
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagnetic hydroxyapatite nanomaterial–cyclodextrin tethered polymer hybrids as anticancer drug carriers†
Sivaraj
Ramasamy
a,
Dinesh
Dhamecha
b,
Kiruthiga
Kaliyamoorthi
c,
Archana Sumohan
Pillai
d,
Aleyamma
Alexander
d,
Premnath
Dhanaraj
e,
Jyothi U.
Menon
 *b and
Israel V. Muthu Vijayan
Enoch
*d
*b and
Israel V. Muthu Vijayan
Enoch
*d
aFunctional polymers and Materials Laboratory, Department of Smart Green Technology and Engineering, Pukyong National University, South Korea
bDepartment of Biomedical and Pharmaceutical Sciences, College of Pharmacy, University of Rhode Island, Kingston, RI 02881, USA. E-mail: jmenon@uri.edu
cDepartment of Chemistry and Department of Chemical Engineering, Hindustan Institute of Technology and Science, Chennai 603103, Tamil Nadu, India
dCentre for Nanoscience and Genomics, Karunya Institute of Technology and Sciences, Coimbatore 641114, Tamil Nadu, India. E-mail: israelenoch@karunya.edu
eDepartment of Biotechnology, Karunya Institute of Technology and Sciences, Coimbatore 641114, Tamil Nadu, India
First published on 23rd March 2021
Abstract
Osteosarcoma, the most common bone cancer, leads to a poor survival rate of patients. Drug targeting employing hydroxyapatite (HAp)-based nanocarriers represents a fascinating choice for non-invasive treatment of osteosarcoma. Herein, we report strontium-doped (Sr-HAp) and iron- and strontium-co-doped (Sr,Fe-HAp) hydroxyapatite nanoparticles as novel materials that deliver doxorubicin to bone cancer cells. A platinum-complexed and cyclodextrin-functionalized chitosan derivative is utilized to coat the NPs. Sr-HAp (aspect ratio ∼20) and Sr,Fe-HAp (aspect ratio ∼3) nanoparticles are formed as nanowhiskers and nanorods, respectively, as revealed by transmission electron microscopy. Strontium ferrite NPs are synthesized and their properties are compared with those of the Sr/Sr,Fe-doped HAp NPs. These ferrite NPs show ferromagnetic behavior, as opposed to Sr-HAp and Sr,Fe-HAp. The latter two respectively display paramagnetic and superparamagnetic behaviors. The loading percentage of the anticancer drug, Doxorubicin (Dox), in the nanocarriers is high and the release of Dox is sustained at physiological pH. The Dox-loaded nanocarriers are tested for their in vitro cytotoxicity against lung, cervical, liver, and bone cancer cell lines. In general, the efficacy of Dox is not diminished on loading in the nanocarriers. In addition, the Dox-carriers demonstrate a time- and dose-dependent cytotoxicity. The efficacy is enhanced in the case of Dox-loaded carriers on MG-63 (osteosarcoma) cell lines. The anticancer activity is tested in vivo on both male and female albino mice. Enhanced chemotherapeutic potential is observed for Dox-loaded Sr,Fe-HAp in a metastatic model of MG-63. The platinum derivative polymer possesses its own therapeutic effect and contributes to the general activity. The novel polymer-HAp nanohybrid represents an effective nanocarrier for the treatment of osteosarcoma.
Introduction
Cancer is a deadly disease capable of invading or spreading from the site of origin to other parts of the body. From the estimated 16 million cancer survivors in the US in 2021, the number is likely to elevate to 22.1 million by 2030.1 Lung, cervical, liver, and bone cancers are a few of the most common ones diagnosed. Finding an effective treatment for cancer depends on improving the efficacy of chemotherapeutic drugs by transporting them using nanocarriers to the target site. It aims at enabling the targeted delivery of anticancer drugs through enhanced bioavailability.2 For enabling targeted delivery of anticancer drugs, smart nanocarriers based on several types of nanomaterials, namely, oxides, ferrites, and composites, have been attempted. The requisite characteristics of the nanomaterials to act as drug carriers are as follows: biocompatibility, response to an external stimulus, suitable size and shape, enhanced uptake, and capacity to accommodate drug molecules.Bone cancer is a prevalent cancer posing a poor long-term survival rate. Osteosarcoma is the most common type of bone cancer. It is necessary to diagnose bone metastasis early because the patients do not sense pain or lump of the tumor. In addition, metastatic bone lesions are driven by breast cancer. This is difficult to overcome with surgical intervention or adjuvant radiotherapy.3 The bone tissue carries the bone-forming cells, known as osteoblasts, abundantly within them and secretes hydroxyapatite (HAp) for the construction of the bone matrix.3 Therefore, targeting osteoblasts through stereotactic techniques utilizing HAp is intriguing in light of their uniqueness and ability to abrogate cancer growth and invasiveness.4
HAp, Ca10(PO4)6(OH)2, is one of the noteworthy choices as a drug carrier, because of its compositional and structural proximity to mineralized tissues.5 It is a material that naturally crystallizes in a sequential process during the process of bone formation.6 HAp NPs in combination with chitosan have been employed as scaffolds for engineering bone tissues.7 HAp has been employed as a non-immunogenic transfection agent owing to its cytophilicity and bio-resorbability.8 Size miniaturization of HAp, modifying the surface area-to-volume ratio and altering the surface roughness lead to materials possessing desired physicochemical properties and mimicking the native tissues in terms of hierarchical structure.9 Therefore, HAp represents a good choice as a biocompatible material that can be utilized for designing drug delivery vehicles.
To employ HAp NPs as smart nanocarriers that respond to a magnetic field, magnetic elements are incorporated into it, substituting Ca2+ ions of the lattice.10 Substituting with elemental iron can serve this purpose. Iron-substituted, engineered HAp NPs do not affect the activity of healthy osteoblasts.11 It is well-known that the presence of Fe2+ in hard tissues does not disturb the structure of HAp. The incorporation of Fe2+ into the crystal of HAp lowers its crystallinity.12 Magnetic Fe-HAp NPs do not show toxicity on the fibroblasts of mouse embryo (3T3).13 Doping Fe2+/Fe3+ with HAp facilitates better cellular internalization and magnetic field-guided transport.14 Furthermore, the incorporation of superparamagnetic NPs into HAp renders them stability in a single phase up to a considerable high temperature and retains their strong magnetism.15 Although there have been attempts by researchers to incorporate Fe into HAp, the mechanisms governing the magnetic characteristics of doped HAps in correlation with their structure are not investigated in-depth.16 Besides iron, another intriguing choice for infusion in HAp as a dopant is strontium. It is an essential and trace element with nutritional values and anticancer properties. Cancers that have spread to the bone can be treated with radioactive strontium.17 Administration of radioactive strontium chloride causes regression of cancer metastasis from breast cancer.18,19
Several nanocarriers consist of an inorganic NP core and a polymer sheath around it. The polymer controls the stability, biocompatibility, and circulation half-life of the nanocarrier. Furthermore, it can be used to encapsulate the therapeutic drug. Value addition to the polymer can be achieved by imparting certain structural provisions to (i) accommodate the drug by means of non-covalent interactions, (ii) pharmacophoric functionalities for added therapeutic efficiency, and (iii) chemical derivatization with targeting ligands like folate units.
To realize the above-mentioned design, the β-cyclodextrin (β-CD)-functionalized polymer is an appreciable choice. β-CD-based polymers have demonstrated pharmaceutical and biomedical applications including the transport of drugs that are complexed by host:guest-type of association.20 β-CD consists of seven α-D-glucopyranose units that form a truncated cone-shaped molecule. Its hydrophobic cavity holds space to engulf organic drug molecules of appropriate size.20 Hydrophobic and van der Waals interactions contribute largely to the driving force for the formation of host:guest complexes by β-CD.20 Therefore, β-CD was chosen as a host molecule and tethered with the biocompatible polymer, i.e., chitosan. Chitosan (Chi) is a cationic polymer derived from natural chitin. It is non-toxic and carries D-glucosamine and N-acetyl-D-gluocosamines. Chi has been employed in drug delivery systems.21 Furthermore, Chi has the ability to form metal complexes.22 These complexes interact with DNA and display suppression of the growth of the tumor. In addition, other apoptotic pathways for cells have been reported.23 Of various types of metal complexes, platinum complexes are noteworthy that are effective against various forms of cancer.23 Therefore, we complexed Pt with the polymer. In addition to the above-mentioned structural design strategies, tethering a folate unit is an additional requirement to enable receptor-mediated cell internalization. Cancer cells are overexpressed with folate receptors and actively take up folate-derivatized molecules.24 Thus, folate modification is considered as a Trojan horse strategy of folate-targeted chemotherapy and involves delivery of therapeutic molecules into living cells through folate-receptor-mediated endocytosis.24 Interlinking β-CD and Chi is a clever strategy to attain the first one of the three-prongs of design-strategy discussed in the previous paragraph. Formation of the Chi–Pt complex meets the second requirement for the foreseen smart carrier. Chemical tethering of a folate unit to the Chi polymer presents the third prong of the designed carrier.
The need for Fe- and Se-incorporated HAp and the design of smart polymeric material in the above line of discussion prompt to develop nanocarriers with a HAp core and a Chi-based polymeric coating. In this paper, we discuss novel hybrids of strontium ferrite (SrFe), Sr-doped HAp (Sr-HAp), and Sr- and Fe-doped HAp (Sr,Fe-HAp) NPs with chitosan-β-CD-platinum-folate as a carrier for doxorubicin (a model anticancer agent) anticancer drug carriers. Doxorubicin (Dox), a popular chemotherapeutic drug, known as anthracycline, is active against cancer cells by slowing or stopping their growth by blocking topoisomerase 2, an enzyme that is essential for cancer cells to divide and grow. Therefore, we apply Dox as a model drug in this work.
Results and discussion
Synthesis of coating polymer
The chemical structure of Chi-CD-fol is presented in Scheme 1. To synthesize the derivative of chitosan, a tosylation reaction between β-CD-folate and chitosan was carried out. This step was followed by the conjugation of the aminoethylamino group of the β-CD with the carboxylic acid group of folate through a coupling aided by dicyclohehylcarbodiimide. The Chi-CD-fol product was purified by gel permeation chromatography (MW: 44.8 kDa). The IR spectra of the Chi-CD-Pt-fol were recorded (see the ESI,† S1). The major and essential information derived from the IR spectra are (i) the appearance of vibration frequencies corresponding to the amide carbonyl group of the folate unit and (ii) the Pt–N bond in the case of Chi-CD-Pt-fol. The amide bond through the conjugation of folate and carboxyl group of Chi-CD shows absorption bands at 1656 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O str.) and 3465 cm−1 (NH str.). These bands get shifted to a shorter wavenumber in the Pt-complexed form, i.e., Chi-CD-Pt-fol. In addition, the Pt–N str. is observed at 540 cm−1. It shows evidence for the formation of the Pt complex. To investigate the structure of the compound Chi-CD-fol in detail, 1H and 13C NMR spectra were recorded. The proton and carbon signals confirm the structure of the compound. The interpretation of the spectra is presented in detail in the ESI,† Fig. S2A and S2B.
O str.) and 3465 cm−1 (NH str.). These bands get shifted to a shorter wavenumber in the Pt-complexed form, i.e., Chi-CD-Pt-fol. In addition, the Pt–N str. is observed at 540 cm−1. It shows evidence for the formation of the Pt complex. To investigate the structure of the compound Chi-CD-fol in detail, 1H and 13C NMR spectra were recorded. The proton and carbon signals confirm the structure of the compound. The interpretation of the spectra is presented in detail in the ESI,† Fig. S2A and S2B.
 | ||
| Scheme 1 The chemical structure of Chi-CD-fol. | ||
Synthesis of magnetic ferrite and hydroxyapatites
The nanomaterials were synthesized, as explained in the ‘Experimental’ section, employing a hydrothermal method from the nitrate precursors of strontium and iron. Fig. 1A(a–c) depicts the XRD patterns of Sr-Fe, Sr-HAp, and Sr,Fe-HAp NPs. The sharp peaks observed for Sr-Fe are due to its pronounced crystallinity. The diffractions corresponding to the crystal planes, namely, (2 2 0), (0 0 2), (2 4 1), (4 2 0), and (5 2 0), appear at 2θ positions of 24.2, 32.8, 33.9, 46.3, and 54.4° respectively. Apparently, these peaks arise from the spinel ferrite structure possessing a hexagonal crystal system. In addition, the XRD represents a pattern similar to PDF No. 48-0156 of JSPDS data. The determined lattice parameters of the SrFe2O4 are a = 8.032, b = 18.209, and c = 5.453 nm. The peaks appear considerably broad, i.e., ∼2° (2θ), suggesting a small size of the crystallites. The crystallite size was calculated employing the Scherrer equation assuming a spherical shape of similar diameter as follows: | (1) |
 | ||
| Fig. 1 A. X-ray diffractograms of (a) Sr–Fe, (b) Sr-Hap, and (c) Sr,Fe-HAp NPs. The TEM images of Sr,Fe-, Sr-HAp-, and Sr,Fe-HAp- NPs are displayed in panels B, C, and D respectively. The morphology of the NPs is altered at the substitution of elements in HAp. | ||
The characteristic peaks of Sr–Fe are observed in the XRD of Sr–Fe-Hap, particularly those due to the reflection planes, namely, (0 0 2), (2 4 1), and (4 2 0). The additional peaks arising from the HAp component are indicated in Fig. 1A(c). The broad (2 1 1) and the accompanying (3 0 0) peaks are the characteristics of the crystal planes of HAp.25 The other obvious peaks arise from the (2 1 0), (2 2 2), and (3 1 0) planes. The doping of magnetic ferrite with HAp is identified from the XRD.
The dimensions and morphologies of the Chi-CD-Pt-fol-coated Sr–Fe, Sr-HAp, and Sr–Fe-HAp were determined using recording bright-field TEM images of the samples (shown in Fig. 1B-D). The Sr–Fe NPs are perceived as single-crystalline distorted hexagonal structures. The size of the NPs is in the range of 90 (±20) nm and 60 (±10) nm across, as observed from the TEM images. Due to the high and low contrasts respectively of the Sr–Fe NPs and Chi-CD-Pt-fol between them and the grid, the polymer coating is not clearly seen on the nanostructures. The size and shape of the Sr-HAp NPs are strikingly different from those of the Sr–Fe NPs as shown in Fig. 1C. The structure is formed as nanowhiskers of length 160 (±20) nm and width 8 (±2) nm. The polymer coating is observed in this nanostructure. Furthermore, the Sr–Fe-HAp appears as straight and thicker nanorods, 120 (±20) nm long and 40 (±10) nm wide, as shown in Fig. 1C. In addition, these nanorods are relatively homogeneous and appreciably regular in shape. Nanoparticles of undoped and Sr-doped HAp have been reported in the literature, as possessing rod shapes of varying lengths and breadths.26–29 In these reports, either the Sr-HAp is larger or the size is of a broader distribution than the NPs reported here. The ∼8 nm wide Sr-HAp in the present case is much smaller than the previously reported ones. The literature reports present various procedures for the synthesis of NPs, namely, sonochemical,26 hydrothermal,27 and reflux28 methods. While different methods can yield various sized NPs, the common formation of rod-shaped structures depends on the precursors and saturation of the reaction systems.30 Similarly, the Sr–Fe-HAp reported here for the first time possesses a width of ∼40 nm. The sizes of all the synthesized NPs, namely, Chi-CD-Pt-fol-coated Sr–Fe, Sr-HAp, and Sr–Fe-Hap, fall in the size range of 10–200 nm that is appropriate for targeted drug delivery.31,32 In addition, rod-shaped NPs, compared to spherical ones, are better suitable for selection as drug-delivery vehicles owing to their high aspect ratio and hence the larger circulation time.33 The Sr-HAp and Sr–Fe-HAp NPs possess aspect ratios of ∼20 and 3, respectively, revealing themselves as good candidates for targeted delivery.
Fig. 2 depicts the FT-IR spectra of various Chi-CD-Pt-fol-coated NPs. In each of the spectra of the coated NPs, the CO str. band is centered at 1626 cm−1 that has shifted to a lower wavenumber compared to that of the as-synthesized polymers (discussed in the ‘Materials’ section). In addition to the bond vibrations corresponding to the Chi-CD-Pt-fol and the Pt–N str. (546–560 cm−1), the bond stretching of CO32− is observed at 1450 cm−1 in the case of Sr-HAp. In both the spectra of Sr-HAp and Sr–Fe-HAp, the PO43− vibration bands2 are observed at 1159 and 1120 cm−1, respectively. The 643 and 662 cm−1 bands corresponding to the PO43− vibrations are apparently seen. These results suggest the formation of the NPs and the intact coating of Chi-CD-Pt-fol on the surface. The IR spectral band of CO str. in the case of Chr-CD-Pt-fol at 1640 cm−1 (S1) shifts to a smaller wavenumber, and a broader band centered at 1626 cm−1 is observed (Fig. 2) for the polymer-coated NPs. This implies that the carbonyl group of chitosan is involved in the physical adsorption of the polymer on the NPs.
 | ||
| Fig. 2 FT-IR spectra of (a) Sr,Fe-HAp, (b) Sr-HAp, and (c) Sr–Fe NPs. | ||
The Chi-CD-Pt-fol-coated Sr–Fe, Sr-HAp, and Sr,Fe-HAp NPs were subjected to thermal degradation at a heating rate of 30 °C per minute. The TG curves are displayed in Fig. 3A–C. The initial loss of weight is due to evaporation and loss of water of hydration on the surface of the NPs up to 150 °C. The weight loss step that extends up to the temperature of 300 °C occurs because of the thermal degradation of the coated polymer. It appears to take place smoothly, indicating the degradation as a one-step reaction. In the case of Sr–Fe, the next weight loss starts at ∼850 °C due to the phase change of the ferrite NPs. For Sr-HAp and Sr,Fe-HAp, the degradation step corresponding to the HAp matrix is observed at around 980 °C. The curve is smooth, assumed to be due to the gradual dehydroxylation of the HAp structures shown as below:
| SrxFeyCa10−x−y(PO4)6(OH)2 → SrxFeyCa10−x−y(PO4)6(OH)2−2zOz + zH2O | (2) |
 | ||
| Fig. 3 A–C show the TG profiles of Sr–Fe, Sr-HAp, and Sr–Fe-HAp NPs, respectively. X-Ray photoelectron spectra of elements present in Sr–Fe NPs (C, O, Sr, and Pt shown, respectively, from D to G); Sr-HAp NPs (C, O, Sr, Pt, P, and Ca presented in order, respectively, from H to M); and Sr-Fe-HAp NPs (Ca, C, O, Sr, Fe, P, and Pt shown, respectively, from N to T). | ||
The percentage weight loss of Chi-CD-Pt-fol in all three samples is around 30%, which indicates the extent of coating of the polymer. A complete degradation of HAp leads to the formation of a graphite-like structure, as reported in the literature.34,35
XPS spectra were recorded to evaluate the chemical state of various elements in the samples, as shown in Fig. 3D–T. In the XPS spectra of polymer-coated Sr–Fe NPs, the C–C, C–O–C, and O–CO bonds show peaks at 284.7, 285.9, and 287.1 eV, respectively (Fig. 3D). These peaks arise because of the 1s photoelectrons of carbon. The O 1s peaks of Fe–O oxygen and the C–O are observed at 529.9 and 532.8 eV, respectively. The Fe–O peak arises from the ferrite core of the NPs. This is further seen in Fig. 3F that shows the binding energy of the Sr electrons of configurations 3d3/2 and 3d5/2. In addition, the corona of the Sr–Fe NPs containing Chi-CD-Pt-fol has a co-ordinate Pt. This is revealed by the 4f electron peaks as shown in Fig. 3G. In the XPS of Sr-HAp, additional peaks corresponding to P (Fig. 3L) and Ca (Fig. 3M) arise due to the HAp. The 2p1/2 and 2p3/2 electrons of P show peaks with a binding energy of 133.8 eV. The Sr 3d and the P 2p peaks are close to each other because of the close proximity of the Sr 3d and P 2p lines that fall in the range of 132–133 eV. As observed in Fig. 3M, the binding energies of Ca 2p appear at 348.6 and 352.5 eV. These peaks have shifted from the respective reported binding energy values of 346.8 and 347.3 eV.36 This result is due to the substitution of Sr in HAp. In the case of Sr,Fe-HAp, the Fe 2p electron shows peaks at 711.1 and 724.4 eV, respectively, for 2p3/2 and 2p1/2. These peaks appear shifted from the typical values of Fe3+ reported for ferrites.37,38 The difference in binding energy is attributed to the variation in the co-ordination of the Fe ion in the lattice of ferrite and HAp. It further leads to the variation in the stabilization of the ion. The Sr 3d peaks of Sr-HAp (134.3 eV) and Sr,Fe-HAp (133.3 eV) differ slightly in binding energies due to the additional substitution of Fe in the latter. Thus, the XP spectra provide clear evidence for the various states of elements in the Sr–Fe, Sr-HAp, and Sr,Fe-HAp samples. Furthermore, the EXD spectra of the as-synthesized NPs were recorded (see the ESI,† Fig. S3). The spectra show the presence of various elements, namely, Sr, Fe, Ca, P, and O, in the samples. Notably, the Fe and Sr peaks appear in the spectrum of Sr–Fe-HAp and are absent in that of Sr-HAp.
Magnetic properties of NPs
The magnetic properties of the Chi-CD-Pt-fol-coated Sr–Fe, Sr-HAp, and Sr–Fe-HAp were investigated using vibrating sample magnetometry. The room temperature coercive field and remanent magnetization were determined from the magnetization vs. field curves (Fig. 4). Sr–Fe shows a broad hysteresis curve with a coercivity of 714 Oe, characteristic of ferromagnetic materials. Ferromagnetic behavior with broad hysteresis loops for strontium hexaferrite has been reported in the literature.39,40 The coercivity values exceed 4000 Oe usually for the ferrites. Nevertheless, in the present case, the Hc value is smaller than usual. Furthermore, the saturation magnetization (MS) is not reached by Sr–Fe NPs up to the maximum applied field of 15 kOe. Therefore, the MS value was determined employing the ‘law of approach to saturation’ (LAS):41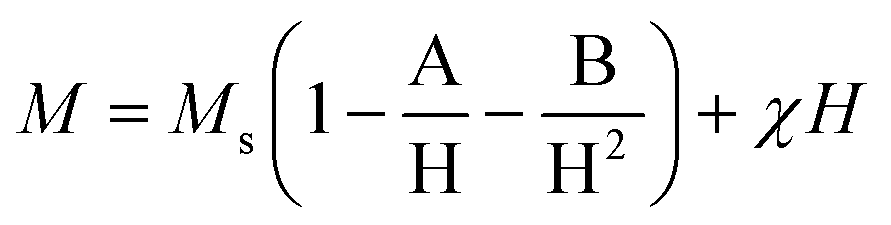 | (3) |
 | ||
| Fig. 4 Magnetization curves of Sr–Fe, Sr-HAp, and Sr–Fe-HAp NPs at room temperature. | ||
The magnetic behavior of Sr-HAp shows a paramagnetic nature as it is observed from the straight line of magnetization passing through the origin. Contrary to the behavior of the Sr–Fe and Sr-HAp, the Sr,Fe-HAp NPs show a sigmoidal curve with an MS value of 4.3 emu g−1. It represents a superparamagnetic behavior suitable for magnetic field-assisted drug delivery.42 The three different magnetic characteristics of the Sr–Fe, Sr-HAp, and Sr,Fe-HAp suggest that the material of choice from them can be utilized for the desired biomedical application. Furthermore, the Sr-HAp attains a magnetic property stronger than paramagnetism with the incorporation of Fe in Sr-HAp, i.e., Sr,Fe-HAp. The smaller saturation magnetization value of Sr,Fe-HAp compared to that of the Sr–Fe is due to the large diamagnetic contribution of the HAp in the former while the Sr–Fe is a typical substituted magnetic iron oxide.
Drug loading and release
The percent encapsulation efficiencies of Dox in various NPs were estimated using equation (1) as explained in the section ‘Doxorubicin–loading and encapsulation efficiency’. The percent adsorption values are 89.2 ± 3.2, 91.1 ± 2.2, and 92.3 ± 2.8, respectively, for Chi-CD-Pt-fol-coated Sr–Fe, Sr-HAp, and Sr-HAp NPs. The drug loading contents were 3.2 ± 0.8, 3.4 ± 0.6, and 3.6 ± 0.9, respectively, for Chi-CD-Pt-fol-coated Sr–Fe, Sr-HAp, and Sr-HAp NPs. These results imply a high amount of drug loaded on the NPs that is enabled by the cyclodextrin-tethered polymer, which aids the inclusion by complex formation with Dox.43 The release profile of loaded Dox from the NPs reveals that it extends well over a period of 45 hours at pH 7.4 and room temperature (Fig. 5). The cumulative release of Dox at 48 hours is 70, 63, and 60%, respectively, from Sr–Fe, Sr-HAp, and Sr,Fe-HAp NPs at pH 7.4. The sustained release is essential for the maintenance of therapeutic efficiency and defying the acquisition of drug resistance by the tumor. The results reveal that NPs are good candidates for drug delivery. | ||
| Fig. 5 Cumulative amount of Dox released in vitro as a function of time from the nanocarriers namely, Sr–Fe, Sr-HAp, and Sr,Fe-HAp NPs. | ||
In vitro cytotoxicity assay
The nanocarriers, namely, the polymer-coated Sr–Fe, Sr-HAp, and Sr,Fe-HAp NPs, (concentration range of 100–2000 μg mL−1) were evaluated for their cyto-compatibility against human cancer cell lines such as A549, HUH7, HeLa, and MG-63 using MTT colorimetric assay. The cytotoxicities are shown in Fig. 6. Overall, Sr–Fe and Sr-HAp NPs were found to be nontoxic when exposed to all cancer cell lines. The Sr–Fe NPs showed greater cyto-compatibility as compared to Sr-HAp NPs. Nevertheless, the difference was not statistically significant. With respect to Sr–Fe-HAp NPs, although the percent cell viability was comparatively nontoxic to the control group at 100 μg mL−1, it displayed a dose-dependent toxicity at higher concentrations against A549, HUH7 and HeLa cancer cells. On the other hand, Sr–Fe-HAp demonstrated a drastic increase in the percent cell viability against MG-63 osteoblast-like cancer cells at 100 μg mL−1, which then gradually reduced with the increase in the concentrations of the NPs. The unique behavior of Sr–Fe-HAp NPs against MG-63 cells as compared to other cancer cells could be attributed to the selective sensitivity towards MG-63 cells.44,45 Denry et al.44 showed that calcium phosphate ceramic scaffolds consisting of hydroxyapatite and beta tri-calcium phosphate have osteointegration potential. Similarly, Pallazo et al.45 have explained that biomimetic HAp-drug conjugates can act as potential bone substitutes with simultaneous anticancer effects. Furthermore, Ortolani et al.46 and Nielsen et al.47 have demonstrated the role of Sr in the treatment of osteoporotic patients. Sr is well known for its therapeutic potential in osteoporotic patients. Kumar et al.48 have demonstrated that strontium-tagged graphene oxide nanoparticles demonstrate increased osteointegration in the in vitro 3D bone tissue model. Sangeetha et al.49 further showed that treatment of osteoblast-like MG-63 cells with Sr-HAp leads to an increase in the cell viability and associated alkaline phosphatase activity.50 These data suggest that Sr, HAp and Fe play a different role when treated with MG-63 osteoblast-like cells. The main skeleton of the developed nanoconjugates is the complex of chitosan, cyclodextrin and folate as shown in Fig. 1. These 3 components are well known for their biocompatible nature51 and are currently in use in drug delivery systems.51 Furthermore, a combination of folate and cyclodextrin allows targeting cancer cells.52 | ||
| Fig. 6 Cytotoxicity of Chi-CD-Pt-fol-coated Sr–Fe, Sr-HAp, and Sr,Fe-HAp NPs on various cell lines, namely, (a) A549 (lung), (b) HeLa (cervical), (c) HUH-7 (liver), and (d) MG63 (bone). All the results are expressed as mean ± SD and the percent cell viability of all the groups (nanoparticles) was compared to that of controls. Outcomes were statistically analyzed by using one-way ANOVA with Dunnett's multiple comparison test, where * represents P < 0.05, # represents P < 0.01 and @ represents P < 0.001. | ||
In vitro anticancer potential of Dox-loaded nanocarriers
The anticancer potential of free Dox and dox-loaded polymer-coated NPs (Sr,Fe-HAp-Dox) in the concentration range of 0.1–2.5 μg mL−1, and equivalent amounts of unloaded Sr,Fe-HAp NPs against A549, HUH7, HeLa and MG-63 human cancer cells was estimated using the MTT assay. The anticancer assays carried out after 48 and 72 hours of administration of Dox-carrying NPs in various cell lines are depicted in Fig. 7. The results indicate that the Dox-loaded carriers and the free Dox displayed a similar anticancer effect in all the concentration ranges, whereas blank NPs remained inert against all cancer cells except for MG-63 cells. In addition, it was observed that the test samples displayed time- and dose-dependent toxicity at 48 and 72 h against A549, HeLa and HUH7 cells. Interestingly, the blank- and Dox-loaded NPs demonstrated enhanced anticancer activity against MG-63 cell lines compared to the free Dox. The reason for this increase in the cytotoxic effect of blank- and Dox-loaded NPs could be attributed to the presence of platinum, shape of the NPs, selective inhibition of bone marrow osteoblast-like cells, and the time of exposure. Clinically, surgical resection of the bone tumor tissue remains the gold standard method for the treatment of bone cancer. Nevertheless, it often results in a huge bone defect following surgery that demands a novel treatment strategy. The strategy incorporates not only an anticancer agent aiming to reduce the recurrence but also an effective osteopromotive agent to regain the lost bone tissue. Based on the experiments, it can be stated that the biomaterial composite consisting of Sr,Fe-HAp may act as an osteopromotive agent and platinum and Dox act as potential anticancer agents.53 | ||
| Fig. 7 Anticancer assay carried out after 48 and 72 hours of administration of Dox-carrying Chi-CD-Pt-fol-coated Sr,Fe-HAp NPs in various cell lines. (a) and (b) A549, (c) and (d) HeLa, (e) and (f) HUH-7, and (g) and (h) MG63. All the results are expressed as mean ± SD and the outcomes were statistically analyzed by using one-way ANOVA with Tukey's multiple comparison test, where * represents P < 0.05, # represents P < 0.01 and @ represents P < 0.001. | ||
In vivo anticancer study of Dox-loaded nanocarriers
Our study shows that MG-63 tumor cells grow in the pleural cavity and invade contiguous structures. Taken together, these data show that the growth patterns of cancer cells implanted intra-medullary were similar to those encountered in bone cancer patients.54 The Dox-loaded nanoformulations were characterized by observing the induced bone tumor cells. In the distal tumor cells, after the administration with formulation of polymer-coated and Dox-loaded NPs, namely, Sr-HAp-Dox and Sr,Fe-HAp-Dox, the observed cells were loosely arranged (Fig. 8). A few red blood cells appeared in the case of Sr-HAp-Dox, Sr-HAp, and Sr,Fe-Hap, respectively. In the cases of Dox, Sr–Fe, and Sr,Fe-HAp-Dox, the red blood cells were found to disappear. In contrast to the histopathology experiment, the standard drug Dox was identified as being moderately active against tumor cells. Finally, the antitumor activity of Sr,Fe-HAp-Dox against highly metastatic models of MG-63 delineates interesting chemotherapeutic potential for this formulation in this disease alone and in combination with other active chemotherapeutic agents.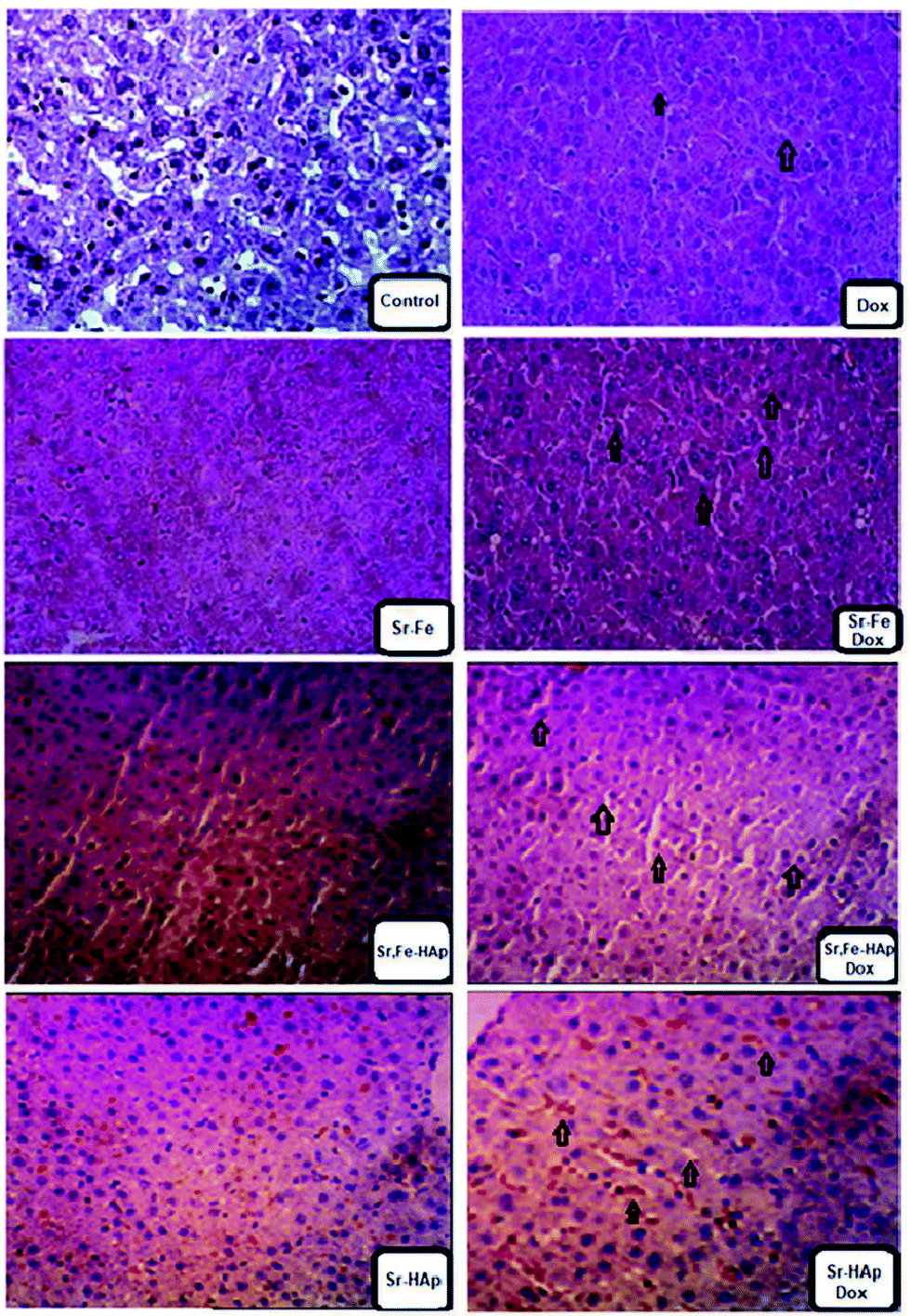 | ||
| Fig. 8 Immunohistochemical controlled bone marrow tissue section of mice. Samples labelled with a Dox tag refer to the Dox-loaded NPs. The others are nanocarriers without the drug in them. The association of NPs with the cancer tissue and the Dox-loaded nanocarriers’ efficacy are indicated with arrow marks. | ||
Experimental
Materials
AnalaR reagents of p-toluenesulfonic chloride, ethylene diamine, and chitosan (MW 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) were procured from Merck, India. Mono-6-deoxy-6-aminoethylamino-β-cyclodextrin and potassium tetrachloroplatinate(II) were purchased from Hi-Media and Alfa Aesar (India), respectively. Analytical reagent (AR) grade solvents were employed for the synthesis and spectral grade for UV–vis spectroscopy. These reagents were obtained from TCI, India. The reagents utilized for preparing strontium ferrite, strontium hydroxyapatite and strontium ferrite- hydroxyapatite composite, namely, strontium nitrate, ferric nitrate, diammonium hydrogen phosphate, and cetyltrimethylammonium bromide (CTAB), were procured from Merck, India. All the chemicals were of above 98% purity. Dialysis membranes were the products of Hi-Media, India (diameter 14.33 mm, LA387–1MT).
000) were procured from Merck, India. Mono-6-deoxy-6-aminoethylamino-β-cyclodextrin and potassium tetrachloroplatinate(II) were purchased from Hi-Media and Alfa Aesar (India), respectively. Analytical reagent (AR) grade solvents were employed for the synthesis and spectral grade for UV–vis spectroscopy. These reagents were obtained from TCI, India. The reagents utilized for preparing strontium ferrite, strontium hydroxyapatite and strontium ferrite- hydroxyapatite composite, namely, strontium nitrate, ferric nitrate, diammonium hydrogen phosphate, and cetyltrimethylammonium bromide (CTAB), were procured from Merck, India. All the chemicals were of above 98% purity. Dialysis membranes were the products of Hi-Media, India (diameter 14.33 mm, LA387–1MT).
Synthesis of compounds
Tosylated β-cyclodextrin folate (CD-fol-Ts)
CD-fol-Ts was synthesized employing a reported method. In a typical procedure, CD-fol (0.5 g) in 3 mL of thrice distilled water (DW) and p-toluenesulfonic chloride (0.059 g) in 3 mL of acetonitrile were mixed. 5 mL of 10% NaOH solution was added to maintain the pH at 8. The mixture was stirred at 35 °C for 2 h. It was then freeze-dried to collect the white color product.Chitosan-β-cyclodextrin folate conjugate (Chi-CD-fol)
Solutions of 1 g of chitosan in 10 mL of hydrofluoric acid and 0.035 g of CD-fol-Ts in 5 mL of DW were prepared. They were then mixed and refluxed at 80 °C for 5 h. The contents of the reaction vessel were then allowed to cool to room temperature, dialyzed with a visking tube (MW cut off:) for 24 h to separate the product from the unreacted CD-fol-Ts, and freeze-dried to collect the product.56Platinum complex of chitosan-β-cyclodextrin folate (Chi-CD-Pt-fol)
To a suspension of Chi-CD-fol (0.5 g) in DW, potassium tetrachloroplatinate (0.004 g) was added. The mixture was stirred for 3 hours using a magnetic stir bar. The solution was then refrigerated for about 48 hours. The brown crystals obtained were separated by recrystallization and dried.57Strontium ferrite nanoparticles (Sr–Fe NPs)
In a reported elegant synthesis approach,58 strontium nitrate (1 M) and iron(III) nitrate (2 M) were mixed. To this nitrate aggregate, 5 mL of ethylene diamine solution was added along with distilled water. The solution was continuously stirred at RT for an hour. Then, the blended solution was transferred into a stainless-steel autoclave containing a Teflon case of 100 mL capacity and maintained at 150 °C for 16 hours. It was allowed to cool down to 35 °C. The brown color precipitate was separated by centrifuging at the rate of 15000 rpm and dried at 80 °C. The product was finally calcined at 700 °C for 3 hours.
Strontium-doped hydroxyapatite nanoparticles (Sr-HAp NPs)
In a fashionable synthesis technique, 0.3 mole of calcium nitrate (0.705 g), 1 mole of CTAB (3.6 g) and 0.1 molar of diammonium hydrogen phosphate (0.131g) were mixed. To this mixture, 0.1 mole of strontium nitrate (0.5 g) was added dropwise under constant stirring at RT for 1 hour. The solution was then transferred to the autoclave and maintained at 150 °C for 16 h. It was then allowed to cool to room temperature when the white colored solid precipitated. The solid was separated by centrifuging (15000 rpm) and dried at 65 °C.
Strontium and iron-doped hydroxyapatite nanoparticles (Sr,Fe-HAp NPs)
The Sr–Fe-HAp NPs were synthesized using a hydrothermal approach. 0.3 mole of calcium nitrate (0.705 g), 1 mole of CTAB (3.6 g), and 0.1 mole of diammonium hydrogen phosphate (0.131 g) were mixed. Then, 0.1 g of Sr–Fe dispersed in 10 mL of distilled water was added drop-wise and stirred at RT for 1 h. Then the solution was transferred to a Teflon stainless-steel autoclave and maintained at 150 °C for 16 h. After cooling down to RT, the brown color precipitate was centrifuged, separated, and dried in an oven at 65 °C.Coating of Chi-CD-Pt-fol on NPs
The NPs and Chi-CD-Pt-fol (10 mg) were mixed together in a 0.5 M NaOH solution. Probe sonication was carried out to obtain the polymer-coated MNPs.Instrumental methods
Gel permeation chromatographic (GPC) analysis was carried out employing a PerkinElmer (USA) equipment. The equipment possesses a Series 200 LC-290 AT pump, Rheodyne injector, and a SPD-6 AV UV-vis detector. Standard pullulans were employed for analysis. An X-stream H2O Mixed Bed 50 × 4.6 mm column was used. Acetate buffer (0.5 M) was the mobile phase. X-ray diffraction patterns were recorded using a Shimadzu XRD 6000 instrument. The radiation source employed was Cu Kα possessing λ = 1.5418 Å. The generator was operated at 40 kV and 30 mA in Bragg-Brentano focusing mode. The size and morphology of the NPs were investigated using a JEOL JSM 2100 HR transmission electron microscope (TEM), with a 200 kV acceleration voltage. A JEOL JSM 6390 device assisted in determining the elements of the NPs by collecting energy-dispersive X-ray (EDX) data. The weight percentages of Sr and Fe in the strontium ferrite sample were estimated using an ARCOS inductively-coupled plasmon resonance–atomic emission spectrometer, ICP-AES. The estimation of various elements in the samples of Sr–Fe, Sr-HAp, and Sr–Fe-HAp NPs was carried out employing a Phi 5000 versa probe II X-ray photoelectron spectrometer. A Shimadzu FT-IR spectrophotometer was utilized to record FT-IR spectra. For recording IR spectra, the samples were made as pellets after mixing with KBr. The spectral range was kept at 450–4000 cm−1. Thermogravimetric analysis (TGA) was carried out employing a Netzsch STA 449 F3 Jupiter thermal analyzer. The thermogravimetric profile was recorded setting up a heating rate of 5 °C per minute under a nitrogen flow (50 mL min−1). The temperature range of measurement was 100–1200 °C. The device was built with a Pt–Rh wire sensor. The magnetic properties of the polymer-coated Sr–Fe, Sr-HAp, and Sr,Fe-HAp NPs were determined using a lake shore 7410 vibrating sample magnetometer (VSM). Room temperature M–H curves were obtained. Chi-CD-Pt-fol was characterized using 1H NMR spectroscopy (Bruker AVANCE III spectrometer, 400 MHz).Doxorubicin–loading and encapsulation efficiency
Doxorubicin (Dox) in Chi-CD-Pt-fol-coated Sr–Fe, Sr-HAp, and Sr,Fe-HAp NPs was loaded by measuring the weight and pre-determining the measure of freeze-dried Dox-loaded NPs using a microbalance and dispersing in a 10 mL buffer of PBS at pH 7.4. The concentration of the loaded Dox was resolved utilizing UV-vis absorption spectrophotometry at 480 nm. The percentage of drug loading and the adsorption efficiency were determined respectively employing the following equations:59 | (4) |
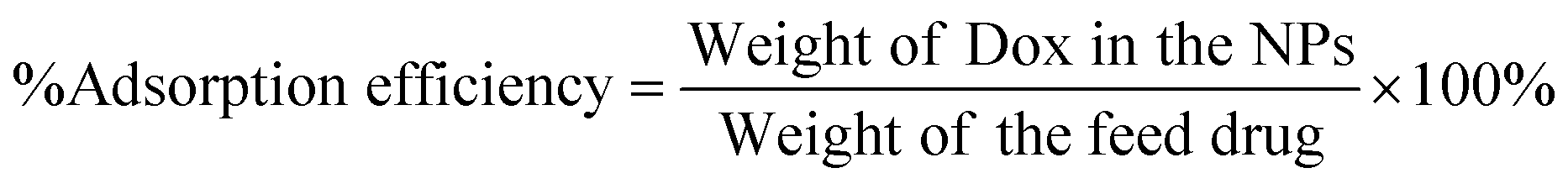 | (5) |
In vitro drug release
Dialysis bags of 20000 molecular weight cut-off were utilized for suspending the Dox-loaded NPs and the in vitro release of Dox was analyzed in PBS solution maintained at pH 7.4. These bags were kept shaking in a water bath at 35 °C. The solutions diffusing out from inside the bags were fetched at regular time intervals and the amount of released Dox was determined. The volume of solutions was kept consistent by adding fresh PBS. The concentration of Dox was determined by measuring UV-vis absorption at 480 nm and the concentration was calculated by standard calibration curve equation. For each time-point, three independent estimations were carried out. The mean values are presented.
Cell culture
Human cancer cell lines such as A549 (lung adenocarcinoma), HUH7 (hepatocellular carcinoma), HeLa (cervical carcinoma) and MG-63 (bone osteosarcoma) were maintained in a T75 flask using DMEM (Dulbecco's Modified Eagle Medium) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin at 37 °C in a CO2 incubator. Cells were trypsinized using 0.25% trypsin–EDTA to seed cells in a 96-well plate for cytotoxicity and anticancer assay.Cell viability assay
Cytocompatibility of the nanocomposites, namely, Sr–Fe-Ch, Sr–HAp-Ch and Sr–Fe-HAp-Ch (concentration range of 100–2000 μg mL−1), was evaluated against cancer cell lines such as A549, HUH7, HeLa and MG-63 using MTT colorimetric assay. Cells were seeded (5000 cells per well) in a flat-bottomed 96-well plate and incubated for 24 h in a CO2 incubator for cell attachment. The sterilized nanoparticle sample was diluted in a 1% dimethylsulfoxide (DMSO)-DMEM mixture and added to the wells followed by incubation for 24 h in the CO2 incubator. After 24 h, the supernatant medium was discarded and the wells were washed thrice with PBS. After washing, 100 μL of media and 10 μL of MTT (5 mg mL−1) were added to all the wells followed by incubation for 1h in the CO2 incubator. After 1h of incubation, the medium was discarded and 100 μL of DMSO was added to all the wells to solubilize the formazan crystals. The plate was protected from light and kept on the shaker for 10 min. The absorbance of the sample was estimated using a microplate reader at 540 nm. Percent cell viability was calculated using a control in comparison with others. A graph was plotted using GraphPad Prism software and the statistical analysis was carried out by applying the paired Student t-test to evaluate the significant differences.The anticancer potential of free Dox and Dox-loaded NPs (Sr–Fe-Hap-Ch-Dox) in the concentration range of 0.1–2.5 μg mL−1, and equivalent amounts of unloaded NPs (Sr–Fe-Hap-Ch) against A549, HUH7, HeLa and MG-63 human cancer cells was estimated by MTT assay. All the steps were similar as mentioned in the above section except for some minor modifications. In this experiment, the samples were incubated for 48 h and 72 h before the addition of MTT to evaluate the anticancer effect over a long time duration.
In vivo anticancer studies
The protocol approved by the Institutional Animal Ethics Committee, Karunya University, Coimbatore, India (Registration No. 158/99/CPCSEA) was employed for the in vivo anticancer studies. Healthy adult albino mice of both sex, aged 6–8 weeks, and weight 22 ± 5 g were used. They were housed in polypropylene cages at 25 ± 30 °C, a humidity of 60 ± 5%, and a 12 h light/dark cycle. During the experimental time, the mice were provided with a standard pellet diet and water. To evaluate oral toxicity, Dox-loaded nanoparticles were intended through intra-medullary administration with an initial dosage of 300 μg kg−1. The mice were observed for 12 days to check the toxicity by monitoring their mortality. Concurrently, the same protocols were followed to confirm acute toxicity by gradually increasing the dosage from 600 to 2000 μg kg−1. The experimental procedure was followed as per the Economic Co-operation and Development (OECD) guidelines.Human osteosarcoma cell lines MG-63 were obtained from NCCS India. Cells were sub-cultured in an RPMI medium with 10% heat-inactivated FBS and 1% of penicillin–streptomycin. The cells were maintained at 37 °C in 5% CO2. The healthy cells at 80% confluence cells were analyzed by their morphology and cell growth pattern. The healthy cells were washed with 1% PBS and revived by using trypsin EDTA buffer solution. The harvested cells were maintained in RPMI medium. To investigate the cell viability, trypan blue assay was performed. More than 96% of cells exhibited good viability. The mice were anesthetized with Zoletil 100 at 30 mg kg−1 and 2% Rompun at 5 mg kg−1. The chemical agents were administered through an intra-peritoneal route. Osteosarcoma cells (105 cells), 50 μL, were intended through the chest wall through the intra-peritoneal route using 26-gauge needles. The mice were returned to their cages until they recovered to their standard conditions under the animal body temperature. The development and growth of the tumor were characterized. As an extension of the in vivo studies, 24 mice were subjected to a similar experimental procedure. The tumor cells were administrated and the mice were sacrificed and autopsied at intervals of 7, 14, 21, and 28 days. The bone-marrow cells were separated and fixed within 5% phosphate buffered formalin and stained in paraffin. The tissues were examined under a microscope.
All Dox-carrying NPs were administered 2 doses through the intra-peritoneal route. The formulation doses causing death and sudden changes in the mice body weight are considered as drug-induced toxicity. The treated and control groups consisted of 5–6 mice. The drug treatment was initiated after tumor developed on the tissues. The femur organ was dissected from the specimens and maintained in a less than 10% formalin buffer solution. The organ specimens were decalcified with hydrochloride buffer solution. Images were obtained using high-resolution micro-imaging computer systems.
Conclusions
The search for agents that effectively deliver anticancer drugs is a special operation against the deadly disease. Strontium- and iron-incorporated hydroxyapatite nanoparticles are suitable candidates for targeted anticancer drug delivery. While the monodoping of either Sr or Fe has been reported in HAp, Sr,Fe-co-doped HAp have not been utilized for the treatment of cancer. In addition, the size, form, and function of these NPs are intriguing and nevertheless poorly studied. We report Sr-doped HAp nanowhiskers and Sr, Fe-co-doped HAp nanorods in this study. These engineered nanomaterials vary in size depending on the presence of iron. On the incorporation of both Sr and Fe, the nanomaterials become superparamagnetic. These nanomaterials are coated with a novel designed polymer, i.e., β-CD, folate, and Pt-containing conjugate. It enables the loading of Dox and thus receptor-mediated drug-targeting. The nanocarriers possess a size in the range suitable for prolonged residence in the circulatory system. The proven enhanced loading and sustained release of Dox is another advantage of nanocarriers. Tested on multiple cell lines, the nanocarrier, i.e., polymer-coated Sr,Fe-Hap, proves itself as a more effective Dox-carrier and demonstrates high activity particularly against bone cancer cells. When the in vivo metastatic anticancer activity is investigated on MG63 cell-infused mice, the Dox-loaded Sr,Fe-HAp reveals enhanced activity. The high aspect ratio of the nanorods, biocompatibility of the modified chitosan, ability of β-CD cavity to encapsulate Dox, and targeting ligand (folate) attachment can work in unison to selectively destroy cancer cells. Thus, the Sr,Fe-HAp nanorods are a promising candidate for targeted delivery of Dox. In addition, since the cavity of β-CD is capable of accommodating several anticancer drugs of appropriate size, the nanocarrier can encapsulate other drugs also. Being superparamagnetic, the Sr,Fe-HAp nanorods respond well to the magnetic field, and can thus be controlled by the application of an externally applied field.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors express their gratitude to the Department of Atomic Energy-Board of Research in Nuclear Sciences, Government of India, for the project 37(2)/14/17/2018-BRNS.Notes and references
- R. L. Siegel, K. D. Miller and A. Jemel, Ca-Cancer J. Clin., 2019, 69, 7–34 CrossRef PubMed.
- H. Yu, J. V. Rowley, D. C. Green and P. D. Thornton, Mater. Adv., 2020, 1, 1293–1300 RSC.
- H.-P. Yu, Y.-J. Zhu, Z.-C. Xiong and B.-Q. Lu, Chem. Eng. J., 2020, 399, 125666 CrossRef CAS.
- Z. Li, J. Tang, H. Wu, Z. Ling, S. Chen, Y. Zhou, B. Guo, X. Yang, X. Zhu, L. Wang, C. Tu and X. Zhang, Nano Res., 2020, 13, 2106–2117 CrossRef CAS.
- W. E. Brown and L. C. Chow, Annu. Rev. Mater. Sci., 1976, 6, 213–236 CrossRef CAS.
- W. E. G. Muller, H. C. Schroder, U. Schlossmacher, V. A. Grebenjuk, H. Ushijima and X. Wang, Biomaterials, 2013, 34, 8671–8780 CrossRef PubMed.
- A. Kumar, K. C. Nune and R. D. Misra, J. Biomed. Mater. Res., Part A, 2016, 104, 1343–1351 CrossRef CAS PubMed.
- V. Uskoković and D. P. Uskoković, J. Biomed. Mater. Res., Part B, 2011, 96, 152–191 CrossRef PubMed.
- T. Dvir, B. P. Timko, D. S. Kohane and R. Langer, Nat. Nanotechnol., 2011, 6, 13–22 CrossRef CAS PubMed.
- S. Mondal, P. Manivasagan, S. Bharathraja, M. S. Moorthy, H. H. Kim, H. Seo, K. D. Lee and J. Oh, Int. J. Nanomed., 2017, 12, 8389–8410 CrossRef CAS PubMed.
- N. Tran and T. J. Webster, Acta Biomater., 2010, 7, 1298–1306 CrossRef PubMed.
- H. C. Wu, T. W. Wang, J. S. Sun, W. H. Wang and F. H. Lin, Nanotechnology, 2007, 18, 165601 CrossRef.
- M. E. Zilm, M. Staruch, M. Jain and M. Wei, J. Mater. Chem. B, 2014, 2, 7176–7185 RSC.
- S. Pernal, V. M. Wu and V. Uskoković, ACS Appl. Mater. Interfaces, 2017, 9, 39283–39302 CrossRef CAS PubMed.
- K. Zhao, X. Liu, C. Jin, F. Yu, A. Rykov, J. Wang and T. Zhang, Hyperfine Interact., 2013, 218, 1–7 CrossRef CAS.
- H. C. Wu, T.-W. Wang, M. C. Bohn, F. H. Lin and M. Spector, Funct. Mater., 2010, 20, 67–77 CrossRef CAS.
- I. G. Finlay, M. D. Mason and M. Shelley, Lancet Oncol., 2005, 6, 392–400 CrossRef CAS.
- K. Yamada, M. Yoshimura and K. Tokuuye, Exp. Ther. Med., 2012, 3, 226–230 CrossRef CAS PubMed.
- S. L. Turner, S. Gruenewald and N. Spry, Br. J. Cancer, 2001, 84, 297–302 CrossRef CAS PubMed.
- N. Sudha, S. Chandrasekaran, S. Yousuf and I. V. M. V. Enoch, Carbohydr. Polym., 2015, 115, 589–597 CrossRef PubMed.
- T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818–1822 CrossRef CAS PubMed.
- E. V. Svirshchevskaya, I. A. Gracheva, A. G. Kuznetsov and E. V. Myrsikova, Med. Chem., 2016, 6, 571–577 Search PubMed.
- M. Coluccia and G. Natile, Adv. Anticancer Agents Med. Chem., 2007, 7, 111–123 CrossRef CAS PubMed.
- Y. S. Yi, W. Ayala-López, S. A. Kularatne and P. S. Low, Mol. Pharm., 2009, 6, 1228–1236 CrossRef CAS PubMed.
- M. M. Klowski, R. J. Friederichs, R. Nichol, N. Antolin, R. Carzaniga, W. Windl, S. M. Best, S. J. Shefelbine, D. W. McComb and A. E. Porter, Acta Biomater., 2015, 20, 129–139 CrossRef PubMed.
- J. Jevtić, M. Mitrić, S. Škapin, B. Jančar, N. Ignjatović and D. Uskoković, Cryst. Growth Des., 2008, 8, 2217–2222 CrossRef.
- Z. Li, Z. Liu, M. Yin, X. Yang, Q. Yuan, J. Ren and X. Qu, Biomacromolecules, 2012, 13, 4257–4263 CrossRef CAS PubMed.
- M. Filippousi, P. I. Siafaka, E. P. Amanatiadou, S. G. Nanaki, M. Nerantzaki, D. N. Bikiaris, I. S. Vizirianakis and G. V. Tendeloo, J. Mater. Chem. B, 2015, 3, 5991–6000 RSC.
- V. S. Chandra, G. Baskar, R. V. Suganthi, K. Elayaraja, M. I. A. Joshy, W. S. Beaula, R. Mythili, G. Venkatraman and S. N. Kalkura, ACS Appl. Mater. Interfaces, 2012, 2, 1200–1210 CrossRef PubMed.
- Y. Yang, Q. Wu, M. Wang, J. Long, Z. Mao and X. Chen, Cryst. Growth Des., 2014, 14, 4864–4871 CrossRef CAS.
- O. Veish, J. W. Gunn and M. Zhang, Adv. Drug Delivery Rev., 2010, 62, 284–304 CrossRef PubMed.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. I. Ipe, M. G. Bawendi and J. V. Fragioni, Nat. Biotehnol., 2007, 25, 1165–1170 CrossRef CAS PubMed.
- Y. Li, M. Kröger and W. K. Liu, Nanoscale, 2015, 7, 16631–16646 RSC.
- M. G. S. Murray, J. Wang, C. B. Pontoon and P. M. Marquis, J. Mater. Sci., 1995, 30, 3061–3074 CrossRef CAS.
- L. Zeng, C. Qin, L. Wang and W. Li, Carbohydr. Polym., 2011, 83, 1553–1557 CrossRef CAS.
- W. Xia, C. Lindahl, J. Lausmaa, P. Borchardt, A. Ballo, P. Thomsen and H. Engqvist, Acta Biomater., 2010, 6, 1591–1600 CrossRef CAS PubMed.
- L. Fu, X. Liu, Y. Zhang, V. P. Dravid and C. A. Mirkin, Nano Lett., 2003, 3, 757–760 CrossRef CAS.
- T. Bala, C. R. Sankar, M. Baidakova, V. Osipov, T. Enoki, P. A. Joy, B. L. V. Prasad and M. Sastry, Langmuir, 2005, 21, 10638–10643 CrossRef CAS PubMed.
- A. A. Farghali, M. H. Khedr and A. F. Moustafa, Adv. Mater. Technol., 2008, 23, 104–109 CrossRef CAS.
- M. M. Hessien, M. M. Rashad and K. El-Barawy, J. Magn. Magn. Mater., 2008, 320, 336–346 CrossRef CAS.
- A. M. Alsmadi, I. Bsoul, S. H. Mahmood, G. Alnawashi, K. Prokeš, K. Siemensmeyer, B. Klemke and H. Nakotte, J. Appl. Phys., 2013, 24, 243910 CrossRef.
- H. Mamiya and B. Jeyadevan, IEEE Trans. Magn., 2014, 50, 2274072 Search PubMed.
- A. S. Braegelman and M. J. Webber, Theranostics, 2019, 9, 3017–3040 CrossRef CAS PubMed.
- I. Denry and L. T. Kuhn, Dent. Mater., 2016, 32, 43–53 CrossRef CAS PubMed.
- B. Palazzo, M. Iafisco, M. Laforgia, N. Margiotta, G. Natile, C. L. Bianchi, D. Walsh, S. Mann and N. Roveri, Adv. Funct. Mater., 2007, 17, 2180–2188 CrossRef CAS.
- S. Ortolani and S. Vai, Bone, 2006, 38, 19–22 CrossRef PubMed.
- S. P. Nielsen, Bone, 2004, 35, 583–588 CrossRef PubMed.
- S. Kumar and K. Chatterjee, Nanoscale, 2015, 7, 2023–2033 RSC.
- K. Sangeetha, M. Ashok, E. K. Girija, G. Vidhya and G. Vasugi, Ceram. Int., 2018, 44, 13782–13789 CrossRef CAS.
- W. F. Zhang, H. Y. Zhou, X. G. Chen, S. H. Tang and J. J. Zhang, J. Mater. Sci.: Mater. Med., 2009, 20, 1321–1330 CrossRef CAS PubMed.
- T. Mortezazadeh, E. Gholibegloo, N. R. Alam, S. Dehghani, S. Haghgoo, H. Ghanaati and M. Khoobi, Magn. Reson. Mater. Phys., Biol. Med., 2019, 32, 487–500 CrossRef CAS PubMed.
- Y. Liu, Y. Pu, L. Sun, H. Yao, B. Zhao, R. Zhang and Y. Zhang, J. Biomed. Nanotechnol., 2016, 12, 1393–1403 CrossRef CAS PubMed.
- M. Iafisco, B. Palazzo, M. Marchetti, N. Margiotta, R. Ostuni, G. Natile, M. Morpurgo, V. Gandin, C. Marzano and N. Roveri, J. Mater. Chem., 2009, 19, 8385–8392 RSC.
- L. Doré-Savard, V. Otis, K. Belleville, M. Lemire, M. Archambault, L. Tremblay and L. Gendron, PLoS One, 2010, 5, e13774 CrossRef PubMed.
- A. R. Vortherms, R. P. Doyle, D. Gao, O. Debrah and P. J. Sinko, Nucleosides, Nucleotides Nucleic Acids, 2008, 27, 173–185 CrossRef CAS PubMed.
- S. Ramasamy, B. Samathanam, H. Reuther, M. S. A. M. Nadar, I. V. M. V. Enoch and K. Potzger, Colloids Surf., B, 2018, 161, 347–355 CrossRef CAS PubMed.
- M. V. de Almeida, J. D. S. Chaves, A. P. S. Fontes, E. T. César and M. Gielen, J. Braz. Chem. Soc., 2006, 17, 1266–1273 CrossRef CAS.
- B. Govindan, B. S. Latha, P. Nagamony, F. Ahmed, M. A. Saifi, A. H. Harrath, S. Alwasel, L. Mansour and E. H. Alsharaeh, Nanomaterials, 2017, 7, 138 CrossRef PubMed.
- A. Zhu, L. Yuan, W. Jin, S. Dai, Q. Wang, Z. Xue and A. Qin, Acta Biomater., 2009, 5, 1489–1498 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: FT-IR, 1H and 13C NMR, and EDX spectra of Ch-CD-fol conjugate; EDX spectra of materials. (PDF). See DOI: 10.1039/d1ma00142f |
| This journal is © The Royal Society of Chemistry 2021 |