
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTailoring of the physical and mechanical properties of biocompatible graphene oxide/gelatin composite nanolaminates via altering the crystal structure and morphology†
Rama K.
Layek
 *ab,
Vijay Singh
Parihar
c,
Mikael
Skrifvars
d,
Farzin
Javanshour
b,
Mart
Kroon
c,
Mikko
Kanerva
b,
Jyrki
Vuorinen
b,
Minna
Kellomäki
c and
Essi
Sarlin
b
*ab,
Vijay Singh
Parihar
c,
Mikael
Skrifvars
d,
Farzin
Javanshour
b,
Mart
Kroon
c,
Mikko
Kanerva
b,
Jyrki
Vuorinen
b,
Minna
Kellomäki
c and
Essi
Sarlin
b
aLUT University, School of Engineering Science, Department of Separation Science, Mukkulankatu 19, 15210 Lahti, Finland. E-mail: rama.layek@lut.fi
bTampere University, Engineering Material Science, P.O. Box 589, 33101 Tampere, Finland
cBiomaterials and Tissue Engineering Group, BioMediTech, Faculty of Medicine and Health Technology, Tampere University, Finland
dSwedish Centre for Resource Recovery, Faculty of Textiles, Engineering and Business, University of Borås, Sweden
First published on 11th June 2021
Abstract
Despite substantial progress being made relating to 2D-nanofiller-based composite nanolaminates, the fabrication of composite nanolaminates with enhanced ductility and toughness is still challenging. In this study, layered structure graphene oxide (GO)/gelatin powder (GP) composites nanolaminates with enhanced ductility and toughness have been achieved by a simple vacuum filtration of aqueous dispersion of GO/GP composite solution containing 5 wt% of GO. The composite film containing 5 wt% GO shows outstanding improvement of 200% in the stress at break value, with simultaneous enhancement of 52% of the strain at break value compared to GP film. A significant improvement in toughness from 2.2 MJ m−3 to 9.5 MJ m−3 is observed in the composite film containing 5 wt% GO. These significant enhancements of the mechanical properties of the composite film are obtained via the formation of an intercalated nanolaminate structure, H-bonding interactions, and the tailoring of the crystal structure of GP in the composite film, as proved via field-emission scanning electron microscopy, Fourier-transform infrared spectroscopy, and wide-angle X-ray diffraction studies. The growing of fibroblast cells on the composite films signifies that they are not cytotoxic. These GO/GP composites with significant mechanical properties and biocompatibility are very promising for various biomedical applications.
Introduction
During recent years, graphene has attracted tremendous attention due to its outstanding mechanical strength, unique electronic, optical, and electrical properties, thermal conductivity, and excellent biocompatibility.1–3 Among these properties, the utilization of the mechanical and biocompatible properties of graphene is very promising for demanding biomedical applications.4 Graphene is a very strong material that exhibits tensile strength of 130 GPa and a Young's modulus of 1 TPa.1,2 The outstanding mechanical properties and large aspect ratio of graphene sheets make graphene an exciting filler for enhancing the mechanical properties of polymer composites. The judicial integration and homogeneous dispersion of graphene sheets into polymer matrices provide a promising way to enhance the mechanical properties of graphene/polymer composites. However, these types of conventional graphene/polymer composites cannot fully exploit the remarkable properties of graphene, and the obtained results are not satisfactory due to a lack of suitable orientation and the presence of low concentrations of graphene sheets (usually less than 5 wt%).5,6 Mostly, the integration of higher concentrations of graphene (usually 5 wt% or more) into conventional graphene/polymer composites cannot enhance the mechanical properties significantly due to the aggregation and inhomogeneous distribution of graphene sheets in the polymer matrix, which can produce a brittle composite film with poor mechanical properties.7,8Conversely, layered structure graphene/polymer composite laminates with high graphene content levels result in unique layered structure nanolaminates with excellent alignment between the graphene sheets and the surface of the composite film.9–12 Thus, to overcome the drawbacks of conventional polymer composites, recently the scientific community has focused significant effort on integrating high concentrations of graphene into the polymer matrix to exploit the mechanical properties of graphene satisfactorily via fabricating layered structure graphene/polymer composite nanolaminates. To fabricate free-standing layered structure graphene-based polymer composite nanolaminate film with excellent performance, a large quantity of graphene or graphene derivative is required as a filler, and it should undergo strong interfacial interactions with the polymer matrix. There are several types of graphene derivatives, such as graphene nanoplatelets with few layers, reduced graphene oxide, and graphene oxide. These graphene derivatives are produced on a large scale and are mainly used for the fabrication of layered structure polymer composites with higher graphene content levels. Among these various types of graphene derivatives, GO has several different oxygen-containing functional groups (epoxide, hydroxyl, and carboxyl groups) through which it can easily interact with the functional groups of hydrophilic polymers. Hence, several studies have reported the fabrication of layered structure GO/polymer composite nanolaminates with enhanced mechanical properties.4,12–16 Putz et al. have fabricated a brick-wall-structure composite nanolaminate with a parallel orientation between the GO sheets and the surface of the composite film via a vacuum-assisted self-assembly (VASA) process, which results in a dramatic enhancement of the mechanical properties.13 Well-ordered layered structure methylcellulose/GO composite nanolaminate films with enhanced mechanical properties have been prepared via solvent-evaporation-assisted assembly by Layek et al.14 The layered GO/PVA composite nanolaminate film produced via simple solvent evaporation exhibits a significant enhancement in mechanical and biocompatible properties.4 GO/triblock copolymer (PDMS–PGMA) composite nanolaminate paper with high mechanical strength, high toughness, and a high dielectric constant have been prepared through a vacuum-assisted self-assembly method.9 Very recently, Al2O3/GO–PVA composite nanolaminate paper with outstanding mechanical properties was fabricated via a layer-by-layer assembly technique.15 Chen et al. fabricated graphene oxide/alginate composite nanolaminate paper and observed synergistic strength/toughness in the composite.16 However, most of these reported layered structure GO/polymer composite nanolaminate films mainly add stiffness to the polymer, causing improvements in the mechanical strength, modulus, and brittleness behaviours of the composite nanolaminates with notable decreases in the elongation at break values.
Although significant efforts have been made by the polymer scientific community to design and fabricate free-standing, layered structure GO/polymer composite nanolaminate films with enhanced mechanical strength and Young's modulus values, layered structure GO/polymer composite nanolaminates with additional functionalities, such as improved elongation at break, ductility, toughness, and enhanced mechanical strength in the same composite, have still rarely been reported. Here, for the first time, we created a dramatic enhancement in the mechanical strength of GO/gelatin powder (GP) composite nanolaminates with a simultaneous improvement of the elongation at the break value via tailoring the morphology and crystal structure of GP in the presence of GO and via interfacial H-bonding interactions between GP molecules and GO sheets through a facile vacuum-filtration-supported assembly process. This leads to the significant enhancement of the ductility and toughness of the layered structure GO/GP composite laminates. In this study, GP is used due to its semi-crystalline nature, very good sustainability, film-forming ability, and outstanding biocompatibility and biodegradability, and the existence of several functional groups that have the ability to form intermolecular H-bonds with a functional nanofiller. It is reported that the elongation at break of a semi-crystalline polymer composite can be tuned via tuning the crystal structure and morphology of the polymer composite.17,18 GP is a very important semi-crystalline biopolymer with a mixture of a triple-helical crystal structure and an amorphous crystal structure, and its crystal structure and morphology can be tuned via a nanocomposite fabrication strategy.19,20 GO has been selected as a filler due to its outstanding mechanical properties and biocompatibility. In addition to this, GO contains numerous oxygen-containing functional groups (hydroxyl, epoxy, and carboxyl groups) that can produce a H-bonding network with GP molecules, powering the changing of the crystal structure and morphology of GP in the composite and governing the formation of layered structure GO/GP composite nanolaminate film with a dramatic enhancement in the mechanical properties. The effective growth of human lung fibroblast cells on the GO/GP composite films shows the absence of cytotoxicity in the GO/GP composite films. Hence, these layered structure GO/GP composites with enhanced mechanical properties and biocompatibility may have potential applications in various demanding biomedical areas, such as the fabrication of biomedical devices, scaffolds for tissue engineering, biomedical packaging, and biotechnology.
Experimental section
Materials
Graphite powder (TIMCAL Ltd, Switzerland), potassium permanganate (Merck, Germany), sodium nitrate (Aldrich, USA), and hydrogen peroxide (Aldrich, USA, 98%) were used as received. Sulphuric acid was used as received from VWR International Oy, Finland. Gelatin powder (Dr Oetker) was used as received. The Milli-Q water used for this study was obtained via purifying tap water using a Milli-Q integral water purification system.Synthesis of GO
Graphite powder was oxidized to synthesize GO using a modified Hummers’ method.21 In a standard procedure, a conical flask containing 46 mL of 98% concentrated sulfuric acid was cooled in an ice bath and stirred for several minutes using a bar magnet. Then, 1 g of sodium nitrate and 2 g of graphite powder were mixed in chronological order with the pre-cooled 98% concentrated H2SO4 under constant stirring using a bar magnet. Next, potassium permanganate (6 g) was steadily mixed into the reaction mixture under constant stirring and during the KMnO4 addition, the temperature of the reaction mixture was maintained between 0 and 5 °C. After that, the ice bath was removed, and the reaction mixture was subjected to stirring for a further 6 h at room temperature, during which time the reaction mixture turned into a thick paste. Later, 92 mL of Milli-Q water was added into this thick paste under vigorous stirring. The temperature of the reaction mixture was abruptly elevated to ∼90 °C upon the addition of water into the reaction mixture. Then the reaction mixture was cooled to room temperature under vigorous stirring for another 30 min. After that, another 280 mL of Milli-Q water was mixed with the resulting solution mixture, followed by the addition of 30% H2O2 (3 mL). With the addition of H2O2, the dark brown colour of the reaction mixture was instantly transformed into a yellow colour. Then, the solution mixture was centrifuged and re-dispersed repeatedly until the pH of the supernatant solution reached ∼7. Finally, the brown-coloured material was collected from the bottom of the centrifuge tube and dried at 40 °C under vacuum to obtain GO.Preparation of nanolaminate GO/GP composites and biocompatibility studies
An aqueous solution of GP produced a sticky gel-like material at room temperature, whereas a warm aqueous solution of GP remains as a sol and does not produce a sticky gel-like material. At first, an aqueous homogeneous GP sol (20 mg mL−1) was prepared via heating GP in 50 mL of water at 70 °C for 15 min, followed by stirring using a bar magnet, and this warm sol was not sticky. Then, various concentrations of aqueous dispersions of GO were prepared via sonicating the desired amount of GO in 50 mL of water. After that, these various concentrations of aqueous GO dispersions were mixed with the warm aqueous GP sol using a bar magnet, followed by sonication for 5 min at 45 °C to produce homogeneous GO/GP sols. Finally, these warm GO/GP sols were poured into a vacuum filtration system and were vacuum filtered through 0.2-μm PTFE membrane filter paper to eliminate water and produce nano-laminated GO/GP composite films. The concentration of GO/GP sol increased and the temperature of the GO/GP sol decreased to room temperature as solvent was eliminated, producing a GO/GP gel-like film. With the further elimination of solvent via vacuum filtration, GO/GP film with uniform thickness was produced. Finally, these films were allowed to dry at room temperature for 2 days and were then pressed using a compression molder at 35 °C and 6 bar to make flat films with a uniform thickness of ∼160 μm. The obtained composite film containing 5 wt% GO is named GO5 and that containing 10 wt% GO is named GO10. Pure GP film was prepared via eliminating the solvent from an aqueous solution of GP. The biocompatibility of the GO/GP composite film was studied via culturing human lung fibroblast cells on the film and using a live/dead viability test kit to assess the cytotoxicity. Details of the procedure are presented in the ESI.†Characterization
Transmission electron microscopy studies of GO were carried out using a Jeol F200 S/TEM instrument. The TEM sample was prepared via placing a drop of an aqueous dispersion of GO (0.05% w/v) on a carbon-coated copper grid, followed by evaporating the solvent at room temperature. Samples for atomic force microscopy (AFM) characterisation were made via casting a drop of an aqueous dispersion of GO (0.01% w/v) on a mica surface, followed by evaporating the solvent at room temperature. Then, AFM studies of GO over a mica surface were carried out in noncontact mode using a tip resonance frequency of 250 KHz. The samples for field-emission scanning electron microscopy studies were prepared via freeze-fracturing film samples in liquid nitrogen, followed by sputtering gold on the fracture surfaces of the sample films. Then, these gold-coated sample film fractures were investigated using a SEM Zeiss ULTRA Plus instrument. FTIR spectroscopy examinations of GO, pure GA, and nano-laminated GO/GA composite films were carried out from 4000–400 cm−1 using a Tensor 27 Bruker instrument. Raman spectra of GO and GO/GP composite film nanolaminates were recorded from 1000 to 2100 cm−1 using a Renishaw InVia Qontor Raman spectrometer with a 532 nm laser as the excitation source. X-ray diffraction (XRD) studies of GO, pure GP, and nano-laminated GO/GP composite films were performed via fixing the sample film over an amorphous sample holder and scanning it in the 2θ range of 5° to 40° at a rate of 2° min−1 using a Panalytical Empyrean multipurpose diffractometer. A Cu Kα (λ = 0.154 nm) radiation source was used and the operating voltage and current of the instrument were 40 kV and 40 mA, respectively. Differential scanning calorimetry (DSC) studies of the composite films were carried out from 20 to 250 °C using 204F1 NETZSCH DSC apparatus under a nitrogen atmosphere at a heating rate of 10 °C min−1. Thermogravimetry (TGA) experiments involving GO, pure GP, and nano-laminated GO/GP composite films were performed under a nitrogen atmosphere at a heating rate of 10 °C min−1 using a simultaneous thermal analyzer instrument (Netzsch STA 409). The mechanical characteristics of pure GP and nanolaminate composite films were measured using specimens with a size of 16 mm × 4 mm × 0.16 mm and at a strain rate of 1 mm min−1 at 30 °C using an Instron 5967 instrument. The biocompatibility of pure GP and nanolaminate GO/GP composite film samples was tested via in vitro fibroblast cell cultures.Results and discussion
Layered structure GP composite nanolaminate with GO
GP is a colourless semi-crystalline protein-based biopolymer composed of the covalent linking of various types of amino acid (glycine, proline, and 4-hydroxy proline) residues.22 Due to the presence of numerous amino and carboxyl groups in gelatin molecules, it is hydrophilic in nature and displays excellent solubility in hot water via the formation of strong H-bonding interactions between the functional groups of GP and water molecules. In hot aqueous solution, the interchain H-bonding interactions between GP molecules disappear and the GP molecules remain as coiled polymer chains.19,23 The removal of water at room temperature from an aqueous solution of GP again increases the interchain GP molecule interactions and produces a solid film with a triple helical structure, as supported by the WAXS results (Fig. 2). GO produced via the modified Hummers’ method (oxidation of graphite powder) contains several hydrophilic oxygen-containing functional groups, such as hydroxyl, epoxy, and carboxyl groups.2,14 Therefore, it undergoes very good dispersion in an aqueous medium and produces a brown-coloured solution. The blending of an aqueous suspension of GO with a hot aqueous solution of GP yields a stable uniform GO/GP composite solution, and this may be due to very good interactions between the functional groups of individual GP molecules and the functional groups of GO in an aqueous medium. As the elimination of solvent from an aqueous suspension of GO/GP composite solution progresses, GP molecules are absorbed over the GO sheets via H-bonding interactions, hindering triple helical structure formation. Then, these GO sheets with absorbed GP produce a gel-like composite film with a coil structure, where the GO sheets with absorbed GP remain oriented with the surface of the film due to the applied force of vacuum filtration. The removal of the maximum amount of solvent from this gel-like GO/GP composite film via vacuum filtration produces a freestanding GO/GP composite nanolaminate film. In the GO/GP composite nanolaminates, the GO molecules interact with GP molecules through H-bonding interactions, hindering GP triple helical structure formation. Therefore, in the GO/GP composites, the number of coil structures is significantly enhanced (as supported by the WAXS results in Fig. 2), and the GO sheets are cemented (FESEM results, Fig. 1(C) and (D)) into the coil-structured GP through H-bonding interactions (FTIR results, Fig. 3) to produce a nanolaminate GO/GP composite film (Scheme 1). 5 wt% and 10 wt% GO reinforced composite nanolaminates (with respect to the GP matrix) were prepared and labelled as GO5 and GO10, respectively. Pure GP film is transparent, whereas the GO/GP composite nanolaminate films are dark brown in colour. The structure–property relationships of the GO/GP composite films were explored via performing structure and morphology analysis, examining the interactions of GP with GO sheets and the mechanical properties, and carrying out biocompatibility studies. | ||
| Fig. 1 (A) An AFM image displaying the sheet-like structure and (B) the corresponding height profile showing the thickness of a single sheet of GO. FESEM images of (C) GO5 and (D) GO10 indicating the formation of the layered structure GO/GP composite nanolaminates. | ||
 | ||
| Fig. 2 WAXS patterns displaying the formation of layered structure GO/GP composite nanolaminates and the changing of triple helical structures to coil structures in the GO/GP composites. | ||
 | ||
| Fig. 3 Enlarged FTIR spectra from (A) 2000 cm−1 to 1000 cm−1 and from (B) 4100 cm−1 to 2400 cm−1, showing the interactions between GP and GO in the GO/GP composite film. | ||
 | ||
| Scheme 1 A schematic representation of the formation of layered structure GO/GP composite nanolaminates. | ||
Structure and morphology studies
Transmission electron microscopy (TEM) images (ESI,† Fig. S1(A)) of GO are obtained via casting a drop of dilute aqueous solution, and the wrinkled morphology of highly exfoliated GO sheets on carbon-coated Cu grids is displayed. This indicates that the GO sheets are highly exfoliated in dilute aqueous solution. The characteristic hexagonal polycrystalline diffraction pattern of graphite is observed in the selected area diffraction pattern (SAED) (ESI,† Fig. S1(B)) of GO and this indicates the presence of graphitic structures in GO.24 An atomic force microscopy (AFM) image of GO and its height profile are shown in Fig. 1(A) and (B), respectively. From the image, it is clear that GO has sheet-like morphology, and the height profile shows that the thickness of a GO sheet is 1.08 ± 0.1 nm, which resembles the height profile of a single-layer sheet.25 As the AFM study of GO was performed via casting a dilute aqueous drop of GO solution onto a mica surface, therefore, the AFM results indicate that in dilute solution GO sheets are present as highly exfoliated GO sheets.In order to examine the morphology, the formation of nanostructures, and the orientation of the GO sheets in the GO/GP composite films, field-emission scanning electron microscopy (FESEM) studies of the cryofracture surfaces (cross-section) of pure GP film and GO/GP composite films were performed, and the resulting FESEM images are shown in Fig. 1(C) and (D) and ESI,† Fig. S2. From ESI,† Fig. S2, it is obvious that there are no GO sheets present in the cross-section of pure GP film, and it displays the lamellar microstructure of GP. In aqueous solution, GP molecules undergo strong H-bonding interactions with water molecules, reducing the interchain interactions between GP molecules. As the removal of water progresses, the GP molecules move closer and the interchain interactions between the GP molecules are enhanced. With the removal of the maximum amount of water, the GP chains are stacked together by intermolecular H-bonding interactions in a layer-by-layer fashion to produce film with a triple helical microstructure, as shown in FESEM images (ESI,† Fig. S2) and wide-angle X-ray scattering data (Fig. 2). From Fig. 1, it is also clear that the GO5 and GO10 composite films exhibit a layered structure nanolaminate morphology, where GP functions as an inter-linker between the GO sheets and as a gluing agent for laminating adjacent GO nanosheets. This leads to the formation of dense and compact layered structure GO/GP composite nanolaminates. A probable mechanism for this formation is presented in Scheme 1. It is reported that GO sheets can interact with hydrophilic polymer chains, like GP, in aqueous media, improving the dispersion of the GO/GP composite solution.26 As the elimination of solvent progresses, some hydrophilic GP starts to absorb onto the GO surface, driven by H-bonding interactions. Then, these GO sheets with absorbed GP are orientated in parallel with the surface of the composite film due to gravitational forces originating from the vacuum filtration of the GO nanosheets with absorbed GP in solution, and these act as inter-linkers between adjacent discrete GO platelets.27 The concentration of the GO/GP composite solution increases as the elimination of solvent progresses. This brings the GO sheets with absorbed GP and matrix GP molecules closer to each other, producing a gel-like composite film. With the elimination of the maximum amount of solvent, the matrix GP molecules cemented the GO sheets with absorbed GP together to give layered structure GO/GP composite film with a nanolaminate microstructure (Scheme 1).
To verify the formation of the layered structure, WAXS studies of GO, pure GP, and GO/GP composite films were performed, as shown in Fig. 2. The WAXS pattern of GO film showed a characteristic diffraction peak at 2θ = 10.9°, and this signifies that the vacuum filtration of an aqueous solution of GO produces a layered structure film with an interlayer spacing of 0.81 nm between the GO layers.28 GP typically crystallizes when forming a solid film upon the elimination of water from an aqueous solution of GP at room temperature, and this shows a typical diffraction peak at 2θ = 8.1° due to the formation of a triple-helical crystalline structure.29 In addition, solid GP film also shows a characteristic diffraction peak at 2θ = 20.5°, which is related to the formation of amorphous coiled structures.29 However, in the GO/GP composite films, the peak intensity at 2θ = 8.1° is decreased with an increase in the GO content. This indicates that the crystallization of GP into triple-helical structures is hampered in the presence of GO sheets. The oxygen-containing functional groups of the GO sheets interact with the functional groups of GP, reducing the interchain interactions between GP molecules and leading to a decrease in the perfect crystallization of GP into triple-helical structures in the GO/GP composites. In addition, the diffraction peak at 2θ = 8.1° is clearly shifted to lower angles (7.8° in GO5 and 7.3° in GO10) in the composite films, which signifies that GO sheets are entering into the GP crystals and increasing the crystal spacing of GP in the GO/GP composites. GP does not have any typical diffraction peaks in the region typical for GO; however, in the GO/GP composites, a similar typical GO diffraction peak appeared. This confirms the formation of a close-packed layered GO structure in the GO/GP composite nanolaminates. The diffraction peaks of the GO/GP composites are slightly shifted to lower 2θ values with respect to the diffraction peak of the GO film. This indicates that GP molecules are integrated in between the GO layers, producing a layered structure GO/GP composite nanolaminate and causing a slight enhancement of the spacing between the GO layers in the GO/GP composite. GO5 and GO10 show shifts of the diffraction peak at 2θ = 10.9° to 2θ = 10.1° and 10.4°, respectively. This indicates that a slightly higher number of GP molecules enter in between the GO platelets in GO5 compared to GO10, causing slightly smaller spacing between the GO layers in the GO10 composite compared to the GO5 composite.
Interactions between GO and the GP matrix
To identify the interactions of GP molecules with GO sheets, Fourier-transform infrared (FTIR) spectra of GO, pure GP, and GO/GP composite films were obtained and are displayed in Fig. 3(A) and (B). The shifting of the peak positions of the functional groups of GP in the GO/GP composite films with respect to pure GP film was observed, allowing the identification of the interactions between the GP molecules and GO sheets. To detect the clear shifts of the peak positions of functional groups of GP in the GO/GP composite films, the FTIR spectra were enlarged in the regions of 2000 cm−1 to 1000 cm−1 (Fig. 3(A)) and 4100 cm−1 to 2400 cm−1 (Fig. 3(B)). The FTIR spectrum of GO exhibits a distinguishing peak at ∼1722 cm−1, which is attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of –COOH groups, and a broad peak at 3415 cm−1, which is attributed to the O–H stretching vibrations of –OH groups of GO and H2O molecules associated with GO molecules.30,31 The peak at ∼1212 cm−1 arises from the stretching vibrations of C–O–C groups, and the peak at ∼1044 cm−1 is from C–O stretching vibrations, both characteristic of epoxy groups. The sharp peak at ∼1620 cm−1 appears due to skeletal vibrations of the oxidized graphitic domain of GO.30,31 The pure GP film exhibits typical bands at 1631 cm−1, 1535 cm−1, and 1235 cm−1 from the –CO stretching vibrations of amide I, the C–N stretching and N–H bending vibrations of amide II, and the C–N stretching and N–H bending vibrations of amide III, respectively.31,32 In GO10, these peaks appear at peak positions of 1626 cm−1, 1525 cm−1, and 1229 cm−1, respectively, and this indicates a shifting of the peak positions of the composite to lower wavenumbers with respect to pure GP film. GP film also displays typical peaks at 3293 cm−1 and 3073 cm−1 from the H-bonded NH-stretching vibrations of amide I and the asymmetric stretching vibrations of C–H and NH3+ of amide II.32,33 It is obvious from the spectra that in the composite films these peaks are shifted to lower wavenumbers compared to pure GP film, and in GO10 these peaks appear at 3289 cm−1 and 3066 cm−1, respectively. This indicates the existence of H-bonding interactions between the –CO, N–H, and C–N groups of GP and the oxygen-containing functional groups of GO. Furthermore, the C–H stretching peaks of the CH2/CH groups of GP and GO appeared at ∼2935/2875 cm−1 and ∼2924/2852 cm−1, respectively.30,33 However in the composites, these two vibration bands merged together, appearing in a new position. The appearance of the C–H stretching peak of GO in the composite films signifies the successful integration of GO into the GP matrix. To investigate further interactions between GO and GP molecules, Raman spectroscopy studies were performed, and the recorded Raman spectra of GO, GO5, and GO10 are presented in ESI,† Fig. S3. The Raman spectrum of GO shows a characteristic G band at 1345 cm−1 and a D band at 1602 cm−1. In both GO5 and GO10 these bands are slightly shifted with respect to pure GO. GO10 shows an upshift of the D band from 1345 cm−1 to 1349 cm−1 and a downshift of the G band from 1602 cm−1 to 1598 cm−1. The shifting of the D band and G band positions in the GO/GP composite films with respect to GO indicates the presence of interactions between GO and GP in the GO/GP composite nanolaminates.34
O stretching vibrations of –COOH groups, and a broad peak at 3415 cm−1, which is attributed to the O–H stretching vibrations of –OH groups of GO and H2O molecules associated with GO molecules.30,31 The peak at ∼1212 cm−1 arises from the stretching vibrations of C–O–C groups, and the peak at ∼1044 cm−1 is from C–O stretching vibrations, both characteristic of epoxy groups. The sharp peak at ∼1620 cm−1 appears due to skeletal vibrations of the oxidized graphitic domain of GO.30,31 The pure GP film exhibits typical bands at 1631 cm−1, 1535 cm−1, and 1235 cm−1 from the –CO stretching vibrations of amide I, the C–N stretching and N–H bending vibrations of amide II, and the C–N stretching and N–H bending vibrations of amide III, respectively.31,32 In GO10, these peaks appear at peak positions of 1626 cm−1, 1525 cm−1, and 1229 cm−1, respectively, and this indicates a shifting of the peak positions of the composite to lower wavenumbers with respect to pure GP film. GP film also displays typical peaks at 3293 cm−1 and 3073 cm−1 from the H-bonded NH-stretching vibrations of amide I and the asymmetric stretching vibrations of C–H and NH3+ of amide II.32,33 It is obvious from the spectra that in the composite films these peaks are shifted to lower wavenumbers compared to pure GP film, and in GO10 these peaks appear at 3289 cm−1 and 3066 cm−1, respectively. This indicates the existence of H-bonding interactions between the –CO, N–H, and C–N groups of GP and the oxygen-containing functional groups of GO. Furthermore, the C–H stretching peaks of the CH2/CH groups of GP and GO appeared at ∼2935/2875 cm−1 and ∼2924/2852 cm−1, respectively.30,33 However in the composites, these two vibration bands merged together, appearing in a new position. The appearance of the C–H stretching peak of GO in the composite films signifies the successful integration of GO into the GP matrix. To investigate further interactions between GO and GP molecules, Raman spectroscopy studies were performed, and the recorded Raman spectra of GO, GO5, and GO10 are presented in ESI,† Fig. S3. The Raman spectrum of GO shows a characteristic G band at 1345 cm−1 and a D band at 1602 cm−1. In both GO5 and GO10 these bands are slightly shifted with respect to pure GO. GO10 shows an upshift of the D band from 1345 cm−1 to 1349 cm−1 and a downshift of the G band from 1602 cm−1 to 1598 cm−1. The shifting of the D band and G band positions in the GO/GP composite films with respect to GO indicates the presence of interactions between GO and GP in the GO/GP composite nanolaminates.34
To explore the interactions between GO and GP, and in order to understand the thermal characteristics of the composites, differential scanning calorimetry (DSC) studies of GP and GO/GP composites were performed, and the resulting DSC traces are displayed in Fig. 4(A). The glass transition temperature (Tg) values of the pure GP and GO/GP composite films were investigated from the DSC thermograms, and the effects of the segmental motion of GP polymer chains in the absence and presence of GO were explored. The shifting of Tg in the GO/GP composite films with respect to pure GP film was analyzed to detect the interactions of GO with GP molecules. From the figure, it is clear that pure GP film shows a Tg value of 63.4 °C.35 Upon the integration of GO into the GP matrix, the Tg values are slightly increased with respect to pure GP, and GO10 shows a Tg value of 64 °C. The presence of significant interfacial H-bonding interactions between GP and GO molecules explains the decrease of the segmental motion of the GP polymer chains in the GO/GP composite. This causes slight enhancements of the Tg values of the GO/GP composites with respect to pure GP film.36 Besides this, pure GP film shows a melting temperature (Tm) of 130 °C, with a melting enthalpy of 171.6 J g−1. This typical endothermic peak is very common in the DSC thermogram of the gelatin and is characterized as the Tm value of GP, as reported in the literature.35 The integration of GO into the GP matrix results in a decrease of the melting temperature and an increase of the melting enthalpy. With an increase in the GO concentration, the melting temperature of the GO/GP composite decreased, with an increase in the melting enthalpy. GP10 shows a melting temperature of 112 °C with a melting enthalpy of 207.7 J g−1. The increase in the melting enthalpy indicates that the crystallinity of the GO/GP composite films is increased upon the integration of GO sheets into the GP matrix, whereas the Tm decrease of the composite films indicates that the GP crystals become less perfect (more defective and less compact) in the composite film compared to GP film.
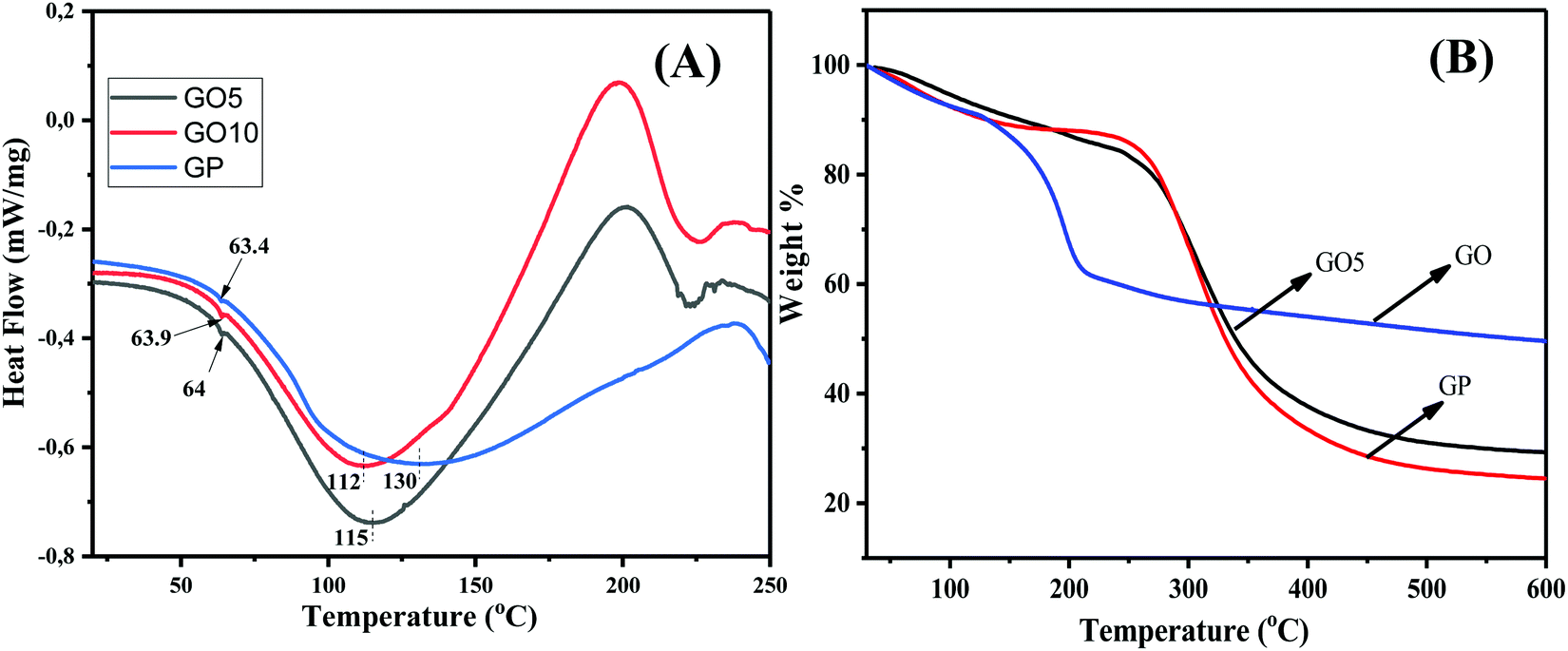 | ||
| Fig. 4 (A) DSC thermograms displaying the changing of the Tg values and the melting temperatures of the GO/GP composite films. (B) TGA thermograms showing the improved thermal stability of the GO/GP composite film. | ||
The thermal stability of the GO, GP, and GO/GP composite laminates was investigated via thermogravimetric analysis (TGA), and the resulting thermograms are displayed in Fig. 4(B). The TGA thermogram of GO indicates that it is thermally unstable. GO starts to degrade below 100 °C, and this is due to the removal of associated water molecules present within the GO sheets. Then, the thermogram of GO displays rapid weight-loss of 32% between 100 and 250 °C. This weight loss is due to the degradation of numerous oxygen-containing functional groups, such as epoxide, hydroxyl, and carboxylic groups, present in GO.37,38 Hence, GO consists of 32 wt% oxygen-containing functional groups. The water molecules are not completely removed from the GP and GO/GP composite films, as minor amounts of water remain associated with the GP and GO/GP composite films due to their hydrophilic nature. The thermogram of pure GP shows minor weight-loss of 7.5 wt% below 100 °C, and this is due to the elimination of associated water molecules from hydrophilic GP.39,40 The thermograms of GO5 and GO 10 (Fig. 4(B) and ESI,† Fig. S4) show lower weight-loss compared to pure GP below 100 °C, which indicates the presence of lower amounts of associated water molecules in the GO/GP composites compared to GP. After that, GP shows a significant amount of weight loss, 56%, between 250 and 400 °C due to the decomposition of GP molecules.39,40 GO10 shows slightly higher weight-loss compared to GP between 200 and 260 °C, and this may be due to the elimination of GO functional groups present in the GO10 composite. Pure GP displays an onset degradation temperature of ∼275 °C, whereas G10 shows an onset degradation temperature of ∼280 °C.39,40 This demonstrates that the onset degradation temperature of GO10 slightly increased with respect to pure GP film. Besides this, the residual mass of G10 is 6% higher compared to pure GA film. A few current studies have shown that high levels of interfacial interactions between GO and the polymer matrix can improve the thermal stability of a polymer composite.41,42 Our GO/GP composite film also exhibits strong interfacial interactions between the GP molecules and GO sheets. Therefore, like other reported GO-based polymer composite systems, the thermal stabilities of our GO/GP composites were enhanced with respect to GP film.
Biocompatibility studies
WI-38 human lung fibroblast cell cultures were used to evaluate the cytotoxicity of GP and GO/GP composite films. The films were washed with ethanol and cut into semi-circles to fit into 24-well polystyrene cell culture plates. The cells were plated directly onto the films and onto a native cell culture plate surface, which acted as a control. Green-fluorescence-emitting calcein-AM was used to stain the live cells, and red-fluorescence-emitting ethidium homodimer-1 was used for the dead cells. The cytotoxicity of the films and cell morphologies were evaluated from fluorescence microscopy images, simultaneously showing both live and dead cells. As shown in Fig. 5, the cell cultures were dominated by live cells. In all samples, only a few dead cells were seen at each time point. The cells proliferate and elongate in contact and in proximity with the tested materials. The cell coverage was higher on the control than in the wells with the GO/GP films, but no apparent differences were seen between the GO5 and GO10 composite films. When in contact with the cell culture medium, the GO/GP films absorbed water. The films were soft during the experiments. This study finds that the GO/GP films were non-cytotoxic towards WI-38 fibroblasts and, therefore, they could be used for tissue-engineering applications. | ||
| Fig. 5 Viability tests with WI-38 fibroblasts on (A) GP film, (B) GO5, and (C) GO10 after 4 days, and on (D) GP film, (E) GO5, and (F) GO10 after 7 days. The green cells are alive and the red cells are dead. The cultures are dominated by live cells, and the cells show normal elongated morphologies after 7 days of culturing. Between days 4 and 7, cell coverage increases noticeably. | ||
Mechanical property studies
The mechanical behaviours of pure GP and GO/GP composite laminates were explored via performing mechanical property measurements using a universal testing instrument, and the resulting stress–strain curves are shown in Fig. 6(A). The typical values of numerous mechanical properties, such as stress at break, elongation at break, and toughness, were directly calculated from the instrumental software after performing tensile tests. Five samples were measured for each set of films, and the statistical average values of the mechanical properties were calculated from the obtained results. The calculated statistical average values of the mechanical properties of pure GP and layered structure GO/GP composite laminate films are displayed in bar-diagram form along with error bars in Fig. 6(B)–(D). From the figure, it is noticeable that pure GP film exhibits a stress at break value of 60 ± 4 MPa and an elongation at break value of 5.8 ± 1.5%. This value of elongation at break specifies that pure GP film is very brittle and shows rapid fracturing at lower applied strains.43 During the solid GP film formation process, the elimination of water from the dilute aqueous solution of GP at room temperature first produces a highly concentrated aqueous solution of GP. In this concentrated GP solution, the coil structures of pure GP are partially transformed into triple helical structures and remain locked. The water molecules are not completely removed from the GP film, as a small amount of water remains associated with the GP film due to its hydrophilic nature. However, upon the elimination of the maximum amount of solvent, the interchain interactions between these locked GP molecules are increased abruptly, leading to the formation of a brittle solid film with a lamellar morphology composed of coil and triple helical structures.44 This phenomenon is supported by the combined results from WAXS, FESEM, and mechanical testing of the GP film. To the contrary, the GO/GP composite laminate films show noteworthy variations with respect to pure GP film in terms of their structure–property relationships. GP film with the integration of 5 wt% GO produced a nanolaminate layered structure composite, and this shows a higher elongation at break value compared to pure GP film. During the GO/GP film drying process, the GO sheets suppress the perfect crystallization of the GP molecules into proper triple helical structures, and with an increase in the GO concentration, this decreases significantly. This phenomenon is further supported by the WAXS and DSC results. In addition to this, we have also shown from the WAXS and DSC results that GO sheets not only decrease the perfect crystallization of GP molecules but they also enhance the spacing of the triple helix crystal structures of the GP molecules. These results indicate that the intermolecular H-bonding interactions between GP molecules decreases abruptly in the GO/GP composite laminate film, causing an increase in the flexibility and elongation at break value of the GO5 composite laminate film compared to pure GP film. Some associated water molecules also remain in the GO/GP composite film due to the hydrophilic nature of the GO/GP composite film; however, the amount of associated water molecules is less compared to GP film. It is reported that water molecules associated with hydrophilic biopolymer films act as a plasticizer.45 It is also reported that the mechanical strength of a biopolymer is reduced and the elongation at break is enhanced with an increase in the plasticizer concentration.46 Though there are less associated water molecules present in the GO5 composite film, the elongation at break increases, with a simultaneous enhancement of the mechanical strength, and this is due to structural changes relating to GP molecules in the composite film and nanolaminate structure formation. Hence, the structural changes of the GP molecules, nanolaminate structure formation, and associated water molecules jointly play a significant role in determining the mechanical properties of the GO/GP composite film. The GO5 composite nanolaminate film shows an increase in the elongation at break value from 5.8 ± 1.5 to 8.8 ± 2%, which is an approximately 50% improvement compared to pure GP film. Upon a further increase of the GO concentration to 10 wt%, GO10 shows a decrease in the strain at break compared to GO5. The molecular mobility and chain flexibility of GP decreases significantly in the presence of a higher concentration of GO, and GO10 shows a lower stress at break value compared to GO5. Both theoretical simulations based on first principles calculations and experimental studies show that the load-bearing capability of an intercalated layered structure nanocomposite film is enhanced significantly in the in-planar direction of the composite film.47,48 In our GO/GP composite nanolaminates, the GP molecules not only cement the neighbouring GO sheets to produce dense and compacted composite nanolaminates, but they also intercalate in between the GO layers, enhancing the interlayer adhesion between adjacent GO sheets via cooperative H-bonding interactions. This causes a significant improvement of the load-bearing capabilities of the intercalated GO sheets, leading to a dramatic enhancement of the tensile strength and toughness of our layered structure GO/GP composite nanolaminates films. With an increase in the GO concentration, the stress at break and toughness of these GO/GP composite laminate films are enhanced with respect to pure GP film. The GO5 composite film shows a substantial improvement in stress at break from 60 ± 4 MPa to 179 ± 3.5 MPa, ∼3 times higher, and the toughness increase from 2.2 ± 0.5 MJ m−3 to 9.5 ± 0.6 MJ m−3, which is 3.6 times higher compared to pure GP film. Therefore, we observed that both the transition of triple-helical crystal structures to coil structures in GP and the formation of an intercalated layered structure morphology in the GO/GP composite nanolaminate has a significant influence on the mechanical properties of the layered structure GO/GP composite nanolaminate films. A majority of previous studies on graphene/polymer composite nanolaminates showed outstanding enhancement of the stress at break with a sharp decrease in the elongation at break with respect to the unfilled polymer.12–16 However, the properties of our layered structure GO5 composite laminate are unique, as the GO5 composite nanolaminate shows simultaneous enhancements in the stress at break and elongation at break values, leading to the fabrication of a GO/GP composite laminate with enhanced ductility and toughness. The GO5 and GO10 composite films show noteworthy toughness enhancements of ∼330% and ∼168%, respectively, with respect to pure GP film. GO10 shows a further enhancement of the stress at break to 196 ± 4.5 MPa. Due to the presence of a higher concentration of GO, more efficient load transfer from the polymer to graphene occurs in GO10 and it shows a higher stress at break compared to GO5. | ||
| Fig. 6 (A) Stress–strain curves showing the simultaneous enhancement of the stress at break and the strain % at break, and bar diagrams of (B) stress at break, (C) strain % at break, and (C) toughness of the GO/GP composite nanolaminates with error bars. | ||
Conclusions
In conclusion, GO/GP polymer composite laminates have been prepared through a facile and eco-friendly process via the vacuum filtration of aqueous solutions of GO/GP composites followed by drying at room temperature. For the first time, we report biocompatible, layered structure GO/GP biopolymer composite laminates that exhibit a significant enhancement of the stress at the break with a simultaneous enhancement of the elongation at the break as a result of controlling the morphology and crystal structure of the GP molecules, and interfacial H-bonding interactions between GP and the GO sheets in the layered structure GO/GP polymer composite laminates. The WAXS and DSC results demonstrate the tailoring of the crystals of GP in the GO/GP composite laminates, whereas the WAXS results and FESEM imaging show the formation of composite nanolaminates with layered structure morphologies, where GP molecules act as bridges between the GO sheets and also cement adjacent individual GO sheets. The WAXS results also show the entering of GO sheets into the GP crystal structure and a decrease in the interchain interactions between GP molecules, leading to the enhancement of the elongation at break. The interactions between GP and GO molecules are supported by H-bonding interactions, as shown in FTIR and Raman studies, causing significant enhancements of the tensile stress at break and toughness. The composite nanolaminate film containing 5 wt% GO shows a significant enhancement of the tensile strength of 200% with concurrent enhancement of the elongation at break of 52%. The ductility and toughness of the composite films are also dramatically improved. The composite films also exhibit good biocompatibility under in vitro conditions. The development of these biocompatible layered structure GO/GP composite nanolaminate films with dramatic enhancements of mechanical properties is very promising for the development of biobased consumer products, biobased structural composites, and biomedical engineering materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by an Academy of Finland postdoctoral project (project no. 345006) and Center of Excellence in Body-On-Chip Research (project no. 312409).References
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906 CrossRef CAS PubMed.
- R. K. Layek and A. K. Nandi, Polymer, 2013, 54, 5087 CrossRef CAS.
- C. J. Bullock and C. Bussy, Adv. Mater. Interfaces, 2019, 6, 1900229 CrossRef.
- Y.-Q. Li, T. Yu, T.-Y. Yang, L.-X. Zheng and K. Liao, Adv. Mater., 2012, 24, 3426 CrossRef CAS PubMed.
- G. W. Jeon, J.-E. An and Y. G. Jeong, Composites, Part B, 2012, 43, 3412 CrossRef CAS.
- K. Hu, D. D. Kulkarni, I. Choi and V. V. Tsukruk, Prog. Polym. Sci., 2014, 39, 1934 CrossRef CAS.
- X. Zhao, Q. Zhang, D. Chen and P. Lu, Macromolecules, 2010, 43, 2357 CrossRef CAS.
- H. Kim, A. A. Abdala and W. Macosko, Macromolecules, 2010, 43, 6515 CrossRef CAS.
- S. Song, Y. Zhai and Y. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 31264 CrossRef CAS.
- S. Wan and Q. Cheng, Adv. Funct. Mater., 2017, 27, 1703459 CrossRef.
- J. Huang, Z. Tang, Z. Yang and B. Guo, Macromol. Rapid Commun., 2016, 37, 1040 CrossRef CAS PubMed.
- H. Lu, Z. Chen and C. Ma, J. Mater. Chem., 2012, 22, 16182 RSC.
- K. W. Putz, O. C. Compton, M. J. Palmeri, S. T. Nguyen and L. Catherine Brinson, Adv. Funct. Mater., 2010, 20, 3322 CrossRef CAS.
- R. K. Layek, K. R. Ramakrishnan, E. Sarlin, O. Orell, M. Kanerva, J. Vuorinen and M. Honkanen, J. Mater. Chem. A, 2018, 6, 13203 RSC.
- J. Wang, J. Qiao, J. Wang, Y. Zhu and L. Jiang, ACS Appl. Mater. Interfaces, 2015, 7, 9281 CrossRef CAS.
- K. Chen, B. Shi, Y. Yue, J. Qi and L. Guo, ACS Nano, 2015, 9, 8165 CrossRef CAS PubMed.
- D. Shah, P. Maiti, E. Gunn, D. F. Schmidt, D. D. Jaing, C. A. Batt and E. P. Giannelis, Adv. Mater., 2004, 16, 1173 CrossRef CAS.
- P. Maiti, P. H. Nam, M. Okamato, N. Hasegawa and A. Usiki, Macromolecules, 2002, 35, 2042 CrossRef CAS.
- J. L. Gornall and E. M. Terentjev, Soft Matter, 2008, 4, 544 RSC.
- Q. He, Y. Zhang, X. Cai and S. Wang, Int. J. Biol. Macromol., 2016, 84, 153 CrossRef CAS PubMed.
- W. S. Hummers Jr. and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- V. Kubyshkin, Org. Biomol. Chem., 2019, 17, 8031 RSC.
- A. Bigi, S. Panzavolta and K. Rubini, Biomaterials, 2004, 25, 5675 CrossRef CAS PubMed.
- K. H. Kim, M. Yang, K. M. Cho, J. Y. L. S. Bok and H. T. Jung, Sci. Rep., 2013, 3, 3251 CrossRef PubMed.
- O. Akhavan, E. Ghaderi, E. Hashemi and R. Rahighi, Nanoscale, 2014, 6, 14810 RSC.
- J. Liang, Y. Huang, L. Zhang, Y. Wang, Y. Ma, T. Guo and Y. Chen, Adv. Funct. Mater., 2009, 19, 2297 CrossRef CAS.
- L. Liu, Y. Gao, Q. Liu, J. Kuang, D. Zhou, S. Ju, B. Han and Z. Zhang, Small, 2013, 9, 2466 CrossRef CAS PubMed.
- J. Wang, T. Tsuzuki, B. Tang, X. Hou, L. Sun and X. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 3084 CrossRef CAS PubMed.
- P. Díaz-Calderon, E. Flores, A. Gonzalez-Munoz, M. Pepczynska and F. Quero, J. Enrione, Food Hydrocolloids, 2017, 71, 118 CrossRef.
- R. K. Layek, A. K. Das, M. U. Park, N. H. Kim and J. H. Lee, J. Mater. Chem. A, 2014, 2, 12158 RSC.
- S. Stankovich, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Carbon, 2006, 44, 3342 CrossRef CAS.
- C. Li, J. Luo, Z. Qin, H. Chen, Q. Gao and J. Li, RSC Adv., 2015, 5, 56518 RSC.
- Y. Wang, R. Zhang, W. Qin, J. Dai, Q. Zhang, K. J. Lee and Y. Liu, Mater. Des., 2020, 185, 108277 CrossRef CAS.
- H. Feng, Y. Li and J. Li, RSC Adv., 2012, 2, 6988 RSC.
- (a) Y. Rao, Polymer, 2007, 48, 5369 CrossRef CAS; (b) S. Ghoshal, S. Stapf and C. Mattea, Appl. Magn. Reson., 2014, 45, 145 CrossRef CAS.
- R. K. Layek, A. Kundu and A. K. Nandi, Macromol. Mater. Eng., 2013, 298, 1166 CrossRef CAS.
- J. Shen, Y. Hu, C. Li, C. Qin, M. Shi and M. Ye, Langmuir, 2009, 25, 6122 CrossRef CAS PubMed.
- L. J. Cote, R. Cruz-Silva and J. Huang, J. Am. Chem. Soc., 2009, 131, 11027 CrossRef CAS PubMed.
- E. Chiellini, P. Cinelli, E. G. Fernandes, E. S. Kenawy and A. Lazzeri, Biomacromolecules, 2001, 2, 806 CrossRef CAS PubMed.
- F. Quero, A. Coveney, A. E. Lewandowska, R. M. Richardson, P. Díaz-Caldero, K. Lee, S. J. Eichhorn, M. Ashraf Alam and J. Enrione, Biomacromolecules, 2015, 16, 1784 CrossRef CAS PubMed.
- H. Zhao, J. Ding and H. Yu, Sci. Rep., 2018, 8, 16560 CrossRef PubMed.
- H. J. Salavagione, G. Martınez and M. A. Gomez, J. Mater. Chem., 2009, 19, 5027 RSC.
- S. Karnnet, P. Potiyaraj and V. Pimpan, Polym. Degrad. Stab., 2005, 90, 106 CrossRef CAS.
- S. Acosta, A. Jimenez, M. Chafer, C. Gonzalez-Martínez and A. Chiralt, Food Hydrocolloids, 2015, 49, 135 CrossRef CAS.
- M. G. A. Vieira, M. A. da Silva, L. O. dos Santos and M. M. Beppu, Eur. Polym. J., 2011, 47, 254 CrossRef CAS.
- M. L. Sanyang, S. M. Sapuan, M. Jawaid, M. R. Ishak and J. Sahari, Polymers, 2015, 7, 1106 CrossRef CAS.
- Y. L. Liu, B. Xie and Z. P. Xu, J. Mater. Chem., 2011, 21, 6707 RSC.
- A. Walther, I. Bjurhager, J. Malho, J. Pere, J. Ruokolainen, L. A. Berglund and O. Ikkala, Nano Lett., 2010, 10, 2742 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00334h |
| This journal is © The Royal Society of Chemistry 2021 |