a/2 + ΔBa | a/2 + ΔBa | a/2 + ΔBa |
| Ti | Δ Ti | Δ Ti | Δ Ti |
| O1 | Δ O1x | 1/2 + ΔO1y | Δ O1x |
For the orthorhombic phase δO2x is perpendicular to the polar axis and makes no contribution to the calculation of the relevant centre of mass, there is also a doubling of the pseudo-cubic unit-cell volume and mass and hence the number of ferroelectric displacements however, these cancel each other out in the determination of K. In the rhombohedral phase the polar axis is along the [111] axis and as a result the determination of the displacements with respect to the polar axis requires taking the scalar product of the displacement vector of the site with the polar axis giving
 | (5) |
 | (6) |
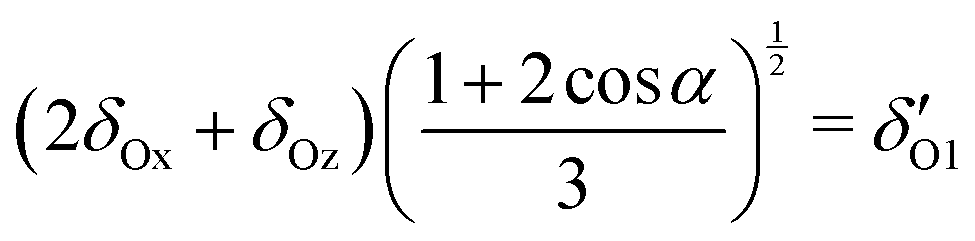 | (7) |
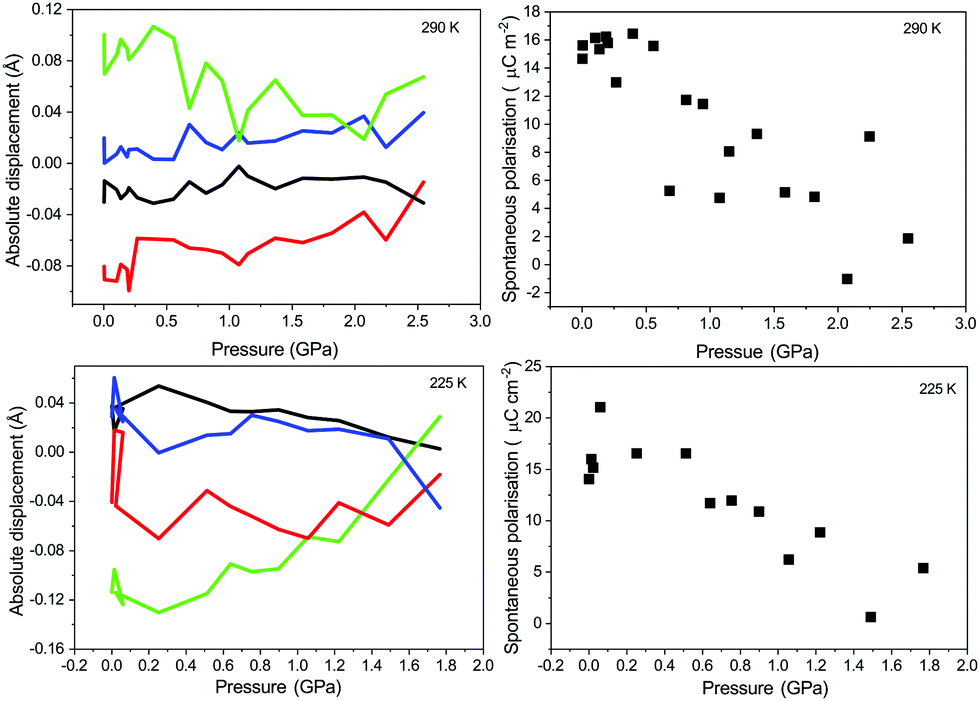 | ||
| Fig. 6 Top left: Variation in absolute displacements of the atoms with BaTiO3 at 290 K in the tetragonal phase with increasing pressure. Bottom left: Variation in absolute displacements of the atoms with BaTiO3 at 225 K in the orthorhombic phase with increasing pressure. In both panels the black line represents absolute displacement of the barium atom, green of the titanium atom, and the displacements of the oxygen atoms O1 and O2 by the red and blue lines respectively. Top left: derived spontaneous polarisation of BaTiO3 with increasing pressure in the tetragonal phase at 290 K. Bottom right: derived spontaneous polarisation of BaTiO3 with increasing pressure in the tetragonal phase at 225 K. | ||
An approximate linear decrease can be seen in the spontaneous polarisation with increasing pressure for the tetragonal and orthorhombic phase at 290 and 225 K respectively. The determined values are in reasonable agreement with those published before and show that the spontaneous polarisation for the tetragonal and orthorhombic phase do not differ significantly in magnitude at ambient pressure. The discrepancy between the values compared to those previously determined by other methods may be a result of the definition of the formal charge and in previous works attempting a similar method to that presented here similar discrepancies were observed.18 The point-charge model only considers the nuclear positions and does not account for the electronic component of the polarization. For polar space groups where the origin is not rigidly defined along the polar axis or plane, as may be the case with a number of previous studies of BaTiO3, the determined atomic displacements will be erroneous, potentially confusing interpretation of the results.
5 Conclusions
The high-pressure behaviour of the archetypal ferroelectric material BaTiO3 has been studied using neutron-powder diffraction. Each of the four known crystallographic phases (found in the temperature range 120–480 K) have been compressed from ambient pressure. Upon compression of the lower-symmetry forms we have been able to observe transitions to the higher symmetry forms and have seen that there is significant first order like behaviour. The compressibilities of each phase have been determined, along with the variation in strains and polarisability which gives rise to the ferroelectric properties of BaTiO3. We see a linear decrease in the strain and polarisability with increasing pressure within each polar phase compressed. Combined, the results presented shed new light on the nature of the pressure driven phase transitions in BaTiO3 and the phase diagram of this archetypal ferroelectric material.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank the ISIS neutron facility (part of the Science and Technology Facilities Council) for access to the PEARL instrument.57 Mara Capone is thanked for her help in collecting some of the data. We also acknowledge the technical support of Chris Goodway (Pressure and Furnace Section, ISIS) and the facilities provided by the Materials Characterisation Laboratory of ISIS and the instrument support provided within by Gavin Stenning and Daniel Nye (MCL, ISIS).References
- A. Devonshire, London Edinburgh Philos. Mag. J. Sci., 1949, 40, 1040–1063 CrossRef CAS.
- A. von Hippel, R. G. Breckenridge, F. G. Chesley and L. Tisza, Ind. Eng. Chem., 1946, 38, 1097–1109 CrossRef CAS.
- M. Lines and A. Glass, Principles and Applications of Ferroelectrics and Related Materials, OUP, Oxford, 2001 Search PubMed.
- J. M. Ballantyne, Phys. Rev., 1964, 136, A429–A436 CrossRef.
- G. Shirane, B. C. Frazer, V. J. Minkiewicz, J. A. Leake and A. Linz, Phys. Rev. Lett., 1967, 19, 234–235 CrossRef.
- P. A. Fleury and P. D. Lazay, Phys. Rev. Lett., 1971, 26, 1331–1334 CrossRef CAS.
- M. Fontana and M. Lambert, Solid State Commun., 1972, 10, 1–4 CrossRef CAS.
- A. Chaves, R. S. Katiyar and S. P. S. Porto, Phys. Rev. B: Condens. Matter Mater. Phys., 1974, 10, 3522–3533 CrossRef CAS.
- A. Scalabrin, A. S. Chaves, D. S. Shim and S. P. S. Porto, Phys. Status Solidi B, 1977, 79, 731–742 CrossRef CAS.
- N. Baskaran, A. Ghule, C. Bhongale, R. Murugan and H. Chang, J. Appl. Phys., 2002, 91, 10038–10043 CrossRef CAS.
- A. S. Barker, Phys. Rev., 1966, 145, 391–399 CrossRef CAS.
- H. D. Megaw and J. D. Bernal, Proc. R. Soc. London, Ser. A, 1947, 189, 261–283 CAS.
- H. T. Stokes, D. M. Hatch and H. M. Nelson, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 9080–9083 CrossRef PubMed.
- R. G. Rhodes, Acta Crystallogr., 1951, 4, 105–110 CrossRef CAS.
- H. Kay and P. Vousden, London Edinburgh Philos. Mag. J. Sci., 1949, 40, 1019–1040 CrossRef CAS.
- G. Shirane, H. Danner and R. Pepinsky, Phys. Rev., 1957, 105, 856–860 CrossRef CAS.
- K. S. Knight, Can. Mineral., 2009, 47, 381–400 CrossRef CAS.
- C. N. W. Darlington, W. I. F. David and K. S. Knight, Phase Transitions, 1994, 48, 217–236 CrossRef CAS.
- Q. Zhang, T. Cagin and W. A. Goddard, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14695–14700 CrossRef CAS PubMed.
- R. Comes, M. Lambert and A. Guinier, Solid State Commun., 1968, 6, 715–719 CrossRef CAS.
- G. H. Kwei, S. J. L. Billinge, S.-W. Cheong and J. G. Saxton, Ferroelectrics, 1995, 164, 57–73 CrossRef CAS.
- B. Ravel, E. A. Stern, R. I. Vedrinskii and V. Kraizman, Ferroelectrics, 1998, 206, 407–430 CrossRef.
- B. Jannot, C. Escribe-Filippini and J. Bouillot, J. Phys. C-Solid State Phys., 1984, 17, 1329–1337 CrossRef CAS.
- M. S. Senn, D. A. Keen, T. C. A. Lucas, J. A. Hriljac and A. L. Goodwin, Phys. Rev. Lett., 2016, 116, 207602 CrossRef CAS PubMed.
- Y.-T. Shao and J.-M. Zuo, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2017, 73, 708–714 CrossRef CAS PubMed.
- P. S. Peercy and G. A. Samara, Phys. Rev. B: Condens. Matter Mater. Phys., 1972, 6, 2748–2751 CrossRef CAS.
- M. Fischer and A. Polian, Phase Transitions, 1987, 9, 205–213 CrossRef CAS.
- A. K. Sood, N. Chandrabhas, D. V. S. Muthu and A. Jayaraman, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 8892–8896 CrossRef CAS PubMed.
- G. A. Samara, Phys. Rev., 1966, 151, 378–386 CrossRef CAS.
- D. L. Decker and Y. X. Zhao, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39, 2432–2438 CrossRef CAS PubMed.
- P. Pruzan, D. Gourdain, J. Chervin, B. Canny, B. Couzinet and M. Hanfland, Solid State Commun., 2002, 123, 21–26 CrossRef CAS.
- E. V. Mejía-Uriarte, R. Y. Sato-Berrú, M. Navarrete, M. Villagrán-Muniz, C. Medina-Gutiérrez, C. Frausto-Reyes and H. M. S, Meas. Sci. Technol., 2006, 17, 1319–1323 CrossRef.
- M. Villagran-Muniz, M. Navarrete and E. V. Mejia-Uriarte, Rev. Sci. Instrum., 2003, 74, 732–734 CrossRef CAS.
- U. D. Venkateswaran, V. M. Naik and R. Naik, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 14256–14260 CrossRef CAS.
- T. Ishidate, S. Abe, H. Takahashi and N. Môri, Phys. Rev. Lett., 1997, 78, 2397–2400 CrossRef CAS.
- S. A. Hayward, S. A. T. Redfern, H. J. Stone, M. G. Tucker, K. R. Whittle and W. G. Marshall, Z. Kristallogr. Cryst. Mater., 2005, 220, 739 Search PubMed.
- D. Gourdain, E. Moya, J. C. Chervin, B. Canny and P. Pruzan, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 3108–3112 CrossRef CAS PubMed.
- S. A. Hayward and E. K. H. Salje, J. Phys.: Condens. Matter, 2002, 14, L599–L604 CrossRef CAS.
- C. L. Bull, N. P. Funnell, M. G. Tucker, S. Hull, D. J. Francis and W. G. Marshall, High Press. Res., 2016, 36, 493–511 CrossRef.
- W. G. Marshall and D. J. Francis, J. Appl. Crystallogr., 2002, 35, 122–125 CrossRef CAS.
- A. D. Fortes, RAL Technical Reports, 2019, RAL-TR-2019-002, year.
- S. Klotz, J. C. Chervin, P. Munsch and G. L. Marchand, J. Phys. D: Appl. Phys., 2009, 42, 075413 CrossRef.
- L. G. Khovstantsev, High Temp. - High Pressures, 1984, 16, 165 Search PubMed.
- J. M. Besson, R. J. Nelmes, G. Hamel, J. S. Loveday, G. Weill and S. Hull, Physica B, 1992, 180, 907–910 CrossRef.
- O. Arnold, J. C. Bilheux, J. M. Borreguero, A. Buts, S. I. Campbell, L. Chapon, M. Doucet, N. Draper, R. Ferraz Leal, M. A. Gigg, V. E. Lynch, A. Markvardsen, D. J. Mikkelson, R. L. Mikkelson, R. Miller, K. Palmen, P. Parker, G. Passos, T. G. Perring, P. F. Peterson, S. Ren, M. A. Reuter, A. T. Savici, J. W. Taylor, R. J. Taylor, R. Tolchenov, W. Zhou and J. Zikovsky, Nucl. Instrum. Methods Phys. Res., Sect. A, 2014, 764, 156–166 CrossRef CAS.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- M. J. Cliffe and A. L. Goodwin, J. Appl. Crystallogr., 2012, 45, 1321–1329 CrossRef CAS.
- M. Fujimoto, Y. Akahama, H. Fukui, N. Hirao and Y. Ohishi, AIP Adv., 2018, 8, 015310 CrossRef.
- T. Ishidate and S. Sasaki, Phys. Rev. Lett., 1989, 62, 67–70 CrossRef CAS PubMed.
- S. Ghose, Min. Soc. Am., 1985, 127–163 CAS.
- G. J. Fischer, Z. Wang and S.-I. Karato, Phys. Chem. Miner., 1993, 20, 97–103 CrossRef CAS.
- J. J. Wang, F. Y. Meng, X. Q. Ma, M. X. Xu and L. Q. Chen, J. Appl. Phys., 2010, 108, 034107 CrossRef.
- M. Uludogan, T. Cagin and W. A. Goddard, MRS Proce., 2002, 718, D10.1 CrossRef.
- M. Malinowski, K. Uukaszewicz and S. Åsbrink, J. Appl. Crystallogr., 1986, 19, 7–9 CrossRef CAS.
- A. W. Hewat, J. Phys. C: Solid State Phys., 1973, 6, 1074–1084 CrossRef CAS.
- W. Cochran, Adv. Phys., 1960, 9, 387–423 CrossRef CAS.
- C. L. Bull, K. S. Knight, C. J. Ridley and A. Gibbs, STFC ISIS Neutron and Muon Source, 2017 DOI:10.5286/ISIS.E.RB1710467.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00651g |
‡ The constant K is required as a zone centre transition requires the centre of mass to be undisplaced i.e.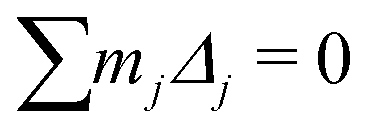 and in the crystallographic measurement we define an origin arbitrarily along the polar axis and hence when Δ is non-zero it is necessary to add a constant displacement term to each atom such that the atom with apparent zero displacement (the origin setting atom) is actually displaced too. and in the crystallographic measurement we define an origin arbitrarily along the polar axis and hence when Δ is non-zero it is necessary to add a constant displacement term to each atom such that the atom with apparent zero displacement (the origin setting atom) is actually displaced too. |
| This journal is © The Royal Society of Chemistry 2021 |