
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInjectable hydrogels self-assembled from oligopeptide-poly(2-methacryloyloxyethyl phosphorylcholine) hybrid graft copolymers for cell scaffolds and controlled release applications†
Tomoyuki
Koga
 *a,
Tomoo
Matsuoka
a,
Yusuke
Morita
b and
Nobuyuki
Higashi
*a
*a,
Tomoo
Matsuoka
a,
Yusuke
Morita
b and
Nobuyuki
Higashi
*a
aDepartment of Molecular Chemistry and Biochemistry, Faculty of Science and Engineering, Doshisha University, Kyotanabe, Kyoto 610-0321, Japan. E-mail: tkoga@mail.doshisha.ac.jp; nhigashi@mail.doshisha.ac.jp
bDepartment of Biomedical Engineering, Faculty of Life and Medical Sciences, Doshisha University, Kyotanabe, Kyoto 610-0321, Japan
First published on 25th May 2021
Abstract
In this paper, we report the synthesis and characterization of novel peptide based hybrid graft copolymers that can form injectable hydrogels. These hybrid graft copolymers are composed of a biocompatible poly(2-methacryloyloxyethyl phosphorylcholine) main chain and self-assembling oligoalanine-block-poly(ethylene glycol) grafts (grafting ratio (m) = 6, 10, 20, 28, 32, and 40%), which are readily synthesized using a macromonomer by combining the solid phase peptide synthesis and conventional radical polymerization methods. Conformational analyses under a wide range of dilute aqueous solutions (pH 3–10 and 4–70 °C) revealed stable β-sheet formation. With an increase in solution concentration, the graft copolymers, except those with a low grafting ratio of m = 6, formed hydrogels displaying shear-thinning and self-healing behaviors because of the reversible self-assembly property of peptide grafts. Interestingly, the mechanical properties of hybrid graft copolymers were strongly dependent on the grafting density of the hybrid copolymers. Thus, it was observed that an increase in grafting ratio strengthened the inter-polymer network. These features enabled the graft-type hybrids to be used as injectable hydrogels for 3D cell scaffolds and controlled-release applications.
Introduction
Extensive effort has gone into designing functional hydrogels with potential utility in the biomedical, cosmetic, and industrial fields. The unique structure of hydrogels traps a large amount of water inside the polymer network, offering biocompatibility, high porosity, and flexibility. Thus, hydrogels resemble the natural extracellular matrix (ECM), making hydrogels a widely accepted biomaterial.1,2 Over the past few decades, a wide variety of hydrogels with unique properties such as stimuli responsiveness,3,4 self-healing ability,5–8 shape memory,9–12 and mechanical toughness13–15 have been developed. Injectable hydrogels,16,17 which flow like a fluid when being injected from a syringe and set rapidly at the target site, are one of the most fascinating variety of hydrogels and are used as a drug and cell carrier. Several injectable hydrogels have been developed using in situ chemical crosslinking,18,19 physical crosslinking in response to external cues including temperature and pH,20–22 and shear-thinning mechanism via self-assembly.23 The constituent polymers of injectable hydrogels are required to be biocompatible and nontoxic as well as have easy tunability of gel mechanics to be used as 3D ECM-mimics and for controlled drug release. Therefore, de novo designed self-assembling proteins and peptides have been employed for synthesizing shear-thinning gels24–30 because of their excellent biocompatibility, easy synthesis, and desirable high-order structure and function based on amino acid sequences.31–34 However, such peptide-based supramolecular hydrogels are often mechanically weak, making them inconvenient to handle. Although solid phase peptide synthesis (SPPS) facilitates the synthesis of monodisperse sequence-controlled oligopeptides, SPPS is difficult to scale up and is unsuitable for the synthesis of high-molecular-weight samples.Our strategy was to hybridize self-assembling peptides and synthetic polymers with appropriate biocompatibility and mechanical strength. The hybridization strategy provided more flexibility to complex material designs by overcoming issues like scaling up and synthesis of high-molecular-weight samples. Many diblock, triblock, multiblock, and graft-type hybrids with α-helix, β-sheet, and coiled-coil motifs have been synthesized using a combination of the SPPS and radical polymerization techniques.35–42 The β-sheet motif is particularly useful for designing robust and shear-thinning peptide-polymer hybrid gels because of the multiple reversible hydrogen bonds, which act as supramolecular crosslinks occurring intermolecularly between strands. In fact, self-assembled hybrid hydrogels have been prepared from β-sheet peptide-grafted poly(N-(2-hydroxy propyl)methacrylamide) and used as an effective bone scaffold.43,44 However, few studies exist on hybrid hydrogel synthesis from β-sheet peptide-grafted vinyl polymers. Therefore, exploring a new β-sheet peptide-biocompatible synthetic polymer system that could function as an injectable hydrogel with controllable physical and mechanical properties in a broad range of biomedical applications has remained a significant challenge.
In this study, we report the synthesis of a novel β-sheet peptide-based graft copolymer. Biocompatible poly(2-methacryloyloxyethyl phosphorylcholine) (PMPC) was grafted with β-sheet-forming oligoalanine (A8)-block-poly(ethylene glycol) (PEG) using a macromonomer. Hybrid copolymers can self-assemble into injectable hydrogels. Detailed analyses of the conformation of the peptide grafts and the mechanical and shear-thinning properties of the resulting hydrogels were conducted, especially with respect to the impact of grafting density. To demonstrate the versatility of this hybrid gel as an injectable biomaterial, potential applications such as 3D scaffolds for chondrocytes and media for controlled release of proteins have been investigated using the hybrid gel.
Experimental
Materials
N,N-Dimethlylformamide (DMF), dichloromethane (DCM), diethylether, N,N′-diisopropylcarbodiimide (DIPC), hydrochloric acid (12 M-HCl), piperidine, 1,1,1,3,3,3-hexafluoroisopropanol (HFIP), dimethyl sulfoxide (DMSO), 2,2′-azobis(2-methylpropionamidine) dihydrochloride (AIBA), Fmoc-βAla-OH, bovine serum albumin (BSA) and D2O were purchased from Wako Pure Chemical. 1-Hydroxybenzotriazole anhydrous (HOBt) and trifluoroacetic acid (TFA) were purchased from Watanabe Chemical Industries. Fmoc-L-Ala-OH was purchased from Peptide Institute. 2-Methacryloyloxyethyl phosphorylcholine (MPC), methacrylic anhydride, 2,5-dihydroxybenzoic acid (DHBA) were purchased from Sigma Aldrich. Fluorescein isothiocyanate (isomer I) (FITC) was purchased from Tokyo Chemical Industries. TentaGel PAP resin was purchased from Rapp Polymere GmbH. All reagents were used as received.1H-NMR and MALDI-TOF MS measurements
1H-NMR spectra were acquired using a JEOL FT-NMR AL400 (JEOL Resonance) spectrometer (400 MHz). Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) analyses were carried out on an Autoflex Speed (Bluker Daltonics) using DHBA as a matrix.Conformational analyses
Circular dichroism (CD) spectra were recorded on a J-820 spectropolarimeter (JASCO Ltd) equipped with a Peltier-type thermostatic cell holder coupled with a PTC-423L controller under an inert atmosphere using nitrogen gas. Experiments were performed in a quartz cell with a path length of 1 mm. The temperature dependency of the CD spectra was observed over the range of 190–400 nm at 4 °C to 70 °C. Transmission-Fourier-transform infrared (FTIR) spectra were measured by the KBr method using a Nexus 470 (Thermo Nicolet Co.) with a Mercury–Cadmium–Tellurium (MCT) detector (resolution: 4 cm−1, number of scans: 512). The samples were prepared by lyophilization of aqueous solutions using liquid N2. In the deconvolution process, the obtained amide I peak was curve-fitted into three Gaussian peaks centered at around 1695 cm−1 (anti-parallel β-sheet), 1660 cm−1 (random coil and/or α-helix) and 1625 cm−1 (β-sheet) using OMNIC software (Thermo Nicolet Co.). The ratio of integrated intensity of each peak was used to assess the β-sheet contents. The pH of the sample solutions for CD and FTIR analyses was adjusted with 0.01 M HCl or 0.01 M NaOH.Rheological study
Rheological characterization was performed using a Discover HR-1 (TA Instruments, New Castle, DE, USA) equipped with a Peltier device for temperature control. During all rheological measurements, a solvent trap was used to minimize evaporation. The measurements were acquired using parallel plates (diameter: 40 mm) at 25 °C. The gap between the plates was adjusted to 300 μm to ensure that the geometry was completely filled.Synthesis of poly(ethylene glycol)-attached self-assembling peptide macromonomer
The poly(ethylene glycol)-attached peptide macromonomer (MA-βA-A8-PEG) used as a graft chain was synthesized via the SPPS method using 9-fluorenylmethoxycarbonyl (Fmoc) chemistry. The oligoalanine block was first synthesized on a TentaGel PAP (0.21 mmol g−1) using Fmoc-L-Ala-OH (3 equiv.), HOBt (3 equiv.), and DIPC (3 equiv.) in DMF for coupling and piperidine (25%)/DMF for Fmoc removal. Fmoc-β-Ala-OH (3 equiv.) was introduced at the N-terminus of the peptide as a spacer. Further, methacrylic anhydride (3 equiv.) was combined with the amino group on the resin in DMF for 12 h. To cleave the peptide macromonomer from the resin, the resin was treated with TFA/DCM (9/1 in vol.). The resultant peptide macromonomer was identified using MALDI-TOF MS and 1H-NMR spectroscopy (Fig. S1, ESI†).MALDI TOF MS: m/z [M + H]+: 3700 (broad).
1H-NMR (DMSO-d6, TMS); 1.3 ppm (CH3-: side chain of Ala), 1.8–2.0 ppm (CH3-:methyl group of methacrylate), 2.4 ppm (-HN-CH2CH2-CO-: βAla), 3.4–4.0 ppm (-CH2CH2O-: main chain of PEG, -HN-CH2CH2-CO-: βAla), 4.2–4.3 ppm (-CH-: Ala), 5.3–5.4 ppm, 5.6–5.7 ppm (CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(CH3)-: methacryloyl).
C(CH3)-: methacryloyl).
Copolymerization of peptide-macromonomer with MPC
Copolymerization of the peptide-macromonomer with MPC was carried out in water at 60 °C for 48 h under a dry N2 atmosphere using AIBA as an initiator. The grafting ratio (m) was controlled by changing the feed ratio of the monomers. All the graft copolymers were purified by dialysis using a membrane of molecular weight cut-off (MWCO) with 6–8 kDa (Fisher Science) in ultra-pure water while changing the water repeatedly and lyophilizing. The grafting ratio was estimated to be m = 6, 10, 20, 28, 32, and 40 by 1H-NMR analyses based on the area ratio of the signal of CH3 in the Ala side chain (1.4–1.6 ppm) to that of CH3 in the main chain (0.9–1.3 ppm) (Fig. S2, ESI†). The isolated graft copolymers were relatively insoluble due to their strong self-assembling nature and were only soluble in highly polar solvents such as HFIP. Therefore, inherent viscosity analyses of 1m HFIP solutions were employed to evaluate the molecular weights of graft copolymers. As a result, the viscosity-average molecular weights (Mv) of all graft copolymers were in the range of Mv = 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–60000 (g mol−1)–for linear PMPC standards, which were separately synthesized by reversible addition fragmentation chain-transfer (RAFT) polymerization.
000–60000 (g mol−1)–for linear PMPC standards, which were separately synthesized by reversible addition fragmentation chain-transfer (RAFT) polymerization.
1H-NMR (D2O/TFA, DSS): 0.9–1.3 ppm (α-CH3- of main chain), 1.4–1.6 ppm (CH3: side chain of Ala), 1.8–2.4 ppm (-CH2C(CH3)-: PMPC main chain and peptide grafts), 2.7–2.8 ppm (-NH-CH2CH2-CO-: βAla), 3.2–3.4 ppm (-N+(CH3)3: side chain of PMPC), 3.6 ppm (-CH2-N+(CH3)3: side chain of PMPC), 3.7–4.0 ppm (-CH2CH2O-: main chain of PEG, -HN-CH2CH2-CO-: βAla), 4.2–4.8 ppm (-CH-: Ala, -COCH2CH2O-, -O-CH2-CH2-N+: side chain of PMPC).
MTT assay
To assess the in vitro biocompatibility of the graft copolymers, an MTT assay was performed. NIH/3T3 cells were cultured to 80% confluence in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% calf serum and 1% penicillin/streptomycin at 37 °C under 5% CO2. The cells were plated onto a 96-well plate (Iwaki) at a density of 1 × 104 cells (100 μL) per well and incubated at 37 °C for 24 h. After the medium was removed, fresh media containing the graft copolymers with various grafting ratios (10 μg mL−1) were added to the wells and incubated for 24 h. After discarding the old media and washing the wells with phosphate buffered saline (PBS), the MTT-containing medium was added to each well and incubated for 2 h. After incubation, the medium was removed, and DMSO was added. The absorbance measured using a microplate reader (Molecular Device FilterMax F5) was 570 nm. Cell viability (%) was calculated relative to the untreated cells (100% viability). The experiments were performed thrice for each sample.Cell assay in injectable hydrogel
Chondrocytes were used for 3D cell culture experiments. The articular cartilage slices were first harvested from the distal femur of the pig knee (6 months, ca. 100 kg weight) purchased from a butcher. The articular cartilage slices were trypsinized (0.25%) at 37 °C for 30 min. The articular cartilage slices were then treated with a sterilized DMEM containing collagenase (0.1%) for 12 h at 37 °C under 100% humidity and 5% CO2. Chondrocytes were isolated using a cell strainer (40 μm, Nunc) by removing the degraded substrates and re-suspending the chondrocytes at a density of 1.0 × 107 cells mL−1 in sterilized DMEM supplemented with 10% bovin serum and 1% penicillin/streptomycin. This cell suspension was then added to lyophilized 1m (m = 28) in a 24 well-plate and gently stirred with a pipette tip. The resultant chondrocyte-encapsulated hydrogel (15 wt%) was cultured at 37 °C under 5% CO2 for 2 days. The hydrogel was loaded into a syringe (Terumo Co. SS-01T) and injected onto a glass slide. During the process, cell viability was evaluated by a live/dead assay using calcein-AM and ethidium homodimer. The 3D imaging of the cells within the hydrogel was performed using a multiphoton excitation microscope (Leica TCS SP8 MP confocal microscope).Controlled release experiment
BSA was used as the drug model for the controlled release of the hydrogels. Fluorescence-labeling of BSA was performed before using it for the controlled release of hydrogels by combining the amino group of the Lys residue with FITC in a buffer (pH 9) containing a small amount of EtOH, which was purified using dialysis at 4 °C for 3 h (MWCO 14 kDa, EIDIA Co. Ltd, UC36-32-100). The drug-loaded hydrogels were prepared by dissolving the polymers (1m) (15 wt%) in PBS containing BSA-FITC (1 wt%) and incubating the solution at 37 °C for 24 h. The in vitro BSA-FITC release experiments were conducted by immersing the above protein-loaded hydrogels in a vial filled with 5 mL of PBS at 37 °C. At a predetermined time, 1 mL of the buffer solution was removed from the system to analyze the released protein, and 1 mL of fresh buffer was added back to maintain the total solution volume. The concentration of BSA-FITC was evaluated by measuring the absorbance at 499 nm using UV-vis spectroscopy (Shimadzu UV-2450). The cumulative BSA-FITC release was calculated according to the following formula: cumulative release (%) = Mt/M0 × 100, where Mt is the amount of BSA released from the hydrogel at time t, and M0 is the amount of BSA-FITC loaded into the hydrogels. Three independent tests were performed for each sample to confirm the reproducibility.Results and discussion
Design and synthesis of poly(2-methacryloyloxyethyl phosphorylcholine) with PEG-attached self-assembling oligopeptides as graft chains (1m)
The synthesis of the novel graft-type peptide-polymer hybrid copolymers (1m) was carried out by combining the SPPS and conventional radical polymerization methods. This hybrid copolymer contained three distinct segments: a hydrophilic PMPC backbone, hydrophobic oligoalanine (A8) acting as a physical crosslink via self-assembly, and a hydrophilic PEG that improved the water solubility of peptide units and enhanced the total water-swollen structure (Fig. 1). Both PMPC and PEG are well-known water-soluble biocompatible synthetic polymers commonly used in biomaterials because of their ability to resist the nonspecific adsorption of proteins and cells.45–47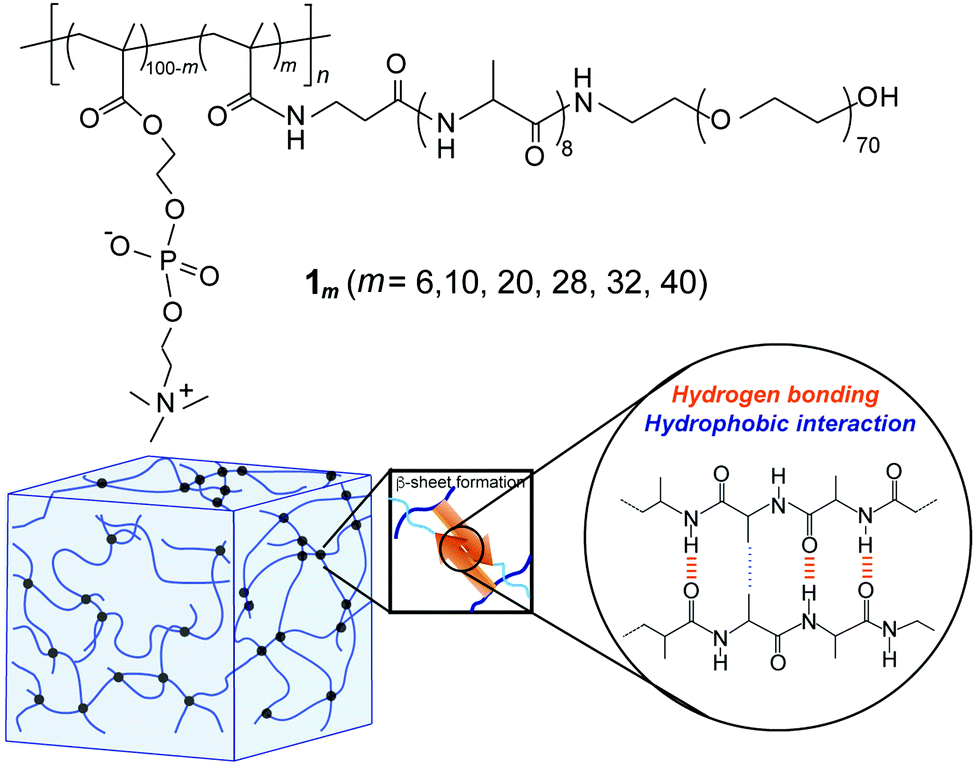 | ||
| Fig. 1 (a) Chemical structure of the PMPC having PEG-attached self-assembling oligopeptide as graft chains (1m). (b) Schematic illustration of the hydrogel formed via self-assembly of 1m. | ||
First, a polymerizable methacryl group (MA)-terminated peptide macromonomer (MA-βA-A8-PEG) was prepared via the SPPS method using Fmoc chemistry followed by a reaction with methacrylic anhydride on TentaGel PAP resin. Cleavage from the resin yielded a linear PEG (average MW of 3000) covalently bound to the C-terminus of the peptide. Radical copolymerization of this peptide macromonomer with MPC was carried out in water at various feed compositions and the obtained copolymers were purified by dialysis against water. The 1H-NMR study revealed that the copolymer composition (i.e., grafting ratio (m)) was readily controlled by changing the feed composition (m = 6, 10, 20, 28, 32, and 40). To assess the biocompatibility of this type of graft copolymer, the cytotoxicity of 1m was evaluated in vitro for NIH/3T3 fibroblast cells using MTT assay (Fig. S3, ESI†). The cell viability of the hybrid copolymers was approximately 90% at a concentration of 10 μg mL−1. This demonstrated that hybrid copolymers were intrinsically nontoxic to NIH/3T3 cells regardless of their grafting ratio.
Conformational property of hybrid graft copolymers (1m) in water
The conformational properties of 1m were studied in dilute aqueous solutions by means of CD and FTIR analyses. Fig. 2a and Fig. S4 (ESI†) showed the CD spectra of 1m at various pH values (3–10) at 25 °C. In a wide pH range, all 1m demonstrated a typical pattern of β-sheet structure with negative and positive maxima at around 217 nm and 196 nm, respectively. In addition, the CD spectra did not exhibit significant variation corresponding to either the increase in temperature from 4 °C to 70 °C or the subsequent decrease in temperature (Fig. 2b and Fig. S5, ESI†). Thus, the hydrophobic A8 grafts, even when conjugated to PMPC and PEG, rapidly self-assembled into a stable β-sheet structure in water. The transmission FTIR spectra of the lyophilized solid state 1m at pH 5.6 agreed with the CD results and suggested that the β-sheets were classified as antiparallel β-strands, as evident from the amide I bands at 1695 and 1625 cm−1 (Fig. 2c).48 An additional CO stretching band arising from the ester groups of the MPC units was also observed at approximately 1720 cm−1. Fig. 2d showed the β-sheet content for 1m (m = 6–40) evaluated from the ratio of integrated peak intensities assigned to the β-sheet conformation to that of the random coil (and/or α-helix) conformation (around 1660 cm−1), which were obtained by peak deconvolution of the amide I band. The data revealed that the β-sheet content varied depending on the grafting density, and low grafting polymers (m = 6 and 10) possessed slightly lower β-sheet values. However, β-sheet value was maintained at over 60% content. The β-sheet-forming ability of the peptide grafts, in addition to good stability over a wide range of pH and temperature values, is favorable for self-assembled hydrogels with significant potential in various fields, such as biomedical materials, cosmetics, and nanotechnology.
 | ||
| Fig. 2 Conformation analyses of hybrid graft copolymer 1m in water. pH (a) and temperature (b) dependencies of CD spectra of 1m (m = 28) at 25 °C. [peptide] = 250 μM. (c) Transmission FTIR spectra of 1m (m = 10, 28, 40). (d) Comparison of β-sheet content for various 1m (m = 6–40) evaluated with respect to relative areas of peaks in the FTIR spectra. | ||
Characterization of the injectable hydrogels self-assembled from hybrid graft copolymers (1m)
The gelation property of the hybrid graft copolymer 1m was subsequently studied in concentrated aqueous solutions (5–15 wt%) at 25 °C using the test tube inversion method (Fig. 3). The hybrid polymers with high grafting densities (m = 28, 32, and 40) formed self-supporting hydrogels at 10 wt% concentration, while the polymers with the lowest grafting ratio (m = 6) were in the fluid solution state even at 15 wt% concentration. Such grafting density-dependent gelation of 1m clearly demonstrated that peptide grafts effectively acted as physical crosslinking points for network formation. | ||
| Fig. 3 Photographs of hybrid graft copolymer solutions at various concentrations at 25 °C and pH = 6.5. | ||
The dynamic mechanical properties of the hydrogels were further studied using rheological tests. The storage modulus (G′) and loss modulus (G′′) of the 1m (m = 28) hydrogel were presented as a function of frequency (0.1–100 rad s−1) at 1% strain, as shown in Fig. 4a. For frequency range considered in this study, the G′ values were greater than the G” values (G′ > G′′) indicating the formation of an elastic solid-like hydrogel. The frequency dependency of all 1m values with different grafting ratios except in the case of m = 6, at which the G′ value was considerably small and lower than that of G′′ (G′ < G′′ at low frequency region), was a characteristic of a typical hydrogel (Fig. S6, ESI†). Interestingly, the stiffness (i.e., G′) of this hydrogel system was grafting density-dependent. For grafting ratios less than m = 32, the stiffness (G′) was found to increase corresponding to the increase in grafting ratios from approximately 100 Pa (for m = 10) to 2500 Pa (for m = 32) under the same conditions. For grafting ratio m = 40, however, the gel stiffness decreased to 1200 Pa (Fig. 4b). As schematically shown in Fig. 4c, the increase in peptide grafts of the hybrid copolymer is considered to enhance the crosslinking density via inter-polymer β-sheet formation. On the other hand, for grafting ratio m = 40, the intra-polymer β-sheet seemed to be more dominant rather than the inter-polymer one preventing the formation of an effective 3D network. Note that the G′ values did not vary significantly for any hybrid gel in the temperature range of 20–40 °C, indicating that their thermal stability was sufficient for biological applications.
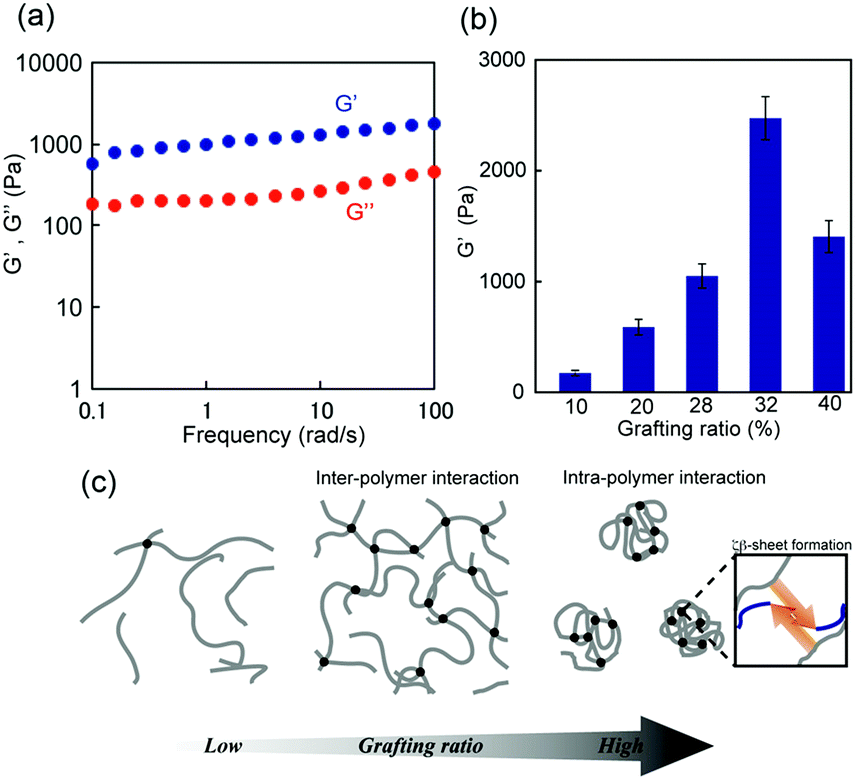 | ||
| Fig. 4 (a) Storage modulus G′ and loss modulus G′′ of self-assembled 1m (m = 28) hydrogel (15 wt%) as a function of frequency at 1% strain. (b) Comparison of G′ values of self-assembled 1m (m = 10–40) hydrogels at 6 rad s−1, 1% strain, and 25 °C. Errors indicate standard deviation. (c) Plausible model of the network structures of 1m hydrogels at different grafting ratios. | ||
The shear-thinning and self-healing properties of the hybrid 1m hydrogels post a mechanical breakdown were also studied. A visible decrease in the G′ value was observed when the 1m hydrogel was treated with a large amplitude strain, resulting in a sol state (G′ < G′′) at approximately 25% strain (Fig. 5a). Upon switching to a low strain amplitude (1%), G′ recovered quickly to approximately 80–90% of the initial value, and the hydrogel returned to its original state in 20–30 minutes (Fig. 5b). It should be noted that the grafting density of 1m did not significantly affect the innate shear-thinning behavior, although the crossover point of G′ and G′′ values of the sample with a low grafting value of m = 10 was somewhat low (ca. 15% strain) compared to that of the other 1m samples with higher grafting values (m ≥ 20) (data not shown). The shear-thinning and rapid self-healing ability of 1m hydrogels can be attributed to the multiple reversible hydrogen bonds among the PEG-block-peptide grafts owing to the strong β-sheet propensity. In fact, the presence of urea in the hydrogel, which is known as a hydrogen bond breaker, led to a significant decrease in both stability over strain amplitude and self-healing efficiency (ca. 50–60%) (Fig. S7, ESI†). Importantly, the 1m hydrogel could be loaded in a standard syringe, injected through the needle manually, and re-formed into a self-supporting structure (Fig. 5c). Overall, a mechanically controllable and injectable hydrogel system was successfully developed by manipulating the grafting density.
 | ||
| Fig. 5 (a) Recovery of stiffness of 1m (m = 28) hydrogel (15 wt%) at 1% strain amplitude after large-amplitude oscillatory breakdown. (b) Rapid recovery of G′ values just after removing strain amplitude (white) and after 30 min healing (black). (c) Photograph taken when the 1m hydrogel (15 wt%) was placed in a Petri dish using a syringe, demonstrating the self-supporting structure. | ||
In vitro assay of the injectable hydrogel toward biomedical applications
Finally, we studied the properties of the 1m hydrogel system as an injectable 3D scaffold for cells and a drug delivery medium. Chen et al. reported that hydrogels rapidly self-assembled from β-hairpin peptides with a G′ of 100 Pa can support 3D cell distribution, but the lack of gel rigidity or strength leads to cell precipitation.49 In addition, Hartgerink et al. reported that β-sheet nanofiber-based hydrogel with ca. 500 Pa stiffness could be attached to a target site in vivo without immediate dissolution into the surrounding body tissue.26 Considering these insights, we chose 1m (m = 28) hydrogel (15 wt%) for its desirable hydrogel stiffness as the most suitable 3D matrix and applied it to dynamic tissue. Chondrocytes (1.0 × 107 cells mL−1) were encapsulated in 1m hydrogel and cultured for 2 days. As shown in Fig. 6a, the live/dead assay demonstrated that the majority of the cells were viable (green signals) (>80%) and distributed homogeneously throughout the hydrogel. The cell viability after injection did not change significantly; that is, cell survival was ca. 95% compared to the live cells in the uninjected control group (Fig. 6b), indicating the effective protection of the gel over entrapped cells against shear stress. In addition, self-healing capability of the 1m hydrogel would be useful in providing an optimal lifetime for 3D cell scaffold, since hydrogels implanted in a dynamic and load-bearing environments, such as cartilage tissue, are prone to structural defects.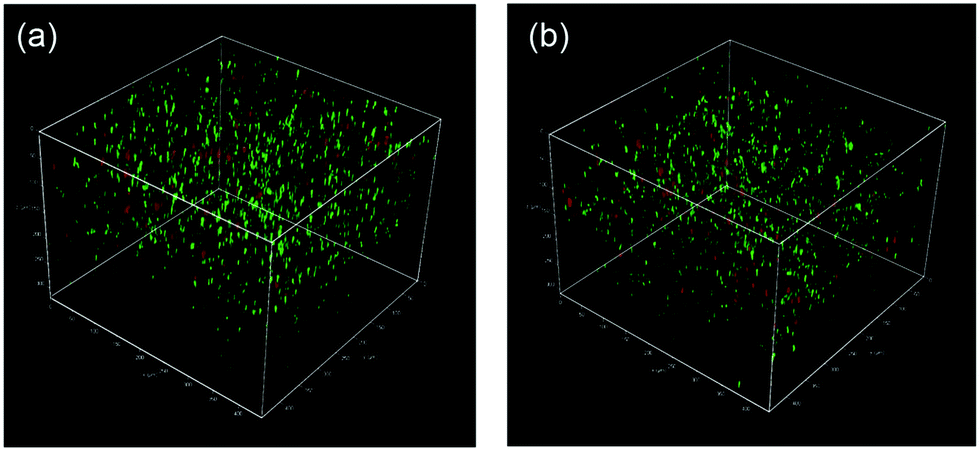 | ||
| Fig. 6 Confocal microscopy images of chondrocytes in 1m (m = 28) hydrogel before (a) and after (b) being injected from a syringe. Note that the cells were cultured for two days in the hydrogel, and were stained using Calcein (green, live cell) and EthD (red, dead cell) prior to imaging. | ||
We further studied the controlled release of 1m hydrogels using BSA as a protein drug model. Fig. 7 shows the cumulative release profiles of BSA-FITC-loaded hydrogels with different grafting densities (m = 10, 28, and 32, 15 wt%) in PBS at 37 °C. Controlled release of BSA-FITC was achieved for up to 5 days, reflecting the increase in grafting density. An increase in the grafting ratio increased both the stiffness and network density, and decreased the release rate, consequently resulting in a longer release time. These controllable features make hydrogels highly desirable for applications in the fields of tissue engineering, regenerative medicine, and drug delivery system.
 | ||
| Fig. 7 Release profiles of BSA-FITC from the 1m hydrogels [m = 10 (blue circle), 28 (red circle), and 32 (orange circle)] (15 wt%) incubated in PBS (pH 7.4) at 37 °C. Errors indicate standard deviation. | ||
Conclusions
In summary, we developed a unique injectable hydrogel material from biocompatible PMPC with PEG-attached self-assembling peptide grafts for various grafting ratios. These graft-type hybrid copolymers formed stable β-sheet structures across a wide range of pH and temperature levels. The β-sheet structures act as reversible supramolecular crosslinks providing hydrogels with self-healing and injectable properties. The mechanical properties of a hydrogel could be controlled easily by manipulating the grafting density. For an increase in the grafting ratio (for m ≤ 32), the inter-polymer network was strengthened leading to an increase in stiffness. However, for m = 40 the inter-polymer network was weakened probably due to the more prominent intramolecular β-sheet formation instead of intermolecular β-sheet formation. Based on such controllable gel characteristics, the hydrogels could be employed as injectable scaffolds for tissue engineering and controlled drug release media. We believe that the findings presented in this study provide relevant insights into the design and synthesis of smart polymer materials with potentials in biomedical, cosmetic, and nanotechnological fields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by Grants-in-Aid for Scientific Research (KAKENHI) (No. 17K04994, 20H02767) from the Japan Society for the Promotion of Science (JSPS), and a MEXT-Supported Program for the Strategic Research Foundation at Private University.Notes and references
- O. Wichterle and D. Lim, Nature, 1960, 185, 117–118 CrossRef.
- D. Seliktar, Science, 2012, 336, 1124–1128 CrossRef CAS PubMed.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2013, 65, 10–16 CrossRef CAS PubMed.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511 CrossRef PubMed.
- C. Bilici and O. Oguz, Macromolecules, 2013, 46, 3125–3131 CrossRef CAS.
- Y. Sun, A. L. Wollenberg, T. M. O’Shea, Y. Cui, Z. H. Zhou, M. V. Sofroniew and T. J. Deming, J. Am. Chem. Soc., 2017, 139, 15114–15121 CrossRef CAS PubMed.
- S. Talebian, M. Mehrali, N. Taebnia, C. P. Pennisi, F. B. Kadumudi, J. Foroughi, M. Hasany, M. Nikkhah, M. Akbari, G. Orive and A. Dolatshahi-Pirouz, Adv. Sci., 2019, 6, 1801664 CrossRef PubMed.
- B. Q. Y. Chan, Z. W. K. Low, S. J. W. Heng, S. Y. Chen, C. Owh and X. J. Loh, ACS Appl. Mater. Interfaces, 2016, 8, 10070–10087 CrossRef CAS PubMed.
- C. Löwenberg, M. Balk, C. Wischke, M. Behl and A. Lendlein, Acc. Chem. Res., 2017, 50, 723–732 CrossRef PubMed.
- J. Shang, X. Le, J. Zhang, T. Chen and P. Theato, Polym. Chem., 2019, 10, 1036–1055 RSC.
- T. Koga, K. Tomimori and N. Higashi, Macromol. Rapid Commun., 2020, 41, 1900650 CrossRef CAS PubMed.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485–487 CrossRef CAS.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef CAS PubMed.
- K. Liang, K. H. Bae and M. Kurisawa, J. Mater. Chem. B, 2019, 7, 3775–3791 RSC.
- Y. Chao, Q. Chen and Z. Liu, Adv. Funct. Mater., 2019, 30, 1902785 CrossRef.
- X. Z. Shu, Y. Liu, F. S. Palumbo, Y. Luo and G. D. Prestwich,, Biomaterials, 2004, 25, 1339–1348 CrossRef CAS PubMed.
- J. S. Temenoff, H. Shin, D. E. Conway, P. S. Engel and A. G. Mikos, Biomacromolecules, 2003, 4, 1605–1613 CrossRef CAS PubMed.
- K. Nagahama, T. Ouchi and Y. Ohya, Adv. Funct. Mater., 2008, 18, 1220–1231 CrossRef CAS.
- Y. L. Chiu, S. C. Chen, C. J. Su, C. W. Hsiao, Y. M. Chen, H. L. Chen and H. W. Sung, Biomaterials, 2009, 30, 4877–4888 CrossRef CAS PubMed.
- N. Higashi, S. Matsubara, S. Nishimura and T. Koga, Materials, 2018, 11, 424 CrossRef PubMed.
- M. Guvendiren, H. D. Lu and J. A. Burdick, Soft Matter, 2012, 8, 260–272 RSC.
- A. P. Nowak, V. Breedveld, L. Pakstis, B. Ozbas, D. J. Pine, D. Pochan and T. J. Deming, Nature, 2002, 417, 424–428 CrossRef CAS PubMed.
- B. D. Olsen, J. A. Kornfield and D. A. Tirrell, Macromolecules, 2010, 43, 9094–9099 CrossRef CAS PubMed.
- E. L. Bakota, Y. Wang, F. R. Danesh and J. D. Hartgerink, Biomacromolecules, 2011, 12, 1651–1657 CrossRef CAS PubMed.
- L. H. Butterick, K. Rajagopal, M. Branco, D. Salick, R. Rughani, M. Pilarz, M. S. Lamm, D. J. Pochan and J. P. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7791–7796 CrossRef PubMed.
- M. C. Giano, D. J. Pochan and J. P. Schneider, Biomaterials, 2011, 32, 6471–6477 CrossRef CAS PubMed.
- H. Song, G. Yang, P. Huang, D. Kong and W. Wang, J. Mater. Chem. B, 2017, 5, 1724–1733 RSC.
- Q. Xu, Z. Zhang, C. Xiao, C. He and X. Chen, Biomacromolecules, 2017, 18, 1411–1418 CrossRef CAS PubMed.
- Q. Luo, C. Hou, Y. Bai, R. Wang and J. Liu, Chem. Rev., 2016, 116, 13571–13632 CrossRef CAS PubMed.
- P. Katyal, M. Meleties and J. K. Montclare, ACS Biomater. Sci. Eng., 2019, 5, 4132–4147 CrossRef CAS PubMed.
- Y. Hirano and D. J. Mooney, Adv. Mater., 2004, 16, 17–25 CrossRef CAS.
- K. Murai, Y. Yamamoto, T. Kinoshita, K. Nagata and M. Higuchi, J. Mater. Chem. B, 2017, 5, 5539–5548 RSC.
- Y. Mei, K. L. Beers, H. C. MichellesByrd, D. L. VanderHart and N. R. Washburn, J. Am. Chem. Soc., 2004, 126, 3472–3476 CrossRef CAS PubMed.
- J. Hentschel and H. G. Börner, J. Am. Chem. Soc., 2006, 128, 14142–14149 CrossRef CAS PubMed.
- H. R. Marsden, A. V. Korobko, E. N. M. van Leeuwen, E. M. Pouget, S. J. Veen, N. A. J. M. Sommerdijk and A. Kros, J. Am. Chem. Soc., 2008, 130, 9386–9393 CrossRef PubMed.
- P. Jing, J. S. Rudra, A. B. Herr and J. H. Collier, Biomacromolecules, 2008, 9, 2438–2446 CrossRef CAS PubMed.
- T. Koga, S. Kamiwatari and N. Higashi, Langmuir, 2013, 29, 15477–15484 CrossRef CAS PubMed.
- S. Nishimura, N. Higashi and T. Koga, Polym. Chem., 2019, 10, 71–76 RSC.
- S. Nishimura, N. Higashi and T. Koga, Chem. Commun., 2019, 55, 1498–1501 RSC.
- S. Nishimura, N. Hokazono, Y. Taki, H. Motoda, Y. Morita, K. Yamamoto, N. Higashi and T. Koga, ACS Appl. Mater. Interfaces, 2019, 11, 24577–24587 CrossRef CAS PubMed.
- L. R. Wu, J. Yang, K. Wu and J. Kopeček, Biomacromolecules, 2009, 10, 2319–2327 CrossRef PubMed.
- L. R. Wu, J. Yang and J. Kopeček, Biomaterials, 2011, 32, 5341–5353 CrossRef CAS PubMed.
- K. Ishihara, Y. Iwasaki, S. Ebihara, Y. Shindo and N. Nakabayashi, Colloids Surf., B, 2000, 18, 325–335 CrossRef CAS PubMed.
- K. Ishihara, Front. Med. Biol. Eng., 2000, 10, 83–95 CAS.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS.
- T. Miyazawa and E. R. Blout, J. Am. Chem. Soc., 1961, 83, 712–719 CrossRef CAS.
- C. Chen, Y. Gu, L. Deng, S. Han, X. Sun, Y. Chen, J. R. Lu and H. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 14360–14368 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S7. See DOI: 10.1039/d1ma00347j |
| This journal is © The Royal Society of Chemistry 2021 |