
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of spatially organized, peptide/peptide derivative containing nanocomposites
Mingyang
Ji
 * and
Zhaoyang
Liu
* and
Zhaoyang
Liu
Research School of Polymeric Materials, School of Materials Science & Engineering, Jiangsu University, Zhenjiang 212013, China. E-mail: jimy@ujs.edu.cn
First published on 11th August 2021
Abstract
The functioning of naturally occurring materials and organisms emerges from the synergistic actions of all involved functional subunits following well-defined spatial hierarchies. Resembling the exquisite structure of biological machinery, the hierarchical organization of synthetic materials over a continuum of length scales and complexity dictates their functional competence. To replicate the structure–function relationships of natural origin materials, peptide-based building blocks capable of spontaneous association have been exploited to construct nanocomposites inherent with precise spatial order and biocompatibility. Moreover, the evolving functionalities of synthetic materials necessitate higher-order chemical diversity and structural complexity, accordingly, additional components have been coupled with peptidic nanoassemblies to achieve hierarchical multicomponent nanocomposites. This review starts with a brief introduction of kinetic and thermodynamic principles of supramolecular polymerization and these principles largely direct the construction of nanostructures containing amphiphilic peptides or peptide derivatives as well. Taking the synergism of kinetic factors and thermodynamic preferences as a guide, we critically review various strategies to construct spatially organized, multicomponent nanocomposites. All these strategies similarly involve amphiphilic building blocks consisting of assembling peptide domains that contribute to the generation of supramolecular frameworks allowing the incorporation of functional chromophores/constituents with nanoscale precision. Nanocomposites integrated with multiple components give access to greater structural tunability and complexity to perform complex tasks, thereby exerting usage in biomedical formulations and energy harvesting and conversion.
1. Introduction
Naturally occurring materials and organisms contain multiple functional subunits whose spatial arrangements dictate their synergistic actions to perform complex tasks.1 To fully replicate the functional competence of biological machinery, emerging research on synthetic materials has been focused on the construction of multicomponent nanocomposites whose diversity in chemical composition may give access to the hierarchical arrangement within resulting materials.2,3 Resembling the sophisticated structural organization of archetypes with natural origin, evolving functionalities of synthetic materials impose unprecedented requirements on their structural regulation with nanoscale precision. Therefore, peptides, the basic sub-segments of proteins that can spontaneously associate into various nanostructures through reversible and directional noncovalent interactions, have been employed to construct supramolecular architectures.4–7 Given the versatile surface of peptidic assemblies capable of various noncovalent interactions, they can serve as supramolecular templates to incorporate additional components and thus to achieve integrative, multicomponent nanostructures with well-defined spatial hierarchy.8 Moreover, with the inherent biocompatibility of peptide frameworks, these peptidic nanocomposites may bridge the gap between organic and inorganic regimes,9 thus exerting usage in disparate fields ranging from biomedical formulations10–12 to energy conversion and storage devices.4,13,14 The purpose of this review is to survey strategies to construct nanostructures from amphiphilic building blocks, particularly from amphiphilic peptides and/or peptide derivatives. In addition, resulting nanostructures can serve as supramolecular frameworks to regulate the incorporation of other functional components in nanoscale precision, thereby producing multicomponent nanocomposites with a well-defined spatial organization. To fully reflect advances in the preparation of well-defined nanostructures involving assembling peptide factors, nanocomposites discussed in this review are not restricted to pure peptide/peptide derivative assemblies but encompass complex hybrids that may also contain proteins, oligonucleotides, polymers, carbon nanotube, even metallic additives. The task execution of these multicomponent nanocomposites that closely correlates with the spatial arrangement of functional constituents is highlighted as well.The desirable functioning of living systems, such as photosynthesis and immune response, depends on the synergism of multiple subunits integrated within biological structures with a finely tuned spatial arrangement.15 For example, adenosine triphosphate (ATP) synthase consists of two subunits, FO and F1, working in a rotational motor mechanism to produce energy storage molecules ATP. Within the ATP synthase, the spinning of its FO region, driven by the proton (H+) concentration gradient in the cellular environment, causes the conformational changes of its F1 region, which propel the dehydration between adenosine diphosphate (ADP) and inorganic phosphate to yield ATP. As FO and F1 regions act in concert, ATP synthase store energy for the consumption of living cells in ATP molecules consecutively.16,17 Inspirations from biological machinery motivates the preparation of functional nanomaterials by spatially integrating monomeric building blocks into well-defined nanostructures.18 However, the structural complexity of current artificial nanoassemblies is still hardly comparable with the exquisite organization of biological machinery, which compromises their functional competence to perform structure-determined complex tasks. Therefore, integrating multiple components into one comprehensive, hierarchical structure with nanoscale precision has garnered growing attention, as the chemical diversity of such multicomponent nanostructures may contribute to a larger structural tunability and complexity to replicate natural processes.
Nanocomposites consisting of multiple components may provide access to emerging properties that cannot be achieved by individual components acting in isolation.8,19 For instance, through the orthogonal assembly of electron donors (D) and acceptors (A), individually continuous electron-sufficient and deficient channels can be produced and brought into intimate contact to construct the supramolecular p-/n-heterojunction (SHJ).20,21 At the D/A interface within SHJ, photo-generated excitons quickly dissociate into charge carriers as electrons and holes that transport to corresponding electrodes to produce the photocurrent.22 While segregated D and A domains in close proximity are indispensable to achieve efficient photo-induced charge separation, strong charge-transfer interactions between D–A pairs can be detrimental for the preferential recognition of self-species during the orthogonal assembly of D–A mixture, thus resulting in the formation of their alternating stacks instead.23,24 To circumvent this undesirable alternating complexation, D and A chromophores are conjugated with additional segments capable of highly tunable association, such as peptides or peptide derivatives,5,25 to mitigate the charge-transfer interactions between D–A pairs. By tailoring the peptide sequence, attractive and repulsive interactions can be programmed into D/A–peptide conjugates, which enable the construction of well-defined nanostructures integrated with segregated domains that synergistically act to produce a higher level of functional competence.26,27
The hierarchical organization of multicomponent structures closely correlates with self-sorting, a phenomenon that produces preferential homo-/hetero-association of monomers within a multi-component mixture (Fig. 1).28 According to the potential energy of the assembling system, self-sorting can be achieved under thermodynamic or kinetic control. While most early self-sorting studies are under thermodynamic control,28,29 kinetic factors such as time, temperature and concentration have been recognized as decisive conditions to achieve heterogeneous microstructures integrated with sorted components.30–33 Given the complex synergism of kinetic and thermodynamic preferences during multi-component assembly, the construction of nanocomposites with well-defined spatial organization necessitates a thorough understanding of the fundamental mechanisms of molecular self-assembly. Multicomponent assembly shares fundamental concepts and well-established theories with single-component systems, although the simultaneous involvement of several types of monomers inevitably diversifies the assembly pathway.19,23 The Gibbs free energy (ΔG) of growing assemblies evolves with the addition of each monomer,32,34–36 and theoretical models deriving from ΔG along with monomer association give rise to assembly mechanisms as isodesmic, cooperative and anti-cooperative.30,34,35,37–41 Although multicomponent systems bear more variabilities on their pathway selection, theories of single component assembly may still be instructional, especially given that each respective component in the assembling mixture can be separately integrated into the growing nanostructure in a stepwise manner.42–46 Studies on thermodynamics and mechanisms of monomer association have deepened our understanding of the intrinsic physical aspects of self-assembly and motivate multicomponent co-assembly toward designable and predictable patterns.
 | ||
| Fig. 1 Co-assembly of amphiphilic building blocks, including peptides and peptide derivatives. A scheme illustrates the positioning of sorted subunits within multicomponent nanostructures that may induce structural synergism, which impacts the functioning of the resulting materials. | ||
Peptides typically comprise 2–50 amino acid residues and serve as the most common sub-segments of proteins.1,47,48 Through well-established solid-phase synthesis, various residues with ionizable or aromatic pendants can be precisely programmed into the peptide sequence, thus imparting positive/negative, polar/non-polar sites to peptides.47,48 The highly designable sequence of peptides renders them versatile building blocks capable of hydrophobic, electrostatic, hydrogen bonding, or van der Waals interactions, and these abundantly available interactions motivate peptides to assemble into α-helices, β-sheets, and other higher-order protein complexes. Also, the integrity of resulting peptide-based constructs is maintained by non-covalent interactions,49,50 which generates the adaptable structure of assembled peptides sensitive to physiological changes including temperature, pH, ionic strength, etc.4,51 The programmability of peptide sequences and the structural flexibility of peptide assemblies toward potential physiological cues permit the versatile applications of peptide derivatives in nanotherapies. Moreover, dimensions of peptide-based constructs cover a continuum of length scales ranging from nanoparticles, nanotubes to micrometer-long fibers and injectable bulk hydrogels, which suggest their applications beyond the cellular level.52 Recently, peptidic assemblies have been utilized to construct the device–tissue interfaces where biocompatibility is highly desirable.4 Given the natural origin of assembling peptides, they have been explored to bridge the gap between innovative bio-products and traditional electronic devices.
This review survey strategies to integrate multiple components, mostly including amphiphilic peptides or peptide derivatives, into one nanostructure to produce spatially organized nanocomposites. The spatial arrangement of functional subunits establishes the sophisticated function of biomolecules, likewise, the precise integration of segregated domains into one comprehensive structure determines the performance of artificial nanomaterials. In order to achieve the fidelity binding between desired species within a complex mixture of monomers, a thorough understanding of the mechanistic aspects of supramolecular self-assembly becomes highly desirable. There are some in-depth reviews that illustrate the assembly mechanisms of synthetic building blocks and the applications of relevant materials across various fields.23,32,34,35,37 Instead of giving an exhaustive compilation of existing works, our discussion starts from the fundamental kinetic and thermodynamic principles of supramolecular polymerization. While these principles arise from studies on pure synthetic systems, they are of use in guiding the assembly of amphiphilic building blocks with natural origins, such as peptides or peptide derivatives. Taking the mechanistic aspects of supramolecular self-assembly into consideration, we critically review some innovative strategies to construct multi-component nanocomposites whose spatial organization is largely influenced or entirely directed by assembling peptide domains. Following these co-assembly strategies wherein every individual component is finely positioned with nanoscale precision, the task execution of resulting nanocomposites that requires the synergistic action of all functional constituents is presented.
2. Mechanistic aspects in supramolecular polymerizations
Supramolecular self-assembly involves spontaneous monomer association that can be either more enthalpy- or entropy-driven. Comparing with synthetic monomers performing organization in apolar environments, the self-assembly of amphiphilic building with natural origin, including amphiphilic peptides and peptide derivatives, usually results from the synergistic action of hydrophilic and hydrophobic domains, which produces its unique thermodynamic preference deviated from typical synthetic systems.31,53–55 While amphiphilic systems discussed in this review exhibit special entropic tendencies during monomer association, their assembly processes are still subjected to the same kinetic factors also encountered by synthetic monomers. Therefore, the growth mechanism of supramolecular polymerization, in particular the cooperative mechanism, may still apply to the description of amphiphilic systems and guide the pathway control of peptide/peptide derivative involved assembly.This section starts from the comparative discussion on various growth mechanisms of supramolecular polymerization. To coordinate with later examples involved, the molecular origin, the rationale of monomer design, available kinetic interventions, and characteristic experimental phenomena of cooperative assembly are highlighted. In addition, our discussion about the cooperative mechanism is coupled with associated pathway complexity, which completes the necessary theoretical context for the following examination on assembling events involving peptide factors. Finally, we combine well-established theories of peptide assembly with the thermodynamic considerations of amphiphilic peptides and peptide derivatives. Despite the differences in intrinsic thermodynamics, the cooperative mechanism arising from synthetic systems can be used to interpret the seeding assembly of peptide derivatives, supported by examples at the end of this section.
2.1 Growth mechanisms of supramolecular polymerization
The self-assembly of peptide-based building blocks shares fundamental concepts and theories with general supramolecular monomer associations. The well-documented, quasi-one-dimensional assembly growth can be simplified as chain propagation to generate theoretical models of monomer association. Due to their resemblance to covalent polymerization, these spontaneous association processes are referred to as supramolecular polymerization and the growth mechanisms are generally categorized as isodesmic, cooperative and anti-cooperative.30,31,34,35 Isodesmic assembly is characterized by the monomer association that is energetically identical and independent of the degree of aggregation (Fig. 2a). For synthetic monomers governed by the isodesmic mechanism, their assembly usually involves a single type of non-covalent interaction and no steric hindrance due to crowded monomers builds up throughout the assembly process.32 In contrast to the identical binding constant of each monomer addition of isodesmic mechanism, cooperative and anti-cooperative processes follow the nucleation–elongation model, which has a smaller (for cooperative mechanism) or larger (for anti-cooperative mechanism) association constant of nucleation than that of elongation.34,56 The anti-cooperative process usually emerges from an energetically favorable dimerization or oligomerization at the initial stage of assembly, which is subsequently coupled with steric congestion among monomers within growing nanostructures.57,58 As a consequence, the energy diagram of anti-cooperative assembly exhibits fast nucleation followed by a relatively slow elongation (Fig. 2a). In contrast, the cooperative assembly shows a decelerated nucleation until the formation of a nucleus with the critical size capable of initiating rapid elongation (Fig. 2a). This energetically unfavourable nucleation phase is commonly encountered in self-assembly processes driven by a combination of multiple non-covalent interactions,30,41,59,60 as such systems need the synergism of these non-covalent interactions, enhanced by growing degree of aggregation, to countervail adverse kinetic factors at an initial stage. The cooperative process is frequently encountered in self-assembly and applies to many cases involving amphiphilic peptides or peptide derivatives,61–71 largely because these amphiphilic building blocks possess both hydrophilic and hydrophobic moieties responsible for monomer association through various non-covalent interactions.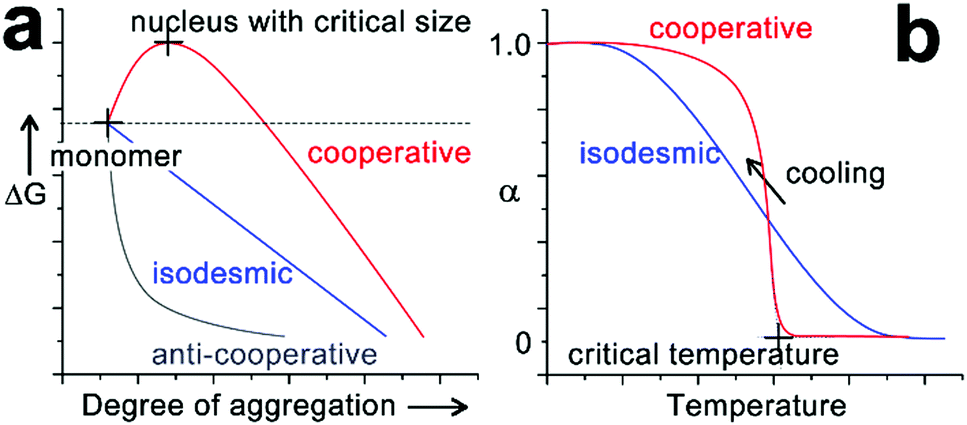 | ||
| Fig. 2 (a) Qualitative energy diagram of cooperative (red), isodesmic (blue) and anti-cooperative (grey) assembly systems, as a function of the degree of aggregation. (b) Description of cooperative and isodesmic assemblies, defined by the fraction of aggregated species (α) upon cooling down from their monomeric hot solutions. | ||
The kinetically suppressed nucleation of cooperative assembly can be identified by the emergence of a thermal hysteresis between the heating- and cooling-cycle during variable temperature spectral studies.32,59,72–74 For most synthetic monomers following the cooperative mechanism, their supramolecular polymerizations are enthalpy-driven processes and thus occur upon cooling.34 Assuming there is no kinetic barrier on the assembly route, heating-aggregated structures and cooling molecularly dissolved monomers at a constant rate should generate the identical temperature-dependent curve. However, the heating and cooling curves of a cooperative system usually fail to overlap each other, as only the heat-induced disassembly normally proceeds under thermodynamic control. As a result, the fraction of aggregated species on the cooling curve corresponding to a specific temperature is smaller than that on the heating curve. Such hysteresis between temperature-dependent curves indicates that opposing reactions fail to equilibrate, due to the existence of a kinetic barrier for the monomer association in the initial stages of cooperative growth.59 As the nucleation–elongation model of cooperative assembly is coupled with a kinetically suppressed initial nucleation, the monomer association to long aggregates is energetically preferred over the monomer self-nucleation. Given the energetically preferred elongation phase, equilibrium aggregates under the thermodynamic control are fragmented through sonication or mechanical agitation, and the resulting structures, relatively longer than the critical nucleus, can serve as seeds to enchain monomers in the cooperative growth. By avoiding the formation of high energy nucleus and pre-nucleus oligomers, this seeding method can divert the cooperative association to the elongation phase directly, thus speeding up the monomer conversion toward the final equilibrium structure.60,74–76
2.2 Cooperativity among multiple non-covalent interactions and associated pathway complexity
The cooperative monomer association arises from the cooperativity of noncovalent interactions, including polarization, electrostatic, and hydrophobic effects, that are dictated by the short- and long-range organization of monomers.30 For example, the isodesmic assembly of perylene-bisimide (PBI) derivative PBI-C1 (Fig. 3a) in methylcyclohexane (MCH) was purely driven by the π–π stacking of PBI cores,77,78 and its degree of aggregation derived from temperature-/concentration-dependent measurements displayed sigmoidal curves which are indicative of the isodesmic mechanism (Fig. 3b).30,32 In contrast, the PBI cores of PBI-C2, PBI-C3, and PBI-C5 were tethered with amide substituents, thus resulting in folded molecular conformations through intramolecular hydrogen bonds (Fig. 3a and b). This H-bonding induced conformational restriction sequestered the planar PBI cores of PBI-C2, C3, and C5 from π–π stacking, and the synergistic effect of intramolecular hydrogen bonds and π–π stacking interactions resulted in the cooperative assembly.41,59,60,79 Folded PBI-C2, C3, and C5 monomers need to break their intramolecular hydrogen bonds to re-expose their aromatic cores for π–π stacking. Accordingly, this energetically unfavored conformational change resulted in the slow nucleation of the cooperative assembly. As nucleation proceeded, however, extended monomers upon assembly formed multiple hydrogen bonds with neighboring units, which promoted a higher order of π–π stacking.80 With the formation of a critical nucleus, the enthalpic gain of monomer addition favored by multiple associations eventually offsetting the concomitant conformational penalty, which shifted the assembly of PBI-C2, C3, and C5 to the elongation regime.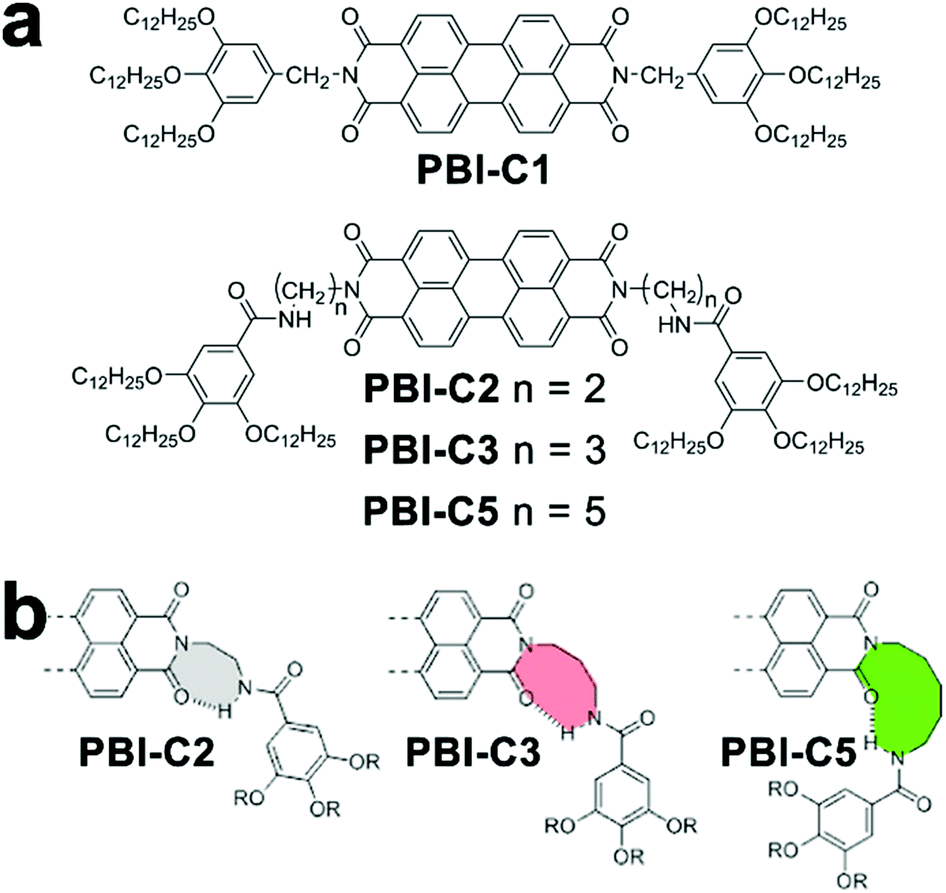 | ||
| Fig. 3 (a) Chemical structures of PBI derivatives following isodesmic (PBI-C1) or cooperative (PBI-C2, C3, and C5) assembly mechanisms. (b) Proposed conformation of folded PBI-C2, C3, and C5 due to intramolecular hydrogen bonds. Adapted with permission from ref. 60. Copyright 2016 American Chemical Society. | ||
In addition to the dictates on the assembly mechanism, the occurrence of cooperativity between assembling monomers may also exert influence on the assembly pathway. Synthetic monomers capable of multiple non-covalent interactions with cooperativity tend to generate kinetically preferred, non-equilibrium structures rather than the equilibrium structure under thermodynamic control.73,81 Due to the inherent lability of a non-equilibrium state, kinetic species always exhibit a conversion trend toward the thermodynamic equilibrium structure over time. However, activation barriers between kinetic and thermodynamic structures can significantly retard this spontaneous conversion, thus resulting in the pathway complexity of self-assembly.36 To overcome the activation barrier, external stimuli such as sonication,75 agitation,72 irradiation, and adding seeds74,76 have been applied on kinetic intermediates to achieve the thermodynamic structure situated in the global minimum of the energy landscape. It is noteworthy that the transformation from kinetic species to the thermodynamic structure can be distinguished in two situations: the consecutive pathway and the competitive pathway (Fig. 4).36 Along the consecutive pathway, non-equilibrium, kinetically trapped species directly build the equilibrium thermodynamic structure by reorganizing initial assemblies into higher-ordered architectures.38 Such kinetic species are on-pathway aggregates toward the final thermodynamic outcome (Fig. 4a). In contrast, kinetic species arising from the competitive pathway must disassemble into monomers before engaging in the thermodynamic state, and such kinetic intermediates are off-pathway aggregates with respect to the thermodynamic structure (Fig. 4b). Given building blocks conjugated with spectroscopic labels, the kinetic/thermodynamic transformation in the competitive pathway will be accompanied by the emergence of isosbestic points in UV-vis spectra and/or isodichroic points in CD spectra, indicating the transformation between structurally defined species.30,82
 | ||
| Fig. 4 Energy landscape of (a) consecutive and (b) competitive pathways. | ||
2.3 Thermodynamic considerations on the assembly of amphiphilic peptides and peptide derivatives
So far, our discussion on self-assembly has largely focused on the mechanistic aspects of synthetic monomers whose π-conjugated domains determine their enthalpy-driven association characterized by a ceiling temperature.24,35 Such cooling-motivated association originates from the enthalpic dispersion of synthetic monomers in apolar environments. Upon cooling, monomer associations driven by non-covalent bond formation are of an enthalpic nature and give off energy in the form of heat (ΔH < 0). The enthalpic gain arising from non-covalent interactions countervail the entropic loss of associated monomers with reduced degrees of freedom, thus resulting in the enthalpically dominated assembly. In contrast to the enthalpic assembly of synthetic monomers, amphiphilic peptides and peptide derivatives often exhibit an entropy-driven aggregation in aqueous media, which is accompanied by a floor temperature.34 In aqueous environments, hydrophobic domains of peptidic amphiphiles cluster together to situate the charged and polar peptide segments toward water. Otherwise, the exposure of hydrophobic domains incapable of forming hydrogen bonds would force surrounding water molecules into highly directional arrangements, which results in significant entropy losses of the system.53–55 As peptide amphiphiles reveal a strong aggregation tendency to minimize the disruptive effect of nonpolar solute on reducing the mobility of water molecules, their assembly is principally driven by the entropic force. Given the entropic nature of amphiphile assembly, heating enhances the contribution of entropic gain (ΔS > 0), therefore promoting the assembly of amphiphilic peptides and peptide conjugates. It is noteworthy that overheating peptidic assemblies breaks hydrogen bonds and destroys the hydration shell maintaining even dispersion, which leads to the precipitation of assembled structures.34Our early thermodynamic interpretations on peptide self-assembly are largely derived from empirical observations on biomolecules in nature.83–88 Peptides in biological contexts go through spontaneous self-association to form secondary structures like α-helices and β-sheets, which constitute the complex tertiary structure of proteins.8,89 β-Sheets are of special importance among other secondary structures, as their presence often indicates the formation of amyloid fibers which closely correlate with protein misfolding diseases such as Alzheimer's disease.4,23,90–92 Canonical β-sheet-forming peptides comprise alternatingly arranged hydrophobic and hydrophilic residues and these peptide segments undergo spontaneous association in water as stretched strands.93–95 Due to the directional constraints from hydrogen bonds along peptide backbones, multiple peptide strands are lined up next to each other to form the extended planar arrangement of peptide strands as β-sheets. Within these β-sheets, the orientation from N- to C-terminus of adjacent peptide strands may adopt the same or opposite directions, thus offering parallel or antiparallel β-sheets, respectively.8,51 β-Sheet peptides have been recognized as the most effective programmable building blocks to construct nanostructures coupled with biocompatibility, biodegradability, as well as ease of large-scale synthesis.51 Previous studies brought reliable strategies to achieve spatial and temporal control over the morphology of peptidic assemblies.6,7,96,97 For example, Grover, Lynn and co-workers demonstrated that changing pH altered the strength of salt bridge between charged termini of Aβ peptide Ac-KLVFFAE-NH2, the nucleating core of Alzheimer's disease, which produced peptide assemblies as nanofibers or nanotubes, respectively.7
Despite advances in modulating the assembly of peptides with natural design, the fundamental relationship between peptide sequence and corresponding assembly outcome remains elusive.98–100 To unambiguously determine essential motifs for peptide assembly, the systematic truncation and sequence modification on assembling peptides, assisted by well-established peptide synthetic approaches, become highly desirable.23,51 Through a reductionist approach, Gazit and coworkers identified diphenylalanine (FF) as the minimal self-assembling motif of β-amyloid, evidenced by the formation of well-defined FF nanotubes upon diluting the monomeric hexafluoroisopropanol (HFIP) solution with water.101–106 Such initial studies on diphenylalanine-involved assembly demonstrate the crucial role of aromatic moieties in peptide/peptide derivative association.107–111 Meanwhile, based on simple and engineerable short peptide sequence, Ulijn and coworkers proposed more guiding principles for peptide assembly through computation and these principles can be experimentally verified as well.98,99,112–116 For instance, tripeptides containing two neighboring aromatic residues exhibit high assembly propensity and this propensity can be further enhanced when the N- and C-terminus of tripeptides are coupled with residues carrying complementary charges.99,117,118 In addition, nuances of peptide structure may cause the inversion of supramolecular chirality of peptide assemblies, despite the intrinsic chirality of amino acid residues. For instance, pH, terminal charges, and the sequence of peptide isomers have been demonstrated to determine the eventual handedness of peptide superstructures.97,119–122
Amphiphilic peptides and peptide derivatives simultaneously contain hydrophilic and hydrophobic domains, both of which contribute toward the spontaneous monomer association synergistically. This synergism usually arises from the cooperativity among multiple non-covalent interactions, which suggests a cooperative mechanism, followed by the assembly of amphiphilic monomers. For instance, taking advantage of the monomer-buffering effect of kinetic intermediates, Hamachi et al. synthesized boronophenyl (BPmoc)-protected triphenylalanine peptide BPmoc-F3 capable of seeding-initiated fiber growth (Fig. 5). The growth of BPmoc-F3 nanofiber followed the cooperative mechanism, as the temperature-dependent CD measurements on the fiber growth revealed a non-sigmoidal curve fitting well with cooperative model. Fully developed nanofibers at the thermodynamic state were fragmented by vortexing and sonication, and the resulting nanofiber segments were demonstrated to be seeds to provide active ends for the subsequent monomer association. As these seeds were fragmented from thermodynamic structures, the monomer addition on seeds avoided energetically disfavored self-nucleation of monomers. Therefore, the seeding assembly eliminated the energy barrier of kinetic-thermodynamic conversion and hence speeded up the nanofiber growth.68 Accordingly, the growing peptidic nanofibers stained by fluorescent probes were visualized using confocal laser scanning microscopy (CLSM) and revealed a seeding-accelerated fiber growth as expected (Fig. 5b).
 | ||
| Fig. 5 (a) Molecular structure of BPmoc-protected triphenylalamine BPmoc-F3 and its aggregation degree (αagg) as a function of temperature, which was calculated from the temperature-dependent CD measurements. (b) Time-lapse CLSM images showing the growth of BPmoc-F3 nanofibrils after adding seeds for 0, 10, 70, 130, and 190 s. As a comparison, the image at 300 s without adding seeds shows hardly any assembled structures. Adapted with permission from Macmillan Publishers Ltd: Nat. Chem., from ref. 68, copyright 2016. | ||
3. Strategies to construct multicomponent nanostructures containing peptides/peptide derivatives
3.1 Programming functional chromophores into nanostructures by pre-linking with peptides
There are twenty amino acids in nature, encoded by a gene, that makeup proteins across all living systems.83,84,123 According to the water affinity of their side chains, these natural amino acids can generally be divided into hydrophilic and hydrophobic.98 Hydrophilic amino acids usually carry polar or ionizable moieties, particularly, Asp, Glu, Lys, Arg, and His are electrically charged at physiological pH, which renders them prevailing constituents in biomedical materials.124,125 Hydrophobic amino acids are featured by the presence of aromatic or aliphatic moieties and peptides consisting of aromatic residues, identified from β-amyloid, have been elucidated as important assembling motifs by extensive early studies.85,98,99Through well-established methods of peptide synthesis, amino acid residues can be integrated into the peptide chain in a predetermined sequence. The programmability of peptide materials establishes their tunable properties, such as solubility, conductivity, and pH/thermal responsiveness,5,23,98 which make them versatile segments in tailoring the intrinsic properties of functional chromophores through peptide modification.126–128 For instance, π-conjugated chromophores are frequently used in organic optoelectronics,129–131 however, the poor water solubility due to aromatic structure hampers their usage in polar environments.5,132 Accordingly, these semiconductive aromatic chromophores are conjugated with hydrophilic peptides to ameliorate their solubility, as well to achieve peptide-assisted self-assembly.133–137 The aqueous self-assembly of resulting amphiphilic conjugates are largely determined by the hydrophobic effects of π-conjugated domains, as hydrogen bonding and permanent dipole–dipole interactions between hydrophilic peptide segments are attenuated in aqueous environments.98,99 However, with the presence of the electrically charged amino acid residues that produce strong electrostatic repulsion, the previously overwhelming attractive hydrophobic effects from π-conjugated chromophores can be significantly mitigated.5,134 As a consequence, the balance between hydrophobic effects and opposing electrostatic repulsion within assembling amphiphilic conjugates can be finely tuned, which enable the positioning of π-conjugated chromophores into well-defined nanoarchitectures.138,139
Despite the success in the self-assembly of D–A conjugates, the complex synthesis of desired peptidic monomers retards the progress of this strategy, which in turn motivates the exploration of the sorted co-assembly of simple peptidic building blocks. For example, Adams et al. demonstrated that by slowly decreasing the solvent pH, the mixture of two naphthalene-functionalized dipeptide hydrogelators, NAPH-AV and NAPH-AA, coupled with different pKa achieved spatially resolved, self-sorted gelation networks (Fig. 6).140 This pH-triggered self-sorting emerged from the gradual acidification on the binary mixture that sequentially approached different pKa values of hydrogelators, thus triggering the gelation of corresponding components in sequence (Fig. 6b and c).140 Following this sequential gelation strategy, D and A chromophores were, respectively, conjugated with various peptides to construct sorted nano-assemblies allowing efficient photoinduced charge separation.20 These studies highlight the great potential of kinetic factors, such as time, temperature, concentration, solvent composition, and mechanical agitation, on spatially engineering multiple components into one global complex integrated with sorted domains.
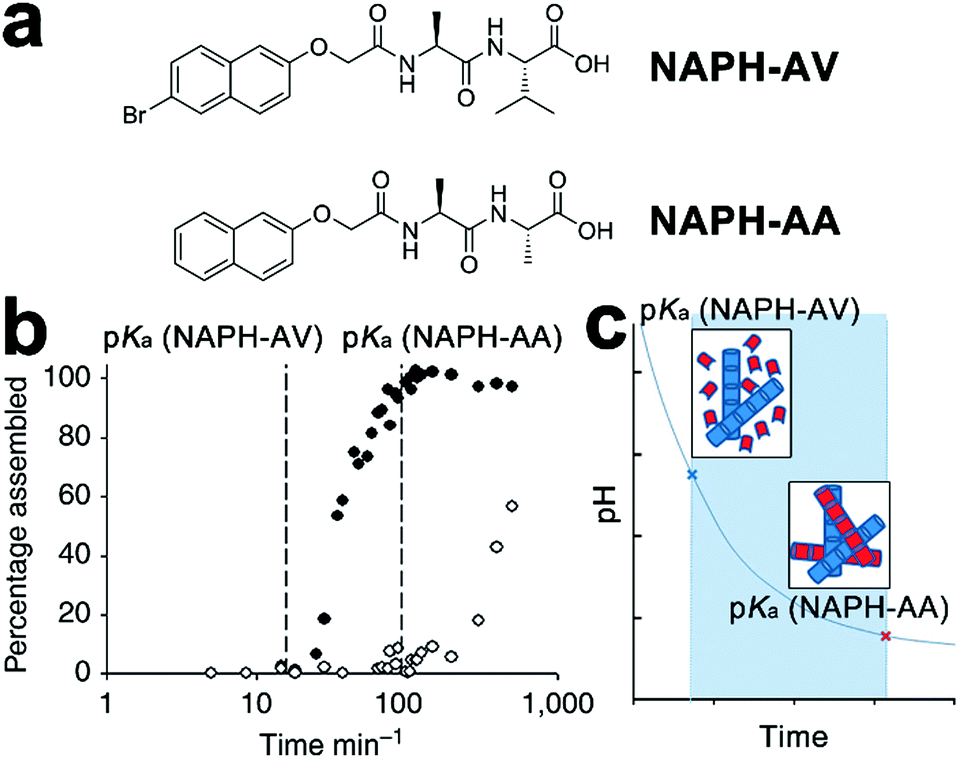 | ||
| Fig. 6 (a) Chemical structures of dipeptide gelators NAPH-AV and NAPH-AA. (b) Degree of assembly derived from integration of NMR data, revealing the sequential gelation of NAPH-AV followed by NAPH-AA. (c) The decreasing pH, due to the hydrolysis of glucono-δ-lactone to gluconic acid, gradually approached the pKa of each component and triggered the assembly of NAPH-AV (blue building blocks) and NAPH-AA (red building blocks) in sequence. Adapted with permission from Macmillan Publishers Ltd: Nat. Commun., from ref. 140, copyright 2013. | ||
3.2 Supramolecular block copolymers through hetero-seeding
On a molecular level, monomers capable of co-assembly should possess geometrical resemblance on their structural motifs and display similar association behavior coupled with comparable free energy gains of homo-association, otherwise, they are likely to produce independently self-sorted homo-aggregates.29,141 Given co-assembly systems whose homo-association of respective monomers is energetically more favorable than hetero-association, the formation of the supramolecular block copolymer as heterogeneous nanostructures integrated with segments of homo-associated monomers may prevail. The construction of supramolecular block copolymer emerges from the development of living supramolecular polymerization.72,73,81,142,143 As an early example, Sugiyasu, Takeuchi, and co-workers synthesized amides and tridodecylbenzene-bearing porphyrin derivatives capable of π–π interactions and intra-/intermolecular hydrogen bonding.72,81 These porphyrin derivatives were prone to form kinetically trapped, off-pathway J-aggregates over thermodynamically equilibrated H-aggregates (Fig. 7), as assembling molecules prefer to form the kinetic intermediate with the lowest activation barrier regarding their predecessor,32,63 the monomeric porphyrin derivatives in this case. For the assumed consecutive pathway, increasing the concentration of starting monomers should shift the assembly equilibrium toward its thermodynamic end, however, it failed to accelerate the formation of H-aggregates here. Because J-aggregates of these porphyrin derivatives are off-pathway species with respect to thermodynamic H-aggregates. Accordingly, larger monomer concentration encouraged the formation of J-aggregates, which entrapped monomers from engaging in the thermodynamic state. Therefore, increasing the monomer concentration enhanced the kinetic preference here on generating off-pathway structures, which in turn retarded the formation of thermodynamic aggregates.30,38 To stimulate the kinetic-thermodynamic transition, the authors added seeds, externally segmented from thermodynamic H-aggregates, to kinetically entrapped J-aggregates, which avoided the energetically suppressed reformation of monomers and nucleation and shifted the divergent assembly pathway to the swift elongation phase directly (Fig. 7). Moreover, this seeding strategy allows structural nuances of monomers, thus applicable to co-assembly systems.72,73,142 Therefore, sequentially feeding the living ends of growing seeds with nonself-monomers (hetero-seeding) produces supramolecular block copolymers (Fig. 7).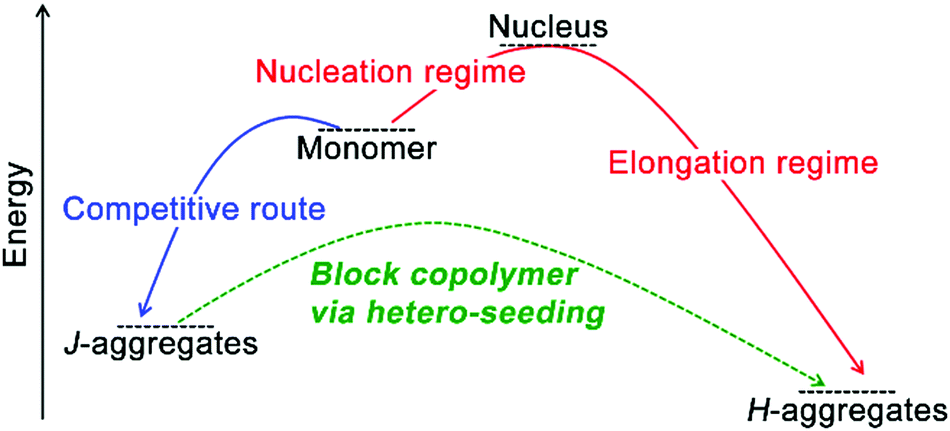 | ||
| Fig. 7 Schematic illustration of the construction of supramolecular block copolymer through hetero-seeding strategy. | ||
Taking advantage of the hetero-seeding strategy, growing assemblies of amphiphilic peptide derivatives can be fed with nonself-monomers and these freshly added monomers may associate with preformed assemblies to produce heterogeneous block architectures.142,143 Otto, van der Gucht et al. studied the cooperative fiber growth of macrocyclic hexamers of SH-Ph and SH-Cy and prepared triblock copolymers by treating the preformed hexamer seeds of SH-Cy with non-assembling trimers or tetramers of SH-Ph (Fig. 8).66 The peptide-decorated dithiol monomers SH-Ph and SH-Cy in this study contained similar peptide segments for the β-sheet formation and used disulfide chemistry to construct the same hexametric core. As peptidic building blocks here possessed geometrical resemblance on their structural motifs, they were able to co-assemble with each other through a hetero-seeding approach. When segmented hexamer nanofibers of SH-Cy were treated with trimers and tetramers of SH-Ph, these non-assembling trimers and tetramers spontaneously rearranged to hexamers to associate with preformed nanofibers, thus generating triblock nanofibers. The formation of hexamer fibers started from the oxidation of peptide-functionalized dithiol-building blocks of SH-Ph or SH-Cy upon air exposure, which yielded macrocyclic disulfides as trimers and tetramers (Fig. 8b). While these metastable trimers and tetramers were incapable of forming nanofibers, applying mechanical agitation on them causing the dynamic exchange of reversible disulfide bonds and thus producing hexamers serving as the starting nuclei to initiate the following hexamer stacking (Fig. 8c). Moreover, continuous agitation caused fragmentation of growing fibers, which produced more active ends for hexamer stacking and resulted in the exponential elongation of hexamer nanofibers.66,67
 | ||
| Fig. 8 (a) Molecular structures of peptide-functionalized dithiol monomers SH-Ph and SH-Cy. (b) Oxidation-triggered cyclization of peptide–dithiol monomers yields a mixture of trimers, tetramers, and hexamers capable of constant disulfide exchange. (c) Hexamers of peptide–dithiol molecule assemble into nanofibers through the β-sheet formation of peptide segments (arrow parts). Upon mechanical agitation, the fragmentation of hexamer fibre generates more active ends for further elongation. Figure adapted with permission from ref. 66. Copyright 2015 Wiley-VCH. | ||
3.3 Peptide assemblies coated by polyelectrolytes
Despite the desirable features of peptidic assemblies such as self-correcting, adaptable, and stimuli-responsive properties, their susceptibility toward environmental changes weakens their structural integrity against adverse environmental circumstances, which compromises their performance in practical utilizations. Stupp and coworkers synthesized a set of peptide amphiphiles (PA) by conjugating β-sheet forming peptides with hydrophobic tails and these PAs self-assembled into one-dimensional fiber-like nanostructures in aqueous environments.70 As long PA nanofibers were superior to shorter ones in supporting cell adhesion, the thermodynamic and kinetic factors regulating fiber growth were explored to preserve their longitudinal dimensions with bioactivity.70,144 For example, above the critical ionic strength (Ic) of 6 mM, the alkyl (C16)-conjugated peptide sequence V3A3K3 spontaneously achieved its thermodynamic structure as ultralong nanofibers.70 Upon dilution, however, the ionic strength dropped down below Ic, which largely removed the previous charge-screening effect on lysine residues of V3A3K3. As weakened charge screening effect, in turn, strengthened the charge repulsion on peptide sequence, the assembly of alkyl-conjugated V3A3K3 was attenuated, which converted the previous thermodynamic structures as ultralong fibers into kinetically trapped species. Accordingly, heating these kinetically trapped ultralong fibers upon dilution resulted in their dissociation and generated monodispersed short segments instead.70 Notably, the energy barrier of this ionic strength-induced kinetic-thermodynamic transition was calculated as 171 kJ mol−1,70 indicating high structural integrity capable of counteracting competitive effects as strong as electrostatic interactions or hydrogen bonds. Therefore, well-defined, peptide-based nano-assemblies may serve as stable frameworks allowing the sequential positioning of distinct components through multiple non-covalent interactions.To improve the structural integrity of peptidic assemblies, Parquette and co-workers created conformal polydopamine (PDA) coating around self-assembled nanofibers of naphthalenediimide-dilysine NDI-KK, which rendered the resulting nanofibers impervious toward adverse changes in pH and temperature (Fig. 9).145 The self-assembly of NDI-KK in water, driven by the β-sheet formation of dilysine and J-type π–π stacking of NDI, generated 1D helically twisted nanofibrils (Fig. 9a).134 Also, the self-assembly of NDI-KK was carried out at pH 8.0 to attenuate the unfavorable electrostatic repulsion for fiber growth due to the protonation of lysine residues. After 12 h of incubation, the resulting nanofibers were treated with dopamine, a common hormone capable of oxidative self-polymerization in alkaline solutions, to produce a crosslinked PDA coating around them,146,147 thus generating the PDA/NDI-KK composite. The starting NDI-KK nanofibers were susceptible to external stimuli and dissociated after heating or under acid conditions, in contrast, the PDA/NDI-KK composite with the protective, conformal PDA coating exhibited much-strengthened resilience toward adverse environmental factors. As demonstrated by circular dichroic (CD) spectra and dynamic light scattering (DLS) measurements, the chiral stacking of NDI chromophores within PDA/NDI-KK withstood acid pH as low as 3.0 and the overall dimension of PDA/NDI-KK remained stable from 25–70 °C (Fig. 9b).145
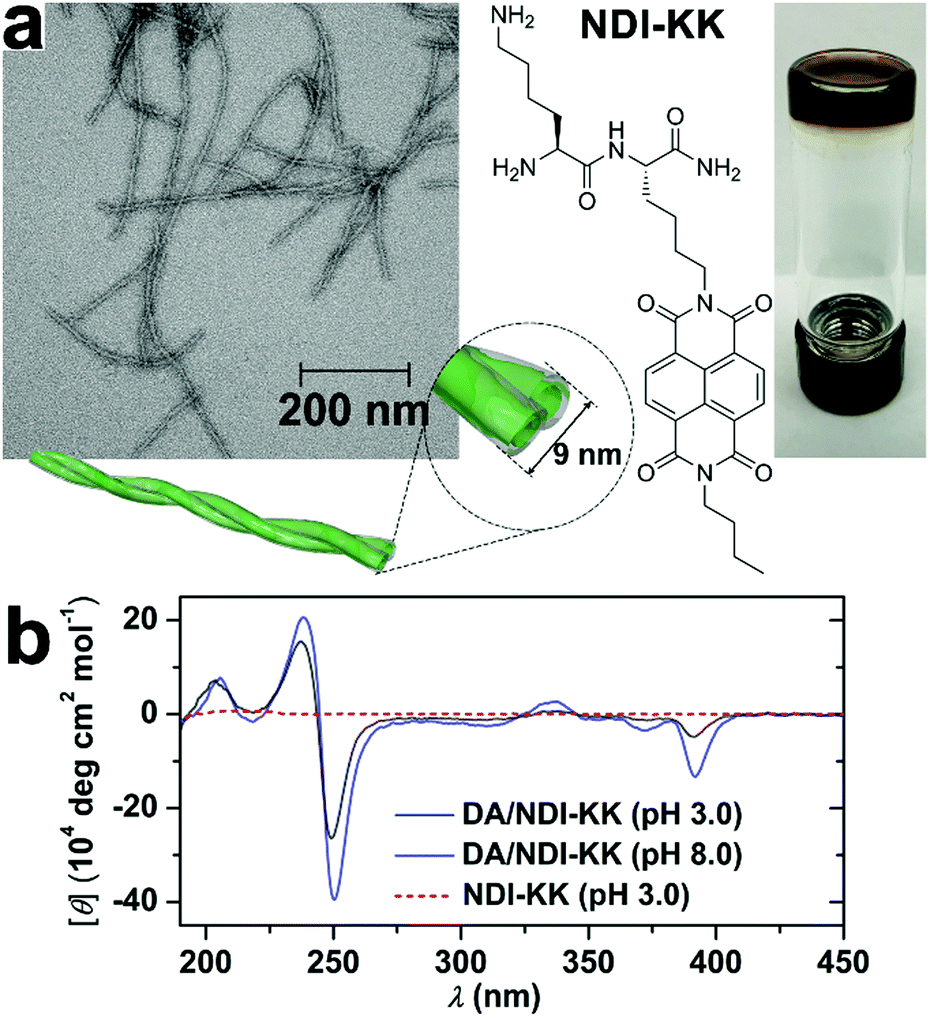 | ||
| Fig. 9 (a) TEM image of PDA/NDI-KK nanofibers and the chemical structure of peptide amphiphile NDI-KK. On the right panel, the photo of PDA/NDI-KK hydrogel after incubation at pH 8.0 for 24 h. (b) CD spectra of PDA/NDI-KK composites at pH 3.0 and 8.0 in water. As a comparison, NDI-KK at pH 3.0 showed no CD signature. Reproduced with permission from ref. 145. Copyright 2020 Wiley-VCH. | ||
3.4 Entrapping carbon nanotubes with amphiphilic peptide derivatives
Single-walled carbon nanotubes (SWNTs) are expected as ideal candidates for optoelectronic applications due to their unique electronic properties emerging from continuous 1D structures.148 However, SWNTs exhibit strong insolubility in polar environments, which causes detrimental bundling of SWNTs and restricts their processability in device fabrication. Early studies demonstrate surfactants and synthetic lipids can form stable and organized coating layers on the surface of SWNTs,149 through a spontaneous process modulated by the interplay between repulsive electrostatic interactions and attractive van der Waals force.150 With the presence of aromatic residues capable of hydrophobic interaction with SWNTs, amphiphilic peptides and peptide derivatives are also employed to exfoliate and stabilize SWNTs under aqueous conditions.151,152 In addition, thiol-containing peptides capable of reversible cyclization around SWNTs153,154 and cyclic peptides appended with pyrene moieties155 have been used to functionalize SWNTs and achieve their aqueous solubilization as well. Recently, Thomas, Kruss and coworkers demonstrated that a coiled-coil peptide rendered SWNTs water-soluble by forming heptamer peptide barrels to encapsulate SWNTs.156 This coiled-coil peptide, by itself, formed peptide barrels with channel cavities accessible to hydrophobic molecules.157 Upon mixing with SWNTs, the coiled-coil peptide barrels by heptamer generated a channel with a pore dimension compatible with the diameter of typical SWNT species. Therefore, multiple peptide barrels encapsulated SWNTs and produced a coaxial peptide coating layer along the length of SWNTs.156 This strategy can tailor the surface of SWNTs via non-destructive modification, thus preserving the optical properties of SWNTs. Moreover, coiled-coil peptides can form well-defined tubular structures with tunable diameters, which envisage their potential in discriminating SWNTs according to the dimension.Parquette and co-workers encapsulated single-walled carbon nanotubes (SWNTs) with self-assembled nanotubes of naphthalenetetracarboxylic acid diimide-lysine bolaamphiphile, NDI-bola (Fig. 10).158 The preformed NDI-bola nanotubes were segmented by sonication prior to mixing with SWNTs to reduce potential steric congestion upon entrapping carbon nanotubes. The resulting nanotube segments encapsulated SWNTs via electrostatic and cation–π interactions, meanwhile, the inherent thermodynamic propensity of supramolecular structures on self-healing159–161 re-combined nanotube segments along the length of SWNTs to form the continuous coaxial coating (Fig. 10b and c).162 Notably, the encapsulation of SWNTs by NDI-bola nanotubes resulted in the overlap of their electrical double layers (EDL), which reduced the surface charge of the outermost layer of lysine residues and redistributed monomeric bola amphiphiles from solvent to aggregates according to the colloidal particle theory.163,164 To mitigate the compromised dispersibility due to reduced surface charge, monomeric NDI-bola formerly dispersed in solvent engaged in the formation of the bilayer wall of SWNT/NDI-bola hybrids. In addition, the surface of SWNT/NDI-bola hybrids were wrapped by various conjugated polyelectrolytes through electrostatic interactions165 to achieve three-component, coaxial tubular nanohybrids for potential optoelectronic applications.
 | ||
| Fig. 10 (a) Chemical structure of the bolaamphiphile NDI-bola, schematic graphics of NDI-bola nanotube and the co-axial tubular nanohybrid of SWNT/NDI-bola. (b) TEM image of the co-axial SWNT/NDI-bola nanohybrid. (c) Schematic presentation of the electrostatic and cation–π interactions between the inner channels of NDI-bola nanotubes and SWNTs after sonication-assisted progressive encapsulation. Adapted from ref. 158 with permission from the Royal Society of Chemistry. | ||
3.5 Welding heterogeneous tubular segments
Non-specific interactions with few directional constraints, such as the electrostatic interaction, may also assemble heterogeneous subunits into hierarchical composites with nanoscale precision. Kameta and co-workers synthesized two undecyl lipids Phe-C11-Gly-COOH and Phe-C11-Gly-NH2 that, respectively, assembled into nanotubes with matched diameters and wall thicknesses in water.46 Both lipids carried an uncharged terminus of phenylalanine derivatives that constituted the neutral exterior of resulting lipid nanotubes; the carboxyl glycine head of Phe-C11-Gly-COOH and the amino glycine head of Phe-C11-Gly-NH2 made up the negatively and positively charged nanotube interiors, respectively (Fig. 11). These oppositely charged interiors arose attractive electrostatic interactions over cross-sections of these two nanotubes, which combined these tubular segments at open ends (Fig. 11a).46 After the electrostatic combination, tubular segments with oppositely charged interiors adopted an alternating arrangement to create heterogeneous nanotubes with blocked inner channels where amine and carboxylic sections were alternately distributed. When a pH-responsive fluorophore CypHer5 with a pKa of 6.1 traversed these blocked nanochannels, the ionization of amine/carboxylic interiors caused the deprotonation/protonation of CypHer5 and thus resulting in its intermittent fluorescence emission.46 | ||
| Fig. 11 (a) Chemical structures of undecyl lipids Phe-C11-Gly-COOH and Phe-C11-Gly-NH2, and their nanotubes with oppositely charged interiors which can be combined at open ends. (b) Scanning electron micrograph (SEM) image of heterogeneous nanotubes after mixing dispersions of Phe-C11-Gly-COOH and Phe-C11-Gly-NH2, and the photograph of the resulting hydrogel. Adapted from ref. 46 with permission from the Chemistry Society of Japan. | ||
3.6 Peptide assembly directed by complementary base pairing
Synthetic oligonucleotides can achieve stoichiometric complexation through complementary base pairing. As supramolecular templates, single-strand oligonucleotides successfully modulated the fine positioning of functional chromophores capable of complementary base pairing with nucleotidic templates to form uniform nanohybrids.166,167 Chau and Ni synthesized a series of tri-segmented peptides which were constituted by an N-terminal oligocation for DNA binding, a middle amyloid-derived segment for β-sheet formation, and a C-terminal hydrophilic segment for water solubility. In aqueous environments, DNAs entrapped by cationic segments were segregated from environmental surroundings by laminated β-sheet layers to form viral capsid-like nanofibrils/nanoribbons. The resulting capsid-like structures are then converted into higher-ordered assemblies through lateral association, which can be useful in constructing gene delivery vehicles.168–170Taking advantage of the reversible Watson–Crick base pairing, Stupp and coworkers studied the reversible intertwining and bundling of alkylated peptide nanofibrils with a surface distribution of complementary oligonucleotides (Fig. 12).171 In water, peptide amphiphile (PA) of alkyl (C16)-conjugated V3A3E3 self-assembled into nanofibers by projecting their peripheral hydrophilic β-sheet segments from their hydrophobically collapsed alkyl cores. Based on this self-assembling PA precursor, complementary oligonucleotides were covalently attached on PA, respectively, thus yielding complementary conjugates PA–DNA-1 and PA–DNA-1′ (Fig. 12a). As PA co-assembled with PA–DNA-1 or PA–DNA-1′ in molar percentages from 0.1% to 10%, the resulting PA/PA–DNA-1 and PA/PA-DNA-1′ nanofibrils were randomly distributed with complementary oligonucleotide sequences on their surfaces. Due to the DNA hybridization between PA/PA–DNA-1 and PA/PA–DNA-1′ fibers, their aqueous dispersions produced gelation after mixing, which generated a nanofibrous network embedded with micrometer-sized bundles of intertwined filaments (Fig. 12b and c).171 Confocal optical microscopy revealed DNA-containing PA–DNA-1 and PA–DNA-1′, labeled with a fluorescent dye Cy3, were enriched in twisted bundles. Considering PA–DNA-1 and PA–DNA-1′ were evenly distributed in the starting PA/PA–DNA-1 and PA/PA–DNA-1′ nanofibers, the author reasoned that the formation of DNA-rich bundles was due to the redistribution of DNA-attached monomers within and among neighboring fibers. To demonstrate how the electrostatic interactions between nucleotides affected the growth of the superstructure, nucleotide sequences in PA–DNA-1 and PA–DNA-1′ were replaced by shorter counterparts to attenuate their charge repulsion. Moreover, one of the short nucleotide sequences was converted to peptide nucleic acid (PNA) sequence to completely remove the charged phosphate-sugar backbone, thus generating PA–PNA conjugate PA–PNA-2 and PA–DNA conjugate PA–DNA-2′ (Fig. 12a). PA–PNA-2 assembled into well-defined nanofibers, in contrast, the overwhelming charge repulsion of PA–DNA-1, PA–DNA-1′ and PA–DNA-2′ precluded the formation of any higher-order nanostructures on their own but simple spherical aggregates. Finally, the hybridization between complementary PNA and DNA sequences produced intertwined PA–PNA-2/PA–DNA-2′ nanofibers with regular pitches, and further hybridization among these intertwined fibers produced twisted bundles over time (Fig. 12d and e).171
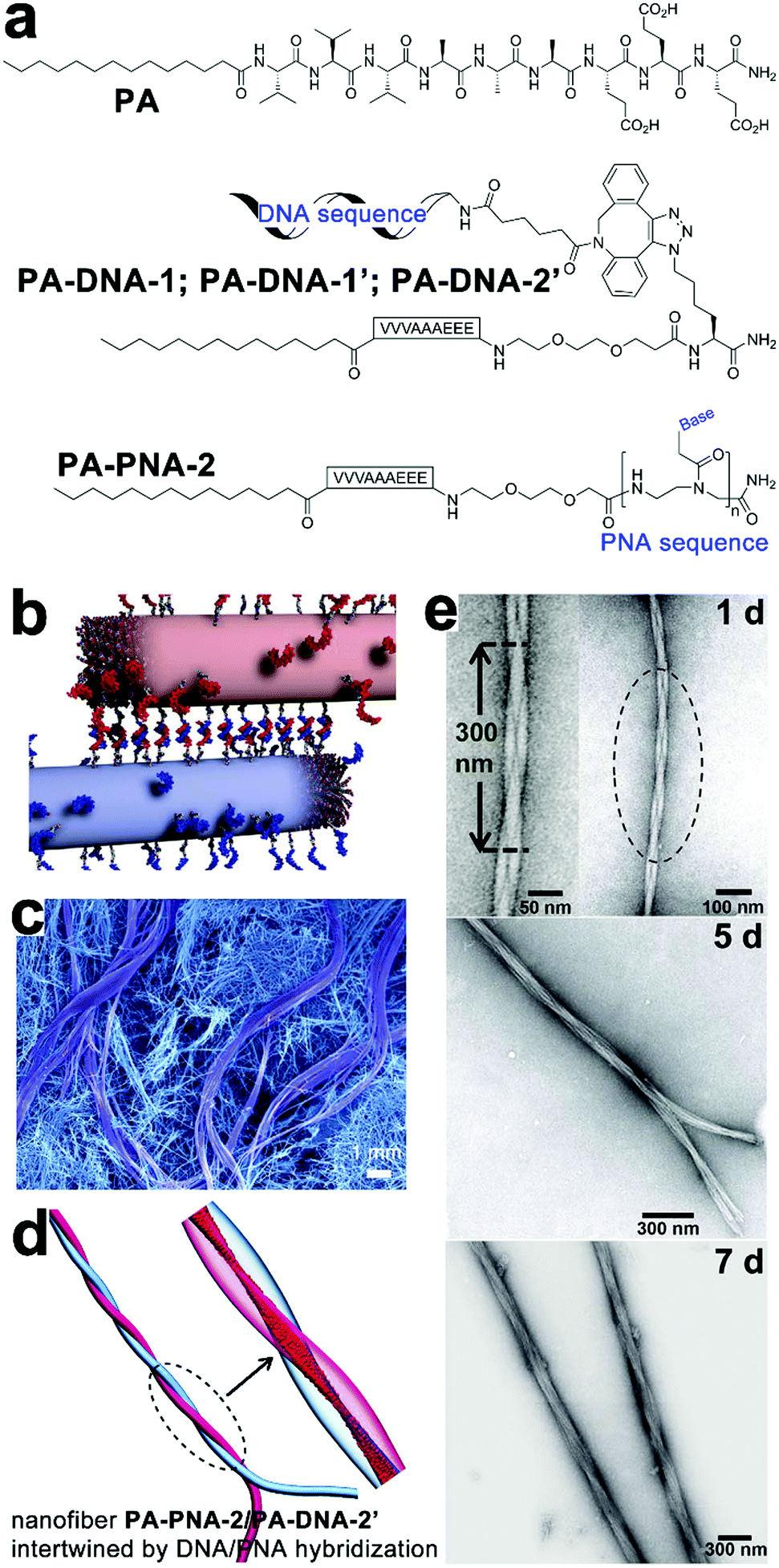 | ||
| Fig. 12 (a) Chemical structures of peptide amphiphile (PA) of alkyl (C16)-conjugated V3A3E3, complementary pairs of conjugates PA–DNA-1/PA–DNA-1′ and PA–PNA-2/PA–DNA-2′. (b) PA/PA–DNA-1 and PA/PA–DNA-1′ nanofibers combined by DNA hybridization. (c) SEM image reveals bundles of intertwined filaments within the gelation network of individual nanofibers. (d) Simulation snapshot of intertwined PA–PNA-2/PA–DNA-2′ nanofibers. (e) TEM images of intertwined PA–PNA-2/PA–PNA-2′ nanofibers evolved into twisted bundles over time. Adapted from ref. 171 with permission from AAAS. | ||
3.7 Nanostructures constituted by cyclic peptides
Studies on cyclic peptides propose well-defined peptidic building blocks to the construction of tubular nanostructures. Early studies by Granja, Ghadiri and co-workers showed that hollow tubular nanostructures could be achieved from the extended ring-stacking of cyclic polypeptides.172–175 For instance, a cyclic octapeptide consisting of alternating D- and L-amino acid residues adopted a free-energy-optimized flat conformation to situate its backbone amides perpendicularly with respect to the cyclic plane. With the protonation of glutamic acid residues, multiple cyclic peptides stacked with each other via β-sheet-like intermolecular hydrogen bonding, therefore producing hundreds nm long tubular structures with internal diameters of 7–8 Å.172Smaller peptide rings, as simple as cyclic dipeptides were demonstrated, by Govindaraju et al., to form various nanoarchitectures, including nanotubes, nanospheres, and nanosheets through tailoring the length of alkyl appendants on dipeptide rings.176,177 The geometry of cyclic dipeptides enables them to project their appended side groups toward the peripheral. Accordingly, the self-assembly of cyclic dipeptides tethered with fluorescent chromophores regulated these chromophores into restricted stacking, which produced strong aggregation-induced emission (AIE).178 Moreover, with the presence of butyloxycarbonyl (tBoc) group capable of host–guest interaction with β-cyclodextrin (β-CD), the self-assembly of tBoc-cyclo-dipeptide was regulated by the addition of a competing guest molecule, adamantane carboxylic acid.179
Recently, Perrier and co-workers demonstrated that polymers conjugated with cyclic peptides allowed further modification/complexation, which generated the tubular nanostructure owing to functional polymeric corona.180–182 For instance, through reversible addition–fragmentation chain transfer (RAFT), the block copolymer poly(acrylic acid)–polyisoprene was grafted onto a cyclic octapeptide.183 Along with the stacking of cyclic peptide cores, this block copolymer-cyclic peptide conjugates assembled into tubular structures consisting of multiple polymer shells. Moreover, through the selective crosslinking of the outer acrylic acid shell and ozonolysis removal of the inner polyisoprene shell, the previous tubular assemblies could be converted into nanotubes with tunable dimensions of the inner core.183 The presence of polymer side chains can also provide extra control on the extent of cyclic peptide stacking.184 For instance, cyclic peptides were conjugated with pH-responsive poly(dimethylamino ethyl methacrylate) (pDMAEMA) to produce cylindrical polymer brushes under basic conditions.184 However, reducing environmental pH enhanced the protonation of pDMAEMA arms, which gave rise to stronger electrostatic repulsion between cyclic peptide–pDMAEMA conjugates, thus attenuating their propensity on aggregation.184 In addition, the cyclic peptide core could be appended with hydrophobic or guest groups, thus providing extra stabilization185 or embedding the host–guest switch186 to the assembly of cyclic peptide–polymer conjugates, respectively.
4. Functioning of peptide/peptide derivative-containing nanocomposites modulated by the spatial organization
4.1 Positioning silver nanoparticles into ordered arrays
Controllable nucleation of heavy metals establishes the preparation of functional metallic nanoparticles applicable to tunable plasmonic resonance, catalysis, antimicrobial, etc. However, metal nanoparticles, with high surface energy, usually exhibit a strong propensity for aggregation, which severely compromise their intrinsic properties.187 Given amino acid residues accessible for further functionalization, peptide derivates carrying nucleation/binding sites are employed to construct well-defined biotemplates for the preparation of stably dispersed metal nanoparticles. Liu and coworkers synthesized a fluorenylmethoxycarbonyl (Fmoc)-peptide amphiphile, which assembled into fibrous structures.10 The surface of the resulting nanofibers was distributed with carboxylic acid and thiol groups. Upon reduction by sodium borohydride, silver ions adsorbed by carboxylic moieties nucleated into silver nanoparticles (AgNPs) and these AgNPs were further stabilized by thiol groups through the formation of coordinate covalent bonds.10 Stupp and co-workers developed an aldehyde-attached peptide-amphiphile capable of forming high-aspect-ratio nanofibers with a complete surface coverage by aldehyde moieties.188 As one aldehyde moiety can stoichiometrically reduce two silver ions to Ag2 cluster via the Tollens’ reaction,189 the aldehyde surface of resulting nanofibers enabled the site-specific nucleation of silver ions along the longitudinal axis, which produced metallic peptide nanofibers exhibiting antimicrobial effect and less toxicity toward eukaryotic cells.188Despite the stable dispersion of AgNPs along with peptide frameworks, the spatial arrangement of nanoparticles largely remains poorly ordered. Employing phenylalanine-based amphipathic self-assemblies of enantiomeric L-NAPH-Phe and D-NAPH-Phe as chiral templates, Srivastava et al. successfully prepared helical tapes of polydopamine (PDA) to position AgNPs into the specific chiral arrangement. (Fig. 13).190 In sodium bicarbonate solution (pH 10), L-NAPH-Phe and D-NAPH-Phe with primary amine residues spontaneously assembled into (P)- and (M)-helical tapes, respectively. Polymerized dopamine contains electrophilic catechol quinones, which create its inherent reactivity toward the nucleophilic amines.191 Therefore, treating amine-contained L-NAPH-Phe and D-NAPH-Phe helical tapes with dopamine resulted in the formation of covalent PDA coatings over helical tapes via the in situ, oxidative self-polymerization of dopamine in alkaline solutions (Fig. 13a). Presumably, due to the pH-dictated ionization of catechol residues, PDA-coated helical tapes, which completely rolled up into nanotubes at pH 7 were re-opened into loose helices as pH increased from 7 to 10 (Fig. 13b). Moreover, the polyphenolic PDA served as mild reducing agents192 for AgNP nucleation and the resulting AgNPs were preferentially positioned along the edge of PDA-coated helical tapes, thus integrating AgNPs into supramolecular architectures with predefined chirality (Fig. 13c).
 | ||
| Fig. 13 (a) Chemical structures of enantiomeric amphiphiles L-NAPH-Phe and D-NAPH-Phe. Self-assembled L-NAPH-Phe and D-NAPH-Phe were treated with dopamine at pH 10, forming (P)-helical PDA/L-NAPH-Phe tapes and (M)-helical PDA/D-NAPH-Phe tapes, respectively. (b) pH-Controlled deformation of PDA-coated helical tapes. (c) The helical arrangement of AgNPs along the edge of helical tapes at pH 10. Reproduced with permission from ref. 190. Copyright 2019 Wiley-VCH. | ||
4.2 Immobilizing multiple functional proteins on peptidic assemblies
The functional competence of synthetic proteinaceous materials highly hinges on the integration of multiple proteins into hierarchical architectures in optimized combinations, as well as in well-defined ratios. Collier and coworkers prepared fluorescent composites by incorporating multiple fluorescent proteins with β-sheet fibrils, and the color of the resulting composites was highly tunable by modulating the ratio of incorporated proteins (Fig. 14).193 The incorporation of fluorescent proteins started with the co-incubation of β-sheet-modified green fluorescent proteins (GFP-βtail) with β-sheet-forming peptides (QQKFQFQFEQQ, Q11) at physiologic pH (Fig. 14a). As the peptide tail of GFP-βtail had slow fibrillization kinetics, GFP-βtail underwent a gradual transition from α-helix to β-sheet upon incubating with Q11. After co-incubation, the resulting GFP-βtail/Q11 nanofibers were sedimented by centrifugation and exhibited tunable fluorescence whose emission intensity was proportional to the concentration of GFP-βtail relative to Q11 at the onset of incubation. | ||
| Fig. 14 (a) Schematic illustration of the incorporation of fusion proteins consisting of β-sheet formation domains into growing β-sheet nanofibers, which generate peptide nanofibers carrying multiple fluorescent proteins in precisely controlled ratios. (b) Modulating the colour of fibrillar microgels by changing the relative molar ratio of various fluorescent protein/Q11 peptide nanofibers. Adapted with permission from Macmillan Publishers Ltd: Nat. Mater., from ref. 193, copyright 2014. | ||
It is noteworthy that due to the non-specific interaction between protein cargos and peptidic fibrils, other β-sheet-forming peptides in addition to Q11 were able to co-assemble with mutated GFP-βtail, whose peptidic tails incapable of inserting into growing fibrils, which suggested the generality, yet empiricism to some extent, of this protein integration strategy. After demonstrating the co-assembly of GFP-βtail and Q11, another two β-sheet-tailed fluorescent proteins, eGFP-βtail and dsRED-βtail, were successfully integrated with growing Q11 nanofibers to achieve blue and red fibrillar composites, respectively. Under mild agitation, resulting nanofibers with different colors were associated into microgels at predetermined ratios to produce composites with various fluorescent colors including pink, orange, and turquoise (Fig. 14b). Finally, statistically mixing β-sheet-forming peptides with β-sheet-modified fluorescent proteins was further applied on the incorporation of other functional fusion proteins to achieve adjustable enzymatic activities and antibody responses.193
The charged surface of some peptide-based nanostructures provides abundant binding sites for multiple enzymes capable of sequential chemical conversions, thereby creating peptidic nano assemblies integrated with multienzyme cascades.194 Tibita, Parquette and co-workers immobilized CO2-fixing enzyme, RubisCO, and the associated assimilatory pathway enzymes of carbon fixation upon self-assembled Ac-KK(CPT)-NH2 or NH2-KK(CPT)-NH2 nanotubes (Fig. 15). These enzyme-loaded peptide nanotubes produced ribulose 1,5-bisphosphate (RuBP), the substrate for RubisCO, from ribose-5-phosphate (R-5-P) or glucose and continued the subsequent CO2-fixation catalyzed by RubisCO to yield the product as 3-phosphoglyceric acid (3-PGA).195 Due to the positively charged surface of nanotubes, form II L2 dimeric RubisCO (from Rhodospirillum rubrum, RubisCO II) or form I L8S8 hexadecameric RubisCO (from Ralstonia eutropha, RubisCO I) were able to be deposited on the nanotube surface through electrostatic interactions.11 The RubisCO enzymes were tagged with gold nanoparticles to render these enzymes observable by TEM imaging (Fig. 15c). In the resulting enzyme–nanotube hybrids, RubisCO II immobilized on NH2-KK(CPT)-NH2 nanotubes exhibited much-enhanced resilience toward proteolysis, as RubisCO II enzymes embedded in nanotubes were sequestered from protease. Moreover, RubisCO I immobilized on NH2-KK(CPT)-NH2 nanotubes exhibited a significantly enhanced selectivity toward CO2 from O2, which was attributed to the electrochemical microenvironment of immobilized RubisCO I preferentially occluded paramagnetic O2 molecules from entering its active sites.195
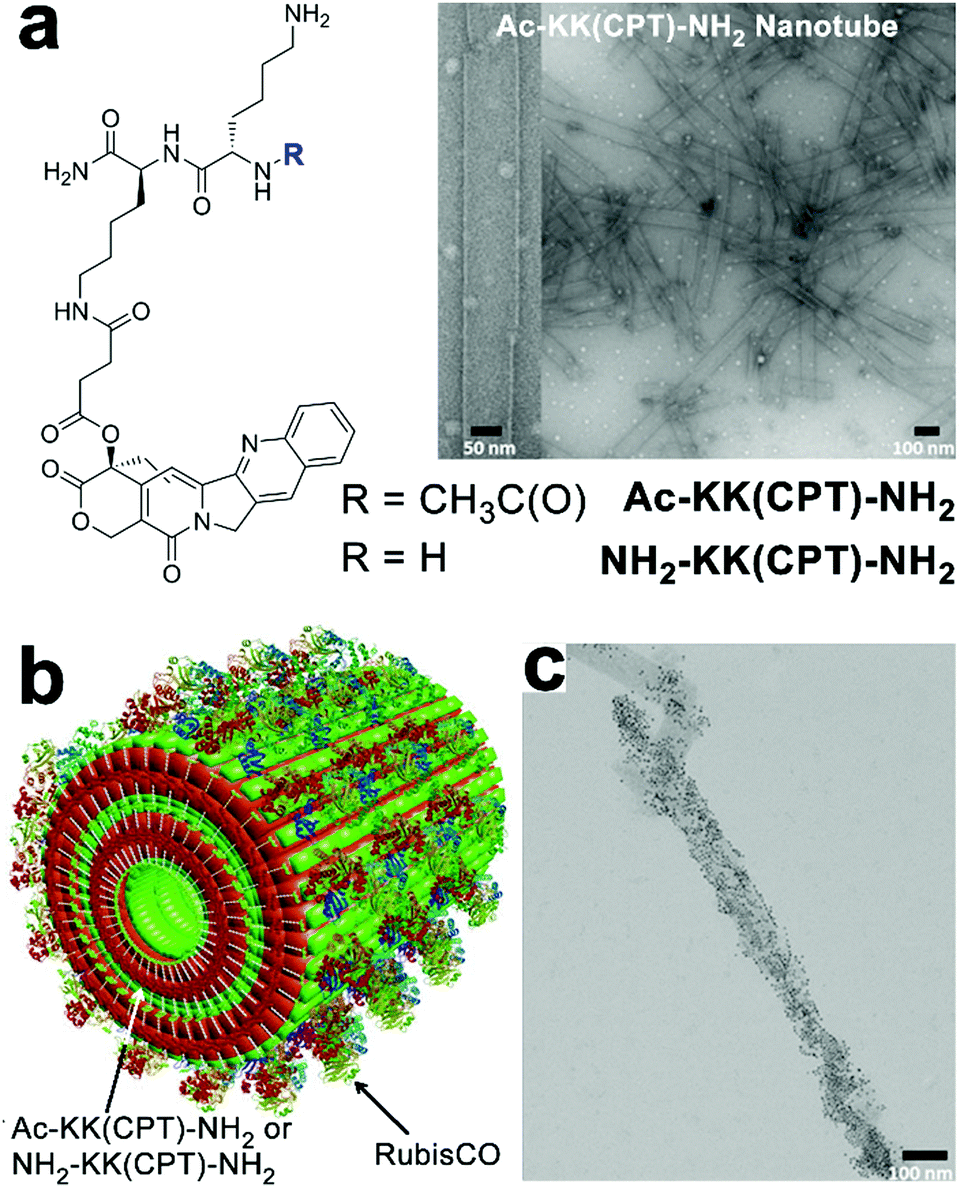 | ||
| Fig. 15 (a) Chemical structures of dipeptide-conjugates Ac-KK(CPT)-NH2 and NH2-KK(CPT)-NH2 and the TEM images of Ac-KK(CPT)-NH2 nanotubes assembled in Bicine buffer. (b) Illustration of RubisCO deposited on Ac-KK(CPT)-NH2/NH2-KK(CPT)-NH2 nanotubes through electrostatic interactions. (c) TEM images of gold-nanoparticle-tagged RubisCO I immobilized on Ac-KK(CPT)-NH2 nanotubes. Adapted from ref. 195 with permission. Copyright 2017, Springer Nature. | ||
4.3 Serving as drug carriers
Owing to the inherent biocompatibility of peptidic assemblies, they have been extensively studied as drug carriers to accommodate the complicate extracellular environment under therapeutic conditions. Natural amphipathic peptide sequences capable of forming β-sheets often comprise alternatingly arranged hydrophobic and hydrophilic residues,8 likewise, many assembling peptide amphiphiles contain conjugated alkyl segments that serve as the hydrophobic drug loading domain. Upon self-assembly, the alkyl segments are embedded in the peptidic periphery and sequestered from water, thus creating a hydrophobic phase to noncovalently entrap drug molecules lacking water solubility.12,196,197 To further improve the structural stability of drug-loaded peptide nanoassemblies and guarantee the reproducibility in their loading contents, drug molecules are covalently attached with assembling peptide amphiphiles to achieve therapeutic peptide conjugates as nanomedicines.11,198–201Cui and co-workers constructed anticancer drug amphiphiles by conjugating four camptothecin (CPT) molecules with various β-sheet-forming sequences (GNNQQNY) bearing diglutamic acid (CPT-Sup35-E2) or dilysine (CPT-Sup35-K2) residues on the C-terminal (Fig. 16).198 The resulting therapeutic amphiphiles CPT-Sup35-E2 and CPT-Sup35-K2 individually assembled into nanofilaments with diameters ∼6 nm. In contrast, mixing oppositely charged CPT-Sup35-E2 and CPT-Sup35-K2 at molar ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (n/n) produced their co-assembled structures as nanotubes with external diameters of ∼123 nm and wall thicknesses of ∼25 nm. The large hydrophobic segments of CPT-Sup35-E2 and CPT-Sup35-K2 containing four CPT molecules were crucial to creating the packing geometry of curved nanotube walls, in contrast, their amphiphilic analogues with only one or two CPT appendants were incapable of forming tubular structures. Additionally, decreasing the concentration of amphiphile mixture revealed not fully developed nanotubes as helical ribbons, which demonstrated that the growth of nanotubes underwent the widening of helical precursors. Although the unfavorable dimension and surface charge of CPT nanotubes in this study prohibited their administration on tumor-bearing mice through tail-vein injection, locally injected CPT-bearing amphiphilic nanotubes exhibited a prolonged retention time on tumor sites, which may support the sustainable CPT release benefitting the local tumor treatment.198
:1 (n/n) produced their co-assembled structures as nanotubes with external diameters of ∼123 nm and wall thicknesses of ∼25 nm. The large hydrophobic segments of CPT-Sup35-E2 and CPT-Sup35-K2 containing four CPT molecules were crucial to creating the packing geometry of curved nanotube walls, in contrast, their amphiphilic analogues with only one or two CPT appendants were incapable of forming tubular structures. Additionally, decreasing the concentration of amphiphile mixture revealed not fully developed nanotubes as helical ribbons, which demonstrated that the growth of nanotubes underwent the widening of helical precursors. Although the unfavorable dimension and surface charge of CPT nanotubes in this study prohibited their administration on tumor-bearing mice through tail-vein injection, locally injected CPT-bearing amphiphilic nanotubes exhibited a prolonged retention time on tumor sites, which may support the sustainable CPT release benefitting the local tumor treatment.198
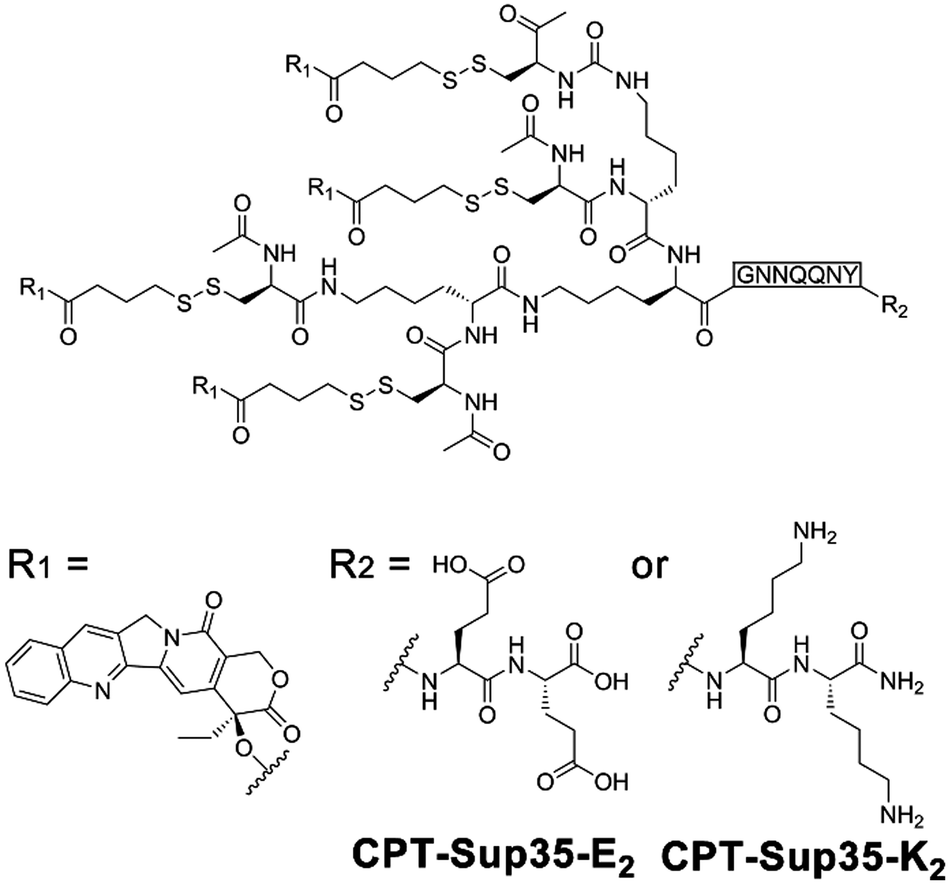 | ||
| Fig. 16 Chemical structures of CPT-bearing amphiphiles CPT-Sup35-E2 and CPT-Sup35-K2. | ||
4.4 Photophysical processes regulated by the spatial arrangement of chromophores
Within photonic/electronic nanostructures, effective and long-range charge delocalization is a direct consequence of the supramolecular arrangement of chromophores. Accordingly, electron donors and/or acceptors have been conjugated with assembling peptides to construct semiconductive hydrogels.26,98 With the formation of peptide frameworks, conjugated donor and acceptor chromophores are positioned into nanostructures with high spatial precision. Resembling the organized light-harvesting chromophores within bacteria and green plants capable of photosynthesis, electron donors (p-type) and acceptors (n-type) organized by assembling peptides can replicate their natural counterparts to support highly efficient photophysical energy-/electron-transfer events.13,20,202,203Ulijn and coworkers synthesized an n-type naphthalenediimide–dipeptide conjugate acceptor, NDI-YF-NH2, thorough the thermolysin-catalyzed condensation between NDI-tyrosine (NDI-Y) and phenylalanine amide (F-NH2) (Fig. 17a).204 This enzymatic amide condensation is reversible, which permits the exchange of F-NH2 by various amino acid amide derivatives and thereby generating a dynamic peptide library capable of self-assembly.205–207 While the peptide bonds within these n-type NDI-appended peptide conjugates were of a dynamic nature, the consequent amide hydrolysis was able to be mitigated by forming alternating stacks with p-type donor chromophores, such as 1,5-dialkoxy naphthalene (1,5-DAN) (Fig. 17a).204,207 Because this alternate donor–acceptor stacking generated strong charge-transfer interactions, which was reasoned to produce favorable free energy change to attenuate the enzymatic amide bond exchange.207 In addition, the closely packed donor–acceptor chromophores in alternating fashion allowed long-range charge delocalization, which resulted in the significant enhancement in conductivity of resulting co-assembled nanostructures.
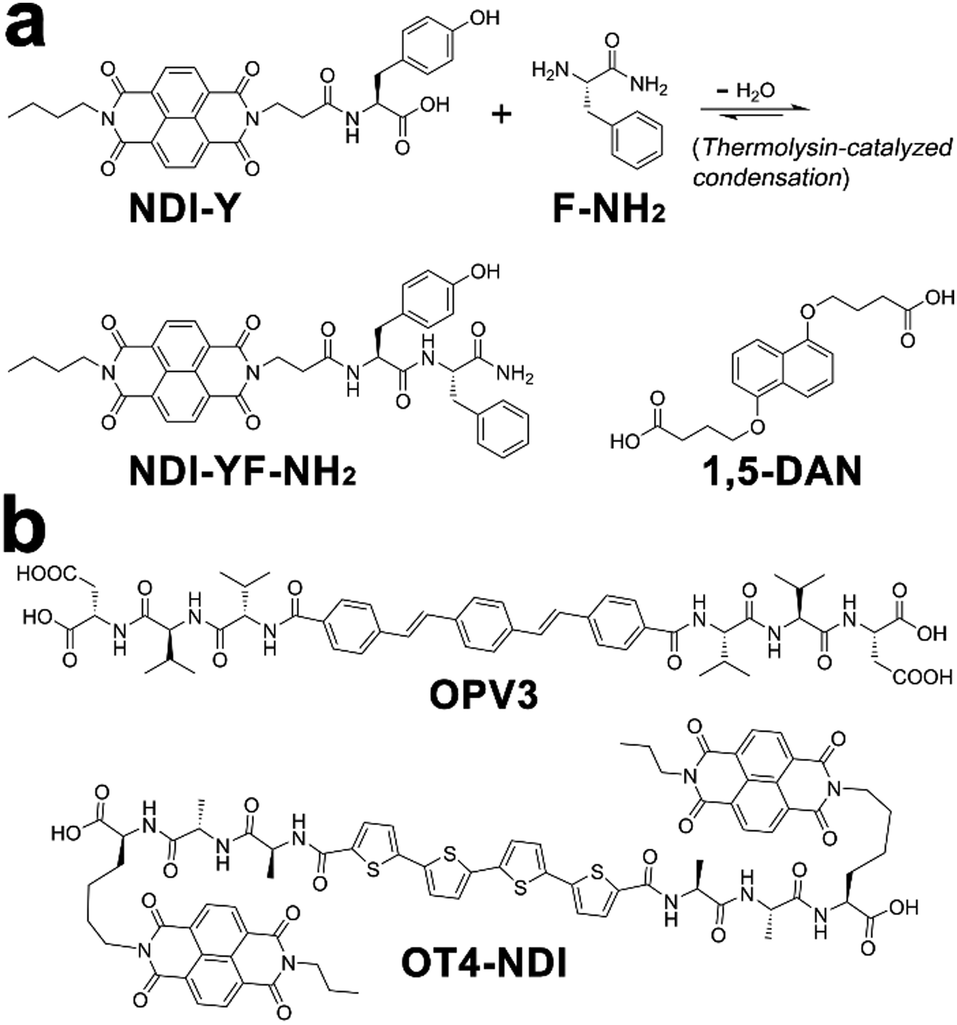 | ||
| Fig. 17 Chemical structures of donor and acceptor peptide conjugate pairs: (a) NDI-YF-NH2/1,5-DAN, (b) OPV3/OT4-NDI. | ||
Tovar, Adams, and coworkers synthesized two peptidic π-electron conjugates: oligo(p-phenylenevinylene) flanked by β-sheet-forming peptides, OPV3, and quaterthiophene flanked by naphthalenediimide-appended peptides, OT4-NDI (Fig. 17b). As these two peptidic π-electron conjugates had different pKa, controlling the rate of acidification on their mixture triggered the gelation of these two conjugates stepwise or simultaneously, which produced self-sorted or co-assembled hydrogel networks, respectively.20 In order to avoid the instant pH decrease by adding HCl, the slow hydrolysis of glucono-δ-lactone (GdL) was employed to achieve gradual acidification on the assembling conjugates. With the hydrolysis of GdL, ionized carboxylic groups on OT4-NDI with a larger pKa were further protonated than OPV3, which accordingly induced the preceding gelation of OT4-NDI. Therefore, GdL hydrolysis-induced acidification triggered the orthogonal assembly of OPV3 and OT4-NDI in the presence of each other, which eventually produced a self-sorted gelation network from their binary mixture. In contrast to hydrolysis-induced acidification, adding HCl caused absolute protonation and charge screening on all peptidic conjugates immediately, which in turn yielded the co-assembled network containing both OPV3 and OT4-NDI in a random arrangement. Comparing with the self-sorted network, co-assembled OPV3 and OT4-NDI supported more efficient energy-/electron-transfer from oligo(p-phenylenevinylene) to quaterthiophene and naphthalenediimide chromophores, demonstrating the influence of spatial organization of chromophores on the photophysical processes within nanoassemblies.20
4.5 Integrating artificial photosynthesis with peptide nanotubes
Diphenylalanine (FF) can spontaneously assemble into high-aspect-ratio nanotubes that may serve as stable supramolecular frameworks to template the deposition of extra-functional constituents on the nanotube surface.208 Park and co-workers created artificial light-harvesting hybrids by depositing J-aggregates of tetra(p-hydroxyphenyl) porphyrin (THPP) and platinum nanoparticles (PtNPs) on the surface of self-assembled FF nanotubes (Fig. 18). Given the high specific surface area of resulting nanohybrids, THPP and PtNPs immobilized on nanotube surface participated in the formation of biomimetic redox gradient, which established the chemical basis of artificial photosynthesis (Fig. 18).209 The preparation of light-harvesting hybrids started from mixing molecularly dissolved FF and THPP in 1,1,1,3,3,3-hexa-fluoro-2-propanol (HFIP), which was then diluted by phosphate buffer (pH 6.0) to induce coaggregation of FF and THPP as a green precipitate. The absorption spectra of the resulting FF/THPP precipitate exhibited a prominent red-shift from monomeric THPP, demonstrating the J-aggregation of protonated THPP (H2THPP2+) in the FF/THPP hybrid which was also consistent with the rough THPP coating on FF nanotubes. Assuming the negatively charged surface of FF nanotubes bound protonated THPP aggregates through electrostatic interactions,210 the metal precursor (K2PtCl4) mixed with FF/THPP was photo-reduced to PtNPs to generate the ternary FF/THPP/PtNPs hybrids. Within the ternary hybrids, THPP aggregates and PtNPs supported strong exciton coupling and efficient charge separation, respectively. Therefore, the FF/THPP/PtNPs hybrid enabled consecutive electron transfer from the starting electron donor, triethanolamine (TEOA), to THPP, then to PtNPs and finally reached the electron mediator ([Cp*Rh(bpy)(H2O)]2+, Cp* = C5Me5, bpy = 2,2′-bipyridine). Moreover, to simulate the photophysical process in photosystem I, the artificial photosystem in this study was coupled with NADH regeneration to drive the enzymatic conversion (glutamate dehydrogenase) of α-ketoglutarate to L-glutamate upon visible light exposure.209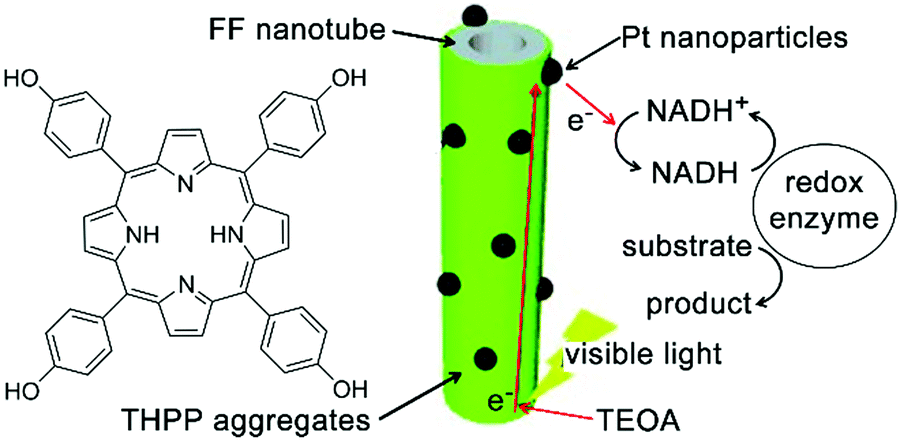 | ||
| Fig. 18 Light-harvesting peptide nanotubes capable of artificial photosynthesis. Figure adapted with permission from ref. 209. Copyright 2012 Wiley-VCH. | ||
4.6 Supercapacitor electrodes constructed by peptide nanotubes
Electron-rich moieties conjugated with peptides can be positioned into densely arranged nanostructures along with the self-assembly of peptides. These organized moieties in proximity may support the delocalization of π-electrons, which underlies the semiconductivity of such peptide assemblies.211,212 Biocompatible diphenylalanine (FF) nanotubes contained quantum-confined regions for electron transfer, accordingly, biosensor electrodes decorated with FF nanotubes owned an enlarged effective surface area, which enabled ultrasensitive detection of ethanol, phenol, and antigen.213–215 Given the great success of peptide nanotubes in the surface modification of high-surface-area electrodes, they were subsequently explored to create a patterned and homogeneous coating on the electrode of the supercapacitor to achieve bio-modified energy storage devices.14,216 Gazit, Rosenman, et al. created a dense coating of vertically aligned peptide nanotubes over supercapacitor electrodes through the vapor deposition of cyclo-diphenylalanine (cyclo-FF) (Fig. 19).216 By modulating the supply of cyclo-FF for deposition, aromatic peptide nanotubes grew into nanoforests with tunable height over electrode surface, thereby producing high-surface-area supercapacitor electrodes (Fig. 19b). The dense and homogeneous alignment of peptide nanotubes significantly enlarged the interface between electrode and electrolyte solution, resulting in a larger capacitance than capacitors constituted by standard or CNT-modified carbon electrodes. Moreover, the highly reproducible electrostatic charge/discharge processes of such nanotube-modified supercapacitors suggested their structural stability and excellent endurance toward potential sweeps.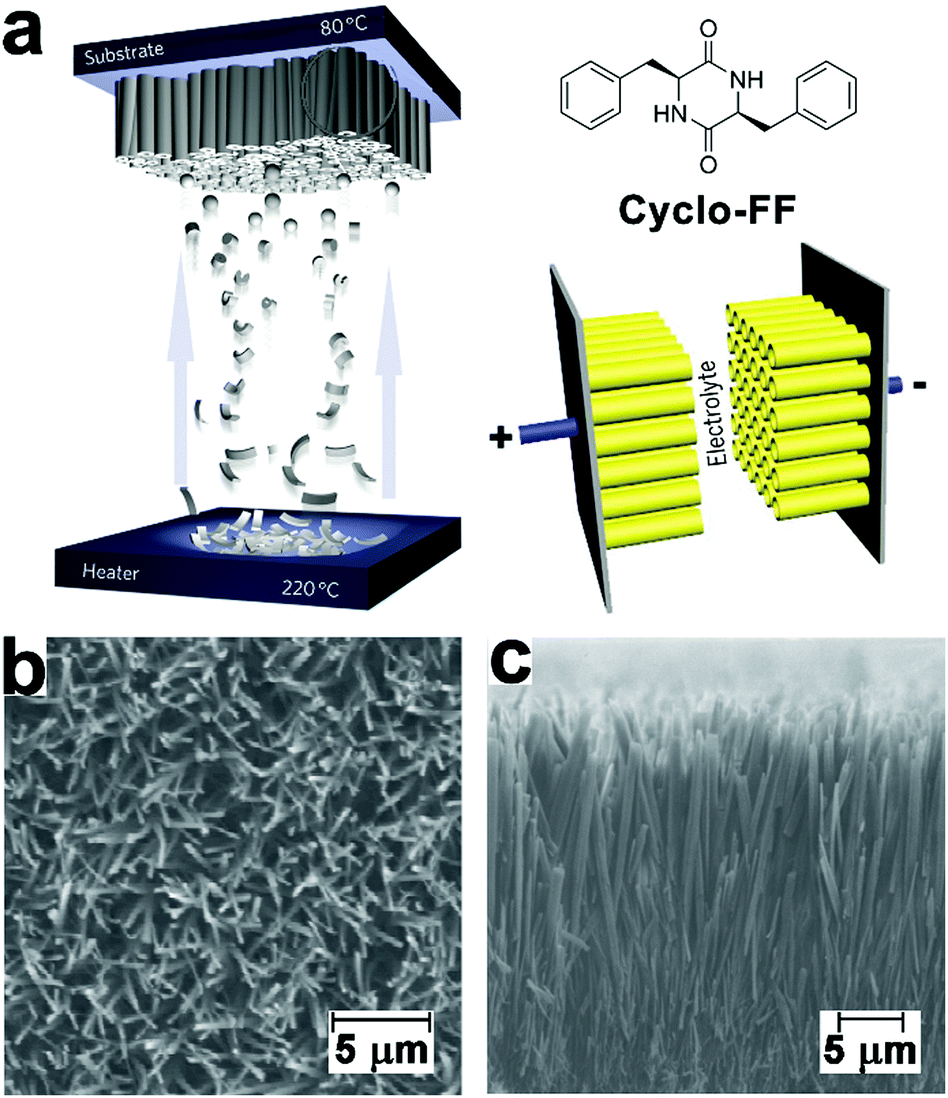 | ||
| Fig. 19 (a) Preparation of supercapacitor electrodes decorated with vertically aligned cyclo-FF nanotubes through vapor deposition. (b) Top-view and (c) side-view SEM images of vertically aligned cyclo-FF nanotubes. Reproduced with permission from Macmillan Publishers Ltd: Nat. Nanotechnol., from ref. 216, copyright 2009. | ||
5. Conclusions and perspectives
Benefiting from the diversity of chemical composition, multicomponent nanocomposites can be imparted with greater tunability and synergism on their hierarchical structures, thus more closely replicate the functional competence of naturally occurring materials and organisms. In the review, we have illustrated the fundamental kinetic and thermodynamic principles of supramolecular polymerization, which establish various strategies to construct multi-component nanostructures from amphiphilic building blocks. Most of these strategies involve monomers consisting of assembling peptide factors, which accordingly generate supramolecular frameworks to regulate the following incorporation of functional components. Moreover, these strategies enable the incorporation of every individual component with high spatial precision, therefore, producing well-defined nanocomposites whose functional competence originates from the synergistic interplay between spatially organized constituents.Despite tremendous advancements in the construction of peptide-based nanohybrids, studies on multicomponent assembly largely remain as semi-empirical exploring. Resembling classic supramolecular systems, peptide-based structures are commonly prepared in solution. Peptidic monomers retain their spatially defined aggregation by one or more noncovalent interactions, including hydrogen bonding, π-stacking, hydrophobic effects, and charge-transfer interactions. The strength of these noncovalent interactions, typically within 10–100 kJ mol−1,217 is similar to those between solute and solvent, which renders the microstructure of peptide-based nanocomposites highly fluxional toward external factors and stimuli.49 Particularly, in contrast to synthetic monomers assembling in organic media, the critical role of hydrophobic collapse in initiating peptide-involved assembly results in larger susceptibility of structural integrity to temperature, concentration, and other instinct properties of aqueous solvents. Therefore, revisiting the knowledge of physical chemistry provides us new insights into the relationship between solute and solvent that correlates with self-assembly.218,219
Moreover, recent progress of supramolecular polymerization brings new theoretical models to unravel the coassembly mechanism of multicomponent system.30–32,38 Quantitative spectroscopic studies, including UV, CD, and fluorescence, combined with morphology imaging through AFM, TEM, and/or super-resolution fluorescence microscopy, set up powerful experimental techniques to resolve the composition of complex coassembly system. Hopefully, future efforts will convert current heuristic studies of peptide-involved assembly to stoichiometric and designable research, which eventually enables us to unambiguously interpret the correlation between monomer structure, arrangement of aggregated species, and functioning of the resulting materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Jiangsu University Grant (No. 20JDG31).Notes and references
- M. J. Webber, E. A. Appel, E. Meijer and R. Langer, Nat. Mater., 2016, 15, 13–26 CrossRef CAS PubMed.
- M. R. Begley, D. S. Gianola and T. R. Ray, Science, 2019, 364, eaav4299 CrossRef CAS PubMed.
- M. Pishvar and R. L. Harne, Adv. Sci., 2020, 7, 2001384 CrossRef CAS PubMed.
- L. Adler-Abramovich and E. Gazit, Chem. Soc. Rev., 2014, 43, 6881–6893 RSC.
- S. H. Kim and J. R. Parquette, Nanoscale, 2012, 4, 6940–6947 RSC.
- C. Liang, R. Ni, J. E. Smith, W. S. Childers, A. K. Mehta and D. G. Lynn, J. Am. Chem. Soc., 2014, 136, 15146–15149 CrossRef CAS PubMed.
- M.-C. Hsieh, C. Liang, A. K. Mehta, D. G. Lynn and M. A. Grover, J. Am. Chem. Soc., 2017, 139, 17007–17010 CrossRef CAS PubMed.
- D. M. Raymond and B. L. Nilsson, Chem. Soc. Rev., 2018, 47, 3659–3720 RSC.
- J. R. Clegg, A. M. Wagner, S. R. Shin, S. Hassan, A. Khademhosseini and N. A. Peppas, Prog. Mater. Sci., 2019, 106, 100589 CrossRef CAS PubMed.
- Y. Wang, L. Cao, S. Guan, G. Shi, Q. Luo, L. Miao, I. Thistlethwaite, Z. Huang, J. Xu and J. Liu, J. Mater. Chem., 2012, 22, 2575–2581 RSC.
- S. H. Kim, J. A. Kaplan, Y. Sun, A. Shieh, H. L. Sun, C. M. Croce, M. W. Grinstaff and J. R. Parquette, Chem. – Eur. J., 2015, 21, 101–105 CrossRef CAS PubMed.
- S. Soukasene, D. J. Toft, T. J. Moyer, H. Lu, H.-K. Lee, S. M. Standley, V. L. Cryns and S. I. Stupp, ACS Nano, 2011, 5, 9113–9121 CrossRef CAS.
- K. J. Channon, G. L. Devlin and C. E. MacPhee, J. Am. Chem. Soc., 2009, 131, 12520–12521 CrossRef CAS.
- P. Beker, I. Koren, N. Amdursky, E. Gazit and G. Rosenman, J. Mater. Sci., 2010, 45, 6374–6378 CrossRef CAS.
- R. N. Perham, Philos. Trans. R. Soc. London, Ser. B, 1975, 272, 123–136 CAS.
- R. E. Blankenship, Chemiosmotic coupling and ATP synthesis, John Wiley & Sons, 2nd edn, 2014, ch. 8, pp. 133–143 Search PubMed.
- J. F. Allen, Cell, 2002, 110, 273–276 CrossRef CAS.
- J.-M. Lehn, Science, 2002, 295, 2400–2403 CrossRef CAS PubMed.
- B. O. Okesola and A. Mata, Chem. Soc. Rev., 2018, 47, 3721–3736 RSC.
- H. A. M. Ardoña, E. R. Draper, F. Citossi, M. Wallace, L. C. Serpell, D. J. Adams and J. D. Tovar, J. Am. Chem. Soc., 2017, 139, 8685–8692 CrossRef.
- J. D. Tovar, Bioinspiration Biomimetics, 2017, 13, 015004 CrossRef PubMed.
- R. Bhosale, J. Míšek, N. Sakai and S. Matile, Chem. Soc. Rev., 2010, 39, 138–149 RSC.
- P. Makam and E. Gazit, Chem. Soc. Rev., 2018, 47, 3406–3420 RSC.
- H. Kar and S. Ghosh, Isr. J. Chem., 2019, 59, 881–891 CrossRef CAS.
- S. Tu, S. H. Kim, J. Joseph, D. A. Modarelli and J. R. Parquette, J. Am. Chem. Soc., 2011, 133, 19125–19130 CrossRef CAS PubMed.
- J. Raeburn and D. J. Adams, Chem. Commun., 2015, 51, 5170–5180 RSC.
- E. R. Draper and D. J. Adams, Chem. Soc. Rev., 2018, 47, 3395–3405 RSC.
- M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS PubMed.
- B. Adelizzi, A. Aloi, A. J. Markvoort, H. M. Ten Eikelder, I. K. Voets, A. R. Palmans and E. Meijer, J. Am. Chem. Soc., 2018, 140, 7168–7175 CrossRef CAS PubMed.
- P. A. Korevaar, T. F. de Greef and E. Meijer, Chem. Mater., 2014, 26, 576–586 CrossRef CAS.
- B. Adelizzi, N. J. Van Zee, L. N. de Windt, A. R. Palmans and E. Meijer, J. Am. Chem. Soc., 2019, 141, 6110–6121 CrossRef CAS PubMed.
- M. Wehner and F. Würthner, Nat. Rev. Chem., 2019, 38–53 Search PubMed.
- G. Ghosh, P. Dey and S. Ghosh, Chem. Commun., 2020, 56, 6757–6769 RSC.
- T. F. De Greef, M. M. Smulders, M. Wolffs, A. P. Schenning, R. P. Sijbesma and E. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- Z. Chen, A. Lohr, C. R. Saha-Möller and F. Würthner, Chem. Soc. Rev., 2009, 38, 564–584 RSC.
- J. Matern, Y. Dorca, L. Sánchez and G. Fernandez, Angew. Chem., Int. Ed., 2019, 58, 16730–16740 CrossRef CAS PubMed.
- D. van der Zwaag, T. F. de Greef and E. Meijer, Angew. Chem., Int. Ed., 2015, 54, 8334–8336 CrossRef CAS.
- P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. Smulders, P. A. Hilbers, A. P. Schenning, T. F. De Greef and E. Meijer, Nature, 2012, 481, 492–496 CrossRef CAS PubMed.
- P. A. Korevaar, C. Schaefer, T. F. de Greef and E. Meijer, J. Am. Chem. Soc., 2012, 134, 13482–13491 CrossRef CAS PubMed.
- G. Fernandez, M. Stolte, V. Stepanenko and F. Wuerthner, Chem. – Eur. J., 2013, 19, 206–217 CrossRef CAS PubMed.
- V. Stepanenko, X. Q. Li, J. Gershberg and F. Würthner, Chem. – Eur. J., 2013, 19, 4176–4183 CrossRef CAS.
- W. Zhang, W. Jin, T. Fukushima, A. Saeki, S. Seki and T. Aida, Science, 2011, 334, 340–343 CrossRef CAS.
- A. Sandeep, V. K. Praveen, K. K. Kartha, V. Karunakaran and A. Ajayaghosh, Chem. Sci., 2016, 7, 4460–4467 RSC.
- W. Zhang, W. Jin, T. Fukushima, T. Mori and T. Aida, J. Am. Chem. Soc., 2015, 137, 13792–13795 CrossRef CAS PubMed.
- Z. Yu, F. Tantakitti, T. Yu, L. C. Palmer, G. C. Schatz and S. I. Stupp, Science, 2016, 351, 497–502 CrossRef CAS PubMed.
- N. Kameta and W. Ding, Bull. Chem. Soc. Jpn., 2019, 92, 1053–1059 CrossRef CAS.
- R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664–675 RSC.
- A. L. Boyle and D. N. Woolfson, Chem. Soc. Rev., 2011, 40, 4295–4306 RSC.
- M. F. Mabesoone, A. R. Palmans and E. Meijer, J. Am. Chem. Soc., 2020, 142, 19781–19798 CrossRef CAS PubMed.
- A. Nandakumar, Y. Ito and M. Ueda, J. Am. Chem. Soc., 2020, 142, 20994–21003 CrossRef CAS PubMed.
- J. Wang, K. Liu, R. Xing and X. Yan, Chem. Soc. Rev., 2016, 45, 5589–5604 RSC.
- Y. Loo, S. Zhang and C. A. Hauser, Biotechnol. Adv., 2012, 30, 593–603 CrossRef CAS PubMed.
- D. Görl and F. Würthner, Angew. Chem., Int. Ed., 2016, 55, 12094–12098 CrossRef PubMed.
- P. P. Syamala, B. Soberats, D. Görl, S. Gekle and F. Würthner, Chem. Sci., 2019, 10, 9358–9366 RSC.
- P. P. Syamala and F. Würthner, Chemistry, 2020, 26, 8426–8434 CrossRef CAS.
- M. Hartlieb, E. D. Mansfield and S. Perrier, Polym. Chem., 2020, 11, 1083–1110 RSC.
- T. Heek, C. Fasting, C. Rest, X. Zhang, F. Würthner and R. Haag, Chem. Commun., 2010, 46, 1884–1886 RSC.
- F. Würthner, Acc. Chem. Res., 2016, 49, 868–876 CrossRef PubMed.
- S. Ogi, V. Stepanenko, K. Sugiyasu, M. Takeuchi and F. Würthner, J. Am. Chem. Soc., 2015, 137, 3300–3307 CrossRef CAS PubMed.
- S. Ogi, V. Stepanenko, J. Thein and F. Würthner, J. Am. Chem. Soc., 2016, 138, 670–678 CrossRef CAS.
- T. P. Knowles, C. A. Waudby, G. L. Devlin, S. I. Cohen, A. Aguzzi, M. Vendruscolo, E. M. Terentjev, M. E. Welland and C. M. Dobson, Science, 2009, 326, 1533–1537 CrossRef CAS PubMed.
- S. I. Cohen, S. Linse, L. M. Luheshi, E. Hellstrand, D. A. White, L. Rajah, D. E. Otzen, M. Vendruscolo, C. M. Dobson and T. P. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9758–9763 CrossRef CAS PubMed.
- A. Levin, T. O. Mason, L. Adler-Abramovich, A. K. Buell, G. Meisl, C. Galvagnion, Y. Bram, S. A. Stratford, C. M. Dobson and T. P. Knowles, Nat. Commun., 2014, 5, 5219 CrossRef CAS PubMed.
- C. Chen, J. Tan, M.-C. Hsieh, T. Pan, J. T. Goodwin, A. K. Mehta, M. A. Grover and D. G. Lynn, Nat. Chem., 2017, 9, 799–804 CrossRef CAS PubMed.
- M.-C. Hsieh, D. G. Lynn and M. A. Grover, J. Phys. Chem. B, 2017, 121, 7401–7411 CrossRef CAS PubMed.
- A. Pal, M. Malakoutikhah, G. Leonetti, M. Tezcan, M. Colomb-Delsuc, V. D. Nguyen, J. van der Gucht and S. Otto, Angew. Chem., Int. Ed., 2015, 54, 7852–7856 CrossRef CAS PubMed.
- M. Colomb-Delsuc, E. Mattia, J. W. Sadownik and S. Otto, Nat. Commun., 2015, 6, 7427 CrossRef CAS.
- S. Onogi, H. Shigemitsu, T. Yoshii, T. Tanida, M. Ikeda, R. Kubota and I. Hamachi, Nat. Chem., 2016, 8, 743 CrossRef CAS PubMed.
- P. A. Korevaar, C. J. Newcomb, E. Meijer and S. I. Stupp, J. Am. Chem. Soc., 2014, 136, 8540–8543 CrossRef CAS PubMed.
- F. Tantakitti, J. Boekhoven, X. Wang, R. V. Kazantsev, T. Yu, J. Li, E. Zhuang, R. Zandi, J. H. Ortony, C. J. Newcomb, L. C. Plamer, G. S. Shekhawat, M. O. de la Cruz, G. C. Schatz and S. I. Stupp, Nat. Mater., 2016, 15, 469–476 CrossRef CAS PubMed.
- C. Yuan, A. Levin, W. Chen, R. Xing, Q. Zou, T. W. Herling, P. K. Challa, T. P. Knowles and X. Yan, Angew. Chem., 2019, 131, 18284–18291 CrossRef.
- S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu and M. Takeuchi, Nat. Chem., 2014, 6, 188–195 CrossRef CAS PubMed.
- W. Wagner, M. Wehner, V. Stepanenko, S. Ogi and F. Würthner, Angew. Chem., Int. Ed., 2017, 56, 16008–16012 CrossRef CAS PubMed.
- S. Ogi, K. Matsumoto and S. Yamaguchi, Angew. Chem., Int. Ed., 2018, 57, 2339–2343 CrossRef CAS PubMed.
- M. Wehner, M. I. S. Röhr, M. Bühler, V. Stepanenko, W. Wagner and F. Würthner, J. Am. Chem. Soc., 2019, 141, 6092–6107 CrossRef CAS PubMed.
- C. Jarrett-Wilkins, X. He, H. E. Symons, R. L. Harniman, C. F. Faul and I. Manners, Chem. – Eur. J., 2018, 24, 15556–15565 CrossRef CAS.
- J. van Herrikhuyzen, A. Syamakumari, A. P. Schenning and E. Meijer, J. Am. Chem. Soc., 2004, 126, 10021–10027 CrossRef CAS PubMed.
- M. M. Smulders, M. M. Nieuwenhuizen, T. F. de Greef, P. van der Schoot, A. P. Schenning and E. A. W. Meijer, Chem. – Eur. J., 2010, 16, 362–367 CrossRef CAS PubMed.
- X.-Q. Li, V. Stepanenko, Z. Chen, P. Prins, L. D. Siebbeles and F. Würthner, Chem. Commun., 2006, 3871–3873 RSC.
- D. Zhao and J. S. Moore, Org. Biomol. Chem., 2003, 1, 3471–3491 RSC.
- S. Ogi, T. Fukui, M. L. Jue, M. Takeuchi and K. Sugiyasu, Angew. Chem., Int. Ed., 2014, 53, 14363–14367 CrossRef CAS PubMed.
- M. M. Bouman and E. Meijer, Adv. Mater., 1995, 7, 385–387 CrossRef CAS.
- W. DeGrado and J. Lear, J. Am. Chem. Soc., 1985, 107, 7684–7689 CrossRef CAS.
- S. Kamtekar, J. M. Schiffer, H. Xiong, J. M. Babik and M. H. Hecht, Science, 1993, 262, 1680–1685 CrossRef CAS.
- E. Gazit, FASEB J., 2002, 16, 77–83 CrossRef CAS PubMed.
- M. Reches, Y. Porat and E. Gazit, J. Biol. Chem., 2002, 277, 35475–35480 CrossRef CAS.
- R. V. Ulijn and D. N. Woolfson, Chem. Soc. Rev., 2010, 39, 3349–3350 RSC.
- L. E. O’leary, J. A. Fallas, E. L. Bakota, M. K. Kang and J. D. Hartgerink, Nat. Chem., 2011, 3, 821–828 CrossRef PubMed.
- A. Saiani, A. Mohammed, H. Frielinghaus, R. Collins, N. Hodson, C. Kielty, M. Sherratt and A. Miller, Soft Matter, 2009, 5, 193–202 RSC.
- L. Adler-Abramovich, L. Vaks, O. Carny, D. Trudler, A. Magno, A. Caflisch, D. Frenkel and E. Gazit, Nat. Chem. Biol., 2012, 8, 701–706 CrossRef CAS PubMed.
- P. C. Ke, M.-A. Sani, F. Ding, A. Kakinen, I. Javed, F. Separovic, T. P. Davis and R. Mezzenga, Chem. Soc. Rev., 2017, 46, 6492–6531 RSC.
- N. L. Truex, Y. Wang and J. S. Nowick, J. Am. Chem. Soc., 2016, 138, 13882–13890 CrossRef CAS PubMed.
- S. Zhang, T. Holmes, C. Lockshin and A. Rich, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 3334–3338 CrossRef CAS.
- J. P. Schneider, D. J. Pochan, B. Ozbas, K. Rajagopal, L. Pakstis and J. Kretsinger, J. Am. Chem. Soc., 2002, 124, 15030–15037 CrossRef CAS PubMed.
- J. Gao, C. Tang, M. A. Elsawy, A. M. Smith, A. F. Miller and A. Saiani, Biomacromolecules, 2017, 18, 826–834 CrossRef CAS PubMed.
- S. Li, A. K. Mehta, A. N. Sidorov, T. M. Orlando, Z. Jiang, N. R. Anthony and D. G. Lynn, J. Am. Chem. Soc., 2016, 138, 3579–3586 CrossRef CAS.
- Y. Hu, R. Lin, P. Zhang, J. Fern, A. G. Cheetham, K. Patel, R. Schulman, C. Kan and H. Cui, ACS Nano, 2016, 10, 880–888 CrossRef CAS PubMed.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- A. Lampel, R. Ulijn and T. Tuttle, Chem. Soc. Rev., 2018, 47, 3737–3758 RSC.
- R. V. Ulijn and A. Lampel, Isr. J. Chem., 2020, 60, 1129–1140 CrossRef CAS.
- M. Reches and E. Gazit, Nano Lett., 2004, 4, 581–585 CrossRef CAS.
- M. Reches and E. Gazit, Isr. J. Chem., 2005, 45, 363–371 CrossRef CAS.
- L. Adler-Abramovich, M. Reches, V. L. Sedman, S. Allen, S. J. Tendler and E. Gazit, Langmuir, 2006, 22, 1313–1320 CrossRef CAS.
- M. Reches and E. Gazit, Phys. Biol., 2006, 3, S10 CrossRef CAS PubMed.
- E. Gazit, Prion, 2007, 1, 32–35 CrossRef.
- P. Tamamis, L. Adler-Abramovich, M. Reches, K. Marshall, P. Sikorski, L. Serpell, E. Gazit and G. Archontis, Biophys. J., 2009, 96, 5020–5029 CrossRef CAS.
- C. H. Görbitz, Chem. Commun., 2006, 2332–2334 RSC.
- N. S. De Groot, T. Parella, F. X. Aviles, J. Vendrell and S. Ventura, Biophys. J., 2007, 92, 1732–1741 CrossRef PubMed.
- M. J. Krysmann, V. Castelletto, A. Kelarakis, I. W. Hamley, R. A. Hule and D. J. Pochan, Biochemistry, 2008, 47, 4597–4605 CrossRef CAS PubMed.
- M. Krysmann, V. Castelletto, J. McKendrick, L. Clifton, P. Harris and S. King, Langmuir, 2008, 24, 8158–8162 CrossRef CAS PubMed.
- S. Marchesan, C. D. Easton, F. Kushkaki, L. Waddington and P. G. Hartley, Chem. Commun., 2012, 48, 2195–2197 RSC.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS.
- C. Tang, A. M. Smith, R. F. Collins, R. V. Ulijn and A. Saiani, Langmuir, 2009, 25, 9447–9453 CrossRef CAS.
- P. W. Frederix, R. V. Ulijn, N. T. Hunt and T. Tuttle, J. Phys. Chem. Lett., 2011, 2, 2380–2384 CrossRef CAS PubMed.
- I. Sasselli, C. Pappas, E. Matthews, T. Wang, N. Hunt, R. Ulijn and T. Tuttle, Soft Matter, 2016, 12, 8307–8315 RSC.
- P. W. Frederix, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2015, 7, 30–37 CrossRef CAS PubMed.
- G. G. Scott, P. J. McKnight, T. Tuttle and R. V. Ulijn, Adv. Mater., 2016, 28, 1381–1386 CrossRef CAS PubMed.
- Y. Fu, B. Li, Z. Huang, Y. Li and Y. Yang, Langmuir, 2013, 29, 6013–6017 CrossRef CAS PubMed.
- M. Wang, P. Zhou, J. Wang, Y. Zhao, H. Ma, J. R. Lu and H. Xu, J. Am. Chem. Soc., 2017, 139, 4185–4194 CrossRef CAS PubMed.
- Q. Xing, J. Zhang, Y. Xie, Y. Wang, W. Qi, H. Rao, R. Su and Z. He, ACS Nano, 2018, 12, 12305–12314 CrossRef CAS PubMed.
- M. L. Mason, R. F. Lalisse, T. J. Finnegan, C. M. Hadad, D. A. Modarelli and J. R. Parquette, Langmuir, 2019, 35, 12460–12468 CrossRef CAS PubMed.
- G. D. Rose, A. R. Geselowitz, G. J. Lesser, R. H. Lee and M. H. Zehfus, Science, 1985, 229, 834–838 CrossRef CAS PubMed.
- M. Avinash and T. Govindaraju, Nanoscale, 2014, 6, 13348–13369 RSC.
- H. Moorthy, L. P. Datta and T. Govindaraju, Chem. – Asian J., 2021, 16, 423–442 CrossRef CAS PubMed.
- M. Pandeeswar, M. Avinash and T. Govindaraju, Chem. – Eur. J., 2012, 18, 4818–4822 CrossRef CAS.
- M. Pandeeswar, H. Khare, S. Ramakumar and T. Govindaraju, RSC Adv., 2014, 4, 20154–20163 RSC.
- M. Avinash, K. Swathi, K. Narayan and T. Govindaraju, ACS Appl. Mater. Interfaces, 2016, 8, 8678–8685 CrossRef CAS PubMed.
- M. Avinash and T. Govindaraju, Acc. Chem. Res., 2018, 51, 414–426 CrossRef CAS PubMed.
- B. Roy and T. Govindaraju, Bull. Chem. Soc. Jpn., 2019, 92, 1883–1901 CrossRef CAS.
- M. Avinash and T. Govindaraju, Adv. Mater., 2012, 24, 3905–3922 CrossRef CAS PubMed.
- B. J. Schwartz, Annu. Rev. Phys. Chem., 2003, 54, 141–172 CrossRef CAS.
- S. R. Diegelmann, J. M. Gorham and J. D. Tovar, J. Am. Chem. Soc., 2008, 130, 13840–13841 CrossRef CAS PubMed.
- H. Shao, T. Nguyen, N. C. Romano, D. A. Modarelli and J. R. Parquette, J. Am. Chem. Soc., 2009, 131, 16374–16376 CrossRef CAS PubMed.
- H. Shao, J. Seifert, N. C. Romano, M. Gao, J. J. Helmus, C. P. Jaroniec, D. A. Modarelli and J. R. Parquette, Angew. Chem., Int. Ed., 2010, 49, 7688–7691 CrossRef CAS PubMed.
- H. Shao and J. R. Parquette, Chem. Commun., 2010, 46, 4285–4287 RSC.
- H. Shao, M. Gao, S. H. Kim, C. P. Jaroniec and J. R. Parquette, Chem. – Eur. J., 2011, 17, 12882–12885 CrossRef CAS PubMed.
- M. Ramanathan, L. K. Shrestha, T. Mori, Q. Ji, J. P. Hill and K. Ariga, Phys. Chem. Chem. Phys., 2013, 15, 10580–10611 RSC.
- K. Ariga, M. Nishikawa, T. Mori, J. Takeya, L. K. Shrestha and J. P. Hill, Sci. Technol. Adv. Mater., 2019, 20, 51–95 CrossRef CAS PubMed.
- K. L. Morris, L. Chen, J. Raeburn, O. R. Sellick, P. Cotanda, A. Paul, P. C. Griffiths, S. M. King, R. K. O’Reilly and L. C. Serpell, Nat. Commun., 2013, 4, 1480 CrossRef PubMed.
- A. Sarkar, T. Behera, R. Sasmal, R. Capelli, C. Empereur-Mot, J. Mahato, S. S. Agasti, G. M. Pavan, A. Chowdhury and S. J. George, J. Am. Chem. Soc., 2020, 142, 11528–11539 CrossRef CAS.
- T. Fukui, S. Kawai, S. Fujinuma, Y. Matsushita, T. Yasuda, T. Sakurai, S. Seki, M. Takeuchi and K. Sugiyasu, Nat. Chem., 2017, 9, 493–499 CrossRef CAS.
- W. Wagner, M. Wehner, V. Stepanenko and F. Würthner, J. Am. Chem. Soc., 2019, 141, 12044–12054 CrossRef CAS PubMed.
- C. H. Chen, L. C. Palmer and S. I. Stupp, Nano Lett., 2018, 18, 6832–6841 CrossRef CAS PubMed.
- J. R. Parquette and M. Ji, Chem. – Eur. J., 2020, 26, 8572–8578 CrossRef.
- Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef CAS PubMed.
- J. Ryu, P. Messersmith and H. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 7523–7540 CrossRef CAS PubMed.
- A. J. Clancy, M. K. Bayazit, S. A. Hodge, N. T. Skipper, C. A. Howard and M. S. Shaffer, Chem. Rev., 2018, 118, 7363–7408 CrossRef CAS PubMed.
- C. Richard, F. Balavoine, P. Schultz, T. W. Ebbesen and C. Mioskowski, Science, 2003, 300, 775–778 CrossRef CAS PubMed.
- B. V. Derjaguin, N. V. Churaev, V. M. Muller and V. Kisin, The Derjaguin-Landau-Verwey-Overbeek (DLVO) Theory of Stability of Lyophobic Colloids, Surface Forces, Springer, Boston, MA, 1987, pp. 293–310 Search PubMed.
- V. Nicolosi, H. Cathcart, A. R. Dalton, D. Aherne, G. R. Dieckmann and J. N. Coleman, Biomacromolecules, 2008, 9, 598–602 CrossRef CAS.
- D. R. Samarajeewa, G. R. Dieckmann, S. O. Nielsen and I. H. Musselman, Carbon, 2013, 57, 88–98 CrossRef CAS.
- A. Ortiz-Acevedo, H. Xie, V. Zorbas, W. M. Sampson, A. B. Dalton, R. H. Baughman, R. K. Draper, I. H. Musselman and G. R. Dieckmann, J. Am. Chem. Soc., 2005, 127, 9512–9517 CrossRef CAS.
- C.-c. Chiu, M. C. Maher, G. R. Dieckmann and S. O. Nielsen, ACS Nano, 2010, 4, 2539–2546 CrossRef CAS PubMed.
- J. Montenegro, C. Vazquez-Vazquez, A. Kalinin, K. E. Geckeler and J. R. Granja, J. Am. Chem. Soc., 2014, 136, 2484–2491 CrossRef CAS.
- F. A. Mann, J. Horlebein, N. F. Meyer, D. Meyer, F. Thomas and S. Kruss, Chem. – Eur. J., 2018, 24, 12241–12245 CrossRef CAS.
- A. R. Thomson, C. W. Wood, A. J. Burton, G. J. Bartlett, R. B. Sessions, R. L. Brady and D. N. Woolfson, Science, 2014, 346, 485–488 CrossRef CAS PubMed.
- M. Ji, M. L. Mason, D. A. Modarelli and J. R. Parquette, Chem. Sci., 2019, 10, 7868–7877 RSC.
- S. Manchineella and T. Govindaraju, RSC Adv., 2012, 2, 5539–5542 RSC.
- S. Manchineella, V. Prathyusha, U. D. Priyakumar and T. Govindaraju, Chem. – Eur. J., 2013, 19, 16615–16624 CrossRef CAS PubMed.
- A. K. Dwivedi, M. Pandeeswar and T. Govindaraju, ACS Appl. Mater. Interfaces, 2014, 6, 21369–21379 CrossRef CAS.
- M. Ji, B. Daniels, A. Shieh, D. A. Modarelli and J. R. Parquette, Chem. Commun., 2017, 53, 12806–12809 RSC.
- J. N. Israelachvili, Intermolecular and Surface Forces, Elsevier, USA, 3rd edn, 2011, pp. 528–530 Search PubMed.
- C. C. Cenker, S. Bucak and U. Olsson, Soft Matter, 2011, 7, 11548 RSC.
- M. Ji, M. B. Dawadi, A. R. LaSalla, Y. Sun, D. A. Modarelli and J. R. Parquette, Langmuir, 2017, 33, 9129–9136 CrossRef CAS.
- P. G. Janssen, J. Vandenbergh, J. L. van Dongen, E. Meijer and A. P. Schenning, J. Am. Chem. Soc., 2007, 129, 6078–6079 CrossRef CAS PubMed.
- P. G. Janssen, S. Jabbari-Farouji, M. Surin, X. Vila, J. C. Gielen, T. F. de Greef, M. R. Vos, P. H. Bomans, N. A. Sommerdijk and P. C. Christianen, J. Am. Chem. Soc., 2009, 131, 1222–1231 CrossRef CAS PubMed.
- R. Ni and Y. Chau, J. Am. Chem. Soc., 2014, 136, 17902–17905 CrossRef CAS.
- R. Ni and Y. Chau, Angew. Chem., Int. Ed., 2017, 56, 9356–9360 CrossRef CAS.
- R. Ni and Y. Chau, Angew. Chem., Int. Ed., 2020, 59, 3578–3584 CrossRef CAS PubMed.
- R. Freeman, M. Han, Z. Álvarez, J. A. Lewis, J. R. Wester, N. Stephanopoulos, M. T. McClendon, C. Lynsky, J. M. Godbe, H. Sangji, E. Luijten and S. I. Stupp, Science, 2018, 362, 808–813 CrossRef CAS.
- M. R. Ghadiri, J. R. Granja, R. A. Milligan, D. E. McRee and N. Khazanovich, Nature, 1993, 366, 324–327 CrossRef CAS PubMed.
- M. R. Ghadiri, J. R. Granja and L. K. Buehler, Nature, 1994, 369, 301–304 CrossRef CAS PubMed.
- M. R. Ghadiri, Adv. Mater., 1995, 7, 675–677 CrossRef.
- J. D. Hartgerink, J. R. Granja, R. A. Milligan and M. R. Ghadiri, J. Am. Chem. Soc., 1996, 118, 43–50 CrossRef CAS.
- K. Pandurangan, B. Roy, K. Rajasekhar, Y. V. Suseela, P. Nagendra, A. Chaturvedi, U. R. Satwik, N. A. Murugan, U. Ramamurty and T. Govindaraju, ACS Appl. Bio Mater., 2020, 3, 3413–3422 CrossRef CAS.
- S. Manchineella and T. Govindaraju, ChemPlusChem, 2017, 82, 88–106 CrossRef CAS PubMed.
- C. Balachandra and T. Govindaraju, J. Org. Chem., 2019, 85, 1525–1536 CrossRef PubMed.
- S. Manchineella, N. A. Murugan and T. Govindaraju, Biomacromolecules, 2017, 18, 3581–3590 CrossRef CAS.
- R. Chapman, M. Danial, M. L. Koh, K. A. Jolliffe and S. Perrier, Chem. Soc. Rev., 2012, 41, 6023–6041 RSC.
- M. Danial, C. M.-N. Tran, P. G. Young, S. Perrier and K. A. Jolliffe, Nat. Commun., 2013, 4, 1–13 Search PubMed.
- S. C. Larnaudie, J. C. Brendel, I. Romero-Canelón, C. Sanchez-Cano, S. Catrouillet, J. Sanchis, J. P. Coverdale, J.-I. Song, A. Habtemariam and P. J. Sadler, Biomacromolecules, 2018, 19, 239–247 CrossRef CAS.
- R. Chapman, K. A. Jolliffe and S. Perrier, Adv. Mater., 2013, 25, 1170–1172 CrossRef CAS PubMed.
- S. Catrouillet, J. C. Brendel, S. Larnaudie, T. Barlow, K. A. Jolliffe and S. Perrier, ACS Macro Lett., 2016, 5, 1119–1123 CrossRef CAS.
- J. Y. Rho, H. Cox, E. D. Mansfield, S. H. Ellacott, R. Peltier, J. C. Brendel, M. Hartlieb, T. A. Waigh and S. Perrier, Nat. Commun., 2019, 10, 1–9 CrossRef PubMed.
- Q. Song, J. Yang, J. Y. Rho and S. Perrier, Chem. Commun., 2019, 55, 5291–5294 RSC.
- N. T. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS PubMed.
- E. Pazos, E. Sleep, C. M. Rubert Perez, S. S. Lee, F. Tantakitti and S. I. Stupp, J. Am. Chem. Soc., 2016, 138, 5507–5510 CrossRef CAS PubMed.
- Y. Yin, Z.-Y. Li, Z. Zhong, B. Gates, Y. Xia and S. Venkateswaran, J. Mater. Chem., 2002, 12, 522–527 RSC.
- A. K. Awasthi, S. D. Bhagat, R. Ramakrishnan and A. Srivastava, Chem. – Eur. J., 2019, 25, 12905–12910 CrossRef CAS.
- J. R. Liebscher, R. Mrówczyński, H. A. Scheidt, C. Filip, N. D. Hădade, R. Turcu, A. Bende and S. Beck, Langmuir, 2013, 29, 10539–10548 CrossRef CAS PubMed.
- H. Xu, X. Liu, G. Su, B. Zhang and D. Wang, Langmuir, 2012, 28, 13060–13065 CrossRef CAS PubMed.
- G. A. Hudalla, T. Sun, J. Z. Gasiorowski, H. Han, Y. F. Tian, A. S. Chong and J. H. Collier, Nat. Mater., 2014, 13, 829–836 CrossRef CAS.
- C. Mahato, A. Chatterjee and D. Das, Angew. Chem., Int. Ed., 2021, 60, 202–207 CrossRef PubMed.
- S. Satagopan, Y. Sun, J. R. Parquette and F. R. Tabita, Biotechnol. Biofuels, 2017, 10, 175 CrossRef.
- N. Wiradharma, Y. W. Tong and Y.-Y. Yang, Biomaterials, 2009, 30, 3100–3109 CrossRef CAS PubMed.
- P. Zhang, A. G. Cheetham, Y.-A. Lin and H. Cui, ACS Nano, 2013, 7, 5965–5977 CrossRef CAS PubMed.
- Y.-A. Lin, A. G. Cheetham, P. Zhang, Y.-C. Ou, Y. Li, G. Liu, D. Hermida-Merino, I. W. Hamley and H. Cui, ACS Nano, 2014, 8, 12690–12700 CrossRef CAS PubMed.
- H. Su, W. Zhang, H. Wang, F. Wang and H. Cui, J. Am. Chem. Soc., 2019, 141, 11997–12004 CrossRef CAS PubMed.
- H. Su, F. Wang, Y. Wang, A. G. Cheetham and H. Cui, J. Am. Chem. Soc., 2019, 141, 17107–17111 CrossRef CAS PubMed.
- Y. Sun, J. A. Kaplan, A. Shieh, H.-L. Sun, C. M. Croce, M. W. Grinstaff and J. R. Parquette, Chem. Commun., 2016, 52, 5254–5257 RSC.
- H. A. M. Ardoña and J. D. Tovar, Chem. Sci., 2015, 6, 1474–1484 RSC.
- J. López-Andarias, M. J. Rodriguez, C. Atienza, J. L. López, T. Mikie, S. Casado, S. Seki, J. L. Carrascosa and N. Martín, J. Am. Chem. Soc., 2015, 137, 893–897 CrossRef PubMed.
- S. K. M. Nalluri, C. Berdugo, N. Javid, P. W. Frederix and R. V. Ulijn, Angew. Chem., 2014, 126, 5992–5997 CrossRef.
- A. R. Hirst, S. Roy, M. Arora, A. K. Das, N. Hodson, P. Murray, S. Marshall, N. Javid, J. Sefcik and J. Boekhoven, Nat. Chem., 2010, 2, 1089–1094 CrossRef CAS PubMed.
- S. Fleming, S. Debnath, P. W. Frederix, N. T. Hunt and R. V. Ulijn, Biomacromolecules, 2014, 15, 1171–1184 CrossRef CAS PubMed.
- C. Berdugo, S. K. M. Nalluri, N. Javid, B. Escuder, J. F. Miravet and R. V. Ulijn, ACS Appl. Mater. Interfaces, 2015, 7, 25946–25954 CrossRef CAS.
- Q. Zou, K. Liu, M. Abbas and X. Yan, Adv. Mater., 2016, 28, 1031–1043 CrossRef CAS PubMed.
- J. H. Kim, M. Lee, J. S. Lee and C. B. Park, Angew. Chem., 2012, 124, 532–535 CrossRef.
- T. H. Han, W. J. Lee, D. H. Lee, J. E. Kim, E. Y. Choi and S. O. Kim, Adv. Mater., 2010, 22, 2060–2064 CrossRef CAS.
- I. Azuri, L. Adler-Abramovich, E. Gazit, O. Hod and L. Kronik, J. Am. Chem. Soc., 2014, 136, 963–969 CrossRef CAS PubMed.
- K. Tao, P. Makam, R. Aizen and E. Gazit, Science, 2017, 358, eaam9756 CrossRef PubMed.
- M. Yemini, M. Reches, J. Rishpon and E. Gazit, Nano Lett., 2005, 5, 183–186 CrossRef CAS PubMed.
- M. Yemini, M. Reches, E. Gazit and J. Rishpon, Anal. Chem., 2005, 77, 5155–5159 CrossRef CAS PubMed.
- L. Adler-Abramovich, M. Badihi-Mossberg, E. Gazit and J. Rishpon, Small, 2010, 6, 825–831. CrossRef CAS PubMed.
- L. Adler-Abramovich, D. Aronov, P. Beker, M. Yevnin, S. Stempler, L. Buzhansky, G. Rosenman and E. Gazit, Nat. Nanotechnol., 2009, 4, 849–854 CrossRef CAS PubMed.
- C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310–5324 CrossRef CAS PubMed.
- F. Biedermann and H.-J. R. Schneider, Chem. Rev., 2016, 116, 5216–5300 CrossRef CAS PubMed.
- M. D. Driver, M. J. Williamson, J. L. Cook and C. A. Hunter, Chem. Sci., 2020, 11, 4456–4466 RSC.
| This journal is © The Royal Society of Chemistry 2021 |