
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the redox decomposition of ethylene carbonate–propylene carbonate in Li-ion batteries†
Jiaxiang
Zhang
a,
Junwen
Yang
a,
Limin
Yang
 b,
Hai
Lu
a,
Huan
Liu
a and
Bin
Zheng
*a
b,
Hai
Lu
a,
Huan
Liu
a and
Bin
Zheng
*a
aSchool of Materials Science and Engineering, Xi’an University of Science and Technology, Xi’an 710054, P. R. China. E-mail: zhengbin@xust.edu.cn
bCollege of Material, Chemistry and Chemical Engineering, Hangzhou Normal University, Hangzhou 310018, P. R. China
First published on 2nd February 2021
Abstract
A fundamental understanding of electrolytes is critical for designing lithium-ion batteries with excellent performance and high safety. The traditional solvents in electrolytes of lithium-ion batteries are mainly ethylene carbonate and propylene carbonate. Despite their similar structures and chemical properties, ethylene carbonate-based electrolytes have been reported to enable the reversible reaction of graphite anodes, whereas propylene carbonate-based electrolytes cause the exfoliation of graphite. Herein, we have investigated the oxidation stability and the reductive decomposition of ethylene carbonate and propylene carbonate from electron-level quantum calculations. While small differences in their oxidation stability were presented, we found disparity in their reductive decomposition. The reductive product of lithium alkyl carbonate exhibits different geometrical and molecular orbitals, which was considered to influence the quality of the ethylene/propylene carbonate solvent-based solid–electrolyte interphase (SEI). This study presents the disparity of redox decomposition of ethylene carbonate–propylene carbonate in Li-ion batteries, which is expected to guide the design of new electrolyte systems, thereby enhancing the performance of Li-ion batteries.
Introduction
Rechargeable batteries are among the most promising candidates to obtain high practical energy density for portable electronics, electric vehicles, and energy-storage devices, in the current, sustainable energy-based economy.2 Electrolytes, as their main component, play an essential role in the critical properties of lithium-ion batteries (LIBs) and lithium-metal batteries (LMBs), such as safety, cycle-life, and power density.3,4 Designing application-oriented electrolytes has become an efficient way to enhance the performance and the safety of LIBs and LMBs.5–7The oxidation stability and reductive inertness are two essential criteria to design electrolytes for improving the efficiency of a battery system. Electrolytes having high oxidation potentials, such as sulfones, ionic liquids, nitriles, and fluorinated compounds, were mainly designed to fit high-voltage electrode materials.8–12 The as-desired high energy density and excellent safety can be achieved by using high voltages and non-flammable electrolytes. On the other hand, due to its low thermodynamic potential (0–0.20 V vs. Li), Li/Li+ is reactive toward most of the electrolytes, which induces the sacrificial decomposition of the electrolytes to form a solid–electrolyte interphase (SEI) or cathode–electrolyte interphase (CEI). The inertness of electrolytes could only be achieved through a robust and stable SEI or CEI film, which prevents the decomposition of the electrolyte and guarantees is insulating to electrons tunneling and conductive for Li+-conductive.13–15 Several efforts have been made on designing electrolytes with additives to form an SEI with enhanced properties, enabling long-term cycling.13,16 Although many new electrolytes were experimentally developed for LIBs and LMBs, the design of better electrolyte and SEI systems is still lack of fundamental guiding. Even for the most commonly used carbonate-based solvents, such as ethylene- or propylene carbonate (EC and PC), and their derivatives, their disparity in forming an SEI on the graphitic anode of LIBs from an electron-level is still not elucidated.4,17,18 Therefore, for developing new electrolytes, more investigations are needed focusing on the mechanisms, which can reduce the consumption of resources originating from a semi-empirical trial-and-error method. Although Borodin et al.19–21 investigated the electrochemical properties of many electrolytes/solvents, direct and comprehensive studies on EC and PC are rare.
In this work, we focused on the insights of the intrinsic disparity of EC and PC in LIBs, using an approach based on electron-level calculations. Fluoroethylene carbonate (FEC) with better oxidative stability and SEI formation capability was considered to highlight the difference between EC and PC (Fig. 1). The three carbonates have similar physical properties (dipole moments and dielectric constants in Table S1, ESI†). The electrochemical properties of EC and PC with a minimal structural difference caused by a single methyl group were revealed. Moreover, we focused not only on single EC and PC molecules but also on their reductive products (lithium alkyl carbonate complexes) as the main SEI components. The current work is expected to guide the design of enhanced electrolyte and SEI systems in LIBs.
 | ||
| Fig. 1 Chemical structures of EC, PC and FEC. | ||
Computational method
Density functional theory (DFT) calculations were performed using the Gaussian 09 program package.22 The M05-2X density functional23 was used to evaluate the oxidation potential due to its advantages in transferability and describing the localized holes in solvent oxidation.24 The B3LYP/6-31G*25–29 level of theory was chosen to investigate the pathway of the reduction reactions. The SMD implicit solvation model30 using water and acetone parameters was also employed to calculate the oxidation potentials. The calculation details can be found in the ESI.†Results and discussion
The oxidation stability of electrolytes is the key feature of Li-ion batteries (LIBs). The cell voltage of common LIBs is ≈3.8 V.31 The computed oxidation potentials of EC, PC, and FEC were above 6.5 V vs. Li/Li+ (Table 1), which is higher than the working potential (3.8 V) of LIBs, also showing excellent oxidation stability. Specifically, the oxidation potentials of EC and FEC were slightly higher than that of PC. The experimental values (EC: 5.5–6.7 V; PC: 6.0–6.8 V),32–35 being sensitive to the experimental conditions, may hide the small difference between the oxidation potentials of EC and PC.| ε = 78.5 (water) | ε = 20.7 (acetone) | |||
|---|---|---|---|---|
| E ad (V vs. Li/Li+) | E vert − Ead (eV) | E ad (V vs. Li+/Li) | E vert − Ead (eV) | |
| EC | 6.78 | 0.27 | 6.84 | 0.36 |
| PC | 6.55 | 0.50 | 6.63 | 0.51 |
| FEC | 7.21 | 0.33 | 7.45 | 0.36 |
Besides the thermodynamic stability of electrolytes, the kinetic stability was also evaluated, based on Marcus electron transfer theory, using the difference between the adiabatic and vertical oxidation potentials (Evert − Ead) (see details in the ESI†).20,21 A large (Evert − Ead) value indicates a high energy barrier and a low oxidation reaction rate and is beneficial for the oxidation stability of the electrolyte.19–21 It was also found from the kinetics study that PC was more stable than EC and FEC in the oxidation reactions (Evert − Ead in Table 1), although PC has a lower oxidation potential. Fluorine could enhance the oxidation potential (EC: 6.78 or 6.84 V; FEC: 7.21 or 7.45 V), but it had a weak effect on the dynamics of the oxidation process. The possible trade-off relationship between the oxidation potential and (Evert − Ead) was observed, also described by Borodin et al.,19,21 which points out that an attractive high-voltage electrolyte has both high oxidation potential and increased (Evert − Ead) value, which is needed to break out the limitation of a traditional materials system.
Another criterion for choosing an appropriate electrolyte for LIBs is the reduction stability. The reduction potential of the solvent molecules mainly depends on the coordinated position of the Li+ cations. It was found that the reduction potentials of EC, PC, and FEC are <1 V, except that in the formation of LiF in FEC (2.25 V).21 Considering the relatively high working potential (3.8 V) of standard Li-ion batteries, it is easy to trigger the single electron reduction of EC, PC, and FEC. However, reduction of electrolytes is only the first step of the electrolyte decomposition to form lithium alkyl carbonates as solid–electrolyte SEIs in LIBs.
The decomposition of EC, PC and FEC corresponded to a regular ring-opening reaction, which usually requires a high driving force (>60 kcal mol−1, Fig. S2 and S3, ESI†). We also found that the coordinated Li+ cations could assist the opening of the five-member ring. The energy barriers of ring-opening in Li+EC (TS-E1, Fig. 2), Li+PC (TS-P1, Fig. 3) and Li+FEC (TS-F1 and TS-F2, Fig. 4) were found to be 5.9, 7.7 and (9.5 and 6.5) kcal mol−1, respectively. A bit higher energy (1.8 kcal mol−1) was required for the Li+PC complex, in comparison with the Li+EC complex. This difference can be attributed to the only methyl group in PC molecules. Also, it was more challenging to break the C–O bond, attached to the terminated F (9.5 kcal mol−1) compared to that which was far from the F atom (6.5 kcal mol−1) in FEC (Fig. 4). This also indicates that the terminated group in PC and FEC could modify the rigidity of the conformation and then the energy barrier of the ring-opening reactions.
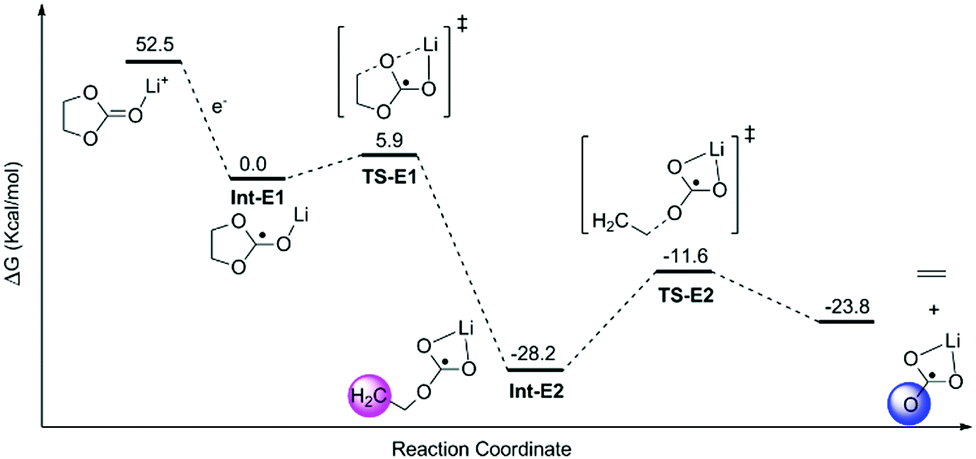 | ||
| Fig. 2 The calculated profile of free energy (ΔG) of EC decomposition assisted by Li ions. The hydration energy of the solvated electron in water was −1.63 eV.1 The shaded areas denote the radical carbon and oxygen. | ||
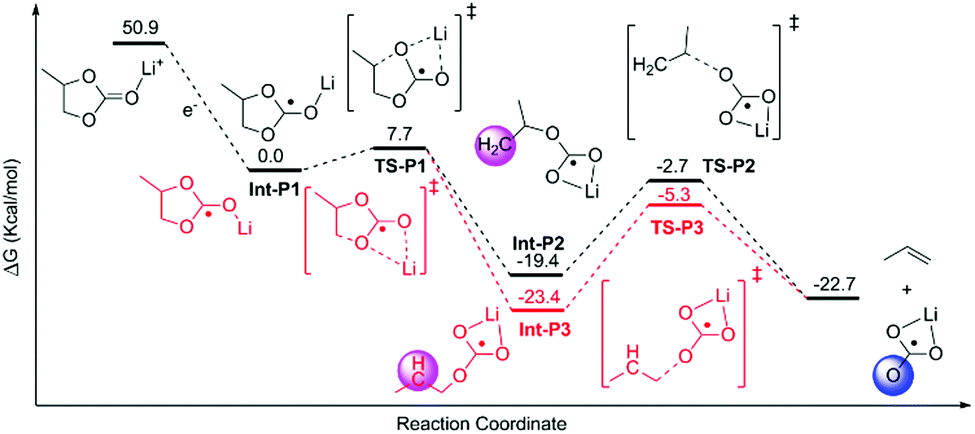 | ||
| Fig. 3 Calculated possible pathways of PC decomposition assisted by Li ions. The hydration energy of the solvated electron in water was −1.63 eV.1 The shaded areas denote the radical carbon and oxygen. | ||
 | ||
| Fig. 4 Calculated possible pathways of FEC decomposition assisted by Li ions. The hydration energy of the solvated electron in water was −1.63 eV.1 The shaded areas denote the radical carbon and oxygen. | ||
The resulting intermediates (Int-E2, Int-P2, Int-P3, Int-F2, and Int-F3) had unsaturated C atoms (pink area in Fig. 2–4). There were two possible ways to saturate these active sites: (i) the adjacent C atoms provided the empty orbitals to form the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond. This pathway required C–O bond breakage, shown in TS-E2, TS-P2, TS-P3, TS-F3, and TS-F4. The energy barrier for cutting the C–O bond is ∼17 kcal mol−1 in these intermediates. The products of LiCO3− anions and small molecules of ethene, propene, and fluorinated propylene-based agents were obtained; (ii) the active LiCO3− anion product of the step mentioned above could directly attract the unsaturated C atoms of Int-E2, Int-P2, Int-P3, Int-F2, and Int-F3 to form lithium alkyl carbonates (Fig. 5).
C double bond. This pathway required C–O bond breakage, shown in TS-E2, TS-P2, TS-P3, TS-F3, and TS-F4. The energy barrier for cutting the C–O bond is ∼17 kcal mol−1 in these intermediates. The products of LiCO3− anions and small molecules of ethene, propene, and fluorinated propylene-based agents were obtained; (ii) the active LiCO3− anion product of the step mentioned above could directly attract the unsaturated C atoms of Int-E2, Int-P2, Int-P3, Int-F2, and Int-F3 to form lithium alkyl carbonates (Fig. 5).
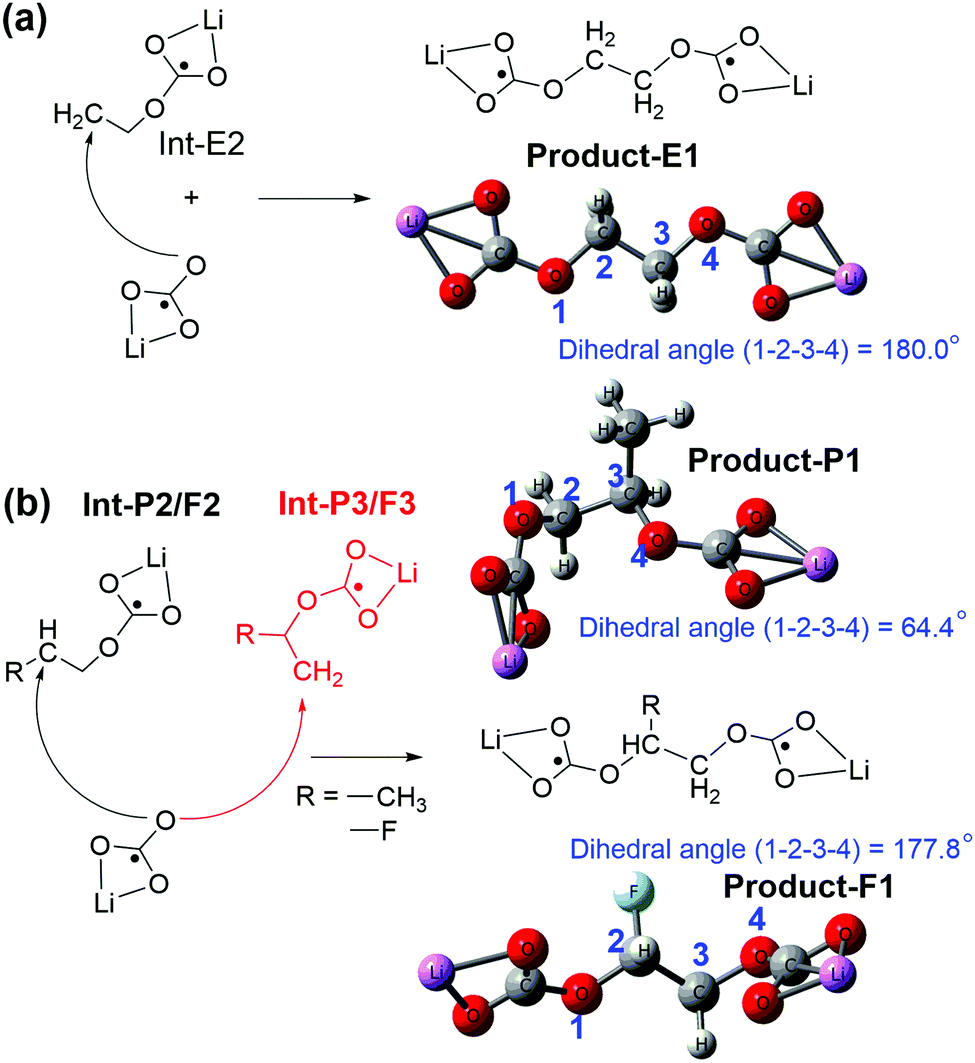 | ||
| Fig. 5 Lithium alkylcarbonates originating from the reactions between lithium carbonate (LiCO3−) and the intermediate state of (a) EC and (b) PC (or FEC) decomposition. | ||
The small structural difference caused by a single methyl group between EC and PC could cause different lithium alkyl carbonates (Fig. 5 and 6). The EC based lithium alkyl carbonate showed centrosymmetric conformation (dihedral angle 1–2–3–4 = 180° of Product-E1 in Fig. 5a). Once one methyl substituent in PC appears, the main chain was distorted to fit the asymmetric conformation (dihedral angle 1–2–3–4 = 64.4° of Product-P1 in Fig. 5b). As a comparison, the strong electron-withdrawing group substituent in FEC showed a similar effect to the EC on the distorted conformation for lithium alkyl carbonates (dihedral angle 1–2–3–4 = 177.8° of Product-F1). This indicates that the geometry of the side termination can be the reason for the symmetry loss of lithium alkyl carbonate molecules (Fig. 5b).
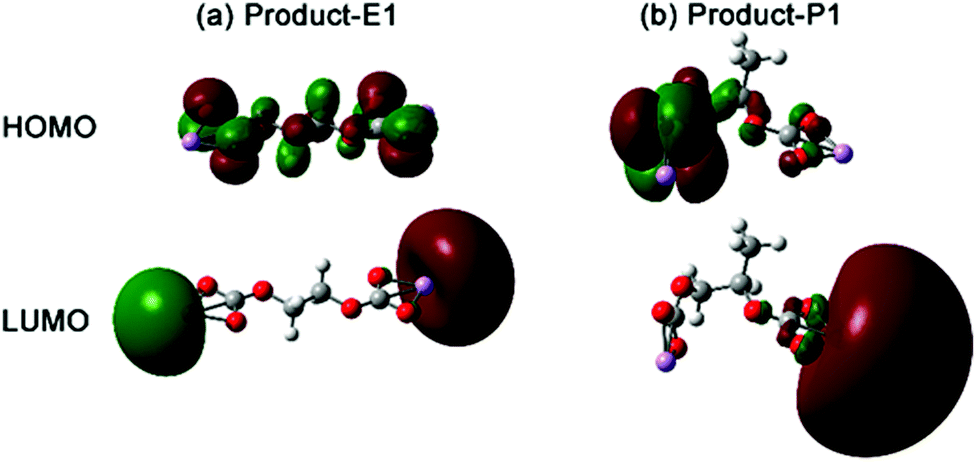 | ||
| Fig. 6 3D representation of the HOMO and LUMO orbitals of the lithium alkyl carbonate complexes. The red and green colors indicate the positive and negative regions. | ||
Besides the difference of the molecule conformation, the EC- (Product-E1) and PC-based (Product-P1) lithium alkyl carbonates showed distinct electronic transition behaviour. The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) can be responsible for the electronic transitions (Fig. 6). The HOMO of Product-E1 was delocalized on the whole organic part of the complex, particularly on the O atoms coordinated with Li. On the other hand, its LUMO was localized on the terminated Li atom sites. However, the molecular orbitals of Product-P1 presented the characteristics of localization and asymmetry. The HOMO and LUMO of Product-P1 were localized on the two sides of lithium alkyl carbonate, respectively, caused by one methyl substituent, present in the structure of PC. The molecular orbitals of Product-F1 also presented the same characteristics as the Product-P1 (Fig. S4, ESI†).
Molecular orbitals affected the chemical reactivity of the lithium alkyl carbonate complex. The HOMO had the priority to provide electrons, while the LUMO accepted the electrons.36 The localized HOMO and LUMO pairs of Product-P1 (Fig. 6) and Product-F1 (Fig. S4, ESI†) provided two side sites for donating and withdrawing electrons. Thus, it was preferential to form a more distorted conformation for a single Product-P1 (ring formation in Fig. S5, ESI†). However, Product-E1 had a delocalized HOMO, and then the electron-donation sites were mainly distributed on the four O atoms of the two sides of the complex. The Li atoms, as the main LUMO distribution, played the role of withdrawing electrons. Thus, it was preferential to form the dimer Product-E1via the Li–O interactions between the two complexes (Fig. 7). Furthermore, this linking mode increased the possibility of extending the dimer complex by coordinating more Product-E1 complexes and then forming a two-dimensional layered structure (Fig. 7 and Fig. S6, ESI†).
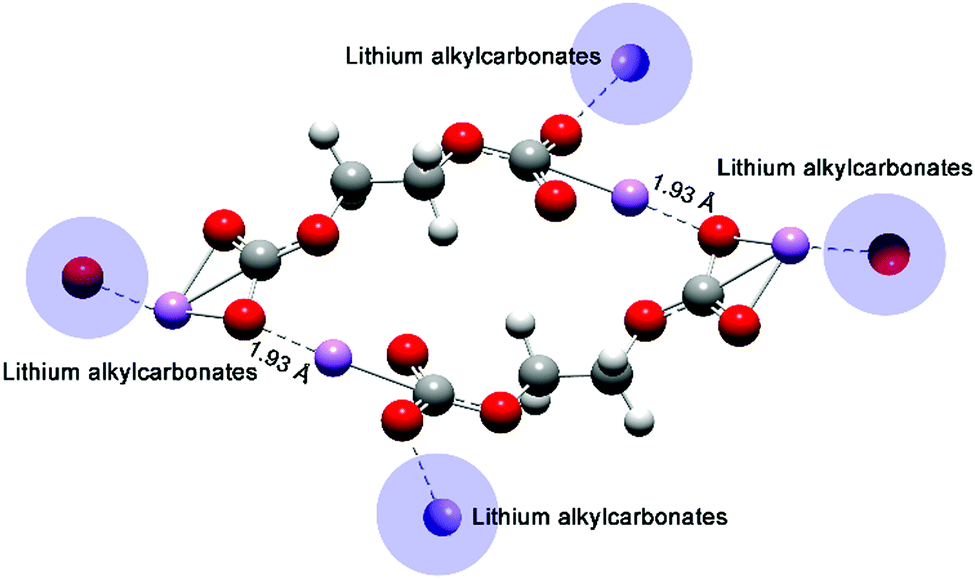 | ||
| Fig. 7 Optimized structure of a dimer Product-E1 complex. The shaded areas denote the assembled complexes. | ||
Experimental results1,18 already showed that EC-based electrolytes could form protective interphases on the anodes of LIBs, while the reversible Li+ intercalation/deintercalation was enabled. The Product-E1 complex had the potential to form an ordered and continuous layered structure, which was a useful component of the protective SEI. The space and the terminated O atoms between adjacent Product-E1 complexes benefit from capturing and transporting Li+ interactions. Also, the possible Product-E1 layer's flexibility could tolerate the volume change due to the intercalation/deintercalation of Li+, while one group substituent in PC would result in the symmetry loss of the lithium alkyl carbonate complexes. The small difference of one methyl group substituent in the PC was further expanded when many complexes were assembled, which then contributed to the two interphase extremities between EC and PC-based electrolytes in LIBs.
The smaller difference (trans–cis) between trans-butylene carbonate (t-BC) and cis-butylene carbonate (c-BC) than that of EC–PC exhibited a similar relationship to the EC–PC mystery.37,38 We found only a small difference (0.5 kcal mol−1) between the energy barrier of the decomposition of single t-BC and c-BC molecules (Fig. S8, ESI†). However, the dimerization of t-BC shows a relatively lower energy barrier than that of c-BC (Fig. 8), which indicates that the former is preferable. The reported experimental results show that the dimerized products are primary components of a robust SEI film.38,39 The current study indicates that a higher number of dimerized t-BC molecules can be obtained, which may contribute to the reversible reaction of graphite anodes (as EC), whereas fewer dimerized c-BC molecules cause the exfoliation of graphite (as PC).
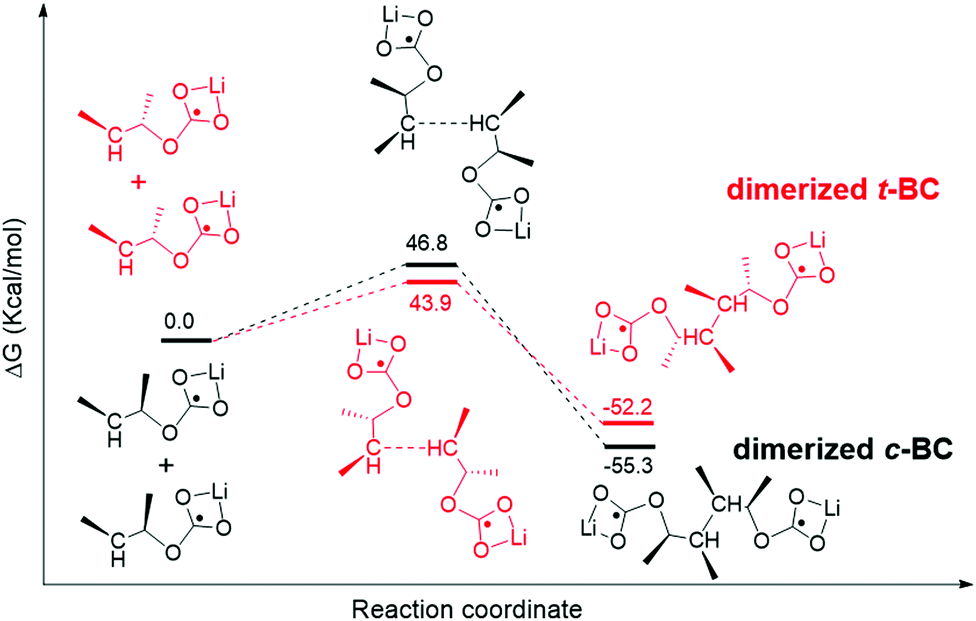 | ||
| Fig. 8 Calculated possible pathways of dimerized trans-2,3-butylene carbonate (t-BC) and cis-2,3-butylene carbonate (c-BC). | ||
The SEI was mainly composed of an inner layer (closer to the solid electrode) and an outer layer (closer to the liquid electrolyte), depending on the reduction state.40 Lithium alkyl carbonate complexes were the nucleus of the outer less-reduced layer of the SEI. As the component of the inner layer, fully reduced inorganic products were possibly missing in EC and PC. However, the LiCO3− anion tended to attract Li+FEC to form the fully reduced inorganic product (LiF) as the nucleus of the inner layer of the SEI (Fig. S7, ESI†). It was also helpful to understand the phenomenon of fluorination, which can significantly improve the formation capability of the SEI, while FEC can form a more robust and stable SEI film than EC.1
Conclusion
In summary, we systematically examined the oxidation stability, the reduction reaction mechanisms, and the reduced complexes of EC and PC in LIBs, using the DFT calculation method. The oxidation of EC was more easily driven than that of PC considering the kinetic stability, based on the fact that the difference of their thermodynamic stability is relatively small. During the reduction reactions, both EC and PC generated active, less-reduced forms of organic intermediates and LiCO3− anions, which could form symmetrical and distorted lithium alkyl carbonates. Analysis of molecular orbitals indicated that the EC-based complex had a delocalized electron distribution and a preference to form multi-complex assemblies, which are the basis of a robust SEI film. However, the PC-based complex with a localized electron distribution tended to form a ring by itself, taking away the opportunity of interacting with another complex. Fluorination could enhance the oxidation potential and improve the SEI formation capability by producing LiF. This electron-level understanding of the intrinsic disparity of EC–PC could help in the design of better electrolytes and interphases for application oriented battery setups.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of China under grant 21503165, the Natural Science Basic Research Plan in Shaanxi Province of China under grant 2020JM-520, and Shaanxi Province 100 plan.References
-
Y. Tabata, CRC Handbook of Radiation Chemistry, CRC Press, 1991 Search PubMed
.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef
- J. Hou, L. Lu, L. Wang, A. Ohma, D. Ren, X. Feng, Y. Li, Y. Li, I. Ootani, X. Han, W. Ren, X. He, Y. Nitta and M. Ouyang, Nat. Commun., 2020, 11, 5100 CrossRef CAS
- Y. Qian, S. Hu, X. Zou, Z. Deng, Y. Xu, Z. Cao, Y. Kang, Y. Deng, Q. Shi, K. Xu and Y. Deng, Energy Storage Mater., 2019, 20, 208–215 CrossRef
- H. Lu, Y. Zhu, B. Zheng, H. Du, X. Zheng, C. Liu, Y. Yuan, J. Fang and K. Zhang, New J. Chem., 2020, 44, 361–368 RSC
- Z. Yu, H. S. Wang, X. Kong, W. Huang, Y. C. Tsao, D. G. Mackanic, K. C. Wang, X. C. Wang, W. X. Huang, S. Choudhury, Y. Zheng, C. V. Amanchukwu, S. T. Hung, Y. T. Ma, E. G. Lomeli, J. Qin, Y. Cui and Z. N. Bao, Nat. Energy, 2020, 5, 526–533 CrossRef CAS
- J. Zhang, J. Yang, Z. Liu and B. Zheng, ACS Omega, 2021 DOI:10.1021/acsomega.0c06067
- C. C. Su, M. He, P. C. Redfern, L. A. Curtiss, I. A. Shkrob and Z. C. Zhang, Energy Environ. Sci., 2017, 10, 900–904 RSC
- Z. L. Hu, F. Xian, Z. Y. Guo, C. L. Lu, X. F. Du, X. Y. Cheng, S. Zhang, S. M. Dong, G. L. Cui and L. Q. Chen, Chem. Mater., 2020, 32, 3405–3413 CrossRef CAS
- Q. F. Zheng, Y. Yamada, R. Shang, S. Ko, Y. Y. Lee, K. Kim, E. Nakamura and A. Yamada, Nat. Energy, 2020, 5, 291–298 CrossRef CAS
- S. Tan, Y. J. Ji, Z. R. Zhang and Y. Yang, ChemPhysChem, 2014, 15, 1956–1969 CrossRef CAS
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS
- Y. F. Zhou, M. Su, X. F. Yu, Y. Y. Zhang, J. G. Wang, X. D. Ren, R. G. Cao, W. Xu, D. R. Baer, Y. G. Du, O. Borodin, Y. T. Wang, X. L. Wang, K. Xu, Z. J. Xu, C. M. Wang and Z. H. Zhu, Nat. Nanotechnol., 2020, 15, 224–230 CrossRef CAS
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS
- M. D. Tikekar, S. Choudhury, Z. Tu and L. A. Archer, Nat. Energy, 2016, 1, 16114 CrossRef CAS
- G. M. Hobold, A. Khurram and B. M. Gallant, Chem. Mater., 2020, 32, 2341–2352 CrossRef CAS
- L. Xing, X. Zheng, M. Schroeder, J. Alvarado, A. von Wald Cresce, K. Xu, Q. Li and W. Li, Acc. Chem. Res., 2018, 51, 282–289 CrossRef CAS
- K. Xu, J. Electrochem. Soc., 2009, 156, A751 CrossRef CAS
- O. Borodin, Curr. Opin. Electrochem., 2019, 13, 86–93 CrossRef CAS
- O. Borodin, W. Behl and T. R. Jow, J. Phys. Chem. C, 2013, 117, 8661–8682 CrossRef CAS
- O. Borodin, X. Ren, J. Vatamanu, A. von Wald Cresce, J. Knap and K. Xu, Acc. Chem. Res., 2017, 50, 2886–2894 CrossRef CAS
-
M. J. T. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision A.01, Gaussian Inc., Wallingford, CT, 2009 Search PubMed
- Y. Zhao, N. E. Schultz and D. G. Truhlar, J. Chem. Theory Comput., 2006, 2, 364–382 CrossRef
- O. Borodin, M. Olguin, C. E. Spear, K. W. Leiter and J. Knap, Nanotechnology, 2015, 26, 354003 CrossRef
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS
- K. Xu, S. P. Ding and T. R. Jow, J. Electrochem. Soc., 1999, 146, 4172–4178 CrossRef CAS
- M. Ue, M. Takeda, M. Takehara and S. Mori, J. Electrochem. Soc., 1997, 144, 2684–2688 CrossRef CAS
- K. Abe, Y. Ushigoe, H. Yoshitake and M. Yoshio, J. Power Sources, 2006, 153, 328–335 CrossRef CAS
- I. Azcarate, W. Yin, C. Méthivier, F. Ribot, C. Laberty-Robert and A. Grimaud, J. Electrochem. Soc., 2020, 167, 080530 CrossRef CAS
- V. Karunakaran and V. Balachandran, Spectrochim. Acta, Part A, 2012, 98, 229–239 CrossRef CAS
- K. Miyazaki, N. Takenaka, T. Fujie, E. Watanabe, Y. Yamada, A. Yamada and M. Nagaoka, ACS Appl. Mater. Interfaces, 2019, 11, 15623–15629 CrossRef CAS
- G.-C. Chung, H.-J. Kim, S.-I. Yu, S.-H. Jun, J.-W. Choi and M.-H. Kim, J. Electrochem. Soc., 2000, 147, 4391 CrossRef CAS
- G.-C. Chung, H.-J. Kim, S.-H. Jun and M.-H. Kim, Electrochem. Commun., 1999, 1, 493–496 CrossRef CAS
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703–A1719 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00847h |
| This journal is © The Royal Society of Chemistry 2021 |