
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA slow and sustained release of methotrexate (MTX) from a new polymeric dicalcium phosphate dehydrate cement (P-DCPD)
Rahul Vaidyaa,
Emily J Rena,
Tong Shib,
Angelica Gardia b and
Weiping Ren*b
b and
Weiping Ren*b
aDepartment of Orthopedics, Detroit Medical Center, Detroit, MI 48201, USA
bDepartment of Biomedical Engineering, Wayne State University, Detroit, MI 48201, USA. E-mail: as7606@wayne.edu
First published on 2nd June 2021
Abstract
A novel and injectable polymeric dicalcium phosphate dehydrate cement (P-DCPD) was developed that is mechanically strong and has excellent cohesion. Methotrexate (MTX) is an anticancer drug for many tumors including bone cancer. The current study assessed the performance of MTX loaded P-DCPD (MTX-P) in vitro. The applied properties of MTX-P include MTX release, effects on cell viability of human breast cancer MCF 7 cells and murine osteoblastic MC3T3 cells. The impacts of MTX-P on the injectability, setting time, material microstructure and mechanical strength were also evaluated. A slow and sustained MTX release from MTX-P was observed. Eluents collected from MTX-P were cytotoxic to MCF7 cells. MT3T3 cells, however, were much less sensitive to MTX-containing eluents. MTX-P was injectable. A remarkable delay of the setting time was observed in 0.5% MTX-P (45.7 ± 1.9 min) and 1% MTX-P (49.5 ± 3.4 min) as compared to control-P (11.8 ± 1.2 min), respectively. The compressive strength was significantly reduced in 0.5% MTX-P (16.49 ± 2.43 MPa) and in 1% MTX-P (10.68 ± 3.97 MPa) as compared to control-P (53.09 ± 8.64 MPa). The inferior performance of MTX-P observed might be due to the insufficient crystal formation and uneven distribution of MTX within P-DCPD matrices. Our data may shed some light on the future application of MTX-P in the treatment of bone defects after tumor excision with a plethora of applications by including other anticancer drugs because of its safety and degradability. Further material optimization and in vivo validation are required to achieve a clinically applicable product.
Introduction
Bone metastases can be found in up to 70% of the cancer patients.1,2 Increased long-term survival of patients with cancer also increases the risk of bone metastases and pathological fractures.3 The success of internal fixation of impending or pathologic fractures following metastasis has a favourable impact on the quality of life of the patient.4 Anticancer drug-loaded bone fillers have been used to fill bone defects after removal surgery,4,5 enhance implant stability, prevent local cancer progression and reconstructive device failure.6–8Poly(methyl methacrylate) (PMMA) bone cements are the most commonly used bone void fillers for the local delivery of anticancer drugs.8–10 While acceptable, PMMA possesses several major limitations,11,12 including limited and burst drug release,6,13 toxic monomer release,14 high exothermic reaction and not degradable.8–10 Calcium phosphate cement (CPC) has been widely used as bone void filler.15 The end products of commercial CPCs can be divided into two types: hydroxyapatite (HA) and dicalcium phosphate dehydrate (DCPD).15 DCPD cements have been proposed as an alternative to PMMA cements for the local delivery of anticancer drugs6,16 because of its injectability and slow degradation.17 Using CPC as local delivery of anticancer drugs is not clinically preferred18 because of some critical weaknesses, including poor cohesion,15 mechanically weak19 and burst drug release.20 Therefore, surgeons are actively looking for new bone void fillers that are biocompatible, mechanical strong, excellent anti-washout, and capable of delivering anticancer drugs in a sustained and controllable manner.
Polyphosphates are polyelectrolytes that play key roles in biological and material science.21 Calcium polyphosphate (CPP) is a polymeric inorganic ceramic composed of linear polyphosphate chains.22 We recently found that CPP hydrogel can be formed in the presence of water at optimized conditions.23 With the CPP gelation technology, we developed a new way to prepare injectable polymeric DCPD-forming cement (P-DCPD) by the reaction of CPP gel with tetracalcium phosphate (TTCP).24 The setting reaction of P-DCPD is initiated by the interaction of numerous ionic binding sites of disentangled polyphosphate chains of CPP gel with ions of calcium and orthophosphate released from TTCP dissolution.24 The setting mechanism of P-DCPD is completely different from that of classical CPCs as described by Brown et al. in 1980s.25 To the best of our knowledge, no ceramic cement using similar technologies has been reported before. P-DCPD represents a new bone void filler with significant advantages, including strong mechanically strength,26 excellent cohesion,24 as well as a mean of sustained drug delivery27,28 because of its unique ionic binding and intrinsic entanglement of polyphosphate chains with embedded drugs.29 The handling properties of P-DCPD is similar to PMMA cement and other commercially available CPCs. P-DCPD is mainly composed of calcium and phosphor and contains no cross-linkers or other additives. P-DCPD is expected to meet the unmet clinical needs as a better anticancer drug-eluting bone void filler for the load-bearing bone defects caused by bone cancer (primary and/or metastatic). Like DCPD, P-DCPD is osteoconductive and has similar mineral composition to that of native bone tissue.17,24,30
Methotrexate (MTX), an inhibitor of DNA synthesis, is one of the most commonly used anticancer drugs for human cancers including bone cancer (primary or secondary).31 However, its clinical efficacy is limited by its short plasma half-life32 and the risk of developing drug resistance.33 PMMA cements are not desirable carrier for local MTX delivery because of unsatisfactory clinical outcome34 caused by limited and burst drug release, non-degradability and the heating reaction during setting.35 Few studies have been conducted for the delivery of MTX via CPC system.36
The aim of this study was to determine the release of MTX from P-DCPD, and the influence of MTX loading on the cell viability of MCF 7 cells and osteoblastic MC3T3 cells. The impacts of MTX loading on the injectability, setting time and mechanical strength were also evaluated. In addition, the mechanisms of MTX release from P-DCPD cement were discussed.
Experimental
Materials
All chemicals used were of analytical grade and purchased from Sigma-Aldrich (USA) if not stated otherwise. Human breast cancer MCF-7 (ATCC® HTB22™) cell line and murine MC3T3-E1 pre-osteoblast cell line were purchased from ATCC (Manassas, VA). Alpha-modified minimum essential medium (α-MEM) and Dulbecco's modified Eagle's medium (DMEM) was purchased from Invitrogen (Carlsbad, CA). Methotrexate (MTX) and MTX assay ELISA kit were from Enzo Life sciences (Farmingdale, NY 11735).Preparation of P-DCPD and MTX loading
CPP gel was prepared as described elsewhere.23 The TTCP powder was prepared by solid-state reaction of dicalcium phosphate anhydrous (DCPA, CaHPO4) and calcium carbonate (CaCO3) (Sigma-Aldrich) by heating at 1500 °C for 18 h. P-DCPD was prepared by mixing CPP gel with TTCP (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.87, w/w) at room temperature for setting.24 P-DCPD was doped with MTX at a ratio of 0%, 0.5% and 1%, respectively (wt/wt), by adding MTX powders into CPP gel prior to mixing with TCPP for setting.
:0.87, w/w) at room temperature for setting.24 P-DCPD was doped with MTX at a ratio of 0%, 0.5% and 1%, respectively (wt/wt), by adding MTX powders into CPP gel prior to mixing with TCPP for setting.
Setting time and injectability
The setting time was recorded using standard Gillmore needle method.24 The injectability was measured using an ASTM F451-08 standard intrusion test mold.24MicroCT analysis
The porosity, pore size and pore size distribution of MTX-P scaffolds were analyzed by MicroCT (Scanco Viva CT 40, Scanco Medical, Switzerland) with a voltage of 70 kVp and a current of 114 µA at 10 µm resolution. A cylindrical volume of interest was selected (54 slices) for each scaffold, and an optimal threshold of 300 was determined. The microstructure of the scaffolds was determined using built-in software to measure the pore wall thickness (Tb.Th), pore size (Tb.Sp), porosity, pore size and distribution.Scanning electron microscopy
The morphology of MTX-P scaffold surface was characterized by scanning electron microscopy (SEM) (Quanta FEG450, FEI, Hillsboro, OR, USA). Scaffolds were coated with gold (Gold Sputter, Effa Coater, USA) before SEM analysis. Morphologies were viewed at 10 kV accelerating voltage.Mechanical testing
Cement cylinders (12 mm × 6 mm) were prepared using a steel mold. The surfaces of cement cylinders were smoothed using sandpaper prior to testing. Specimen were loaded into a universal servo-hydraulic test machine (Model 8521, Instron, Norwood, MA) and tested in axial compression at a speed of 0.1 mm s−1 until failure. The maximum compressive stress and Young's elastic modulus were calculated. Each cement group was measured in triplicate.Release of MTX and degradation of cement discs
Following curing, cement discs (5 mm × 6 mm) were submerged into 1.5 mL of deionized distilled water in sealed tubes and maintained at 37 °C. At predetermined time points, eluents were collected and replaced with the same amount of deionized distilled water. Eluents collected were frozen at −80 °C till use. At the completion of the release study, cement disc residues were dried, and total mass loss calculated. The disc weight before soaking was measured as W1, the disc weight after soaking was measured as W2, the weight loss: ΔW = (W1 − W2)/W2. The released MTX was measured by MTX ELISA kit (Endo Life) following manufacturer's instruction. The total percentage released was calculated based on the known initial MTX content. Each group consisted of 3 discs.Cell viability
MCF7 cell line was derived from a patient with breast cancer. MCF7 cell line has bone metastasis capability and is sensitive to MTX treatment. MC3T3 is a murine pre-osteoblastic cell line that has been used for cell biocompatibility evaluation. MCF 7 cells were cultured in DMEM medium, supplemented with 5% fetal bovine serum (FBS, Invitrogen), insulin and penicillin and streptomycin (Life Technologies, Gaithersburg, MD, USA). MC3T3 cells were cultured in α-MEM medium supplemented with 10% FBS, 10 mM β-Glycerophosphate (Sigma Aldrich), and antibiotics. Cells were seeded into 48-well plates at concentration of 1.0 × 105 per well. After cell attachment, eluents collected at different time points were passed through a syringe filter for sterilization, and 30 µl of the eluent was added into 270 µl culture medium (final 10%, v/v) and continue cell culture for three days. Cells cultured in the absence of eluent were included as controls. At the end of the culture, cell viability was evaluated by MTT (3-(4,5-Dimethylthiazol-2-yl)- 2,5-diphenyltetrazolium bromide) activity in cell lysates.Statistical analysis
All values are expressed as mean ± standard deviation. Data were analyzed by One-way Analysis of Variance (ANOVA) with post-hoc Tukey–Kramer test. Statistical significance was set to be p < 0.05.Results
Setting time, injectability and cement degradation
As shown in Table 1, the setting time was significantly extended in 0.5% MTX-P (45.7 ± 1.9 min) and 1% MTX-P (49.5 ± 3.4 min) as compared to control-P (11.8 ± 1.2 min), respectively (p < 0.05). Through MTX-P was injectable, but the injectability was reduced as compared to P-DCPD (p < 0.05). In addition, the loading of MTX at current concentration (0.5% and 1%, respectively) has little impacts on cement degradation rate (% of weight loss) 28 days after MTX elution study.SEM analysis
SEM was used to analyze the surface morphology of MTX-P scaffolds (Fig. 1D–F). As compared to control-P, MTX loading increased the surface roughness and microcracks.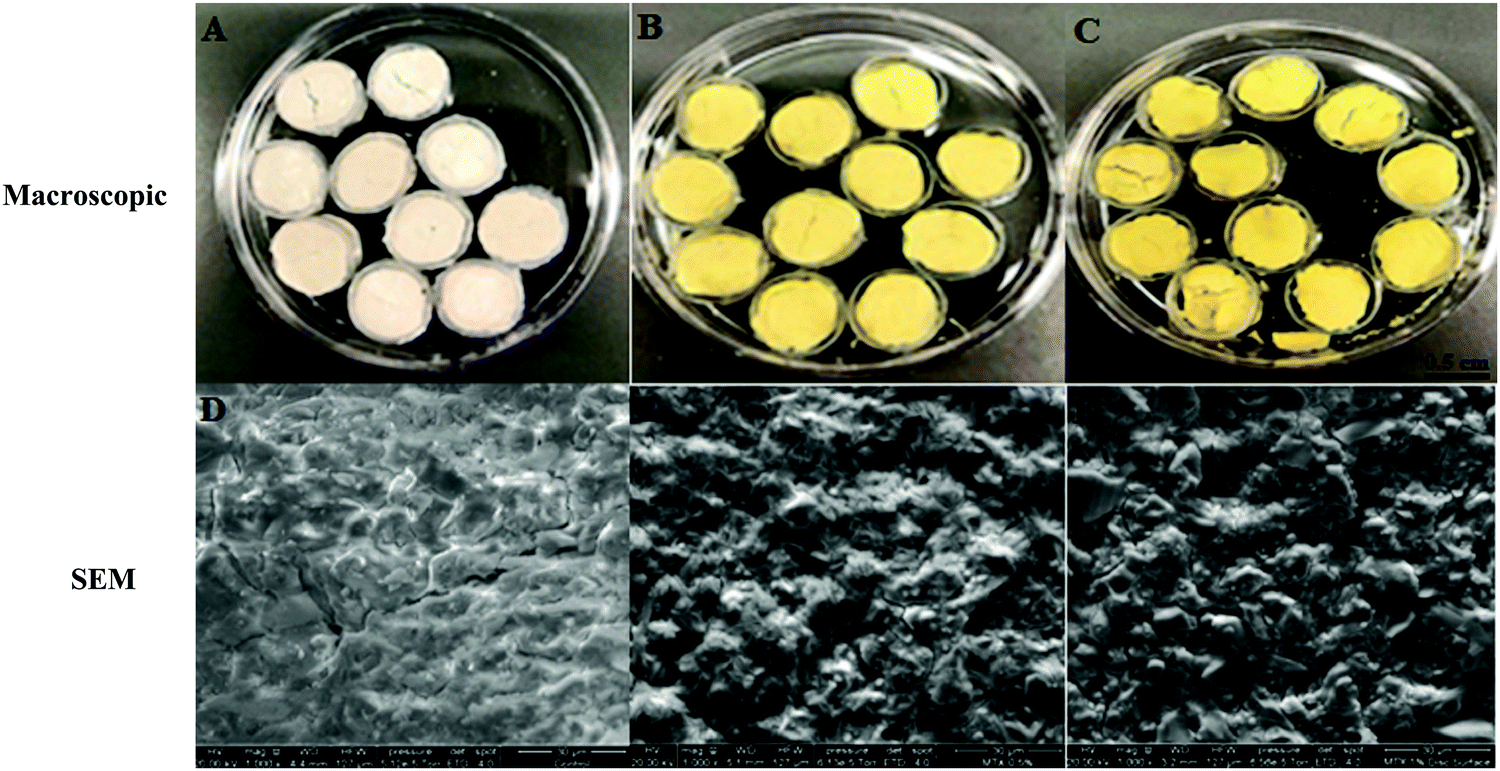 | ||
| Fig. 1 Microscopic and SEM (×1000) appearance of MTX-P scaffolds (A and D) Control-P; (B and E) 0.5% MTX-P, and (C and F) 1% MTX-P. | ||
MicroCT analysis
The representative images of 3D reconstructed P-DCPD scaffolds were shown in Fig. 6. Scaffolds were fabricated with an interconnective porous structure, with a pore size distribution ranging from <10–120 µm. MTX-loading changed both the PDCPD scaffold surface morphology (Fig. 1) and the inner pore characteristics. As shown in Table 2, the average pore sizes of 0.5% MTX-P (70.8 ± 38.3 µm) and 1% MTX-P (71.8 ± 21.1 µm) were slightly reduced, as compared with that of control-P (110 ± 60 µm). MTX loading also changed the pattern of pore size distribution (Fig. 2). There was a significantly lower percentage of pore size in the range of >90 µm for the MTX-P scaffold (29.84% and 29.14% for 0.5% MTX-P and 1% MTX-P, respectively) than control-P (59.43%, p < 0.05).| Sample | Pore size (µm) | Porosity (%) | Tb.Th (mm) | Tb.Sp (mm) |
|---|---|---|---|---|
| Control-P | 110 ± 60 | 1.62 ± 2.9 | 0.3 ± 0.27 | 0.06 ± 0.12 |
| 0.5% MTX-P | 70.8 ± 38.3 | 2.71 ± 1.71 | 0.27 ± 0.28 | 0.08 ± 0.07 |
| 1% MTX-P | 71.8 ± 21.1 | 2.66 ± 3.0 | 0.27 ± 0.02 | 0.07 ± 0.03 |
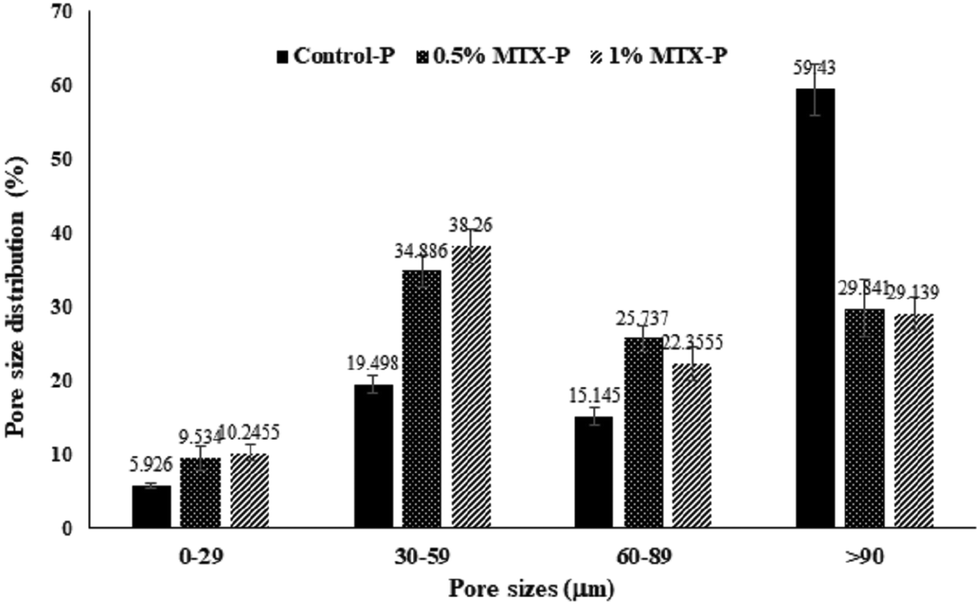 | ||
| Fig. 2 Pore size distribution of MTX-P scaffold from MicroCT analysis. The 3-D images of scaffolds were formed by overlapping 54 slices of scanned images. | ||
Mechanical strength
A significant reduction of the mechanical strength (16.49 ± 2.43 MPa) and Young's modulus (1700.75 ± 351.22 MPa) was observed in 0.5% MTX-P, as compared to control-P (53.09 ± 8.64 MPa and 3170.72 ± 165.37 MPa, respectively, p < 0.05), that was more apparent in 1% MTX-P (8.68 ± 6.98 MPa and 918.63 ± 609.34 MPa, respectively, Fig. 3).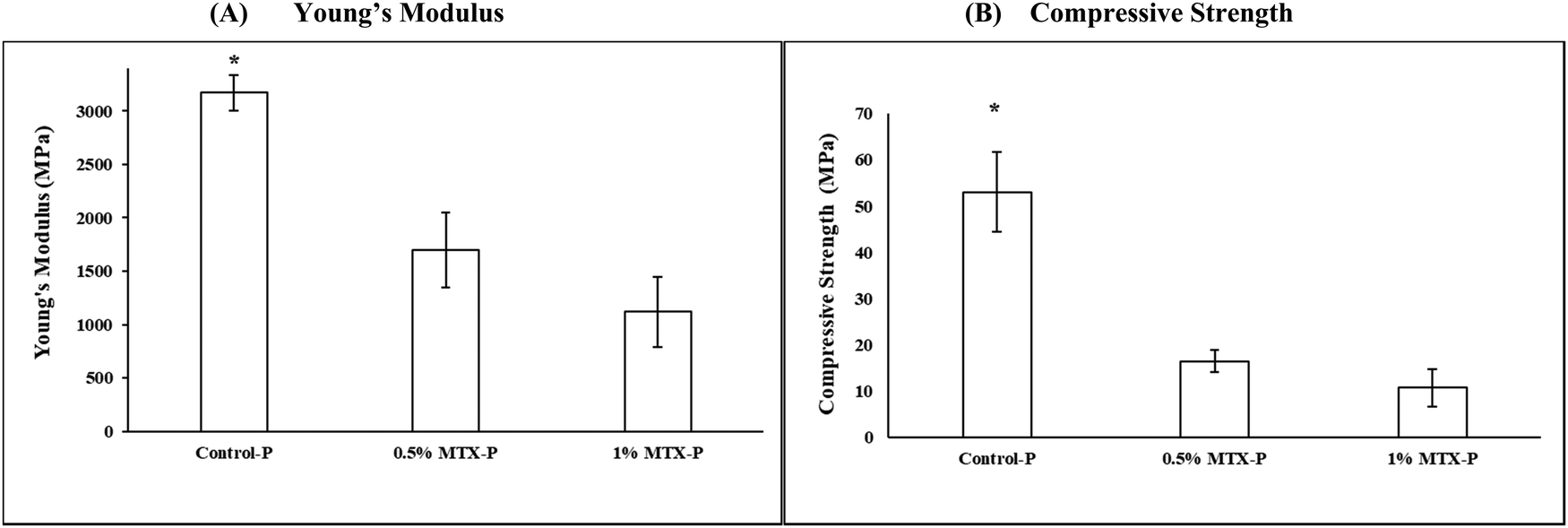 | ||
| Fig. 3 Mechanical strength measurement of (A) Young's Modulus and (B) Compressive strength. Statistical differences between drug free control (P) and both 0.5%MTX-P and 1%MTX-P were indicated by (*) (n = 3, p < 0.05). No statistical difference between 0.5%MTX-P and 1%MTX-P was found. | ||
MTX release
The release of MTX from P-DCPD up to 28 days was studied and the results shown in Fig. 4. A very low percentage (%) of MTX loaded was released from 0.5% MTX-P (1.03 ± 0.06%) that was higher than that from 1% MTX-P (0.52 ± 0.03%, p < 0.05). There was a statistically significant difference of the between the % of release of MTX between 0.5%MTX-P and 1%MTX-P at all given time points (Fig. 4A, p < 0.05). The absolute concentration of MTX released from 0.5%MTX-P was higher at the time of 24 h (3877 ± 170 ng ml−1) than that of 1% MTX-P (2394± 120 ng ml−1, p < 0.05). Starting from 48 h, the concentration of MTX released from 1% MTX-P was gradually increased, and a much higher MTX release was observed in 1%MTX- P in day 21 (553.38 ± 147.03 ng ml−1) and day 28 (526.21 ± 81.93 ng ml−1) as compared to 0.5%MTX-P (21.44 ± 3.48 ng ml−1 and 17.24 ± 4.02 ng ml−1, respectively, Fig. 4B, p < 0.05).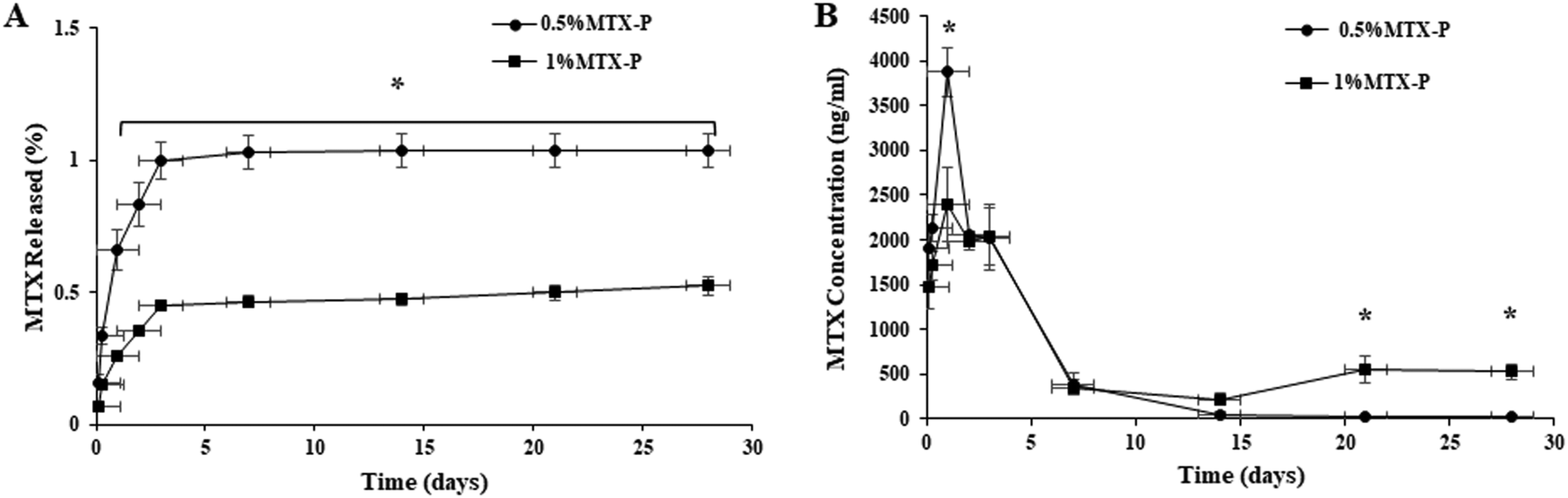 | ||
| Fig. 4 MTX release profiles (A) cumulative release (% of initial load) of MTX from MTX-P cement matrices over the course of 28 days. A statistical difference was found between 0.5%MTX-P and 1% MTX-P cement at all given time points, and (B) cumulative release (absolute concentration of MTX released) from MTX-P cement matrices over the course of 28 days. Statistical differences were found between 0.5%MTX-P and 1% MTX-P cement at 1 day, 21 and 28 days, respectively (n = 3, * p < 0.05). | ||
Cell viability
Eluents from control-P had no negative impacts on cell viability of both MCF 7 and MC3T3 cells. As shown in Fig. 5, eluents from both 0.5% MTX-P and 1% MTX-P significantly reduced cell viability of MCF 7 cells as compared to control-P (p < 0.05). However, the significant reduction of cell viability at day 21 and day 28 was only observed in eluents from 1% MTX-P because of much higher amount of MTX released (Fig. 4). Interestingly, MT3T3 cells were less sensitive to MTX-containing eluents. Eluents from MTX-P (both 0.5% and 1%) had no effects on cell viability of MC3T3 cells except on the time of day 1 and 2. The reduction of cell viability observed in these two time points might be due to the higher MTX concentration in the eluents (>2 µg ml−1, Fig. 4).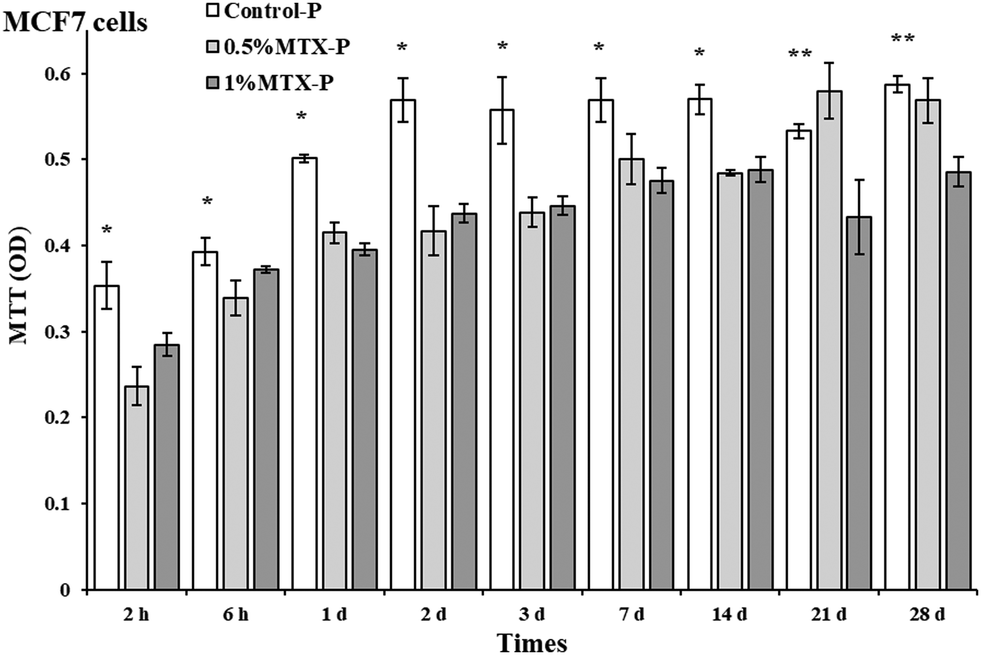 | ||
| Fig. 5 Cell viability (MTT) of MCF 7 cells and MC3T3 cells when exposed to 10% eluents collected from MTX-loaded P-DCPD cements at given times; control: eluents from MTX-free P-DCPD cements. Cells were cultured three days before tests. (n = 3, *p < 0.05 between control and MTX-P, ** between 0.5%MTX and 1%MTX). | ||
 | ||
| Fig. 6 Representative 3D rendering images of the Control-P, 0.5%MTX-P and 1%MTX-P scaffolds determined by micro-CT (voltage: 70 kVp; current: 114 µA). | ||
Discussion
A new injectable P-DCPD was developed in our lab that is mechanically strong and with excellent anti-washout capability.24 The key molecular structural difference between P-DCPD and classical DCPD is that P-DCPD is composed of interconnected P-DCPD crystals by interlocking to the polyphosphate chains, while DCPD is composed of a package of DCPD crystal particles with weak mutual bonding.24DCPD has been proposed as an alternative to PMMA for local drug delivery.18 Many studies have been aimed at improving DCPD properties capable of delivering drugs in a sustained pattern but with limited success.37 Yang et al.38 reported an initial burst release of MTX from CPCs doped with 0–1% MTX, and the release rate was faster in vivo than in vitro. An understanding of the elution profile of MTX from P-DCPD is essential for the prediction of its future in vivo performance. In this study, we showed a unique slow and sustained release of MTX from P-DCPD (Fig. 4), which is rarely seen in ceramic drug delivery system.18,39 The intermolecular interaction of entangled polyphosphate chains of CPP gel with loaded MTX plays a critical role in the MTX release profile observed.40 We propose that a slow and sustained MTX release observed might be due to an exclusive distribution of MTX within the P-DCPD matrix.41 Instead of reacting with CPP polyphosphate chains, MTX, with a small size (C20H22N8O5·H2O, MW: 454.5), is absorbed with calcium and phosphate ions dissolved and released from TTCP and then easily penetrated and integrated into formed crystalline lattice of P-DCPD crystals during setting.41 In addition, MTX is soluble only in alkaline solution and insoluble in weak acidic (pH 6.5–7.0 during setting) P-DCPD matrix. Therefore, a slow and sustained MTX release was mainly driven by the slower cement degradation accompanied by the opening of the pores within cement matrix. MTX embedded deep in cement matrix become more available for elution after dissolution of the P-DCPD in the presence of water penetration and when the pores or channels are formed during cement degradation. A sustained release of MTX for months at effective concentration right after MTX-P implantation is clinically desirable and is expected to provide a constant inhibition of tumor growth during bone remolding and repair. As a bone void filler, MTX-P is biodegradable and provides a dynamic interaction between degrading cement with surrounding new bone formation, that was not seen in PMMA cement.9 The potential therapeutic efficacy of MTX-P will be further tested in animal tumor model before clinical application.
We also noticed that the net MTX release was much higher from 0.5%MTX-P than that of 1% MTX-P, especially at the time of 24 h, followed by a gradually increased MTX release from 1% MTX-P that is more substantial after 14 days (Fig. 4). In vitro antibiotic release profiles can be affected by many factors including the dose of drug loaded.42 We proposed that the observed earlier “higher” release from 0.5% MTX-P might be due to the differences of surface roughness (Fig. 1), particle size distribution (Fig. 2) and degradation rate (Table 1) caused by different amount of MTX added.
Our data showed that eluents from control-P had no negative impacts on cell viability of both MCF 7 and MC3T3 cells. Eluents collected from both 0.5% MTX-P and 1% MTX-P were cytotoxic on MCF 7 cells (Fig. 5). However, only eluents from 1% MTX-P remained the cytotoxicity to MCF 7 cells after 14 days, indicating that the cytotoxicity observed was dose-dependent that was supported by the data of MTX release profile (Fig. 4). MT3T3 cells were less sensitive to MTX-containing eluents. The cell viability of MC3T3 cells were not affected by eluents from MTX-P unless in the eluents collected at day 1 and 2 (with higher MTX concentration >2 µg ml−1). Decker et al. had a similar finding for the MTX released from PMMA cement.43 The different cytotoxic sensitivity between two cell lines might be due to their difference of proliferation rate since MTX acts as an inhibitor of DNA synthesis.34 We propose that MTX-P is clinically desirable for the eradication of residual cancer cells at the margin of surgery resection on the very beginning, while has much less impacts on new bone formation. The potential therapeutic efficacy of MTX-P will be further tested in animal tumor model.
The unique distribution of MTX had inferior impacts on the setting time and mechanical strength of P-DCPD (Table 1). The setting time was significantly delayed in both 0.5% MTX-P (45.7 ± 1.9 min) and 1% MTX-P (49.5 ± 3.4 min). The mechanism might be due to that MTX reduced the diffusion efficiency of calcium and orthophosphates dissolved from TTCP, the first step for setting.24 It also can be caused by the integration of MTX within the P-DCPD crystals and/or the inhibition of P-DCPD crystal growth and morphological change of the formed crystals.24,44 It is critical to prepare a cement with a suitable setting time (around 10–20 minutes) so that it can set slowly enough for the surgeon to perform implantation but fast enough to prevent delaying the operation time. The cement should be moldable and injectable that enables minimally invasive application.45 Therefore, more efforts are needed to tailor many processing parameters such as cement composition, additives, porogens, particle size, drug loading amount, adjustment of gel/powder ratio, among others.15
Few studies have been conducted on the impacts of loading MTX on the mechanical strength of CPCs. Usually the mechanical strength of DCPD cements tends to decrease when increasing drug loading does.46–48 Yang et al.38 reported that the compressive strength of CPC was significantly decreased when 1% MTX was added, but Liao et al.36 described that adding 0.1% or 0.2% MTX to CPC had no impacts on the mechanical strength. In current study, a significant reduction of the mechanical strength (16.49 ± 2.43 MPa) was observed in 0.5% MTX-P, as compared to control-P (53.09 ± 8.64 MPa, p < 0.05), that was more obvious when the concentration of MTX was increased in 1% MTX-P (8.68 ± 6.98 MPa). We propose that the inferior mechanical strength observed in MTX-P was mainly caused by the insufficient crystal formation and uneven distribution of solid MTX within P-DCPD matrix.24 The inferior impacts of MTX loading on the mechanical strength of P-DCPD remains a key obstacle for the clinical application of load-bearing bone defects. Our current studies demonstrated that the property of the loaded drugs (molecular weight, functional groups and water solubility, etc.) plays a critical role in the mechanical performance of P-DCPD. For example, loading of erythromycin28 and/or tobramycin27 at high concentration (10% wt/wt) had little impacts on the mechanical performance of P-DCPD. Doxorubicin is one of the effective and broad spectrum anticancer drugs. Data from our current study showed that the handling property and mechanical performance of P-DCPD were negligibly changed by adding doxorubicin (5% wt/wt, unpublished data). Taken together, we propose that the performance improvements of P-DCPD should be achievable by screening of drug candidates with similar anticancer pharmacology besides the modification of cement processing parameters.15
As shown in Table 1, the in vitro degradation rate of control-P (15.54 ± 1.33%) was only marginally changed as compared to either 0.5%MTX-P (16.31 ± 1.72%) or 1% MTX-P (13.31 ± 2.15%). DCPD is biodegradable and known to possess the highest solubility among all CPCs at physiological conditions.49 Many studies have demonstrated that the degradation rate of DCPD was within a period of 8–52 weeks both in vitro and in vivo based on experiment settings.50–53 We49 currently demonstrated that P-DCPD has a more stable chemical structure than DCPD as evidenced by much less transformation to HA during setting and evolution. Importantly, P-DCPD has lower zeta potential and less hydrophilicity than that of DCPD because of its entangled and interconnected polyphosphate chains. It is expected that superhydrophilic DCPD undergoes faster dissolution than P-DCPD in an aqueous environment.
There are some limitations to this study. First, MTX-loading significantly reduced the mechanical strength of PDCPD and thus limited its potential application for the load-bearing bone defect filling. Second, MTX loading reduced the pore size. More than 70% of MTX-P was composed of pore size <90 µM, as defined by MicroCT measurement (Fig. 2 and Table 2). The potential impact(s) of MTX loading on cellular behaviors and bone ingrowths warrants further investigation. In addition, more extensive studies are needed to optimize MTX loading, setting time optimization, and drug release adjustment.
Conclusions
A slow and sustained MTX release from MTX-P was observed. MTX-P is biocompatible and biodegradable. Incorporation of MTX at current amount had inferior impacts on the setting time and mechanical strength of P-DCPD. We propose that the detrimental performance of P-DCPD observed in MTX-P was mainly caused by the insufficient crystal formation and uneven distribution of MTX within P-DCPD matrix. Our data may reveal the future application of MTX-P in the treatment of bone defects after tumor excision. Further material optimization and in vivo validation are required to achieve a clinically applicable product.Author contributions
Rahul Vaidya: Conceptualization, methodology; Emily J Ren: Investigation, formal analysis; Tong Shi: Investigation; Angelica Gardia: Investigation, writing–original draft; Weiping Ren: Supervision, validation, writing-review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Mrs Paula Dietz and Dr Yawen Li for the assistance of MicroCT and SEM data analysis.References
- A. S. Gdowski, A. Ranjan and J. K. Vishwanatha, J. Exp. Clin. Cancer Res., 2017, 36, 108 Search PubMed.
- E. Verron, H. Schmid-Antomarchi, H. Pascal-Mousselard, A. Schmid-Alliana and J.-C. Scimeca, Drug Discovery Today, 2014, 19, 1419–1426 Search PubMed.
- A. Schmid-Alliana, H. Schmid-Antomarchi, R. Al-Sahlanee, P. Lagadec, J.-C. Scimeca and E. Verron, Int. J. Mol. Sci., 2018, 19, 148 Search PubMed.
- M. Levy, T. Smith, A. Alvarez-Perez, A. Back and J. N. Baker, J. Natl. Compr. Cancer Network, 2016, 14, 82–113 Search PubMed.
- A. Myoui and H. Yoshikawa, Clinical calcium, 2008, 18, 1767–1773 Search PubMed.
- I. Pountos and P. V. Giannoudis, Expert Rev. Med. Devices, 2018, 15, 301–311 Search PubMed.
- H. Futani, S. Kamae, K. Atsui, K. Yoh, H. Tateishi and S. Maruo, J. Orthop. Sci., 2002, 7, 262–266 Search PubMed.
- H. Futani, S. Kamae, K. Atsui, K. Yoh and H. Tateishi, J. Orthop. Sci., 2002, 7, 262–266 Search PubMed.
- G. Maccauro, A. Cittadini, M. Casarci, F. Muratori, D. De Angelis, C. Piconi, M. A. Rosa, A. Spadoni, M. Braden and A. Sgambato, J. Mater. Sci.: Mater. Med., 2007, 18, 839–844 Search PubMed.
- G. W. Fröschle, J. Mahlitz, H. U. Langendorff, E. Achilles and J. Pollock, Anticancer Res., 1997, 17, 995–1002 Search PubMed.
- H. Katagiri, K. Sato, M. Takahashi, H. Sugiura, S. Yamamura and H. Iwata, Archives of Orthopaedic and Trauma Surgery, 1997, 116, 329–333 Search PubMed.
- R. Król and S. Radomski, Ortopedia, traumatologia, rehabilitacja, 2003, 5, 319–326 Search PubMed.
- S. Wasserlauf, A. Warshawsky, R. Arad-Yelin, Y. Mazur and R. Salama, Bull. Hosp. Jt. Dis., 1993, 53, 68–74 Search PubMed.
- W. Song, J. Seta, M. K. Eichler, J. J. Arts and B. M. Boszczyk, J. Biomed. Mater. Res., Part B, 2018, 106, 2693–2699 Search PubMed.
- J. Zhang, W. Liu, V. Schnitzler, F. Tancret and J. M. Bouler, Acta Biomater., 2014, 10, 1035–1049 Search PubMed.
- Y. Tanzawa, H. Tsuchiya, T. Shirai, H. Nishida and K. Hayashi, J. Orthop. Sci., 2011, 16, 77–84 Search PubMed.
- M. Gallo, S. Tadier, S. Meille, L. Gremillard and J. Chevalier, Acta Biomater., 2017, 53, 515–525 Search PubMed.
- D. Arcos and M. A. Vallet-Regí, Acta Mater., 2013, 61, 890–911 Search PubMed.
- E. Van Lieshout, G. Van Kralingen, Y. El-Massoudi, H. Weinans and P. Patka, BMC Musculoskeletal Disord., 2011, 12, 34 Search PubMed.
- P. J. Jiang, S. Patel, U. Gbureck, R. Caley and L. M. Grover, J. Biomed. Mater. Res., Part B, 2010, 93, 51–58 Search PubMed.
- X. Wang, H. C. Schroeder and W. E. G. Mueller, Biotechnol. J., 2016, 11, 11–30 Search PubMed.
- R. M. Pilliar, M. J. Filiaggi, J. D. Wells, M. D. Grynpas and R. A. Kandel, Biomaterials, 2001, 22, 963–972 Search PubMed.
- W. Song, J. Seta, R. Kast, G. Auner, L. Chen, D. Markel and W. Ren, J. Am. Ceram. Soc, 2015, 98, 3758–3769 Search PubMed.
- W. Ren, W. Song, S. Yurgelevic and D. C. Markel, J. Mech. Behav. Biomed. Mater., 2018, 79, 226–234 Search PubMed.
- W. E. Brown and L. C. Chow, Int. Am. Assoc. Dent. Res, 1983, 62, 672 Search PubMed.
- A. Criado, S. Yokhana, T. Rahman, S. McCarty, C. Andrecovich, W. Ren and W. K. Yassir, Spine Deformity, 2020, 8, 165–170 Search PubMed.
- E. J. Ren, A. Guardia, T. Shi, P. Begeman, W. Ren and R. Vaidya, Biomed. Mater., 2021, 16, 025019 Search PubMed.
- A. Guardia, T. Shi, T. Bou-Akl, P. Dietz, B. Wu, W. Ren and D. Markel, J. Orthop. Res., 2021, 1–9 Search PubMed.
- Z. Zhou, J. Seta, D. C. Markel, W. Song, S. M. Yurgelevic, X. W. Yu and W. Ren, J. Biomed. Mater. Res., Part B, 2018, 106, 2827–2840 Search PubMed.
- U. Tariq, R. Hussain, K. Tufail, Z. Haider and R. Tariq, Mater. Sci. Eng., C, 2019, 103, 109863 Search PubMed.
- S. S. Abolmaali, A. M. Tamaddon and R. Dinarvand, Cancer Chemother. Pharmacol., 2013, 71, 1115–1130 Search PubMed.
- N. Limelette, M. Ferry, S. Branger, A. Thuillier and C. Fernandez, Ther. Drug Monit., 2003, 25, 81–87 Search PubMed.
- J.-X. Zheng, H. Jiang, L.-P. Xiao, S.-S. Wang and X.-D. Li, Int. J. Clin. Exp. Pathol., 2017, 10, 368–376 Search PubMed.
- E. Prochazka, T. Soukup, M. Hroch, L. Fuksa, E. Brcakova, J. Cermanova, G. Kolouchova, K. Urban, J. Mokry and S. Micuda, International Orthopaedics, 2010, 34, 137–142 Search PubMed.
- K. Anagnostakos, O. Furst and J. Kelm, Acta Orthop., 2006, 77, 628–637 Search PubMed.
- G. Liao, D. Sun, J. Han and J. Tan, Orthopedics, 2014, 37, e906–e910 Search PubMed.
- M. P. Hofmann, A. R. Mohammed, Y. Perrie, U. Gbureck and J. E. Barralet, Acta Biomater., 2009, 5, 43–49 Search PubMed.
- Z. Yang, J. Han, J. Li, X. Li and Z. Li, Orthopedics, 2009, 32, 27 Search PubMed.
- S. M. H. Dabiri, A. Lagazzo, B. Aliakbarian, M. Mehrjoo, E. Finocchio and L. Pastorino, J. Biomed. Mater. Res., Part A, 2019, 107, 2063–2075 Search PubMed.
- Y. Chehreghanianzabi, R. Barua, T. Shi, S. Yurgelevic, G. Auner, D. C. Markel and W. Ren, J. Biomed. Mater. Res., Part B, 2020, 108, 475–483 Search PubMed.
- M. P. Ginebra, C. Canal, M. Espanol, D. Pastorino and E. B. Montufar, Adv. Drug Delivery Rev., 2012, 64, 1090–1110 Search PubMed.
- H. van de Belt, D. Neut, D. R. A. Uges, W. Schenk and H. van, Biomaterials, 2000, 21, 1981–1987 Search PubMed.
- S. Decker, W. Winkelmann, B. Nies and F. van Valen, J. Bone Jt. Surg., Br. Vol., 1999, 81-B, 545–551 Search PubMed.
- M. P. Ginebra, T. Traykova and J. A. Planell, J. Controlled Release, 2006, 113, 102–110 Search PubMed.
- K. L. Low, S. H. Tan, S. H. S. Zein, J. A. Roether, V. Mourio and A. R. Boccaccini, J. Biomed. Mater. Res., Part B, 2010, 94, 273–286 Search PubMed.
- M. H. Alkhraisat, C. Rueda, J. Cabrejos-Azama, J. Lucas-Aparicio, F. T. Marino, J. Torres Garcia-Denche, L. B. Jerez, U. Gbureck and E. L. Cabarcos, Acta Biomater., 2010, 6, 1522–1528 Search PubMed.
- M. Takechi, Y. Miyamoto, K. Ishikawa, M. Nagayama and M. Kon, J. Biomed. Mater. Res., 1998, 39, 308–316 Search PubMed.
- Y. Huang, C. S. Liu, H. F. Shao and Z. J. Liu, Key Eng. Mater., 2000, 192–195, 853–860 Search PubMed.
- R. Barua, C. S. Daly-Seiler, Y. Chenreghanianzabi, D. Markel, Y. Li, M. Zhou and W. Ren, J. Biomed. Mater. Res., Part B, 2021, 1–12 Search PubMed.
- J. M. Kuemmerle, A. Oberle, C. Oechslin, M. Bohner and C. Frei, J. Cranio-Maxillofacial Surg., 2005, 33, 37–44 Search PubMed.
- S. Shadanbaz, J. Walker, T. Woodfield, M. Staiger and G. Dias, J. Mater. Sci.: Mater. Med., 2014, 25, 173–183 Search PubMed.
- F. Tamimi, Z. Sheikh and J. Barralet, Acta Biomater., 2012, 8, 474–487 Search PubMed.
- Z. Xia, L. M. Grover, Y. Huang, L. E. Adamopoulos and U. Gbureck, Biomaterials, 2006, 27, 4557–4565 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |