
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBifunctional electrochemical properties of La0.8Sr0.2Co0.8M0.2O3−δ (M = Ni, Fe, Mn, and Cu): efficient elemental doping based on a structural and pH-dependent study†
J.
Cheng
 *ab,
P.
Ganesan
a,
Z.
Wang
b,
M.
Zhang
c,
G.
Zhang
d,
N.
Maeda
a,
J.
Matsuda
ae,
M.
Yamauchi
af,
B.
Chi
b and
N.
Nakashima
*a
*ab,
P.
Ganesan
a,
Z.
Wang
b,
M.
Zhang
c,
G.
Zhang
d,
N.
Maeda
a,
J.
Matsuda
ae,
M.
Yamauchi
af,
B.
Chi
b and
N.
Nakashima
*a
aInternational Institute for Carbon-Neutral Energy Research (I2CNER), Kyushu University, 744 Motooka, Nishi-ku, Fukuoka, 819-0395, Japan. E-mail: cheng.junfang.423@m.kyushu-u.ac.jp; pandian.ganesan.631@m.kyushu-u.ac.jp; nakashima.naotoshi.614@m.kyushu-u.ac.jp
bCenter for Fuel Cell Innovation, State Key Laboratory of Material Processing and Die & Mould Technology, School of Materials Science and Engineering, Huazhong University of Science & Technology, Wuhan, 430074, China
cCAS Key Laboratory of Design and Assembly of Functional Nanostructures, and Fujian Provincial Key Laboratory of Nanomaterials, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, China
dDepartment of Applied Chemistry, Graduate School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
eInternational Research Center for Hydrogen Energy, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka, 819-0395, Japan
fAdvanced Institute for Materials Research (WPI-AIMR), Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai, 980-8577, Japan
First published on 13th October 2021
Abstract
Perovskite oxides with a low cost and high catalytic activity are considered as suitable candidates for the oxygen evolution reaction (OER)/oxygen reduction reaction (ORR), but most of them favour only either the ORR or the OER. Besides, their underlying catalytic mechanisms are subject of an ongoing debate. Herein, La0.8Sr0.2CoO3−δ (LSC) was selected as a base perovskite oxide for doping different elements into its B-site to fabricate four different La0.8Sr0.2Co0.8M0.2O3−δ (LSCM; M = Ni, Fe, Mn, and Cu) perovskite oxides. Among the catalysts tested with and without multi-walled carbon nanotubes (MWNTs), La0.8Sr0.2Co0.8Fe0.2O3−δ (LSCF) outperformed any other catalysts in terms of both OER and ORR activity. The OER/ORR activity enhancement with LSCF is also discussed based on spectroscopic and microscopic analyses and lattice oxygen transportation.
1 Introduction
The oxygen evolution reaction (OER) and the oxygen reduction reaction (ORR) with a sluggish kinetic rate and high overpotential often cause a low overall efficiency and poor power capability for metal–air batteries and regenerative fuel cells. Precious-metal catalysts are highly active for the OER/ORR; however, their high cost imposes a limitation on their commercial applications. In addition, Ir- and Ru-based catalysts have been known to be the most active catalysts for the OER but suffer from poor ORR activity.1–3 Conversely, Pt-based catalysts are widely used for ORR but provide unsatisfactory OER performances.4,5 In response, extensive research efforts have been made to explore non-precious metal-based bifunctional catalysts for the OER/ORR with high performances.6,7 Perovskite oxides (ABO3) have the potential of being bifunctional catalysts or efficient oxygen electrodes due to their excellent oxygen mobility, low cost and their structures with atomic-level defects that play important roles in the OER/ORR.8–10 One way to improve the OER/ORR of perovskite oxides is to increase their specific surface area and porosity of the catalysts, which have been reported to be beneficial for exposing more accessible active sites and facilitating the mass transfer and electron transfer during the OER/ORR processes.11,12 However, the morphology optimization is difficult to break through the limitation of material's intrinsic properties. Thus, we explored another efficient approach that is to optimize the intrinsic structures of the perovskite oxides by elemental doping or nonstoichiometric substitution.For the ABO3 perovskite oxides, an important advantage is their flexibility in composition; namely both the A- and B-sites can systematically vary by using metal cations with different valences or ionic radii.13–15 Substitution of the A- and/or B-site by other elements or doping at these sites is also relatively straightforward and affords numerous degrees of freedom to tailor the physicochemical properties for enhancing the catalytic activity. Owing to the flexibility in composition, extensive research has been carried out in a series of derived perovskite oxides among which La0.8Sr0.2CoO3 and its doped perovskite oxides have acted as excellent cathode materials in the solid oxide fuel cells (SOFC) and used as efficient catalysts for metal air batteries.16–18
However, two main problems exist in the research of the La0.8Sr0.2CoO3-based perovskite oxides. One is that most perovskite oxides have electronic structures that favor only the ORR or OER and their bifunctionality should be carefully combined by tailoring the electronic environment of metal cations and oxygen anions based on a systematic study and design principles.19,20 Another problem is that the mechanism of electrochemical improvement is still controversial and has attracted the attention of researchers. The conventional adsorbate evolution mechanism proceeds at the transition metal active sites, where the adsorbed oxygen intermediates should be neither too strong nor too weak according to the Sabatier principle.21 The balance of the adsorbed oxygen intermediates is influenced by the covalency of the B–O bond that efficiently tunes the affinity between the B-site and the intermediate. It has also been demonstrated by S.-Horn et al. that the filling of electrons in the σ-antibonding states (eg orbital) of transition metals serves as the descriptor for the ORR activity based on the molecular orbital theory.22 The eg orbital strongly overlaps with the O-2pσ orbital determining the B–O bond strength during the chemisorption of oxygenated species in the ORR.8,23 They also proposed a volcano-type relationship describing the OER activity as a function of eg filling of the B-site cations and demonstrated that the perovskite oxides can achieve an enhanced intrinsic OER activity when the value of the eg orbital-filling electrons is close to unity.24
However, other electronic structural factors of the transition metal sites have also been reported to affect the ORR/OER activity. Koper et al. showed a correlation between the bulk oxide formation energy and the OER activity, in which they described the physicochemical properties of perovskites from different families and suggested the significant effect of the B-site stoichiometry and doping variation on the ORR/OER activity, which requires a further systematic study.15 More recently, a new proposed lattice oxygen-mediated mechanism,25–29 which involves the direct participation of perovskite lattice oxygen anions for the OER, was supported by the 18O isotope detection of the reaction product as well as DFT calculations. Research on this lattice oxygen mechanism pathway is still limited and it needs more perovskite oxides to support it.
Based on the structure flexibility of the perovskites, La0.8Sr0.2CoO3−δ (LSC) was selected as the base perovskite oxide and different elements were doped at its B-site to obtain the LSCM (M = Ni, Fe, Mn, and Cu) series of perovskite oxides. The structures of the obtained perovskite oxides were analyzed by X-ray diffraction (XRD) refinement, high-resolution transmission electron microscopy (HRTEM) results and X-ray absorption spectroscopy (XAS) measurements. The nanoparticle size and the surface oxygen species of these perovskite oxides were also compared. The OER activity measurements were conducted in KOH media of different pH values. Based on the results, the perovskite catalyst with the best OER activity was selected. Similarly, the ORR activity and electrochemical surface area (ESCA) were also compared. Overall, the improvement of the element doping for OER and ORR of these perovskite oxides are discussed and the most possible mechanism for the LSCF catalytic improvement was proposed.
2 Experimental
2.1 Preparation of perovskite oxides
The perovskite oxides, La0.8Sr0.2CoO3−δ (LSC) and LSCM (M = Ni, Fe, Mn, and Cu), were prepared according to a conventional sol–gel method.30 Briefly, La(NO3)3·6H2O, Co(NO3)2·6H2O, (Aladdin Industrial Corp.), Fe(NO3)2·9H2O, Cu(NO3)2·6H2O Sr(NO3)2, Ni(NO3)3·6H2O and 50% Mn(NO3)2 solution (Guoyao Chemical Reagent Co., Ltd) were dissolved in deionized water based on the stoichiometric amounts of the different elements in different perovskite oxides. D-Glucose monohydrate and acrylamide (Guoyao Chemical Reagent Co. Ltd) were used as chelating agents and added to the nitrate mixed solution, which was stirred at 80 °C for 5 h. The pH of the solution was maintained between 8 and 10 by adding a moderate amount of NH4OH. After stirring and drying, the obtained gels were heated to 350 °C for 3 h, then ground into powders. Finally, the powders were calcined in air at 750 °C for 3 h to obtain the perovskite oxides, which were used as the ORR and OER catalysts in this study.2.2 Physical characterizations of perovskite oxides
The powder XRD measurements were performed using an X′Pert PRO X-ray diffractometer with Cu Kα radiation, and the data were collected from 20 to 80° in the 2θ range. The SEM images were obtained using an SU9000 (Hitachi High-Technologies) at the accelerating voltage of 20 kV. HRTEM, scanning transmission electron microscopy (STEM), high-angle annular dark-field (HAADF) TEM and energy-dispersive spectroscopy (EDS) elemental mapping images were obtained on a JEM-ARM200F instrument (JEOL Ltd) at 200 kV. The X-ray photoelectron spectroscopy (XPS) spectra were obtained by an AXIS-ULTRA DLD (Shimadzu, Co., Japan) and XAS measurements were carried out using the Kyushu University beamline (SAGA-LS/BL06 in Kyushu Synchrotron Light Research Center) equipped with a storage ring operating at the energy of 1.4 GeV. A monochromator with two Si(111) crystals was used to scan the energy of the X-ray beam. The photon flux was 2 × 1010 photons per second. The Co K-edge of the perovskite samples was measured in the fluorescence mode by a silicon drift detector. To examine the local structural properties around the Co atoms, the X-ray absorption near edge structure (XANES) spectroscopy data were extracted from the full XAS spectra. The EXANES data were analyzed using Athena software following a standard procedure.2.3 Electrochemical measurements
The electrochemical activities of the catalysts were measured by a three-electrode system, which consisted of a rotating ring-disk electrode (RRDE), Pt wire counter electrode and Hg/HgO reference electrode saturated in 1 M NaOH solution. The electrochemical characterization was conducted on a Model 760E Electrochemical Analyzer (ALS Co., Ltd) at 25 °C in 0.1 M and 1 M KOH electrolyte solutions. All the potentials were normalized with respect to the RHE.The catalyst ink without carbon was prepared by dispersing 2 mg of the perovskite oxide in 105 μL of DI water, 405 μL of isopropyl alcohol and 15 μL of Nafion (5%), then ultrasonication for 20 min to obtain a homogeneous ink. A 10 μL ink solution was cast on a glassy carbon (GC) electrode, resulting in a catalyst loading of 0.3 mg cm−2. The catalyst ink with carbon was prepared by dispersing 2 mg of the perovskite oxide and 1 mg of multi-walled carbon nanotubes (MWNTs, φ ∼ 20 nm, Nikkiso Co., Ltd) in 210 μL of DI water, 910 μL of isopropyl alcohol and 30 μL of Nafion (5%), then ultrasonication for 30 min to obtain a homogeneous ink. A 20 μL ink solution was cast on the glassy carbon (GC) electrode, resulting in a perovskite catalyst loading of 0.3 mg cm−2 and carbon loading of 1.5 mg cm−2.
For the ORR tests, the electrochemical accessibility of the catalyst was determined by performing cyclic voltammetry (CV) cycling between 1.1 and 0.4 V vs. RHE at 50 mV s−1 in N2- and O2-saturated 0.1 M KOH electrolytes. Linear sweep voltammetry (LSV) curves for the ORR data were then collected at the scan rate of 10 mV s−1 in the O2-saturated 0.1 M KOH and 1 M KOH electrolytes at 1600 rpm controlled by a rotation speed controller (BAS Inc.). The LSV ORR curves were also collected at 400, 800, 1200 and 2000 rpm in order to obtain their corresponding Koutecky–Levich (K–L) plots.
For the OER tests, the LSV measurements were performed in the range of 1.1 and 1.8 V vs. RHE at 10 mV s−1 in N2-saturated KOH electrolytes with different pH values at the rotation speed of 1600 rpm. The concentrations of the KOH electrolyte were 0.01 M, 0.0316 M, 0.1 M, 0.316 M and 1 M KOH, and their corresponding pH values were 12, 12.5, 13, 13.5 and 14, respectively. The CV cycling tests at potentials between 1.1 and 1.8 V vs. RHE were conducted to evaluate the OER stability of the catalysts. The double layer capacitance was determined by the CV measurements at the scan rates of 20, 40, 60, 80 and 100 mV s−1 in order to compare the ESCA of the different perovskite oxide catalysts.
2.4 XRD refinement and TEM analysis
For the Rietveld refinement analysis of the XRD pattern, Y (Yobs) is the value obtained from a Rigaku Smart Lab instrument, and Y calculated (Ycal) is either refinement with the PDXL2 database card number or simulated from the CrystalMaker, PDXL2 and Crystal Diffract software. Crystal Maker (software ver.10.4) was used to obtain the crystal structures shown in Fig. 1c and Fig. S1b, S2b, S3b, S4b (ESI†) based on the analysis of their crystallographic information files (CIF) from the final refinement results from Fig. 1b and Fig. S1a, S2a, S3a, S4a (ESI†) and aligned in the direction of planes which exhibited 100% peak intensities of each catalyst. The d-spacing of the LSCM in the high-angle annular dark-field (HAADF)-STEM images were calculated using Gatan software. The hkl indices were attributed based on the single crystal structure.31 | ||
| Fig. 1 (a) XRD patterns of LSC, LSCN, LSCF, LSCM and LSCC perovskite oxides compared to the standard PDF patterns; (b) the XRD rietveld refinement results, (c) crystal structure, (d) high-angle annular dark-field imaging (HAADF)-STEM image and (e) diffraction pattern of La0.8Sr0.2Co0.8Fe0.2O3−δ; (f) the color contrasted images in the red dotted cube area of (d). Note: the d-spacing (d110) of crystal structure in (c) and diffraction patterns in (e) are inter-related. | ||
3 Results and discussion
3.1 Characterization of materials
The XRD patterns of these synthesized samples (LSCC, LSCM, LSCF, LSCN, and LSC) are shown in Fig. 1a, together with standard perovskite oxides including La0.4Sr0.6CoO3 (cubic), La0.8Sr0.2CoO3 (orthorhombic), and LaCoO3 (hexagonal). All main peaks of the obtained materials are assigned to perovskite oxides, while the peaks of LSCN, LSCF, LSCM and LSCC shift toward a lower angle compared with the peaks of LSC, which is due to lattice relaxation and can be viewed as evidence of successful doping. Besides, the unsmooth burr peaks indicated some impurity phases, which is most likely metal hydroxide. Although there are suitable standard patterns for the synthesized samples, some are not in complete agreement with the synthesized samples because some shifted peaks were observed. Hence, we simulated new phases and refined them based on these standard patterns using Crystal Maker, Crystal Diffract and Rigaku PDF2.In order to analyze the structure on an atomic level, we also studied the perovskite oxides using the HAADF images of the TEM. The Rietveld refinement and TEM atomic pattern analysis results of all the perovskite oxides are shown in Fig. 1 (LSCF) and Fig. S1–S4 (ESI†) (LSC, LSCN, LSCM and LSCC), and the obtained results are listed in Table 1, in which ‘Y observed (Yobs)’ and ‘Y calculated (Ycal)’ are the observed XRD raw data and the simulated results obtained by using Crystal Maker and Crystal Diffract software, respectively. The weighted profile R-factor (RWP) is the factor of agreement between the observed and calculated peak intensities. The value of Rwp of less than 7% means the refinement accuracy is sufficient.
| Catalysts | PDXL2 card number | Space group | Cell parameter | S value |
|---|---|---|---|---|
| a See Cartesian co-ordinates in Table S2 (ESI). | ||||
| La0.8Sr0.2CoO3 | 01-075-8569 |
R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c c |
a = b = 5.4467 Å, c = 13.1781 Å, α = β = 90°, γ = 120° | 0.86 |
| La0.8Sr0.2Ni0.2Co0.8O3 | Simulateda | Cmc21 | a = b = 5.457 Å, c = 7.704 Å, α = β = γ = 90° | 1.19 |
| La0.9Sr0.1Fe0.5Co0.5O3 | 01-078-6202 | P23 | a = b = c = 3.853 Å, α = β = γ = 90° | 1.00 |
| La0.72Sr0.28Mn0.92Co0.08O3 | 01-070-8677 |
Rc |
a = b = 5.5356 Å, c = 13.449 Å, α = β = 90°, γ = 120° | 1.15 |
| La3.2Sr0.8Cu0.4Co0.6O3 | 01-071-6496 |
Pmm |
a = b = c = 3.8944 Å, α = β = γ = 90° | 1.36 |
The XRD refinement results of LSCF are shown in Fig. 1b, in which the Yobs completely agrees with the Ycal that corresponds to La0.9Sr0.1Co0.5Fe0.5O3. The difference between Yobs and Yobs of 1 can be confirmed by the blue curve in Fig. 1b and the value of S listed in Table 1. The lower value of S means the higher accuracy of the simulated results. The slight shift in the blue curve and low value of S (S = 1) indicate the high accuracy of the LSCF refinement results. Similarly, the XRD refinement results of LSC, LSCN, LSCM and LSCC are shown in Fig. S1a–S4a (ESI†) and the corresponding simulated perovskites are La0.8Sr0.2CoO3, La0.8Sr0.2Co0.2Ni0.8O3, La0.72Sr0.28Co0.08Mn0.92O3 and La3.2Sr0.8Co0.6Cu0.4O3, respectively. For LSC, LSCN and LSCM, the blue curves shifted slightly and the values of S were around 1, which indicated that the refinement results of LSC, LSCN and LSCM were in good agreement with the reference data. For LSCC shown in Fig. S4a (ESI†), the peak of carbon and another oxide, Co2.54O4, are observed in addition to the perovskite main peaks. In order to understand the lattice position of each atom in the perovskite, the crystal structures from the CIF of the refined XRD data of LSCF are displayed in Fig. 1c. The HAADF images from TEM of LSCF (Fig. 1d) showed its clear atomic lattice arrangement and its selected area electron diffraction pattern is shown in Fig. 1e.
To distinguish the difference between each plane of the HAADF image of LSCF and the crystal structure obtained from the XRD refinement, we constructed a color contrast image using the single crystal software and calculated the d-spacing of LSCF from the major intensity planes. The color contrast image of LSCF is shown in Fig. 1f and the calculated d-spacing of its 110 lattice plane is 2.724 Å, which is in good agreement with the XRD refinement results. In the same way, the corresponding results of LSC, LSCN, LSCM and LSCC are respectively shown in Fig. S1b–d, S2b–d, S3b–d and S4b–d (ESI†), which indicates that the d-spacing of all the perovskite oxides from the major intensity planes completely agrees with the crystal structure obtained from the XRD refinement. For LSCC, the impurity phase of the Co2.54O4 (card no. 01-078-5623) and carbon impurity are traced in Fig. S4 (ESI†). In addition, the cell volumes are tabulated in Table 2. It is notable that the cell volumes of LSCF and LSCC are 57.187 Å3 and 57.192 Å3, respectively, which are much lower than those of LSC, LSCN and LSCM. The small cell volume is beneficial for oxygen transportation and better kinetics for the ORR/OER.
| Catalysts | Planes of high intensity peak | d-Spacing from XRD refinement (Å) | d-Spacing from TEM (HAADF pattern) (Å) | Cell volumes (Å3) |
|---|---|---|---|---|
| La0.8Sr0.2CoO3 | 110 | 3.83 | 3.83 | 338.571 |
| La0.8Sr0.2Ni0.2Co0.8O3 | 112 | 2.70 | 2.70 | 227.230 |
| La0.9Sr0.1Fe0.5Co0.5O3 | 110 | 2.72 | 2.72 | 57.187 |
| La0.72Sr0.28Mn0.92Co0.08O3 | 104 | 2.76 | 2.76 | 358.230 |
| La3.2Sr0.8Cu0.4Co0.6O3 | 110 | 2.75 | 2.75 | 57.192 |
The SEM images of the obtained perovskite oxides are shown in Fig. S5 (ESI†). Compared to LSC (Fig. S5a, ESI†), the grains of LSCN, LSCF, LSCM and LSCC (Fig. S5b–e, ESI†) are refined by elemental doping. A more specific microstructure has been observed by TEM measurements and the results are shown in Fig. 2 and Fig. S6–S9 (ESI†). As shown in Fig. 2a, the particle diameter range of LSCF was ∼40–70 nm. In the HRTEM image shown in Fig. 2b, the adjacent fringes of LSCF are 2.72 Å, which matches the results of the diffraction patterns of 2.71 Å (Fig. 1f) and the XRD refinement results. The STEM image and EDS mapping results are shown in Fig. 2c–g, in which the homogeneous distribution of the different elements (La, Sr, Co and Fe) in the LSCF is observed. Similarly, as shown in Fig. S6a, S7a, S8a and S9a (ESI†), the particle diameter ranges of LSC, LSCN, LSCM and LSCC are found to be ∼80–100, 20–40, 40–70 and 20–60 nm, respectively. The obtained lattice spaces match well the diffraction patterns and XRD refinement results. The EDS mapping results demonstrated that the distribution of the different elements in these perovskite oxides were homogeneous.
 | ||
| Fig. 2 (a) TEM image, (b) HRTEM image, (c) STEM image and (d–g) EDS Elemental mappings (lanthanum-La, strontium-Sr, cobalt-Co, iron-Fe) of the La0.8Sr0.2Co0.8Fe0.2O3−δ perovskite oxide. | ||
The XPS measurements were conducted in order to investigate the surface element chemical states of the synthesized perovskite oxides. As shown in Fig. S10a (ESI†), the full XPS spectra of the synthesized perovskite oxides resemble each other. The XPS narrow scans of La, Sr, Co are shown in Fig. S10b–d (ESI†), respectively. For La (Fig. S10b, ESI†) in LSCN, LSCF, LSCM and LSCC, the binding energy (BE) peaks of 3d3/2 (850.3 V and 854.9 V) and 3d5/2 (833.6 V and 837.4 V) appear at similar peak positions to those of LSC.32,33 The Sr 3d XPS spectra of the five perovskite oxides showed a complex peak shape with two distinct 3d3/2 and 3d5/2 doublets as shown in Fig. S10c (ESI†). Some parts of these two pairs overlapped with each other, which was common for Sr-containing complex oxides.34 The XPS spectrum of Co 2p (Fig. S10d, ESI†) showed two main peaks at ∼780 eV (2p3/2) and 795.3 eV (2p1/2), suggesting that the Co3+ state is predominant.35
The XPS narrow scans of Ni in LSCN, Fe in LSCF, Mn in LSCM and Cu in LSCC are shown in Fig. 3a, b, c and d, respectively. The Ni 2p XPS spectra (Fig. 3a) provide complexed, multiple-split peaks due to the overlapping of the Ni 2p3/2 and La 3d3/2 peaks. The two main peaks of the Ni 2p3/2 and Ni 2p1/2 appeared at ∼855.3 V and ∼872 V, respectively. We also observed two satellite (sat.) peaks suggesting the existence of Ni(OH)2 on the surface of LSCN.33,36 For the Fe 2p (Fig. 3b), the peak positions that appeared at ∼711 eV and ∼724 eV are assigned to the Fe 2p3/2 and Fe 2p1/2, respectively. In addition, the two observed sat. peaks also indicated the existence of Fe(OH)x on the surface of LSCF.37 For LSCM, the Mn 2p3/2 and Mn 2p1/2 peaks are located at ∼643 eV and ∼654 eV (Fig. 3c).38 The two main peaks of Cu 2p3/2 and Cu 2p1/2 are also shown in Fig. 3d, together with two sat. peaks that suggested the existence of Cu(OH)x on the surface of LSCC.39 The previous XRD results also indicated the existence of metal hydroxides, which can either directly contribute to catalytic performances or facilitate OH− absorption and subsequently improve the activities. Fig. 3e shows the O 1s XPS spectra of the specified perovskite oxides, which can be deconvoluted into four characteristic peaks including lattice oxygen species (O2−, denoted as OI) at 528.4–528.7 eV, chemisorbed oxygen species (O−/O2−, denoted as OII) at 529.3–529.7 eV, hydroxyl groups or carbonate species (OH−/CO32−, denoted as OIII) at 531.2–531.45 eV, and absorbed water species (H2O, denoted as OIV) at 533.3–533.5 eV.40–42Table 3 lists the relative contents of these different kinds of oxygen species, which were calculated from the relative area of their deconvoluted peaks. Among these four oxygen species, OII (O−/O2−) is highly oxidative and closely related to the surface oxygen species, which are key for oxygen transportation, and can provide active sites for the ORR/OER processes.37,43,44 In addition, a recently proposed mechanism for the perovskite oxides was associated with the redox chemistry of lattice oxygen anions,25,29 indicating that OI (lattice oxygen) is also vital for the OER. Comparing the five perovskite oxides, LSCF was found to show the highest amount of OI and OII, which are both beneficial for ORR/OER.
 | ||
| Fig. 3 XPS spectra of perovskite oxides: (a) Ni 2p in LSCN, (b) Fe 2p in LSCF, (c) Mn 2p in LSCM, (d) Cu 2p in LSCC; (e) XPS peaking fitting results of O 1s for these perovskite oxides. | ||
| Relative content of oxygen species (%) | LSC | LSCN | LSCF | LSCM | LSCC |
|---|---|---|---|---|---|
| OI | 29.33 | 29.82 | 33.59 | 30.38 | 26.91 |
| OII | 4.23 | 5.94 | 9.24 | 5.10 | 4.76 |
| OIII | 59.10 | 53.64 | 47.04 | 40.12 | 54.23 |
| OIV | 7.34 | 10.58 | 10.13 | 24.40 | 14.10 |
To clarify the effect of element substitutions on the oxidation state of Co in La0.8Sr0.2CoO3−δ, we carried out XAS measurements of LSC, LSCN, LSCF, LSCM and LSCC. Fig. 4 shows the Co K edge XANES spectra of the perovskite samples. The bottom-right inset displaying details of while line indicates its slight shift toward lower energies upon doping of the different elements. The white line of the Co K-edge was reported to shift toward a higher energy, indicating a higher oxidation state of the Co.45 Thus, the XANES results demonstrated that the average oxidation state of the Co slightly changed by element doping into the B site of the perovskite.
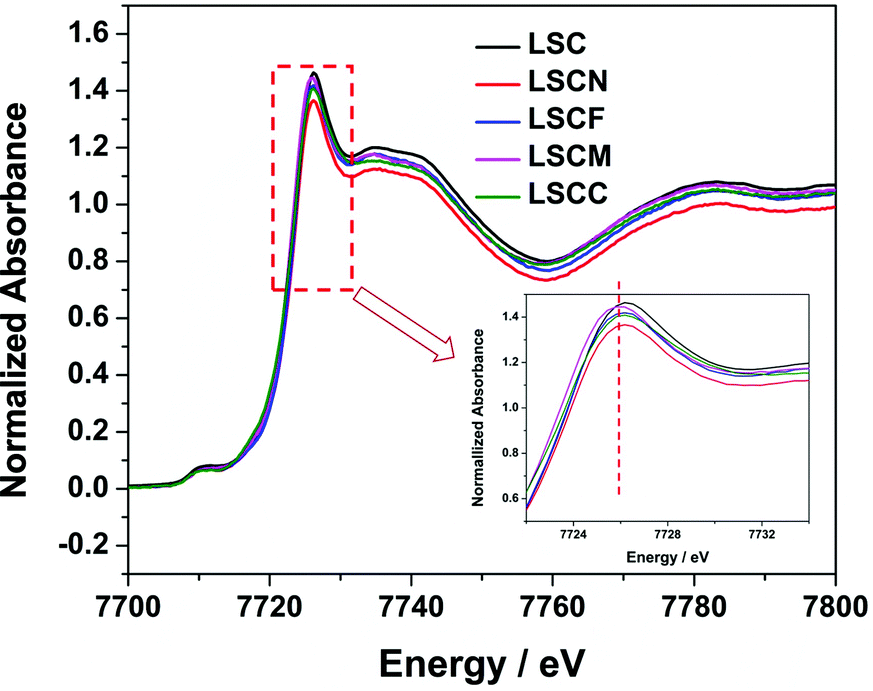 | ||
| Fig. 4 Co K-edge XANES spectra of LSC, LSCN, LSCF, LSCM and LSCC perovskite oxides at room temperature; the inset shows the main peak region. | ||
3.2 OER activities of perovskite oxides
The OER activity of the perovskite oxides was measured by linear sweep voltammetry (LSV) in KOH media with different pH values. We first carried out the OER activity measurements of the perovskite oxides including LSC, LSCN, LSCF, LSCM and LSCC not containing the MWNTs together with GC. The obtained LSV OER curves of the various perovskite oxides measured in 0.1 M and 1 M KOH media and their corresponding Tafel plots are shown in Fig. S11 and S12 (ESI†), which show that element doping is beneficial for the OER, and among the five catalysts, LSCF showed the best OER activity in the 0.1 M and 1 M KOH media.MWNTs provide a superior electrochemical stability compared to other conductive carbons,46–48 because the oxidation potential of the sp2 carbon is much higher than those of the sp3 carbons and possess a very high electrical conductivity.49 Thus, we selected this material as a carbon support material.50,51 The IR-compensated LSV curves of the different perovskite oxides with MWNTs are shown in Fig. 5, in which Fig. 5c (0.1 M KOH) and e (1 M KOH) show that the current density and OER activity of the perovskite oxides combined with the MWNTs increased compared to those of the pure perovskite oxides shown in Fig. S11a and S12a (ESI†). It should be noted that a slight pH dependence in Fig. 5a (pH = 12) and Fig. 5b (pH = 12.5) was observed; that is, the OER activity of these samples slightly improved, while this pH dependence is not very evident. However, as shown in Fig. 5c, when pH = 13, the OER activity shows a strong pH dependence: namely, we recognized much improved OER activity in the LSCF-MWNT compared to other four perovskite catalysts. Increasing the pH to 13.5 (Fig. 5d) and 14 (Fig. 5e) resulted in further improved OER activities of the provided catalysts. The pH sensibility of the perovskite catalysts was more intuitive when all the LSV curves in the different pH media were merged into one figure as shown in Fig. S13 (ESI†). Based on the corresponding current densities at the potential of 1.6 V vs. RHE in the different pH media, we plotted the curves in Fig. 5f, which obviously show that for all perovskite catalysts, the current density and OER activity increased with an increase in the pH, especially LSCF-MWNT showed the most evident pH dependent-OER improvement. These results suggested that the OER activity of the perovskite oxides is influenced by the OH concentration of the electrolyte, which is associated with the circuit conductivity and the surface OH species of the perovskite catalysts. Table S1 (ESI†) lists the OER activities of the obtained perovskite catalysts with the MWNTs and the reported state-of-art perovskite oxides with different carbon materials. At the current density of 10 mA cm−2, the potentials of LSC-MWNT, LSCN-MWNT, LSCF-MWNT, LSCM-MWNT and LSCC-MWNT in 0.1 M and 1 M KOH media were 1.66, 1.65, 1.59, 1.67, 1.66 V and 1.60, 1.58, 1.56, 1.59, and 1.58 V, respectively. It was determined that the LSCF-MWNT showed the highest OER activity among the synthesized catalysts although their performances were not very different.
 | ||
| Fig. 5 OER activity of LSC, LSCN, LSCF, LSCM and LSCC perovskite oxides with MWNTs in KOH media at 1600 rpm with different pH value of (a) 12, (b) 12.5, (c) 13, (d) 13.5 and (e) 14; (f) pH-dependant OER activity of different perovskite oxides at the working potential of 1.6 V vs. RHE. | ||
The ECSA can be determined by the electrochemical double-layer capacitance (Cdl) method, which requires the specific capacitance, a value from an ideal flat surface of the catalyst. In this research, the catalysts are a series of perovskite oxides with similar surface morphology, so we can use the values of Cdl to evaluate the level of ECSA. Cdl can be measured from the double layer charging curves using cyclic voltammetry (CV) at different scan rates in the non-faradaic region. The CV curves of the obtained perovskite oxides in 1 M KOH at different scan rates from 20 to 100 mV s−1 in the potential range of 1.1–1.35 V vs. RHE were shown in Fig. S14a–e (ESI†) and 1.00–1.15 V vs. RHE were shown in Fig. S15a–e (ESI†), respectively. The corresponding plots of the current density as the function of the scan rate were shown in Fig. S14f (at 1.20 V) (ESI†) and Fig. S15f (ESI†) (at 1.08 V), indicating a linear relationship between the current density and the scan rate. The value of the slope is Cdl. At 1.20 V, the obtained slope values (Cdl) of LSC, LSCN, LSCF, LSCM and LSCC were 2.88, 9.60, 5.23, 4.58 and 13.74 mF cm−2, respectively. At 1.08 V, the Cdl values of LSC, LSCN, LSCF, LSCM and LSCC were 1.80, 6.48, 3.11, 2.93 and 9.15 mF cm−2, respectively. Although the Cdl values at different potential were not the same, the ECSA level trends among the obtained perovskite oxides was the same: LSCC > LSCN > LSCF > LSCM > LSC. It is seen that LSCC shows the highest Cdl value but inferior OER activity while LSCF shows the third highest Cdl value but the best OER activity, which indicates that ECSA is not a major factor for determining the OER activity.
3.3 ORR activities of perovskite oxides
The ORR activities of the obtained perovskite catalysts that are combined with the MWNTs were carried out in 0.1 M and 1 M KOH media at 1600 rpm. Fig. 6 compares the LSV curves for the ORR in the O2 saturated 0.1 M and 1 M KOH solutions of the pristine MWNTs and the perovskite catalysts combined with the MWNTs. It has been found that the half-wave potential (E1/2) of the MWNTs was about 0.61 V, while it changed to 0.63, 0.64, 0.66, 0.67 and 0.63 V for LSC-MWNT, LSCN-MWNT, LSCF-MWNT, LSCM-MWNT and LSCC-MWNT in the 0.1 M KOH, respectively (Fig. 6a). Similarly, the E1/2 of LSC-MWNT, LSCN-MWNT, LSCF-MWNT, LSCM-MWNT and LSCC-MWNT in 1 M KOH medium was much improved to 0.69, 0.71, 0.71, 0.74 and 0.71 V as shown in Fig. 6b. These results indicated that element doping at the B-site in the LSC is efficient for the ORR improvement, and Mn doping shows the highest efficiency, which is consistent with the reported results.52–54 In addition, LSCF also showed an ORR activity improvement. | ||
| Fig. 6 ORR linear sweep voltammetry curves of different perovskite oxide catalysts combined with MWNTs in (a) 0.1 M and (b) 1 M KOH electrolytes at 1600 rpm. | ||
The ORR activities of the perovskite oxides without the MWNTs were also measured in 0.1 M KOH. Fig. S16 (ESI†) shows the LSV curves of the obtained perovskite oxides for the ORR and their corresponding Tafel plots. It was determined that LSCN, LSCF and LSCM showed similar ORR activities. In order to further investigate the ORR pathway, we measured the LSV curves of LSCF at rotation speeds ranging from 400 rpm to 2000 rpm, and the obtained Koutecky–Levich (K–L) curves are shown in Fig. S17a (ESI†). The current was plotted as a function of the ω−1/2 (ω is the angular velocity of the RDE) at 0.20, 0.25, 0.30, 0.35 and 0.4 V vs. RHE (Fig. S17b, ESI†). The average slope, 1 K−1, was 19.68 and the electron-transfer-number (n) was calculated as 3.76 based on the method described in the ESI.† Noticeably, the electron-transfer-numbers of the LSCF catalyst during the ORR were approximately 4, implying that most of the O2 was directly and completely reduced to OH−.20,55
3.4 OER and ORR improvement mechanism of LSCF
The ORR activity of the obtained perovskite catalysts that are combined with MWNTs were carried out in 0.1 M and 1 M KOH at 1600 rpm. According to the above OER and ORR comparison results, LSCM showed the best ORR activity while its OER performance was not high. LSCF was thus selected as the bifunctional catalyst for the mechanism study because it showed the best OER activity and considerable ORR activity.Although the B–O bond strength, which is associated with the eg orbital, was reported to play an important role in the OER for many perovskite oxides, the XANES results indicated that the Co–O bonds of the LSC almost remained the same value even after different element doping. The occurrence of the lattice oxygen transportation mechanism for the OER in LSCF is associated with the pH-dependent OER kinetics on the RHE scale, which can be deduced from eqn (1):27
| i = θ·cOH·e−ΔG/RT | (1) |
Further evidence supporting the participation of lattice oxygen transportation for the OER was the highest amount of OI (lattice oxygen) of the LSCF among the obtained perovskite oxides according to the XPS results. As schematically illustrated in Fig. 7, one possible lattice oxygen transportation pathway involves five evolution procedures I–V, which corresponds to eqn (2)–(6):27,56
 | (2) |
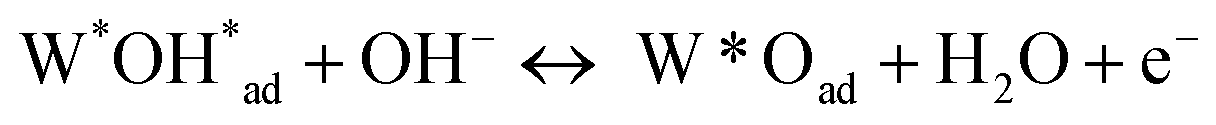 | (3) |
| W*Oad + OH− ↔ W*OOHad + e− | (4) |
| W*OOHad + OH− ↔ W*OO + H2O + e− | (5) |
| W*OO ↔ W* + O2 | (6) |
 | ||
| Fig. 7 A proposed OER and ORR mechanism of LSCF perovskite oxide. | ||
Similarly, Fig. 7 also reveals the catalytic mechanism of the LSCF for the ORR. The calculated electron-transfer-numbers of the LSCF catalyst during the ORR was 3.76, indicating that the ORR process of LSCF was very close to the direct 4-electron transfer processes. Therefore, the proposed ORR pathway involves two evolution procedures I and II, which corresponds to eqn (7) and (8):55,57
| W* + O2 ↔ W*OO | (7) |
| W*OO + 2H2O + 4e− ↔ W* + 4OH− | (8) |
4 Conclusions
Four different La0.8Sr0.2Co0.8M0.2O3−δ (M = Ni, Fe, Mn, Cu) perovskite oxides were synthesized using La0.8Sr0.2CoO3 as a base perovskite oxide whose B-site was doped with different elements. LSCF was the most efficient bifunctional catalyst showing the best OER activity and a considerably high ORR activity. The XRD Rietveld refinement and TEM atomic pattern analysis evidenced that the smaller cell volume of LSCF supplies more perovskite lattice cells for the lattice oxygen transportation during the OER/ORR processes. The XPS spectra also support the evidence that the higher relative concentration of OI (lattice oxygen) in LSCF is beneficial for the OER/ORR processes. Furthermore, the pH-dependent OER performance of LSCF demonstrated the occurrence of a certain proton–electron transfer step, which is associated with oxygen vacancies upon the evolution of a lattice oxygen-containing oxygen molecule. This indicates the participation of the lattice oxygen of LSCF in the OER. The ORR process of LSCF was almost a direct 4-electron transfer process based on the calculated electron-transfer-numbers of the LSCF. The proposed OER/ORR mechanism for the LSCF is the extension of the lattice oxygen-mediated mechanism for perovskite oxides. These findings only not deepen our understanding of the OER mechanism of perovskite oxide catalysts, but also have important significance for the design of a new perovskite catalyst with a low cost and high bifunctional efficiency.Conflicts of interest
There is no conflict to declare.Acknowledgements
The authors would like to thank the Materials Characterization Center of Huazhong University of Science and Technology and Ultramicroscopy Research Center (URC) of Kyushu University for characterization assistance. This study was supported by the Core Research for Evolutional Science and Technology (CREST), Japan Science and Technology Agency (JST) and JSPS KAKENHI Grant Number JP16H02083 (for N. N.).References
- J. Sun, L. Du, B. Sun, G. Han, Y. Ma, J. Wang, H. Huo, C. Du and G. Yin, ACS Appl. Mater. Interfaces, 2020, 12, 24717–24725 CrossRef CAS.
- Y. Zhu, Z. He, Y. Choi, H. Chen, X. Li, B. Zhao, Y. Yu, H. Zhang, K. A. Stoerzinger and Z. Feng, Nat. Commun., 2020, 11, 1–10 Search PubMed.
- Z. Li, L. Lv, J. Wang, X. Ao, Y. Ruan, D. Zha, G. Hong, Q. Wu, Y. Lan and C. Wang, Nano Energy, 2018, 47, 199–209 CrossRef CAS.
- L. Yu, N. Xu, T. Zhu, Z. Xu, M. Sun and D. Geng, Int. J. Hydrogen Energy, 2020, 45, 30583–30591 CrossRef CAS.
- H. Wang, W. Xu, S. Richins, K. Liaw, L. Yan, M. Zhou and H. Luo, Electrochim. Acta, 2019, 296, 945–953 CrossRef CAS.
- X. Xu, W. Wang, W. Zhou and Z. Shao, Small Methods, 2018, 2, 1800071 CrossRef.
- M.-J. Choi, T. L. Kim, J. K. Kim, T. H. Lee, S. A. Lee, C. Kim, K. Hong, C. W. Bark, K.-T. Ko and H. W. Jang, Nano Lett., 2020, 20, 8040–8045 CrossRef CAS PubMed.
- L. Yan, Y. Lin, X. Yu, W. Xu, T. Salas, H. Smallidge, M. Zhou and H. Luo, ACS Appl. Mater. Interfaces, 2017, 9, 23820–23827 CrossRef CAS PubMed.
- R. Yuan, Y. He, W. He, M. Ni and M. K. H. Leung, Appl. Energy, 2019, 251, 113406 CrossRef.
- S. Gupta, W. Kellogg, H. Xu, X. Liu, J. Cho and G. Wu, Chem. – Asian J., 2016, 11, 10–21 CrossRef CAS PubMed.
- G. Li, S. Hou, L. Gui, F. Feng, D. Zhang, B. He and L. Zhao, Appl. Catal., B, 2019, 257, 117919 CrossRef CAS.
- J. Cheng, Y. Jiang, L. Zou, M. Zhang, G. Zhang, Z. Wang, Y. Huang, B. Chi, J. Pu and L. Jian, ChemistryOpen, 2019, 8, 206–209 CrossRef CAS PubMed.
- Q. Guo, X. Li, H. Wei, Y. Liu, L. Li, X. Yang, X. Zhang, H. Liu and Z. Lu, Front. Chem., 2019, 7, 224 CrossRef CAS PubMed.
- H. Sun, J. Dai, W. Zhou and Z. Shao, Energy Fuels, 2020, 34, 10547–10567 CrossRef CAS.
- X. Cheng, E. Fabbri, M. Nachtegaal, I. E. Castelli, M. El Kazzi, R. Haumont, N. Marzari and T. J. Schmidt, Chem. Mater., 2015, 27, 7662–7672 CrossRef CAS.
- C. Zhao, N. Li, R. Zhang, Z. Zhu, J. Lin, K. Zhang and C. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 47858–47867 CrossRef CAS.
- J. Cheng, Z. Wang, L. Zou, M. Zhang, G. Zhang, Y. Dong, Y. Jiang, Y. Huang, N. Nakashima and B. Chi, J. Alloys Compd., 2020, 831, 154728 CrossRef CAS.
- J. Cheng, Y. Jiang, M. Zhang, Y. Sun, L. Zou, B. Chi, J. Pu and L. Jian, ChemCatChem, 2018, 10, 1635–1642 CrossRef CAS.
- J. Cheng, Y. Jiang, M. Zhang, L. Zou, Y. Huang, Z. Wang, B. Chi, J. Pu and J. Li, Phys. Chem. Chem. Phys., 2017, 19, 10227–10230 RSC.
- A. Safakas, G. Bampos and S. Bebelis, Appl. Catal., B, 2019, 244, 225–232 CrossRef CAS.
- J. Rossmeisl, Z.-W. Qu, H. Zhu, G.-J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS.
- P. Kolla, G. Nasymov, R. Rajappagowda and A. Smirnova, J. Power Sources, 2020, 446, 227234 CrossRef CAS.
- W. T. Hong, K. A. Stoerzinger, Y.-L. Lee, L. Giordano, A. Grimaud, A. M. Johnson, J. Hwang, E. J. Crumlin, W. Yang and Y. Shao-Horn, Energy Environ. Sci., 2017, 10, 2190–2200 RSC.
- J. S. Yoo, X. Rong, Y. Liu and A. M. Kolpak, ACS Catal., 2018, 8, 4628–4636 CrossRef CAS.
- J. T. Mefford, X. Rong, A. M. Abakumov, W. G. Hardin, S. Dai, A. M. Kolpak, K. P. Johnston and K. J. Stevenson, Nat. Commun., 2016, 7, 1–11 Search PubMed.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS.
- J. Liu, E. Jia, K. A. Stoerzinger, L. Wang, Y. Wang, Z. Yang, D. Shen, M. H. Engelhard, M. E. Bowden and Z. Zhu, J. Phys. Chem. C, 2020, 124, 15386–15390 CrossRef CAS.
- Y. Pan, X. Xu, Y. Zhong, L. Ge, Y. Chen, J.-P. M. Veder, D. Guan, R. O’Hayre, M. Li and G. Wang, Nat. Commun., 2020, 11, 1–10 Search PubMed.
- J. Cheng, M. Zhang, Y. Jiang, L. Zou, Y. Gong, B. Chi, J. Pu and L. Jian, Electrochim. Acta, 2016, 191, 106–115 CrossRef CAS.
- P. Ganesan, A. Staykov, H. Shu, M. Uejima and N. Nakashima, ACS Appl. Energy Mater., 2020, 3, 10961–10975 CrossRef CAS.
- G. Zhang, S. Zhang, L. Yang, Z. Zou, D. Zeng and C. Xie, Sens. Actuators, B, 2013, 188, 137–146 CrossRef CAS.
- R. C. Rabelo-Neto, H. B. E. Sales, C. V. M. Inocêncio, E. Varga, A. Oszko, A. Erdohelyi, F. B. Noronha and L. V. Mattos, Appl. Catal., B, 2018, 221, 349–361 CrossRef CAS.
- D. H. Prasad, S. Y. Park, E.-O. Oh, H. Ji, H.-R. Kim, K.-J. Yoon, J.-W. Son and J.-H. Lee, Appl. Catal., A, 2012, 447, 100–106 CrossRef.
- M. M. Natile, E. Ugel, C. Maccato and A. Glisenti, Appl. Catal., B, 2007, 72, 351–362 CrossRef CAS.
- T. D. Thanh, N. D. Chuong, J. Balamurugan, H. Van Hien, N. H. Kim and J. H. Lee, Small, 2017, 13, 1701884 CrossRef PubMed.
- S. She, J. Yu, W. Tang, Y. Zhu, Y. Chen, J. Sunarso, W. Zhou and Z. Shao, ACS Appl. Mater. Interfaces, 2018, 10, 11715–11721 CrossRef CAS PubMed.
- W. Xu, N. Apodaca, H. Wang, L. Yan, G. Chen, M. Zhou, D. Ding, P. Choudhury and H. Luo, ACS Catal., 2019, 9, 5074–5083 CrossRef CAS.
- Y. Huang, Y. Jiang, L. Zou, J. Cheng, B. Chi, J. Pu and J. Li, J. Electrochem. Soc., 2017, 164, A3896–A3902 CrossRef CAS.
- J. Zhang, D. Tan, Q. Meng, X. Weng and Z. Wu, Appl. Catal., B, 2015, 172–173, 18–26 CrossRef CAS.
- Y. Zhu, W. Zhou, J. Yu, Y. Chen, M. Liu and Z. Shao, Chem. Mater., 2016, 28, 1691–1697 CrossRef CAS.
- N. A. Merino, B. P. Barbero, P. Eloy and L. E. Cadús, Appl. Surf. Sci., 2006, 253, 1489–1493 CrossRef CAS.
- J. Zhu, H. Li, L. Zhong, P. Xiao, X. Xu, X. Yang, Z. Zhao and J. Li, ACS Catal., 2014, 4, 2917–2940 CrossRef CAS.
- Y. Lu, A. Ma, Y. Yu, R. Tan, C. Liu, P. Zhang, D. Liu and J. Gui, ACS Sustainable Chem. Eng., 2019, 7, 2906–2910 CrossRef CAS.
- X. Cheng, E. Fabbri, M. Nachtegaal, I. E. Castelli, M. El Kazzi, R. Haumont, N. Marzari and T. J. Schmidt, Chem. Mater., 2015, 27, 7662–7672 CrossRef CAS.
- Y. Shao, G. Yin, Y. Gao and P. Shi, J. Electrochem. Soc., 2006, 153, A1093 CrossRef CAS.
- Z. Chen, W. Deng, X. Wang and Y. Yan, ECS Trans., 2007, 11, 1289 CrossRef CAS.
- S. M. Andersen, M. Borghei, P. Lund, Y.-R. Elina, A. Pasanen, E. Kauppinen, V. Ruiz, P. Kauranen and E. M. Skou, Solid State Ionics, 2013, 231, 94–101 CrossRef CAS.
- M. Tominaga, Y. Yatsugi and N. Watanabe, RSC Adv., 2014, 4, 27224–27227 RSC.
- J. Yang, T. Fujigaya and N. Nakashima, Sci. Rep., 2017, 7, 1–9 CrossRef.
- J. Yang, P. Ganesan, A. Ishihara and N. Nakashima, ChemCatChem, 2019, 11, 5929–5944 CrossRef CAS.
- A. Ashok, A. Kumar, R. R. Bhosale, F. Almomani, S. S. Malik, S. Suslov and F. Tarlochan, J. Electroanal. Chem., 2018, 809, 22–30 CrossRef CAS.
- J. Sunarso, A. A. J. Torriero, W. Zhou, P. C. Howlett and M. Forsyth, J. Phys. Chem. C, 2012, 116, 5827–5834 CrossRef CAS.
- Y.-L. Lee, M. J. Gadre, Y. Shao-Horn and D. Morgan, Phys. Chem. Chem. Phys., 2015, 17, 21643–21663 RSC.
- Y. Aoki, E. Tsuji, T. Motohashi, D. Kowalski and H. Habazaki, J. Phys. Chem. C, 2018, 122, 22301–22308 CrossRef CAS.
- Z. Wu, L.-P. Sun, T. Xia, L.-H. Huo, H. Zhao, A. Rougier and J.-C. Grenier, J. Power Sources, 2016, 334, 86–93 CrossRef CAS.
- D.-G. Lee, S. H. Kim, S. H. Joo, H.-I. Ji, H. Tavassol, Y. Jeon, S. Choi, M.-H. Lee, C. Kim, S. K. Kwak, G. Kim and H.-K. Song, Energy Environ. Sci., 2017, 10, 523–527 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00632k |
| This journal is © The Royal Society of Chemistry 2022 |