
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSubstitutional effects in TiFe for hydrogen storage: a comprehensive review
Erika M.
Dematteis
 ab,
Nicola
Berti
b,
Fermin
Cuevas
*a,
Michel
Latroche
a and
Marcello
Baricco
b
ab,
Nicola
Berti
b,
Fermin
Cuevas
*a,
Michel
Latroche
a and
Marcello
Baricco
b
aUniv Paris Est Creteil, CNRS, ICMPE, 2 rue Henri Dunant, 94320 Thiais, France. E-mail: cuevas@icmpe.cnrs.fr; Tel: +33 149781225
bDepartment of Chemistry, Inter-departmental Center Nanostructured Interfaces and Surfaces (NIS), and INSTM, University of Turin, Via Pietro Giuria 7, 10125 Torino, Italy
First published on 18th March 2021
Abstract
The search for suitable materials for solid-state stationary storage of green hydrogen is pushing the implementation of efficient renewable energy systems. This involves rational design and modification of cheap alloys for effective storage in mild conditions of temperature and pressure. Among many intermetallic compounds described in the literature, TiFe-based systems have recently regained vivid interest as materials for practical applications since they are low-cost and they can be tuned to match required pressure and operation conditions. This work aims to provide a comprehensive review of publications involving chemical substitution in TiFe-based compounds for guiding compound design and materials selection in current and future hydrogen storage applications. Mono- and multi-substituted compounds modify TiFe thermodynamics and are beneficial for many hydrogenation properties. They will be reviewed and deeply discussed, with a focus on manganese substitution.
 Erika M. Dematteis | Dr Erika M. Dematteis obtained her PhD cum Laude in Chemistry and Material Science at the University of Turin (Italy) in 2018, defending a thesis titled “Thermodynamics of Boron-based Complex Hydrides for Energy Storage”. She has been PostDoc at the CNRS (France) and University of Turin (Italy) involved in the FCH-JU HyCARE project. Her research interests focus on the experimental and theoretical determination of thermodynamics and applications of complex hydrides (i.e. borohydrides) and intermetallic (FeTi-based) as solid-state hydrogen storage material, solid electrolyte in batteries and energy storage, towards industrial applications. |
 Nicola Berti | Dr Nicola Berti obtained his master in Material Science at the University of Padua (Italy) and the PhD at the University Paris-Est (France) working at the ICMPE-CNRS within the European project ECOSTORE for energy storage, funded by the Marie Curie Association. He spent one year as PostDoc at the University of Turin (Italy). His research work focuses on the development of new negative electrodes and solid-state electrolytes for Li-ion batteries, and metal hydrides for hydrogen storage for practical applications. |
 Fermin Cuevas | Dr Fermin Cuevas obtained in 1996 his PhD in Physics at UAM University of Madrid (Spain). After postdoctoral studies at MPI-Stuttgart (Germany) and CNRS-Thiais (France), he became in 2002 permanent researcher at CNRS. His main research activities are hydrogen storage in solids as well as electrode and electrolyte materials for Ni-MH and Li-ion batteries. Currently, he is leader of the Interaction of Hydrogen and Matter group at the East Paris Institute of Chemistry and Materials Science, co-head of the hydrogen storage axis at the French Research Network on Hydrogen Energy and French coordinator of the International Research Network FACES. |
 Michel Latroche | Dr Michel Latroche is currently appointed as Director of Research at CNRS in France. He studied at Nantes' University and obtained a PhD at the Institut Jean Rouxel in Solid State Chemistry. After a post-doctoral position at Northwestern University in USA, he joined the CNRS to work in the field of metal hydrides. He was the head of the East Paris Institute of Chemistry and Materials Science and is now Representative Officer for the Energy Research Coordination at the Institute of Chemistry (CNRS). He is an Expert member at the IEA-HIA Task 40: Energy storage and conversion based on hydrogen. |
 Marcello Baricco | Prof. Marcello Baricco is, at the Department of Chemistry of the University of Turin, full professor in Materials Science and Technology. He obtained the PhD in Chemistry in 1987 and joined the University of Turin in 1990. He coordinated the SSH2S European project on hydrogen storage and now coordinates the EU H2020 FCH-JU project HyCARE. He is an expert in the Task 40 of the IEA-HIA and member of Scientific Committee of FCH-JU. He is coordinator of SP7 on Hydrogen Storage of JP on Fuel Cells and Hydrogen of the European Energy Research Alliance (EERA). |
Introduction
The hydrogen energy chain is foreseen as one of the key technologies to face the issues of climate change and scarce oil resources. Hydrogen can be worldwide and cleanly produced through electrolysis of water using renewable primary energies. If not consumed on-site, it can be transported by gas pipelines, trucks and ships. Finally, hydrogen can be used to feed fuel cells and generate electricity (and heat) on demand, releasing only water as a by-product and then closing the hydrogen cycle. Such an electricity–hydrogen–electricity conversion process is only sustainable if electricity is produced from renewable energies and cannot be directly injected in the grid, then it can be used later on with a fuel cell. Therefore, it is mandatory to add a storage step in the hydrogen chain for the time-management of hydrogen production and use. The intrinsic intermittency of most renewable energy sources makes unavoidable the implementation of efficient hydrogen storage systems.Efficient hydrogen storage can be achieved as dihydrogen molecules in high-pressure tanks (typically 350 or 700 bar) or in liquid state (at temperature lower than −252 °C [21 K]).1–3 In addition, dihydrogen can be physically adsorbed in high-surface-area solids such as MOFs and activated carbons, typically at liquid nitrogen temperature (−196 °C [77 K]).4–8 As an alternative, hydrogen molecules can be chemisorbed at the surface of solid compounds and diffused in atomic form to form hydrogen-containing compounds.9–11 In some cases, after suitable activation, these compounds can reversibly absorb and desorb hydrogen close to normal conditions of pressure and temperature (i.e. 1 bar, 25 °C).12–16 These materials are typically intermetallic compounds of general formula ABn, which are commonly named metallic hydrides, from the facts that both hydrogen-metal bonding and electronic conductivity have a metallic character.17 A is an element that forms very stable metallic hydrides (e.g. rare earths and early transition metals) and B an element that only forms hydrides at very high pressure (e.g. late transition metals), as reported in Fig. 1. Their combination in stoichiometric ratio n = B/A allows for the formation of hydrides with intermediate stability. Representative intermetallic compounds suitable for hydrogen storage are LaNi5, CeNi3, TiMn2 and TiFe for n = 5, 3, 2 and 1, respectively.
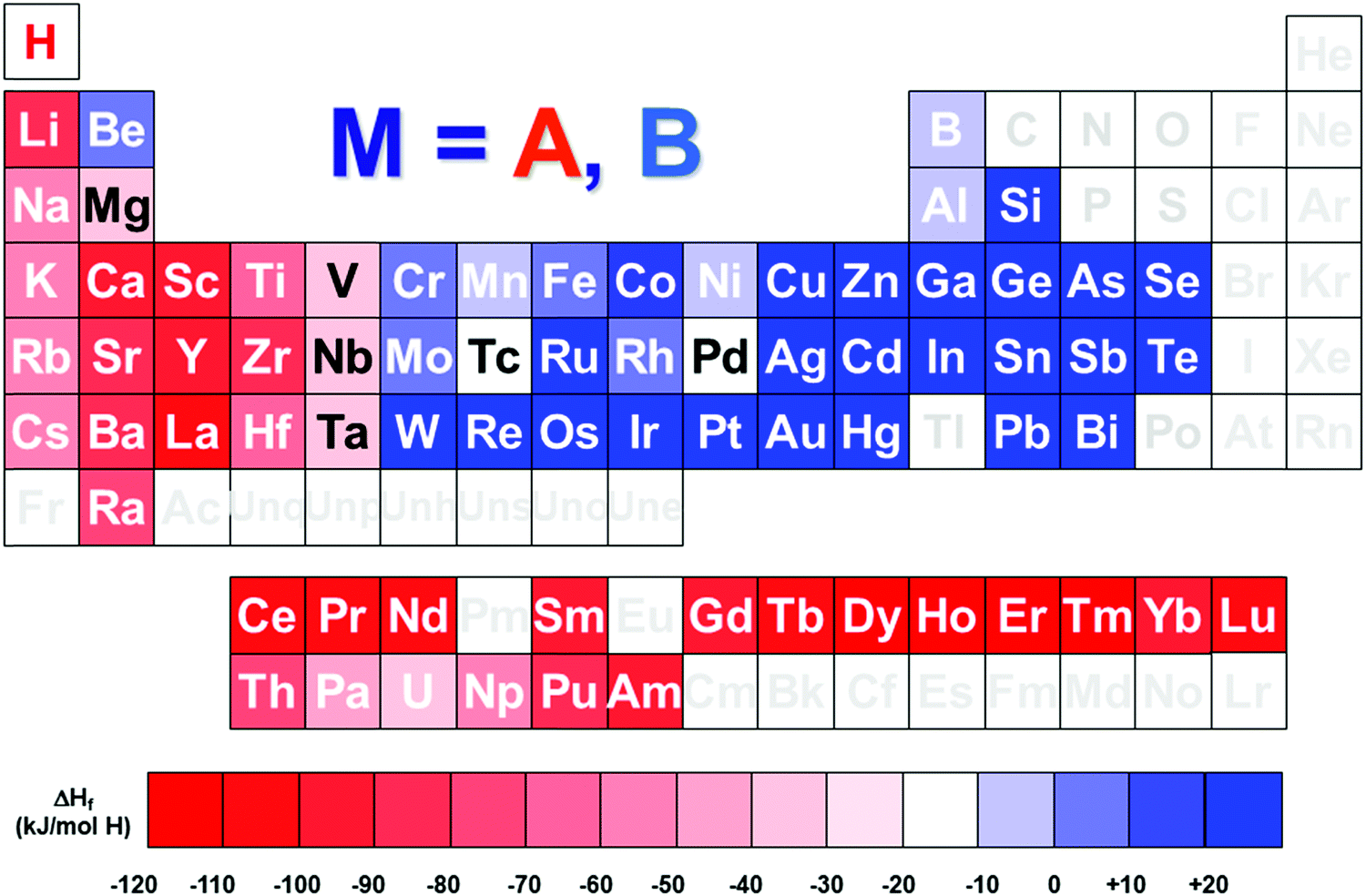 | ||
| Fig. 1 Periodic table of the elements showing the formation enthalpy of binary M–H metal hydrides, and the relative classification of A (in red) or B-type (in blue) M elements. Formation enthalpy was chosen from literature values.60,206,207,234,235 | ||
Intermetallic compounds, being formed by heavy elements, offer modest mass storage capacities (i.e. 1–2 wt%). Hydrogen systems based on this technology have low gravimetric capacities, due to the weight of the reservoir and ancillary equipment.3 When compared to classical molecular methods (5–6 wt% system basis, for both pressurized and liquid storage), their typical operation conditions (0–80 °C, 1–50 bar) guarantee higher safety conditions. This is a key property when hydrogen tanks must be installed close to domestic facilities or in confined space. Moreover, in the case of stationary applications, footprint instead of mass capacity is the most relevant performance indicator for the hydrogen storage system. The volumetric capacity of intermetallic compounds, i.e. 100–120 kgH2 m−3, is significantly higher than that of pressurized or liquid hydrogen: 39 (at 700 bar) and 70 kgH2 m−3, respectively. Furthermore, if the required tank is considered, the system volumetric density decreases significantly in the case of gas and liquid storage.
Finally, yet importantly, intermetallic compounds are highly versatile materials as their operation temperature and pressure can be tuned at will, through suitable chemical substitutions of both A and B-type elements. As an example, the EU-funded HyCARE (Hydrogen CArrier for Renewable Energy storage) project, kicked off in January 2019, aims to develop a prototype large-scale hydrogen storage tank using a solid-state hydrogen carrier based on metal powder, operating at low pressure and temperature.18,19 The project involves the production of almost 4 tons of metal powder, which will be placed in stainless steel containers. The thermal management of the plant will follow an innovative approach, making use of phase-change materials, significantly increasing the efficiency of the process. The aim is to store about 50 kg of hydrogen, which is a rather high quantity to be stored using this technique.
Among many intermetallic compounds described in the literature for reversible hydrogen storage at room temperature (RT), TiFe-based systems have recently regained vivid interest. With a mass and volumetric capacity for the binary compound of 1.87 wt% and 105 kgH2 m−3, respectively,20 the relevance of this system is mainly driven by its low cost, as compared to other intermetallics. However, in 2020, the European Union's (EU) has updated a list of 30 critical raw materials (CRMs), including titanium, considering their supply risk and economical importance.21 In fact, titanium is widely exploited in aeronautics and medical applications, and its processing is making EU strongly dependent on import from main global producers (45% China, 22% Russia, 22% Japan).21 Titanium End of Life Recycling Input Rate (EoL-RIR) is still limited (reported as 19%),21 however, for applications as solid state hydrogen storage material, it could be recycled and reused effectively. Fig. 2 shows the CRMs, highlighted with an orange frame, which should thus be avoided or limited in alloy formulation towards large-scale production. Economic and supply indicators demonstrate that TiFe-based compounds are today target materials for practical applications, as shown by the implementation of R&D projects worldwide, and especially in Japan. Intermetallic TiFe compound is promising for hydrogen storage tanks thank to its high volumetric density, good sorption kinetic, reversibility and because it can work in mild temperature and pressure conditions. Moreover, the hydrogenation thermodynamics of TiFe must be tuned to the required conditions of pressure and temperature imposed by each specific hydrogen storage application. As stated above, this can be achieved through suitable atomic substitutions. Indeed, titanium (Ti) and iron (Fe) can be substituted by other elements within certain homogeneity composition ranges, as reported in ternary phase diagrams.22 Numerous mono and multi-substituted alloys have been explored in the literature as highlighted in green in Fig. 2. These substitutions have not only a noticeable effect on hydrogen sorption thermodynamics, but also on other key properties, such as alloy activation, reaction kinetics and cycle life.
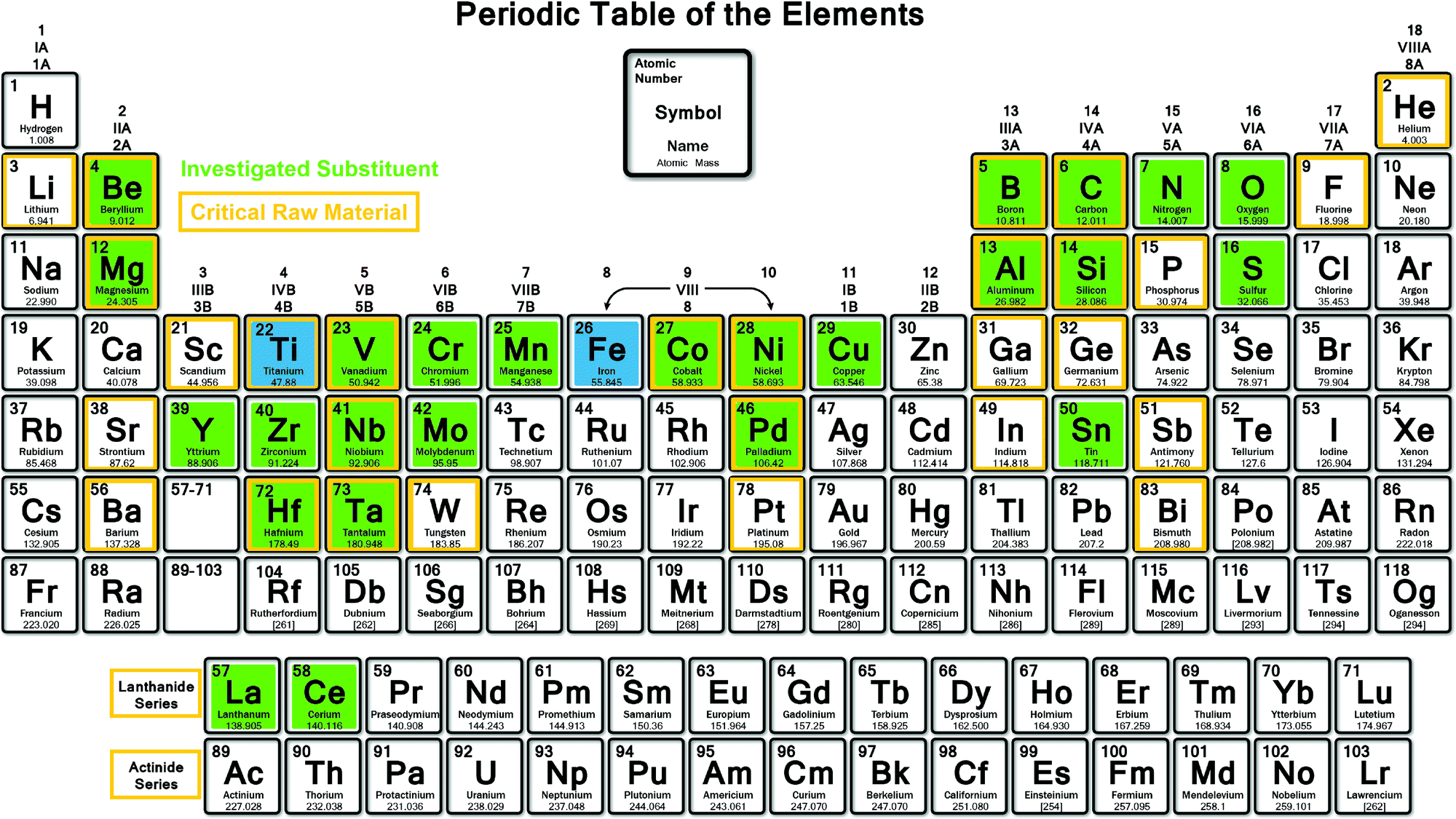 | ||
| Fig. 2 Periodic table showing the investigated substituting elements (highlighted in green) in TiFe (highlighted in blue). Orange frame highlight the critical raw materials for Europe.21 | ||
Recently, Sujan et al. provided a review focused on binary TiFe compound and its hydrogenation properties,23 while Lys et al. reported in a short review the effect of substitution on the hydrogenation properties of AxBy alloys.24 Here, after a short overview of binary TiFe, we focus our attention on reviewing the literature on substitutional effects, which are fundamental for practical applications, aiming at extending the previous reviews work in a comprehensive manner. Mono-substituted compounds are discussed with a focus on manganese substitution, which has been proved to be a key element. Some examples of prominent multi-substituted alloys are also reported here. This work aims to provide a comprehensive analysis of the many publications involving chemical substitution in TiFe-based compounds. As a conclusion, some correlations between compositions and hydrogen sorption properties are drawn, for guiding compound design and selection in current and future hydrogen storage applications.
TiFe
Synthesis and crystal structures of TiFe and its hydrides
TiFe exhibits a narrow homogeneity range, with the largest domain extending from 49.7 to 52.5 at% Ti at the eutectic temperature of 1085 °C.25,26. TiFe neighbouring phases are TiFe2, at the Fe-rich side, and β-Ti with a maximum solubility of 21 at% Fe, at the Ti-rich one. The hydrogen storage properties of this intermetallic compound are strictly linked to the composition and to the presence of secondary phases. In fact, due to the composition range in which TiFe phase can be formed, different properties can be observed for stoichiometric TiFe, Fe-rich (TiFe1.012) or Ti-rich (TiFe0.905) alloys. In addition, for Ti-rich alloys, the formation of β-Ti precipitates enables the hydrogen sorption at RT without activation. The TiFe heat of formation measured at 1167 °C is ΔfH = −31.0 kJ mol−1.27TiFe is usually produced by melting the elements in a high temperature furnace.28 As shown in the titanium-iron phase diagram (Fig. 3), the binary compound is obtained from the melt through a liquid + TiFe2 → TiFe peritectic reaction at 1317 °C. As an alternative to melting, TiFe can also be obtained and processed by Severe Plastic Deformation (SPD) techniques, such as ball milling29–45 and high pressure torsion46–50, as well as by self-ignition.51–53. SPD techniques lead to fresh and defective surfaces, that help alloy activation and also nanostructuration, but reducing nominal capacity.41,54,55

The crystal structure of TiFe was first identified as CsCl-type (B2, space group Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) by Laves et al.56. Further studies confirmed a cubic lattice, but contrary to bcc alloys, with distinct ordered atoms at the cube vertices, 1a sites (0,0,0), and its center, 1b sites (1/2,1/2,1/2).57 The lattice constant of the stoichiometric compound is reported to be 2.9763 Å.58 As stated before, the phase diagram exhibits a small homogeneity domain showing that the crystal structure of TiFe can accommodate some defects, such as partial substitution of Ti on the Fe sites.59,60
m) by Laves et al.56. Further studies confirmed a cubic lattice, but contrary to bcc alloys, with distinct ordered atoms at the cube vertices, 1a sites (0,0,0), and its center, 1b sites (1/2,1/2,1/2).57 The lattice constant of the stoichiometric compound is reported to be 2.9763 Å.58 As stated before, the phase diagram exhibits a small homogeneity domain showing that the crystal structure of TiFe can accommodate some defects, such as partial substitution of Ti on the Fe sites.59,60
Regarding the hydrides, four different phases have been reported for the TiFe-H system: α-solid solution, two monohydrides (β1 and β2) and γ-dihydride. The solid solution retains the CsCl-type structure with minor changes in the lattice parameters.61 Neutron diffraction experiments showed that H atoms occupy the octahedral 3d sites (1/2,0,0) along the cube edges with Ti4Fe2 coordination, exhibiting a shorter distance from Fe atoms than Ti atoms (1.49 Å vs. 2.11 Å). Even though Ti is known for its stronger hydrogen affinity compared to iron, this feature is common for all phases in the TiFe–H system.62–65 The maximum solubility of hydrogen in the α-solid solution is TiFeH0.1. Both β-phases crystallize in an orthorhombic structure, with minor structural differences between them except for hydrogen content, being TiFeH and TiFeH1.4 for β1 and β2, respectively. The most advanced studies by neutron diffraction suggest a P2221 space group,66 though Pmc21 cannot be completely ruled out.67 In both monohydrides, H atoms partially occupy the octahedral sites H1 and H2, both with coordination Ti4Fe2, whereas Fe is found at site 2c (0,0.294,1/4) and Ti at site 2d (1/2,0.757,1/4). In the β1-phase, hydrogen shows occupancies of 88% and 12% for sites H1 and H2, respectively, whereas in β2-phase, it exhibits occupancies of 92% and 45%, respectively. Due to the difficulties in achieving the fully hydrogenated phase, and the broadening of diffraction peaks because of strains, the crystal structure of the γ-phase has been subject of debate. In a first study of Reilly et al.,68 a cubic structure was suggested. Subsequent analyses proposed a monoclinic structure.62,63,69,70 Later, Fischer et al.64 found an orthorhombic structure (space group Cmmm), which was confirmed by both experimental works and theoretical calculations.71,72 Here, Ti atoms occupy site 4h (0.223,0,1/2), Fe site 4i (0,0.2887,0), while H atoms occupy three different octahedral sites, two of them fully occupied with Ti4Fe2 coordination and the third one, with Ti2Fe4 coordination, partially occupied at 91%.
Activation
The main drawback for practical application of TiFe is probably the laborious treatment required after synthesis to promote the first hydrogen absorption. This treatment, usually named “activation”, has been the subject of extensive work and controversy. The fact that TiFe does not readily absorb hydrogen at RT has been attributed to a native passivating layer, which forms at its surface. Indeed, TiFe is sensitive to air moisture and might react with it, forming oxides and hydroxides and then hindering the reaction with hydrogen. Consequently, one needs to apply harsh conditions to induce hydrogen penetration, to break the passivating surface layer or to avoid its formation at the alloy surface.The first description of an activation process for TiFe was reported in the pioneering work of Reilly et al.68. The authors performed a sequence of absorption (up to 65 bar) and desorption (under vacuum) cycles at both high (400–450 °C) and room temperatures. Upon triggering hydrogen absorption/desorption cycles, TiFe undergoes expansion and contraction, respectively, leading to volume changes and inducing the crack of the passivating layer. Since TiFe is a brittle material, fresh and clean TiFe surfaces are uncovered, where hydrogen can promptly be absorbed.
Subsequently, several authors tried to identify the species formed during the oxidation and after activation, with the purpose of better understanding the mechanisms involved in this treatment. Pande et al. investigated the surface of oxidized TiFe by means of electron microscopy.73 In the electron diffraction patterns, they found a phase claimed to be Ti3Fe3O, because this oxide was found unable to absorb hydrogen, making it a relevant candidate as passivating layer.74 Bläsius et al. studied the surface of activated TiFe by Mössbauer spectroscopy, revealing the presence of small Fe clusters.75 Because the signal of iron oxide was not detected, they inferred that only titanium was oxidized. Fe precipitates at the alloy surface were also found by other authors, and this free Fe was claimed to play a catalytic role in the dissociation of hydrogen.76–79 However, Schober et al. studied the activation process by TEM and detected TiO2, TiFe2 and suboxide TiFeOx as surface species.80 They did not observe any elemental Fe and concluded that Fe clusters are only formed after severe oxygen contamination, following the TiFe2 + O2 → TiO2 + 2Fe reaction. Hiebl et al. demonstrated that Ti2FeOx can absorb hydrogen81 and other authors detected this compound during annealing of oxidized TiFe,82,83 casting doubts on the catalytic effect of Fe. Later on, Schlapbach et al. identified an oxide layer mainly consisting of FeIII and Ti<IV on the surface of passivated TiFe.84 After heating, they noticed the formation of Fe and TiO2, suggesting that TiO2 is not an effective catalyst for the reaction, but rather a support for Fe clusters which might split hydrogen molecules.
Reilly et al.58 showed that the composition of the surface layer strongly depends on the annealing conditions and the quantity of oxygen that could be present in the raw materials, synthesis atmosphere or thermal treatment atmosphere. This observation partially explains the controversies in the literature, where each research group followed different treatments. Nonetheless, the procedure suggested by Reilly et al. to ensure TiFe activation remained highly laborious for practical activation.
To simplify the activation, Chu et al.85 synthesized TiFe by means of mechanical alloying, starting from elemental powders of Ti and Fe. The authors prepared an equiatomic TiFe mixture, ball-milled for different duration times. They got amorphous materials that required one hour annealing at 300 °C under 7 bar H2 for activation. Hotta et al.29 also produced TiFe by ball milling pure Ti and Fe, which required an activation at 300 °C and 150 bar of H2. However, compared to the work of Chu et al., Hotta et al. obtained crystalline TiFe that absorbed ∼1 wt% H2. Zaluski et al.32 ball milled Ti and Fe, noticing that the final structure of the composite strongly depends on oxygen contamination. For an oxygen content below 3 at%, TiFe crystallized in the expected CsCl-type structure, whereas at higher oxygen content it became amorphous. Still, both samples required a high temperature to get activated: 300 and 400 °C in vacuum for 0.5 hour for amorphous and crystalline materials, respectively.
Instead of synthesizing TiFe from elemental powders, Emami et al.47 crushed and then ball milled a commercial TiFe ingot. Then, they exposed the powder sample to air for one month and before PCI analysis, activated it in vacuum at 150 °C for 2 hours. Readily, the sample absorbed 1.5 wt% H2. In comparison, the same crushed ingot exposed to air and only annealed did not absorb hydrogen, clearly showing the activation effect induced by ball milling.
Instead of ball milling, Edalati et al.46 used mechanically activated TiFe by high pressure torsion. Small TiFe disks were pressed under 60 kbar in air, and then annealed in vacuum at 150 °C, for 2 hours. The resulting sample stored 1.7 wt% H2 during the first hydrogenation. Later, the same group investigated the effect of groove rolling on TiFe previously activated by high pressure torsion.49 This latter sample required a few absorption/desorption cycles before reaching a capacity of 1.7 wt% H2. However, after air exposure for one day, it remains activated showing the same hydrogen uptake characteristics in the subsequent cycling.
In conclusion, easy activation in TiFe intermetallic compound can be promoted by a mechanical treatment or by the formation of secondary phases. The latter can be attained varying the Ti/Fe ratio with the precipitation of β-Ti or TiFe2 for Ti-rich and Fe-rich alloys, respectively.86–89
Thermodynamics of hydrogen sorption
The first Pressure Composition Isotherm (PCI) curves of the TiFe-H system were monitored by Reilly et al.68. An example of absorption/desorption PCI isotherm at 40 °C is displayed in Fig. 4. Three different regions were observed during the absorption of hydrogen: a steep pressure increase at low H-content (<TiFeH0.1), followed by two pressure plateaus located at PH2 = 1.5 and ∼40 bar and extending from 0.1 < H f.u.−1 < 1, and 1 < H f.u.−1 < 2, respectively. The initial branch (0.1< H f.u.−1) was associated with the formation of the α-solid solution. The first plateau was attributed to the phase transition from the α-phase into the β-monohydrides. The second plateau, which is rather sloppy, was ascribed to the γ-dihydride formation.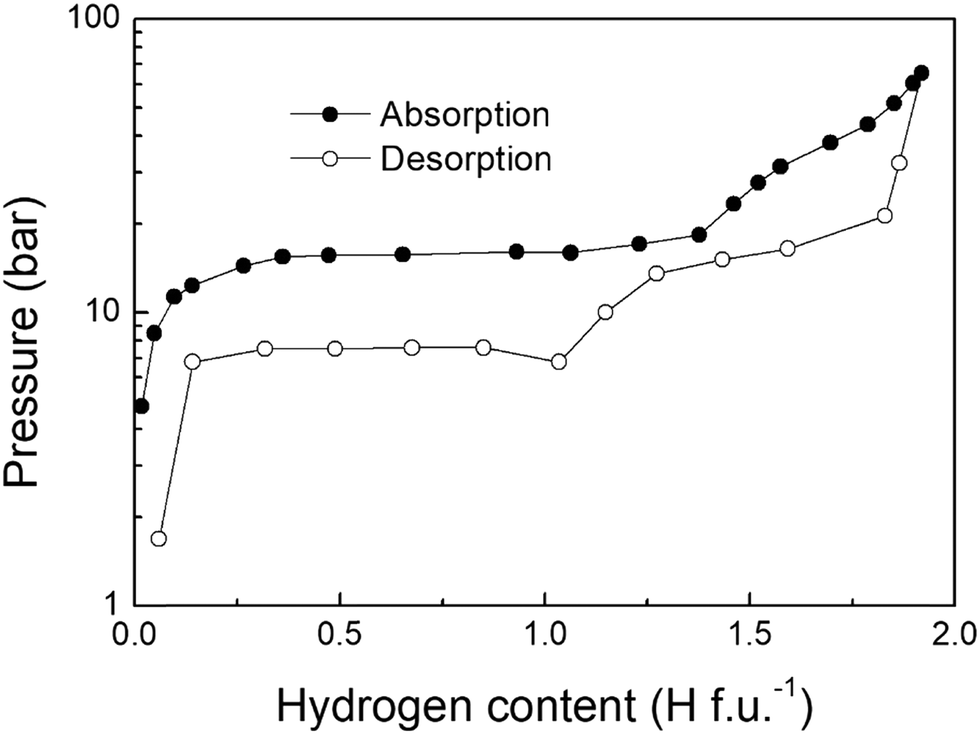 | ||
| Fig. 4 Pressure-composition-isotherm of TiFe performed at 40 °C. | ||
In Fig. 4, the length of the first plateau differs between absorption and desorption, suggesting different hydrogen contents for the intermediate β-phases. As mentioned above, Schefer et al.62 proposed the existence of two different β1 and β2 phases, with similar crystal structures, except for small differences in the occupancy of the octahedral sites. The occurrence of these phases has been further investigated with volumetric measurements by Reidinger et al.90. On absorption, only β2-TiFeH1.4 was observed, while during desorption both β1-TiFeH1.0 and β2-TiFeH1.4 were detected. Based on these results, they suggested that the formation of the β-phases is related to the presence of strains induced by the absorption of hydrogen. This assumption was later confirmed by Reilly et al.91, who, after activation, obtained a free-strain sample by annealing overnight at 800 °C under helium, and they achieved the full hydrogenation state in a single α → γ step, without detecting any β-phase during absorption. However, after the formation of the γ-phase, which induces a volume expansion and thus lattice strains, β-phases appeared again during desorption. In addition, they demonstrated the strain effect on the overall performance of TiFe while cycling. They observed a decrease of the quantity of absorbed hydrogen with the increase of cycle number, mainly due to the disappearance of the upper γ-phase plateau, which shifts to higher pressures. On the other hand, the lower β-phase plateau seems unaffected. After several cycles, the quantity of hydrogen reaches a steady state value, suggesting a saturation of the internal strain. By annealing the samples for 2 days at various temperatures (from 230 to 350 °C), thus reducing the strain, some capacity was recovered, observing again the formation of the γ-phase.
Hydrogenation of amorphous TiFe showed no plateau pressure and low quantity of absorbed H2 (0.3 wt%) while nanocrystalline (5 nm size) TiFe displayed a single plateau with higher hydrogen content (0.9 wt%).32 Haraki et al. prepared TiFe from the elements by two different techniques: mechanical alloying and radio frequency melting.38 After synthesis, the melted sample was later ball milled for 5 hours, and both TiFe specimens were annealed in vacuum for 2 hours at 300 °C before hydrogen absorption analysis. Interestingly, both samples exhibited absorption/desorption plateaus at lower pressures compared to TiFe produced by conventional arc melting. However, the PCI curves differ in shape and quantity of absorbed H2. TiFe prepared by ball milling absorbed 1.3 wt% exhibiting a single plateau, whereas the one prepared by radio frequency melting clearly showed two plateaus, reaching a content of 1.7 wt% H2, and suggesting the formation of both β and γ phases. The disappearance of the γ-phase formation in ball milled TiFe was confirmed by Zadorozhnyy et al.92. After an activation at 400 °C under 10 bar H2 for 0.5 hour, a single plateau was found for absorption. X-ray diffraction analysis after hydrogenation (at 1.1 wt% H2) showed that only the monohydride β was formed.
By monitoring PCI curves at different temperatures, thermodynamic parameters can be determined thanks to the Van’t Hoff equation:
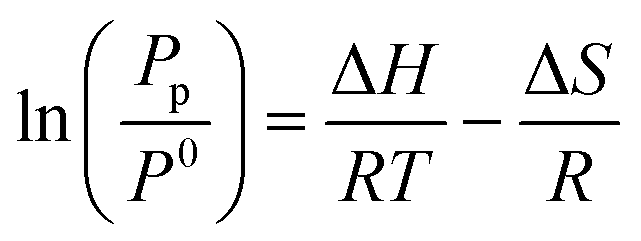
The first thermodynamic data for hydrogen sorption in TiFe were reported by Reilly et al.,68 providing values of ΔHd1st = 28.1 kJ molH2−1 and ΔSd1st = 106 J molH2−1 K−1, during β → α and ΔHd2nd = 33.7 kJ molH2−1 and ΔSd2nd = 132 J molH2−1 K−1, during γ → β desorption reactions, respectively.
Later, a more detailed investigation of the thermodynamics of both hydrogen absorption and desorption reactions in TiFe was performed by Wenzl et al.93 Slight differences were found between absorption/desorption due to hysteresis loop. It is interesting to notice that, during the hydrogen absorption, the transformation α to β for the first plateau is less exothermic than that of β to γ for the second plateau (i.e. −25.4 kJ molH2−1 and −29.8 kJ molH2−1, respectively),93 which is unusual in multi-plateau systems.54,66,68,93. In fact, if the entropy change is assumed to be constant (typically 130 J molH2−1 K−1 as result of the entropy change of hydrogen from the gas phase into the solid state of the hydride). However, enthalpy evaluation from PCI data were confirmed by calorimetric analyses, which allow a direct measurement of the heat of reaction (Q), hence the enthalpy changes (ΔH = −ΔQ).43,93 Results of thermodynamic analyses of hydrogen sorption reactions in TiFe, performed by both PCI measurements and calorimetric experiments, are summarized in Table 1. It is observed that the entropy change in TiFe is anomalously low (99 J molH2−1 K−1) for the first plateau. Likely, this is linked to the high strains that stabilize the beta phase as mentioned above.
| Plateau | −ΔHabs, kJ mol−1 | −ΔSabs, J mol−1 K−1 | ΔHdes, kJ mol−1 | ΔSdes, J mol−1 K−1 | T, °C | References and notes, technique: van’t Hoff |
|---|---|---|---|---|---|---|
| First | 28.1 | 106 | Ref. 68, prepared from zone-refined melting of Ti and Fe in an arc furnace | |||
| Second | 33.7 | 132 | ||||
| First | 25.4 | 104 | 25.6 | 97 | Ref. 93, prepared by induction melting of Ti and Fe | |
| Second | 33.2 | 137 | 31.6 | 125 | ||
| First | 26.4 | 113 | 27.8 | 107 | Ref. 54, nanocrystalline TiFe prepared by mechanical alloying of Ti and Fe powders | |
| Second | ||||||
| First | 31.0 | 118 | Ref. 66, prepared by arc melting of Ti and Fe and loaded with deuterium | |||
| Second | ||||||
| First | 24.3 | 100 | 27.4 | 103 | Ref. 178, prepared by induction melting of Ti and Fe, annealed at 1000 °C for 1 week | |
| Second |
| Plateau | −ΔHabs, kJ mol−1 | −ΔSabs, J mol−1 K−1 | ΔHdes, kJ mol−1 | ΔSdes, J mol−1 K−1 | T, °C | References and notes, technique: calorimetry |
|---|---|---|---|---|---|---|
| First | 24.6 | 92 | 24.8 | 85 | 1 | Ref. 93, prepared by induction melting of Ti and Fe |
| Second | 29.4 | 114 | 30.6 | 111 | 1 | |
| First | 24.2 | 99 | 24.2 | 90 | 25 | |
| Second | 29.8 | 128 | 32.0 | 126 | 25 | |
| First | 23.4 | 100 | 23.4 | 92 | 41 | |
| Second | 26.6 | 122 | 27.8 | 117 | 41 | |
| First | 22.8 | 104 | 23.4 | 98 | 71 | |
| Second | 28.4 | 134 | 28.0 | 126 | 71 | |
| First | 24.9 | 27.4 | 35 | Ref. 43, prepared by mechanical alloying of Ti and Fe powders | ||
| Second | 35 | |||||
| First | 23.0 | 27.2 | 35 | Ref. 43, as received commercial TiFe powder | ||
| Second | 33.9 | 35.2 | 35 |
Kinetics of hydrogen sorption
The kinetics of hydrogen sorption in TiFe was first investigated by Park et al.,94 to determine reaction rates, mechanisms and rate-limiting steps. As shown in Fig. 5, hydrogen absorption rates were determined as a function of the reacted fraction, showing a maximum at ∼25% of reaction that evidences two different mechanisms. They were initially ascribed to nucleation and growth, at the start of reaction, followed by hydrogen diffusion through an enveloping hydride layer after the rate maximum. However, the authors doubted about the first step assignment due to too fast absorption rates. Through a careful analysis of the absorption rate as a function of the gas pressure, they noticed that, before the maximum, it increases linearly, suggesting a step controlled either by H2 mass transfer through cracks or surface chemisorption. SEM images of activated TiFe showed very large cracks facilitating hydrogen transport; therefore, chemisorption was suggested as initial rate-controlling step. Park et al.94 proposed a core–shell model to explain observed kinetics, where the hydrogenation reaction proceeds as follows: hydrogen is chemisorbed on TiFe surface, from which the nucleation and growth of the hydride occurs, and then hydrogen slowly diffuses through the hydride layer in the last step of hydrogenation. Furthermore, Bowman et al. studied hydrogen sorption kinetics by NMR measurement to determine hydrogen diffusivites and activation energies at a local (microscopic) scale.95–98 Compared to other metallic hydrides, which generally exhibit at room temperature a hydrogen diffusion coefficient in the range 10−6–10−8 cm2 s−1,99 Bowman et al. found a value of the order of 10−12 cm2 s−1,95, for β-TiFeH. This slow diffusion was attributed to the ordered structure restricting possible diffusion path, since H1 sites are almost fully occupied, while only a few H atoms are located in H2 sites.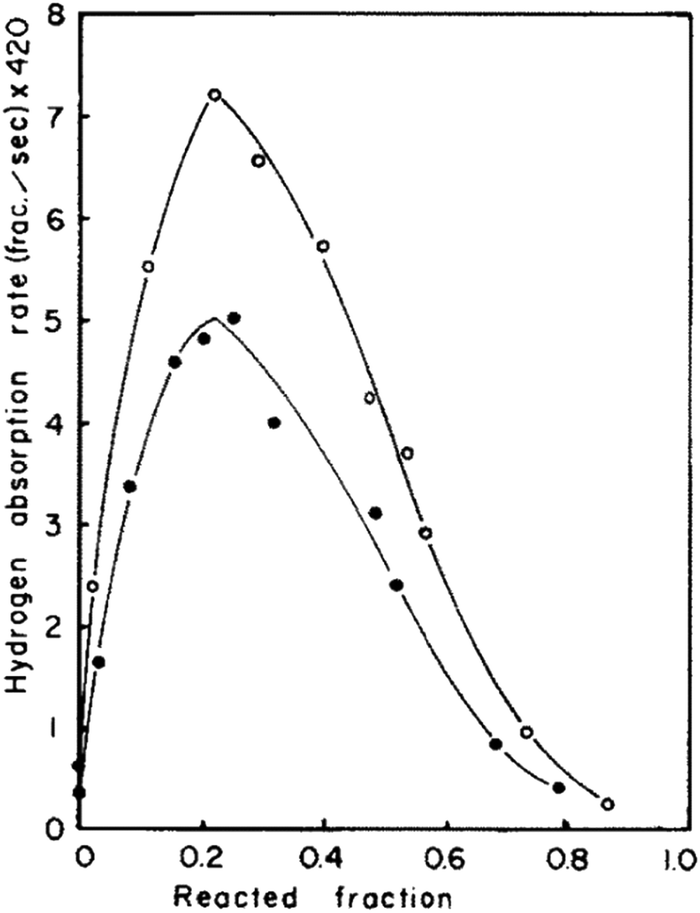 | ||
| Fig. 5 Hydrogen absorption rate as a function of reacted fraction at 20 °C. The curve with black dots was obtain under 20 bar of hydrogen, whereas the one with white dots under 24 bar. Reproduced with permission from ref. 94. | ||
Cycling and resistance to poisoning
One major fact to consider for practical application is the alloy degradation when cycled for long periods. Changes in the PCI curves of TiFe after cycling were reported by Goodell et al.100. Freshly activated TiFe exhibited two plateaus for the formation of β-monohydrides and γ-dihydride, with large hysteresis between absorption and desorption. However, with the increase of cycle numbers, the hysteresis gap decreases, but the γ-phase plateau shifted towards higher-pressure values, until it disappeared. Similar results were found also by Reilly et al.,91 who showed that the PCI curves change in shape during several cycles, until they stabilize becoming almost independent on the cycle number. The authors supposed that, until lattice strain and defects do not reach saturation, the isotherms keep changing. This implies that the disappearance of the γ-phase is due to the presence of internal stress and defects, due to an expansion and shrinking of the unit cell during hydrogenation and dehydrogenation, respectively. Further analysis performed by Ahn et al.101 confirmed the reduction of hydrogen stored due to the disappearance of γ-dihydride because of stress. Moreover, they observed a decrease also in the hydrogenation rate with the number of cycles. The authors suggested that, besides lattice distortion, also the formation of stable hydrides (TiHx) due to alloy disproportionation during cycling could be a cause of the degradation. Indeed, stable hydrides do not release hydrogen, and their formation hinders hydrogenation on the TiFe surface due to rearrangements of neighbour atoms and the introduction of lattice strain.Besides cycling-induced degradation, contaminants in the hydrogen gas such as H2O, O2, CO2 and CO have a prominent influence. Adsorption of impurities at active sites on TiFe surface will prevent hydrogen molecules to dissociate during the chemisorption step. As demonstrated by Sandrock et al.102, this passivation is generally manifested as a decrease in the reaction rate or a reduction in the storage capacity. These authors have investigated the effect of H2 containing 300 ppm of H2O, O2 and CO on the cycling of TiFe. H2O and O2 split on the surface, forming a thick passivating layer composed by complex oxides. The effect of this layer is similar in both cases, exhibiting a continuous decrease in the quantity of hydrogen stored during cycling. The main observed difference is that O2 reacts faster than H2O at the surface. In both cases, TiFe could be partially reactivated cycling at moderate temperature (80 °C) with pure H2. On the other hand, CO has shown to be more detrimental than H2O and O2. It is adsorbed in less than one minute, completely deactivating TiFe in a few cycles. However, TiFe poisoned by CO was easily reactivated by simply cycling at room temperature under pure H2. Additional information was provided by Block et al.,103 who investigated also the effect of CO2, CH4 and H2S in various concentrations. The presence of 10 vol% CH4 showed a stable and slight decrease in the capacity and reaction rate of TiFe. Surprisingly, when pure H2 was provided again, the active material exhibited a reaction rate even faster than before, restoring also its hydrogen absorption capacity. The authors suggested that CH4 does not passivate TiFe, and the decrease during cycling was probably due to the lower H2 partial pressure in presence of methane. In the presence of CO2 there is a constant decrease in the storage capacity during cycling and, moreover, the absorption rate decreases with the increase of impurity concentration into H2. A concentration of 1 vol% CO2 in the gas stream was enough to fully passivate TiFe after two cycles. The sample was reactivated by cycling with pure hydrogen at 127 °C. Introducing 0.2 vol% H2S did not affect the reaction rate, but it strongly reduced the quantity of hydrogen absorbed upon cycling, so that few cycles were enough to completely deactivate TiFe. Even performing intensive heat treatments, the authors were not able to reactivate the sample due to the presence of a stable sulphur layer on the surface of TiFe, which inhibited the absorption of hydrogen.
From these results, it can be concluded that TiFe hydrogenation properties easily deteriorate in the presence of contaminants. To face this issue, two main strategies were suggested: the design and implementation of reactivation systems or the enhancement of TiFe resistance to poisoning. Resistance to passivation reaction might be induced by adding a secondary phase, but still no complete resistance to contamination has been reported in the literature for TiFe. Hence, leaks and gas purity must be carefully checked for long-cycling applications for hydrogen storage.
All properties mentioned above for binary TiFe can be tailored by chemical substitutions and this topic will be discussed in detail in the following sections.
Modifications of TiFe properties by substitutions
Extensive studies have been performed to synthetize and characterize substituted TiFe intermetallic compounds with many elements, as it can be visualized in the periodic table reported in Fig. 2.The substitution of Fe or of Ti has been the subject of recent papers that evidence the role of Ti-substitution or Fe-substitution and their effect on hydrogen storage properties.104 Substitution can include either or both A-type (Ti) or B-type (Fe) site substitution, as it will be discussed later on, depending on each element. Optimization of operational pressure range, a theoretical understanding of alloy thermodynamics, the role of secondary phases’ formation or TiFe single phase domain compositional stretching need to be better considered in a full picture of available studies. Substitution can significantly lower plateau pressure or make full hydrogenation more difficult, decreasing the usable capacity. On the other hand, for example, Mn can change equilibrium pressure introducing a smoothing effect, levering plateau pressure in a narrow pressure range and maximizing the reversible capacity.
Vivid literature studies on substitutional effects have been carried out aiming to tailor hydrogenation properties of TiFe, indicating that substitution for Fe is dominant. In the following, mono-substituted system will be considered first, then we will specifically focus on the manganese-substituted system and finally prominent examples of Ti(Fe,Mn) multi-substituted alloys will be presented. Throughout the description of literature results, when studied, quaternary alloy are reported as well, while a focus on substitutional effect and Ti or Fe substitution are commented in detail in the discussion section. Few examples of reported additives or catalysts (as nanoparticles or oxides) will be cited and discussed too when relevant.
Substitutional elements are classified according to their location in the periodic table. Investigated TiFe–M systems, their hydrogen storage properties and thermodynamics are summarized in Table 2.
| TiFe–M | Ti, at% | Fe, at% | M, at% | Secondary, phases | TiFe, a (Å) | Capacity (wt%), conditions | PCI, thermodynamics | Ref. |
|---|---|---|---|---|---|---|---|---|
| a: absorption, d: desorption, 1st: 1st plateau, 2nd: 2nd plateau, RT: room temperature. | ||||||||
| –Mg | ||||||||
| TiFeMg0.04 | 49.0 | 49.0 | 2.0 | 2.982 | 1.10 | 22 °C | 108 | |
| 22 °C, 20 bar | ||||||||
| TiFeMg0.08 | 48.0 | 48.0 | 4.0 | α-Fe | 2.990 | 108 | ||
| TiFeMg0.13 | 47.0 | 47.0 | 6.0 | α-Fe | 2.998 | 108 | ||
| –Be | ||||||||
| TiFe0.90Be0.10 | 50.0 | 45.0 | 5.0 | 2.977 | 1.05 | 21,d 50, 80 °C | 110 | |
| 50 °C, 10 bar | ΔH = 29.9 kJ mol−1 | |||||||
| ΔS = 108 J mol−1 K−1 | ||||||||
| TiFe0.80Be0.20 | 50.0 | 40.0 | 10.0 | 2.977 | 1.00 | 21,d 50, 80, 125d °C | 109 | |
| 2.979 | 50 °C, 10 bar | 21,d 50, 80, 125d °C | 110 | |||||
| ΔH = 31.2 kJ mol−1 | ||||||||
| ΔS = 105 J mol−1 K−1 | ||||||||
| TiFe0.70Be0.30 | 50.0 | 35.0 | 15.0 | 2.980 | 0.95 | 50, 80, 125d °C | 110 | |
| 50 °C, 10 bar | ΔH = 35.4 kJ mol−1 | |||||||
| ΔS = 108 J mol−1 K−1 | ||||||||
| –Zr | ||||||||
| TiFeZr0.04 | 49.0 | 49.0 | 2.0 | 2.979 | 1.10 | 22 °C | 117 | |
| 22 °C, 37 bar | ||||||||
| TiFeZr0.04 | 48.9 | 48.9 | 2.2 | TiFe2 | 1.60 | 40 °C | 119 | |
| 40 °C, 20 bar | ||||||||
| (Ti2Fe) | 1.60 | 111 | ||||||
| RT, 45 bar | ||||||||
| (Ti2Fe, TiFe2) | 2.983 | 115, 116 | ||||||
| (TiFe2) | 2.980 | |||||||
| 120 | ||||||||
| TiFeZr0.05 | 48.8 | 48.8 | 2.4 | 2.983 | RT | 114 | ||
| TiFeZr0.08 | 48.0 | 48.0 | 4.0 | 2.995 | 117 | |||
| TiFeZr0.09 | 47.9 | 47.8 | 4.3 | (Ti2Fe, TiFe2) | 2.986 | 116 | ||
| TiFeZr0.10 | 47.7 | 47.7 | 4.6 | (Ti2Fe, TiFe2) | 2.980 | 115 | ||
| TiFeZr0.13 | 47.0 | 47.0 | 6.0 | 2.995 | 117 | |||
| TiFeZr0.16 | 46.4 | 46.4 | 7.2 | (Ti2Fe, TiFe2) | 2.984 | 115 | ||
| TiFeZr0.17 | 46.0 | 46.0 | 8.0 | Fe | 2.997 | 117 | ||
| TiFeZr0.22 | 45.1 | 45.1 | 9.8 | (Ti2Fe, TiFe2) | 2.981 | 115 | ||
| TiFe0.95Zr0.05 | 50.0 | 47.5 | 2.5 | 2.980 | RT | 114 | ||
| Ti1.3Fe0.80Zr0.20 | 56.5 | 34.8 | 8.7 | Ti2Fe, TiFe2, Ti | 1.20 | 100 °C | 112 | |
| 200 °C, 20 bar | ||||||||
| Ti0.99FeZr0.01 | 49.5 | 50.0 | 0.5 | 30 °Cd | 118 | |||
| Ti0.95FeZr0.05 | 47.5 | 50.0 | 2.5 | 2.985 | RT | 114 | ||
| Ti0.90FeZr0.10 | 45.0 | 50.0 | 5.0 | 20, 40 °C d | 113 | |||
| −ΔHa1st = 21.8 kJ mol−1 | 118 | |||||||
| 30,d 45, 60 °C | ||||||||
| ΔHd = 28.9 kJ mol−1 | ||||||||
| ΔSd = 105 J mol−1 K−1 | ||||||||
| −ΔHa = 26.2 kJ mol−1 | ||||||||
| −ΔSa = 102 J mol−1 K−1 | ||||||||
| Ti0.84FeZr0.16 | 42.0 | 50.0 | 8.0 | 50, 100, 150, 200 °Ca | 236 | |||
| Ti0.80FeZr0.20 | 40.0 | 50.0 | 10.0 | 40 °Cd | 113 | |||
| 30 °Cd | 118 | |||||||
| Ti0.50FeZr0.50 | 25.0 | 50.0 | 25.0 | 40 °C d | 113 | |||
| –Hf | ||||||||
| TiFeHf0.02 | 49.4 | 49.4 | 1.2 | TiFe2 | 2.982 | RT | 121 | |
| TiFeHf0.04 | 48.9 | 48.9 | 2.2 | TiFe2 | 2.986 | RT | 121 | |
| TiFeHf0.07 | 48.3 | 48.3 | 3.4 | TiFe2 | 2.989 | RT | 121 | |
| TiFeHf0.09 | 47.8 | 47.8 | 4.4 | TiFe2 | 2.990 | RT | 121 | |
| –V | ||||||||
| Ti0.95FeV0.06 | 47.1 | 49.8 | 3.1 | 1.10 | 126 | |||
| 54 °C, 20 bar | ||||||||
| Ti0.94FeV0.06 | 47.0 | 50.0 | 3.0 | TiFe2, Ti4Fe2O | 2.976 | 30 °Cd | 104 | |
| Ti0.96FeV0.04 | 48.0 | 50.0 | 2.0 | 25 °Cd | 237 | |||
| Ti0.90FeV0.10 | 45.0 | 50.0 | 5.0 | TiFe2, Ti4Fe2O | 2.976 | 30 °Cd | 104 | |
| Ti0.80FeV0.20 | 40.0 | 50.0 | 10.0 | TiFe2 | 2.971 | 125 | ||
| Ti0.96Fe0.94V0.10 | 48.0 | 47.0 | 5.0 | 20, 40, 60 °Cd | 124 | |||
| Ti0.94Fe0.96V0.10 | 47.0 | 48.0 | 5.0 | Ti4Fe2O | 2.980 | 30 °Cd | 104 | |
| 2.979 | 25, 65, 100 °C | 125 | ||||||
| ΔHd = 27.0 kJ mol−1 | ||||||||
| ΔSd = 99 J mol−1 K−1 | ||||||||
| −ΔHa = 29.2 kJ mol−1 | ||||||||
| −ΔSa = 111 J mol−1 K−1 | ||||||||
| Ti0.96Fe0.68V0.36 | 48.0 | 34.0 | 18.0 | Bcc, TiFe2 | 3.000 | 125 | ||
| TiFe0.90V0.05 | 51.3 | 46.2 | 2.6 | TiFe2, β-Ti | 2.987 | 1.96 | 25 °C | 238 |
| 25 °C, 25 bar | ||||||||
| TiFe0.94V0.06 | 50.0 | 47.0 | 3.0 | β-Ti, Ti4Fe2O | 2.989 | 30 °Cd | 104 | |
| TiFe0.90V0.10 | 50.0 | 45.0 | 5.0 | TiFe2, β-Ti | 2.989 | 1.68 | 25 °C | 238 |
| 25 °C, 25 bar | ||||||||
| β-Ti, Ti4Fe2O | 2.995 | 30 °Cd | 104 | |||||
| TiFe0.80V0.20 | 50.0 | 40.0 | 10.0 | 54, 79, 102 °Cd | 212 | |||
| TiFe0.50V0.17 | 60.0 | 30.0 | 10.0 | Bcc | 2.999 | 125 | ||
| –Nb | ||||||||
| TiFeNb0.04 | 49.0 | 49.0 | 2.0 | 2.979 | 35 °C | 129 | ||
| −ΔHa1st = 28.2 kJ mol−1 | ||||||||
| ΔHd1st = 28.5 kJ mol−1 | ||||||||
| −ΔHa2nd = 35.5 kJ mol−1 | ||||||||
| ΔHd2nd = 38.2 kJ mol−1 | ||||||||
| α-Fe, TiFe2 | 2.984 | 35 °C | 129 | |||||
| −ΔHa1st = 23.2 kJ mol−1 | ||||||||
| ΔHd1st = 25.3 kJ mol−1 | ||||||||
| TiFeNb0.08 | 48.0 | 48.0 | 4.0 | α-Fe | 129 | |||
| TiFe0.90Nb0.10 | 50.0 | 45.0 | 5.0 | β-Ti | 2.989 | 87 | ||
| Ti0.96FeNb0.04 + 1.0 wt% Fe2O3 | TiFe2, Ti10Fe7O3 | 30 °C | 127 | |||||
| Ti0.92FeNb0.08 + 1.0 wt% Fe2O3 | TiFe2, Ti10Fe7O3 | 30 °C | 127 | |||||
| Ti0.90FeNb0.10 + 1.0 wt% Fe2O3 | TiFe2, Ti10Fe7O3 | 30 °C | 127 | |||||
| Ti0.88FeNb0.12 + 1.0 wt% Fe2O3 | TiFe2, Ti10Fe7O3 | 30 °C | 127 | |||||
| –Ta | ||||||||
| TiFe0.90Ta0.10 | 50.0 | 45.0 | 5.0 | 2.990 | 40 °C | 130 | ||
| TiFe0.80Ta0.20 | 50.0 | 40.0 | 10.0 | 2.994 | 40 °C | 130 | ||
| –Cr | ||||||||
| TiFeCr0.04 | 49.0 | 49.0 | 2.0 | 2.973 | 134 | |||
| TiFeCr0.08 | 48.0 | 48.0 | 4.0 | 2.974 | 1.00 | 22 °C | 134 | |
| 22 °C, 60 bar | ||||||||
| TiFeCr0.13 | 47.0 | 47.0 | 6.0 | 2.972 | 134 | |||
| TiFeCr0.17 | 46.0 | 46.0 | 8.0 | α-Fe | 2.972 | 134 | ||
| TiFe0.95Cr0.05 | 50.0 | 47.5 | 2.5 | TiCr2 | 50 °Cd | 131 | ||
| 40 °Cd | 136 | |||||||
| TiFe0.90Cr0.10 | 50.0 | 45.0 | 5.0 | ΔHd = 30.1 kJ mol−1 | 215 | |||
| ΔSd = 101 J mol−1 K−1 | ||||||||
| TiCr2 | 50 °Cd | 131 | ||||||
| 40 °Cd | 136 | |||||||
| Ti | 2.989 | 10, 30, 50 °C | 132 | |||||
| ΔHd = 34.2 kJ mol−1 | ||||||||
| ΔSd = 110 J mol−1 K−1 | ||||||||
| −ΔHa = 27.2 kJ mol−1 | ||||||||
| −ΔSa = 92 J mol−1 K−1 | ||||||||
| TiFe0.80Cr0.20 | 50.0 | 40.0 | 10.0 | ΔHd = 35.6 kJ mol−1 | 215 | |||
| ΔSd = 108 J mol−1 K−1 | ||||||||
| −ΔHa = 33.1 kJ mol−1 | ||||||||
| −ΔSa = 103 J mol−1 K−1 | ||||||||
| 24, 40, 60 °C d | 212 | |||||||
| TiFe0.70Cr0.16 | 53.8 | 37.6 | 8.6 | TiCr2 | 2.993 | 1.80 | 141 | |
| 25 °C, 20 bar | ||||||||
| –Mo | ||||||||
| TiFe0.90Mo0.10 | 50.0 | 45.0 | 5.0 | 40 °Cd | 136 | |||
| β-Ti | 2.989 | 87 | ||||||
| 2.989 | 40 °C | 130 | ||||||
| TiFe0.80Mo0.20 | 50.0 | 40.0 | 10.0 | 2.992 | 40 °C | 130 | ||
| –Co | ||||||||
| TiFeCo0.04 | 49.0 | 49.0 | 2.0 | TiFe2 | 2.988 | 35 °C | 129 | |
| −ΔHa1st = 26.0 kJ mol−1 | 129 | |||||||
| ΔHd1st = 29.1 kJ mol−1 | ||||||||
| −ΔHa2nd = 28.4 kJ mol−1 | ||||||||
| ΔHd2nd = 30.5 kJ mol−1 | ||||||||
| α-Fe, TiFe2 | 2.975 | 35 °C | ||||||
| −ΔHa1st = 25.7 kJ mol−1 | ||||||||
| ΔHd1st = 26.7 kJ mol−1 | ||||||||
| TiFeCo0.08 | 48.0 | 48.0 | 4.0 | α-Fe | 129 | |||
| TiFe0.90Co0.10 | 50.0 | 45.0 | 5.0 | ΔHd = 30.6 kJ mol−1 | 215 | |||
| ΔSd = 106 J mol−1 K−1 | ||||||||
| 40 °C d | 136 | |||||||
| 20, 30, 40, 50 °C | 138 | |||||||
| TiFe0.80Co0.20 | 50.0 | 40.0 | 10.0 | ΔHd = 32.7 kJ mol−1 | 215 | |||
| ΔSd = 109 J mol−1 K−1 | ||||||||
| 52.5, 82, 110 °C d | 212 | |||||||
| −ΔHa = 31.4 kJ mol−1 | ||||||||
| −ΔSa = 102 J mol−1 K−1 | ||||||||
| TiFe0.50Co0.50 | 50.0 | 25.0 | 25.0 | 1.10 | 80, 100, 120 °C d | 137 | ||
| RT, 30 bar | ΔHd = 42.3 kJ mol−1 | |||||||
| ΔSd = 123 J mol−1 K−1 | ||||||||
| 80, 100, 120 °C | 139 | |||||||
| ΔHd = 49.1 kJ mol−1 | ||||||||
| ΔSd = 126 J mol−1 K−1 | ||||||||
| −ΔHa = 45.3 kJ mol−1 | ||||||||
| −ΔSa = 151 J mol−1 K−1 | ||||||||
| TiFe0.30Co0.70 | 50.0 | 15.0 | 35.0 | 1.10 | 137 | |||
| RT, 30 bar | ||||||||
| TiFe0.05Co0.95 | 50.0 | 2.5 | 47.5 | 1.10 | 137 | |||
| RT, 30 bar | ||||||||
| –Ni | ||||||||
| Ti1.10Fe0.90Ni0.10 | 52.4 | 42.9 | 4.8 | Ti2Ni, TiNi3 | 40, 60, 80 °C | 146 | ||
| ΔHd = 17.9 kJ mol−1 | ||||||||
| ΔSd = 57 J mol−1 K−1 | ||||||||
| −ΔHa = 16.6 kJ mol−1 | ||||||||
| −ΔSa = 59 J mol−1 K−1 | ||||||||
| TiFeNi0.07 | 48.3 | 48.3 | 3.4 | TiFe2 | 40 °C | 119 | ||
| TiFeNi0.50 | 40.0 | 40.0 | 20.0 | C14 | 60 °C | 145 | ||
| TiFe0.90Ni0.10 | 50.0 | 45.0 | 5.0 | 2.981 | ΔHd = 34.8 kJ mol−1 | 215 | ||
| ΔSd = 112 J mol−1 K−1 | ||||||||
| −ΔHa = 31.4 kJ mol−1 | ||||||||
| −ΔSa = 106 J mol−1 K−1 | ||||||||
| −ΔHa = 31.8 kJ mol−1 | 142 | |||||||
| 60 °C | 145 | |||||||
| 20, 30, 40, 50 °C | 138 | |||||||
| TiFe2, Ti2Fe | 2.977 | 40, 60, 80, 100 °C | 147 | |||||
| ΔH = 22.3 kJ mol−1 | ||||||||
| ΔS = 85 J mol−1 K−1 | ||||||||
| TiFe0.85Ni0.15 | 50.0 | 42.5 | 7.5 | 2.984 | ΔHd = 37.7 kJ mol−1 | 215 | ||
| ΔSd = 118 J mol−1 K−1 | ||||||||
| −ΔHa = 32.2 kJ mol−1 | ||||||||
| −ΔSa = 103 J mol−1 K−1 | ||||||||
| 50 °C | 138 | |||||||
| TiFe0.80Ni0.20 | 50.0 | 40.0 | 10.0 | 1.30 | ΔH1st = 41.2 kJ mol−1 | 186 | ||
| ΔS1st = 119 J mol−1 K−1 | ||||||||
| 50, 80 °C | 215 | |||||||
| ΔHd = 41.9 kJ mol−1 | ||||||||
| ΔSd = 118 J mol−1 K−1 | ||||||||
| −ΔHa = 39.4 kJ mol−1 | ||||||||
| −ΔSa = 113 J mol−1 K−1 | ||||||||
| 62.5, 91, 122 °Cd | 212 | |||||||
| −ΔHa = 39.7 kJ mol−1 | ||||||||
| −ΔSa = 116 J mol−1 K−1 | ||||||||
| 55 °C,a 30 cycles | 148 | |||||||
| −ΔHa = 37.2 kJ mol−1 | ||||||||
| 60 °C | 145 | |||||||
| 1.50 | 150 °C | 150 | ||||||
| 28 °C, 20 bar | −ΔHa = 41.0 kJ mol−1 | |||||||
| 50 °C | 138 | |||||||
| TiFe2, Ti2Fe | 2.984 | 40, 60, 80, 100 °C | 147 | |||||
| ΔH = 25.1 kJ mol−1 | ||||||||
| ΔS = 76 J mol−1 K−1 | ||||||||
| TiFe0.60Ni0.40 | 50.0 | 30.0 | 20.0 | 60 °C | 145 | |||
| TiFe2, Ti2Fe | 2.992 | 40, 60, 80, 100 °C | 147 | |||||
| ΔH = 26.4 kJ mol−1 | ||||||||
| ΔS = 58 J mol−1 K−1 | ||||||||
| TiFe0.50Ni0.50 | 50.0 | 25.0 | 25.0 | 3.001 | 239 | |||
| ΔHd = 45.2 kJ mol−1 | 215 | |||||||
| ΔSd = 107 kJ mol−1 K−1 | ||||||||
| −ΔHa = 43.1 kJ mol−1 | ||||||||
| −ΔSa = 102 J mol−1 K−1 | ||||||||
| TiFe0.40Ni0.60 | 50.0 | 20.0 | 30.0 | 60 °C | 145 | |||
| TiFe0.25Ni0.75 | 50.0 | 12.5 | 37.5 | 3.010 | 144,239,240 | |||
| Ti0.90FeNi0.10 | 45.0 | 50.0 | 5.0 | C14 | 60 °C | 145 | ||
| Ti0.80FeNi0.20 | 40.0 | 50.0 | 10.0 | C14 | 60 °C | 145 | ||
| –Pd | ||||||||
| TiFe + <1 wt% Pd | RT | 156 | ||||||
| TiFe0.90Pd0.05 | 51.3 | 46.2 | 2.7 | 2.980 | 0 °Cd | 151 | ||
| TiFe0.90Pd0.10 | 50.0 | 45.0 | 5.0 | 3.000 | 0 °Cd | 151 | ||
| TiFe0.80Pd0.20 | 50.0 | 40.0 | 10.0 | 3.030 | 0 °Cd | 151 | ||
| –Cu | ||||||||
| TiFeCu0.04 | 49.0 | 49.0 | 2.0 | 2.978 | 1.46 | 22 °C | 117 | |
| 22 °C, 40 bar | ||||||||
| TiFeCu0.10 | 47.6 | 47.6 | 4.8 | 40 °Cd | 136 | |||
| TiFeCu0.11 | 47.5 | 47.5 | 5.0 | 2.982 | 117 | |||
| TiFeCu0.17 | 46.0 | 46.0 | 8.0 | 2.982 | 117 | |||
| TiFeCu0.22 | 45.0 | 45.0 | 10.0 | TiFe2 | 2.980 | 117 | ||
| TiFe0.80Cu0.20 | 50.0 | 40.0 | 10.0 | 55, 87, 116 °Cd | 212 | |||
| −ΔHa = 37.2 kJ mol−1 | ||||||||
| −ΔSa = 111 J mol−1 K−1 | ||||||||
| Ti0.98FeCu0.02 + 0.5 wt% Fe2O3 | TiFe2, Ti10Fe7O3 | 30 °C | 127 | |||||
| Ti0.96FeCu0.04 + 0.5 wt% Fe2O3 | TiFe2, Ti10Fe7O3 | 30 °C | 127 | |||||
| Ti0.94FeCu0.06 + 0.5 wt% Fe2O3 | TiFe2, Ti10Fe7O3 | 30 °C | 127 | |||||
| Ti0.92FeCu0.08 + 0.5 wt% Fe2O3 | TiFe2, Ti10Fe7O3 | 30 °C | 127 | |||||
| Ti0.90FeCu0.10 + 0.5 wt% Fe2O3 | TiFe2, Ti10Fe7O3 | 30 °C | 127 | |||||
| –Y | ||||||||
| TiFeY0.04 | 48.9 | 48.9 | 2.2 | TiFe2, Y | 1.79 | 157 | ||
| RT, 25 bar | ||||||||
| TiFeY0.07 | 48.3 | 48.3 | 3.4 | TiFe2, Y | 1.75 | 157 | ||
| RT, 25 bar | ||||||||
| TiFeY0.09 | 47.8 | 47.8 | 4.4 | β-Ti, Y | 1.71 | 157 | ||
| RT, 25 bar | ||||||||
| –La | ||||||||
| TiFe0.78La0.03 | 55.4 | 43.2 | 1.4 | Ti, La | 20, 30, 65 °C | 158 | ||
| ΔHd = 26.7 kJ mol−1 | ||||||||
| −ΔHa = 24.8 kJ mol−1 | ||||||||
| –Ce | ||||||||
| TiFe0.94Ce0.06 | 50.0 | 47.0 | 3.0 | 2.983 | 159 | |||
| –Mm | ||||||||
| TiFe + 4 wt% Mm | 27 °Cd | 160 | ||||||
| Ti1.3Fe + 1.5 wt% Mm | β-Ti | 25, 45, 60 °Cd | 161 | |||||
| Ti1.3Fe + 4.0 wt% Mm | β-Ti | 25, 45, 60 °Cd | 161 | |||||
| Ti1.3Fe + 4.5 wt% Mm | Ti, Ti2Fe | 2.978 | 1.71, RT | RT, 100 °Cd | 162 | |||
| 1.90, 100 °C | ||||||||
| 20 bar | ||||||||
| Ti1.4Fe + 4.5 wt% Mm | Ti, Ti2Fe | 2.978 | 1.85 | 100 °Cd | 162 | |||
| 100 °C, 20 bar | ||||||||
| Ti1.5Fe + 4.5 wt% Mm | Ti, Ti2Fe | 2.978 | 1.17 | 100 °Cd | 162 | |||
| 100 °C, 20 bar | ||||||||
| –Al | ||||||||
| TiFeAl0.11 | 47.5 | 47.5 | 5.0 | 2.980 | 0.99 | 22 °C | 134 | |
| 22 °C, 60 bar | ||||||||
| TiFeAl0.22 | 45.0 | 45.0 | 10.0 | 2.982 | 134 | |||
| TiFeAl0.35 | 42.5 | 42.5 | 15.0 | 2.988 | 134 | |||
| TiFeAl0.50 | 40.0 | 40.0 | 20.0 | 2.991 | 134 | |||
| TiFe0.98Al0.02 | 50.0 | 49.0 | 1.0 | 30 °C | 241 | |||
| ΔHd1st = 26.4 kJ mol−1 | ||||||||
| ΔSd1st = 100 J mol−1 K−1 | ||||||||
| −ΔHa1st = 23.8 kJ mol−1 | ||||||||
| −ΔSa1st = 98 J mol−1 K−1 | ||||||||
| 2.977 | 50 °Cd | 138 | ||||||
| TiFe0.96Al0.04 | 50.0 | 48.0 | 2.0 | 2.988 | 40 °Cd | 122 | ||
| 30 °C | 241 | |||||||
| ΔHd1st = 23.4 kJ mol−1 | ||||||||
| ΔSd1st = 95 J mol−1 K−1 | ||||||||
| −ΔHa1st = 21.9 kJ mol−1 | ||||||||
| −ΔSa1st = 97 J mol−1 K−1 | ||||||||
| 2.986 | 1.10 | 25,d 50, 80 °C | 110 | |||||
| 50 °C, 10 bar | ΔH = 29.9 kJ mol−1 | |||||||
| ΔS = 106 J mol−1 K−1 | ||||||||
| TiFe0.95Al0.05 | 50.0 | 47.5 | 2.5 | 2.985 | 50 °Cd | 138 | ||
| TiFe0.94Al0.06 | 50.0 | 47.0 | 3.0 | 30, 40, 50 °C | 164 | |||
| 30 °C | 241 | |||||||
| ΔHd1st = 21.4 kJ mol−1 | ||||||||
| ΔSd1st = 89 J mol−1 K−1 | ||||||||
| −ΔHa1st = 19.6 kJ mol−1 | ||||||||
| −ΔSa1st = 88 J mol−1 K−1 | ||||||||
| TiFe0.92Al0.08 | 50.0 | 46.0 | 4.0 | 30 °C | 241 | |||
| ΔHd1st = 29.5 kJ mol−1 | ||||||||
| ΔSd1st = 109 J mol−1 K−1 | ||||||||
| −ΔHa1st = 22.7 kJ mol−1 | ||||||||
| −ΔSa1st = 91 J mol−1 K−1 | ||||||||
| TiFe0.90Al0.10 | 50.0 | 45.0 | 5.0 | 2.989 | 40 °Cd | 122 | ||
| 30 °C | 241 | |||||||
| ΔHd1st = 20.8 kJ mol−1 | 138 | |||||||
| ΔSd1st = 78 J mol−1 K−1 | 110 | |||||||
| −ΔHa1st = 18.3 kJ mol−1 | ||||||||
| −ΔSa1st = 73 J mol−1 K−1 | ||||||||
| 2.997 | 50 °Cd | |||||||
| 2.996 | 1.00 | 25,d 50, 80 °C | ||||||
| 50 °C, 10 bar | ΔH = 31.2 kJ mol−1 | |||||||
| ΔS = 105 J mol−1 K−1 | ||||||||
| TiFe0.80Al0.20 | 50.0 | 40.0 | 10.0 | 3.015 | 0.90 | 50, 80, 120 °Cd | 110 | |
| 50 °C, 10 bar | ΔH = 35.0 kJ mol−1 | |||||||
| ΔS = 100 J mol−1 K−1 | ||||||||
| TiFe0.76Al0.24 | 50.0 | 38.0 | 12.0 | 3.030 | 40 °Cd | 122 | ||
| TiFe0.60Al0.40 | 50.0 | 30.0 | 20.0 | 3.070 | 122 | |||
| –Si | ||||||||
| TiFe0.94Si0.02 | 51.0 | 48.0 | 1.0 | TiFe2, β-Ti | 40 °Cd | 122 | ||
| TiFe0.87Si0.03 | 52.0 | 45.0 | 3.0 | TiFe2, β-Ti | 40 °Cd | 122 | ||
| TiFe0.83Si0.10 | 52.0 | 43.0 | 5.0 | TiFe2, β-Ti | 40 °Cd | 122 | ||
| –Sn | ||||||||
| TiFe0.98Sn0.02 | 50.0 | 49.0 | 1.0 | TiFe2 | 1.60 | RT,a,d 30, 40, 50, 60 °C d | 165 | |
| ΔH = 27.0 kJ mol−1 | ||||||||
| ΔS = 103 J mol−1 K−1 | ||||||||
| TiFe0.95Sn0.05 | 50.0 | 47.5 | 2.5 | TiFe2 | 1.40 | RT,a,d 30, 40, 50, 60 °Cd | 165 | |
| ΔH = 26.6 kJ mol−1 | ||||||||
| ΔS = 103 J mol−1 K−1 | ||||||||
| –B | ||||||||
| TiFeB0.001 | 49.98 | 49.97 | 0.05 | TiFe2, Ti | 50 °C | 166 | ||
| Ti1.1FeB0.001 | 52.35 | 47.60 | 0.05 | TiFe2, Ti | 50 °C | 166 | ||
| –C | ||||||||
| TiFeC0.001 | 49.98 | 49.97 | 0.05 | TiFe2, Ti | 50 °C | 166 | ||
| Ti1.1FeC0.001 | 52.35 | 47.60 | 0.05 | TiFe2, Ti | 50 °C | 166 | ||
| –S | ||||||||
| TiFe0.95S0.02 | 50.7 | 48.3 | 1.0 | Ti2S | 25, 50 °C | 176 | ||
| −ΔHa = 21.8 kJ mol−1 | ||||||||
| TiFeS0.02 | 49.5 | 49.5 | 1.0 | Ti2S | 2.978 | 0.95 | 22 °C | 108 |
| 22 °C, 40 bar | ||||||||
In the case of single elemental substitution, an empirical geometric model was proposed by Lundin et al.105 and Achard et al.106, reporting that by enlarging the unit-cell volume of TiFe, interstitial holes size increases and plateau pressures in PCI curves shift to lower values. This empirical law, to which many intermetallic systems obey, can differ from that observed for some substitutions, therefore, electronic band structure should be considered and implemented with ab initio studies, as demonstrated by Jung et al.104
Alkaline earths (Mg, Be)
Magnesium (Mg, rMg = 0.16013 nm, radius values reported from ref. 107, for comparison rTi = 0.14615 nm and rFe = 0.12412 nm) can be substituted up to 2 at% by ball milling, while up to 6 at% the precipitation of Fe as secondary phase is observed.108 It induces an easier activation compared to pure TiFe, an enlargement of the cell parameter and a concomitant decrease of equilibrium pressure in the PCI, which presents a single plateau related to the formation of the monohydride.108 So, in the case of Mg substitution, the formation of the γ phase is suppressed thus reducing the reversible capacity of the material.108The substitution of Fe with beryllium (Be, rBe = 0.1128 nm), a smaller element with respect to Fe (rFe = 0.12412 nm), up to 15 at%, evidences that geometrical factors alone fail to explain the variation of hydride stability. Although the TiFe unit-cell shrinks with Be substitution, the plateau pressures decrease as reported by Bruzzone et al.109. Furthermore, lower capacity but narrower hysteresis and sufficiently good kinetic were evidenced.109 Besides, the thermodynamics are modified, evidencing higher values of ΔH introducing Be.110
Early transition metals (Zr, Hf, V, Nb, Ta)
Zirconium (Zr, rZr = 0.16025 nm) substitution for Ti (rTi = 0.14615 nm) has a positive effect on activation.111,112 Following the geometric model, it increases the cell parameter of TiFe and decreases the plateau pressures.113–117 However, a decrease of reversible capacity was observed and related to the enlarged solubility of hydrogen in the solid solution (α phase) at high Zr content.113 Zr substitution leads to slopping plateaus, no variation in hysteresis and fast kinetics.118 Jain et al. studied the effect of 4 wt% Zr addition to TiFe, which confirms the positive effect of this substitution for activation (no need of thermal treatment), fast kinetics, a good maximum capacity of 1.60 wt% at 20 bar and 40 °C, and a good resistance to air.119 However, an increase in hysteresis was observed as well, in contrast with previous findings.119 Mechanical treatment (i.e. ball milling and cold rolling) can easily recover hydrogen capacity of this material after air exposure.120Hafnium (Hf, rHf = 0.15775 nm) can be introduced into TiFe up to 2 at%, causing an increase of the cell parameter and a subsequent decrease of plateau pressures. The formation of secondary phases have also been observed, improving activation (possible at room temperature and 20 bar) and kinetics, but slightly reducing the hydrogen capacity of the material.121
Vanadium (V, rV = 0.1316 nm) can substitute both Ti and Fe in TiFe.104,122 Furthermore, it has been reported that the addition of V to TiFe0.90 decreases the total capacity of the material, but on the other hand it decreases hysteresis between absorption and desorption, even if slopped plateaus are observed.123. The addition of V levers the difference between the two plateaus introducing a smoothing effect towards a single plateau that has been widely discussed by Jung et al. combining DFT calculations and experiments.104 They evidenced a stronger effect in lowering both plateau pressure when V substitutes Fe. While V substitution for Ti increases the first plateau pressure and decreases the second one.104 However, V substitution does not improve kinetics, neither cycling stability or resistance to poisoning and oxidation.124,125 Furthermore, it enlarges the cell parameter of the TiFe phase and promotes the formation of smaller crystallite sizes.126
Niobium (Nb, rNb = 0.1429 nm) substitution was studied by in situ X-ray diffraction in TiFe0.90Nb0.10, possibly substituting Ti. It evidenced the formation of β-Ti as secondary phase that starts absorbing hydrogen upon first hydrogenation, allowing easy activation at 22 °C and under 50 bar after 5000 s of incubation time.87 The incubation time can be shorten by the combined substitution of Ti by Nb and the addition of Fe2O3. This mixture results in the formation of secondary phases that improve the activation process (making the material more brittle). In addition, a slight shift of PCI curves towards lower plateau pressure values was observed.127 The lower plateau pressure can be linked to the increasing lattice constant of TiFe introducing Nb.128 Similar results have been observed recently in Nb-substituted materials by mechanical alloying, but evidencing the suppression of the γ phase, thus decreasing the total capacity.129 The suppression of the second plateau by ball milling has been justified by nanostructuration, defects and possibly oxygen contamination, influencing and deforming the coordination site for hydrogen.129 Improved resistance to poisoning was mentioned as well.129
Finally, Tantalum (Ta, rTa = 0.1430 nm) substitution was recently studied by Kuziora et al.130. It enlarges the cell parameter of TiFe and lowers the equilibrium pressure in PCI curves.130
Late transition metals (Cr, Mo, Co, Ni, Pd, Cu)
Chromium (Cr, rCr = 0.12491 nm) substitution in TiFe0.90Cr0.10 and TiFe0.95Cr0.05 forms TiCr2 as a secondary phase, which helps in accelerating activation process of the alloys.122,131 The Cr substitution stabilized as well the first plateau, while reducing the length of the second one. Cr-Substituted TiFe alloys have higher hardness, are more brittle and easier to pulverise with respect to the non-substituted compound. This can be the reason for improved kinetics and reduced hysteresis due to Cr-substitutions.132 On the other hand, this conclusion is in contrast with the higher strain claimed due to Cr substitution. As a matter of fact, usually hysteresis is generated either by elastic strain or by plastic deformation (dislocations, slip bands), and it increases with hardening.133 By mechanochemistry, up to 6 at% of Cr can be included into TiFe, enlarging the cell parameter with a small expansion of the cell volume, lowering crystallite size, and simplifying the activation process.134 Differently to what is expected from geometric considerations, both plateau pressures are shifted to higher value compared to TiFe, and the gamma phase is also destabilised.134The combined substitution of chromium and yttrium in TiFe evidenced an enlargement of the cell constant, the formation of secondary phases (Ti, Cr–Fe solid solution, α-Y), improved kinetic and sloped PCI curves, with a lowering of the plateau pressures and hysteresis.132 On the other hand, the combined substitution of Cr and Zr in TiFe evidenced the formation of TiFe2 as secondary phase, which acts as gateway for hydrogen, easing the activation process of the material.135 The material was activated at 28 °C under 31 bar, and it did not lose any capacity after 50 cycles.135
Molybdenum (Mo, rMo = 0.13626 nm) substitution was reported to lower plateau pressures and to introduce sloppy plateaus.130,136
The substitution of Fe with cobalt (Co, rCo = 0.1251 nm) linearly decreases the first plateau pressure and also reduces the capacity of the material, since only the monohydride is formed.137–139 Recently, improved resistance to poisoning and the suppression of the γ phase was also observed for Co-substituted materials by mechanical alloying, while, in the as-cast conditions, the second plateau was observed to increase the equilibrium pressure.129
Nickel (Ni, rNi = 0.12459 nm) substitutes Fe with no significant changes in the microstructure,122 improves activation and lowers hysteresis between absorption and desorption curves.140 However, it increases the pressure gap between the first and second plateau, reducing the reversible capacity of the material in a narrow pressure range.141 Owing to Ni substitution, improved kinetics is observed because of promoted surface sorption. In fact, the rate determining step in hydrogen sorption is the bulk reaction.142 Furthermore, the use of catalysts like Ni nanoparticles at TiFe surface has been reported to considerably enhance the rate of hydrogenation process, even if it cannot be consider as a substituent.138,143 Additionally, increasing the Ni content, the cell parameter of TiFe increases, decreasing plateaus pressures, decreasing capacity and increasing the decomposition temperature and the cohesive energy of the hydride.144,145 Modified thermodynamics have been reported as well, with lower value of enthalpy and entropy for Ni-substituted TiFe.146,147 Distorted γ region has been observed.138
Addition of Ni stabilized the monohydride as observed in mono-substituted TiFe1−xNix compounds.138,145 It improves cyclability of the material up to 65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles and the reduced loss in capacity was related to possible hydrogen trapped or deactivated reaction site in the material (TiFe0.80Ni0.20).148 Nevertheless, Jain et al. reported a general negative impact on hydrogen storage properties of Ni substitution for Fe.119
000 cycles and the reduced loss in capacity was related to possible hydrogen trapped or deactivated reaction site in the material (TiFe0.80Ni0.20).148 Nevertheless, Jain et al. reported a general negative impact on hydrogen storage properties of Ni substitution for Fe.119
In the literature, few examples of multi-substituted TiFe–Ni alloys are reported. In the same paper cited before, Jain et al. reported a beneficial improvement of activation and kinetics by introducing 4 wt% of Zr7Ni10. It reduces the plateaus pressure but decreases as well the capacity, down to 1.34 wt% at 40 °C and 20 bar, thus with negative effect on hydrogenation properties.119,149
The simultaneous addition of Ni and V, or Ni and Nb, to TiFe was beneficial for the activation process, possible at 28 °C and under 20 bar with a short incubation time (30–40 minutes), reducing hysteresis, lowering the plateau pressures, and granting good capacity and kinetics with no sensible variation of the thermodynamics.150 In contrast, a significant variation of the thermodynamics has been evidenced in the case of combined Ni and Mg substitution.146
Substitution in TiFe by alloying Palladium (Pd, rPd = 0.13754 nm) mitigates the activation process, lowers the plateau pressure of the monohydride (enlarging the cell parameter of TiFe) but it has no effect on the stability of the γ phase.151 Beside, the addition of free Pd as nano-catalyst nanostructured with TiFe by milling has been reported in many studies to improve air resistance to poisoning and facile activation at room temperature.152–156 Equally to Ni, the use of Pd nanoparticles as catalyst considerably enhances the rate of hydrogenation process.138,143 Mechanochemical synthesis can introduce copper (Cu, rCu = 0.1278 nm) into TiFe, enlarging the cell parameter, thus reducing the first plateau pressure of the binary compound.117 The combination of Cu substitution for Fe in TiFe and the addition of Fe2O3 has a positive impact on activation process, that is promoted thanks to a more brittle TiFe matrix, an enhanced formation of active surface by cracking, and the lowering of the plateau pressure.127
Rare-earths (Y, La, Ce, Mm)
Yttrium substitution (Y, rY = 0.18015 nm) for Ti into TiFe modifies the properties of the material. The increase in Y content linearly increases the cell parameter of TiFe and reduces the crystallite size, without changing significantly the microstructure.157 A fast kinetics and an easy activation process are observed, with no incubation time at room temperature and 25 bar.157 However, on increasing Y content, the capacity of the material decreases, due to the formation of secondary phases (TiFe2, Y and Ti precipitates).157The addition of 5 wt% of Lanthanum (La, rLa = 0.1879 nm) to TiFe faces the issue of La immiscibility in the intermetallic compound, but still improves the activation process, thanks to crack formation. Indeed, the incubation time is reduced increasing La content.158 The capacity is claimed to be improved and small hysteresis is observed.158
Cerium (Ce, rCe = 0.18247 nm) substitution for Fe improves activation process and kinetics, because of the formation of small crystallite sizes, that induce high surface reactivity, while increasing the cell parameter of TiFe.159
The addition of mischmetal (Mm, containing La, Ce, Pr, Nd) to TiFe allows easy activation at room temperature after a short incubation time, owing to the cracking of the material caused by Mm inclusions. No evidence of Mm substitution in TiFe were clearly reported.160,161 In these materials, TEM analysis evidenced the formation of channels that could be depicted by electron micrograph in the hydrogenated sample, and that improves absorption kinetic.162 Xin-Nan et al. demonstrated that the addition of Mischmetal to TiFe enhances the resistance towards impurities (mainly O2 and CO2).163 In their work, TiFe with 3 wt% Mm was cycled under hydrogen with a purity below 99%. Moreover, they claimed that the material exhibits a lower decrease in capacity during cycling compared to pure TiFe. When exposed to pure H2, it recovers its capacity within a few cycles without any annealing treatment.
p-block elements (Al, Si, Sn)
The effect of aluminium (Al, rAl = 0.14317 nm) substitution in TiFe has been extensively studied, evidencing an improvement of the kinetics, but the generation of sloped PCI curves.164 Aluminium substitutes Fe increasing TiFe cell parameter.122 It causes negative impact on hydrogenation properties. An increase of the Al content increases the slope of the PCIs, which, compared to TiFe, are shifted to higher pressure values, even if larger cell parameters are observed.138 The second plateau either disappears or moves to high pressures, inhibiting the formation of the γ hydride, with a consequent drastic fall of the hydrogen capacity of the material.110,164 The generation of a sloped plateau has been related compositional inhomogeneity in as cast samples and the formation of octahedral interstice's size gradient.138 Increasing Al substitution for Fe also modifies the thermodynamics and reduces hysteresis, due to the difference in valence electrons between Al and Fe.164 In the same study of Zadorozhnyy et al. cited before, up to 20 at% of Al were substituted to Fe in TiFe by mechanochemistry, with results similar to those observed for Cr substitution.134Silicon (Si, rSi = 0.1153 nm) can substitute Fe, causing the formation of secondary phases such as TiFe2 and Ti, depending on the stoichiometry, and diminishing the hydrogen storage capacity.122 Si has deleterious influence on the capacity because the second plateau is shifted to high pressures, while the first plateau becomes very sloppy and shifted to lower pressure as compared to TiFe, as it occurs for Cu and Ni substitutions.122
Kulshreshtha et al. studied Tin (Sn, rSn = 0.162 nm) that substitutes both Ti and Fe in TiFe. Activation and kinetics are improved, while capacity is decreased.165 The improvement of activation has been related to the formation of TiFe2 as secondary precipitates (in a critical minimum size), which causes strain induced micro cracks owing to different thermal expansion. Surprisingly, the Sn substitution causes a shrink in cell volume of TiFe, leading to an increase of pressure for both plateaus, together with an increase of corresponding ΔH and ΔS values.165
Reactive non-metal elements (B, C, N, O, S)
In general, reactive non-metal elements are present as interstitial atoms or promote the formation of secondary phases.Small quantities of boron (B, rB = 0.082 nm) and carbon (C, rC = 0.0773 nm) induced the formation of secondary phases (i.e. TiFe2 and Ti), promoting easy activation, but reducing drastically the storage capacity.166 Sloped plateaus at high equilibrium pressures were observed, with no formation of the γ phase.166 Furthermore, carbon and nitrogen (N, rN = 0.075 nm) form carbides and nitrides lowering the total capacity of the material.122
In 1977, Sandrock et al. discussed the effect of element contamination by Al, Si, C, N, O during material production and processing to phase homogeneity and microstructure of TiFe.122 Oxygen (O, rO = 0.073 nm) mostly forms oxides that causes capacity deterioration but could also help in activation.122 Extensive studies have been dedicated to the understanding of oxygen influence in TiFe materials and the role of oxide phases in activation process, which however are not the focus of this review. Some related studies can be found in the references cited hereafter.79,80,83,84,102,122,153,154,167–175
The addition of sulphur (S, rS = 0.102 nm) to TiFe affects cycling properties, avoiding pulverisation of the material, improving activation at moderate temperature and reducing the incubation time, thanks to the formation of Ti2S as a secondary phase at the grain boundaries.176 With the increase of S-content, a slight rise of plateau pressure in PCI is observed.176 However, the addition of 1 at% of S by ball milling evidenced an enlargement of the TiFe cell parameter, resulting in a decreased plateau pressure, and reduced reversible capacity, because of the suppression of the dihydride.108 Furthermore, it has been reported that the introduction of small amount of sulphur in pure TiFe can improve the resistance to poisoning.176
Manganese-substituted TiFe alloys
Mn-substitution is of paramount importance in the design of TiFe alloys for large-scale storage application due to the improvement of the main hydrogenation properties. In addition, under the European strategy, Manganese (Mn, rMn = 0.135 nm), such as Fe, is inexpensive and is not listed as CRMs. Many studies can be found in the literature regarding Mn substitutions in TiFe, together with determined thermodynamic properties as reported in Table 3.| Stoichiometry | Ti, at% | Fe, at% | Mn, at% | Secondary phases | TiFe a (Å) | Capacity (wt%) conditions | PCI thermodynamics | Ref. |
|---|---|---|---|---|---|---|---|---|
| a: absorption, d: desorption, 1st: 1st plateau, 2nd: 2nd plateau, RT: room temperature. | ||||||||
| Ti:Fe 1:1 |
||||||||
| TiFe0.95Mn0.05 | 50.0 | 47.5 | 2.5 | 5, 50 °Cd | 215 | |||
| ΔHd = 29.3 kJ mol−1 | ||||||||
| ΔSd = 108 J mol−1 K−1 | ||||||||
| −ΔHa = 25.1 kJ mol−1 | ||||||||
| −ΔSa = 101 J mol−1 K−1 | ||||||||
| 2.977 | 1.73 | 25, 55, 85 °C | 178 | |||||
| 25 °C, 80 bar | ΔHd1st = 28.2 kJ mol−1 | |||||||
| ΔS1std = 105 J mol−1 K−1 | ||||||||
| −ΔH1sta = 24.7 kJ mol−1 | ||||||||
| −ΔS1sta = 100 J mol−1 K−1 | ||||||||
| TiFe0.90Mn0.10 | 50.0 | 45.0 | 5.0 | 1.68 | 8, 22, 45 °Cd | 188 | ||
| 25 °C, 40 bar | −ΔHa = 21.8 kJ mol−1 | |||||||
| −ΔSa = 89 J mol−1 K−1 | ||||||||
| ΔHd = 30.3 kJ mol−1 | ||||||||
| ΔSd = 110 J mol−1 K−1 | ||||||||
| 2.980 | ΔHd = 27.6 kJ mol−1 | 215 | ||||||
| ΔSd = 100 J mol−1 K−1 | ||||||||
| −ΔHa = 23.9 kJ mol−1 | ||||||||
| −ΔSa = 95 J mol−1 K−1 | ||||||||
| 50 °Cd | 131 | |||||||
| 40 °Cd | 136 | |||||||
| 25 °C | 52 | |||||||
| −ΔHa = 23.9 kJ mol−1 | ||||||||
| 30, 40, 50 °C | 242 | |||||||
| 25, 40 °C | 202 | |||||||
| 1.70 | 184 | |||||||
| 40 °C | 243 | |||||||
| Ti-type | 2.979 | 10, 30, 50 °C | 132 | |||||
| ΔHd = 29.3 kJ mol−1 | ||||||||
| ΔSd = 105 kJ mol−1 K−1 | ||||||||
| −ΔHa = 24.2 kJ mol−1 | ||||||||
| −ΔSa = 96 J mol−1 K−1 | ||||||||
| 2.979 | 1.75 | 25, 55, 85 °C | 178 | |||||
| 25 °C, 79 bar | ΔHd1st = 28.1 kJ mol−1 | |||||||
| ΔSd1st = 102 J mol−1 K−1 | ||||||||
| −ΔHa1st = 24.7 kJ mol−1 | ||||||||
| −ΔSa1st = 97 J mol−1 K−1 | ||||||||
| ΔHd2nd = 38.9 kJ mol−1 | ||||||||
| ΔSd2nd = 144 J mol−1 K−1 | ||||||||
| −ΔHa2nd = 29.7 kJ mol−1 | ||||||||
| −ΔSa2nd = 120 J mol−1 K−1 | ||||||||
| Ti1.5(Fe,Mn), Ti(Fe,Mn)1.5 | 30 °C | 179 | ||||||
| TiFe0.885Mn0.115 | 50.0 | 44.2 | 5.8 | 2.985 | 70 | |||
| TiFe0.85Mn0.15 | 50.0 | 42.5 | 7.5 | 1.90 | ΔH1st = 29.5 kJ mol−1 | 186 | ||
| ΔS1st = 107 J mol−1 K−1 | ||||||||
| 1.70 | 40, 50,d 60,d 70 °Cd | 136, 184 | ||||||
| 30 °C | 48 | |||||||
| −ΔHa = 32.6 kJ mol−1 | 142 | |||||||
| 20, 30, 40, 50 °Cd | 187 | |||||||
| TiFe0.80Mn0.20 | 50.0 | 40.0 | 10.0 | 5 °C | 215 | |||
| ΔHd = 31.8 kJ mol−1 | ||||||||
| ΔSd = 105 J mol−1 K−1 | ||||||||
| −ΔHa = 27.6 kJ mol−1 | ||||||||
| −ΔSa = 95 J mol−1 K−1 | ||||||||
| 18, 45, 80 °C | 212 | |||||||
| −ΔH = 21.8 kJ mol−1 | ||||||||
| −ΔS = 75 J mol−1 K−1 | ||||||||
| TiMn1.5 | 50 °Cd | 131 | ||||||
| 2.983 | 22 °C d | 196 | ||||||
| 61 °Cd | 182 | |||||||
| 40 °Cd | 136 | |||||||
| 1.65 | 25 °C | 52 | ||||||
| 25 °C, 41 bar | −ΔHa = 28.4 kJ mol−1 | |||||||
| Ti1.5(Fe,Mn), Ti(Fe,Mn)1.5 | 30 °C | 179 | ||||||
| TiFe0.70Mn0.30 | 50.0 | 35.0 | 15.0 | 2.994 | 22 °C | 196 | ||
| 40 °Cd | 136, 182 | |||||||
| 30 °C | 48 | |||||||
| 25 °C | 52 | |||||||
| −ΔHa = 29.3 kJ mol−1 | ||||||||
| Ti1.5(Fe,Mn), Ti(Fe,Mn)1.5 | 30 °C | 179 | ||||||
| Ti-Rich side | ||||||||
| TiFe0.88Mn0.02 | 52.6 | 46.3 | 1.1 | β-Ti, Ti4Fe2O | 2.986 | 1.86 | 25, 40, 55 °C | 201 |
| 25 °C, 55 bar | −ΔHa1st = 29.8 kJ mol−1 | |||||||
| −ΔSa1st = 106 J mol−1 K−1 | ||||||||
| ΔHd1st = 33.3 kJ mol−1 | ||||||||
| ΔSd1st = 112 J mol−1 K−1 | ||||||||
| −ΔHa2nd = 30.3 kJ mol−1 | ||||||||
| −ΔSa2nd = 112 J mol−1 K−1 | ||||||||
| ΔHd2nd = 27.2 kJ mol−1 | ||||||||
| ΔSd2nd = 106 J mol−1 K−1 | ||||||||
| TiFe0.85Mn0.05 | 52.6 | 44.7 | 2.6 | β-Ti, Ti4Fe2O | 2.985 | 1.73 | 25, 40, 55, 70 °C | 178 |
| 25 °C, 24 bar | ΔHd1st = 30.6 kJ mol−1 | |||||||
| ΔSd1st = 103 J mol−1 K−1 | ||||||||
| −ΔHa1st = 27.8 kJ mol−1 | ||||||||
| −ΔSa1st = 99 J mol−1 K−1 | ||||||||
| ΔHd2nd = 35.2 kJ mol−1 | ||||||||
| ΔSd2nd = 126 J mol−1 K−1 | ||||||||
| −ΔHa2nd = 32.5 kJ mol−1 | ||||||||
| −ΔSa2nd = 121 J mol−1 K−1 | ||||||||
| TiFe0.80Mn0.10 | 52.6 | 42.1 | 5.3 | TiFe2, β-Ti | 2.988 | 1.92 | 25, 65 °C | 141 |
| 25 °C, 20 bar | ||||||||
| TiFe2, β-Ti | 2.989 | 1.68 | 25 °C | 238 | ||||
| 25 °C, 25 bar | ||||||||
| β-Ti, Ti4Fe2O | 2.987 | 1.77 | 25, 40, 55 °C | 178 | ||||
| 25 °C, 55 bar | ΔHd1st = 29.2 kJ mol−1 | |||||||
| ΔSd1st = 96 J mol−1 K−1 | ||||||||
| −ΔHa1st = 28.3 kJ mol−1 | ||||||||
| −ΔSa1st = 98 J mol−1 K−1 | ||||||||
| ΔHd2nd = 35.4 kJ mol−1 | ||||||||
| ΔSd2nd = 124 J mol−1 K−1 | ||||||||
| −ΔHa2nd = 31.1 kJ mol−1 | ||||||||
| −ΔSa2nd = 114 J mol−1 K−1 | ||||||||
| TiFe0.70Mn0.20 | 52.6 | 36.9 | 10.5 | TiFe2, β-Ti | 2.993 | 1.98 | 25, 45,d 65 °Cd | 141 |
| 25 °C, 20 bar | ||||||||
| 30 °C | 105, 136, 190 | |||||||
| −ΔHa1st = 36.3 kJ mol−1 | ||||||||
| −ΔSa1st = 106 J mol−1 K−1 | ||||||||
| −ΔHa2nd = 32.1 kJ mol−1 | ||||||||
| −ΔSa2nd = 111 J mol−1 K−1 | ||||||||
| TiFe0.90Mn0.05 | 51.3 | 46.2 | 2.6 | Ti4Fe2O | 2.982 | 1.84 | 25, 40, 55 °C | 178 |
| 25 °C, 55 bar | ΔHSd1st = 28.2 kJ mol−1 | |||||||
| ΔSd1st = 98 J mol−1 K−1 | ||||||||
| −ΔHa1st = 26.2 kJ mol−1 | ||||||||
| −ΔSa1st = 97 J mol−1 K−1 | ||||||||
| ΔHd2nd = 35.3 kJ mol−1 | ||||||||
| ΔSd2nd = 128 J mol−1 K−1 | ||||||||
| −ΔHa2nd = 31.1 kJ mol−1 | ||||||||
| −ΔSa2nd = 120 J mol−1 K−1 | ||||||||
| TiFe0.80Mn0.05 | 54.1 | 43.243.9 | 2.7 | β-Ti, Ti4Fe2O | 2.986 | 1.55 | 25, 40, 55 °C | 178 |
| 25 °C, 57 bar | ΔHd1st = 32.4 kJ mol−1 | |||||||
| ΔSd1st = 107 J mol−1 K−1 | ||||||||
| −ΔHa1st = 27.6 kJ mol−1 | ||||||||
| −ΔSa1st = 98 J mol−1 K−1 | ||||||||
| ΔHd2nd = 38.9 kJ mol−1 | ||||||||
| ΔSd2nd = 138 J mol−1 K−1 | ||||||||
| −ΔHa2nd = 35.4 kJ mol−1 | ||||||||
| −ΔSa2nd = 132 J mol−1 K−1 | ||||||||
| TiFe0.86Mn0.10 | 51.0 | 5.1 | 2.984 | 1.94 | 45, 60, 80 °Cd | 199 | ||
| 0 °C, 50 bar | ΔHd = 29.5 kJ mol−1 | |||||||
| ΔSd = 106 J mol−1 K−1 | ||||||||
| 2.978 | 1.83 | 60 °C | 244 | |||||
| TiFe0.76Mn0.13 | 52.9 | 40.2 | 6.9 | 40 °Cd | 136 | |||
| Ti1.10Fe0.80Mn0.20 | 52.4 | 38.1 | 9.5 | 40, 60, 80 °C | 245 | |||
| ΔHd = 23.6 kJ mol−1 | ||||||||
| Fe-Rich side | ||||||||
| TiFeMn0.04 | 49.0 | 49.0 | 2.0 | 2.970 | 117 | |||
| TiFeMn0.05 | 48.8 | 48.8 | 2.4 | TiFe2 | 2.976 | 1.55 | 25, 55, 85 °C | 178 |
| 25 °C, 58 bar | ΔHd1st = 28.2 kJ mol−1 | |||||||
| ΔSd1st = 105 J mol−1 K−1 | ||||||||
| −ΔHa1st = 24.3 kJ mol−1 | ||||||||
| −ΔSa1st = 99 J mol−1 K−1 | ||||||||
| ΔHd2nd = 35.3 kJ mol−1 | ||||||||
| ΔSd2nd = 139 J mol−1 K−1 | ||||||||
| −ΔHa2nd = 26.2 kJ mol−1 | ||||||||
| −ΔSa2nd = 116 J mol−1 K−1 | ||||||||
| TiFeMn0.08 | 48.0 | 48.0 | 4.0 | 2.976 | 117 | |||
| TiFeMn0.11 | 47.5 | 47.5 | 5.0 | Ti(Fe,Mn)1.5 | 30 °C | 179 | ||
| TiFeMn0.13 | 47.0 | 47.0 | 6.0 | 2.977 | 1.14 | 22 °C | 117 | |
| 22 °C, 32 bar | ||||||||
| TiFeMn0.17 | 46.0 | 46.0 | 8.0 | 2.977 | 117 | |||
| TiFeMn0.22 | 45.0 | 45.0 | 10.0 | Ti(Fe,Mn)1.5 | 30 °C | 179 | ||
| TiFeMn0.36 | 42.0 | 42.0 | 15.0 | Ti(Fe,Mn)1.5 | 30 °C | 179 | ||
| TiFe1.25Mn0.25 | 40.0 | 50.0 | 10.0 | Ti(Fe,Mn)1.5 | 30 °C | 179 | ||
| TiFe1.18Mn0.18 | 42.5 | 50.0 | 7.5 | Ti(Fe,Mn)1.5 | 30 °C | 179 | ||
| TiFe1.11Mn0.11 | 45.0 | 50.0 | 5.0 | Ti(Fe,Mn)1.5 | 30 °C | 179 | ||
| TiFe0.94Mn0.10 | 49.0 | 46.0 | 5.0 | 2.976 | 22 °C d | 196 | ||
| TiFe0.83Mn0.25 | 48.0 | 40.0 | 12.0 | 2.990 | 22 °C d | 196 | ||
The ternary Ti–Fe–Mn phase diagram presents many phases at 1000 °C, as reported in Fig. 6. The β-Ti solid solution region, as well as the Ti(Fe,Mn)2 C14 laves phases, have large homogeneity domains. At 1000 °C, the intermetallic compound TiFe exists in the range of 49.7 to 52.5 at% Ti. Mn can substitute Fe in this compound up to 27 atomic percent in a narrow region of Ti composition, as reported by Dew-Hughes et al.177. For this reasons, the authors recently investigated Mn-substituted alloys in a wide range of composition, variating both Mn and Ti content.178
 | ||
| Fig. 6 Isotherm section at 1000 °C of the Ti–Fe–Mn phase diagram. Reproduced with permission from ref. 22. | ||
Manganese substitution for Fe enables the hydrogen sorption at lower pressure by enlarging the cell volume of TiFe.70 The higher the Mn content, the lower is the hydrogen sorption pressure for both plateaus.179 Furthermore, easy activation of Mn-substituted TiFe has been related to highly reactive grain boundaries induced by segregation of metal atoms or cluster-like precipitates formation, that can deviate the concentration ratio of components especially at the surface.70,84,180,181
Reilly et al. were the first group in the 70's studying TiFe for hydrogen storage. They have published several reports on Mn substitution either in equiatomic or Ti-rich TiFe alloys.79,88,105,136,182,183 They evidenced many positive improvements compared to pure TiFe. Mn modifies the microstructure, reduces the hysteresis,140,184 improves the activation.122 In addition, Mn substitutions promote the presence of secondary phases, such as β-Ti solid solution or Ti(Fe,Mn)2, that facilitate the alloy cracking and the creation of fresh clean surfaces during the first hydrogen absorption. Furthermore, good long-term cycling performances (without disproportionation or phase separation) and improved hydrogen capacity are reported.105,136,182 The latter probably relates to the thermodynamic stabilisation (i.e. decreased plateau pressure) of the dihydride, so that hydrogen saturation in the γ-phase can be easily reached at low applied pressures.105,136,182 It was also evidenced that a high amount of Mn (e.g. TiFe0.70Mn0.30) actually decreases the total capacity of the material, due to the significant formation of secondary phases, i.e. Ti(Fe,Mn)2, which are not reactive to hydrogen in mild condition of temperature and pressure.136
Lee et al. reported better kinetics and activation when Mn is introduced into TiFe, thanks to the presence of secondary phases, improving capacity as well.131 In their study, the PCI of TiFe0.90Mn0.10 and TiFe0.80Mn0.20 reported at 50 °C showed lower plateau pressures and reduced dihydride region due to the substitution, compared to TiFe.131 Since, by increasing the Mn content, an improvement of kinetics is observed, activation is realized in short incubation time under moderate conditions (room temperature and low pressure). Furthermore the hysteresis between absorption and desorption is reduced.52 The good kinetics seems not to be related to hydride stability nor to particle size, but to the formation of Mn clusters that enhances a faster hydrogenation compared to TiFe.185
Sandrock et al. reported the thermodynamics of TiFe0.85Mn0.15, where the incorporation of Mn in the compound generates higher enthalpy and entropy of reaction, but also higher capacity, compared to TiFe.186 TiFe0.85Mn0.15 was also recently investigated, evidencing an easy single-step reactivation at 300 °C after oxidation in air.187
Milling effect in Ti(Fe,Mn) alloys evidenced that a reduction of size and microstructure promotes an easy activation and an enhanced kinetics, slightly modifying hydrogenation properties and generating a sloping plateau, which is stabilized at lower pressure compared to the pristine alloy.117,188
Severe plastic deformation, such as high pressure torsion, has been used to improve activation and air resistivity on Mn-substituted TiFe alloys, owing to the formation of lattice defects at the grain boundaries and amorphous regions, that are claimed to act as channels for fast hydrogen diffusion facilitating activation.48
Lee et al. stated that the addition of manganese to TiFe increases the hydriding rate.189 In TiFe0.80Mn0.20, at low reacted fraction, the rate-determining step is chemisorption, while, towards the end of sorption, it is the chemical reaction at the metal–hydride interface.189 The hydriding reaction rate increases with increasing pressure at constant temperature and with decreasing temperature at constant pressure. In fact, if the temperature increases at constant pressure, the exponential term of the rate equation, which includes the activation energy term, increases, but the equilibrium pressure or the driving force term decreases. Thus, since a relatively small activation energy is compared to a rather drastic pressure change with temperature, it results in a decrease in the reaction rate with temperature.189 Lee et al. calculated the rate constant for TiFe and TiFe0.80Mn0.20 and the obtained values suggest chemisorption as main rate-determining step. The rate constant increases through Mn for Fe substitution.189
Another study confirmed that the rate determining step is the reaction of hydrogenation at the surface, which is followed by that in the bulk.142 The latter becomes dominant at lower temperatures and at the later stages of reaction.142 The study considered also Ni-substituted TiFe alloy (TiFe0.90Ni0.10), in which the surface reaction is no longer found to be the dominant kinetic mechanism.142
Reilly et al.105,190 and Challet et al.141 reported some studies on Mn-substituted TiFe for Ti-rich alloys (i.e. TiFe0.70Mn0.20). Reilly et al. monitored a PCI curve at 30 °C with strongly slopped and short plateaus and with reduced hysteresis as well105,190. The shape of PCI curve differs from that reported by Challet et al.,141 which presents the typical double flat plateaus. The materials from Challet et al. were annealed at 1000 °C for one week, thus, possible differences in PCI curves could be related to different homogeneity in chemical composition.
Guéguen et al.123 investigated as well the Ti-rich TiFe0.80Mn0.10 composition. In their samples, Ti-type and Ti2Fe-type precipitates were observed as secondary phases favouring alloy activation without any thermal treatment.
Recently Mn substituted TiFe materials have been scaled-up and produced by some industrial companies. Bellosta Von Colbe et al. demonstrated that 6 kg of an industrial Ti(Fe,Mn) alloy, containing 6 at% Mn, could be easily activated at a large scale after a short ball-milling treatment. This treatment reduced the particle and crystallite size, improving activation without thermal treatment (at RT and PH2 = 20 bar), kinetics, cyclability and hydrogenation properties.191
However, even if activation is improved and is reproducible in Mn-substituted TiFe compounds, they still suffer from sensitivity to contaminants such as O2, CO, CO2, H2S, H2O136,168 and Cl2.184 Shwartz et al. reported that Ti is oxidised by O2 to form TiO2 in two steps. H2O directly reacts with Ti(Fe,Mn) alloy to form TiO2, whereas Fe is not oxidised and Mn is oxidised only by oxygen and not by water.175 Mn acts as preferable oxidation element.175,180
Another example was provided in the work of Sandrock et al.,102 where iron was partially substituted by manganese, forming the TiFe0.85Mn0.15 alloy by arc melting. This compound has a similar behaviour to pure TiFe in presence of H2O and O2, but a higher resistance towards CO, with a lower decrease in the storage capacity. Moreover, its reactivation after being exposed to CO was completed within the first cycle under pure H2 at room temperature, whereas TiFe needed several absorption/desorption cycles.102
Multi-substituted Ti(Fe,Mn) alloys
Thanks to the good properties of Mn-substituted Ti(Fe,Mn) pseudo-binary intermetallics, further research has been focused on their tailoring adding further elements and studying the synergic effect with Mn on hydrogenation thermodynamics. Main results are summarized in Table 4.| TiFeMn–M | Ti, at% | Fe, at% | Mn, at% | M, at% | Secondary phases | TiFe a (Å) | Capacity (wt%) conditions | PCI thermodynamics | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a: absorption, d: desorption, 1st: 1st plateau, 2nd: 2nd plateau, RT: room temperature. | |||||||||
| −Zr | |||||||||
| TiFeMn0.02Zr0.01 | 49.2 | 49.2 | 1.0 | 0.6 | (TiFe2) | 2.980 | 195 | ||
| TiFeMn0.04Zr0.02 | 48.5 | 48.5 | 2.0 | 1.0 | 2.980 | 1.0 | 192 | ||
| 22 °C, 40 bar | |||||||||
| (TiFe2) | 2.980 | RT | 195 | ||||||
| TiFeMn0.09Zr0.04 | 47.0 | 47.0 | 4.0 | 2.0 | (TiFe2) | 2.980 | RT | 195 | |
| TiFeMn0.12Zr0.06 | 45.8 | 45.8 | 5.6 | 2.8 | (TiFe2) | 2.986 | RT | 195 | |
| −V | |||||||||
| Ti–Fe–Mn–V | 45.7 | 44.7 | 8.4 | 1.2 | C14 | 2.971 | 22 °Cd | 196 | |
| Ti–Fe–Mn–V | 45.7 | 44.7 | 7.2 | 2.4 | C14 | 2.978 | 22 °Cd | 196 | |
| Ti–Fe–Mn–V | 45.7 | 44.7 | 4.8 | 4.8 | C14 | 2.981 | 22, 30, 50, 80 °Cd | 196 and 197 | |
| ΔHd1st = 28.6 kJ mol−1 | |||||||||
| ΔSd1st = 104 J mol−1 K−1 | |||||||||
| ΔHd2nd = 44.9 kJ mol−1 | |||||||||
| ΔSd2nd = 159 J mol−1 K−1 | |||||||||
| Ti–Fe–Mn–V | 45.7 | 44.7 | 1.2 | 8.4 | C14 | 2.983 | 22 °Cd | 196 | |
| Ti–Fe–Mn–V | 41.7 | 41.0 | 13.0 | 4.3 | C14 | 2.987 | 22 °Cd | 196 | |
| Ti–Fe–Mn–V | 48.0 | 37.0 | 10.0 | 5.0 | C14 | 2.988 | 22 °Cd | 196 | |
| Ti–Fe–Mn–V | 43.6 | 42.8 | 9.1 | 4.5 | C14 | 2.990 | 22 °Cd | 196 | |
| Ti–Fe–Mn–V | 40.0 | 39.1 | 16.7 | 4.2 | C14 | 2.994 | 22 °Cd | 196 | |
| TiFe0.91Mn0.10V0.01 | 49.5 | 45.0 | 5.0 | 0.5 | 25 °Cd | 237 | |||
| TiFe0.82Mn0.20V0.02 | 49.0 | 40.0 | 10.0 | 1.0 | 25 °Cd | 237 | |||
| TiFe0.92Mn0.10V0.02 | 49.0 | 45.0 | 5.0 | 1.0 | 25 °Cd | 237 | |||
| TiFe0.80Mn0.10V0.05 | 51.3 | 41.0 | 5.1 | 2.6 | TiFe2, β-Ti | 2.994 | 1.71 | 25 °C | 238 |
| 25 °C, 25 bar | |||||||||
| TiFe0.80Mn0.10V0.10 | 50.0 | 40.0 | 5.0 | 5.0 | TiFe2, β-Ti | 2.996 | 1.76 | 25 °C | 238 |
| 25 °C, 25 bar | |||||||||
| TiFe0.80Mn0.20 + V | 50, 60, 70 °C | 198 | |||||||
| ΔH = 33.4 kJ mol−1 | |||||||||
| ΔH = 31.8 kJ mol−1 | |||||||||
| −Co | |||||||||
| TiFe0.86Mn0.06Co0.04 | 51.0 | 43.9 | 3.1 | 2.0 | 2.983 | 1.97 | 45, 60, 80 °Cd | 199 | |
| 0 °C, 50 bar | ΔHd = 28.5 kJ mol−1 | ||||||||
| ΔSd = 102 J mol−1 K−1 | |||||||||
| TiFe0.86Mn0.05Co0.05 | 51.0 | 43.9 | 2.6 | 2.6 | 2.983 | 1.98 | 45, 60, 80 °Cd | 199 | |
| 0 °C, 50 bar | ΔHd = 29.3 kJ mol−1 | ||||||||
| ΔSd = 104 J mol−1 K−1 | |||||||||
| TiFe0.86Mn0.04Co0.06 | 51.0 | 43.9 | 2.0 | 3.1 | 2.983 | 1.96 | 45, 60, 80 °Cd | 199 | |
| 0 °C, 50 bar | ΔHd = 29.8 kJ mol−1 | ||||||||
| ΔSd = 106 J mol−1 K−1 | |||||||||
| TiFe0.86Mn0.05Co0.08 | 50.3 | 43.2 | 2.5 | 4.0 | 2.982 | 45 °Cd | 199 | ||
| TiFe0.86Mn0.04Co0.10 | 50.0 | 43.0 | 2.0 | 5.0 | 2.980 | 30, 45,d 50, 55 °C | 199 | ||
| TiFe0.86Mn0.05Co0.12 | 49.3 | 42.4 | 2.5 | 5.9 | 2.981 | 45 °Cd | 199 | ||
| −Ni | |||||||||
| TiFe0.70Mn0.16Ni0.04 | 52.6 | 36.9 | 8.4 | 2.1 | 2.993 | 1.8 | 141 | ||
| 25 °C, 20 bar | |||||||||
| TiFe0.70Mn0.12Ni0.08 | 52.6 | 36.9 | 6.3 | 4.2 | 2.994 | 1.66 | 25 °Ca | 141 | |
| 25 °C, 20 bar | |||||||||
| −Cu | |||||||||
| TiFe0.86Mn0.02Cu0.02 | 52.6 | 45.2 | 1.1 | 1.1 | β-Ti, Ti4Fe2O | 2.988 | 1.83 | 25, 40, 55 °C | 201 |
| 25 °C, 55 bar | −ΔHa1st = 34.1 kJ mol−1 | ||||||||
| −ΔSa1st = 115 J mol−1 K−1 | |||||||||
| ΔHd1st = 37.6 kJ mol−1 | |||||||||
| ΔSd1st = 122 J mol−1 K−1 | |||||||||
| −ΔHa2nd = 29.3 kJ mol−1 | |||||||||
| −ΔSa2nd = 115 J mol−1 K−1 | |||||||||
| ΔHd2nd = 32.5 kJ mol−1 | |||||||||
| ΔSd2nd = 122 J mol−1 K−1 | |||||||||
| TiFe0.84Mn0.02Cu0.04 | 52.6 | 44.2 | 1.1 | 2.1 | β-Ti, Ti4Fe2O | 2.992 | 1.68 | 25, 40, 55 °C | 201 |
| 25 °C, 55 bar | −ΔHa1st = 33.4 kJ mol−1 | ||||||||
| −ΔSa1st = 107 J mol−1 K−1 | |||||||||
| ΔHd1st = 34.7 kJ mol−1 | |||||||||
| ΔSd1st = 107 J mol−1 K−1 | |||||||||
| −ΔHa2nd = 25.1 kJ mol−1 | |||||||||
| −ΔSa2nd = 107 J mol−1 K−1 | |||||||||
| ΔHd2nd = 26.9 kJ mol−1 | |||||||||
| ΔSd2nd = 107 J mol−1 K−1 | |||||||||
| −Y | |||||||||
| TiFe0.90Mn0.10Y0.05 | 48.8 | 43.9 | 4.9 | 2.4 | α-Y | 2.981 | 10, 30, 50 °C | 132 | |
| ΔHd = 31.2 kJ mol−1 | |||||||||
| ΔSd = 109 J mol−1 K−1 | |||||||||
| −ΔHa = 26.7 kJ mol−1 | |||||||||
| −ΔSa = 102 J mol−1 K−1 | |||||||||
| −Ce | |||||||||
| TiFe0.90Mn0.10Ce0.02 | 49.5 | 44.6 | 5.0 | 1.0 | Ce, CeO2 | 25 °C | 202 | ||
| TiFe0.90Mn0.10Ce0.04 | 49 | 44.1 | 4.9 | 2.0 | Ce, CeO2 | 25, 40 °C | 202 | ||
| TiFe0.90Mn0.10Ce0.06 | 48.5 | 43.7 | 4.9 | 2.9 | Ce, CeO2 | 25 °C | 202 | ||
| −Mm | |||||||||
| Til+xFe1−yMnyMmz | β-Ti | 2.15 | 20, 49, 77, 99 °Cd | 203 | |||||
| (x = 0.0–0.9, y = 0.04–0.2, z = 0.002–0.028) | |||||||||
Ball milling has been used to incorporate both Zr and Mn into TiFe intermetallic compound, enhancing the activation of the material, and making it possible at room temperature and 40 bar.192 By increasing the content of Mn and Zr, the equilibrium pressure is lowered, while activation, kinetics and resistance to air are improved.193,194 In contrast, capacity is reduced, because only the monohydride can be formed by Zr substitution.195
The introduction of both V and Mn results in lower plateaus pressures, decreased hysteresis, flatter plateaus, good activation and capacity, showing a synergic effect of both Mn and V.123 The effect of the double substitution in Fe-rich alloys (detailed composition reported in Table 4) was explored by Mitrokhin et al. evidencing the formation of a secondary C14 Laves phase.196,197 Combined Mn and V substitution reduces the pressure gap (V effect) between the two plateaus, that merge into one at lower pressure compared to pure TiFe, while enthalpy of reaction increases (Mn effect).196 Even if resistance to contaminants was not improved, easy activation was observed but with very slow kinetics.196,197 TiFe0.80Mn0.20 with the addition of V was scaled up to 55 kg by Japan Metals & Chemicals Co. Ltd and studied by Endo et al., showing easy activation under 10 bar and 80 °C.198
Substitution effect of Co over Mn has been explored by Qu et al.199 An increase of the Co content decreases the cell volume of TiFe, while improving activation, resistance to pulverisation upon cycling and increasing capacity. As a drawback, PCI curves are sloped and the second plateau is observed at an increased equilibrium pressure.199
Another example of bi-substituted compounds is the Ni addition to pseudobinary Ti(Fe,Mn) compounds. Ni for Mn substitution has been evaluated, evidencing no significant variation of the cell parameter. Challet et al.141 introduced both Mn and Ni into Ti-rich TiFe0.90 observing that Ni addition improved activation process but strongly affects the hydrogenation properties as well. A decrease of the first plateau pressure and an increase of the second one was observed, stabilising the monohydride and destabilising the dihydride. As a consequence, the adjustment of both plateau pressures in a narrow pressure range is much more difficult to achieve when substituting Mn by Ni. Finally, the reversible capacity decreases with Ni content because of the increase of the pressure gap between the two plateaus.
Furthermore, the process history of the sample influences the shape of the PCI curves. For example, milling under argon with small amount of Ni, dispersed as a catalyst, allows to synthetize a material that does not need activation and has a longer cycle lifetime compared to pure TiFe.200
The combined substitution of Cu and Mn for Fe in TiFe0.90 was recently evaluated by Dematteis et al.201. Easy activation and fast kinetics were granted thanks to the Mn substitution and the formation of small amount of secondary phases (β-Ti and Ti4Fe2O-type as precipitates). Cu augments the secondary phase amounts while increasing the TiFe cell parameter and decreasing the first plateau pressure. Similarly to Ni, a negative effect of Cu substitution is that it rises the second plateau pressure, revealing the predominance of electronic effects associated with this substitution that should be verified and deepen by ab initio calculations of their electronic structure (i.e. analysis of density of states).
The combined substitution of Mn and Y causes an enlargement of the cell constant, thus lowering the plateau pressure in PCI curves, which are still flat and shows double plateaus only in desorption.132 Y is generally not highly soluble into TiFe, so α-Y precipitates are formed.132
Addition of Ce to TiFe0.90Mn0.10 evidenced no effect on the thermodynamics and cycling properties of the material, while, increasing Ce content improves kinetics but slightly lower the hydrogen capacity.202 The activation process is remarkably improved as well, requiring no annealing at high temperature and no incubation time at 80 °C and 40 bar.202
The study of Til+xFe1−yMnyMmz (x = 0.0–0.9, y = 0.04–0.2, z = 0.002–0.028) system evidenced that 0.5–1.0 wt% of mischmetal is the optimum amount for improving activation at room temperature in this system, preventing oxidation during processing, and slightly affecting the hydrogenation properties. In fact, at 49 °C the PCI curve presents a single slightly sloppy plateau.203
Recently, even three-substituted alloys have been explored as in the case of TiFe0.86Mn0.1Y0.10−xCux (0.01 ≤ x ≤ 0.09).204,205 Ali et al. showed that by increasing Y content, while decreasing Cu, the cell parameter of the cubic phase and the capacity are increased, while the plateaus pressure decreased. The formation of secondary phases (α-Y, CuY, Cu4Y, Cu2Y) likely allows easy activation. In their study, a complete determination of the PCI at different temperatures allowed the determination of the thermodynamics of these alloys.204,205
Discussion and correlations
The definition of A or B-type atoms in terms of formation enthalpy of binary hydrides60,206,207 is shown in Fig. 1. In addition, the definitions of A- and B-type elements from the structural point of view can include geometric and electronic parameters. For instance, sigma phases in binary systems typically have an A and B element. The A element is poor in d-electrons, it has a bcc crystal structure, a large atomic radius and a preference for sites with large coordination numbers. Whereas the B element is rich in d-electrons, it has fcc or hcp crystal structure, a small atomic radius and a preference for sites with limited coordination number.208Based on these definitions, elemental substitution for either Ti (A-type) or Fe (B-type) in TiFe intermetallic compound can be explained and the definition based on binary hydride enthalpies of formation is particularly effective (Fig. 1). Unfortunately, in the performed literature survey, the location of the substitution element either at Ti or Fe sites is not always described and verified experimentally. Table 5 shows an overview of elemental substitutional effects and it reports Ti or Fe substitution when known. Generally, B-type substitution for Fe are more numerous as compared to A-type substitution for Ti, as evidenced in Table 5 and from this literature survey as reported for each elements in the previous chapter. In the following general trend by group of elements will be described.
| Element(s) | Ti Sub. | Fe Sub. | Act. | Kin. | TiFe, a | β Peq (1st) | γ Peq (2nd) | Plat. | Cap. | Hys. | ΔH | ΔS | Cyc. | Res. to Pois. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mg | ✓ | ↑ | ↓ | ✗ | ↓ | |||||||||
| Be | ✓ | ✓ | ↓ | ↓ | ✗ | ↓ | ↓ | ↑ | = | |||||
| Zr | ✓ | ✓ | ✓ | ↑ | ↓ | ✗ | Sloped | ↓ | = | = | = | ✓ Air | ||
| Hf | ✓ II ph. | ✓ | ↑ | ↓ | ↓ | |||||||||
| V | ✓ | ✓ | ✓ II ph. | ✓ | ↑ | ↓ | ↓ | Smoothed | ↑ | ↓ | ✗ | ✗ | ||
| Nb | ✓ | ✓ II ph., brittle | ↑ | ↓ | ↓ | ✓ | ||||||||
| Ta | ↑ | ↓ | ||||||||||||
| Cr | ✓ | ✓ II ph., brittle | ✓ | ↑ | ↓ | ✗ | Sloped | ↓ | ↓ | ↑ | ||||
| CrY | ✓ | ✓ | ✓ II ph., brittle | ✓ | ↑ | ↓ | ↓ | Sloped | ↓ | ↑ | ||||
| CrZr | ✓ II ph. | |||||||||||||
| Mo | ✓ | ↓ | Sloped | |||||||||||
| Co | ✓ | ✓ II ph. | ↑ | ↓ | ↑ | ↓ | ↓ | ✓ | ||||||
| Ni | ✓ | ✓ II ph., reactive surface | ✓ | ↑ | ↓ | ↑ | 2nd Sloped | ↓ | ↓ | ✓ | ||||
| NiZr | ✓ | ✓ | ↑ | ↓ | ↓ | |||||||||
| NiV | ✓ | ✓ | ✓ | ✓ | ↑ | ↓ | ↑ | ↓ | = | = | ||||
| NiNb | ✓ | ✓ | ✓ | ✓ | ↑ | ↓ | ↑ | ↓ | = | = | ||||
| Pd | ✓ | ✓ | ↑ | ↓ | = | ✓ Air | ||||||||
| Cu | ✓ | ✓ II ph., brittle | ✓ | ↑ | ↓ | ↑ | ↓ | |||||||
| Y | ✓ | ✓ II ph. | ✓ | ↑ | ↓ | ↓ | ||||||||
| La | ✗ | ✗ | ✓ cracks | = | ↑ | ↓ | ||||||||
| Ce | ✓ | ✓ reactive surface | ✓ | ↑ | ||||||||||
| Mm | ✗ | ✗ | ✓ II ph., cracks | ✓ | = | ✓ O2, CO2 | ||||||||
| Al | ✓ | ✓ | ↑ | ↑ | ✗ | Sloped | ↓ | ↓ | ↑ | = | ||||
| Si | ✓ | ✓ II ph., | ↑ | ↓ | ↑ | Sloped | ↓ | |||||||
| Sn | ✓ | ✓ | ✓ II ph. | ✓ | ↓ | ↑ | ↓ | ↑ | ↑ | |||||
| B | ✓ II ph. | ↑ | ✗ | Sloped | ↓ | |||||||||
| C | ✓ II ph. | ↑ | ✗ | Sloped | ↓ | |||||||||
| N | ✓ II ph. | ↓ | ||||||||||||
| S | ✓ II ph. | ↑ | ↓ | ✗ | ↓ | ✓ | ✓ | |||||||
| Mn | ✓ | ✓ II ph., reactive surface | ✓ | ↑ | ↓ | ↓ | Smoothed | ↑ | ↓ | ↑ | ↑ | ✓ | ✓ CO | |
| MnZr | ✓ | ✓ | ✓ II ph. | ✓ | ↑ | ↓ | ✗ | ↓ | ✓ Air | |||||
| MnV | ✓ | ✓ | ✓ II ph. | ✗ | ↑ | ↓ | Smoothed | ↑ | ↓ | ↑ | ✗ | |||
| MnCo | ✓ | ✓ | ↑ | ↓ | ↑ | Sloped | ↓ | ✓ | ||||||
| MnNi | ✓ | ✓ | = | ↓ | ↑ | ↓ | ✓ | |||||||
| MnCu | ✓ | ✓ II ph. | ✓ | ↑ | ↓ | ↑ | ||||||||
| MnCuY | ✓ | ✓ | ✓ II ph. | ↑ | ↓ | ↑ | ||||||||
| MnY | ✓ | ✓ | ✓ II ph. | ↑ | ↓ | ↓ | ||||||||
| MnCe | ✓ | ✓ II ph. | ✓ | ↑ | ↓ | ↓ | = | = | = | |||||
| MnMm | ✓ | ✓ II ph. | ↑ | ↓ | Sloped | ↑ | ✓ O2 | |||||||
As a matter of fact, alkaline earths (Mg, Be) are expected to substitute Ti in the case of Mg (not verified experimentally), and Fe in the case of Be. Early transition metals possibly substitute Ti as they are A-type elements. However, hafnium and tantalum substitutions were not clearly investigated. Late transition metals substitute Fe as they are B-type elements. This behaviour has been verified for all elements except for Pd, which either has been added as a catalyst or substituted without further details on atomic site location. Rare earths possibly substitute Ti as they are A-type compounds. Though this is the case for yttrium, lanthanum and mischmetal are reported to have no solubility in TiFe, and Cerium probably substitutes Fe. p-block elements are expected to substitute Fe, as they are B-type elements. However, Tin has been reported to have both A and B-type behaviour (as it was described for vanadium), as regards to substitution for Ti and Fe.
The extensive literature survey performed in this review evidences that, generally, the addition of a third element to TiFe intermetallic compound has beneficial effects on its activation, hydrogenation kinetics, cycling and resistance to poisoning properties. However, also some negative effects have been reported.
Addition of elements in the formulation of TiFe can lead either to a substitution (i.e. entering into the TiFe structure at Ti or Fe sites) or to an addition (i.e. forming precipitates like second intermetallic phases or oxides). An element can present both behaviours, depending on its limit of solubility in the TiFe phase. For the first case (substitution), the lattice parameter of TiFe phase is modified according to the change in its composition. It leads to modifications of the thermodynamic properties, such as enthalpy, entropy, PCI shape and number of plateaus. For the second one (addition), secondary phases are formed, often accompanied by microstructural changes and increased brittleness. It leads to modification on dynamic and ageing properties like activation, kinetics, poisoning, and cycling.
In this section, the review focuses on the effects of an elemental substitution or addition on properties of the TiFe intermetallic compound, which are essential to be known for efficient tailoring of this material towards real applications. The chemistry behind TiFe-substituted compound will be described together with some useful correlations and guidelines, highlighted by the extensive study of the literature.
Activation and kinetics
The introduction of a substitutional element in TiFe alloys always brings the positive effect of improving activation process and kinetics. It is reported that the TiFe activation is facilitated by adding Al, Si, Mn and Mg, which prevent oxidation, hence allowing the hydrogen absorption process. Cu and Ni substitution seems to be less effective compared to the previous cited elements.169 The prevented TiFe oxidation by Al, Si, Mn and Mg may be related to the fact that these elements form more stable oxides than Cu and Ni, getting preferentially oxidized compared to TiFe, and behaving as oxygen getters.Improved activation can be related to a catalytic behaviour of the additive or the formation of secondary phases at the grain boundaries, with enough size and abundance. Surface solid–gas interactions are enhanced by the presence of secondary phases or elemental clusters, which are highly reactive towards hydrogen, thus promoting hydrogen chemisorption and causing cracks by expansion during hydriding reaction. This process creates preferential channels for hydrogen towards the TiFe-phase or fresh oxide-free surfaces accessible for hydrogenation. Even if secondary phases have different mechanical behaviours, the activation process can be improved owing to the cracking process caused by different thermal expansion compared to TiFe. Alternatively, activation can also be facilitated by the creation of lattice defects (such as vacancies, stacking faults) or amorphous regions, generated by severe plastic deformation processes. Mechanical properties of the material are thus important to facilitate activation properties. For instance, brittle materials can be easily pulverized and activated because they expose fresh surfaces. The effect of substitution on modifying microstructure is thus important to be characterized and understood.
TiFe has poor absorption and desorption kinetic properties, however the partial substitution of Fe (e.g. with Ni, Mn, Cu) can improve the rate of the processes. This effect can be explained by the highly catalytic effect of Ni towards hydrogen chemisorption or, in the case of Mn, by the lowering of plateaus pressure, which increases the driven force for a given applied pressure. Kinetics can also be influenced by the type of synthesis (e.g. ball milling)209 or thermal treatment, mainly due to microstructural modifications (nanostructuration, defects formation, etc.) that will not be discussed here in detail.
Thermodynamics
General correlations have been evidenced regarding geometric models stating that a linear relationship exists between the logarithm of the plateau pressure and the volume of the unit-cell or interstitial sites of the alloy. The larger the volume, the more stable the hydride is.105,136,210,211Shinar et al. collected different PCI curves at different temperatures and the related thermodynamics of TiFe0.80M0.20 alloys substituted by M = Mn, Cr, V, Co, Ni and Cu, evidencing that only the Mn-substituted hydrides showed clearly two flat plateaus, while, Cr- and V-substituted ones present two sloped plateaus and the formation of secondary phases.212 For Co, Ni and Cu, only one plateau was observed.212 Slopped plateaus can be caused by chemical inhomogeneity in substituted TiFe-alloys. Furthermore, size distribution of interstices caused by inhomogeneous distribution of substituents is claimed to cause sloping plateaus.138,213 Another reason related to sloped plateaus is derived from continuous re-distribution of metallic elements during hydrogenation to form the fully hydrogenated compound, if fast diffusion and full local equilibrium (i.e. ortho-equilibrium) occur, instead of equilibrium limited to hydrogen as fast diffuser (i.e. para-equilibrium).214 The addition of a ternary substituent can reduce as well the hysteresis effect.215
Many different parameters should be considered and can be correlated to the substitution effects of different elements to hydrogenation properties of the TiFe intermetallic compound. As cited before, the logarithm of the first plateau pressure can be inversely correlated with the unit cell volume of the TiFe phase. During the structural transition from the intermetallic compound to the hydride, the lattice expansion/distortion is related to the thermodynamic stability of the hydrides and to the dimension of the octahedral interstices where hydrogen is hosted. Geometrical features can also explain why, in some cases, the formation of the γ phase is hindered, together with electronic effects associated to the difference in valence electrons between the substituted element and Ti or Fe. Substitution does not only modify the volume of the interstices, but also the bond strength between hydrogen and metal atoms. The strength of M–H bonds determines the hydrogenation properties too. Yukawa et al. demonstrated that molecular orbital method and the study of electronic structure can clarify the relationship between bond order and strength, and their relation to hydrogen storage properties in substituted TiFe materials.216 They reported that the hydrogen atoms interact stronger with Fe rather than with Ti, thus the chemical bond energy or the type of interaction with this atom or any substitutional elements modify the stability of the hydride phase.216
Different substitutional elements’ properties (e.g. electronegativity and hydrogen affinity) can also influence hydrogenation properties, leading to different stabilities or binding energies (i.e. electronic structure) of hydrogen in the interstitial site. The role and influence of these properties on hydrogenation should be further studied and understood, possibly combining DFT calculations to determine electronic properties, density of states and to further understand their role and relation to hydrogenation properties.
Moreover, substitution influences the hydrogenation properties of the material, modifying its Gibbs free energy. Thus, hydrogenation properties can be related to the entropy of mixing of hydrogen to enter the interstitial voids or the affinity of hydrogen with the substitutional elements.217,218 This can explain the correlation between different elements and the variation of enthalpy or entropy of hydrides formation.217,218 In 1981, Mintz et al. already discussed the influence of substitution in ternary TiFe alloys, reporting a sequence of hydride stability (from more to less stable Cr > Mn > Ni > Co > Fe).138,215 The authors showed that a linear correlation of hydrogenation enthalpy as a function of substituent content is experimentally verified.215
In Fig. 7, enthalpy and entropy values (a and b, respectively) for the first and second plateau in TiFe-type alloy collected in this review are reported as a function of values of the TiFe cell volume. Dashed lines for ΔH (39 kJ mol−1) required at 25 °C to obtain 0.1 MPa plateau pressure with an expected entropy for gas–solid transition (130 J mol−1 K−1) are reported as reference values.
 | ||
| Fig. 7 Correlation between cell volumes of substituted TiFe phase with absolute (a) enthalpy and (b) entropy values of hydrogenation. Black dashed lines for ΔH (39 kJ mol−1) required at 25 °C to obtain 0.1 MPa plateau pressure with expected entropy for gas–solid transition (130 J mol−1 K−1). Values related to the first plateau in absorption are reported as red circle, blue triangle (up) for first plateau values in desorption, pink triangle (down) for second plateau values in absorption, green diamond for second plateau values in desorption, and black square when the plateau was not defined. | ||
As a rule, it can be observed that the hydrogenation enthalpy of both plateaus correlates linearly with the cell volume of the TiFe phase (Fig. 7a). However, some highly dispersed value of enthalpy can be found in the literature for the same alloy or same cell volume, as it can be observed at a fixed cell volume value in Fig. 7a. Value dispersion can be related to different methodologies, different conditions of PCI curves determination or material processing and experimental errors.
A linear trend of hydrogenation enthalpy related to the first plateau in absorption as a function of TiFe cell volume is evidenced in Fig. 7a. The same trend can be visualised for the first plateau desorption enthalpy values, which however are higher with respect to the first plateau absorption enthalpies. A clear trend for the second plateau enthalpies, both in absorption and desorption, cannot be visualized, even if a linear proportion as a function of TiFe cell volume could be suggested.
On the other hand, hydrogenation entropy does not significantly variate with cell volume (Fig. 7b). In fact, entropy change is mostly related to the gas to solid transition of hydrogen. Nevertheless, it must be underlined that even if entropy values are rather constant, they are higher for the second than for the first plateau, being both below the expected entropy change for the gas–solid transition (130 J mol−1 K−1). Entropy values for the first plateau in absorption results lower than the one in desorption. Entropy values for the second plateau evidence higher values with respect to the first plateau, with higher values in desorption than absorption, as already reported for the first plateau.
Cycling and resistance to poisoning
Cycling and poisoning of the material can cause a drop or regular decrease in capacity, thus reducing the amount of hydrogen that can be stored. Two main causes have been argued for this fact. First, the formation of defects because of induced stress during hydriding and dehydriding reactions and, second, the formation of unreactive sites due to oxidation by low purity hydrogen, coming from commercial electrolyser mainly containing H2O as contaminant. In addition, intrinsic degradation on cycling could be caused by alloy disproportion into TiH2 and Fe, which is driven by thermodynamics.219 Moreover, hydrogenation capacity can be reduced on cycling by decomposition of TiFe into TiO2 and Fe due to oxygen (or moisture) uptake. In this sense, the addition of a third element to the alloy can be beneficial if it acts as a getter element for oxidation and if TiFe can be easily recovered and oxides removed in mild conditions of annealing under hydrogen atmosphere or by leaching.Application of TiFe for hydrogen storage
A recent review from Lototskyy et al.220 reports some examples of MH-tank and FC system developed at the lab-scale using metal hydrides. Most of systems are based on AB2 ((TiZr)(Mn,Fe,V)2-type) and AB5 (LaNi5-type) intermetallic compounds, which are usually preferred thanks to their low pressure and temperature working conditions, despite their moderate gravimetric capacity. In the following applications of TiFe-based alloys will be reported and reviewed.Back in the 70s, the Brookhaven National Laboratory studied and developed bulk storage techniques for hydrogen using TiFe. The program consisted of a variety of activities which include engineering analysis and design of a large bulk hydrogen storage facility, engineering-scale tests, work on the selection and development of suitable iron–titanium alloys, and the construction of a large prototype energy storage system.221 Strickland et al. implemented a small test bed of 38 kg of iron titanium hydride as storage media. The maximum hydrogen storage capacity, under the studied operating conditions, was 1.19 wt% for the FeTi alloy and a uniform hydrogen flow rate of 9 normal litre per min was sustained for a 10 hour transfer period.222 Vessels of different dimensions were considered and tested for volumetric expansion with different alloy loading.223 In the study, TiFe0.70Mn0.18 alloy was also considered.223 Finally, the reservoir has been scaled up to store 6 kg hydrogen using 405 kg of TiFe.224 This latter prototype system was built for the Public Service Electric and Gas Company of New Jersey to study the feasibility of storing off-peak electrical energy through the use of a water electrolyser, a hydride reservoir and a fuel cell stack.224
The use of TiFe in hydride beds has been developed for storing and supplying hydrogen fuel in power plant and automotive applications.225 An hydride bed was built at Brookhaven National Laboratory for the Public Service Electric and Gas Co., with a release rate roughly constant of approximately 0.036 Ib ft−1 h (0.000015 Pa s).224 Finally, a techno-economic assessment was performed for a hydrogen–chlorine energy storage system for electric utility load levelling and peak shaving applications that involved hydrogen storage in TiFe-alloy.226
Recently, a conventional bench-scale tank based on TiFe carriers have been developed in Japan.198 Endo et al. developed a TiFe-based material to be used in an integrated system with an electrolyser and a fuel cell. The material is a modified TiFe0.80Mn0.20 alloy, with the introduction of some V and annealed at 1100 °C for 24 h.198 The activation of the material was performed at modest temperature and low pressure, according to Japanese safety regulations (i.e. approx. 80 °C and 2 bar). Vacuum condition at the bench scale are crucial for decreasing incubation time during activation.198 Furthermore, a similar material was later used in a recent study for a hydrogen energy utilization system in a zero emission building.227 The integrated system used 520 kg of MH for 80 Nm3 of total hydrogen storage and it has been tested under different weather conditions.228
They later on demonstrate that limited use of auxiliaries for cooling can be implemented reducing power consumption in a bench-scale hydrogen system (Hydro Q-BiC), reaching an overall energy efficiency of 60% (power-to-hydrogen-to-power/heat).229
In Europe, the only demonstrative energy storage system based on TiFe carrier is under development in South Tirol by GKN. It will demonstrate the use of hydrogen carrier for about 10 kg H2, working close to room temperature and pressure.
Finally, the European HyCARE project is willing to develop an integrated system for stationary storage of hydrogen in TiFe-based material efficiently coupled with a electrolyser, fuel cell and heat storage system based on phase change material.18,19 The total amount of hydrogen that will be stored is expected to be 50 kg, in approx. 4 tons of TiFe-based carrier.
In conclusion, it can be stated that among the several potential materials that can be generated by elemental substitution on TiFe intermetallic compound, the definition of an optimal material is strictly connected with pressure and temperature ranges required for the specific application. As highlighted by the examples of applications developed and reviewed above, Mn and V substitution resulted particularly effective in the case of stationary storage in mild conditions of temperature and pressure.
Conclusions
As a conclusion, TiFe remains an attractive alloy for hydrogen storage regarding its low cost and significant capacity. However, it is worth noting that the binary compound has some drawbacks to be overcome by suitable substitutions, strongly improving activation and kinetics. However, thermodynamics and hydrogenation properties are also influenced, and often a drop in capacity can be observed. This review evidences that Mn is a key element as substituent in TiFe system. In addition, V, and the synergic effect of Mn and V can be exploited. However, it should be kept in mind that V is a CRMs for Europe. On the other hand, many other elements such as Mg, Ta, Zr, Cr, Co, Ni, Cu and S are not suitable for industrial upscaled materials. Zr, Cr and S lead to sensible reduction of storage capacity. Mg, Ta, Co, Ni are CRMs. Cu increases sensibly the pressure gap between the first and second plateau. Effect of contaminants during synthesis (such as B, C, N, O) should be studied and understood with care because they can negatively influence hydrogenation properties of the material. As a general assertion, bi- and multi-substituted alloys can combine some synergic and beneficial effects, which however are not drastically better than for mono-substituted cases.The study performed in this work evidenced that systematic studies on TiFe-substituted alloys are scarce in the literature. In the last years, a high number of reports have been published with only partial characterisation of the studied alloys, making hard the understanding of the full picture concerning activation, thermodynamics, kinetics and chemistry of the studied systems. To further understand the role of elemental substitution in TiFe on the modification of hydride stability and hydrogenation properties, there is a strong need of complete studies including compositional, structural, microstructural, activation, kinetic, and thermodynamic data. More attention should be focused on the determination of elemental substitution in TiFe, perhaps by electron microprobe analysis (EMPA) or by coupling scanning electron microscope (SEM) and energy-dispersive X-ray spectroscopy (EDX) mapping to analytically determine the alloy (TiFe-phase) exact composition. Structural studies should also include experimental or computational evidences of possible elemental substitution in Ti or Fe position. The determination of cell parameter of TiFe, and relative abundances of secondary phases determined by microscopy and X-ray diffraction (coupled to Rietveld analysis) methods are useful information to understand the geometrical implication of substitution. Neutron studies are complementary for the full solution of crystal structure of substituted systems. The review was not focused on neutron studies on TiFe systems, but there is a big lack of these determinations. Some studies can be found in the following ref. 61–63, 66, 67, 69, 70, 74 and 230–233.
The study and assessment of ternary and higher phase diagrams can also help in the definition of phase boundaries, thus elucidating the formation of secondary phases and their possible role in enhancing activation properties. Mechanical properties and microstructural studies of these materials are also of great interest because they can be related to enhanced activation process and easy handling during industrial processing such as crushing process or pellet preparation.
In conclusion, the determination of thermodynamics in these systems must be conducted with care. Annealed samples can be representative of equilibrium phases, and the study of PCI curves with sufficient waiting time for the determination of equilibrium pressure points both in absorption and desorption is essential. PCI curves should also be determined in an appropriately large temperature range to guarantee a correct determination of enthalpy and entropy by the Van’t Hoff plot. Coupling calorimetric determinations can confirm and complete the thermodynamic study. Cycling properties and resistance to poisoning are not frequent in literature studies, but they are of great interest for real applications of these materials. More efforts should focus on these studies and the definition of the mechanisms involved, in order to solve dropping capacity problems in case of not pure hydrogen produced by commercial electrolysers. Few other properties, which were not included in this review, are also of great interest for real applications. They include thermal properties (thermal conductivity, heat capacity), density and porosity determination. The definition of these parameters will help the upscale and development of prototype tanks for solid-state hydrogen storage based on TiFe systems, which nowadays are limited to few example that exploit Mn and V substitution for stationary hydrogen storage in mild condition of temperature and pressure.
Author contributions
E. M. D.: conceptualization, data curation, formal analysis, investigation, writing – original draft, writing – review & editing; N. B.: bata curation, formal analysis, investigation, writing – original draft; F. C.: conceptualization, formal analysis, supervision, writing – review & editing; M. L.: conceptualization, funding acquisition, formal analysis, supervision, writing – review & editing; M. B.: conceptualization, funding acquisition, project administration, formal analysis, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The project leading to this publication has received funding from the Fuel Cells and Hydrogen 2 Joint Undertaking (JU) under grant agreement no. 826352, HyCARE project. The JU receives support from the European Union's Horizon 2020 research, Hydrogen Europe, Hydrogen Europe Research, innovation programme and Italy, France, Germany, Norway, which are all thankfully acknowledged.References
- A. Züttel, A. Remhof, A. Borgschulte and O. Friedrichs, Hydrogen: the future energy carrier, Philos. Trans. R. Soc., A, 2010, 368, 3329–3342, DOI:10.1098/rsta.2010.0113.
- A. Züttel, Materials for hydrogen storage, Mater. Today, 2003, 6, 24–33, DOI:10.1016/S1369-7021(03)00922-2.
- J. Bellosta von Colbe, J.-R. Ares, J. Barale, M. Baricco, C. Buckley, G. Capurso, N. Gallandat, D. M. Grant, M. N. Guzik, I. Jacob, E. H. Jensen, T. Jensen, J. Jepsen, T. Klassen, M. V. Lototskyy, K. Manickam, A. Montone, J. Puszkiel, S. Sartori, D. A. Sheppard, A. Stuart, G. Walker, C. J. Webb, H. Yang, V. Yartys, A. Züttel and M. Dornheim, Application of hydrides in hydrogen storage and compression: Achievements, outlook and perspectives, Int. J. Hydrogen Energy, 2019, 44, 7780–7808, DOI:10.1016/j.ijhydene.2019.01.104.
- M. Schlichtenmayer and M. Hirscher, The usable capacity of porous materials for hydrogen storage, Appl. Phys. A: Mater. Sci. Process., 2016, 122, 379, DOI:10.1007/s00339-016-9864-6.
- E. Callini, K.-F. Aguey-Zinsou, R. Ahuja, J. R. Ares, S. Bals, N. Biliškov, S. Chakraborty, G. Charalambopoulou, A.-L. Chaudhary, F. Cuevas, B. Dam, P. de Jongh, M. Dornheim, Y. Filinchuk, J. Grbović Novaković, M. Hirscher, T. R. Jensen, P. B. Jensen, N. Novaković, Q. Lai, F. Leardini, D. M. Gattia, L. Pasquini, T. Steriotis, S. Turner, T. Vegge, A. Züttel and A. Montone, Nanostructured materials for solid-state hydrogen storage: A review of the achievement of COST Action MP1103, Int. J. Hydrogen Energy, 2016, 41, 14404–14428, DOI:10.1016/j.ijhydene.2016.04.025.
- D. P. Broom, C. J. Webb, G. S. Fanourgakis, G. E. Froudakis, P. N. Trikalitis and M. Hirscher, Concepts for improving hydrogen storage in nanoporous materials, Int. J. Hydrogen Energy, 2019, 1, 7768–7779, DOI:10.1016/j.ijhydene.2019.01.224.
- P. Á. Szilágyi, D. M. Rogers, I. Zaiser, E. Callini, S. Turner, A. Borgschulte, A. Züttel, H. Geerlings, M. Hirscher and B. Dam, Functionalised metal–organic frameworks: a novel approach to stabilising single metal atoms., J. Mater. Chem. A, 2017, 5, 15559–15566, 10.1039/C7TA03134C.
- D. P. Broom, C. J. Webb, K. E. Hurst, P. A. Parilla, T. Gennett, C. M. Brown, R. Zacharia, E. Tylianakis, E. Klontzas, G. E. Froudakis, T. A. Steriotis, P. N. Trikalitis, D. L. Anton, B. Hardy, D. Tamburello, C. Corgnale, B. A. van Hassel, D. Cossement, R. Chahine and M. Hirscher, Outlook and challenges for hydrogen storage in nanoporous materials, Appl. Phys. A: Mater. Sci. Process., 2016, 122, 151, DOI:10.1007/s00339-016-9651-4.
- C. Milanese, T. R. Jensen, B. C. Hauback, C. Pistidda, M. Dornheim, H. Yang, L. Lombardo, A. Zuettel, Y. Filinchuk, P. Ngene, P. E. de Jongh, C. E. Buckley, E. M. Dematteis and M. Baricco, Complex hydrides for energy storage, Int. J. Hydrogen Energy, 2019, 44, 7860–7874, DOI:10.1016/j.ijhydene.2018.11.208.
- J. Puszkiel, S. Garroni, C. Milanese, F. Gennari, T. Klassen, M. Dornheim and C. Pistidda, Tetrahydroborates: Development and Potential as Hydrogen Storage Medium, Inorganics, 2017, 5, 74, DOI:10.3390/inorganics5040074.
- E. Hadjixenophontos, E. M. Dematteis, N. Berti, A. R. Wołczyk, P. Huen, M. Brighi, T. T. Le, A. Santoru, S. Payandeh, F. Peru, A. H. Dao, Y. Liu and M. Heere, A Review of the MSCA ITN ECOSTORE—Novel Complex Metal Hydrides for Efficient and Compact Storage of Renewable Energy as Hydrogen and Electricity, Inorganics, 2020, 8, 17, DOI:10.3390/inorganics8030017.
- B. Sakintunaa, F. Lamari-Darkrimb and M. Hirscher, Metal hydride materials for solid hydrogen storage: A review, Int. J. Hydrogen Energy, 2007, 32, 1121–1140, DOI:10.1016/j.ijhydene.2006.11.022.
- P. Dantzer, Metal-Hydride technology: A critical review, Hydrogen in Metals III, Springer, Berlin, Heidelberg, 1997, vol. 73, pp. 279–340 Search PubMed.
- J. J. Reilly, Metal Hydride Technology, Z. Phys. Chem., 1979, 117, 155–184, DOI:10.1524/zpch.1979.117.117.155.
- A. El Kharbachi, E. M. Dematteis, K. Shinzato, S. C. Stevenson, L. J. Bannenberg, M. Heere, C. Zlotea, P. Á. Szilágyi, J.-P. Bonnet, W. Grochala, D. H. Gregory, T. Ichikawa, M. Baricco and B. C. Hauback, Metal Hydrides and Related Materials. Energy Carriers for Novel Hydrogen and Electrochemical Storage, J. Phys. Chem. C, 2020, 124, 7599–7607, DOI:10.1021/acs.jpcc.0c01806.
- M. Latroche, Structural and thermodynamic properties of metallic hydrides used for energy storage, J. Phys. Chem. Solids, 2004, 65, 517–522, DOI:10.1016/j.jpcs.2003.08.037.
- F. Cuevas, J.-M. Joubert, M. Latroche and A. Percheron-Guégan, Intermetallic compounds as negative electrodes of Ni/MH batteries, Appl. Phys. A: Mater. Sci. Process., 2001, 72, 225–238, DOI:10.1007/s003390100775.
- HyCARE focuses on large-scale, solid-state hydrogen storage Fuel Cells Bull. 2019 2019 11 DOI:10.1016/S1464-2859(19)30068-9 .
- HyCARE project Available online: http://www.hycare-project.eu.
- F. Cuevas and E. Burzo, in Hydrogen Storage Materials, ed. E. Burzo, Berlin, Springer Berlin Heidelberg, Berlin, Heidelberg, 2018 Search PubMed.
- EU Critical Raw Materials Resilience: Charting a Path towards greater Security and Sustainability. Communication from the commission to the European parliament, the council, the European Economic and Social Committee and the committee of the regions. COM 2020, 474.
- P. Villars, H. Okamoto and K. Cenzual, A-B-C Phase Diagram, ASM Alloy Phase Diagrams Database – http://www.asminternational.org. ASM Int. – Mater. Park. OH 2016.
- G. K. Sujan, Z. Pan, H. Li, D. Liang and N. Alam, An overview on TiFe intermetallic for solid-state hydrogen storage: microstructure, hydrogenation and fabrication processes, Crit. Rev. Solid State Mater. Sci., 2019, 1–18, DOI:10.1080/10408436.2019.1652143.
- A. Lys, J. O. Fadonougbo, M. Faisal, J.-Y. Suh, Y.-S. Lee, J.-H. Shim, J. Park and Y. W. Cho, Enhancing the Hydrogen Storage Properties of AxBy Intermetallic Compounds by Partial Substitution: A Short Review, Hydrogen, 2020, 1, 38–63, DOI:10.3390/hydrogen1010004.
- J.-L. Murray, Phase Diagrams of Binary Titanium Alloys, ASM Int., 1987 Search PubMed.
- G. Cacciamani, J. De Keyzer, R. Ferro, U. E. Klotz, J. Lacaze and P. Wollants, Critical evaluation of the Fe–Ni, Fe–Ti and Fe–Ni–Ti alloy systems, Intermetallics, 2006, 14, 1312–1325, DOI:10.1016/j.intermet.2005.11.028.
- J. C. Gachon and J. Hertz, Enthalpies of formation of binary phases in the systems FeTi, FeZr, CoTi, CoZr, NiTi, and NiZr, by direct reaction calorimetry, CALPHAD, 1983, 7, 1–12, DOI:10.1016/0364-5916(83)90024-X.
- A. Percheron-Guégan and J.-M. Welter, Preparation of intermetallics and hydrides, 1988, pp. 11–48 Search PubMed.
- H. Hotta, M. Abe, T. Kuji and H. Uchida, Synthesis of Ti–Fe alloys by mechanical alloying, J. Alloys Compd., 2007, 439, 221–226, DOI:10.1016/j.jallcom.2006.05.137.
- M. Abe and T. Kuji, Hydrogen absorption of TiFe alloy synthesized by ball milling and post-annealing, J. Alloys Compd., 2007, 446–447, 200–203, DOI:10.1016/j.jallcom.2006.12.063.
- T. Bratanich, S. Solonin and V. Skorokhod, Mechanical activation of hydrogen sorption with intermetallic compounds LaNi5 and TiFe in powder systems, Int. J. Hydrogen Energy, 1995, 20, 353–355, DOI:10.1016/0360-3199(94)00062-5.
- L. Zaluski, P. Tessier, D. H. Ryan, C. B. Doner, A. Zaluska, J. O. Ström-Olsen, M. L. Trudeau and R. Schulz, Amorphous and nanocrystalline Fe–Ti prepared by ball milling, J. Mater. Res., 1993, 8, 3059–3068, DOI:10.1557/JMR.1993.3059.
- L. Zaluski, S. Hosatte, P. Tessier, D. H. Ryan, J. O. Ström-Olsen, M. L. Trudeau and R. Schulz, Hydrogen Absorption in Amorphous and Nano-Crystalline FeTi, Z. Phys. Chem., 1994, 183, 45–49, DOI:10.1524/zpch.1994.183.Part_1_2.045.
- E. Jankowska and M. Jurczyk, Electrochemical behaviour of high-energy ball-milled TiFe alloy, J. Alloys Compd., 2002, 346, L1–L3, DOI:10.1016/S0925-8388(02)00492-9.
- V. Y. Zadorozhnyy, S. N. Klyamkin, S. D. Kaloshkin and Y. A. Skakov, Production of intermetallic compound of FeTi by means of mechanical-chemical synthesis and its interaction with hydrogen, Inorg. Mater. Appl. Res., 2010, 1, 41–45, DOI:10.1134/S2075113310010065.
- P. Tessier, R. Schulz and J. O. Ström-Olsen, Elastic stress in composite FeTi hydrogen storage materials, J. Mater. Res., 1998, 13, 1538–1547, DOI:10.1557/JMR.1998.0214.
- V. Y. Zadorozhnyi, Y. A. Skakov and G. S. Milovzorov, Appearance of metastable states in Fe-Ti and Ni-Ti systems in the process of mechanochemical synthesis, Met. Sci. Heat Treat., 2008, 50, 404–410, DOI:10.1007/s11041-008-9078-4.
- T. Haraki, K. Oishi, H. Uchida, Y. Miyamoto, M. Abe, T. Kokaji and S. Uchida, Properties of hydrogen absorption by nano-structured FeTi alloys, Int. J. Mater. Res., 2008, 99, 507–512, DOI:10.3139/146.101669.
- I. Tajima, M. Abe, H. Uchida, M. Hattori, Y. Miyamoto and T. Haraki, Hydrogen sorption kinetics of FeTi alloy with nano-structured surface layers, J. Alloys Compd., 2013, 580, S33–S35, DOI:10.1016/j.jallcom.2013.03.011.
- A. A. Novakova, O. V. Agladze, S. V. Sveshnikov and B. P. Tarasov, Supersaturated solid solutions and metastable phases formation through different stages of mechanical alloying of FeTi, Nanostruct. Mater., 1998, 10, 365–374, DOI:10.1016/S0965-9773(98)00077-4.
- H. Aoyagi, K. Aoki and T. Masumoto, Effect of ball milling on hydrogen absorption properties of FeTi, Mg2Ni and LaNi5, J. Alloys Compd., 1995, 231, 804–809, DOI:10.1016/0925-8388(95)01721-6.
- T. Kondo, K. Shindo, M. Arakawa and Y. Sakurai, Microstructure and hydrogen absorption–desorption properties of Mg–TiFe0.92Mn0.08 composites prepared by wet mechanical milling, J. Alloys Compd., 2004, 375, 283–291, DOI:10.1016/j.jallcom.2003.11.152.
- E. A. Berdonosova, S. N. Klyamkin, V. Y. Zadorozhnyy, M. Y. Zadorozhnyy, K. V. Geodakian, M. V. Gorshenkov and S. D. Kaloshkin, Calorimetric study of peculiar hydrogenation behavior of nanocrystalline TiFe, J. Alloys Compd., 2016, 688, 1181–1185, DOI:10.1016/j.jallcom.2016.07.145.
- V. Zadorozhnyy, S. Klyamkin, M. Zadorozhnyy, O. Bermesheva and S. Kaloshkin, Hydrogen storage nanocrystalline TiFe intermetallic compound: Synthesis by mechanical alloying and compacting, Int. J. Hydrogen Energy, 2012, 37, 17131–17136, DOI:10.1016/j.ijhydene.2012.08.078.
- L. Sun, H. Liu, D. H. Bradhurst and S. Dou, Formation of FeTi hydrogen storage alloys by ball-milling, J. Mater. Sci. Lett., 1998, 17, 1825–1830, DOI:10.1023/A:1006622023563.
- K. Edalati, J. Matsuda, H. Iwaoka, S. Toh, E. Akiba and Z. Horita, High-pressure torsion of TiFe intermetallics for activation of hydrogen storage at room temperature with heterogeneous nanostructure, Int. J. Hydrogen Energy, 2013, 38, 4622–4627, DOI:10.1016/j.ijhydene.2013.01.185.
- H. Emami, K. Edalati, J. Matsuda, E. Akiba and Z. Horita, Hydrogen storage performance of TiFe after processing by ball milling, Acta Mater., 2015, 88, 190–195, DOI:10.1016/j.actamat.2014.12.052.
- K. Edalati, M. Matsuo, H. Emami, S. Itano, A. Alhamidi, A. Staykov, D. J. Smith, S. I. Orimo, E. Akiba and Z. Horita, Impact of severe plastic deformation on microstructure and hydrogen storage of titanium-iron-manganese intermetallics, Scr. Mater., 2016, 124, 108–111, DOI:10.1016/j.scriptamat.2016.07.007.
- K. Edalati, J. Matsuda, A. Yanagida, E. Akiba and Z. Horita, Activation of TiFe for hydrogen storage by plastic deformation using groove rolling and high-pressure torsion: Similarities and differences, Int. J. Hydrogen Energy, 2014, 39, 15589–15594, DOI:10.1016/j.ijhydene.2014.07.124.
- K. Edalati, J. Matsuda, M. Arita, T. Daio, E. Akiba and Z. Horita, Mechanism of activation of TiFe intermetallics for hydrogen storage by severe plastic deformation using high-pressure torsion, Appl. Phys. Lett., 2013, 103, 143902, DOI:10.1063/1.4823555.
- R. Wakabayashi, S. Sasaki, I. Saita, M. Sato, H. Uesugi and T. Akiyama, Self-ignition combustion synthesis of TiFe in hydrogen atmosphere, J. Alloys Compd., 2009, 480, 592–595, DOI:10.1016/j.jallcom.2009.02.008.
- N. Yasuda, R. Wakabayashi, S. Sasaki, N. Okinaka and T. Akiyama, Self-ignition combustion synthesis of TiFe1−xMnx hydrogen storage alloy, Int. J. Hydrogen Energy, 2009, 34, 9122–9127, DOI:10.1016/j.ijhydene.2009.08.102.
- I. Saita, M. Sato, H. Uesugi and T. Akiyama, Hydriding combustion synthesis of TiFe, J. Alloys Compd., 2007, 446–447, 195–199, DOI:10.1016/j.jallcom.2007.02.150.
- V. Y. Zadorozhnyy, G. S. Milovzorov, S. N. Klyamkin, M. Y. Zadorozhnyy, D. V. Strugova, M. V. Gorshenkov and S. D. Kaloshkin, Preparation and hydrogen storage properties of nanocrystalline TiFe synthesized by mechanical alloying, Prog. Nat. Sci.: Mater. Int., 2017, 27, 149–155, DOI:10.1016/j.pnsc.2016.12.008.
- L. Zaluski, A. Zaluska and J. Ström-Olsen, Nanocrystalline metal hydrides, J. Alloys Compd., 1997, 253–254, 70–79, DOI:10.1016/S0925-8388(96)02985-4.
- F. Laves and H. J. Wallbaum, Zur Kristallchemie von Titan-Legierungen, Naturwissenschaften, 1939, 27, 674–675, DOI:10.1007/BF01494992.
- A. E. Dwight, CsCl-type equiatomic phases in binary alloys of transition elements, Trans. Am. Inst. Min., Metall. Pet. Eng., 1959, 215, 283–286 CAS.
- J. Reilly, J. Johnson, F. Reidinger, J. Lynch, J. Tanaka and R. Wiswall, Lattice expansion as a measure of surface segregation and the solubility of hydrogen in α-FeTiHx, J. Less-Common Met., 1980, 73, 175–182, DOI:10.1016/0022-5088(80)90358-6.
- H. Bo, J. Wang, L. Duarte, C. Leinenbach, L. Liu, H. Liu and Z. Jin, Thermodynamic re-assessment of Fe–Ti binary system, Trans. Nonferrous Met. Soc. China, 2012, 22, 2204–2211, DOI:10.1016/S1003-6326(11)61450-7.
- H. Züchner, in Hydrogen in Intermetallic Compounds I, ed. L. Schlapbach, Topics in Applied Physics, Springer Berlin Heidelberg, Berlin, Heidelberg, 1988, vol. 63 Search PubMed.
- P. Thompson, F. Reidinger, J. J. Reilly, L. M. Corliss and J. M. Hastings, Neutron diffraction study of α-iron titanium deuteride, J. Phys. F: Met. Phys., 1980, 10, L57–L59, DOI:10.1088/0305-4608/10/2/001.
- J. Schefer, P. Fischer, W. Hälg, F. Stucki, L. Schlapbach and A. F. Andresen, Structural phase transitions of FeTi-deuterides, Mater. Res. Bull., 1979, 14, 1281–1294, DOI:10.1016/0025-5408(79)90005-9.
- W. Schäfer, G. Will and T. Schober, Neutron and electron diffraction of the FeTi - D(H) - γ - phase, Mater. Res. Bull., 1980, 15, 627–634, DOI:10.1016/0025-5408(80)90143-9.
- P. Fischer, J. Schefer, K. Yvon, L. Schlapbach and T. Riesterer, Orthorhombic structure of γ-TiFeD ≈ 2, J. Less-Common Met., 1987, 129, 39–45, DOI:10.1016/0022-5088(87)90031-2.
- T. Nambu, H. Ezaki, H. Yukawa and M. Morinaga, Electronic structure and hydriding property of titanium compounds with CsCl-type structure, J. Alloys Compd., 1999, 293–295, 213–216, DOI:10.1016/S0925-8388(99)00421-1.
- P. Fischer, W. Hälg, L. Schlapbach, F. Stucki and A. F. Andresen, Deuterium storage in FeTi. Measurement of desorption isotherms and structural studies by means of neutron diffraction, Mater. Res. Bull., 1978, 13, 931–946, DOI:10.1016/0025-5408(78)90105-8.
- P. Thompson, M. A. Pick, F. Reidinger, L. M. Corliss, J. M. Hastings and J. J. Reilly, Neutron diffraction study of β iron titanium deuteride, J. Phys. F: Met. Phys., 1978, 8, L75–L80, DOI:10.1088/0305-4608/8/4/001.
- J. J. Reilly and R. H. Wiswall, Formation and properties of iron titanium hydride, Inorg. Chem., 1974, 13, 218–222, DOI:10.1021/ic50131a042.
- P. Thompson, J. J. Reilly, F. Reidinger, J. M. Hastings and L. M. Corliss, Neutron diffraction study of gamma iron titanium deuteride, J. Phys. F: Met. Phys., 1979, 9, L61–L66, DOI:10.1088/0305-4608/9/4/001.
- D. Fruchart, M. Commandré, D. Sauvage, A. Rouault and R. Tellgren, Structural and activation process studies of FeTi-like hydride compounds, J. Less-Common Met., 1980, 74, 55–63, DOI:10.1016/0022-5088(80)90073-9.
- P. Thompson, J. J. Reilly and J. M. Hastings, The application of the Rietveld method to a highly strained material with microtwins: TiFeD1.9, J. Appl. Crystallogr., 1989, 22, 256–260, DOI:10.1107/S002188988801430X.
- A. Kinaci and M. Aydinol, Ab initio investigation of FeTi–H system, Int. J. Hydrogen Energy, 2007, 32, 2466–2474, DOI:10.1016/j.ijhydene.2006.10.006.
- C. S. Pande, M. A. Pick and R. L. Sabatini, The “activation” of FeTi for hydrogen absorption; An electron microscopic study, Scr. Metall., 1980, 14, 899–903, DOI:10.1016/0036-9748(80)90317-8.
- M. Pick and H. Wenzl, Physical metallurgy of FeTi-hydride and its behaviour in a hydrogen storage container, Int. J. Hydrogen Energy, 1977, 1, 413–420, DOI:10.1016/0360-3199(77)90094-5.
- A. Bläsius and U. Gonster, Mössbauer surface studies on Tife hydrogen storage material, Appl. Phys., 1980, 22, 331–332, DOI:10.1007/BF00899887.
- L. Schlapbach, A. Seiler and F. Stucki, Surface segregation in FeTi and its catalytic effect on the hydrogenation II: AES and XPS studies, Mater. Res. Bull., 1978, 13, 1031–1037, DOI:10.1016/0025-5408(78)90168-X.
- F. Stucki and L. Schlapbach, Magnetic properties of LaNi5, FeTi, Mg2Ni and their hydrides, J. Less-Common Met., 1980, 74, 143–151, DOI:10.1016/0022-5088(80)90084-3.
- G. K. Shenoy, D. Niarchos, P. J. Viccaro, B. D. Dunlap, A. T. Aldred and G. D. Sandrock, Surface segregation of iron by hydrogenation of FeTi: An 57Fe mössbauer conversion electron study, J. Less-Common Met., 1980, 73, 171–173, DOI:10.1016/0022-5088(80)90357-4.
- H. Züchner and G. Kirch, Auger electron spectroscopy investigation of the activation of TiFe for hydrogen uptake, J. Less-Common Met., 1984, 99, 143–150, DOI:10.1016/0022-5088(84)90344-8.
- T. Schober, On the activation of FeTi for hydrogen storage, J. Less-Common Met., 1983, 89, 63–70, DOI:10.1016/0022-5088(83)90249-7.
- K. Hiebl, E. Tuscher and H. Bittner, Untersuchungen an Hydriden im Bereich der η-Phase Ti4Fe2O, Monatsh. Chem., 1979, 110, 9–19, DOI:10.1007/BF00903742.
- M. Mintz, Z. Hadari and M. Dariel, Hydrogenation of oxygen-stabilized Ti2MOx (M = Fe, Co, Ni; 0 ⩽ x < 0.5) compounds, J. Less-Common Met., 1980, 74, 287–294, DOI:10.1016/0022-5088(80)90164-2.
- C. Stioui, D. Fruchart, A. Rouault, R. Fruchart, E. Roudaut and J. Rebiére, Absorption d’hydrogene par Ti4Fe2O et diverses phases M6O, Mater. Res. Bull., 1981, 16, 869–876, DOI:10.1016/0025-5408(81)90162-8.
- L. Schlapbach and T. Riesterer, The activation of FeTi for hydrogen absorption, Appl. Phys. A: Solids Surf., 1983, 32, 169–182, DOI:10.1007/BF00820257.
- B. Chu, S. Lee and T. Perng, Preparation and hydrogen absorption property of amorphous Ti50Fe50, Int. J. Hydrogen Energy, 1991, 16, 413–416, DOI:10.1016/0360-3199(91)90141-5.
- T. Mizuno and T. Morozumi, Titanium concentration in FeTix (1 ⩽ x ⩽ 2) alloys and its effect on hydrogen storage properties, J. Less-Common Met., 1982, 84, 237–244, DOI:10.1016/0022-5088(82)90148-5.
- T. Matsumoto, M. Amano and Y. Sasaki, Hydrogenation of FeTi-based alloys containing β-Ti, J. Less-Common Met., 1982, 88, 443–449, DOI:10.1016/0022-5088(82)90255-7.
- S.-M. Lee and T.-P. Perng, Microstructural correlations with the hydrogenation kinetics of FeTi1 + x alloys, J. Alloys Compd., 1991, 177, 107–118, DOI:10.1016/0925-8388(91)90061-Y.
- M. Amano, T. Hirata, T. Kimura and Y. Sasaki, Hydriding Process in Fe-Ti Alloys Consisting of the FeTi and β Ti Phases, Trans. Jpn. Inst. Met., 1984, 25, 657–661, DOI:10.2320/matertrans1960.25.657.
- F. Reidinger, J. F. Lynch and J. J. Reilly, An X-ray diffraction examination of the FeTi-H 2 system, J. Phys. F: Met. Phys., 1982, 12, L49–L55, DOI:10.1088/0305-4608/12/3/007.
- J. Reilly, J. Johnson, J. Lynch and F. Reidinger, Irreversible effects in the FeTi-H system, J. Less-Common Met., 1983, 89, 505–512, DOI:10.1016/0022-5088(83)90362-4.
- V. Y. Zadorozhnyy, S. N. Klyamkin, S. D. Kaloshkin, M. Y. Zadorozhnyy and O. V. Bermesheva, Mechanochemical synthesis and hydrogen sorption properties of nanocrystalline TiFe, Inorg. Mater., 2011, 47, 1081–1086, DOI:10.1134/S0020168511100232.
- H. Wenzl and E. Lebsanft, Phase diagram and thermodynamic parameters of the quasibinary interstitial alloy Fe0.5Ti0.5Hx in equilibrium with hydrogen gas, J. Phys. F: Met. Phys., 1980, 10, 2147–2156, DOI:10.1088/0305-4608/10/10/012.
- C.-N. Park and J.-Y. Lee, Kinetic properties of the hydrogenation of the FeTi intermetallic compound, J. Less-Common Met., 1983, 91, 189–201, DOI:10.1016/0022-5088(83)90312-0.
- R. C. Bowman and W. E. Tadlock, Hydrogen diffusion in β-phase titanium iron hydride, Solid State Commun., 1979, 32, 313–318, DOI:10.1016/0038-1098(79)90954-2.
- R. C. Bowman, A. Attalla, G. C. Carter and Y. Chabre, NMR studies of hydrogen relaxation and diffusion in TiFeHx and TiFe1-yMnyHx, Hydrides for Energy Storage, Elsevier, 1978, vol. 1, pp. 97–118 Search PubMed.
- R. C. Bowman, A. Attalla, G. C. Carter and Y. Chabre, Nuclear Magnetic Resonance Studies of Diffusion in FeTiHx, Second Int. Congr. Hydrogen Met., 1977, 1–8 Search PubMed.
- R. C. Bowman, A. Attalla and W. E. Tadlock, NMR studies of structure and diffusion in metal hydrides, Int. J. Hydrogen Energy, 1977, 1, 421–426, DOI:10.1016/0360-3199(77)90095-7.
- G. Alefeld and J. Voelkl, Hydrogen in metals I – basic properties, Springer, Berlin, New York, 1978, vol. 28 Search PubMed.
- P. D. Goodell, G. D. Sandrock and E. L. Huston, Kinetic and dynamic aspects of rechargeable metal hydrides, J. Less-Common Met., 1980, 73, 135–142, DOI:10.1016/0022-5088(80)90352-5.
- H. J. Ahn, S. M. Lee and L. Jai-Young, Intrinsic degradation of FeTi by thermally induced hydrogen absorption-desorption cycling, J. Less-Common Met., 1988, 142, 253–261, DOI:10.1016/0022-5088(88)90183-X.
- G. D. Sandrock and P. D. Goodell, Surface poisoning of LaNi5, FeTi and (Fe,Mn)Ti by O2, Co and H2O, J. Less-Common Met., 1980, 73, 161–168, DOI:10.1016/0022-5088(80)90355-0.
- F. R. Block and H.-J. Bahs, Investigation of selective absorption of hydrogen by LaNi5 and FeTi, J. Less-Common Met., 1983, 89, 77–84, DOI:10.1016/0022-5088(83)90251-5.
- J. Y. Jung, Y.-S. Lee, J.-Y. Suh, J.-Y. Huh and Y. W. Cho, Tailoring the equilibrium hydrogen pressure of TiFe via vanadium substitution, J. Alloys Compd., 2021, 854, 157263, DOI:10.1016/j.jallcom.2020.157263.
- C. E. Lundin, F. E. Lynch and C. B. Magee, A correlation between the interstitial hole sizes in intermetallic compounds and the thermodynamic properties of the hydrides formed from those compounds, J. Less-Common Met., 1977, 56, 19–37, DOI:10.1016/0022-5088(77)90215-6.
- J. C. Achard, A. Percheron-Guégan, H. Diaz, F. Briaucourt and F. Demany, Rare earth ternary hydrides. Hydrogen storage applications, Second Int. Congr. Hydrogen Met. Paris, 1977, 1–12 Search PubMed.
- Y. Zhang, T. T. Zuo, Z. Tang, M. C. Gao, K. A. Dahmen, P. K. Liaw and Z. P. Lu, Microstructures and properties of high-entropy alloys, Prog. Mater. Sci., 2014, 61, 1–93, DOI:10.1016/j.pmatsci.2013.10.001.
- V. Y. Zadorozhnyy, S. N. Klyamkin, M. Yu. Zadorozhnyy, M. V. Gorshenkov and S. D. Kaloshkin, Mechanical alloying of nanocrystalline intermetallic compound TiFe doped with sulfur and magnesium, J. Alloys Compd., 2014, 615, S569–S572, DOI:10.1016/j.jallcom.2013.12.144.
- G. Bruzzone, G. Costa, M. Ferretti and G. L. Olcese, Hydrogen storage in a beryllium substituted TiFe compound, Int. J. Hydrogen Energy, 1980, 5, 317–322, DOI:10.1016/0360-3199(80)90075-0.
- G. Bruzzone, G. Costa, M. Ferretti and G. L. Olcese, Hydrogen storage properties of Titanium-Iron-Beryllium and Titanium-Iron-Aluminum alloys, Metal–Hydrogen Systems, Elsevier, 1982, pp. 539–549 Search PubMed.
- C. Gosselin and J. Huot, Hydrogenation Properties of TiFe Doped with Zirconium, Materials, 2015, 8, 7864–7872, DOI:10.3390/ma8115423.
- B. K. Singh, A. K. Singh, C. S. Pandey and O. N. Srivastava, Investigation on synthesis, characterization and hydrogenation behaviour of hydrogen storage material: Fe1−xZrxTi1.3 (x = 0.2), Int. J. Hydrogen Energy, 1999, 24, 1077–1082, DOI:10.1016/S0360-3199(98)00145-1.
- N. Nishimiya, T. Wada, A. Matsumoto and K. Tsutsumi, Hydriding characteristics of zirconium-substituted FeTi, J. Alloys Compd., 2000, 313, 53–58, DOI:10.1016/S0925-8388(00)01181-6.
- P. Lv and J. Huot, Hydrogen storage properties of Ti0.95FeZr0.05, TiFe0.95Zr0.05 and TiFeZr0.05 alloys, Int. J. Hydrogen Energy, 2016, 41, 22128–22133, DOI:10.1016/j.ijhydene.2016.07.091.
- C. Gosselin, D. Santos and J. Huot, First hydrogenation enhancement in TiFe alloys for hydrogen storage, J. Phys. D: Appl. Phys., 2017, 50, 375303, DOI:10.1088/1361-6463/aa7d6a.
- A. K. Patel, P. Sharma and J. Huot, Effect of annealing on microstructure and hydrogenation properties of TiFe + X wt% Zr (X = 4, 8), Int. J. Hydrogen Energy, 2018, 43, 6238–6243, DOI:10.1016/j.ijhydene.2018.02.029.
- M. Y. Zadorozhnyi, S. D. Kaloshkin, S. N. Klyamkin, O. V. Bermesheva and V. Y. Zadorozhnyi, Mechanochemical synthesis of a TiFe nanocrystalline intermetallic compound and its mechanical alloying with third component, Met. Sci. Heat Treat., 2013, 54, 461–465, DOI:10.1007/s11041-013-9531-x.
- T. H. Jang, J. I. Han and L. Jai-Young, Effect of substitution of titanium by zirconium in TiFe on hydrogenation properties, J. Less-Common Met., 1986, 119, 237–246, DOI:10.1016/0022-5088(86)90684-3.
- P. Jain, C. Gosselin and J. Huot, Effect of Zr, Ni and Zr7Ni10 alloy on hydrogen storage characteristics of TiFe alloy, Int. J. Hydrogen Energy, 2015, 40, 16921–16927, DOI:10.1016/j.ijhydene.2015.06.007.
- J. Manna, B. Tougas and J. Huot, Mechanical activation of air exposed TiFe + 4 wt% Zr alloy for hydrogenation by cold rolling and ball milling, Int. J. Hydrogen Energy, 2018, 43, 20795–20800, DOI:10.1016/j.ijhydene.2018.09.096.
- V. Razafindramanana, S. Gorsse, J. Huot and J. L. Bobet, Effect of Hafnium Addition on the Hydrogenation Process of TiFe Alloy, Energies, 2019, 12, 3477, DOI:10.3390/en12183477.
- G. D. Sandrock, The Metallurgy and Production of Rechargeable Hydrides, Hydrides for Energy Storage, Elsevier, 1978, pp. 353–393 Search PubMed.
- A. Guéguen and M. Latroche, Influence of the addition of vanadium on the hydrogenation properties of the compounds TiFe0.9Vx and TiFe0.8Mn0.1Vx (x = 0, 0.05 and 0.1), J. Alloys Compd., 2011, 509, 5562–5566, DOI:10.1016/j.jallcom.2011.02.036.
- S. V. Mitrokhin, V. N. Verbetskii and K. N. Semenenko, Influence of impurities on the hydrogen-absorption characteristics of the alloy Ti0.96Fe0.94V0.1, Zh. Obshch. Khim., 1991, 61, 785–790 CAS.
- B. Massicot, Étude du système Fe–Ti–V et de ses applications au stockage de l’hydrogène, Université Paris-Est, 2009 Search PubMed.
- S. Kumar, G. P. Tiwari, S. Sonak, U. Jain and N. Krishnamurthy, High performance FeTi – 3.1 mass % V alloy for on board hydrogen storage solution, Energy, 2014, 75, 520–524, DOI:10.1016/j.energy.2014.08.011.
- H. Nagai, M. Nakatsu, K. Shoji and H. Tamura, Effect of simultaneous addition of oxygen with copper or niobium on the hydriding characteristics of FeTi for hydrogen storage, J. Less-Common Met., 1986, 119, 131–142, DOI:10.1016/0022-5088(86)90203-1.
- G. H. Cao, Y. F. Peng, N. Liu, X. Li, Z. S. Lei, Z. M. Ren, D. Gerthsen and A. M. Russell, Formation of a bimodal structure in ultrafine Ti–Fe–Nb alloys with high-strength and enhanced ductility, Mater. Sci. Eng., A, 2014, 609, 60–64, DOI:10.1016/j.msea.2014.04.088.
- E. A. Berdonosova, V. Y. Zadorozhnyy, M. Y. Zadorozhnyy, K. V. Geodakian, M. V. Zheleznyi, A. A. Tsarkov, S. D. Kaloshkin and S. N. Klyamkin, Hydrogen storage properties of TiFe-based ternary mechanical alloys with cobalt and niobium. A thermochemical approach, Int. J. Hydrogen Energy, 2019, 44, 29159–29165, DOI:10.1016/j.ijhydene.2019.03.057.
- P. Kuziora, I. Kunce, S. McCain, N. J. E. Adkins and M. Polański, The influence of refractory metals on the hydrogen storage characteristics of FeTi-based alloys prepared by suspended droplet alloying, Int. J. Hydrogen Energy, 2020, 45, 21635–21645, DOI:10.1016/j.ijhydene.2020.05.216.
- S.-M. Lee and T.-P. Perng, Effect of the second phase on the initiation of hydrogenation of TiFe1−xMx (M= Cr, Mn) alloys, Int. J. Hydrogen Energy, 1994, 19, 259–263, DOI:10.1016/0360-3199(94)90095-7.
- T. Yang, P. Wang, C. Xia, N. Liu, C. Liang, F. Yin and Q. Li, Effect of chromium, manganese and yttrium on microstructure and hydrogen storage properties of TiFe-based alloy, Int. J. Hydrogen Energy, 2020, 45, 12071–12081, DOI:10.1016/j.ijhydene.2020.02.086.
- P. J. Cappillino, E. J. Lavernia, M. D. Ong, W. G. Wolfer and N. Y. Yang, Plastic deformation and hysteresis for hydrogen storage in Pd–Rh alloys, J. Alloys Compd., 2014, 586, 59–65, DOI:10.1016/j.jallcom.2013.10.033.
- V. Y. Zadorozhnyy, S. N. Klyamkin, M. Y. Zadorozhnyy, O. V. Bermesheva and S. D. Kaloshkin, Mechanical alloying of nanocrystalline intermetallic compound TiFe doped by aluminum and chromium, J. Alloys Compd., 2014, 586, S56–S60, DOI:10.1016/j.jallcom.2013.01.138.
- T. Ha, S.-I. Lee, J. Hong, Y.-S. Lee, D.-I. Kim, J.-Y. Suh, Y. W. Cho, B. Hwang, J. Lee and J.-H. Shim, Hydrogen storage behavior and microstructural feature of a TiFe–ZrCr2 alloy, J. Alloys Compd., 2021, 853, 157099, DOI:10.1016/j.jallcom.2020.157099.
- J. R. Johnson and J. J. Reilly, The Use of Manganese Substituted Ferrotitanium Alloys for Energy Storage, Altern. Energy Sources, 1978, 8, 3739–3769 CAS.
- H. Suzuki, Y. Osumi, A. Kato, K. Oguro and M. Nakane, Hydrogen absorption-desorption characteristics of Ti-Co-Fe Alloys, J. Less-Common Met., 1981, 80, 179–185, DOI:10.1016/0022-5088(81)90091-6.
- S.-M. Lee and T.-P. Perng, Correlation of substitutional solid solution with hydrogenation properties of TiFe1−xMx (M = Ni, Co, Al) alloys, J. Alloys Compd., 1999, 291, 254–261, DOI:10.1016/S0925-8388(99)00262-5.
- M. Boulghallat, A. Jouaiti and L. Lâallam, Effect of Substitution of Iron by Cobalt on Hydrogenation Properties of TiFe, Int. J. Sci. Res., 2014, 3, 904–908 Search PubMed.
- S. L. Isaacka, E. A. Karakisha, M. Solimanb and E. El-Rafey, A new approach to deal with the hysteresis phenomenon in hydride systems TiFe1-xMx-H2(M = Mn,Ni), Int. J. Hydrogen Energy, 2004, 29, 1637–1642, DOI:10.1016/j.ijhydene.2004.03.014.
- S. Challet, M. Latroche and F. Heurtaux, Hydrogen Storage in TiFe(0.70 + x)Mn(0.20-x) (0 < x < 0.15) and TiFe(0.70)Mn(0.20-y)Ni(y) (0 < y < 0.08) Metallic Alloys, Mater. Sci. Technol., 2005, 13–21 Search PubMed.
- H. Chung and J. Lee, Effect of partial substitution of Mn and Ni for Fe in FeTi on hydriding kinetics, Int. J. Hydrogen Energy, 1986, 11, 335–339, DOI:10.1016/0360-3199(86)90153-9.
- M. Bououdina, D. Fruchart, S. Jacquet, L. Pontonnier and J. L. Soubeyroux, Effect of nickel alloying by using ball milling on the hydrogen absorption properties of TiFe, Int. J. Hydrogen Energy, 1999, 24, 885–890, DOI:10.1016/S0360-3199(98)00163-3.
- K. D. Ćirić, A. Kocjan, A. Gradišek, V. J. Koteski, A. M. Kalijadis, V. N. Ivanovski, Z. V. Laušević and D. L. Stojić, A study on crystal structure, bonding and hydriding properties of Ti–Fe–Ni intermetallics – Behind substitution of iron by nickel, Int. J. Hydrogen Energy, 2012, 37, 8408–8417, DOI:10.1016/j.ijhydene.2012.02.047.
- H. Miyamura, M. Takada, K. Hirose and S. Kikuchi, Metal hydride electrodes using titanium–iron-based alloys, J. Alloys Compd., 2003, 356–357, 755–758, DOI:10.1016/S0925-8388(03)00084-7.
- H. Shang, Y. Zhang, Y. Li, Y. Qi, S. Guo and D. Zhao, Investigation on gaseous and electrochemical hydrogen storage performances of as-cast and milled Ti1.1Fe0.9Ni0.1 and Ti1.09Mg0.01Fe0.9Ni0.1 alloys, Int. J. Hydrogen Energy, 2018, 43, 1691–1701, DOI:10.1016/j.ijhydene.2017.11.163.
- Y. Li, H. Shang, Y. Zhang, P. Li, Y. Qi and D. Zhao, Investigations on gaseous hydrogen storage performances and reactivation ability of as-cast TiFe1-Ni (x = 0, 0.1, 0.2 and 0.4) alloys, Int. J. Hydrogen Energy, 2019, 44, 4240–4252, DOI:10.1016/j.ijhydene.2018.12.144.
- E. Bershadsky, Y. Josephy and M. Ron, Investigation of kinetics and structural changes in TiFe0.8 Ni0.2 after prolonged cycling, J. Less-Common Met., 1991, 172–174, 1036–1043, DOI:10.1016/S0022-5088(06)80009-3.
- P. Jain, C. Gosselin, N. Skryabina, D. Fruchart and J. Huot, Hydrogenation properties of TiFe with Zr7Ni10 alloy as additive, J. Alloys Compd., 2015, 636, 375–380, DOI:10.1016/j.jallcom.2015.02.104.
- K. Oguro, Y. Osumi, H. Suzuki, A. Kato, Y. Imamura and H. Tanaka, Hydrogen storage properties of TiFe1−xNiyMz alloys, J. Less-Common Met., 1983, 89, 275–279, DOI:10.1016/0022-5088(83)90280-1.
- I. Yamashita, H. Tanaka, H. Takeshita, N. Kuriyama, T. Sakai and I. Uehara, Hydrogenation characteristics of TiFe1−xPdx (0.05 ≤ x ≤ 0.30) alloys, J. Alloys Compd., 1997, 253–254, 238–240, DOI:10.1016/S0925-8388(96)02925-8.
- L. Zaluski, A. Zaluska, P. Tessier, J. O. Ström-Olsen and R. Schulz, Catalytic effect of Pd on hydrogen absorption in mechanically alloyed Mg2Ni, LaNi5 and FeTi, J. Alloys Compd., 1995, 217, 295–300, DOI:10.1016/0925-8388(94)01358-6.
- M. Williams, M. V. Lototsky, M. W. Davids, V. Linkov, V. A. Yartys and J. K. Solberg, Chemical surface modification for the improvement of the hydrogenation kinetics and poisoning resistance of TiFe, J. Alloys Compd., 2011, 509, S770–S774, DOI:10.1016/j.jallcom.2010.11.063.
- M. W. Davids, M. Lototskyy, A. Nechaev, Q. Naidoo, M. Williams and Y. Klochko, Surface modification of TiFe hydrogen storage alloy by metal-organic chemical vapour deposition of palladium, Int. J. Hydrogen Energy, 2011, 36, 9743–9750, DOI:10.1016/j.ijhydene.2011.05.036.
- L. Zaluski, A. Zaluska, P. Tessier, J. O. Ström-Olsen and R. Schulz, Hydrogen absorption by nanocrystalline and amorphous Fe-Ti with palladium catalyst, produced by ball milling, J. Mater. Sci., 1996, 31, 695–698, DOI:10.1007/BF00367887.
- L. Zaluski, A. Zaluska, P. Tessier, J. O. Ström-Olsen and R. Schulz, Effects of relaxation on hydrogen absorption in Fe-Ti produced by ball-milling, J. Alloys Compd., 1995, 227, 53–57, DOI:10.1016/0925-8388(95)01623-6.
- C. Gosselin and J. Huot, First Hydrogenation Enhancement in TiFe Alloys for Hydrogen Storage Doped with Yttrium, Metals, 2019, 9, 242, DOI:10.3390/met9020242.
- X. Wang, R. Chen, C. Chen and Q. Wang, Hydrogen storage properties of TixFe + ywt.% La and its use in metal hydride hydrogen compressor, J. Alloys Compd., 2006, 425, 291–295, DOI:10.1016/j.jallcom.2006.01.025.
- L. Hong, N. Shiwen, G. Cheng, J. Longhuan, T. Fang, B. Deyou and S. Lanying, Neutron diffraction study of α-iron titanium cerium hydride, Chin. Phys. Lett., 1987, 4, 105–108, DOI:10.1088/0256-307X/4/3/003.
- V. Bronca, P. Bergman, V. Ghaemmaghami, D. Khatamian and F. D. Manchester, Hydrogen absorption characteristics of an FeTi + misch metal alloy, J. Less-Common Met., 1985, 108, 313–325, DOI:10.1016/0022-5088(85)90226-7.
- J. Ma, Hydrogen storage properties of FeTi1.3 + x wt%Mm (x = 0.0, 1.5, 3.0, 4.5, 6.0) hydrogen storage alloys, Int. J. Hydrogen Energy, 2000, 25, 779–782, DOI:10.1016/S0360-3199(99)00100-7.
- B. K. Singh, A. K. Singh and O. N. Srivastava, Improved hydrogen sorption characteristics in FeTi1 + xMm material, Int. J. Hydrogen Energy, 1996, 21, 111–117, DOI:10.1016/0360-3199(95)00024-0.
- Y. Xin-Nan and L. Zhangda, Surface properties on hydrogen storage material TiFeMm, J. Less-Common Met., 1987, 130, 535–540, DOI:10.1016/0022-5088(87)90153-6.
- S. H. Lim and L. Jai-Young, The effects of the addition of aluminium on the kinetic properties of the intermetallic compound TiFe, J. Less-Common Met., 1984, 97, 59–64, DOI:10.1016/0022-5088(84)90008-0.
- S. K. Kulshreshtha, R. Sasikala, P. Suryanarayana, A. J. Singh and R. M. Iyer, Studies on hydrogen storage material FeTi: Effect of Sn substitution, Mater. Res. Bull., 1988, 23, 333–340, DOI:10.1016/0025-5408(88)90006-2.
- S. Lee, Effects of boron and carbon on the hydrogenation properties of TiFe and Ti1.1Fe, Int. J. Hydrogen Energy, 2000, 25, 831–836, DOI:10.1016/S0360-3199(99)00107-X.
- M. L. Trudeau, L. Dignard-Bailey, R. Schulz, P. Tessier, L. Zaluski, D. H. Ryan and J. O. Strom-Olsen, The oxidation of nanocrystalline FeTi hydrogen storage compounds, Nanostruct. Mater., 1992, 1, 457–464, DOI:10.1016/0965-9773(92)90078-C.
- G. D. Sandrock and P. D. Goodell, Cyclic life of metal hydrides with impure hydrogen: Overview and engineering considerations, J. Less-Common Met., 1984, 104, 159–173, DOI:10.1016/0022-5088(84)90452-1.
- H. C. Kim and L. Jai-Young, The effect of surface conditions on the activation of FeTi, J. Less-Common Met., 1985, 105, 247–253, DOI:10.1016/0022-5088(85)90411-4.
- B. Rupp, On the change in physical properties of Ti4−xFe2 + xOy during hydrogenation I: Activation behaviour of ternary oxides Ti4−xFe2 + xOy and β-Ti, J. Less-Common Met., 1984, 104, 51–63, DOI:10.1016/0022-5088(84)90435-1.
- L. Schlapbach and T. Riesterer, The comparison of the surface properties of FeTi and Fe2Ti4Ox in view of the different hydrogen sorption behaviours, J. Less-Common Met., 1984, 101, 453–458, DOI:10.1016/0022-5088(84)90121-8.
- T. Schober and D. G. Westlake, The activation of FeTi for hydrogen storage: A different view, Scr. Metall., 1981, 15, 913–918, DOI:10.1016/0036-9748(81)90277-5.
- P. Selvam, B. Viswanathan, C. S. Swamy and V. Srinivasan, X-ray photoelectron spectroscopic investigations of the activation of FeTi for hydrogen uptake, Int. J. Hydrogen Energy, 1987, 12, 245–250, DOI:10.1016/0360-3199(87)90028-0.
- J. J. Reilly and F. Reidinger, Surface aggregates produced on activated FeTi as determined by X-ray diffraction, J. Less-Common Met., 1982, 85, 145–153, DOI:10.1016/0022-5088(82)90066-2.
- A. Shwartz, N. Shamir, N. Froumin, S. Zalkind, I. Edry, A. Haim and M. H. Mintz, Initial oxidation of TiFe1−xMnx (x = 0–0.3) by low dose exposures to H2O and O2, J. Alloys Compd., 2014, 610, 6–10, DOI:10.1016/j.jallcom.2014.04.196.
- R. Suzuki, J. Ohno and H. Gondoh, Effect of sulphur addition on the properties of Fe-Ti alloy for hydrogen storage, J. Less-Common Met., 1984, 104, 199–206, DOI:10.1016/0022-5088(84)90455-7.
- D. Dew-Hughes, The addition of Mn and Al to the hydriding compound FeTi: Range of homogeneity and lattice parameters, Metall. Trans. A, 1980, 11, 1219–1225, DOI:10.1007/BF02668146.
- E. M. Dematteis, D. M. Dreistadt, G. Capurso, J. Jepsen, F. Cuevas and M. Latroche, Fundamental hydrogen storage properties of TiFe-alloy with partial substitution of Fe by Ti and Mn, 2020 Search PubMed , submitted.
- H. Nagai, K. Kitagaki and K. Shoji, Microstructure and hydriding characteristics of FeTi alloys containing manganese, J. Less-Common Met., 1987, 134, 275–286, DOI:10.1016/0022-5088(87)90567-4.
- A. Seiler, F. Stucki and P. Charpié, How additives of Mn improve the hydrogenation characteristics of FeTi – and the role of its subsurface, Solid State Commun., 1982, 42, 337–341, DOI:10.1016/0038-1098(82)90149-1.
- Y. Shenzhong, Y. Rong, H. Tiesheng, Z. Shilong and C. Bingzhao, A study of the activation of FeTi and Fe0.9 TiMn0.1, Int. J. Hydrogen Energy, 1988, 13, 433–437, DOI:10.1016/0360-3199(88)90129-2.
- J. J. Reilly and R. H. Wiswall, Iron Titanium Manganase Alloy hydrogen storage, US Pat., 3922872, 1975 Search PubMed.
- K. Ikeda, Kondo Effect in the Transport Properties of the CsCl-Type Compounds Fe1−xTi1+x. I. Their Anomalous Behaviors in the Titanium-Rich Compositions, Phys. Status Solidi, 1974, 62, 655–664, DOI:10.1002/pssb.2220620235.
- Proceedings of the DOE Chemical/Hydrogen Energy Systems Contractor Review, 1978.
- H. I. Miller, J. Murray, E. Laury, J. Reinhardt and A. J. Goudy, The hydriding and dehydriding kinetics of FeTi and Fe0.9TiMn0.1, J. Alloys Compd., 1995, 231, 670–674, DOI:10.1016/0925-8388(95)01750-X.
- G. Sandrock, A panoramic overview of hydrogen storage alloys from a gas reaction point of view, J. Alloys Compd., 1999, 293–295, 877–888, DOI:10.1016/S0925-8388(99)00384-9.
- P. Modi and K.-F. Aguey-Zinsou, Titanium-iron-manganese (TiFe0.85Mn0.15) alloy for hydrogen storage: Reactivation upon oxidation, Int. J. Hydrogen Energy, 2019, 44, 16757–16764, DOI:10.1016/j.ijhydene.2019.05.005.
- A. Zeaiter, D. Chapelle, F. Cuevas, A. Maynadier and M. Latroche, Milling effect on the microstructural and hydrogenation properties of TiFe0.9Mn0.1 alloy, Powder Technol., 2018, 339, 903–910, DOI:10.1016/j.powtec.2018.08.085.
- J. Y. Lee, S. Byun, C. Park and J. Park, A study of the hydriding kinetics of TiFe and its alloys, J. Less-Common Met., 1982, 87, 149–164, DOI:10.1016/0022-5088(82)90050-9.
- J. J. Reilly, Synthesis and properties of useful metal hydrides, A review of recent work at Brookhaven National Laboratory, in Hydrides for Energy Storage. Proceedings of an International Symposium Held in Geilo, Norway, 14–19 August 1977, 1977, pp. 301–322.
- J. M. Bellosta von Colbe, J. Puszkiel, G. Capurso, A. Franz, H. U. Benz, H. Zoz, T. Klassen and M. Dornheim, Scale-up of milling in a 100 L device for processing of TiFeMn alloy for hydrogen storage applications: Procedure and characterization, Int. J. Hydrogen Energy, 2019, 44, 29282–29290, DOI:10.1016/j.ijhydene.2019.01.174.
- G. Romero, P. Lv and J. Huot, Effect of ball milling on the first hydrogenation of TiFe alloy doped with 4 wt% (Zr + 2Mn) additive, J. Mater. Sci., 2018, 53, 13751–13757, DOI:10.1007/s10853-018-2301-9.
- A. K. Patel, A. Duguay, B. Tougas, C. Schade, P. Sharma and J. Huot, Microstructure and first hydrogenation properties of TiFe alloy with Zr and Mn as additives, Int. J. Hydrogen Energy, 2020, 45, 787–797, DOI:10.1016/j.ijhydene.2019.10.239.
- J. Manna, B. Tougas and J. Huot, First hydrogenation kinetics of Zr and Mn doped TiFe alloy after air exposure and reactivation by mechanical treatment, Int. J. Hydrogen Energy, 2020, 45, 11625–11631, DOI:10.1016/j.ijhydene.2020.02.043.
- P. Lv and J. Huot, Hydrogenation improvement of TiFe by adding ZrMn2, Energy, 2017, 138, 375–382, DOI:10.1016/j.energy.2017.07.072.
- S. V. Mitrokhin, V. N. Verbetsky, R. R. Kajumov, H. Cunmao and Z. Yufen, Hydrogen sorption peculiarities in FeTi-type Ti-Fe-V-Mn alloys, J. Alloys Compd., 1993, 199, 155–160, DOI:10.1016/0925-8388(93)90443-Q.
- S. V. Mitrokhin, V. N. Verbetsky, H. Cunmao and Z. Yufen, Hydriding Characteristics of FeTi-Based Ti-Fe-V-Mn Alloy, Z. Phys. Chem., 1993, 181, 283–287, DOI:10.1524/zpch.1993.181.Part_1_2.283.
- N. Endo, S. Suzuki, K. Goshome and T. Maeda, Operation of a bench-scale TiFe-based alloy tank under mild conditions for low-cost stationary hydrogen storage, Int. J. Hydrogen Energy, 2017, 42, 5246–5251, DOI:10.1016/j.ijhydene.2016.11.088.
- H. Qu, J. Du, C. Pu, Y. Niu, T. Huang, Z. Li, Y. Lou and Z. Wu, Effects of Co introduction on hydrogen storage properties of Ti–Fe–Mn alloys, Int. J. Hydrogen Energy, 2015, 40, 2729–2735, DOI:10.1016/j.ijhydene.2014.12.089.
- J. J. Reilly and R. H. J. Wiswall, Iron Titanium hydride: its formation, properties, and application, Am. Chem. Soc., Div. Fuel Chem., Prepr., 1973, 11973, 53–77 Search PubMed.
- E. M. Dematteis, F. Cuevas and M. Latroche, Hydrogen storage properties of Mn and Cu for Fe substitution in TiFe0.9 intermetallic compound, J. Alloys Compd., 2021, 851, 156075, DOI:10.1016/j.jallcom.2020.156075.
- H. Leng, Z. Yu, J. Yin, Q. Li, Z. Wu and K.-C. Chou, Effects of Ce on the hydrogen storage properties of TiFe 0.9 Mn 0.1 alloy, Int. J. Hydrogen Energy, 2017, 42, 23731–23736, DOI:10.1016/j.ijhydene.2017.01.194.
- S. Lanyin, L. Fangjie and B. Deyou, An advanced TiFe series hydrogen storage material with high hydrogen capacity and easily activated properties, Int. J. Hydrogen Energy, 1990, 15, 259–262, DOI:10.1016/0360-3199(90)90045-Z.
- W. Ali, Z. Hao, Z. Li, G. Chen, Z. Wu, X. Lu and C. Li, Effects of Cu and Y substitution on hydrogen storage performance of TiFe0.86Mn0.1Y0.1−xCux, Int. J. Hydrogen Energy, 2017, 42, 16620–16631, DOI:10.1016/j.ijhydene.2017.04.247.
- W. Ali, M. Li, P. Gao, C. Wu, Q. Li, X. Lu and C. Li, Hydrogenation properties of Ti-Fe-Mn alloy with Cu and Y as additives, Int. J. Hydrogen Energy, 2017, 42, 2229–2238, DOI:10.1016/j.ijhydene.2016.09.037.
- R. Griessen and A. Driessen, Heat of formation and band structure of binary and ternary metal hydrides, Phys. Rev. B: Condens. Matter Mater. Phys., 1984, 30, 4372–4381, DOI:10.1103/PhysRevB.30.4372.
- R. Griessen and T. Riesterer, Heat of formation models, Hydrog. Intermet. Compd. I Electron. Thermodyn. Crystallogr. Prop. Prep., 1988, vol. 63, pp. 219–284 Search PubMed.
- J.-M. Joubert, Crystal chemistry and Calphad modeling of the σ phase, Prog. Mater. Sci., 2008, 53, 528–583, DOI:10.1016/j.pmatsci.2007.04.001.
- M. Bououdina, D. Grant and G. Walker, Review on hydrogen absorbing materials—structure, microstructure, and thermodynamic properties, Int. J. Hydrogen Energy, 2006, 31, 177–182, DOI:10.1016/j.ijhydene.2005.04.049.
- D. G. Westlake, Site occupancies and stoichiometries in hydrides of intermetallic compounds: Geometric considerations, J. Less-Common Met., 1983, 90, 251–273, DOI:10.1016/0022-5088(83)90075-9.
- G. Sandrock, The Interrelations Among Compositions, Microstructure, and Hydriding Behavior for Alloys Based on the Intermetallic Compound FeTi, Final Report for Contract BNL 352410S, The International Nickel Company, Inc., Suffern, NY. 1976, 10901.
- J. Shinar, I. Jacob, D. Davidov and D. Shaltiel, Hydrogen sorption properties in binary and pseudobinary intermetallic compounds, Hydrides for Energy Storage, Elsevier, 1978, vol. proceeding, pp. 337–352 Search PubMed.
- D. G. Ivey and D. O. Northwood, Storing Hydrogen in AB 2 Laves-Type Compounds, Z. Phys. Chem., 1986, 147, 191–209, DOI:10.1524/zpch.1986.147.1_2.191.
- E. R. Pinatel, M. Palumbo, F. Massimino, P. Rizzi and M. Baricco, Hydrogen sorption in the LaNi5-xAlx-H system (0 ≤ x ≤ 1), Intermetallics, 2015, 62, 7–16, DOI:10.1016/j.intermet.2015.03.002.
- M. H. Mintz, S. Vaknin, S. Biderman and Z. Hadari, Hydrides of ternary TiFexM1−x (M= Cr, Mn, Co, Ni) intermetallics, J. Appl. Phys., 1981, 52, 463–467, DOI:10.1063/1.329808.
- H. Yukawa, Y. Takahashi and M. Morinaga, Electronic structures of hydrogen storage compound, TiFe, Comput. Mater. Sci., 1999, 14, 291–294, DOI:10.1016/S0927-0256(98)00121-9.
- R. Burch and N. B. Mason, Absorption of hydrogen by titanium–cobalt and titanium–nickel intermetallic alloys. Part 2.—Thermodynamic parameters and theoretical models, J. Chem. Soc., Faraday Trans. 1, 1979, 75, 578, 10.1039/f19797500578.
- R. Burch and N. B. Mason, Absorption of hydrogen by titanium–cobalt and titanium–nickel intermetallic alloys. Part 1—Experimental results, J. Chem. Soc., Faraday Trans. 1, 1979, 75, 561, 10.1039/f19797500561.
- V. N. Fokin, E. E. Fokina and B. P. Tarasov, Study of the Interaction with Hydrogen and Ammonia of Titanium and Its Alloys with Iron, Russ. J. Appl. Chem., 2019, 92, 35–44, DOI:10.1134/S1070427219010051.
- M. V. Lototskyy, I. Tolj, L. Pickering, C. Sita, F. Barbir and V. Yartys, The use of metal hydrides in fuel cell applications, Prog. Nat. Sci.: Mater. Int., 2017, 27, 3–20, DOI:10.1016/j.pnsc.2017.01.008.
- A. H. Beaufrere, F. J. Salzano, R. J. Isler and W. S. Yu, Hydrogen storage via iron-titanium for a 26 MW(e) peaking electric plant, Int. J. Hydrogen Energy, 1976, 1, 307–319, DOI:10.1016/0360-3199(76)90025-2.
- G. Strickland and W. S. Yu, Some rate and modeling studies on the use of iron-titanium hydride as an energy storage medium for electric utility companies, Upton, NY (United States), 1977 Search PubMed.
- G. Strickland, J. Milau and M. Rosso, Some observations on the effects of the volumetric expansion of iron-titanium hydride on vessels built at BNL, Upton, NY (United States), 1977 Search PubMed.
- T. N. Veziroglu, in Hydrogen Energy, ed. T. N. Veziroğlu, Springer US, Boston, MA, 1975 Search PubMed.
- F. J. Salzano; C. Braun, A. Beaufrere, S. Srinivasan, G. Strickland, J. J. Reilly and C. Waide, Hydrogen storage via metal hydrides for utility and automotive energy storage applications, HCl electrolysis H/sub 2/-Cl/sub 2/fuel cells, United States, 1976.
- A. Beaufrere, R. S. Yeo, S. Srinivasan, J. McElroy and G. Hart, Hydrogen-halogen energy storage system for electric utility applications. Conf. 12. Intersoc. energy Convers. Eng. Conf. Washington, Dist. Columbia, United States Am. (USA), 28 Aug 1977 1977.
- N. Endo, E. Shimoda, K. Goshome, T. Yamane, T. Nozu and T. Maeda, Construction and operation of hydrogen energy utilization system for a zero emission building, Int. J. Hydrogen Energy, 2019, 44, 14596–14604, DOI:10.1016/j.ijhydene.2019.04.107.
- N. Endo, E. Shimoda, K. Goshome, T. Yamane, T. Nozu and T. Maeda, Operation of a stationary hydrogen energy system using TiFe-based alloy tanks under various weather conditions, Int. J. Hydrogen Energy, 2020, 45, 207–215, DOI:10.1016/j.ijhydene.2019.10.240.
- N. Endo, K. Goshome, M. Tetsuhiko, Y. Segawa, E. Shimoda and T. Nozu, Thermal management and power saving operations for improved energy efficiency within a renewable hydrogen energy system utilizing metal hydride hydrogen storage, Int. J. Hydrogen Energy, 2020, 1–10, DOI:10.1016/j.ijhydene.2020.10.002.
- L. Mohammedi, B. Daoudi and A. Boukraa, Ab-initio structural and electronic properties of the intermetallic compound TiFeH2, Comput. Condens. Matter, 2015, 2, 11–15, DOI:10.1016/j.cocom.2014.12.001.
- E. Lebsanft, D. Richter and J. Töpler, Investigation of the hydrogen diffusion in FeTiHx by means of quasielastic neutron scattering, J. Phys. F: Met. Phys., 1979, 9, 1057–1064, DOI:10.1088/0305-4608/9/6/012.
- G. Busch, L. Schlapbach, F. Stucki, P. Fischer and A. F. Andresen, Hydrogen storage in FeTi: Surface segregation and its catalytic effect on hydrogenation and structural studies by means of neutron diffraction, Int. J. Hydrogen Energy, 1979, 4, 29–39, DOI:10.1016/0360-3199(79)90127-7.
- T. Schober and W. Schäfer, Transmission electron microscopy and neutron diffraction studies of Feti-H(D), J. Less-Common Met., 1980, 74, 23–31, DOI:10.1016/0022-5088(80)90070-3.
- M. Dornheim, N. Eigen, G. Barkhordarian, T. Klassen and R. Bormann, Tailoring Hydrogen Storage Materials Towards Application, Adv. Eng. Mater., 2006, 8, 377–385, DOI:10.1002/adem.200600018.
- B. Łosiewicz, M. Popczyk, I. Napłoszek and A. Budniok, Intermetallic Compounds as Catalysts in the Reaction of Electroevolution/Absorption of Hydrogen, Solid State Phenom., 2015, 228, 16–22, DOI:10.4028/www.scientific.net/SSP.228.16.
- N. Rajalakshmi, Hydrogen solubility properties of Ti0.42Zr0.08Fe0.50 alloy, Int. J. Hydrogen Energy, 1999, 24, 625–629, DOI:10.1016/S0360-3199(98)00121-9.
- J. Liu, N. J. Ramsey and C. E. Lundin, Alloys for hydrogen storage, US Pat., 4358316, 1982 Search PubMed.
- A. Guéguen and M. Latroche, Influence of the addition of vanadium on the hydrogenation properties of the compounds TiFe0.9Vx and TiFe0.8Mn0.1Vx (x = 0, 0.05 and 0.1), J. Alloys Compd., 2011, 509, 5562–5566, DOI:10.1016/j.jallcom.2011.02.036.
- A. Szajek, M. Jurczyk, M. Nowak and M. Makowiecka, The electronic and electrochemical properties of the LaNi5-based alloys, Phys. Status Solidi, 2003, 196, 252–255, DOI:10.1002/pssa.200306399.
- M. Jurczyk, E. Jankowska, M. Makowiecka and I. Wieczorek, Electrode characteristics of nanocrystalline TiFe-type alloys, J. Alloys Compd., 2003, 354, L1–L4, DOI:10.1016/S0925-8388(02)01347-6.
- S. H. Lim and L. Jai-Young, The effects of aluminium substitution in TiFe on its hydrogen absorption properties, J. Less-Common Met., 1984, 97, 65–71, DOI:10.1016/0022-5088(84)90009-2.
- E. Schmidt-Ihn, O. Bernauer, H. Buchner and U. Hildebrandt, Storage on Hydrides for Electrical Load-Equilibration, in Hydrogen as an Energy Vector, Intergovernmental Panel on Climate Change, Springer Netherlands, Dordrecht, 1980, vol. 53, pp. 516–530 Search PubMed.
- G. Strickland, Hydrogen storage technology for metal hydrides. Conf. Hydrog. energy Distrib. Chicago, IL, USA, 24 Jul 1978, 1978.
- C. Li, J. He, Z. Zhang, B. Yang, H. Leng, X. Lu, Z. Li, Z. Wu and H. Wang, Preparation of TiFe based alloys melted by CaO crucible and its hydrogen storage properties, J. Alloys Compd., 2015, 618, 679–684, DOI:10.1016/j.jallcom.2014.08.154.
- H. Shang, Y. Zhang, Y. Li, Y. Qi, S. Guo and D. Zhao, Effects of adding over-stoichiometrical Ti and substituting Fe with Mn partly on structure and hydrogen storage performances of TiFe alloy, Renewable Energy, 2019, 135, 1481–1498, DOI:10.1016/j.renene.2018.09.072.
| This journal is © The Royal Society of Chemistry 2021 |