
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanostructured copper selenide as an ultrasensitive and selective non-enzymatic glucose sensor†
Siddesh
Umapathi
 a,
Harish
Singh
a,
Jahangir
Masud
b and
Manashi
Nath
*a
a,
Harish
Singh
a,
Jahangir
Masud
b and
Manashi
Nath
*a
aDepartment of Chemistry, Missouri University of Science and Technology, Rolla, USA. E-mail: nathm@mst.edu
bEnergy and Environmental Research centre, University of North Dakota, Grand Forks, USA
First published on 18th November 2020
Abstract
CuSe nanostructures exhibit high-efficiency for glucose detection with high sensitivity (19.419 mA mM−1 cm−2) and selectivity at a low applied potential of +0.15 V vs. Ag|AgCl, a low detection limit of 0.196 μM and a linear range of glucose detection from 100 nM to 40 μM.
Diabetes caused by the imbalance of glucose level in blood has been a severe concern lately, leading to 1.5 million deaths across the globe according to World Health Organization reports. It has also been predicted that diabetes will become the 7th leading cause of mortality by 2030.1–4 Diabetes is a silent killer wherein the symptoms may not be expressed until very advanced stage leading to more fatality. Hence, continuous monitoring of blood glucose levels in susceptible as well as healthy individuals is very important to detect the onset of diabetes at an early stage and minimize progression of the disease by taking preventive measures. Although commercially available enzyme-based glucose sensing strips are widely used for measuring blood glucose levels, their limited shelf life, low sensitivity, non-reusability, and high cost make it desirable to seek alternate solutions for glucose sensing.5–7 Moreover, non-enzymatic glucose sensors are also lucrative as long-term continuous blood glucose monitoring systems that can be implanted in the peripheral tissue including sub-dermis or tooth enamel. Electrochemical glucose sensors work on the principle of direct glucose oxidation on electrocatalytic surface, and can be categorized into two types: enzymatic and non-enzymatic glucose sensors.8–10 Among these, non-enzymatic glucose sensors have attracted considerable attention over the last few years due to their advantages such as high stability and sensitivity, low cost, and simple preparation.11–13
Over the last several years, various non-enzymatic glucose sensors based on different kinds of materials have been reported, such as metal nanoparticles and carbon materials, where polymer binders have been used to immobilize these nanoparticles. Such non-conductive polymeric binders add inactive components in the catalytic composite which may hinder the ability for quick electron transfer within the catalytic composite and reduce sensitivity.14–17 On the other hand, transition metals consisting of Ni, Co and Fe have been demonstrated as promising materials for glucose oxidation which also have the advantage of being earth abundant, low cost and environmentally friendly.18–22 Multi-metallic alloys and multi-metallic compounds such as Co–Ni, Ni–Fe and Ni–Cu have also shown good electrochemical glucose sensing.23–26
In recent years, transition metal chalcogenides (TMCs) have gained considerable attention in electrochemical devices such as water electrolyzers, fuel cells, and supercapacitors, owing to their unprecedented high electrocatalytic activity. This improvement of the electrochemical activity of TMC is primarily caused by the reduced anion electronegativity and high degree of covalency in the lattice which leads to better electrochemical tunability and reduced bandgap in the materials. The electrochemical tunability aids in adsorption of reactive intermediates on the catalyst surface through local oxidation/reduction of the transition metal active site, while a reduced bandgap also enhances charge transport at the catalyst–electrolyte interface as well as through the catalyst composite.27–29 The effect of decreasing anion electronegativity on electrocatalytic activity has been recently observed in a series of Ni–chalcogenide water oxidation catalysts where it was observed that the catalytic efficiency progressively improves from Ni–oxide to Ni–telluride.30–32 Copper has been studied recently for its electrochemical activity in various systems, and presents an attractive case for further expansion attributed to its abundancy on earth's surface and low-cost.33–35 These attributes have led to the usage of Cu in various catalytic processes.36 However, reports of copper chalcogenides in electrochemical devices are still limited. As explained above, decreasing the anion electronegativity is expected to improve the electrochemical tunability of the catalytically active transition metal center leading to better electrocatalytic activity.
In this communication, we have reported a high efficiency, non-enzymatic, direct glucose electrochemical sensor based on CuSe nanostructures synthesized by one step electrodeposition directly on the electrode surface as well as by hydrothermal techniques. Such direct growth on the electrode surface avoids the use of any adhesive or polymeric binder which can reduce sensing performance. The as-prepared CuSe shows excellent sensitivity and low limit of detection for glucose. The developed sensor was also applied successfully for the detection of glucose in human blood samples.
D-Glucose, copper chloride, selenium oxide and NaOH were purchased from Arcos chemicals. Uric acid (UA), l-ascorbic acid (AA), dopamine (DA), NaCl and KCl were obtained from Alfa Aesar. All chemicals were used as received without any further purification. Deionized water was used in all experiments.
The CuSe thin film was prepared directly on a carbon cloth electrode through direct electrodeposition using a conventional three electrode set-up, where Ag|AgCl was used as the reference electrode, graphite rod as the counter electrode and commercial carbon cloth as the working electrode. The deposition area of CuSe was pre-defined by using a masking tape exposing a 0.08 cm2 hole on the electrode surface. The electrolyte contained 2 mM of copper chloride, 4.5 mM of SeO2 and 0.1 M of KCl in deionized water. The pH of the electrolyte was adjusted to 2 using dilute HCl. This solution was purged with N2 gas for 20 minutes prior to the electrodeposition to reduce the amount of dissolved air. Electrodeposition was carried out at an applied voltage of −0.16 V vs. Ag|AgCl for 300 seconds. Following electrodeposition, the substrate was mildly washed with DI water and dried naturally. CuSe powder was also synthesized through hydrothermal techniques as described in the ESI.†
The composition, phase, and morphology of the copper selenide catalyst composite were identified through powder X-ray diffraction (PXRD), scanning electron microscopy (SEM) (FEI Helios Nanolab 600) using 10 kV accelerated voltage, and energy dispersive spectroscopy (EDS). The composition of the film was also analyzed through X-ray photoelectron spectroscopy (XPS) using a KRATOS AXIS 165 spectrometer with an Al source. Transmission electron microscopy (Tecnai F20 with an accelerating voltage of 200 kV) was also performed to investigate the nanostructure details of the morphology. Electrochemical measurements were performed using an Iviumstat electrochemical workstation using a three-electrode system with CuSe on carbon cloth as the working electrode, saturated Ag|AgCl as the reference electrode and a graphite rod served as the counter electrode.
The PXRD pattern collected from hydrothermally synthesized copper selenide powder showed a highly crystalline pattern as shown in Fig. 1A confirming the phase purity and composition of the catalyst. The diffraction pattern matched with the standard diffraction pattern for CuSe (PDF# 00-086-1239) confirming the phase. CuSe crystallizes in a hexagonal structure with Cu in two different coordination geometries, trigonal planar and tetrahedra. Such low coordination geometries around the active sites are expected to enhance the adsorption of oxygenated reactive intermediates on the surface through coordination expansion leading to an improved electrocatalytic performance as has been observed previously in other transition metal selenide based electrocatalysts.32 The composition of the as-deposited film was confirmed through XPS, which also provides details of the local bonding environment and the oxidation states of the elements. As shown in Fig. 1B, the Cu 2p spectrum shows peaks centered at 932.2 and 952.3 eV for Cu+ 2p3/2 and 2p1/2 and 934.4 and 954.6 eV for Cu2+ 2p3/2 and 2p1/2, respectively. This also suggested that Cu was present in mixed oxidation states, while the satellite peaks are observed at 942.4 and 962.6 eV. The deconvoluted Se 3d spectra of the electrodeposited CuSe (inset of Fig. 1B) show peaks at 54.4 and 55.4 eV for Se 3d5/2 and 3d3/2, respectively, which is in accordance with those of the previously reported copper selenide.37
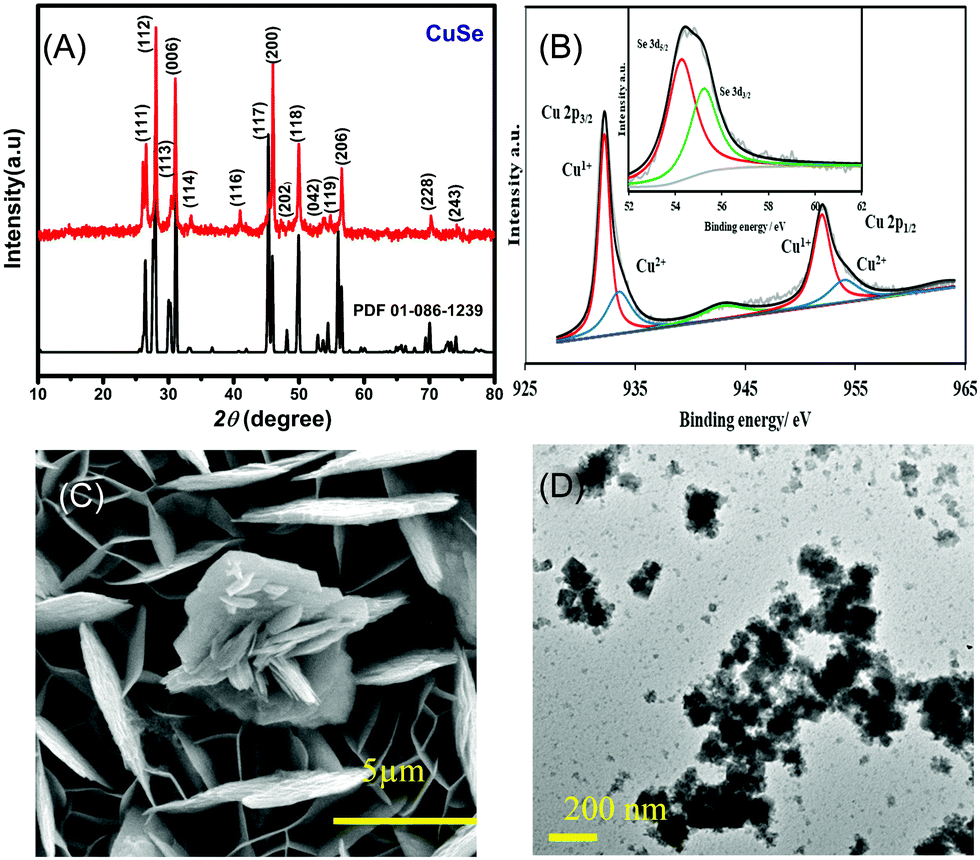 | ||
| Fig. 1 (A) PXRD pattern of hydrothermally synthesized CuSe, compared with the reference pattern (PDF#00-086-1239) (* denotes Au peaks). (B) Deconvoluted XPS spectra of CuSe showing Cu 2p peaks. Inset in (b) shows the corresponding Se 3d signals. (C) SEM and (D) TEM images of CuSe. | ||
The SEM images of the as-deposited CuSe thin film as depicted in Fig. 1C showed that CuSe had a rough surface topology comprising a nanoflake-like morphology. The nanoflakes are randomly oriented leading to a porous film which provides a high surface area for glucose adsorption. The elemental mapping through EDS showed a uniform distribution of Cu and Se throughout the composite, while quantification of the EDS data showed an elemental ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for Cu:Se (Fig. S1, ESI†). TEM studies (Fig. 1D) showed similar flake-like nanostructures, while HRTEM images showed lattice fringes corresponding to a d-spacing of 3.31 Å which could be matched to the 101 lattice spacing of CuSe (Fig. S2, ESI†).
:1 for Cu:Se (Fig. S1, ESI†). TEM studies (Fig. 1D) showed similar flake-like nanostructures, while HRTEM images showed lattice fringes corresponding to a d-spacing of 3.31 Å which could be matched to the 101 lattice spacing of CuSe (Fig. S2, ESI†).
The electrocatalytic performance of the CuSe thin film towards the oxidation of glucose was studied by cyclic voltammetry (CV). Fig. 2A shows the CV of the CuSe thin film on carbon cloth measured in the presence and absence of 0.1 mM glucose in 0.1 M NaOH electrolyte at 10 mV s−1 scan rate. The current response was moderate in a blank 0.1 M NaOH electrolyte, but upon the addition of 0.1 mM of glucose into the alkaline electrolyte, there was a substantial increase in the anodic current, indicating oxidation of glucose on the CuSe-coated electrode. This oxidation was also observed in the reverse sweep of CV, which further confirmed the electrochemical process to be analyte-oxidation, i.e. glucose oxidation on the electrode surface. To further evaluate the electrocatalytic performance of CuSe towards glucose oxidation, the scan rates were varied from 5 mV s−1 to 75 mV s−1 as shown in Fig. 2B. The glucose oxidation peaks show an obvious trend in the increase of current with respect to the scan rate in addition to a positive shift of the anodic oxidation potential. The redox peak current showed a linear correlation (R2 = 0.9965) with the square root of the scan rate, which is typical for a diffusion-controlled process for any electrochemical oxidation. On addition of 0.25, 0.5, 1, 2 and 4 mM of glucose to 0.1 M NaOH solution, the CuSe composite electrode showed an increase in the current density corresponding to the increase in glucose concentration (Fig. 2C), indicating that the oxidation current is mainly due to the availability of increased glucose content in the electrolyte.
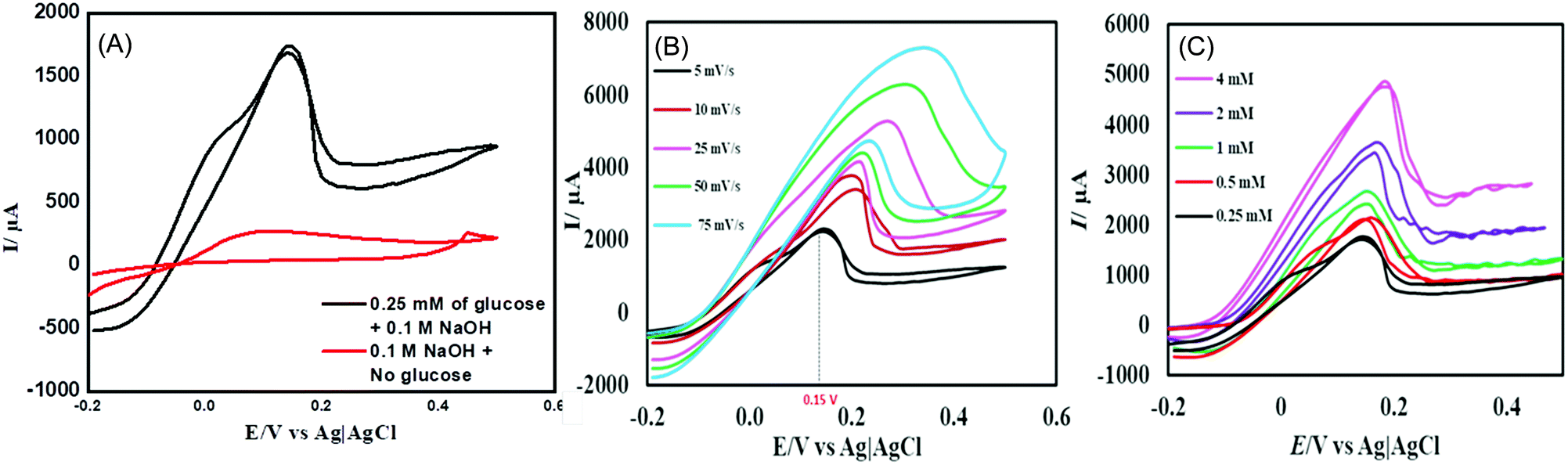 | ||
| Fig. 2 (A) CV curves of CuSe with 0.25 mM glucose and no glucose in 0.1 M NaOH solution. (B) CV plots with scan rates ranging from 5 to 75 mV s−1. (C) CV curves of CuSe with varying concentrations of glucose ranging from 0.25 mM to 4 mM. | ||
In order to determine the optimal applied potential for glucose sensing, oxidation current was measured by scanning the potential ranging from 0.05 V to 0.3 V vs. Ag|AgCl using amperometric technique with successive addition of 0.1 mM glucose to 0.1 M NaOH electrolyte under constant stirring. Fig. S3 (ESI†) shows that the ratio of oxidation current vs. potential ranges from 0.05 V to 0.30 V, where the highest oxidation current was achieved at 0.15 V, after which it begins to decay. Hence, the ideal working potential for the oxidation of glucose at the electrodeposited CuSe thin film was selected to be +0.15 V vs. Ag|AgCl for the rest of the study.
Similar electrochemical measurements were also performed for hydrothermally synthesized CuSe powder assembled on the electrode as described in the ESI.† The hydrothermally synthesized CuSe powder showed enhanced glucose oxidation at 0.25 V vs. Ag|AgCl as shown in Fig. S4 (ESI†).
Chronoamperometric technique was used to measure the response of CuSe composite electrode upon successive injection of glucose in a homogenously stirred NaOH solution. As a control experiment, the current response upon successive addition of glucose was also measured from a bare carbon cloth electrode to confirm that the current increments observed are not due to accidental jumps due to changes in experimental conditions. The limit of detection and linear range were also determined using the above method. As shown in Fig. 3A, a constant potential of +0.15 V vs. Ag|AgCl was applied, when the CuSe-modified electrodes showed a rapid and significant response of increasing anodic current upon addition of glucose ranging from 100 nM to 5 mM, which indicates the high sensitivity of CuSe towards glucose sensing. The bare carbon electrode on the other hand did not show any response on successive addition of glucose as shown in Fig. S5(a) in the ESI.† Additional control experiments were also performed by adding 100 μL of PBS (0.1 M) and 100 μL of DI water separately to the electrolyte for checking the instantaneous current response in absence of glucose. As shown in Fig. S5(b) (ESI†), there was no instantaneous current response upon addition of PBS or DI water in the same electrolyte, indicating that the current response upon addition of glucose solution is the actual sensing current. A calibration curve was obtained by plotting the peak anodic current vs. concentration of glucose from the amperometric experiment described above. Fig. 3(D–F) shows the calibration curve from 100 nM to 5 mM where the corresponding regression equation can be described as I (mA) = 19.419 C (mM) + 0.0231 (R2 = 0.9998) having a high sensitivity of 19.419 mA mM−1 cm−2. Further the linear detection range of CuSe towards glucose was 100 nM to 40 μM and a second linear region for higher concentrations from 80 μM to 5 mM was observed, with a limit of detection of 196 nM. Fig. S6 (ESI†) shows the response time of CuSe upon addition of glucose. The catalyst achieves a steady state current within 2 s of glucose addition, which shows that these CuSe-modified electrodes are capable of real-time monitoring of glucose in the body. Chronoamperometric measurements were also performed with hydrothermally synthesized CuSe powder as shown in Fig. S7a (ESI†), which showed a sensitivity of 8.341 mA mM−1 cm−2, and a first and second linear detection range of 10 μM–80 μM and 320 μM–2 mM of glucose detection, respectively (Fig. S8 (a–c), ESI†). The slightly lower sensitivity for the hydrothermally synthesized powder can be attributed to the fact that the composite electrode contains Nafion which restricts exposure of catalytic site to glucose in the electrolyte. Nevertheless, the CuSe-based electrodes show high sensitivity for glucose detection with a low LOD compared to other non-enzymatic glucose sensors as shown in Table 1.
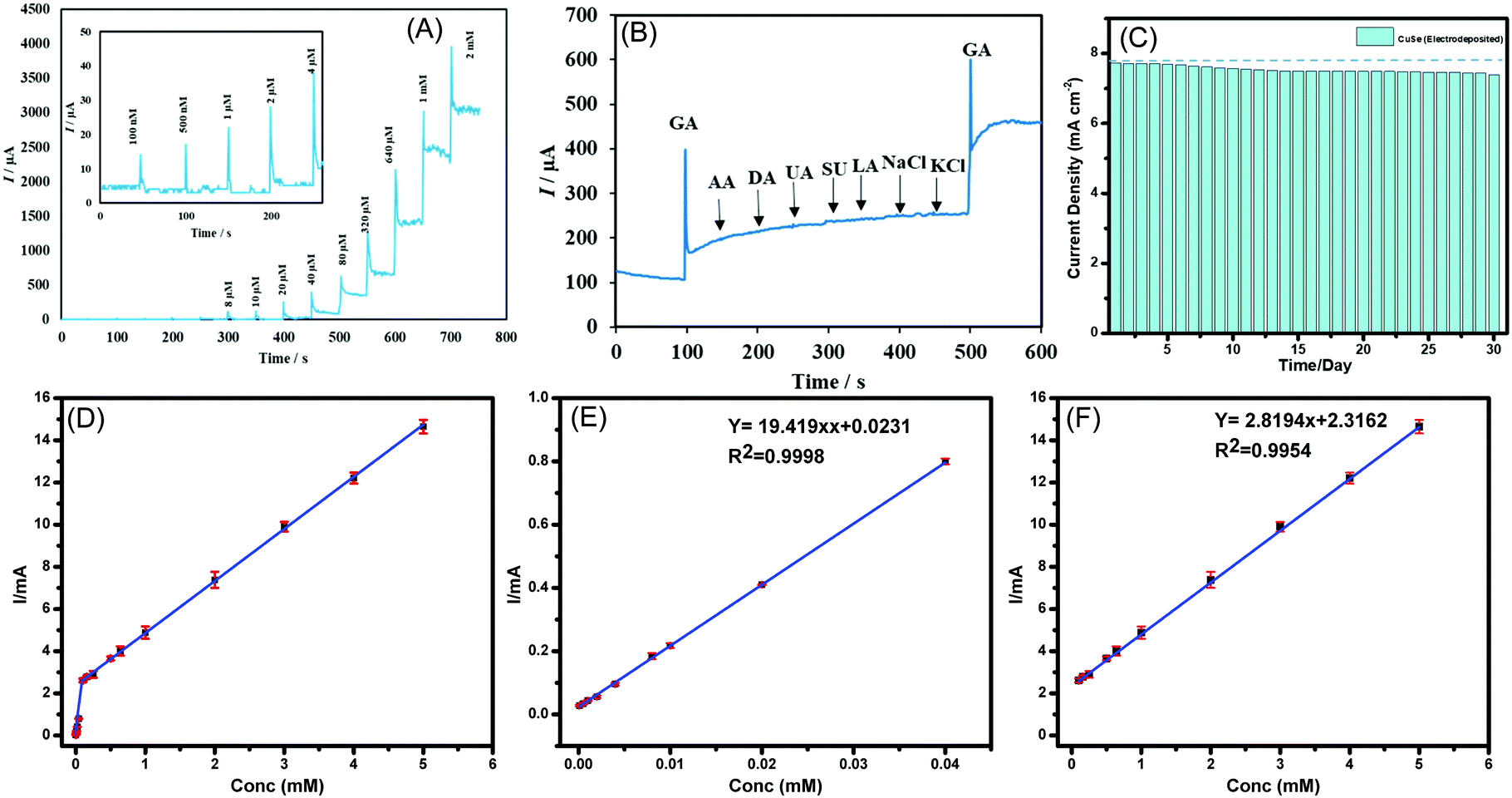 | ||
| Fig. 3 (A) Chronoamperometric response of the CuSe-modified electrode to successive additions of glucose into the stirred 0.1 M NaOH electrolyte. The working potential was set at +0.15 V vs. Ag|AgCl, and the glucose concentrations ranged from 100 nM to 2 mM for sequential addition. Inset shows magnified portion of amperometric response for lower concentrations of glucose addition. (B) Amperometric response of the CuSe-modified electrode measured in 0.1 M NaOH with successive addition of glucose (0.1 mM), AA (0.5 mM), DA (0.5 mM), UA (0.5 mM), Sucrose (0.1 mM), Lactose (0.1 mM), NaCl (0.5 mM), KCl (0.5 mM), and glucose (0.1 mM) at an applied potential of +0.15 V vs. Ag|AgCl. (C) Long-term stability check of CuSe electrodes showing glucose sensing current by addition of 1 mM glucose solution each day for over 30 days. (D) Peak current versus the concentration of glucose at low and high concentration regions for electrodeposited CuSe. (E) Linear range from 100 nM to 40 μM and (F) linear range from 100 μM to 5 mM. | ||
| Electrode | Applied potential (V vs. Ag|AgCl) | Sensitivity (mA mM−1 cm−2) | Linear range | LOD (μM) | Ref. |
|---|---|---|---|---|---|
| CuSe (electrodeposition) | 0.15 | 19.41 | 100 nM–80 μM; 100 μM–5 mM | 0.196 | This work |
| CuSe (hydrothermal) | 0.25 | 8.314 | 10–80 μM; 320 μM–2 mM | 0.391 | This work |
| CuO nanowires | 0.55 | 0.648 | — | 2 | 12 |
| Cu2Se SPs/CF | 0.50 | 18.66 | 0.25 μM–0.237 mM | 0.25 | 35 |
| CuO NWA/CF | 0.50 | 32.33 | 0.10–0.50 mM | 0.02 | 41 |
| CuNi/C nanosheet | 0.54 | 17.12 | 0.2 μM–2.72 mM | 0.066 | 42 |
| Cu@porous carbon | 0.55 | 10.1 | 1 μM–6.0 mM | 0.6 | 43 |
| CuS/RGO/CuS/Cu | 0.65 | 22.67 | 0.001–0.655 mM | 0.5 | 44 |
| CuO NPs | 0.50 | 1.430 | 0.04–6.0 mM | 5 | 45 |
| CuCo2O4 NWAs/CC | 0.55 | 3.930 | 0.001–0.93 mM | 0.5 | 46 |
| CuO/rGO/CNT | 0.60 | 9.278 | 0.01–1 mM | 1 | 47 |
| CuO/NiO/PANI/GCE | 0.60 | 3.402 | 20 μM–2.5 mM | 2 | 48 |
| CuO–ZnO NRs/FTO | 0.62 | 2.961 | Up to 8.45 mM | 0.4 | 49 |
Several biomolecules with similar oxidation profiles are known to interfere with the detection of glucose which makes development of nonenzymatic glucose sensors very challenging. Species such as ascorbic acid (AA), dopamine (DA), lactose, NaCl and KCl commonly available in a lower concentration in bodily fluids can exhibit interference by undergoing electro-oxidation. Therefore, the selectivity of CuSe towards glucose oxidation was confirmed by measuring the amperometric response of CuSe composite electrode upon consecutive injection of glucose and other interferents as mentioned above. A constant potential of +0.15 V vs. Ag|AgCl was applied to an evenly stirred 0.1 M NaOH solution wherein the addition of 0.1 mM of glucose showed a rapid increase of anodic current. Addition of sucrose and lactose (0.1 mM) and AA, DA, LC, NaCl, and KCl (0.5 mM) did not show any appreciable oxidation current. However, the second addition of 0.1 mM glucose showed similar jump in the anodic current density as observed from the 1st addition which validated the functionality and selectivity of the CuSe based composite electrode (Fig. 3B). Interference studies were also conducted with hydrothermally synthesized CuSe powder which shows similar selectivity for glucose oxidation at a low applied potential as shown in Fig. S7b (ESI†). Thus, it was confirmed that CuSe exhibits high sensitivity and selectivity for non-enzymatic glucose sensing at an extremely low working potential.
To confirm the long-term functional stability of CuSe electrodes, glucose oxidation currents were measured by exposing the same CuSe-coated electrode repeatedly to 1 mM glucose solution each day for over 30 days. The electrode used was stored under ambient conditions. It was observed that even after being exposed to air for 30 days, both electrodeposited and hydrothermally synthesized CuSe-modified electrodes retained more that 90% of their original current response as shown in Fig. 3C and Fig. S7c (ESI†) for electrodeposited and hydrothermally synthesized CuSe, respectively. Such studies confirmed the long-term stability of these electrodes.
The practical applicability of the fabricated non-enzymatic glucose sensor was investigated by the determination of glucose in human blood samples following standard method38 and comparing it with the commercially available enzymatic glucometer kit (ReliOn). Specifically, the experiment comprised of first stabilizing current response of the electrode by adding 1 mM of glucose two times. The blood sample was then injected directly into the NaOH electrolyte in the vicinity of the CuSe-modified electrode. 1 mM of glucose was added again and the current response was recorded. The glucose level in the blood samples was measured from the linear fit of the plot obtained by plotting current density vs. glucose concentration of standard glucose additions as shown in Fig. S9 (ESI†). The range of chronoamperometry measurements was also extended to a much higher glucose concentration, specifically, from 0.1 mM to 5 mM in order to match the human blood glucose concentration (Fig. S10, ESI†). Table 2 lists the glucose concentration as detected using a standard glucometer and a CuSe based sensor. Each sample was tested three times and the calculated relative standard deviation of less than 3% suggests robustness and reliability of CuSe towards glucose sensing in physiological samples.
| Sample | Glucometer (mM) | CuSe (mM) | RSD (%, n = 3) |
|---|---|---|---|
| 1st glucose | 6.37 | 6.39 | 1.4 |
| 2nd glucose | 4.72 | 4.84 | 3.59 |
| 3d glucose | 5.7 | 5.55 | 2.4 |
| Blood 1 | 5.45 | 5.56 | 2.6 |
| Blood 2 | 5.48 | 5.5 | 2.1 |
| Blood 3 | 5.18 | 5.25 | 2.5 |
| Blood 4 | 4.66 | 4.75 | 2.4 |
Owing to its high sensitivity, the short response time and low detection limit electrodeposited CuSe is a potential candidate as a continuous glucose monitoring system for commercial applications. Additionally, CuSe has a low working potential and selectivity to sense glucose and not the other biomolecules commonly present in bodily fluids which is an advantage in wearable biosensors. Other than biosensing, CuSe has also been reported for electrochemical energy conversion.39 The superior electrochemical performance of CuSe especially towards glucose oxidation can be attributed to several factors. The initial step of glucose oxidation is the activation of the catalyst achieved by attachment of the molecule on electrode surface through coordination of the –OH functional group on catalytically active transition metal site (Cu). Such –OH attachment proceeds through local site oxidation of the active site. Previously, we have shown that –OH adsorption can be facilitated by controlling the ligand environment, typically by decreasing anion electronegativity,31 which reduces the required potential for catalyst activation, thereby increasing the efficiency.40 Moreover, Cu in copper selenide has mixed oxidation states. In the case of Cu+ and Se2− we can expect a certain degree of polarization due to charge imbalance. However, in the case of Cu2+ there is an increase in the covalency between Cu–Se bonds. This mixed oxidation states leads to an inductive effect and redistribution of the electron density at metal sites through d–d interactions, which is favorable for –OH groups to adsorb. Additionally, replacing oxides with less electronegative selenides also leads to increased covalency in the lattice and enhances the redox activity at the Cu site which consequently has an effect on the reversible electrochemical response. The low potential required for glucose oxidation is advantageous for making affordable and energy efficient non-enzymatic glucose sensors.
In conclusion, simple, binary copper selenide has been identified as a highly efficient, non-enzymatic, electrochemical glucose biosensor with a low limit of detection and high sensitivity. CuSe was synthesized directly on the electrodes by electrodeposition producing a porous morphology comprising flake-like nanostructures as well as solvothermal method. The electrocatalytic activity for glucose oxidation was studied under alkaline conditions. The electrodeposited CuSe exhibited superior efficiency for glucose oxidation with a sensitivity of 19.419 mA mM−1 cm−2 and a low detection limit of 0.196 μM, with a wide linear range of 100 nM–40 μM, a fast response time of less than 2 s, long term stability and excellent selectivity. These attributes ensure that this system will be able to reliably detect very small fluctuation in the glucose level in other bodily fluids such as urine, sweat, tears, and tissue fluids, which have a very low concentration of glucose. Additionally, the glucose oxidation at CuSe-modified electrodes occurs at a very low working potential of +0.15 V vs. Ag|AgCl, which increases energy efficiency of the system. These results reveal great potential of electrodeposited CuSe as a high-efficiency glucose sensor with practical applicability.
Conflicts of interest
The authors declared they have no conflict of interest in this work.Acknowledgements
This work was partially supported by the NSF DMR-1710313.Notes and references
- K. M. Bullard, C. C. Cowie, S. E. Lessem, S. H. Saydah, A. Menke, L. S. Geiss, T. J. Orchard, D. B. Rolka and G. Imperatore, Morb. Mortal. Wkly. Rep., 2018, 67, 359–361 CrossRef.
- A. L. Galant, R. C. Kaufman and J. D. Wilson, Food Chem., 2015, 188, 149–160 CrossRef CAS.
- A. T. Kharroubi and H. M. Darwish, World J. Diabetes, 2015, 6, 850–867 CrossRef.
- A. Stokes and S. H. Preston, PLoS One, 2017, 12, e0170219 CrossRef.
- G. Rocchitta, A. Spanu, S. Babudieri, G. Latte, G. Madeddu, G. Galleri, S. Nuvoli, P. Bagella, M. I. Demartis, V. Fiore, R. Manetti and P. A. Serra, Sensors, 2016, 16, 780 CrossRef.
- R. Gaia, S. Angela, B. Sergio, L. Gavinella, M. Giordano, G. Grazia, N. Susanna, B. Paola, D. Maria Ilaria, F. Vito, M. Roberto and S. Pier Andrea, Sensors, 2016, 16, 780 CrossRef.
- R. Wilson and A. P. F. Turner, Biosens. Bioelectron., 1992, 7, 165–185 CrossRef CAS.
- E.-H. Yoo and S.-Y. Lee, Sensors, 2010, 10, 4558–4576 CrossRef.
- J. Wang, Chem. Rev., 2008, 108, 814–825 CrossRef CAS.
- A. Harper and M. R. Anderson, Sensors, 2010, 10, 8248–8274 CrossRef CAS.
- P. Si, Y. Huang, T. Wang and J. Ma, RSC Adv., 2013, 3, 3487–3502 RSC.
- Y. Zhang, Y. Liu, L. Su, Z. Zhang, D. Huo, C. Hou and Y. Lei, Sens. Actuators, B, 2014, 191, 86–93 CrossRef CAS.
- Y. Mu, D. Jia, Y. He, Y. Miao and H.-L. Wu, Biosens. Bioelectron., 2011, 26, 2948–2952 CrossRef CAS.
- A. A. Saei, J. E. N. Dolatabadi, P. Najafi-Marandi, A. Abhari and M. de la Guardia, TrAC, Trends Anal. Chem., 2013, 42, 216–227 CrossRef CAS.
- J. Luo, S. Jiang, H. Zhang, J. Jiang and X. Liu, Anal. Chim. Acta, 2012, 709, 47–53 CrossRef CAS.
- H.-X. Wu, W.-M. Cao, Y. Li, G. Liu, Y. Wen, H.-F. Yang and S.-P. Yang, Electrochim. Acta, 2010, 55, 3734–3740 CrossRef CAS.
- Z. Zhu, L. Garcia-Gancedo, A. J. Flewitt, H. Xie, F. Moussy and W. I. Milne, Sensors, 2012, 12, 5996–6022 CrossRef.
- Y. Zhang, Y. Wang, J. Jia and J. Wang, Sens. Actuators, B, 2012, 171–172, 580–587 CrossRef CAS.
- P. Vennila, D. J. Yoo, A. R. Kim and G. G. kumar, J. Alloys Compd., 2017, 703, 633–642 CrossRef CAS.
- T. Chen, D. Liu, W. Lu, K. Wang, G. Du, A. M. Asiri and X. Sun, Anal. Chem., 2016, 88, 7885–7889 CrossRef CAS.
- P. K. Kannan and C. S. Rout, Chem. – Eur. J., 2015, 21, 9355–9359 CrossRef CAS.
- X. Niu, M. Lan, H. Zhao and C. Chen, Anal. Chem., 2013, 85, 3561–3569 CrossRef CAS.
- K. Ramachandran, T. Raj kumar, K. J. Babu and G. Gnana kumar, Sci. Rep., 2016, 6, 36583 CrossRef CAS.
- J. Yang, X. Liang, L. Cui, H. Liu, J. Xie and W. Liu, Biosens. Bioelectron., 2016, 80, 171–174 CrossRef CAS.
- M. Ranjani, Y. Sathishkumar, Y. S. Lee, D. Jin Yoo, A. R. Kim and G. Gnana kumar, RSC Adv., 2015, 5, 57804–57814 RSC.
- P. V. Suneesh, V. Sara Vargis, T. Ramachandran, B. G. Nair and T. G. Satheesh Babu, Sens. Actuators, B, 2015, 215, 337–344 CrossRef CAS.
- A. T. Swesi, J. Masud, W. P. R. Liyanage, S. Umapathi, E. Bohannan, J. Medvedeva and M. Nath, Sci. Rep., 2017, 7, 2401 CrossRef.
- M. Pumera, Z. Sofer and A. Ambrosi, J. Mater. Chem. A, 2014, 2, 8981–8987 RSC.
- M.-R. Gao, J. Jiang and S.-H. Yu, Small, 2012, 8, 13–27 CrossRef CAS.
- S. Umapathi, J. Masud, A. T. Swesi and M. Nath, Adv. Sustainable Syst., 2017, 1, 1700086 CrossRef.
- U. D. Silva, J. Masud, N. Zhang, Y. Hong, W. P. R. Liyanage, M. A. Zaeem and M. Nath, J. Mater. Chem. A, 2018, 6, 7608–7622 RSC.
- J. Masud, P.-C. Ioannou, N. Levesanos, P. Kyritsis and M. Nath, ChemSusChem, 2016, 9, 3128–3132 CrossRef CAS.
- J. Masud, W. P. R. Liyanage, X. Cao, A. Saxena and M. Nath, ACS Appl. Energy Mater., 2018, 1, 4075–4083 CrossRef CAS.
- Y. Wang, S. Liu, Y. Lai, Y. Zhu, R. Guo, Y. Xia, W. Huang and Z. Li, Sens. Actuators, B, 2018, 262, 801–809 CrossRef CAS.
- W. Zhu, J. Wang, W. Zhang, N. Hu, J. Wang, L. Huang, R. Wang, Y. Suo and J. Wang, J. Mater. Chem. B, 2018, 6, 718–724 RSC.
- M. B. Gawande, A. Goswami, F.-X. Felpin, T. Asefa, X. Huang, R. Silva, X. Zou, R. Zboril and R. S. Varma, Chem. Rev., 2016, 116, 3722–3811 CrossRef CAS.
- X. Liu, X. Duan, P. Peng and W. Zheng, Nanoscale, 2011, 3, 5090–5095 RSC.
- K. G. Schick, V. G. Magearu and C. O. Huber, Clin. Chem., 1978, 24, 448–450 CrossRef CAS.
- X. Cao, E. Johnson and M. Nath, ACS Sustainable Chem. Eng., 2019, 7, 9588–9600 CrossRef CAS.
- X. Cao, Y. Hong, N. Zhang, Q. Chen, J. Masud, M. A. Zaeem and M. Nath, ACS Catal., 2018, 8, 8273–8289 CrossRef CAS.
- X. Liu, W. Yang, L. Chen and J. Jia, Electrochim. Acta, 2017, 235, 519–526 CrossRef CAS.
- L. Zhang, C. Ye, X. Li, Y. Ding, H. Liang, G. Zhao and Y. Wang, Nano-Micro Lett., 2017, 10, 28 CrossRef.
- X. Zhang, J. Luo, P. Tang, J. R. Morante, J. Arbiol, C. Xu, Q. Li and J. Fransaer, Sens. Actuators, B, 2018, 254, 272–281 CrossRef CAS.
- C. Zhao, X. Wu, X. Zhang, P. Li and X. Qian, J. Electroanal. Chem., 2017, 785, 172–179 CrossRef CAS.
- F. Huang, Y. Zhong, J. Chen, S. Li, Y. Li, F. Wang and S. Feng, Anal. Methods, 2013, 5, 3050–3055 RSC.
- X. Luo, M. Huang, L. Bie, D. He, Y. Zhang and P. Jiang, RSC Adv., 2017, 7, 23093–23101 RSC.
- C. Lee, S. H. Lee, M. Cho and Y. Lee, Microchim. Acta, 2016, 183, 3285–3292 CrossRef CAS.
- K. Ghanbari and Z. Babaei, Anal. Biochem., 2016, 498, 37–46 CrossRef CAS.
- R. Ahmad, N. Tripathy, M.-S. Ahn, K. S. Bhat, T. Mahmoudi, Y. Wang, J.-Y. Yoo, D.-W. Kwon, H.-Y. Yang and Y.-B. Hahn, Sci. Rep., 2017, 7, 5715 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: SEM and HRTEM images, i–V plots and i–t plot. See DOI: 10.1039/d0ma00890g |
| This journal is © The Royal Society of Chemistry 2021 |