
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Review of the current state of protein aggregation inhibition from a materials chemistry perspective: special focus on polymeric materials
Robin
Rajan
 *a,
Sana
Ahmed
a,
Neha
Sharma
b,
Nishant
Kumar
a,
Alisha
Debas
a and
Kazuaki
Matsumura
*a
*a,
Sana
Ahmed
a,
Neha
Sharma
b,
Nishant
Kumar
a,
Alisha
Debas
a and
Kazuaki
Matsumura
*a
aSchool of Materials Science, Japan Advanced Institute of Science and Technology, 1-1 Asahidai, Nomi, Ishikawa, 923-1292, Japan. E-mail: robin@jaist.ac.jp; mkazuaki@jaist.ac.jp
bResearch Center for Bioscience and Nanoscience, Japan Agency for Marine-Earth Science and Technology, 2-15 Natsushima-cho, Yokosuka 237-0061, Japan
First published on 15th January 2021
Abstract
Protein instability caused by exposure to external additives or severe stress may result in different diseases. Of these diseases, many are triggered by protein misfolding and denaturation, leading to neurodegenerative disorders. Additionally, many other diseases are treated by biopharmaceutical approaches using proteins as drugs, which is again prone to denaturation. Numerous reagents and methods have been developed to understand protein instability and protection. However, the development of polymer-based protein protection agents as well as small molecules and peptides for combatting protein instability by inhibiting aggregation and facilitating protein refolding only gained attention in the last decade. In this review, we discuss protein aggregation inhibition and the role of polymers in protein protection. Further, we outline the significance of chemical additives in facilitating protein refolding and their importance in the use of recombinant proteins for treating neurodegenerative disorders.
 Robin Rajan | Robin Rajan completed his PhD from the Japan Advanced Institute of Science and Technology (JAIST) in 2016 in Materials Science. He carried out his first postdoctoral stint at JAIST before moving to Nanyang Technological University, Singapore for another postdoctoral tenure. Since 2018, he has been working as an Assistant Professor at JAIST. His research interests include polymer chemistry, organic synthesis, and biomaterials, and is focused on the effect of polymers on cells, proteins, and other biologically relevant molecules. |
 Kazuaki Matsumura | Kazuaki Matsumura is a Professor at the Japan Advanced Institute of Science and Technology (JAIST). He received his PhD from Kyoto University in the field of Polymer Chemistry in 2004. He moved to JAIST in 2011 to set up a biomaterials laboratory and continues to investigate polymeric biomaterials and biomedical engineering applications of polymers. His research interests include the molecular design of protective materials for biomolecules and cells, drug delivery system carriers, and tissue engineering materials. |
1. Introduction
Protein aggregation occurs when misfolded/unfolded proteins physically aggregate due to a series of sequential and parallel events, which may be accompanied by changes in the native state of the protein, structural perturbations, and protein–protein interactions. Protein aggregation, which causes various diseases including neurodegenerative diseases, is a major challenge to the development of protein-based biopharmaceuticals.1–5Protein drugs are an indispensable class of biopharmaceutical products used to treat severe health conditions that cannot be treated using conventional drugs. As promising biotherapeutic agents, proteins offer advantages such as selectivity, specificity, and potency. A common example of protein drugs is insulin, which is used for the treatment of type 1 and type 2 diabetes. According to a recent report by Syngene Research, the global market for protein-based therapeutics is expected to reach almost 303 billion USD by 2026.6 However, protein instability is a challenge for biopharmaceutical formulations, and overcoming it requires in-depth understanding of the underlying physical process and the methods that can be used to protect proteins.
Neurodegenerative disorders, such as Parkinson's disease, Alzheimer's disease, amyotrophic lateral sclerosis, and Huntington's disease are caused by protein aggregation.1,2 Globally, ∼10 million people suffer from Parkinson's disease, and in the USA, ∼5.4 million individuals have Alzheimer's disease.7 These disorders do not have definite cures; however, multiple strategies for disease management have been reported, including administering cholinesterase inhibitors for cognitive disorders,8 drugs, and suppressing the effects of genetic mutations.9–13 For combating neurodegenerative disorders, strategies to remove amyloid fibrils or refold misfolded proteins from the brain may be effective. A brief schematic illustration of protein aggregation and inhibition strategies is shown in Fig. 1.
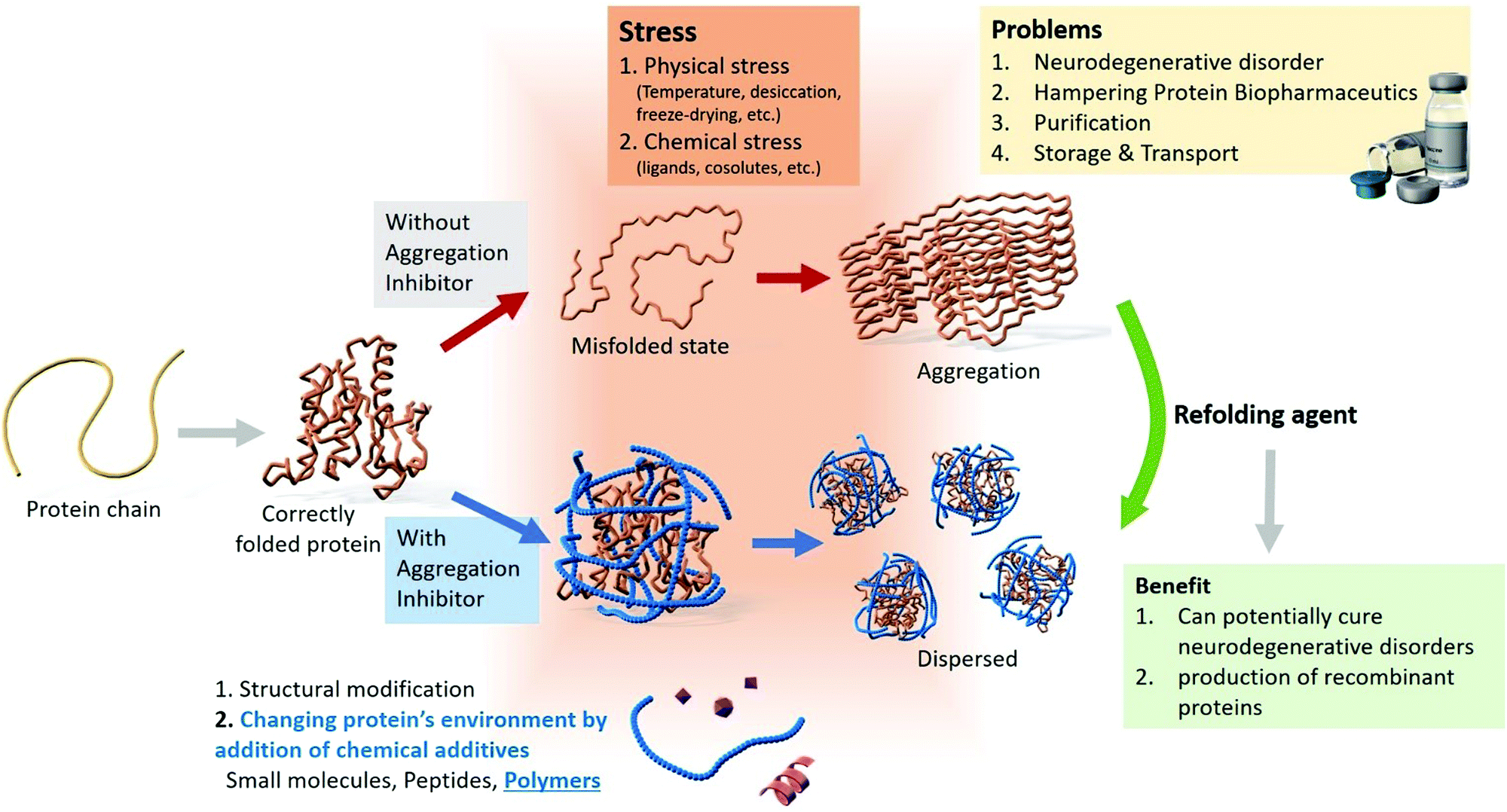 | ||
| Fig. 1 Overview of the modes of protein aggregation, problems associated with aggregation, and the various inhibition strategies. | ||
Several processes and compounds that suppress protein aggregation inhibition with relative success have been developed. Several reviews on protein aggregation, it's mechanistic aspects, and strategies for suppressing aggregation have been previously published,4,14–20 focusing on small molecule inhibitors of protein aggregation. For example, W. Wang has reviewed protein aggregation phenomenon with emphasis on small molecule compounds used for the inhibition of this process.4 Similarly, Estrada and Soto17 and Dhouafli et al.16 have described protein aggregation inhibition by short peptides and phenolic compounds, respectively. Polymers have recently been recognized as efficient alternatives to small molecules for the inhibition of protein aggregation, as these have great potential to combat aggregation-induced issues and are effective in inhibiting protein denaturation and facilitating refolding. An exhaustive review detailing the problems, inducements, consequences, and available inhibitors of protein aggregation, with focus on polymers is required. Moreover, a review focusing on the refolding of denatured/misfolded proteins using chemical additives is unavailable in the literature.
In this review, we summarize recent advances in protein studies and discuss various mechanistic aspects and strategies to overcome protein aggregation. Further, we emphasize the development of polymeric compounds for stabilizing proteins both in vitro and in vivo and discuss the removal of aggregated species (refolding) using chemical additives. This review aims at providing an understanding of the mechanisms underlying protein aggregation and ways to overcome it, with particular focus on polymer-based inhibitors of protein aggregation. These inhibitors have the potential to be game-changers in the field of protein-based biopharmaceuticals owing to their greater flexibility and tunability compared to small-molecule analogues.
2. Protein aggregation
Under normal conditions, newly synthesized proteins fold into distinct three-dimensional structures and occasionally assemble into multimeric protein complexes. Self-assembly is a folding process through which proteins attain a specific conformation (Fig. 2). However, self-assembly is successful only under optimal conditions and in the absence of any external factors.21 At times, correctly folded proteins are exposed to cellular and physical stress, resulting in aggregation, misfolding, and unfolding.22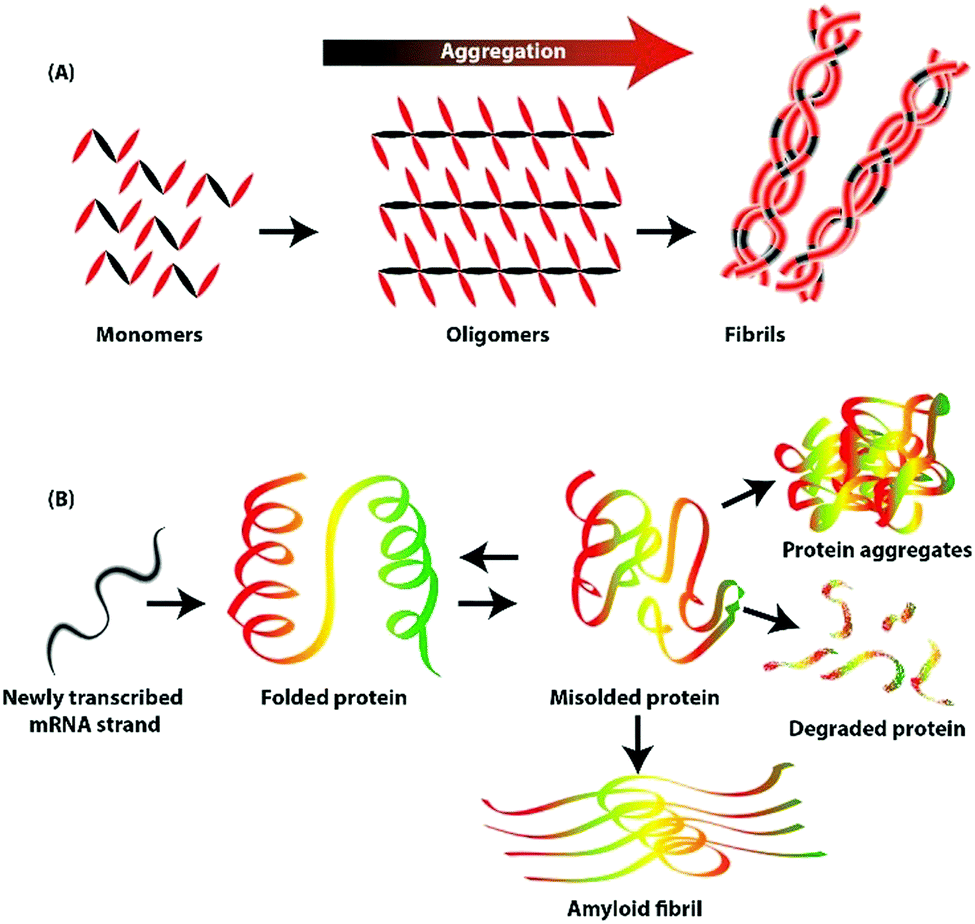 | ||
| Fig. 2 (A) Diagrammatic representation of the steps for the formation of aggregation. (B) Depicted events shows the misfolded proteins or amyloid fibrils can form protein aggregate or refold back to the native structure. | ||
Many factors, including thermodynamic behaviour, affect the durability and strength of proteins.23 There is high probability for the loss of protein function and impairment of critical function under stresses such as heat shock and oxidative stress, resulting in protein misfolding or unfolding.24 Misfolding results in the transformation of proteins into non-native structures and further into organized fibrils.25 Over 35 human diseases, including Alzheimer's disease, Parkinson's disease, Huntington's disease, and sickle cell anaemia, are reportedly associated with protein aggregation and misfolding, presenting challenges for medical technology and industries.26 Additionally, the possibility of formation of intermediates such as protofibrils and oligomers cannot be ignored.27
There are 20 homologous proteins and a few associated proteins such as Aβ1–40,28 lysozymes,29 and insulin,30 which are known to form amyloid fibrils that trigger incurable diseases such as Parkinson's disease.31 Amyloid fibrils comprise 3–6 filaments that change form when the unfolded monomers are assembled into oligomers, followed by transformation into the metastable β-sheet conformation and subsequently into β-sheet rich structures.32 These oligomers can function as nucleation seeds to form further oligomers and fibrils; however, the dimension of the nucleus depends on the reaction kinetics and conditions.33,34
Several factors, such as covalent and non-covalent interactions, are involved in the formation of clusters that lead to the loss of biological activity, immunogenicity, and other effects.35 The initiation of aggregation may occur due to the interaction between hydrophobic patches on proteins, resulting in a highly flexible folded state and insolubility.36 Non-covalent interactions such as van der Waals forces and electrostatic interactions are responsible for self-aggregation.37,38 Insulin is a perfect example for studying oligomers in the native state, and it forms amyloid fibrils in a non-physiological state under reversible aggregation conditions.39 Similarly, covalent aggregates are formed through disulphide linkages via free thiol-groups where disulphide-bonded proteins aggregate due to interchange via β-elimination.40 Dityrosine formation, oxidation, and the Maillard reaction are a few examples of covalent interactions that induce protein aggregation.41 Thus, only a few aggregate types have been characterized so far, viz. irreversible or reversible,42 non-covalent43 and covalent,44 and small soluble aggregates;45 where the nucleation–propagation mechanism46 is commonly involved in the formation of aggregates.
3. Consequences of protein aggregation
Changes in protein structure associated with aggregation result in the formation of oligomers, followed by the growth of linear aggregates, and eventually, visible particles with low solubility are formed. In 1931, Hsien Wu outlined the issue of protein instability47 and postulated a theory on protein denaturation. Protein aggregation in addition to triggering neurodegenerative diseases, causes challenges in the development and commercialization of biotechnology products and is observed in all stages of protein product development and manufacturing processes, such as protein expression,48 formulation,49 purification,50 product filling,51 lyophilization,52 and storage (Fig. 3).53 Protein purification poses different challenges for the solubilization of insoluble particles or aggregates, and the requirement of additional steps may result in reduced final yield of proteins, which is not economically viable. Moreover, other techniques steps such as vial filtration (purification method), inactivation, and chromatography can cause protein aggregation. In addition, conditions such as high protein concentrations, shear stress, changes in pH, and ionic strength may also trigger protein instability. For instance, chromatography, one of the most used purification methods that involves the presence of stationary and mobile phases (solvent), requires the addition of protein stabilizers to avoid aggregation. Another example is the addition of glutathione and ascorbic acid to prevent the generation of reactive oxygen species, which may promote aggregation.54,55 Moreover, proteins may adsorb on the stationary phase, leading to protein unfolding and increased aggregation. In addition, chromatography may require low pH, resulting in conformational or colloidal instability of the protein and causing aggregation.50 Preparation of protein samples also involve centrifugation and filtration, which expose proteins to shear stress. Furthermore, the proteins at the air–liquid interface may form soluble or insoluble aggregates. Thus, conditions resulting in protein aggregation during purification need to be mitigated. Processes such as product filling, lyophilization, and long/short-term storage are discussed in the next section.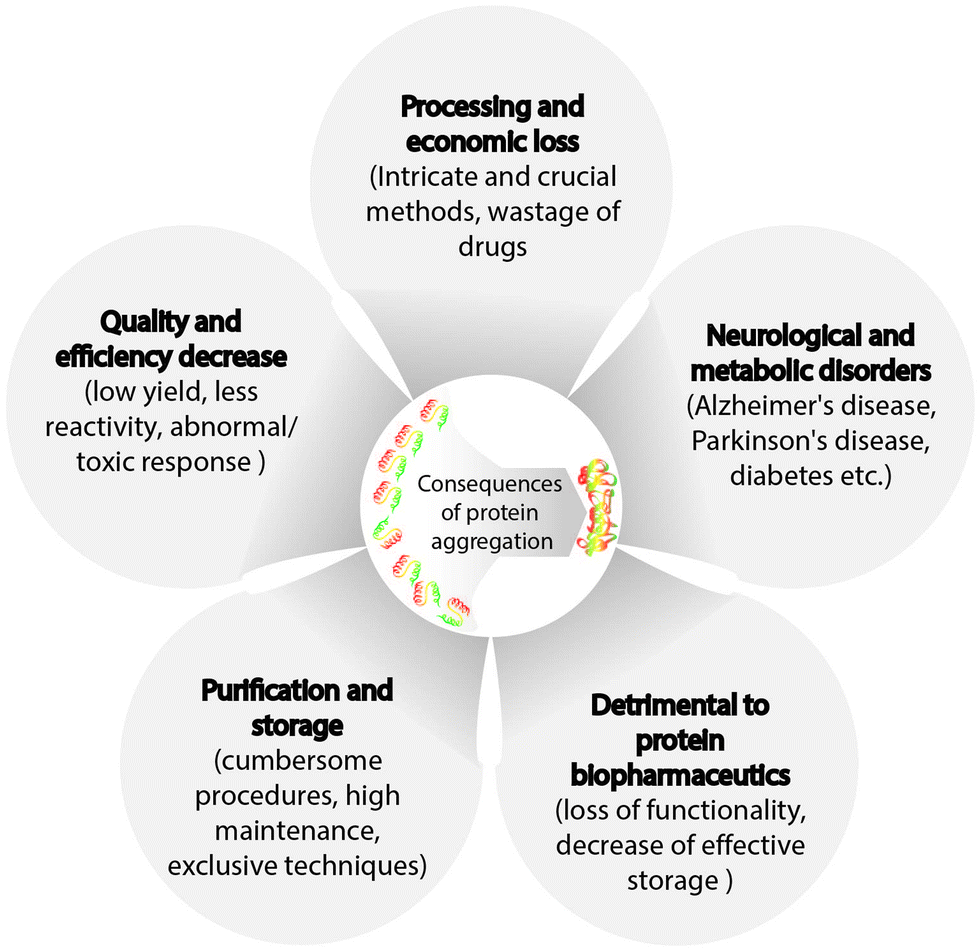 | ||
| Fig. 3 An overview of various problems associated with protein aggregation. | ||
Additionally, protein aggregation can occur during or after the administration of protein in a therapeutic context.56,57 Multiple disorders and ailments such as myocardial infarction, diabetes, and cancer can be cured using commercially available protein-based products. At present, hundreds of such products are in the preclinical and clinical development stages. After production, the pharmaceutical protein must be stabilized and ideally requires a shelf life of 1.5–2 years.58 This task is challenging as proteins are prone to aggregation, leading to their chemical and physical degradation and subsequent loss of activity.59,60
Physical degradation occurs during the unfolding, accumulation, aggregation, and surface adsorption of the protein.59–61 Chemical degradation involves the oxidation, deamidation, and movement of the disulphide bond. Aggregation can occur under the influence of stresses and even under favourable conditions for native state/conformation. The formation of small aggregates is reversible, but denatured protein aggregation or the formation of non-native β-sheet structures is an irreversible process.62 Protein drugs that are currently available have aggregation issues; for example, insulin tends to precipitate and cause blockages in implantable and portable delivery systems. The most popular delivery method for insulin is via syringe, but its improper storage could lead to insulin aggregation. The use of lysine or arginine as additives may also induce the aggregation of insulin, as arginine changes the overall charge and hydrophobicity of insulin by binding to partially unfolded intermediates. The induced structural changes do not depend on the native structure of the protein or the final morphology of the aggregates. Further, the delivery of insulin via the gastrointestinal tract is not feasible owing to the presence of acid and enzymes. Multiple factors such as protein stabilizers, additives, and pH can affect the adsorption, aggregation, and retention of insulin in the gastrointestinal tract.63
Glucagon is another synthesizable hormone that is used to maintain blood glucose levels in diabetic patients, and it facilitates glycogenolysis and gluconeogenesis by the liver. Glucagon helps treat acute hypoglycaemia, and it quickly restores normal levels of glucose. Its crystalline form is dissolved in diluted acid buffer and injected intramuscularly. In aqueous solutions, glucagon forms aggregates and precipitates owing to its low solubility64 and is known to form amyloid-like fibrils in alkaline and acidic conditions.65–67 Therefore, it is important to have an alternative or analogue of glucagon with similar biological activity that is sufficiently soluble and stable under relevant physiological conditions.
Monoclonal antibodies (mAbs) are multidomain glycoproteins used by the immune system to protect the body against various invaders. They are used in treating infectious ailments, autoimmunity/inflammation, and metabolic disorders, and in oncological studies. Presently, more than 30 mAbs are commercially available. The preferred method of delivery, i.e., subcutaneous injection, needs relatively high concentrations of antibodies. However, antibodies can aggregate irreversibly and form amyloid structures enriched in β-sheets,68,69 because of change in temperature, pH, salt concentration, type of buffer, and storage conditions.70,71 Bansal et al. analysed the aggregation of two antibodies (with pI of around 8.5 and 8.0) at different temperatures using acetate and citrate buffers (100 mM concentrations and pH 3.0 with 100 mM NaCl). The rate of aggregation was prominently higher in the presence of citrate relative to acetate buffer.72 Dispersed antibodies in citrate buffer have low stability, which leads to high aggregation. Moreover, there is less protein–protein repulsion due to decrease in protein charge, as citrate molecules possess higher charge. Hence, electrolytes in citrate buffer shield the electric double layer around the protein, thus decreasing the energy barrier. It is possible that faster aggregation in citrate buffer results because of the decrease in the net energy barrier for protein–protein interaction leading to antibody instability. To function, proteins must be stably folded into their three-dimensional framework. In certain circumstances, this may not occur due to the presence of mutations, failure of the proteostasis network, pathological conditions, or the prevalence of conditions such as temperature fluctuations, high pressure and agitation, or extreme pH, resulting in unfolded/misfolded proteins and aggregates.
More than forty human diseases are caused by the accumulation of distinct proteins with a unique clinical profile. For example, Alzheimer's disease (AD) is characterized by β-amyloid and tau protein aggregates, Parkinson's disease (PD) by α-synuclein (αSyn), and Huntington's disease by Huntington protein (Htt) aggregates. Alzheimer's disease (AD), which was first detected in 1906, affects millions of lives worldwide,73 and is a major cause of dementia with no substantial treatment. The pathological hallmark of AD is the extracellular plaque deposition in the brain, which consists primarily of the β-amyloid peptide. The most toxic species of this peptide comprises 42 amino acids with a molecular weight of ∼4.5 kDa. In solution, β-amyloid can form filamentous β-sheet aggregates without any change in physiological conditions.74
In contrast, protein aggregates reportedly have a positive effect on stressed striatal neurons.75,76 Leitman et al. have proposed that protein aggregation is a defensive mechanism to protect, isolate, and segregate faulty proteins.75 In this context, Arrasate et al. have shown that inclusion bodies (IB) protect neural cells by regulating the diffusion of toxic Htt.77 Elevated β-amyloid protein levels are a marker for AD; however, in C. elegans, toxic aggregates of β-amyloid 42 were reduced under the influence of insulin/insulin growth factor-1-like signalling pathway (IIS).78 The IIS pathway is responsible for determining the lifespan of multiple model organisms such as C. elegans, flies, mice, and humans as it regulates aging, stress, and resistance to disease.
Protein purification requires the solubilization of insoluble particles or aggregates, and it involves the use of conditions such as high protein concentrations, shear stress, changes in pH, and ionic strength, which may cause protein instability. Further, it involves the use of procedures that cause losses in the final yield that are not economical. Purification steps such as viral filtration, inactivation, and chromatography can cause protein aggregation. For instance, chromatography is an important filtration method and uses stationary and mobile phases (solvent) requiring the addition of protein stabilizers such as glutathione and ascorbic acid. This is done to prevent the generation of reactive oxygen species, which may provide hot spots for aggregation.54,55 The stationary phase may also cause adsorption of protein to the surface, leading to protein unfolding and increased aggregation. In addition, chromatography may require the presence of low pH, leading to conformational or colloidal instability in the protein, causing aggregation.50 Protein preparations also require centrifugation and filtration, which expose proteins to shear stress (high speed of centrifugation). Further, exposure of proteins at the air–liquid interface may lead to the formation of soluble or insoluble aggregates. Thus, conditions leading to protein aggregation during purification need to be mitigated.
Protein aggregation is an inevitable challenge at every stage of protein synthesis, processing, and administration, which needs to be overcome using appropriate measures. Moreover, protein aggregation triggers numerous complications in vivo. Hence, developing strategies to both protect proteins from aggregation as well as remove it post-aggregation is extremely crucial in many fields.
4. Factors affecting protein aggregation
Proteins can aggregate under various environmental conditions, and the extent of aggregation depends on intrinsic (indirect) and extrinsic (direct) factors. Extrinsic factors include environmental factors, genetic factors, metal ions, and oxidative substances that promote aggregation.79 In this review, we focus on environmental factors and explain the role of temperature, pH, ionic strength, salt type and concentration, co-solutes, ligands, and various processes in protein handling that affect aggregation (summarized in Table 1).| Factors | Protein/antibodies | Condition of protein aggregation | Literature |
|---|---|---|---|
| Temperature | β-Lactoglobulin | Rapid increase in aggregates from 30 °C to 50 °C. | 80 |
| pH | Bovine-serum albumin, β-galactosidase glucagon-like peptide-1 | pH 7 to 1 caused freezing-induced aggregation (20 to −25 °C). | 81 |
| β-Lactoglobulin | pH 8.2 to 7.5 resulted in the formation of amyloid-like fibrils. | 82 | |
| Building blocks vary at pH 2 and 3.5. | 83 and 84 | ||
| Ionic strength | Soy protein | Decrease in surface charge and hydrophobicity when strength was increased from 0–500 mM. | 85 |
| Ligands | Insulin | Small peptide prevented the early formation of a critical nucleus. | 86 |
| Cosolutes | Lysozyme, insulin | Trehalose inhibits aggregation. | 87 |
| Salt type | Myofibrillar protein | MgCl2 and CaCl2 induced higher disulphide bonding than NaCl. | 88 |
| Salt concentration | Potato protein | Increase in NaCl affects surface dilatational elasticity and surface activity; also facilitates protein–protein interactions within surface film. | 89 |
| Pumping | Intravenous Ig | Electrostatic interactions between pump surfaces (negatively charged) and antibodies (positively charged). | 90 |
| Agitation | Whey protein | At 5% protein concentration, aggregation increased at a high shear rate. | 91 |
| Drying | Blood plasma | Sugar addition checks freeze-drying-induced aggregation. | 92 |
| Light exposure | α-Lactalbumin | Disulphide-mediated aggregation on exposure to UV-B (0–24 h). | 93 |
4.1 Temperature
Temperature affects protein aggregation in multiple ways by influencing protein–protein interactions, rate of protein diffusion in solutions and the solid-state, conformational stability, solubility of proteins/aggregates, and chemical degradation. The thermodynamic stability of native protein conformation ranges from ∼5–20 kcal mol−1 in free energy.94,95 In physiological conditions, the native state of a protein is stable relative to the unfolded and biologically inactive conformations.96–98 Electrostatic forces, hydrophobic interactions, hydrogen bonding, van der Waals forces, and local peptide interactions are responsible for the free energy of the folding state.99 Conformational entropy is the driving force that induces protein unfolding, and it is sensitive to changes in salt, solution pH, and temperature.The thermodynamic stability of the native protein state as a function of temperature shows a typical parabolic profile.100 An increase in temperature is likely to accelerate the aggregation process due to an increased rate of protein diffusion and the amount of partially unfolded states for various proteins.101,102 In terms of Gibbs free energy (ΔG = ΔH − TΔS) for the transition between folded and unfolded state, ΔG decreases as temperature increases and eventually becomes zero and subsequently the free energy of the unfolded state of proteins becomes lower than that of the folded state. Thus protein unfolding becomes thermodynamically favourable at elevated temperatures.99,103,104 At high temperatures, proteins denature or degrade physically, in what is mostly an irreversible aggregation, as seen in the recombinant human Flt2 ligand,105 streptokinase,106 ribonuclease A107 and the recombinant human keratinocyte growth factor.108 Completely folded protein molecules do not form aggregates because aggregation is initiated at temperatures well below the equilibrium melting temperatures of proteins; at these temperatures, the amounts of native and denatured proteins are at equilibrium.62 Meanwhile, decreasing the temperature can cause cold-folding of the protein leading to its aggregation. Luan et al. have shown that the ribosomal protein L9 exhibited 76% aggregation at 4 °C and ∼36% aggregation at 25 °C.109 For antibodies, a decrease in temperature could induce precipitation as seen for the monoclonal IgM cryoglobulin at 14–15 °C.110 A recent report by Wälchli et al. using two immunoglobulins showed various types and mechanisms of aggregate formation between 40 °C and 5 °C.111 Further, reversible oligomerization has been demonstrated in mAbs.112,113 Wang et al. have shown that antibodies with overall attractive protein–protein interactions at low temperatures could exhibit liquid–liquid phase separation, which is a type of protein condensation that involves separation of proteins into two phases: protein rich (bottom) and protein-poor (top).114
Temperature plays a crucial role in reaction kinetics as rate constants increase exponentially with temperature, i.e., the rate of protein aggregation increases at high temperatures because of the rise in global molecular mobility (kinetic energy). Therefore, an increase in temperature accompanies an increase in the probability and number of collisions with enough energy to overcome activation energies.115 Moreover, these phenomena work in a cycle, with a rise in temperature enhancing the rate of monomer diffusion, accelerating the rate of the reaction. Proteins such as bevacizumab may follow the traditional Arrhenius relationship within a narrow temperature range between 25 °C and 50 °C;116 however, most proteins do not behave this way for a wide range of temperatures.117,118 Therefore, the rate of aggregation might be different at 25 °C than at 40 °C70 due to the change in protein aggregate solubility,119 various aggregation mechanisms,120 conformational stability,121 and the reversibility of protein aggregation122 at different temperatures. Thus, prescribed storage temperatures for proteins based on accelerated stability data,121 calculated using a modified Arrhenius equation121 and the extended Lumry–Eyring (ELE) aggregation model should be followed.123
4.2 pH and ionic strength
Protein stability changes with alterations in solution conditions. One such condition is the solution pH, which affects protein aggregation by causing changes in protein conformation and stability.124–126 Filipe et al. showed that immunoglobulin G1 (IgG1) transformed from dimers to micrometer-sized aggregates in a few hours when the pH decreased from 6 to pH 1.127 The pH can cause differences in the growth of the nucleus and the rate of aggregation128 by influencing protein–protein interactions,129,130 the solubility of protein aggregates,131 and the reactivity of protein cross-linking reactions.132 Proteins can be very sensitive to a change in pH outside a narrow pH range, e.g., insulin,133 relaxin,134 ribonuclease A,135 rhGCSF,136 and the recombinant factor VIII SQ.137 pH affects electrostatic interactions and consequently protein stability in two ways; e.g., when the solution pH is significantly different from the isoelectric point of the protein, it causes the protein to become highly charged, giving rise to repulsions.99 These repulsions can destabilize the protein conformation via unfolding due to a change in charge density, and the unfolding decreases the free energy of the protein. Second, conformational stability changes while ion pairing, and instead of destabilizing the folded protein conformation, ion-pairing stabilizes it.99,138 Therefore, ionic strength influences protein aggregation.139,140 Ionic strength may first change the nature and extent of protein–protein interactions by altering the conformational stability of the protein.141 Anions and cations affect protein aggregation differently by binding or screening the protein molecules,129,142 and anions affect protein aggregation significantly143 and for a wider range of pH due to their association with positively charged protein groups.144 The effect of ionic strength greatly relies on the solution pH. Further, the ability of anions to affect aggregation varies with the electroselectivity and polarizability of the protein and solution conditions.142,1454.3 Cosolutes and ligands
Strongly binding ligands play a crucial role in the physical stability of a protein. According to the Wyman linkage function, in a two-state equilibrium during ligand binding, equilibrium always shifts towards the state with greater binding. Between the native and unfolded states of the acidic fibroblast growth factor, polyanions bind to and shift the equilibrium towards the native state.107 Similarly, zinc ions bind to the human growth hormone leading to a rise in the free energy of unfolding.146 Compounds such as ammonium sulphate, sugars, and polyols provide stability to the native state of proteins, whereas guanidine hydrochloride and urea are known denaturants.147,148 This may occur due to a difference in solute binding to a protein in which some solutes prefer the folded (native) state over the unfolded state, and vice versa.Sugars and polyols, which act as protein stabilizers and are added as co-solutes to solutions, inhibit aggregation and impart physiological osmolality in drug formulations. Exclusion of co-solutes from protein surfaces is preferential and varies with the ratio of solvent concentration to protein surface area.147,149 Preferential exclusion refers to the repulsive force of protein molecules observed when interactions between solvent and protein molecules are unfavourable. Exclusion increases as the surface area of the protein increases with protein unfolding, indicating a preference for the native folded state of proteins. Nicoud et al. has shown that the addition of sorbitol to glycosylated IgG1, in absence of a polyol sugar, delays the depletion of monomers corresponding to the protein unfolding.150 Furthermore, solutes that bind weakly to the protein surface negatively impact the rate of protein aggregation.
4.4 Salt type and concentration
Salts can affect the physical properties and stability of a protein by altering the protein conformation, solubility, and the rate of aggregation.151–153 The addition of NaCl alters the solubility equilibrium for the recombinant human tissue factor pathway154 and causes a decrease in the aggregation speed of the recombinant factor VIII SQ.137 In contrast, NaCl addition causes the rate of aggregation to increase for rhGCSF.155 As explained earlier, cations and anions bind protein surfaces preferentially via unpaired charged side chains. When multivalent ions are present, they can bind to side chains and crosslink charged residues on the protein surface, stabilizing the native state of the protein.156 However, ions can destabilize the native state of the protein via binding to peptide bonds, which have a partial negative charge on the carbonyl oxygen, and a partial positive charge on the amino group.153 Electrolytes modulate the strength of electrostatic interactions between charged groups, both within the protein and between protein molecules. Thus, whereas intramolecular charge–charge interactions affect conformational stability, intermolecular electrostatic interactions affect equilibrium and the rate of aggregate formation.Salt concentration is another vital parameter required for protein stability. A salt affects protein–protein interactions while also interacting with protein molecules. At high salt concentrations, ions primarily interact with a protein to shield and bind to the protein surface. This results in higher protein solubility while decreasing the thermodynamic stability,152 leading to destabilized protein folding and denaturation.157 Metal salts can denature proteins and form insoluble metal–protein salts; for instance, silver nitrate was shown to cause fluorescence quenching of tryptophan in BSA.158 Silver ion is a hydrophobic quencher that induces protein misfolding and unfolding, thereby distorting the native state of the protein. In contrast, low concentrations of salts reduce electrostatic interactions as the ions shield the charges instead of binding them. Thus, the type, number, and distribution of charges in a protein depend on the pH. Hence, the effects of salts vary greatly depending on changes in pH.
4.5 Processes
Various processes for manufacturing drug products (proteins) may influence the aggregation behaviour of proteins, as explained below.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s−1,160 but insulin forms fibrils at a minimal shear of 150 s−1.161 Another procedure in drug manufacture is filling, which requires prior mixing of proteins to ensure homogeneity, using methods like stirring and suspended impellers that may cause protein damage and aggregation.162 However, multiple methods can cause agitation while mixing protein solutions such as vortexing, shaking, stirring, and rotating,163 leading to aggregation in different proteins or peptides.140,164–166
000 s−1,160 but insulin forms fibrils at a minimal shear of 150 s−1.161 Another procedure in drug manufacture is filling, which requires prior mixing of proteins to ensure homogeneity, using methods like stirring and suspended impellers that may cause protein damage and aggregation.162 However, multiple methods can cause agitation while mixing protein solutions such as vortexing, shaking, stirring, and rotating,163 leading to aggregation in different proteins or peptides.140,164–166
Sonication affects protein aggregation and causes partial unfolding due to an increase in temperature, pressure, and void formation.173,174 Additionally, the long-term storage of protein-containing syringes could produce particles and aggregates of protein due to the formation of radicals on exposure to radiation during the sterilization process.175
5. Mechanism of protein aggregation
In 1954, Lumry and Eyring outlined a theory that laid the foundation for protein structure, stability, folding, and aggregation.176 Aggregation is a common and critical issue during the synthesis and storage of proteins. The classic Lumry–Eyring model describes an irreversible protein denaturation in two steps:N ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) U → F, U → F, |
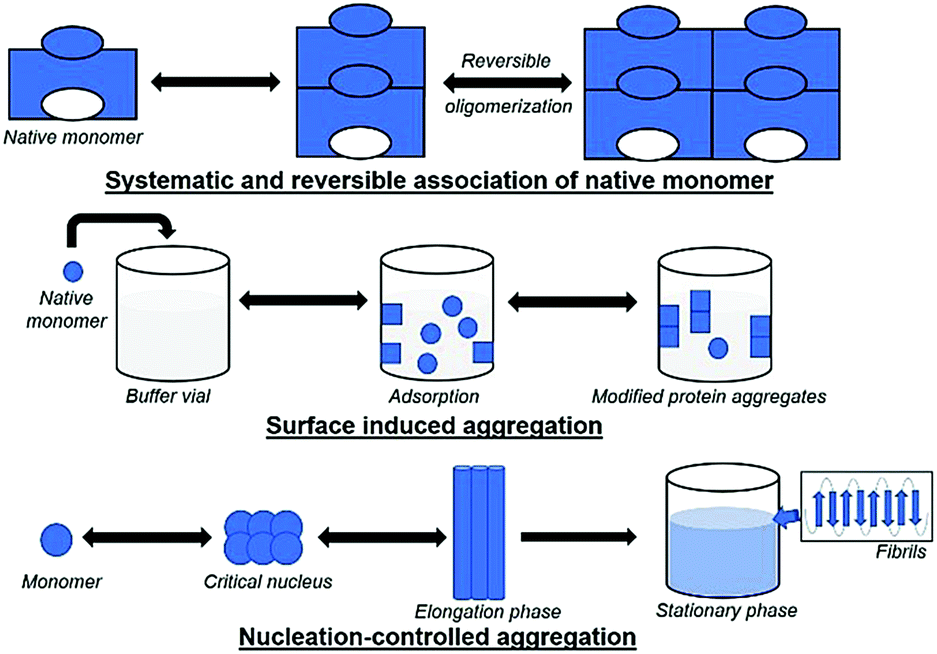 | ||
| Fig. 4 Schematic illustration of the different modes of protein aggregation. | ||
5.1 Systematic and reversible association of the native monomer
The reversible association hypothesis is valid for native and denatured proteins and starts with the aggregation of monomers into the growing aggregate. The hypothesis postulates that the tendency for reversible aggregation of a protein is intrinsic to its native form. In native monomers, self-association takes place and results in small reversible oligomers. The surface of protein monomers may have various complementary regions, which may provide multiple interface types with different patterns for the growth of oligomers, and multiple conformations for oligomers with identical stoichiometry. Insulin is an example of a therapeutic protein that readily associates to form reversible oligomers,178 which can greatly affect its bioactivity; additionally, structural or confirmational rearrangement in the aggregates could produce new entities.179 An increase in the concentration of protein and the growth of oligomers may eventually cause aggregates to turn into irreversible forms; e.g., interleukin-1 receptor antagonist (rhIL-1RA) forms reversible dimers that convert to irreversible dimers and trimers at high concentrations.180 This mechanism has two types of variants, where (a) the conformation of monomers is altered, and (b) the monomer is modified chemically.5.2 Conformationally altered monomer
In this mechanism type, the monomer does not associate reversibly. However, after partial refolding or owing to a change in conformation, monomers tend to associate strongly. Monomer changes to a non-native state usually occur when the amount of protein monomer is quite small. Conformational changes occur mostly by heat or shear stress, which facilitates protein aggregation. The conditions that stabilize or favour the native state inhibit the process of aggregation. G-CSF181 and interferon182 have been shown to exhibit this mechanism. This mechanism is predominant for many proteins and has been extensively discussed in multiple reviews.183,1845.3 Chemically modified products
In this mechanism, changes in the covalent structure of proteins due to chemical degradation such as proteolysis, deamidation, or oxidation of methionine occur, which may lead to the formation of new regions on the protein surface or result in a decrease in electrostatic repulsion between monomers. For example, the un-glycosylation or under-glycosylation of glycoproteins causes aggregation. Unlike the conformationally-altered monomer, modified protein monomers can be present in large amounts and are a diagnostic feature of this mechanism. However, aggregates do not contain modified monomers exclusively and harbour non-modified monomers as well. Therefore, the enhancement of chemical stability can reduce aggregation. Importantly, chemically modified protein aggregates can be precisely immunogenic.1855.4 Surface-induced aggregation
Here, native monomers first bind to a surface to initiate the process of aggregation. Next, a conformational change in the monomer (e.g., to increase the contact area with the surface) may occur. The driving forces could be hydrophobic or electrostatic interactions, based on the nature of the surface. The mechanism is different from those described previously for conformationally-altered monomers, as aggregation takes place at the surface or after the detachment of monomers into the solution. Freeze/thaw damage can also lead to protein aggregation due to instability resulting from temperature changes which can occur via surface-induced aggregation. Aggregate formation at the surface of ice crystals or due to a change in pH may also occur. Processes involving induction of shear forces or agitation can cause long exposure to surfaces and lead to changes in protein conformational stability (as discussed in the case of conformationally altered monomers). Thus, it is difficult to identify the actual stress that causes protein aggregation.5.5 Nucleation-controlled aggregation
In general, aggregation initiates via formation of a nucleus, which grows and accumulates with time, leading to insolubility and precipitation of the protein. The accumulation of insoluble aggregates results in lower or no bioactivity of a protein, loss of cellular function, and high toxicity due to disturbed intracellular transport. This mechanism is well-studied, and the formation of aggregates takes place slowly in a lag phase, followed by an elongation (exponential) phase, and a final plateau. For some proteins, the lag phase arises due to an energy barrier for nucleation resulting from the free energy required to create an interface between a solid particle of protein and a liquid (solvent), and it varies with the size of the aggregates. Here, the probability of production of small or mid-sized oligomers is low. Therefore, the formation of moderate-sized aggregates is called the “critical nucleus/seed”, which probably converts to larger species by the addition of monomers at a fast rate. The lag phase is fairly slow, with no visible changes over a long period, followed by sudden changes in the size of aggregates leading to precipitation. Moreover, the formation of nuclei may vary with orientation and may prefer a lower free energy for initiation. Fibril formation is an example of nucleation-controlled aggregation. In some cases, the rate-limiting step can involve the formation of a pre-nucleus species. In αSyn, the pre-nucleus entity formed on protein oxidation is a dityrosine-crosslinked dimer,186 and the rate of nucleation increases rapidly when the dimer concentration is ∼1–2% of the total protein population. Starting with preformed dimer species (called seeding) could quickly result in fibril formation. Nucleation can be of homogenous or heterogeneous type. In the former, as described above, the critical nucleus is itself a product aggregate. In the latter type, the nucleus/seed can be a contaminant instead of a protein. For example, silica (shed by vials) or steel particles (shed by piston pumps during the filling process) act as the nucleus/seed for aggregation.187,188 After a lag phase, monomers convert to oligomers, leading to β-sheet formation, which are intermediate structures known as protofibrils, eventually forming fibrils till the final plateau phase occurs.6. Characterization of protein aggregation
Protein aggregates induce immunogenic responses that can be fatal. Several techniques are widely used for the qualitative and quantitative analysis of protein aggregates and amyloid particles. In this section, we discuss the diverse analytical methods used for the characterization and identification of aggregates, ranging from simple investigations to sophisticated analytical methods that require expertise. Changes in molecular weight, conformational state, shape, and size are some of the physicochemical properties studied to investigate aggregation. A few commonly used techniques for the detection and characterization of protein aggregates are outlined in Table 2.| Classification | Method | Aggregate size | Pros | Cons | Ref. |
|---|---|---|---|---|---|
| Separation-based methods | SEC | 1–50 nm | Short-run and quantitative analysis | Large sub-visible aggregates cannot be analysed | 189 and 190 |
| AUC | 1–100 nm | High resolution, quantification of proteins | High concentration required, expensive | 191 and 192 | |
| FFF | 1 nm to few μm | No stationary phase requirement, broad size range possible | Less robust, concentration dependence | 191, 193 and 194 | |
| Scattering-based methods | SLS | 1 nm to few μm | Detection of several absolute parameters (radius of gyration, molecular weight) | Not applied for low molecular weight solutes | 195 and 196 |
| DLS | 0.1–5 μm | Less sample required, high resolution | Highly sensitive to temperature, presence of any large particle or dust can interfere | 197 and 198 | |
| Imaging-based methods | TEM | 0.1 nm to few cm | High powerful magnification and resolution | Not quantitative, expensive, time-consuming | 199 and 200 |
| AFM | 0.1 nm–100 μm | Non-destructive imaging, easy sample preparation | Limited magnification range | 199, 201 and 202 | |
| Calorimetric-based methods | DSC | N/A | Specific for thermal stability assay of proteins | Low accuracy and precision | 203–205 |
| ITC | N/A | Enables quantitative determination | Large sample volume required | 206–209 | |
| Spectroscopy-based techniques | UV-vis | N/A | Non-destructive, quick analysis, easy to use | Lack of sensitivity and selectivity | 210–213 |
| CD | N/A | Simple and quick technique, secondary structure detection | Interference by buffer components | 214–217 | |
| FT-IR | N/A | Fast solid-state analysis possible | Low sensitivity | 218–220 | |
| NMR | N/A | High reproducibility, useful for protein folding | Requires concentrated samples, which may lead to aggregation | 221–223 | |
| X-ray scattering techniques | XRD | N/A | Provides information about 3D protein aggregate structure | Crystal production required, time-consuming | 224 and 225 |
| SAXS | 1–100 nm | Provides information about various stages of aggregation | Expensive, limited availability | 226 and 227 |
6.1 Separation-based methods
Protein-based pharmaceuticals such as interferons and human growth hormones are genetically engineered. At times, the resultant proteins are unsteady, resulting in aggregation that affects their biological activity. Chromatography is a convenient and versatile technique to precisely monitor aggregation.6.2 Scattering-based methods
The size of protein aggregates can range from microscopic to macroscopic in scale, which can be analysed using simple techniques. Often, they can be detected with the naked eye because of the change in turbidity or large aggregates clumping together. Various promising technologies using visual inspection and quantification methods can detect the aggregation of therapeutic proteins in a reproducible manner, as discussed below.6.2.1.1 Static light scattering. Static light scattering is used for investigating parameters such as molar mass and the radii of biological macromolecules such as protein aggregates in solution.252,253 It measures the intensity of scattered light and using Rayleigh's theory, helps estimate the molecular weight of particles, which is proportional to the intensity of scattered light, with larger particles scattering more light than smaller particles. SLS measurements are generally taken at different angles to determine parameters such as the radius of gyration, which is used to predict the compactness of protein structure.254 Further, SLS can be used in combination with other methods like FFF to provide better resolution and help determine the molecular weight of different species in a solution to determine the extent of aggregation.
6.2.1.2 Dynamic light scattering (DLS). DLS is a scattering method used for investigating the distribution of particle size with a diameter range of 1 nm to 5 μm.255,256 In contrast to SLS, which measures the intensity of scattered light depending on angle or concentration, DLS measures fluctuations of scattered light in time. An autocorrelation function helps data interpretation to yield parameters such as size distribution, mean hydrodynamic radius, and polydispersity. DLS is used to monitor the aggregation process as small aggregates can be detected, and has helped determine the aggregation kinetics of Aβ, αSyn, and huntingtin.257,258 DLS is a simple technique and provides fast and accurate information; however, it cannot reliably provide accurate quantitative information as it cannot differentiate between the various stages of aggregation.
Scanning transmission electron microscopy (STEM) is a method similar to TEM but incorporates certain features of SEM and can be used to decipher the organization of amyloid aggregates. Like cryo-TEM, STEM does not require sample staining and is used in molecular biology to analyse the structure of biological molecules. It also provides better spatial resolution. Scattered electrons can be observed to obtain intensities that are proportional to the mass of the irradiated region.268 Subsequently, the mass per length (MPL) of the sample can be investigated by observing the incident electron beam flux under certain conditions. STEM has been used for the determination of the secondary structure of amyloid fibrils.269–272 The MPL calculation has been used to characterize the β-helix-like structure of the HET-s prion (HET-s218–289) protein fibrils, in which peptide molecules spanned 2 turns of the β-helix.273
AFM is an imaging technique used to visualize sample topography. It provides high-resolution information about the size, structure, stage, and distribution of aggregates, especially amyloid aggregates. AFM has been used to characterize low molecular weight oligomers,274,275 insulin fibrils,276,277 conjugated IgG aggregates,278 and other fibrillar systems such as collagens,279–281 fibrillogenesis,282 and silk protein fibrils.283 Additionally, it provides information on the height, contour length, and periodicity of the fibril structure. AFM has multiple advantages, such as enabling work in normal atmospheric pressure and room temperature conditions, high-resolution imaging at the molecular level, and the ease of sample preparation. Moreover, liquid samples can also be analysed using AFM at the physiological temperature.284,285 However, the slow speed scan rate and small scan area are a drawback of this technique. Still, AFM is a powerful and robust technique for investigating protein aggregation.
6.2.3.1 Differential scanning calorimetry (DSC). DSC is a cost-effective method used for determining the thermodynamic aspects of protein stability and unfolding in terms of the thermal transition temperature (melting temperature, Tm), and the energy needed to disturb the interactions stabilizing the tertiary structure (enthalpy, ΔH) of proteins.204 Protein samples are subjected to temperature changes, and the native state of the proteins undergo conformational alterations and the heat change is recorded. DSC can measure the ΔH associated with protein unfolding and the heat capacity (Cp) for denaturation. The water molecules next to the hydrophobic domains of the protein's surfaces are usually more ordered and strongly bound, resulting in greater hydrogen bonding between water molecules and the formation of a clathrate-like structure.70 On increasing the temperature, the highly ordered water molecules tend to reorganize and become more disordered, generating a large heat capacity.
The unfolding of the protein due to temperature yields the enthalpy (ΔH) due to the disturbance of hydrophobic interactions, and can be determined by the heat capacity of the sample relative to temperature.286
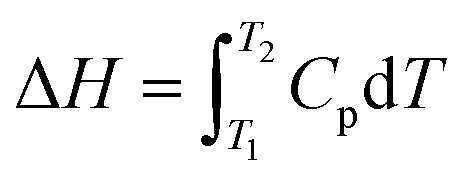
In one study, the thermodynamic characterization of lysozyme was investigated via stabilizing and destabilizing interactions using the DSC method.286 DSC has several advantages such as high precision temperature control and the direct observation of protein folding. However, this method may provide misleading information about misfolding and aggregation due to the large size and heterogeneity of aggregates, since larger sticky aggregates may stick on DSC cell surfaces and produce noise in the DSC thermogram.
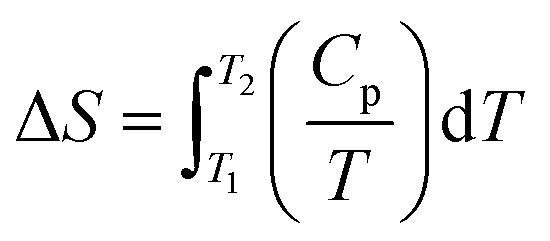
6.2.3.2 Isothermal titration calorimetry (ITC). ITC is used to determine the binding and kinetic events associated with ligand or excipients induced aggregation. ITC was initially used for determining Gibbs free energy, and is used for monitoring biomolecular interactions such as between enzymes and substrates.287,288 In contrast to DSC, which uses a constant temperature between the sample and the reference while the power used is variable, ITC measures the variation in temperature between the sample and the reference when using the same power. During the process of heat release or absorption, a signal can be detected, indicating binding between the two reactants. The variation in heat is represented as the change in Gibbs free energy (ΔG = RT
lnKD), where KD is the dissociation constant indicating the binding affinity between the interacting molecules. ITC is a quantitative technique used specifically for studying intermolecular interactions for proteomics and pharmacogenomics applications.287 Additionally, ITC can determine the binding affinity and binding stoichiometry of molecules. In ITC, sample concentration is an important parameter for obtaining reliable data, and care needs to be taken to avoid small molecule impurities and changes in pH, which can generate non-reliable thermograms.
6.2.4.1 UV-vis spectroscopy. UV-absorption spectroscopy is a rapid, safe, high-resolution technique which is used extensively to investigate protein aggregation. The thermodynamic and kinetic modes of protein aggregation, changes in protein conformation, and dynamic fluctuations can be investigated via observing the change in turbidity using UV-vis spectroscopy.289,290 An increase in absorbance in the non-absorbing region is used to predict the extent of aggregation.291 The measurement of turbidity or cloud point is used to uncover details of protein unfolding.292 The standard equation for determination of the turbidity is:
| Turbidity (τ) = −ln(I/I0), |
| AI = A350/(A280 − A350) × 100, |
The AI parameter can be straightforwardly used for determining protein content.293–295 However, this method may not be universally applicable for all conditions of proteins, with or without excipients, though the kinetic study of turbidity is often utilized in the field of protein pharmaceuticals to characterize the inhibitory effect of protein aggregation.293
6.2.4.2 Dyes and extrinsic fluorescent probes. Certain extrinsic dyes can provide details about the folding and unfolding of proteins.293 Dyes have been used for the determination of amyloid fibrils and to study the extent of aggregation (Fig. 5).293
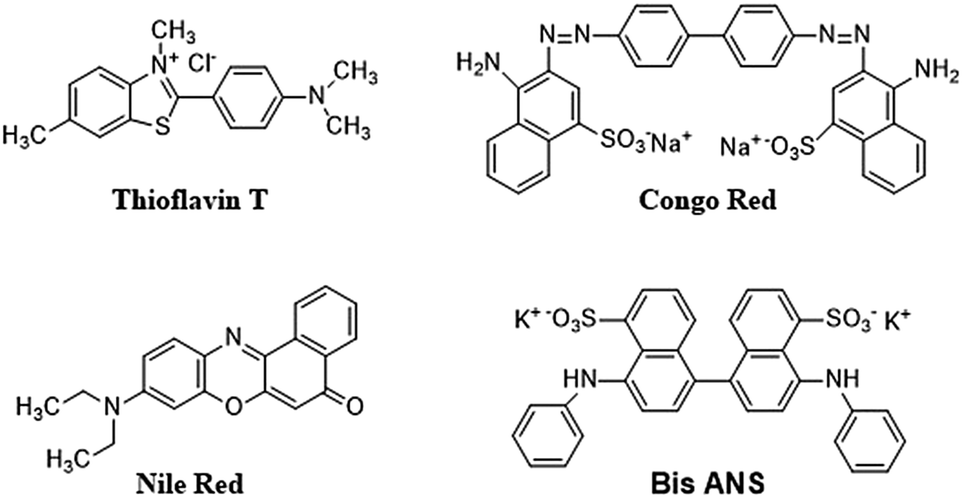 | ||
| Fig. 5 Chemical structure of extrinsic dyes used for the detection of aggregated proteins. | ||
The fluorescence of the protein sample mixed with dye is measured and any change in the fluorescence signal in terms of wavelength shift (blue shift: hydrophobic environment; red shift: hydrophilic environment) or intensity change is recorded. A protein undergoing structural change will interact with dyes differently than proteins in their native state, and this information can assist in quantitatively determining protein aggregation. One such dye is Congo Red (CR), which is often used for investigating amyloid fibrils that are rich in β-sheet content, yielding a characteristic apple-green birefringence under polarized light.240 Although this measurement using CR is easy and gives quick results, it poses certain challenges, as reports show that CR can interfere with protein structure. A recent study showed that CR triggered the β-sheet formation and peptide aggregation of Aβ1–40, and induces a β-sheet rich conformation in other amyloidogenic peptides.296,297 However, CR has also demonstrated a positive effect by suppressing the formation of fibrils, and has been found to be effective for modelling certain neurodegenerative diseases.298
Thioflavin T (4-(3,6-dimethylbenzothiazol-2yl)-N,N-dimethyl-aniline; ThT) is another extensively used dye as a fluorescent probe for the analysis of fibril formation. It can be used for in situ observation and does not affect amyloid formation kinetics.299 Usually, ThT imparts a strong fluorescence signal after binding with amyloid fibrils at 482 nm, after excitation at 450 nm.300 An increase in fluorescence intensity or a red shift is observed on binding with β-sheet rich structures such as amyloid fibrils. The exact mechanism of interaction between CR or ThT molecules with amyloid fibrils is not completely understood. A widely believed theory is that the intercalation of ThT molecules takes place between the grooves of the amyloid fibril parallel to the fibril axis.301–303 The extension of the torsional angle between the benzothiazole and benzene rings varies from 37 to 90 degrees.304 ThT-fibril binding and steady-state ThT fluorescence emission is observed at 490 nm when excited at 440 nm when the sample contains fibril structures and the aggregated material can be estimated using a standard curve showing linearity between fluorescence intensity and protein concentration. At high concentrations of amyloid fibril, a saturation effect may take place where ThT cannot be used reliably.305 Further, a few amyloid fibres do not interact with ThT and in a few other cases, ThT has been found to interact with the excipients, generating false results. Thus, the results should be verified by investigating the ultrastructure of the aggregate.
Other dyes such Nile Red and Bis-ANS are frequently used for the evaluation of protein aggregation. Bis-ANS does not exhibit fluorescence in aqueous solution and shows fluorescence only in the presence of hydrophobic moieties.306 Hence, misfolded and aggregated proteins can be detected using ANS. However, in a few cases, the ANS dye does not exhibit fluorescence due the large size of Bis-ANS, which prevent its inclusion into protein aggregates.
These studies demonstrate that extrinsic fluorescent dyes are extremely versatile and can be used for monitoring aggregation. However, they have their limitations and cannot be used universally to investigate protein aggregation. Therefore, proper care needs to be taken to observe the presence of any unwanted interactions (either between dye and protein, or dye, and excipients), and the results need to be correlated with other methods discussed in this review.
6.2.4.3 Circular dichroism (CD) spectroscopy. CD is a powerful technique used in the investigation of different orders of protein structure, including the α-helix, β-sheet, β-turns, or random structures. CD employs circularly polarized light which is made to pass through an optically active solution like those containing proteins. This induces differences in the 2 components of circularly polarized light (left and right) in terms of speed, wavelength, and the extent of absorption. The difference in molar absorptivity of proteins for the 2 components is referred to as molar CD. CD has various advantages such as simple operation, high precision, effectiveness, investigation of the absolute configuration, and it can reveal secondary protein structure.307 Consequently, CD has been extensively used for the analysis of protein folding and unfolding.308 Measurements are usually taken between the far UV and near UV wavelength range (180–320 nm), which enables the efficient characterization of distinctive secondary structural elements of proteins. Commonly, the secondary structure of proteins is affected by temperature, pH, ligand, denaturation, heat, mutation, and binding interactions.309 Investigating the resultant CD signals and observing the difference between the native and the aggregated state can assist in understanding aggregation at the molecular level. The contents of higher-order protein structures can also be evaluated by various methods.310,311 Further, measuring CD at a particular wavelength as a function of temperature can be used to estimate the degree of unfolding.312 An advanced version of CD known as vibration circular dichroism which is related to infrared spectroscopy can be used to investigate the sensitivity and chirality of the supramolecular architecture of fibrils.313 Additionally, far UV-CD (FUV-CD) can be used to investigate the kinetic and thermodynamic information for changes in protein secondary structure.313 However, the sensitivity of CD is low for data interpretation, and therefore, appropriate controls are needed for obtaining structural information.
6.2.4.4 Fourier transform infrared spectroscopy (FTIR). FTIR is usually employed for analysing changes in the secondary structure of proteins post denaturation or aggregation. It is responsive to the amide I band (∼1650 cm−1) and is usually responsive and dependent on the secondary structure.314 This method is relatively sensitive to the secondary structure of insoluble proteins such as inclusion bodies,313 heat-gelled proteins,62 and amyloid fibrils.315 FTIR is used extensively for the characterization of the alignment of β-strands in Aβ oligomers, amyloid fibrils in vitro, and the study of conformational dynamics.316 A detailed discussion on the use of FTIR for the analysis of protein structure can be referred to in the review by Haris and Severcan.314
6.2.4.5 Nuclear magnetic resonance (NMR) spectroscopy. NMR spectroscopy is widely used for the analysis of protein aggregation, as it can reveal information on dynamic properties such as relaxation and diffusion.317 The motility of proteins and internuclear distances are a few components that can provide details of dynamics and structure. A study of relaxation times can reveal information about the exchange kinetics of monomeric Aβ and oligomeric Aβ protofibrils.318
Using the NMR method, information on β-strand segments, backbone torsional angles, arrangement of β-strands in parallel or anti-parallel β-sheets, and the relative orientation of β-sheets can be obtained. In one study, fibril formation for αSyn was measured using paramagnetic relaxation enhancement (PRE) NMR to investigate the heterogeneously disordered monomers.319 Further, NMR can be used to investigate changes in amino acid residues, which can reveal the possible mode of denaturation.320 Folded proteins yield a broad range of chemical shifts, whereas denatured proteins exhibit a relatively narrow range of chemical shifts.321 NMR can be used to examine the interaction of any excipient with the protein by observing the individual amino acid residues to reveal the mode of aggregation, or the protection from aggregation.322 In a recent study by Taraban et al., water NMR was compared with other characterization techniques such as SEC, DLS, and others, and NMR outperformed them, proving to be more effective and sensitive for detecting changes in soluble and insoluble protein aggregates.221 Time-consuming investigation of the relaxation analysis, peak assignments, and the inability to characterize larger aggregates are some of the limitations of solution NMR. However, solid-state NMR (ssNMR) can elucidate the structure, dynamics, and kinetic aspects of protein folding and aggregation, and can determine the intermediate states of amyloid proteins323–325 and mutation of proteins.326 Moreover, ssNMR can be used to characterize the large insoluble aggregates. Tycko et al. have demonstrated the use of solid-state NMR for the characterization of the structure of Aβ fibrils in brain tissues.327
7. Protein aggregation inhibitors
Multiple strategies have been developed for inhibiting aggregation. One method to change the protein structure uses site-directed mutagenesis, which is effective under certain conditions.328–330 Tirrell et al. have shown that insulin denaturation can be prevented by the modifying it via replacement of the proline residue at position 28 (ProB28) with different groups.331,332 However, this strategy cannot be applied universally for the stabilization of proteins as the modification may result in proteins losing their activity.328 Another method is to add external additives (excipients) which change the environment around the protein to enable them to retain their native structure even when subjected to severe stress. In the following sections, we describe excipients as small molecules and polymeric molecules and discuss the various sub-categories and mode of action of these molecules in protecting the proteins' structure.7.1 Small molecules
For inhibiting protein aggregation in vitro, several techniques have been developed such as the use of molecular chaperones, pH control, temperature, ionic strength, and protein concentration. However, a simple and practical approach is the use of small molecules as additives that can act as protein aggregating inhibitors. Additives such as proline,333 polyamines,334 guanidine,335,336 and polyphenols have been used and are discussed in this review. Some of the well-known classes of small molecule inhibitors are briefly discussed in Table 3. For detailed discussions, refer to other reviews.16–18,20,337| Category | Inhibitors | Mechanism of action | Target protein | Ref. |
|---|---|---|---|---|
| Molecular tweezers | CLR01 | Fibrillogenesis inhibition | Aβ, IAPP, transthyretin, insulin | 338 and 339 |
| Metal chelators | Pentosan polysulphate | Important role in protein misfolding pathologies | Prions | 340 and 341 |
| Deferrioxamine, clioquinol | α-Synuclein | |||
| Nanoparticles | BAM (N-isopropylacrylamide:N-tert-butylacrylamide) Fe3O4 magnetic CDTe | Fibrillogenesis inhibition | Aβ, IAPP, lysozyme | 342–344 |
| Aggregates disintegration | ||||
| Peptide | Polyglutamine-binding peptide, Aβ peptide (KLVFF) | Fibrillogenesis inhibition | Aβ, polyglutamine | 345–347 |
| Phthalocyanines | Zn(II) and Ni(II) derivatives of phthalocyanine | Fibrillogenesis inhibition | α-Synuclein, Aβ | 348 and 349 |
| Aggregate disintegration | ||||
| Polyamines | Arginine, spermine, spermidine, putrescine, monoamines, diamines | Conformational change prevention | Immunotoxin, lysozyme | 334–336 and 350–354 |
| Fibrillogenesis inhibition | ||||
| Aggregate disintegration | ||||
| Polyphenols | Curcumin, EGCG, apigenin, resveratrol | Conformational change prevention | Aβ, α-synuclein, HEWL, IAPP, insulin | 355–361 |
| Fibrillogenesis inhibition | ||||
| Aggregate disintegration | ||||
| Vitamins | Vitamin E | Conformational change prevention | Aβ, α-synuclein | 362–366 |
| Vitamin C | Fibrillogenesis inhibition | Insulin | ||
| Vitamin B12 | Aggregate disintegration | |||
| Non-detergent sulfobetaine | NDSBs, choline-O-sulphate | Interacting with early folding intermediates | Aβ, β2 subunit (Tryptophan synthase), lysozyme | 367–371 |
| Fibrillogenesis inhibition | ||||
| Osmolytes | Raffinose, betaine, hydroxyproline, | Disintegration of amyloid fibrils | TGFBIp, insulin, BSA | 372–374 |
| Preferential hydration and polar interaction, disrupting inter-oligomer interactions |
Kudou et al. focused on some naturally occurring polyamines [spermine, NH2(CH2)3NH(CH2)4NH(CH2)3NH2; spermidine, NH2(CH2)3NH(CH2)4NH2; putrescine, NH2(CH2)4NH2] as small molecules for preventing the heat-induced aggregation and inactivation of proteins. Further, they studied the effects of some additives on the heat-induced aggregation and denaturation of lysozyme.334 Monoamines, diamines, and diols were selected to examine the effects of multivalent amines, chain length, and charge on the inhibition of protein. The protein thermal aggregation inhibiting curve of lysozyme was found to be almost identical to lysozyme with 1,5-diaminopentane, although diols and monoamines did not prevent the thermal inactivation of lysozyme.
In 1998 T. K. S. Kumar et al., proposed that proline with a concentration >3 M behaves as an enzyme stabilizer as well as a protein solubilizing solute.350 The amino acid, proline, shows solubility in water, and at higher concentrations, it behaves like a hydrotrope.351 Due to these properties, proline was investigated for its role as a protein-folding chaperone. It has been reported that proline at concentrations >3 M forms an amphipathic supramolecular assembly and successfully thwarts the aggregation associated with the refolding of bovine carbonic anhydrase. In another study, Choudhary et al. reported the inhibition of the protein fibrillation of lysozyme and insulin by proline and sorbitol.376 Proline contains a closed ring structure in its side chain which has a hydrophobic surface, which enables it to interact with proteins through hydrophobic interactions.
Curcumin is a yellow substance present in turmeric ((1E,6E)-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione). It has many beneficial anticancer, antioxidant and antiviral properties, and has also been investigated for its anti-aggregation properties in proteins. Curcumin has been reported to inhibit the aggregation of proteins such as Aβ, insulin, lysozyme, synuclein, prion protein, and lysozyme from hen egg white (HEWL).355,356 Owing to its lipophilic nature, curcumin can easily cross the blood–brain barrier and binds with amyloids, thereby destabilizing their oligomeric forms and inhibiting amyloid formation.357 Curcumin can have different modes of action for different proteins, such as preventing the conformational changes in the protein, disaggregating the formed aggregates, and inhibiting fibrillogenesis. Unfortunately, its wide application is limited due to its pharmacokinetics, which disfavours its bioavailability, and several derivatives have been synthesized to overcome this issue.378,379
Another polyphenol, (−)-epigallocatechin-gallate (EGCG), which is mainly found in green tea358 has been shown to have antibacterial, antioxidant, neuroprotective, and antitumor activity both in vitro and in vivo.359,360 EGCG shows promising results for the inhibition of proteins such as Aβ, tau, αSyn, IAPP, TTR, and Htt; however, the mechanism involved is still unclear.
Another compound, rosmarinic acid has the potential to inhibit amyloid fibril generation and can destabilize preformed fibrils similar to curcumin.361
A large number of polyphenols have been reported, and some of their structures are shown in Fig. 6.
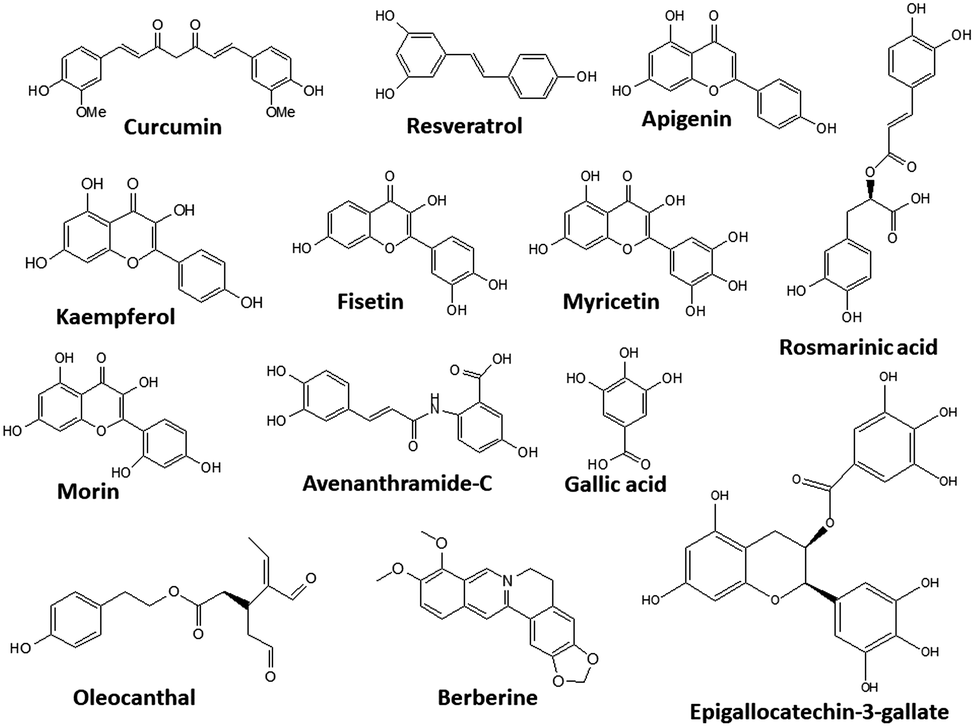 | ||
| Fig. 6 Chemical structures of some polyphenols. | ||
7.2 Polymeric inhibitors
The use of polymeric compounds in protein aggregation inhibition has generated interest only in the last decade, despite their tremendous potential. The molecular weight of polymeric inhibitors can be easily manipulated according to requirements, and the compounds can have various useful properties like flexible conformations (which may affect its interactions with biological molecules), higher viscosity, surface charge, and the incorporation of additional functionalities. In the next section, we describe polymer-based inhibitors of protein aggregation and have been summarized in Table 4.| Type of polymer | Probable mode of action | Key results | Ref. |
|---|---|---|---|
| PEG based | Strongly interacts with unfolded proteins due to dehydration of PEG | Nearly 80% enzymatic activity of lysozyme retained at 30 mM triangular PEG | 320 and 395 |
| Pullulan-based nanogels | Act as molecular chaperones | Nearly 20% ThT fluorescence at glucose unit/Aβ ratio of 250 | 396–398 |
| Late embryogenesis abundant (LEA) proteins | Act as molecular shield | Nearly 70% activity of LDH during desiccation at PvLEA-22/LDH molar ratio | 399–402 |
| Zwitterionic polymers | Act as molecular shield | 0.42% ThT fluorescence of insulin after 12 incubation with 1.5% poly-SPB and 30% BuMA | 322, 403 and 404 |
| Glycopolymers | Combines action of sugars (vitrification, water replacement, and water entrapment) and surfactant (molecular chaperones) | Almost 100% activity of β-Gal at 10 wt% equiv. of trehalose glycopolymers | 405 and 406 |
| Conjugated polymers | Strong hydrophobic interaction between polymer and proteins | Reduction in preformed amyloid plaques in brain slice culture (∼60%) | 407 and 408 |
In an important study by Kinbara and co-workers, the group synthesized monodispersed triangular PEG which suppressed the thermal aggregation of HEWL (Fig. 7).320 The polymer was synthesized using pentaerythritol and substituted oligoethylene glycol as the junction units using a multi-step reaction scheme. The addition of the resultant polymer (30 mM) helped in the retention of ∼75% enzymatic activity. Owing to the higher dimensional structure, triangular PEG undergoes dehydration and switches conformational state from gauche to anti-form on increasing the temperature, suggesting that dynamic changes take place in the molecule on heating. This results in increased hydrophobicity of PEG, as seen by the occurrence of dehydration at a lower temperature compared to linear PEG. The authors hypothesized that triangular PEG interacts strongly with thermally unfolded proteins, thus protecting them from aggregation-inducing collisions.
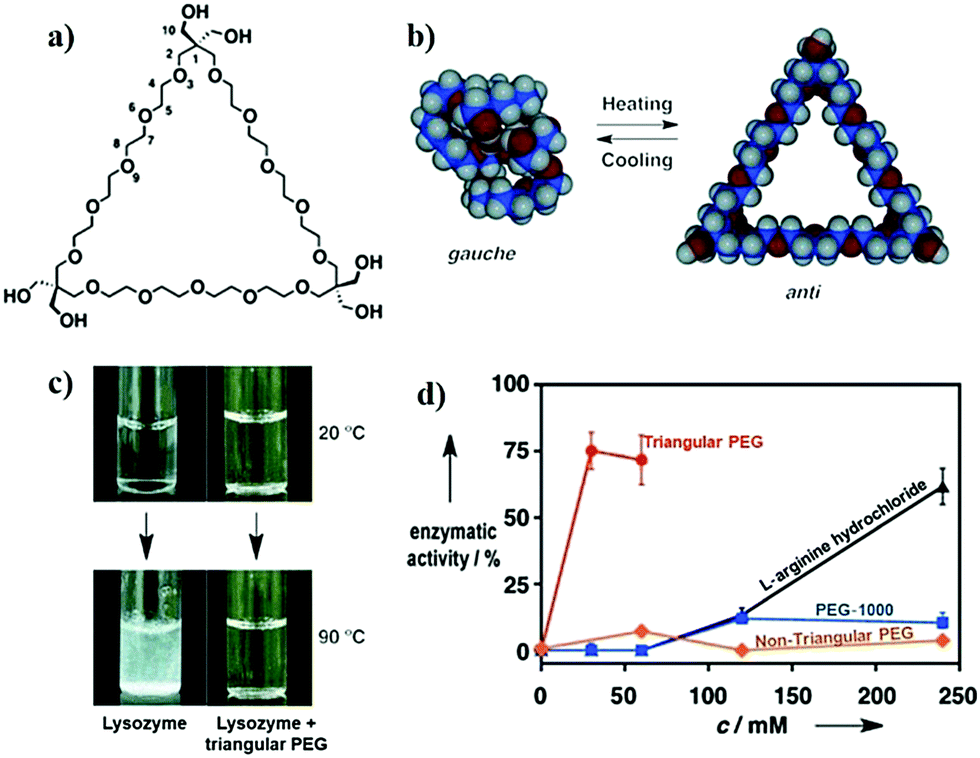 | ||
| Fig. 7 Protein aggregation inhibition by structured monodisperse PEG. (a) Molecular structures of triangular PEG; (b) schematic illustration of the heat-triggered conformational change from gauche to anti-form; (c) photographs of lysozyme and lysozyme + triangular PEG at 20 °C (upper panel), and after heating at 90 °C for 30 min (lower panel). Photographs clearly show aggregation of protein when heated in the absence of polymer, and the presence of polymer suppresses aggregation; (d) residual enzymatic activity of lysozyme heated in the presence of different additives, demonstrating that the topology of PEG significantly affects their properties. Reproduced with permission from ref. 320, copyright (2013), Wiley. | ||
In another study, Kameta et al. developed the functionalization of nanochannels of soft nanotubes with PEG.395 The resulting systems showed remarkable suppression of thermal aggregation of HEWL. Further, the denatured proteins could also be refolded by this system. Kameta et al. demonstrated that the activity results due to the hydrophobic interactions between the short PEG chains that have been dehydrated after heating at elevated temperature and the surface-exposed hydrophobic amino acid domains of HEWL. In the same year, Breydo and his team developed a hyper-branched PEG-based polymer which contained dopamine.409 The polymer synthesis was carried out using a one-pot RAFT polymerization of dopamine methacrylamide, PEG diacrylate, and PEG methyl ether acrylate. The resulting polymer suppressed fibril formation in αSyn, and the activity was attributed to the presence of dopamine which promotes the formation of oligomeric aggregates. These aggregates are resistant to further conversion into mature fibrils as they can form covalent adducts with αSyn, thus stabilizing the oligomeric form. Although the activity of the hyper-branched polymer was less than that of dopamine alone, the authors hypothesized that the ability to change polymer structure and composition may assist future studies examining the interaction between the polymer and proteins.
These studies show the immense potential of PEG-based polymers in suppressing the aggregation of proteins. Owing to their biocompatible nature, PEG-based polymers, particularly high molecular weight PEG, and PEG functionalized with known inhibitors may show remarkable properties and have potential use in clinical applications.
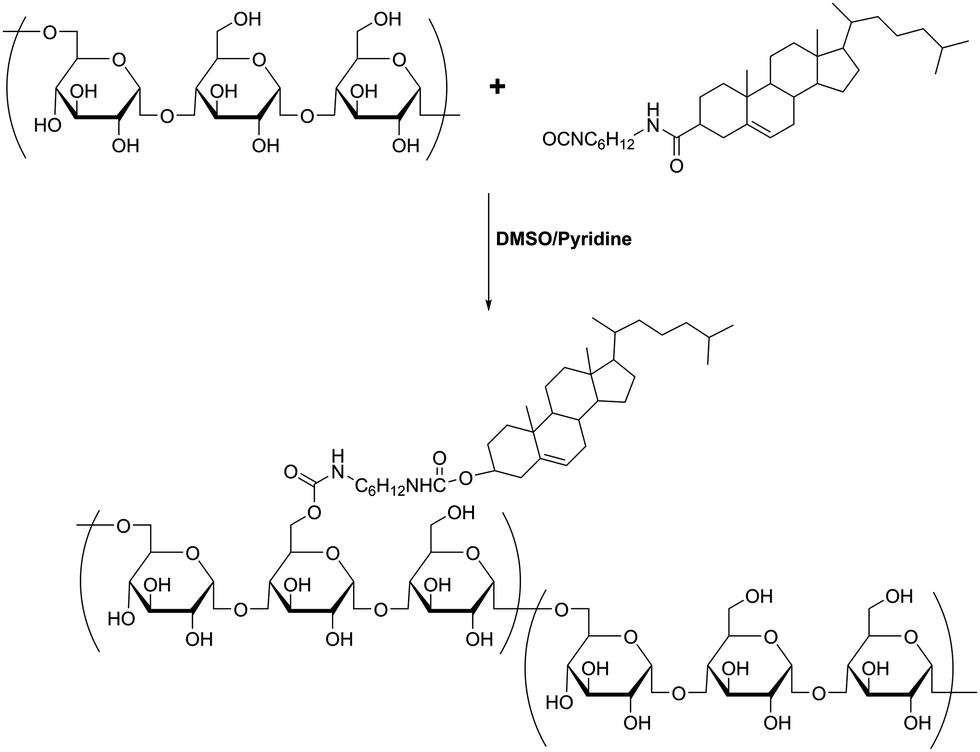 | ||
| Scheme 1 Synthetic route to prepare cholesterol pullulan (CHP). | ||
In subsequent studies, the authors demonstrated that these nanogels can encapsulate protein molecules.411,412 Studies with – CHP nanogels revealed that they can function as artificial molecular chaperones and prevent the aggregation of denatured proteins (Fig. 8a).398,413,414 In one study, Ikeda et al. showed that CHP can inhibit the formation of amyloid-like fibrils of Aβ-(1–42).396 The authors synthesized a positively charged CHP nanogel (CHPNH2) and compared its efficiency with neutral CHP nanogels. The results showed that the positively charged nanogel yielded greater activity compared to CHP, indicating that electrostatic interactions affect the activity to a great extent. Further, the proteins can be released from the nanogels by the addition of a cyclodextrin derivative (methyl-b-cyclodextrin; MβCD), which can bind to the cholesterol part of CHP, inducing the disassociation of CHP nanogels (Fig. 8b). The recovered proteins were examined and showed that fibril formation was completely suppressed due to the interaction of Aβ with the nanogels. In another study, Sawada et al. reported that CHP nanogels can act as artificial chaperones for horseradish peroxidase (HRP) and protects it against thermal denaturation.397 They also showed that CHP can assist in the refolding of denatured HRP, indicating that CHPs may be useful for both the protection and post-denaturation stages. Nochi et al. demonstrated that CHPNH2 can be used as a delivery vehicle for intranasal vaccine-delivery.415 To show its effectiveness, the authors administered a prototype vaccine antigen (subunit fragment of Clostridium botulinum type-A neurotoxin BoHc/A) incorporated into CHP. The neurotoxin was successfully absorbed by the mucosal dendritic cells after it was released from CHP nanogels and also induced the required antibody response. Thus, the study by Nochi et al. highlights the potential for the development of protein delivery vehicles using polymeric materials.
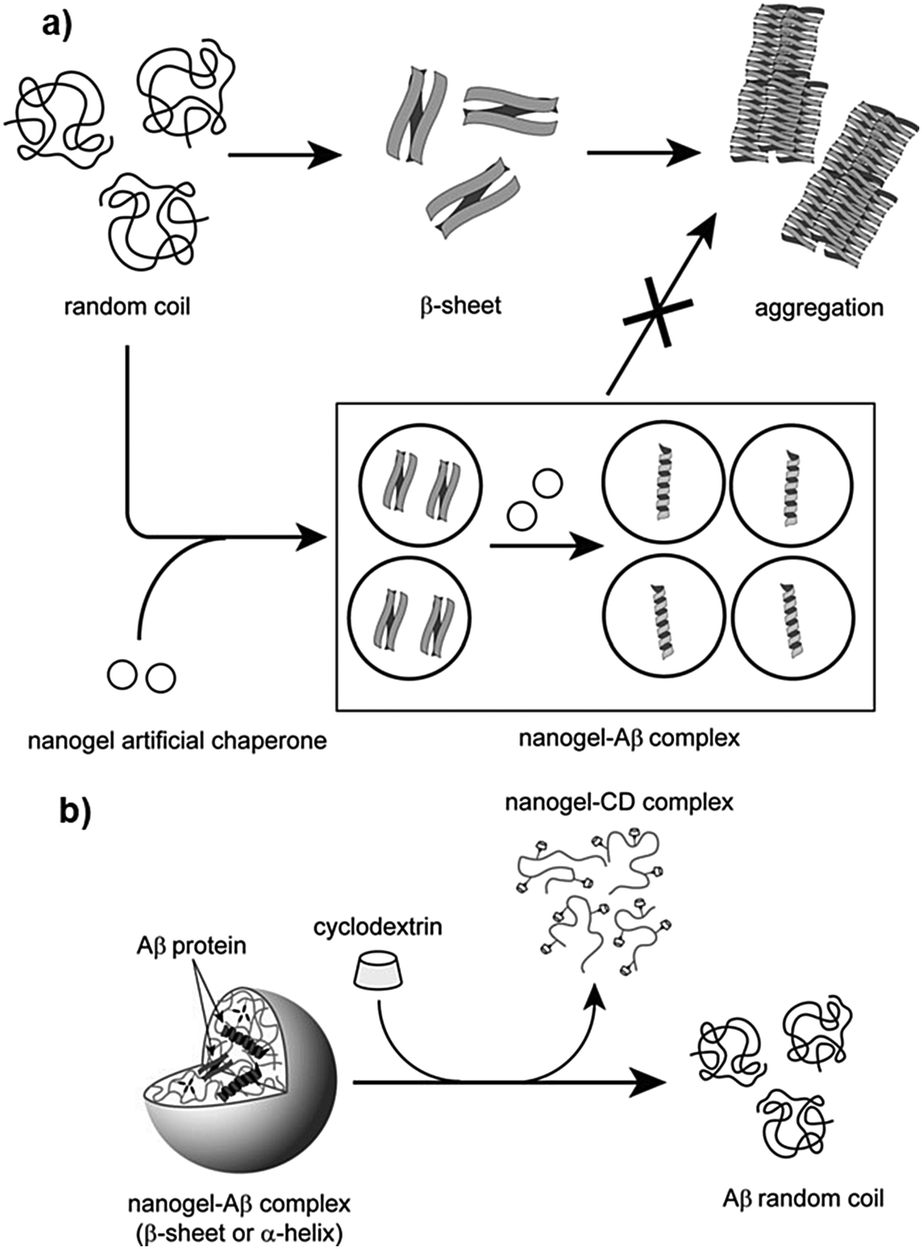 | ||
| Fig. 8 Schematic illustration of the interaction between CHP and proteins; (a) probable mechanism of protection of proteins by CHP nanogels via molecular chaperone function, and (b) releasing of proteins from CHP nanogels by MβCD. Reproduced with permission from ref. 396, copyright (2006), Elsevier. | ||
Multiple studies have demonstrated the ability of LEA proteins to protect biological entities such as proteins and membranes in desiccating environments.400,418–422 In one such study, Goyal et al. reported that recombinant forms of AavLEA1, a group 3 LEA protein, can protect against desiccation induced aggregation of citrate synthase (CS), and helps retain its activity.401 They hypothesized that owing to an unordered and flexible structure, LEA proteins can act as a molecular shield, thus forming a physical barrier to suppress any contact between CS molecules. Interestingly, they demonstrated that in the presence of trehalose, LEA proteins can protect CS against heat-induced aggregation (Fig. 9). Although LEA proteins alone do not possess such properties, a synergistic effect occurs in the presence of trehalose, causing LEA proteins to affect the unfolding pathway. In a similar study, Furuki and Sakurai showed that the group 3 LEA protein model peptide PvLEA-22 enables the retention of ∼70% activity of lactate dehydrogenase during desiccation.399
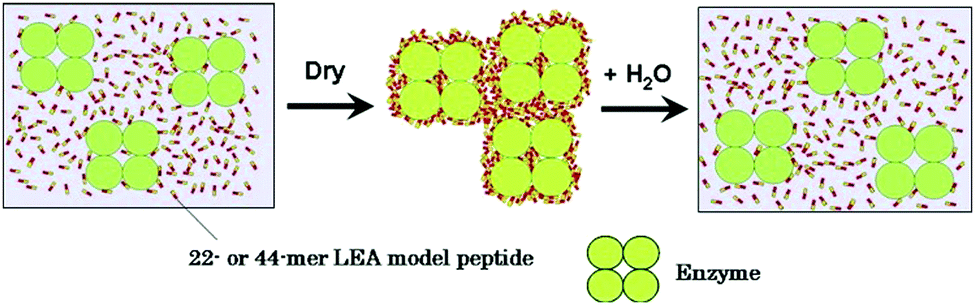 | ||
| Fig. 9 Schematic illustration of the chaperone-like activity of LEA proteins. Reproduced with permission from ref. 402, copyright (2016), Elsevier. | ||
The studies described above illustrate that LEA proteins decrease molecular collisions by acting as a molecular shield, but do not completely arrest the collisions, indicating that proteins will aggregate eventually, albeit at a slower rate. To corroborate this, Liu et al. studied the kinetics of polyglutamine (polyQ)-dependent protein aggregation (polyQ sequences Q23 or Q37) in the presence of LEA proteins. Their results showed that the anti-aggregation activity of LEA proteins in cells is time dependent. Further, by using the time-dependent Thioflavin T (ThT) analysis, Liu et al. observed that polyQ37 showed an increase in ThT intensity in 1 h, indicating the formation of polyQ37 aggregates, followed by a gradual increase in intensity till 5 hours, after which a plateau was reached (Fig. 10). In contrast, no increase was observed in the case of polyQ23. The incubation of polyQ37 with LEA proteins resulted in a significant reduction in polyQ aggregation in the initial phase, which later increased with a longer incubation time. These results suggested that LEA proteins reduce the rate of aggregation alone, and act as kinetic stabilizers of aggregating proteins.
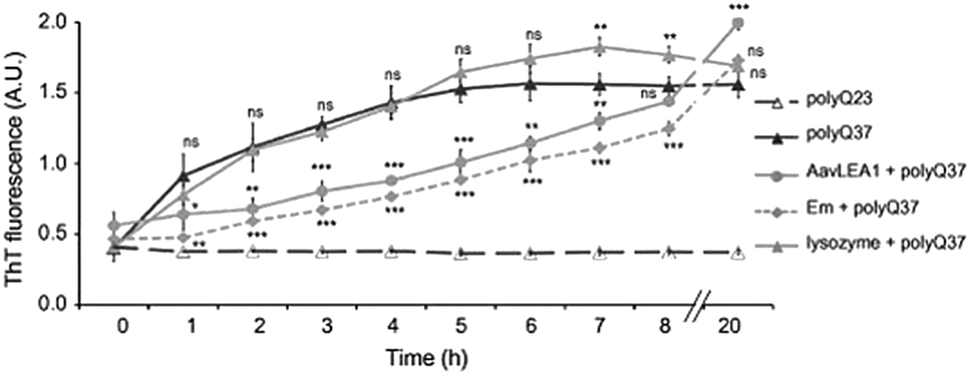 | ||
| Fig. 10 Time dependent ThT assay of polyQ37 aggregation in the presence of various proteins. polyQ23 and polyQ37 indicate different polyQ sequences. Em and AavLEA1 indicates group 1 and group 3 LEA proteins respectively. Lysozyme was used as a negative control. Reproduced with permission from ref. 416, copyright (2011), Elsevier. | ||
A study by Takao et al. showed that PvLEA-22 suppresses the heat-induced aggregation of lysozyme.417 Importantly, the peptide could facilitate the refolding of lysozyme on cooling, and ∼80% native catalytic activity was restored. Based on simulation studies, Takao et al. established that these peptide molecules act as a physical barrier and shield the collision of denatured lysozyme molecules, thus preventing aggregation. This is in good agreement with the previous studies which suggested molecular chaperone-like functions for LEA proteins.401,423,424 Interestingly, they suggested that a rapid exchange takes place between the peptide molecules attached to denatured proteins and they remain in contact with the protein for a short period of ∼200 ns, enabling the retention of protein properties and promoting refolding on the removal of heat-induced stress.
Since LEA proteins contain charged groups, they can be used as polyelectrolytes or charged macromolecules in mechanistic studies. Their ability to protect proteins under severe stress suggests that they can act as potential models to help develop synthetic polyampholytes or polyelectrolytes in the future for protein stabilization.
Zwitterionic polymers are excellent anti-biofouling agents, as they can maintain the water structure at the polymer-material interface.428,429 They have also been conjugated with proteins to improve the stability of proteins.430 In 2015, our group reported that poly-sulfobetaine (poly-SPB), synthesized via reversible addition-fragmentation chain transfer (RAFT) polymerization (Fig. 11a), shows a remarkably high efficiency in suppressing the thermal aggregation of HEWL (Fig. 12a).403 Additionally, the zwitterionic polymer could protect the higher-order structure of lysozyme from changing its conformation, and ∼85% activity was retained even after heating at elevated temperatures in the presence of poly-SPB (15% w/v). We hypothesized that owing to the anti-biofouling property of poly-SPB, zwitterionic polymers behave as molecular shields and suppress aggregation-inducing collisions between the hydrophobic domains of misfolded proteins, thus protecting proteins against aggregation. Increasing the molecular weight resulted in increased efficiency of aggregation inhibition, which could be attributed to the greater anti-biofouling ability compared to polymers with lower molecular weight.431
 | ||
| Fig. 11 Structure of the various polymers prepared with sulfobetaine via RAFT polymerization; (a) poly-SPB homopolymer; (b) poly-(SPB-r-BuMA) copolymer; (c) core–shell nanogel; (d) graft polymer. | ||
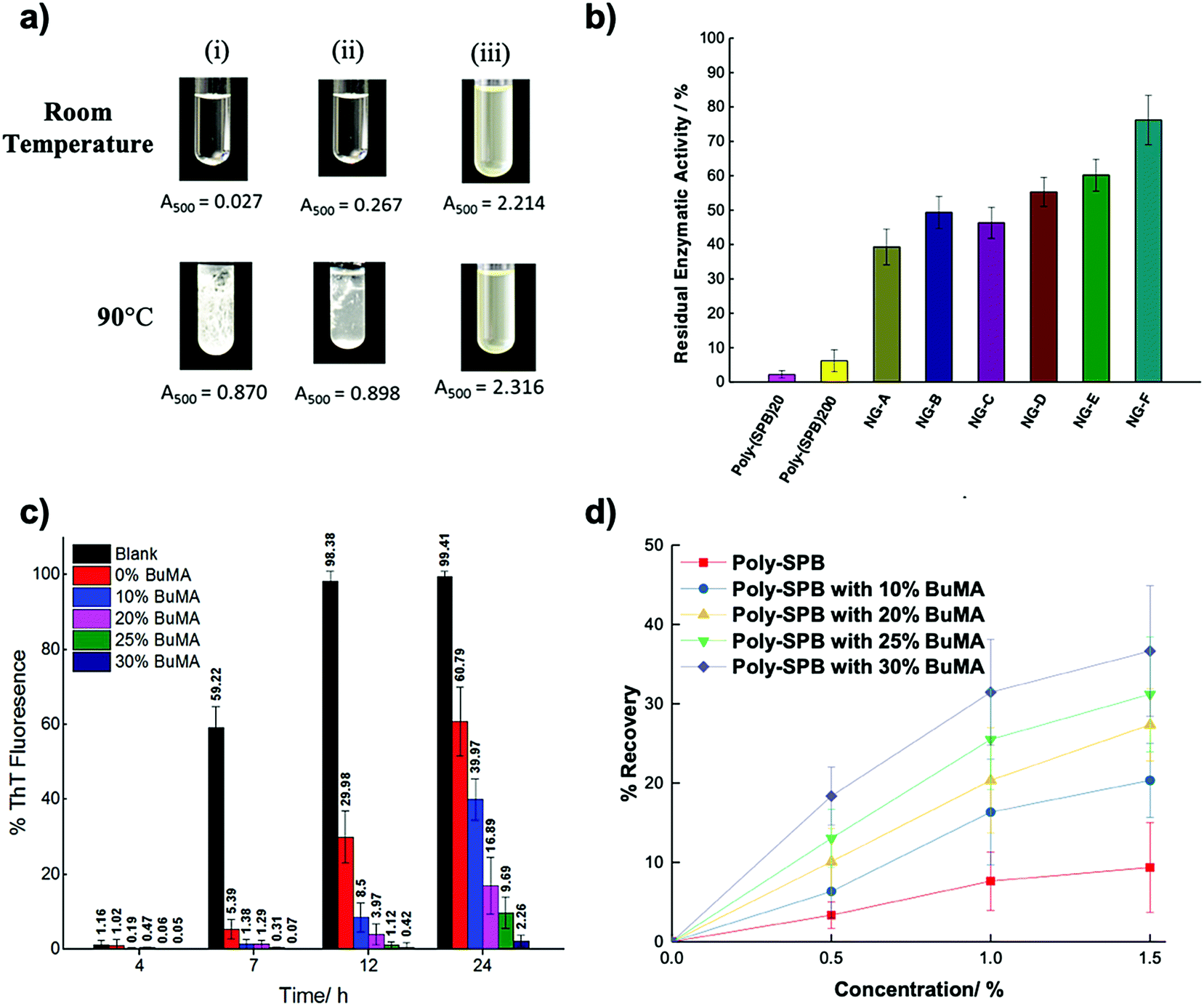 | ||
| Fig. 12 Protein aggregation inhibition by zwitterionic polymers. (a) Photographs of lysozyme (i) without any additive at room temperature (top) and after heating at 90 °C for 30 min, (ii) with poly-SPB. Numbers below indicate absorbance at 500 nm; (b) residual enzymatic activity of lysozyme after heating at 90 °C for 30 min in presence of various additives. NG-A to F indicates various nanogels with degree of hydrophobicity and shell size; (c) ThT assay of insulin with poly-SPB derivates; and (d) recovery yield of insulin calculated from ThT assays for analysing refolding efficiency. (a) Was reproduced from ref. 403 with permission from The Royal Society of Chemistry, (b) was reproduced from ref. 404 under Creative Commons CC BY; (c) and (d) were reproduced with permission from ref. 322, Copyright (2018), Wiley. | ||
Although the results were satisfactory, a high polymer concentration was required to obtain high protein protection. To further improve the efficiency of these copolymers, we transformed the linear polymer to a core–shell nanogel. The nanogels were prepared by utilizing the end group of the RAFT agent and using poly-SPB as a macro-RAFT Agent (macro-CTA). Next, a radical cross-linker and SPB monomers were polymerized in the presence of macro-CTA to yield a core–shell nanogel (Fig. 11b). These nanogels were synthesized with different degrees of hydrophobicity in the core, and had different shell sizes (NG-A to NG-F, where the molecular weight of the polymer in the shell and the degree of hydrophobicity increases from A–F). The nanogel was found to protect lysozyme with a higher efficiency relative to the linear polymer, possibly because of the smaller size (Fig. 12b), resulting in greater shielding from aggregation-inducing intermolecular interactions.404 Increasing the incorporation of hydrophobic monomers also yielded higher protection efficiencies, probably due to the proclivity of hydrophobic moieties to cover the hydrophobic domains of proteins. Preliminary mechanistic investigations revealed that lysozyme loses its higher-order structure on prolonged heating at elevated temperatures, as indicated by the disappearance of signals representing various amino acid residues (Fig. 13a). In contrast, when nanogels were added to lysozyme before heat treatment, all the peaks identified had nearly equal intensity, suggesting that the nanogel allowed the protein to retain its higher-order structure under severe stress.
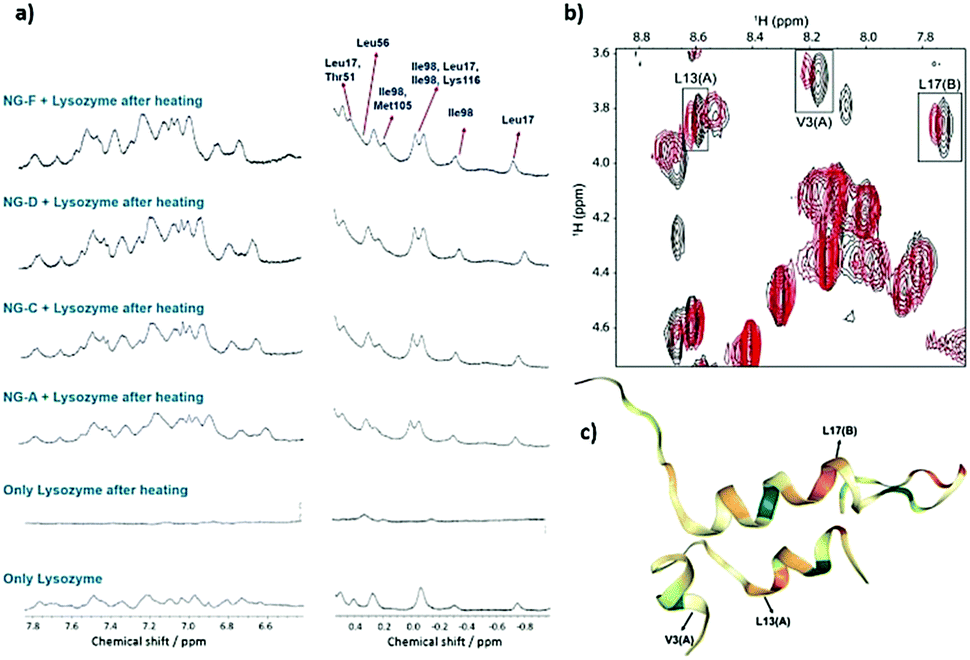 | ||
| Fig. 13 Mechanistic investigation of protein aggregation inhibition by zwitterionic polymers by NMR. (a) 1H-NMR investigation of lysozyme with and without additives. NG-A to F indicates various nanogels with degree of hydrophobicity and shell size; (b) 1H–1H TOCSY for native insulin (black) and insulin and hydrophobic derivative of poly-SPB; (c) MD ensemble of insulin by hydrophobicity. (a) Was reproduced from ref. 404 under Creative Commons CC BY; (b) and (c) were reproduced with permission from ref. 322, Copyright (2018), Wiley. | ||
This was followed by another study where poly-SPB was copolymerized with a hydrophobic monomer (butyl methacrylate; BuMA) to obtain random copolymers with different degrees of hydrophobicity (Fig. 11c).322 The aggregation of insulin was suppressed by the addition of the copolymers, and no turbidity could be seen even after prolonged incubation at 37 °C for 24 h, which would otherwise aggregate within 7 hours of incubation. The addition of small doses of hydrophobic monomers to the homopolymer of poly-SPB increased the protection efficiency significantly. Further, a ThT assay revealed that the formation of amyloid-like fibrils was prevented, and less than 3% fibrillation was observed after a 24 h incubation (Fig. 12c). CD spectroscopy revealed that secondary structural elements of insulin were retained by the addition of zwitterionic polymers. To investigate the underlying mechanism, 1H–1H TOCSY spectra were recorded using WATER suppression by GrAdient Tailored Excitation (WATERGATE), to suppress the resonance signals from the solvent. The spectra revealed that these hydrophobic poly-SPB polymers interact with the hydrophobic domains of insulin (Valine-3 and Leucine-13), possibly preventing intermolecular interactions between these domains of structural subunits that may lead to the formation of aggregates (Fig. 13b and c). These polymers were also found to facilitate the refolding of insulin by ∼40% (Fig. 12d). This opens new avenues for the development of efficient protein protection and refolding agents, which may be used to prevent and cure numerous neurodegenerative disorders.
Further, we developed a graft polymer where poly-SPB was grafted onto the backbone of carboxylated ε-poly-L-lysine (PLL-SA).432 This was done in a multi-step reaction scheme where PLL-SA was first synthesized by adding succinic anhydride to ε-PLL, followed by the controlled substitution of a RAFT agent on PLLSA by acid–amine coupling to obtain the ε-PLL-SA-macro RAFT agent. Finally, this macro RAFT agent was used to polymerize the sulfobetaine monomer to obtain the graft polymer (Fig. 11d). ε-PLL-SA is a well-known cryoprotectant and was selected due to its membrane-stabilizing property.433 The graft polymer showed a greater propensity to protect lysozyme against thermal aggregation compared to the homopolymer, and it could efficiently bind to proteins electrostatically and release it on a change in the pH; i.e., it was capable of exhibiting pH responsive protein delivery. Thus, these systems may have potential to be used for the safe delivery of therapeutic proteins and protect these proteins against denaturation.
In a study by Zhang et al., a zwitterionic gel was developed which encapsulated proteins and provided them stability.434 Proteins were encapsulated in a hydrogel nanoparticle by first introducing an acryloyl group on their surface, followed by an in situ free radical polymerization reaction using carboxybetaine monomer and a carboxybetaine cross linker. A highly immunogenic fungal-derived uricase was used for studying the encapsulation efficiency, and the results showed that encapsulated proteins had better thermal stability and showed greater enzymatic activity after being subjected to heat treatment. Moreover, its efficiency was compared with PEGylated uricase and it was found that the zwitterionic polymer encapsulated enzyme had better stability and showed a superior pharmacokinetic profile.
Certain polymers like polyvinyl alcohol (PVA) and polyampholytes exhibit ice re-crystallization inhibition (IRI activity) (Fig. 14a) and are effective in cryopreservation of cells.427,435,436 Towards this end, Mitchell et al. showed that polymers exhibiting IRI can also protect proteins against freeze-induced stress.437 They analysed different IRI active and inactive polymers (PEG, hydroxyethyl starch (HES), and polyvinylpyrrolidone (PVP)) for their ability to protect proteins against stress (Fig. 14b). Trehalose was used as a control, owing to its well-known efficiency in protecting proteins,438 whereas PEG was also selected due to its cryoprotective property. The results showed that polymers alone did not exhibit any protecting activity; however, the mixture of PEG and PVA yielded higher protein recovery, similar to that to trehalose (Fig. 14c). Similarly, polyampholytes mixed with PEG also yielded very high protein recovery (Fig. 12d). These results suggest that the high efficiency of these polymers to stabilize proteins during freezing is linked to their high IRI activity, which in turn is due to the inhibition of irreversible aggregation.
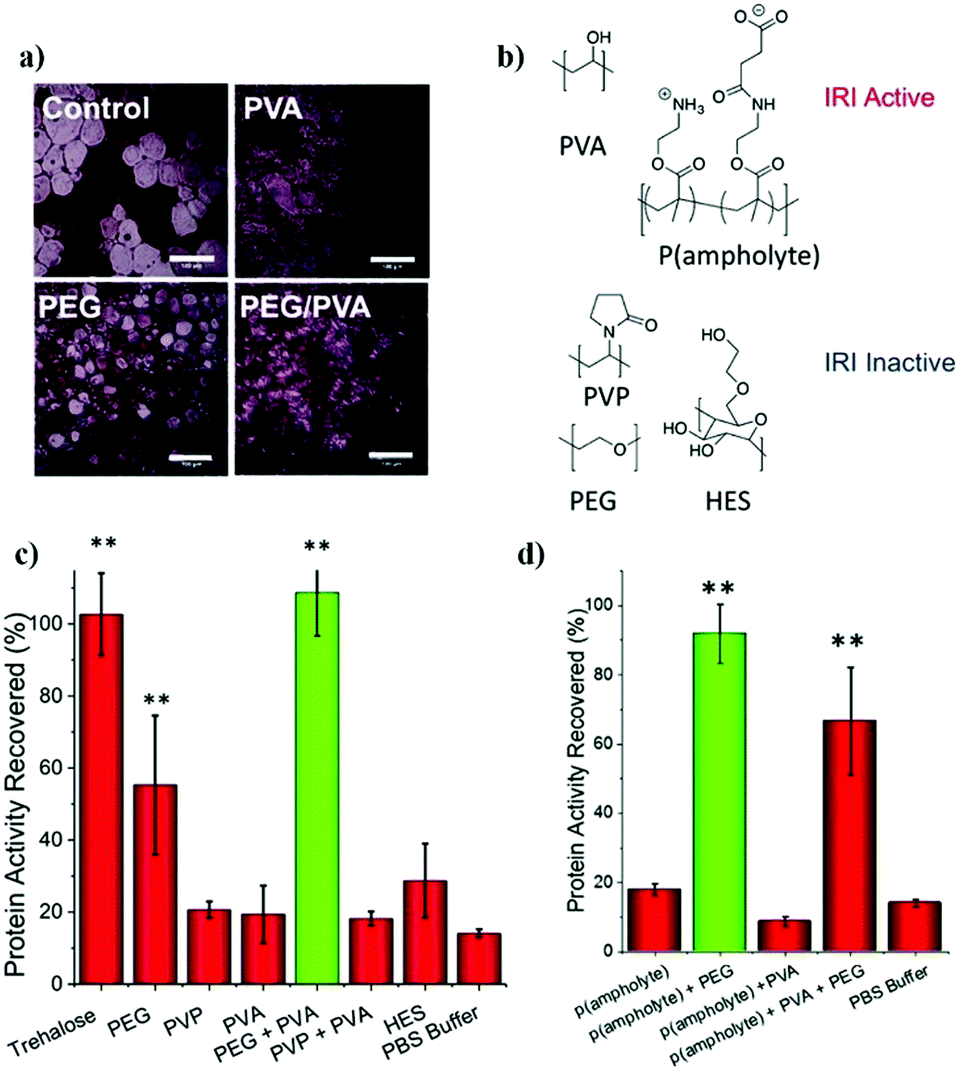 | ||
| Fig. 14 Protein aggregation inhibition with IRI polymers. (a) IRI activity of different polymers (scale bar 100 μm), (b) chemical structures of various polymers, (c) and (d) residual activity (recovery) of β-galactosidase after freezing at −20 °C for 3 days in the presence of various polymers. Reproduced with permission from ref. 437 under Creative Commons Attribution 3.0 Unported Licence. | ||
In another study, Sofronova et al. showed that polyelectrolytes bearing the same charge as the protein (at a particular pH) suppress protein aggregation to the greatest extent.439 They studied a polycation and a polyanion along with different proteins at different pH values (higher and lower than the isoelectric point of the proteins). The results showed that suppression of aggregation was most effective when the charge of the polymer was the same as the surface charge of the protein. When the charge of the polymer was opposite to that of the protein, it resulted in the formation of large protein aggregates. Through molecular dynamic simulation studies, the authors postulated that protein protection is maximal when the electrostatic binding was the least (i.e., only a small part of the polymer chain interacts with the protein surface). The remaining part of the polyelectrolyte forms charged loops and tails around the protein surface, and the loop size determines the aggregation inhibition propensity. Sofronova et al. hypothesized that this loop provides solubility to the complex. However, concerns regarding the toxicity of the polycations remain, which may create hurdles for in vitro or in vivo studies. Further, it remains to be seen whether the hypothesis can be applied to all kinds of polyelectrolytes.
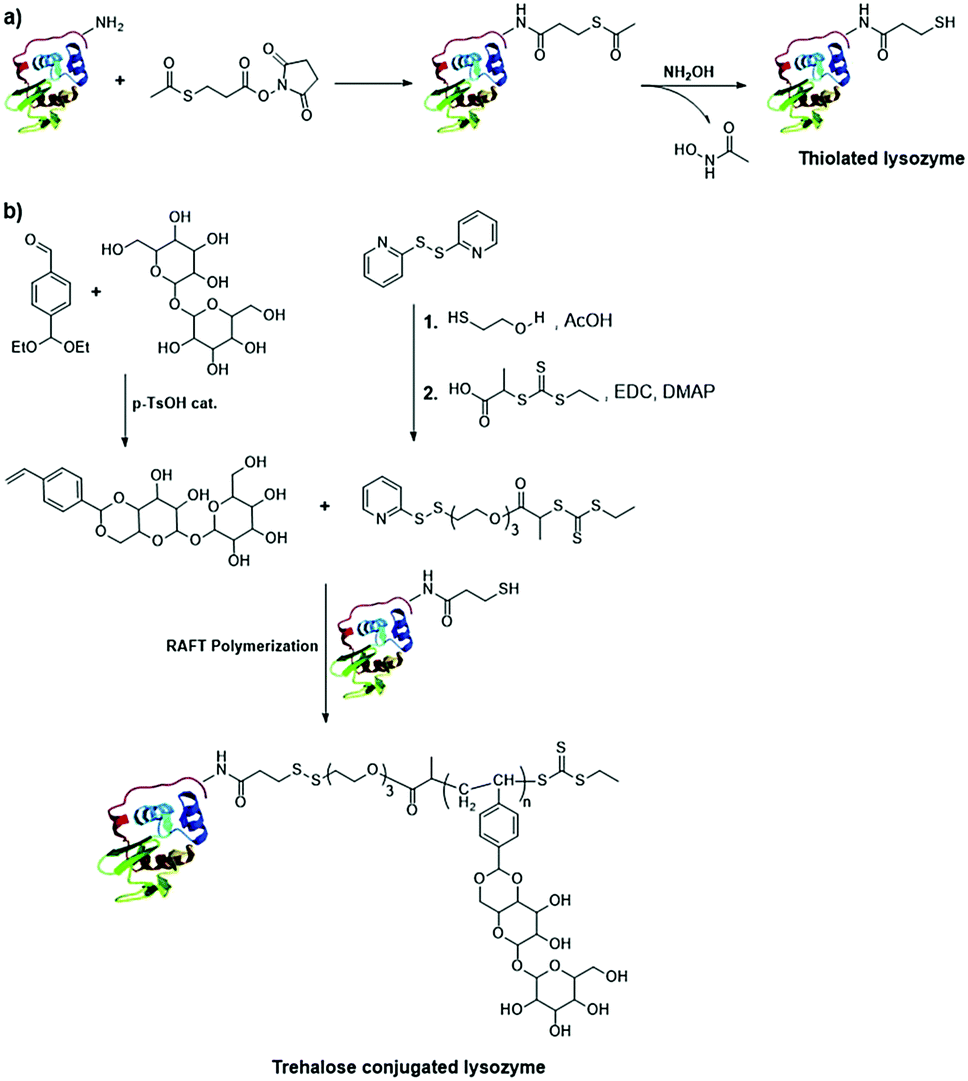 | ||
| Scheme 2 Schematic representation of the conjugation of trehalose with lysozyme. (a) Preparation of thiolated lysozyme by treatment with SATP, and (b) RAFT polymerization to produce trehalose-conjugated lysozyme. | ||
In another study, the authors used trehalose glycopolymers as excipients for stabilizing proteins.406 Here, trehalose was modified with reagents such as styrenyl acetal (P1), methacrylate acetal (P2), styrenyl ether (P3), and methacrylate (P4), and further subjected to free-radical polymerization. To analyse the activity, horseradish peroxidase (HRP) and β-galactosidase (β-Gal) were used, and the enzymes were subjected to heating and multiple lyophilization cycles, respectively. The protein activity in the presence of additive was found to be much higher in both cases, relative to no additive controls. These polymers were found to be non-toxic when used with several cell lines, indicating their potential as additives to proteins to protect them from denaturation under various stresses.
A study by Debnath et al. demonstrated that poly-trehalose nanoparticles suppress amyloid aggregation.440 The nanoparticles used consisted of an iron oxide core and a zwitterionic shell, which was introduced by adding amino and acrylate monomers, which were then polymerized to form polymer-coated nanoparticles. Trehalose was introduced next by conjugating the amine groups with trehalose monocarboxylic acid via EDC coupling to yield poly(trehalose) nanoparticles (Fig. 15a). They tested the efficiency of these nanoparticles in inhibiting Aβ fibrillation, and their results showed that these nanoparticles were 1000–10000 times more effective in suppressing fibrillation compared to trehalose (Fig. 15b–d). The efficacy of the nanoparticles was also tested on a transgenic mouse with Huntington's disease. Treatment with the poly(trehalose) nanoparticles reduced the number of mutant huntingtin aggregates in different regions of the brain. Further, there was no lethal effect of these nanoparticles on mice. The authors studied the iron content in the brain after administration of the nanoparticles and found a 3.5× increase in the iron content, indicating that these nanoparticles can cross the blood–brain barrier. These findings indicate that zwitterionic nanoparticles may be effective in overcoming amyloid-derived neurodegenerative diseases.
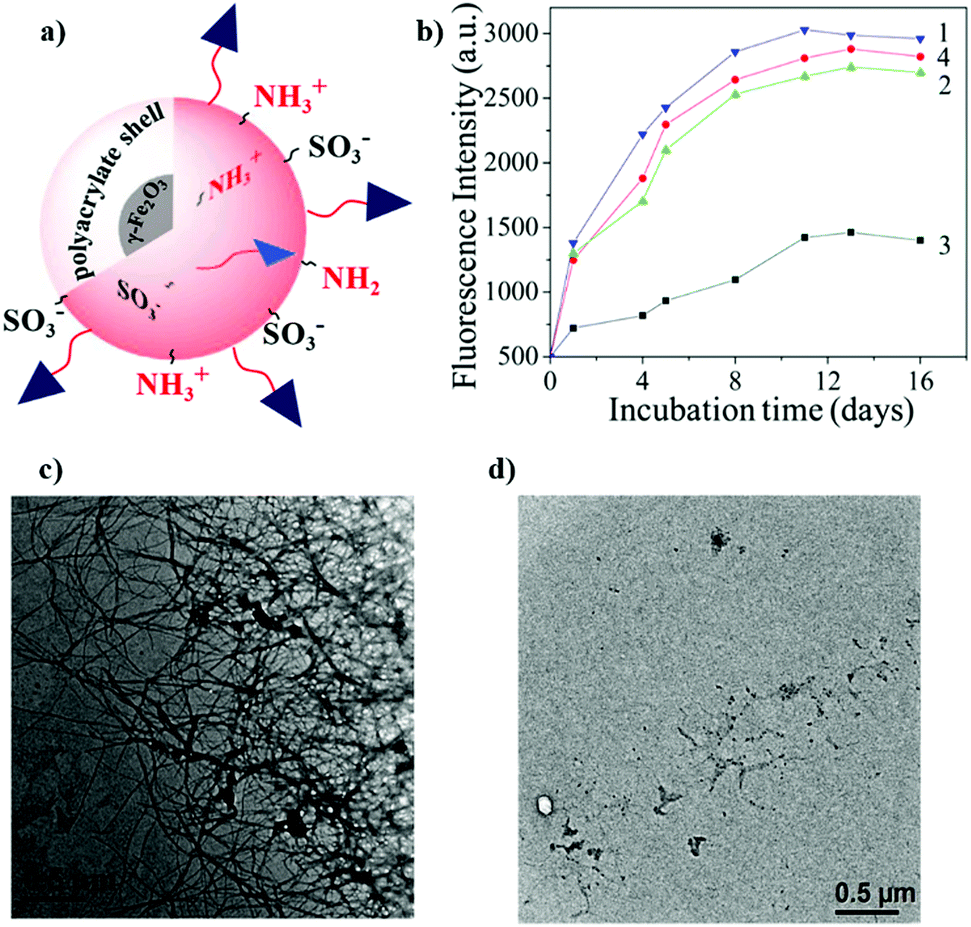 | ||
| Fig. 15 Inhibition of protein aggregation. (a) Schematic representation of poly(trehalose) nanoparticles, (b) ThT assay for inhibition of Aβ aggregation in the presence and absence of various reagents; 1, 2, 3 and 4 indicate the absence of additives, the presence of nanoparticles with 5 μm trehalose, 5 μm molecular trehalose, and 5 mm molecular trehalose, respectively; (c) and (d) show the TEM image of the Aβ fibrils in the absence and presence of nanoparticles, respectively. Reproduced with permission from ref. 440 copyright (2017), American Chemical Society. | ||
In a study by Mantovani et al., linear and star-shaped (four-arm) glycopolymers with different saccharides like galactose, trehalose, mannose, arabinose, and trehalose,441 were used as excipients. The results showed that these glycopolymers could modulate the colloidal and conformational stability of a model mAb.
Chai et al. prepared water-soluble conjugated polymers to detect and inhibit the UV-light induced aggregation of lysozyme.408 The polymer (PPV-NMe3+) was synthesized using a divinylbenzene derivative and a diiodobenzene derivate using a Heck coupling reaction, followed by quaternization with trimethylamine (Scheme 3).
 | ||
| Scheme 3 Synthesis of PPV-NMe3+ from conjugated diiodo and divinyl compounds via a Heck coupling reaction. | ||
The authors reported that upon UV illumination, HEWL initially forms granular aggregates, followed by self-assembly into globule-like aggregates. However, when PPV-NMe3+ is added to HEWL prior to UV illumination, the polymer attaches to the surface of the aggregates formed and disrupts the π–π stacking of PPV-NMe3+, resulting in increased fluorescence intensity. As the size of the aggregates grows, the hydrophobic interaction between the polymer and HEWL increases, resulting in the enhancement of fluorescence intensity which can be visualized by a change in colour. They argued that the technique can be used to probe the aggregation of proteins. Additionally, owing to the strong hydrophobic interaction between HEWL and the protein, the aggregation of HEWL is suppressed by the polymer (Fig. 16).
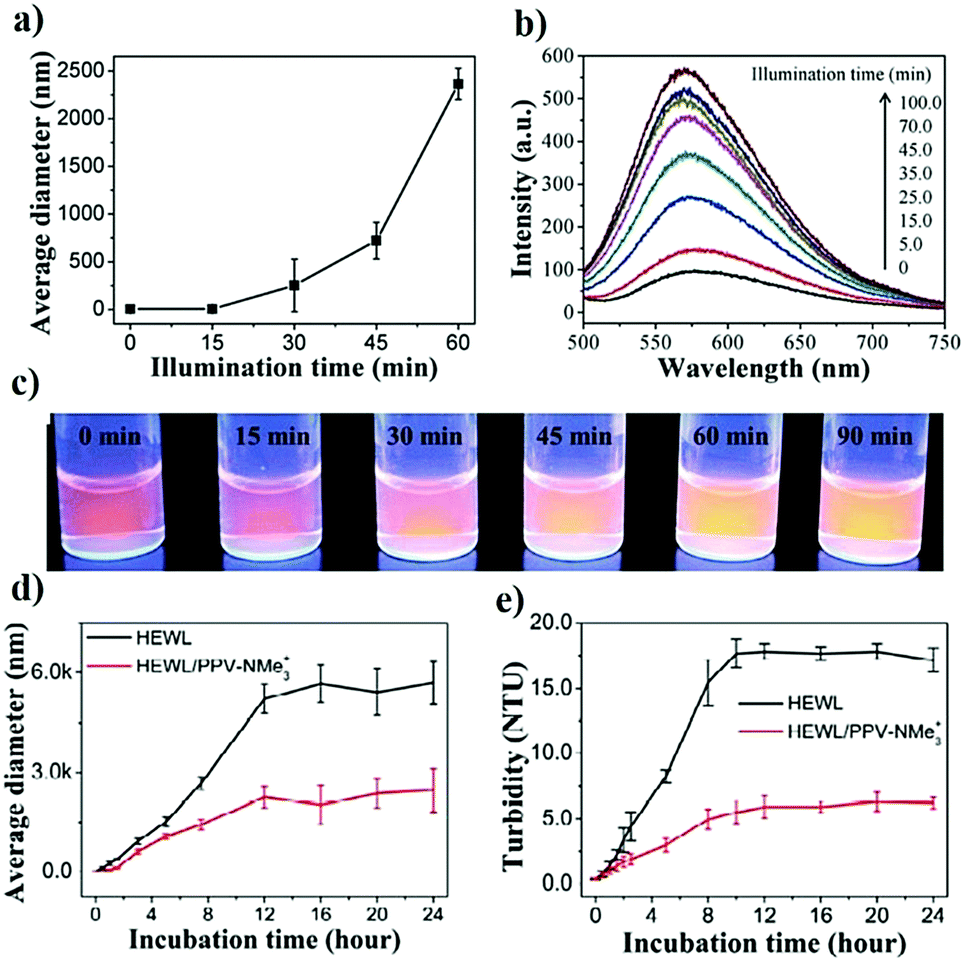 | ||
| Fig. 16 Detection and inhibition of protein aggregation by the addition of PPV-NMe3+. Detection of aggregation after UV illumination by (a) DLS analysis of HEWL alone, (b) fluorescence spectra of PPV-NMe3+ in the presence of lysozyme samples, (c) fluorescence imaging of PPV-NMe3+ in the presence of UV illuminated lysozyme samples. The suppression of aggregation by the addition of PPV-NMe3+ to HEWL prior to UV illumination was studied by (d) DLS, and (e) turbidity analysis. Reproduced with permission from ref. 408 copyright (2016), Wiley. | ||
In another study, Sun et al. developed similar amphiphilic conjugated polymers for suppressing amyloid formation.407 They functionalized the polymer using p-nitrophenyl esters (PPV-NP), which allows the attachment of the polymer to lysine residues of the protein. The authors reported that PPV-NP selectively reacts with amyloid species over other proteins due to the synergistic effects of hydrophobic and covalent reactions. They suggested that PPV-NP inhibits Aβ42 aggregation because of the covalent linkage between the polymer and protein, which blocks the hydrophobic cores of Aβ42 and prevents secondary structure conversion and polymerization of Aβ42. PPV-NP suppresses the formation of aggregates and also eliminates the existing amyloid plaques (Fig. 17a). They checked its efficiency using brain slices from transgenic mice and found that brain vibrosections treated with the polymer showed fewer Aβ plaques and decreased the plaque area (Fig. 17b). This result highlighted the potential of conjugated polymers to suppress aggregation and allow the removal of already aggregated species.
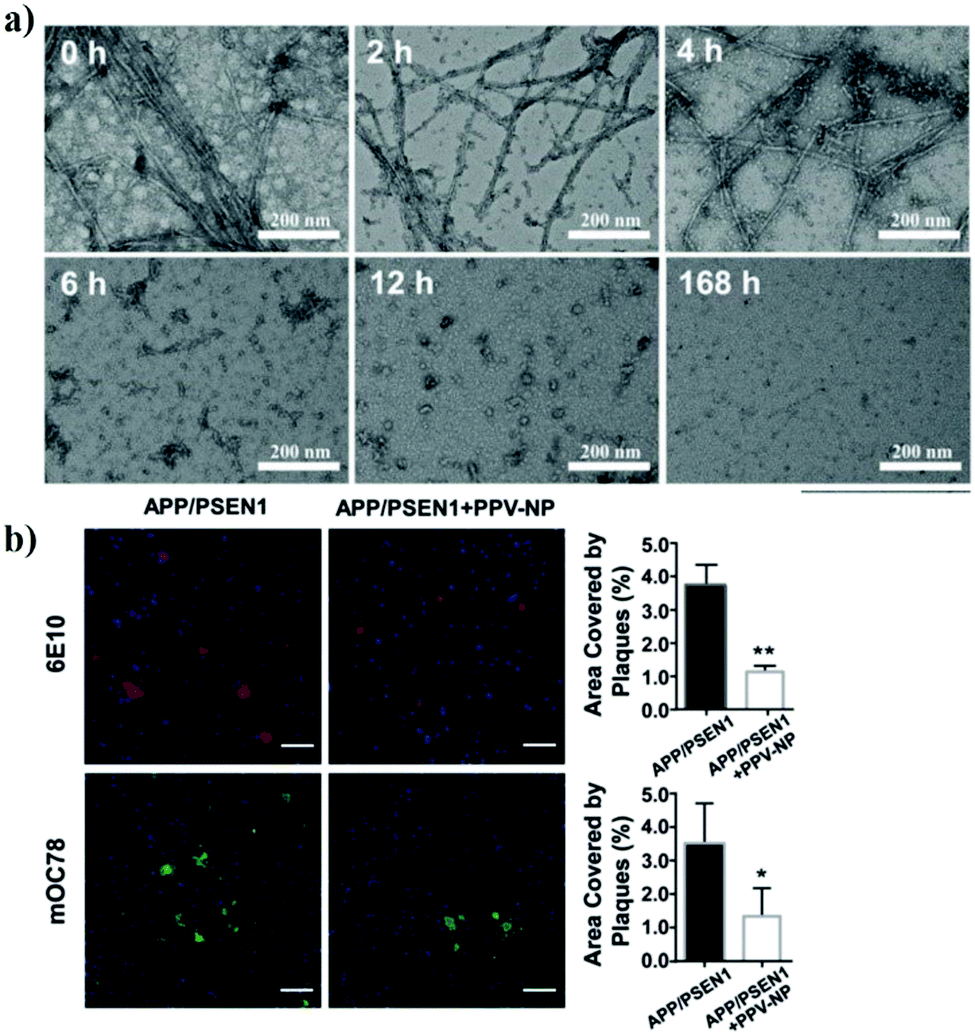 | ||
| Fig. 17 Elimination of existing amyloid plaques. (a) TEM analysis of preformed Aβ42 fibrils mixed with PPV-NP for different times, and (b) representative images of 6E10 (red) and mOC78 (green) immunoreactive Aβ plaques and their quantification in the cortical fields of transgenic mice after treating with and without PPV-NP for 10 h by ex vivo cultures. Reproduced with permission from ref. 407 copyright (2019), Wiley. | ||
In another study, the same group developed similarly conjugated polymers functionalized with an N-hydroxysuccinimide ester and a pentafluorophenol ester.446 These polymers suppressed the aggregation of islet amyloid polypeptide (IAPP). Further, these polymers also could irreversibly remove preformed IAPP fibrils, via the mechanism proposed in their previous study.
These studies clearly show the potential of conjugated polymers in inhibiting protein aggregation and enabling the refolding of proteins. Additional studies need to be conducted with the conjugated polymers, using different functional groups or by attaching other known inhibitors to obtain a synergistic effect. However, a drawback these systems is their inherent non-biodegradability. Studies to effect degradation should also be carried out, which will enable the use of these systems for clinical applications.
8. Refolding
Protein refolding is a process by which proteins can attain their native structure, and it is established by preventing the stabilization of misfolded proteins or by destabilizing monomeric intermediates and β-sheet oligomers. Protein refolding is as important as suppressing protein misfolding, and is a vital step in the production of recombinant proteins.447 Refolding of misfolded/unfolded/solubilized proteins into their native conformation is a pre-requisite to procure a biologically active form.Another key application of refolding proteins is for the cure of neurodegenerative diseases. Certain neurodegenerative diseases such as Alzheimer's are associated with the formation of amyloid plaques,448 that are formed by the aggregation of proteins. It is believed that the deposition of amyloid plaques damages the nerve cells by a mechanism that is not well understood. However, the removal/solubilization of amyloid plaques can aid in the cure of multiple neurodegenerative diseases.
In protein misfolding disorders (PMDs), the secondary and tertiary structure of a typical protein is altered without changes in its primary structure. The conformational change may promote the occurrence of diseases due to the toxic activity of the protein or the abrogation of biological functions of the natively folded proteins.449,450 In most PMDs, the misfolded protein consists of β-sheet conformation.449,451 In α-helices, the hydrogen bonds are present between groups that are within the same strand, while in β-sheets, the bonds occur between two different strands. Since the second β-strand belongs to different regions of the same protein or a different molecule, the formation of these β-sheets is generally stabilized via protein oligomerization or aggregation. In most of PMDs, the misfolded protein undergoes self-association and is deposited as amyloid-like aggregates in different organs, leading to tissue damage and organ dysfunction.452
Thus, materials that can stabilize the native structure of a protein by suppressing the aggregation of proteins and facilitating the refolding process of denatured proteins are of great interest. Therapies directed towards diseases caused by misfolded proteins should aim to reverse or inhibit the conformational changes in the structure of proteins. Therefore, compounds or methods that assist protein folding systems, the destruction of misfolded aggregates, or those which prevent the misfolding of proteins may prove to be efficient in treating such diseases.
8.1 Refolding kinetics
Proteins undergo different unstable conformations before reaching their final native structure. In the absence of chaotropic operators, these intermediate conformations may show intermolecular communication, leading to the accumulation and precipitation of proteins and difficulties in refolding them. In refolding models that approximate the final yield from the renaturation procedure, these side reactions are high order reactions, while the folding reaction is estimated by a first-order reaction.453A refolding reaction can be expressed as follows:
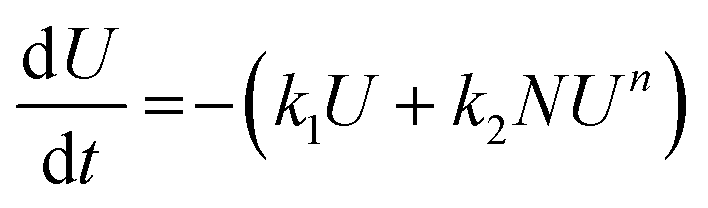
where Y(t) is the yield of the refolding reaction, U0 is the initial concentration of the denatured protein, and K2 is the rate constant of aggregation k2N.
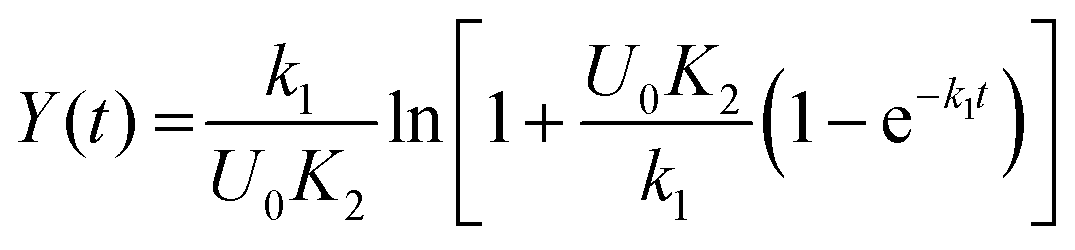
| Y(t) = Ψ{tan−1[(1 + Ψ2)e2k2t − 1]1/2 − tan−1Ψ} |
8.2 Determination of refolding conditions
For the isolation of recombinant proteins, enzymes are used to lyse cells, leading to the degradation of cell walls. Inclusion bodies are isolated through mechanical disruption of cells, followed by centrifugation, suspension of the cell debris and protein products via high-pressure homogenization, and sedimentation of inclusion bodies.456 A conical plate centrifuge is used for the centrifugal process to perform separation of inclusion bodies based on their spectrophotometric properties. In 1997, Wong et al. showed that repeated homogenizer passes increase the purity of the inclusion bodies upon better fractionation of cell debris and inclusion bodies.457Falconer et al. later suggested a chemical method through which the selective extraction of inclusion bodies can be achieved to produce recombinant proteins. The method involves the use of a urea and EDTA solution to permeabilize cell membranes for extraction of proteins from host cells, and the use of reagents that promote disulphide linkages to keep the inclusion bodies insoluble. Once the extraction compounds are removed, chaotropic and reducing conditions are used to solubilize the inclusion body protein.428 In another approach, a solution of EDTA and Triton X-100 was used to disintegrate cells while the inclusion bodies remained insoluble.458
8.3 Refolding techniques
A solubilization strategy must be formulated before proceeding with the refolding reaction. Starting from the isolation of inclusion bodies, most techniques aim at achieving the native structure of the protein, i.e. the unfolded state by using chaotropic agents such as guanidinium chloride (GdmCl) or urea at high concentrations in combination with reducing agents. Depending on the protein, the presence of some amount of native structure within the inclusion bodies could lead to higher yields. Additionally, high yields can be attained using detergents,459 buffers (at high pH),460 GdmCl, arginine,461 and sodium hydroxide462 as solvents.Further, refolding must be initiated by the removal of denaturants and providing conditions that allow intramolecular interactions to occur, leading to the formation of the correct protein structure. However, this strategy has no general applicability as it varies with different proteins. Some of the compounds widely used for protein refolding are presented in Table 5.
| No. | Additive | Protein | Recovery yield (%) | Conditions | Ref. |
|---|---|---|---|---|---|
| (1) Small molecules | |||||
| (i) | Guanidinium chloride | P. fluorescence lipase | 90 | 0.70 M with no optimum protein conc. | 470 |
| HEWL | 95 | 1.25 M with 1 mg mL−1 HEWL | 455 | ||
| Carbonic Anhydrase II | 96 | 1.0 mM with 0.50 mg mL−1 CA II | 483 | ||
| (ii) | Urea | Porcine growth hormone | 85 | 3.5 M with 05 mg mL−1 PGH | 484 |
| HEWL | 40 | 4 M with 20 μg mL−1 HEWL | 485 | ||
| (iii) | L-Arginine | HEWL | 94 | 0.75 M with 1 mg mL−1 HEWL | 455 |
| (iv) | Sulfobetaines | HEWL | 35 | 1.8 M with 11.2 mg mL−1 HEWL | 367 |
| (2) Sugars | |||||
| (v) | Glycerol | HEWL | ≥80 | In presence of 50% glycerol with 20 μg mL−1 HEWL | 485 |
| (vi) | N-Acetyl glucosamine, glucose, sarcosine | HEWL | 60–80 | 0.5 M, 1 M, 4 M resp. | 485 |
| (3) Short chains alcohols | |||||
| (vii) | n-Pentanol, n-hexanol, cyclohexanol | Carbonic anhydrase II | 60–70 | 0.40 mM with 0.20 mg mL−1 CAII | 483 |
| (4) Salts | |||||
| (viii) | Ammonium sulphate | HEWL | ≥70 | 1 M with 20 μg mL−1 HEWL | 485 |
| (5) Detergents and surfactants | |||||
| (ix) | Sodium lauryl sarcosine (SLS) | Single-chain Fv (SFv) | 80 | SFv solubilized in 2% SLS solution mixed with 10% Dowex resin | 486 |
| (x) | Chaps (derivative of cholic acid) | Carbonic Anhydrase II | 80 | 31 mM with 0.50 mg mL−1 CA II | 483 |
| (xi) | Phospholipids | HEWL | 95 | 0.19 mg mL−1 conc. with 0.1 mg mL−1 HEWL | 487 |
| (6) Polymers | |||||
| (xii) | PvLEA-22 (peptide; group 3 LEA protein) | HEWL | ∼80 | Refolding probability calculated with DSC | 417 |
| (xiii) | N-tert-Butylcrylamide (TBAm) & acrylic acid (AAc) hydrogel | Lysozyme | 75 | 50 μg mL−1 nanoparticle consisting of 70% TBAm & 10% Aac | 479 |
| (xiv) | Polymeric nanochaperones | HEWL | 97 | 10:1 ratio of nanochaperone/HEWL |
488 |
| (xv) | Poly(p-phenylene vinylene) with p-nitrophenyl esters | Aβ42 | — | Aβ plaques removed from mice brain slice after treatment (200 mm) | 446 |
| (xvi) | Poly sulfobetaine with butyl methacrylate (BuMA) | Insulin from bovine pancreas | 40 | 1.5% polymer consisting of 30% BuMA | 322 |
| (xvii) | Poly(N-isopropylacrylamide) (PNIPAAm) | β-Lactamase | 85 | 4 mg mL−1 polymer | 473 |
| Carbonic anhydrase B | 98.2 | PNIPAAm with a molecular weight of 23000 |
474 | ||
| (xviii) | Eudragit S-100 | α-Chymotrypsin | — | 100% recovery of activity at a polymer; enzyme ratio of 1:1 |
475 |
| HEWL | 81.3 | 0.4% (w/v) polymer with 1 mg mL−1 denatured lysozyme | 476 | ||
To achieve an optimal yield, it is essential to study the appropriate refolding conditions for every protein which is possible only empirically. Refolding additives463 include denaturants (at low concentration), ionic and non-ionic detergents, and polyols such as sugars or alcohols, which may either promote the refolding of the protein or inhibit the process of aggregation.
At small scales, refolding efficiencies can be improved using artificial chaperones,466 and redox pairs such as GSH–GSSG, since the use of chaperones is not feasible at large scales due to their requirement in stoichiometric proportions. Mini chaperones,467 GroEL and GroES,468 and oxidoreductases that shuffle disulphide bond formation469 have been used for immobilizing folding aids. Mannen et al. in 2001 reported the use of immobilized cyclodextrin to remove detergent from a denatured protein to allow refolding to occur.470
Aggregation can be suppressed by interference with the intermolecular hydrophobic interactions by using additives that can be easily removed once refolding is done. These additives may affect the solubility or the stability of the native, denatured, and intermediate states by changing the ratios of folded and aggregated proteins.
The addition of L-arginine to proteins undergoing folding increases the yield of correctly folded proteins as it decreases the formation of aggregates. GdmCl enhances the solubility and stability of partially structured folding intermediates, which further leads to a decrease in the formation of aggregates. Polyethylene glycol (PEG) follows a similar mechanism to that of GdmCl. It interacts with the denatured states of the protein. Due to the partial hydrophobic character of PEG, it interacts with the hydrophobic side chains present in unfolded or partially folded polypeptides.
Alcohols, carbonic acid amides, and alkyl urea can also improve the efficiency of folding reactions in vitro. Adding cyclodextrin to the refolding buffer solution enhances refolding for some proteins.471 It solubilizes the folding intermediates by interchelating the aromatic amino acid and hydrophobic side chains.472 Additionally, surfactants and detergents promote proper folding by binding to the folding intermediates and reduce self-association. The detergents bind to the hydrophobic sites, which change the tertiary structure and dampen the fluctuations of amino acid side chains.
000 effectively refolds carbonic anhydrase B with a refolding yield of 98.2%. PNIPAAm has high surface hydrophobicity owing to its high molecular weight and therefore, it exhibits higher efficacy in forming complexes with the aggregation-prone species.474
Roy et al. have demonstrated the refolding efficacy of Eudragit S-100, a pH-sensitive polymer poly(methyl methacrylate), which can recover 100% activity of α-chymotrypsin with a high refolding rate.475 The addition of Eudragit to the refolding solution also enhances the refolding of denatured lysozyme, increasing the yield up to 81%. Notably, Eudragit S-100 at pH ≥ 7 consists of carboxylate moieties, which are negatively charged and form complexes with the denatured protein through electrostatic interactions that further shield the hydrophobic surfaces of unfolded protein molecules, promoting hydrophobic-prone aggregation. Moreover, owing to the reversible nature of Eudragit polymer–protein interactions, the refolded lysozyme structure remains unaltered. Eudragit S-100 has also been shown to successfully recover the activities of transforming growth factor (TGF)-β1 and keratinocyte growth factor (KGF)-2, even at lower concentrations.476
The naturally occurring anionic polymer alginate has been reported to be highly effective in recovering the activities of denatured urea and thermally denatured α-amylase.477
In 2008, Doyle et al. reported that the heat-shock protein 104 (Hsp104) along with caseinolytic peptidase B (ClpB) acts as a protein disaggregating machine.478 The two chaperones catalyse the disaggregation of proteins by extracting the polypeptides from the protein aggregates via unfolding and translocation through Hsp104/ClpB cavities. Further, in 2016, Nakamoto et al. showed that acrylamide-based hydrogel nanoparticles can facilitate the refolding of positively charged denatured lysozymes.479 These nanoparticles showed high affinity towards the denatured protein and had weak interactions with the native protein. These features help in the refolding of aggregated proteins and enable the dissociation of the nanoparticles from the refolded proteins (Fig. 18).
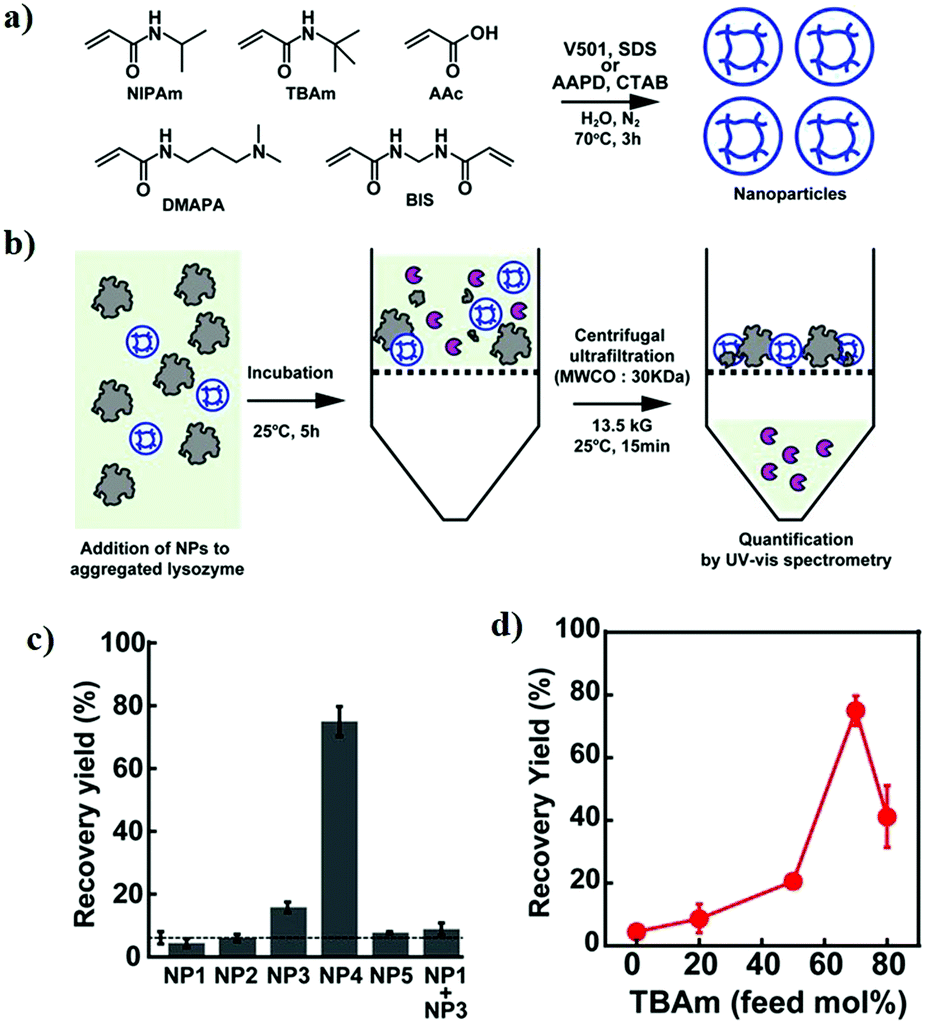 | ||
| Fig. 18 Refolding of proteins by nanoparticles. (a) Preparation of hydrogel nanoparticles. (b) Schematic illustration of the yield of aggregated lysozyme. (c) Facilitated recovery yield of lysozyme by NP1–NP5 hydrogels, where NP1–NP5 indicates nanoparticles with different monomer ratios and (d) effect of hydrophobicity of TBAm in facilitating the refolding activity. Reproduced with permissions from ref. 479. | ||
Chaperones show a high affinity towards denatured proteins rather than native proteins. Similarly, the hydrogel nanoparticles encapsulate the denatured lysozymes. Nanoparticles bind with the lysozyme due to their high dimensional structure and functional monomer sequencing. The hydrophobic N-tert-butyl acrylamide (TBAm) derivative and the negatively charged acrylic lysozyme acid (AAc) hydrogel on polymerization (NP4) have shown high recovery yield indicating that the combination of both hydrophobic and anionic groups may facilitate the resolubilization of lysozyme aggregates. Due to the weak interaction of NP4 and native lysozyme, the refolded lysozyme is released from the nanoparticle, resulting in the potential refolding of the aggregated proteins.
In a recent study, we reported that the sulfobetaine-based zwitterionic polymer poly-SPB, which harbours the attached hydrophobic moiety BuMA suppressed insulin aggregation and further facilitated the refolding of denatured insulin via interactions with the hydrophobic domains of the denatured proteins.322
| Reo = Dωxo/v |
High yields of final refolded protein can be achieved using pulse renaturation or fed-batch dilution,482 whose applicability is based on the stability of native proteins.
In 2015, Schoner et al. showed that bacterial inclusion bodies were activated through pressure refolding without the use of any denaturant, demonstrating that proteins prone to aggregation can be refolded using pressure treatments.493 Further, this technique enables the refolding of proteins at higher concentrations while reducing the use of chaotropic reagents.
In one study, renaturation of lysozyme was successfully implemented using simulated moving bed (SMB) chromatography.496 This process was structured by utilizing the lysozyme and denaturant partition coefficients, which are obtained from batch column experiments. However, the applicability of this process for refolding crude denatured protein is not known. In 2001, Cho et al. reported the use of expanded bed chromatography for crude samples.497 An ion exchange matrix was used for adsorption of the recombinant fusion protein, washing out of the cell debris and unbound constituents, and to exchange the buffer for the initiation of the refolding. The stability of the bed can be maintained by dilution of a high molarity urea buffer for reducing the applied sample volume.
9. Conclusions and outlook
Over the years, progress has been made to further develop efficient inhibitors of protein aggregation. Although several methods are in use, breakthroughs are required to enable the creation of a near-perfect technology for the protection of proteins from denaturation, and the development of more sensitive and easier characterization and separation techniques. Powerful and robust methods are required for characterization of protein aggregates. The size factor is crucial for detection of aggregates ranging from nanometers to microns in size. Therefore, prediction of aggregate formation is critical because of the possibility of artefact generation. Additionally, experimental approaches for solubilizing protein aggregates are necessary to identify quick fixes for the various stages of biopharmaceutical design. Assessment of biophysical and dynamic features of fibrils can be explored by the amalgamation of various techniques. The innovation of such technologies can pave the way for advanced clinical-grade approaches to observe aggregation for early diagnosis. Additionally, such techniques could help obtain insight into amyloidosis at the individual level.Understanding the detailed molecular mechanism and the various steps involved in protein aggregation not only has academic significance but also increases the likelihood that efficient protecting strategies can be developed easily. As different proteins misfold via different pathways, uncovering mechanistic information is challenging. However, the consensus opinion is that when proteins misfold, hydrophobic domains are exposed and can interact with each other, leading to aggregation-inducing collisions, which result in the formation of large aggregates. Therefore, additives to inhibit aggregation should work in either of 3 ways; prevention of misfolding, suppression of collisions between the misfolded monomers/oligomers, or dissolution of the formed aggregates. In the first case, the protein retains all its functions and activities, and in the second case, a part of the activity is lost. However, the misfolded state is still reversible, and proteins can be made to undergo refolding under optimum conditions. In the third case, considerable activity is lost, and it is extremely difficult to return the protein to its native structure, necessitating the removal of insoluble aggregates to circumvent aggregation-induced toxicity or occlusion of automated pumps used to deliver therapeutic proteins. Though difficult, an ideal strategy would be to prevent protein misfolding even when subjected to stress and the use of compounds to prevent aggregation-inducing collisions.
Over the last 3–4 decades, numerous small molecules and macromolecular aggregation inhibitors have been identified or developed that show relatively high efficiency in preventing misfolding or enabling refolding. Better therapeutic strategies could also be developed by using a cocktail of small molecule inhibitors for the treatment of protein aggregation-related diseases.
The utilization of polymers for inhibiting protein aggregation and facilitating refolding has ushered in a new era of protein-based therapeutics holding great promise. The field of polymer-based inhibitors is in its nascent stage and warrants further interest and dedicated research to overcome the current shortcomings, which would speed up the widespread application of protein-based therapeutics. In the various polymers and different categories described in this review, there appears to be no correlation between the mode of action between the different groups. The current mechanistic studies and details for polymeric inhibitors are inadequate, and additional analyses are required using different kinds of polymeric inhibitors, to reveal a common mode of action and to facilitate the rational design of more efficient polymeric materials in the future. Among the polymers described, polyelectrolytes, especially zwitterionic polymers and polyampholytes, are extremely effective in suppressing stress-induced aggregation of proteins. However, further research is needed to achieve the required efficacy for clinical applications. Drawbacks for their use include the high concentration required to suppress aggregation; therefore, the polymers should be further modified by modifying the functional group and architecture. Second, these polymers are non-biodegradable, which can pose serious challenges to their in vivo administration. Moreover, because polymer-based protein aggregation inhibitors have been developed only recently and hence, these agents have not yet been approved by the respective regulatory bodies. However, PEG- or polysaccharides-based compounds (already approved) can be employed for clinical applications if their activities are improved further.
Further, development of biocompatible protein delivery systems to protect therapeutic proteins from denaturation before delivery to the targeted area is required. The delivery vehicle should be able to specifically target the affected area and release the protein in a controlled manner. This can be achieved by transforming polymer-based protein aggregation inhibitors into a micelle/nanoparticle that can deliver as well as protect the proteins. It would be interesting to see some of the polymeric candidates discussed above being used or new classes being developed to deliver therapeutic proteins as well as aid the treatment of neurodegenerative diseases.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported in part by research grant Grant-in-Aid, KAKENHI (20K20197 and 20H04532), for scientific research from the Japan Society for the Promotion of Science and Adaptable and Seamless Technology transfer Program through Target-driven R&D from Japan Science and Technology Agency (JPMJTM19FB).References
- C. Ross and M. Poirier, Nat. Med., 2004, 10, S10–S17 CrossRef.
- C. Soto and S. Pritzkow, Nat. Neurosci., 2018, 21, 1332–1340 CrossRef CAS.
- L. R. C. Vasconcellos, F. F. Dutra, M. S. Siqueira, H. A. Paula-Neto, J. Dahan, E. Kiarely, L. A. M. Carneiro, M. T. Bozza and L. H. Travassos, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E7474–E7482 CrossRef CAS.
- W. Wang, Int. J. Pharm., 2005, 289, 1–30 CrossRef CAS.
- C. J. Roberts, Trends Biotechnol., 2014, 32, 372–380 CrossRef CAS.
- Global Protein Therapeutics Market Analysis 2019, https://www.researchandmarkets.com/reports/4857810/global-plasma-protein-therapeutics-market#rela0-4857806.
- Neurodegenerative Disease Market – Growth, Trends, and Forecast (2019–2024), https://www.reportlinker.com/p05865942/Neurodegenerative-Disease-Market-Growth-Trends-and-Forecast.html?utm_source=GNW.
- P. K. Crane and R. S. Doody, Neurology, 2009, 73, 1514–1516 CrossRef.
- Y. Tizabi, L. L. Hurley, Z. Qualls and L. Akinfiresoye, Molecules, 2014, 19, 20864–20879 CrossRef.
- K. R. Chaudhuri and A. H. V. Schapira, Lancet Neurol., 2009, 8, 464–474 CrossRef CAS.
- A. K. Desai and G. T. Grossberg, Neurology, 2005, 64, S34–S39 CrossRef CAS.
- Y. Mizuno, Integr. Med. Int., 2014, 1, 67–79 CrossRef.
- S. J. Tabrizi, B. R. Leavitt, G. B. Landwehrmeyer, E. J. Wild, C. Saft, R. A. Barker, N. F. Blair, D. Craufurd, J. Priller, H. Rickards, A. Rosser, H. B. Kordasiewicz, C. Czech, E. E. Swayze, D. A. Norris, T. Baumann, I. Gerlach, S. A. Schobel, E. Paz, A. V. Smith, C. F. Bennett and R. M. Lane, N. Engl. J. Med., 2019, 380, 2307–2316 CrossRef CAS.
- T. I. Chandel, M. Zaman, M. V. Khan, M. Ali, G. Rabbani, M. Ishtikhar and R. H. Khan, Int. J. Biol. Macromol., 2018, 106, 1115–1129 CrossRef CAS.
- E. De Bernardez Clark, E. Schwarz and R. Rudolph, Methods Enzymol., 1999, 309, 217–236 CAS.
- Z. Dhouafli, K. Cuanalo-Contreras, E. A. Hayouni, C. E. Mays, C. Soto and I. Moreno-Gonzalez, Cell. Mol. Life Sci., 2018, 75, 3521–3538 CrossRef CAS.
- L. Estrada and C. Soto, Curr. Pharm. Des., 2006, 12, 2557–2567 CrossRef CAS.
- J. F. Carpenter, B. S. Kendrick, B. S. Chang, M. C. Manning and T. W. Randolph, Methods Enzymol., 1999, 309, 236–255 CAS.
- P. Alam, K. Siddiqi, S. K. Chturvedi and R. H. Khan, Int. J. Biol. Macromol., 2017, 103, 208–219 CrossRef CAS.
- M. Bartolini and V. Andrisano, ChemBioChem, 2010, 11, 1018–1035 CrossRef CAS.
- G. M. Cooper, The Cell: A Molecular Approach, Sinauer Associates, Sunderland (MA), 2nd edn, 2000 Search PubMed.
- J. T. Marinko, H. Huang, W. D. Penn, J. A. Capra, J. P. Schlebach and C. R. Sanders, Chem. Rev., 2019, 119, 5537–5606 CrossRef CAS.
- L. L. Porter and G. D. Rose, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9420–9425 CrossRef CAS.
- J. Verghese, J. Abrams, Y. Wang and K. A. Morano, Microbiol. Mol. Biol. Rev., 2012, 76, 115–158 CrossRef CAS.
- J. Tyedmers, A. Mogk and B. Bukau, Nat. Rev. Mol. Cell Biol., 2010, 11, 777–788 CrossRef CAS.
- E. Gaggelli, H. Kozlowski, D. Valensin and G. Valensin, Chem. Rev., 2006, 106, 1995–2044 CrossRef CAS.
- A. Zhang, W. Qi, T. A. Good and E. J. Fernandez, Biophys. J., 2009, 96, 1091–1104 CrossRef CAS.
- M. A. Findeis, G. M. Musso, C. C. Arico-Muendel, H. W. Benjamin, A. M. Hundal, J.-J. Lee, J. Chin, M. Kelley, J. Wakefield, N. J. Hayward and S. M. Molineaux, Biochemistry, 1999, 38, 6791–6800 CrossRef CAS.
- A. Cao, D. Hu and L. Lai, Protein Sci., 2004, 13, 319–324 CrossRef CAS.
- A. I. Sulatskaya, E. A. Volova, Y. Y. Komissarchik, E. S. Snigirevskaya, A. A. Maskevich, E. A. Drobchenko, I. M. Kuznetsova and K. K. Turoverov, Cell Tissue Biol., 2014, 8, 186–191 CrossRef.
- K. Araki, N. Yagi, K. Aoyama, C.-J. Choong, H. Hayakawa, H. Fujimura, Y. Nagai, Y. Goto and H. Mochizuki, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 17963–17969 CrossRef CAS.
- J. C. Sacchettini and J. W. Kelly, Nat. Rev. Drug Discovery, 2002, 1, 267–275 CrossRef CAS.
- F. Hasecke, T. Miti, C. Perez, J. Barton, D. Schölzel, L. Gremer, C. S. R. Grüning, G. Matthews, G. Meisl, T. P. J. Knowles, D. Willbold, P. Neudecker, H. Heise, G. Ullah, W. Hoyer and M. Muschol, Chem. Sci., 2018, 9, 5937–5948 RSC.
- L. F. Pease 3rd, M. Sorci, S. Guha, D.-H. Tsai, M. R. Zachariah, M. J. Tarlov and G. Belfort, Biophys. J., 2010, 99, 3979–3985 CrossRef.
- W. Wang, S. Nema and D. Teagarden, Int. J. Pharm., 2010, 390, 89–99 CrossRef CAS.
- T. R. Jahn and S. E. Radford, Arch. Biochem. Biophys., 2008, 469, 100–117 CrossRef CAS.
- H.-X. Zhou and X. Pang, Chem. Rev., 2018, 118, 1691–1741 CrossRef CAS.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2016, 116, 2775–2825 CrossRef CAS.
- E. J. Nettleton, P. Tito, M. Sunde, M. Bouchard, C. M. Dobson and C. V. Robinson, Biophys. J., 2000, 79, 1053–1065 CrossRef CAS.
- M. V. Trivedi, J. S. Laurence and T. J. Siahaan, Curr. Protein Pept. Sci., 2009, 10, 614–625 CrossRef CAS.
- H. B. Cardoso, P. A. Wierenga, H. Gruppen and H. A. Schols, Food Chem., 2019, 276, 652–661 CrossRef CAS.
- O. V. Galzitskaya, Curr. Protein Pept. Sci., 2016, 17, 319–331 CrossRef CAS.
- A. L. P. Chapman, C. C. Winterbourn, S. O. Brennan, T. W. Jordan and A. J. Kettle, Biochem. J., 2003, 375, 33–40 CrossRef CAS.
- E. Lévy, N. El Banna, D. Baïlle, A. Heneman-Masurel, S. Truchet, H. Rezaei, M.-E. Huang, V. Béringue, D. Martin and L. Vernis, Int. J. Mol. Sci., 2019, 20, 3896 CrossRef.
- A. L. Fink, Folding Des., 1998, 3, R9–R23 CrossRef CAS.
- W.-F. Xue, S. W. Homans and S. E. Radford, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8926–8931 CrossRef CAS.
- H. Wu, Adv. Protein Chem., 1995, 46, 6–26 CrossRef CAS.
- A. J. Paul, K. Schwab, N. Prokoph, E. Haas, R. Handrick and F. Hesse, Anal. Bioanal. Chem., 2015, 407, 4849–4856 CrossRef CAS.
- M. Pindrus, S. J. Shire, R. F. Kelley, B. Demeule, R. Wong, Y. Xu and S. Yadav, Mol. Pharmaceutics, 2015, 12, 3896–3907 CrossRef CAS.
- T. Arakawa, D. Ejima and T. Akuta, Int. J. Biol. Macromol., 2017, 95, 1153–1158 CrossRef CAS.
- A. Nayak, J. Colandene, V. Bradford and M. Perkins, J. Pharm. Sci., 2011, 100, 4198–4204 CrossRef CAS.
- B. C. Roughton, L. K. Iyer, E. Bertelsen, E. M. Topp and K. V. Camarda, Comput. Chem. Eng., 2013, 58, 369–377 CrossRef CAS.
- B. D. Connolly, L. Le, T. W. Patapoff, M. E. M. Cromwell, J. M. R. Moore and P. Lam, J. Pharm. Sci., 2015, 104, 4170–4184 CrossRef CAS.
- S. Hasanbašić, A. Jahić, S. Berbić, M. T. Znidarič and E. Zerovnik, Oxid. Med. Cell. Longevity, 2018, 2018, 8613209 Search PubMed.
- V. H. Guaiquil, J. C. Vera and D. W. Golde, J. Biol. Chem., 2001, 276, 40955–40961 CrossRef CAS.
- S. A. Giannos, E. R. Kraft, Z.-Y. Zhao, K. H. Merkley and J. Cai, Pharm. Res., 2018, 35, 78 CrossRef.
- M. R. Nilsson, Amyloid, 2016, 23, 139–147 CrossRef CAS.
- J. L. Cleland, M. F. Powell and S. J. Shire, Crit. Rev. Ther. Drug Carrier Syst., 1993, 10, 307–377 CAS.
- D. B. Volkin and C. R. Middaugh, in Stability of Protein Pharmaceuticals: Part A: Chemical and Physical Pathways of Protein Degradation, ed. T. J. Ahern and M. C. Manning, Plenum, New York, 1992, pp. 215–247 Search PubMed.
- M. C. Manning, K. Patel and R. T. Borchardt, Pharm. Res., 1989, 6, 903–918 CrossRef CAS.
- J. F. Carpenter, B. S. W. Chang, W. Garzon-Rodriguez and T. W. Randolph, Rational Design of Stable Lyophilized Protein Formulations: Theory and Practice, in Rational Design of Stable Protein Formulations, ed. J. F. Carpenter and M. C. Manning, Springer US, Boston, MA, 2002, pp. 109–133 Search PubMed.
- A. Dong, S. J. Prestrelski, S. D. Allison and J. F. Carpenter, J. Pharm. Sci., 1995, 84, 415–424 CrossRef CAS.
- C. Tang, C.-H. Yin, Y.-Y. Pei, L. Yu and F.-Y. Gui, Chin. Pharm. J., 2004, 39, 764–768 CAS.
- A. B. Joshi, E. Rus and L. E. Kirsch, Int. J. Pharm., 2000, 203, 115–125 CrossRef CAS.
- S. Onoue, K. Ohshima, K. Debari, K. Koh, S. Shioda, S. Iwasa, K. Kashimoto and T. Yajima, Pharm. Res., 2004, 21, 1274–1283 CrossRef CAS.
- R. E. Johnson, V. J. Hruby and J. A. Rupley, Biochemistry, 1979, 18, 1176–1179 CrossRef CAS.
- J. S. Pedersen, D. Dikov, J. L. Flink, H. A. Hjuler, G. Christiansen and D. E. Otzen, J. Mol. Biol., 2006, 355, 501–523 CrossRef CAS.
- M. K. Joubert, Q. Luo, Y. Nashed-Samuel, J. Wypych and L. O. Narhi, J. Biol. Chem., 2011, 286, 25118–25133 CrossRef CAS.
- P. Arosio, S. Rima and M. Morbidelli, Pharm. Res., 2013, 30, 641–654 CrossRef CAS.
- M. L. Brader, T. Estey, S. Bai, R. W. Alston, K. K. Lucas, S. Lantz, P. Landsman and K. M. Maloney, Mol. Pharmaceutics, 2015, 12, 1005–1017 CrossRef CAS.
- D. S. Goldberg, R. A. Lewus, R. Esfandiary, D. C. Farkas, N. Mody, K. J. Day, P. Mallik, M. B. Tracka, S. K. Sealey and H. S. Samra, J. Pharm. Sci., 2017, 106, 1971–1977 CrossRef CAS.
- R. Bansal, P. Srivastava, A. S. Rathore and P. Chokshi, Chem. Eng. Sci., 2020, 216, 115479 CrossRef CAS.
- N. C. Berchtold and C. W. Cotman, Neurobiol. Aging, 1998, 19, 173–189 CrossRef CAS.
- J. D. Harper and P. T. Lansbury, Annu. Rev. Biochem., 1997, 66, 385–407 CrossRef CAS.
- J. Leitman, F. Ulrich Hartl and G. Z. Lederkremer, Nat. Commun., 2013, 4, 2753 CrossRef.
- C. Haass and D. J. Selkoe, Nat. Rev. Mol. Cell Biol., 2007, 8, 101–112 CrossRef CAS.
- M. Arrasate, S. Mitra, E. S. Schweitzer, M. R. Segal and S. Finkbeiner, Nature, 2004, 431, 805–810 CrossRef CAS.
- E. Cohen, J. Bieschke, R. M. Perciavalle, J. W. Kelly and A. Dillin, Science, 2006, 313, 1604–1610 CrossRef CAS.
- I. W. Hamley, Chem. Rev., 2012, 112, 5147–5192 CrossRef CAS.
- J. J. Kayser, P. Arnold, A. Steffen-Heins, K. Schwarz and J. K. Keppler, J. Food Eng., 2020, 270, 109764 CrossRef CAS.
- A. A. Thorat, B. Munjal, T. W. Geders and R. Suryanarayanan, J. Controlled Release, 2020, 323, 591–599 CrossRef CAS.
- K. L. Zapadka, F. J. Becher, S. Uddin, P. G. Varley, S. Bishop, A. L. Gomes dos Santos and S. E. Jackson, J. Am. Chem. Soc., 2016, 138, 16259–16265 CrossRef CAS.
- J. K. Keppler, T. R. Heyn, P. M. Meissner, K. Schrader and K. Schwarz, Food Chem., 2019, 289, 223–231 CrossRef CAS.
- T. R. Heyn, V. M. Garamus, H. R. Neumann, M. J. Uttinger, T. Guckeisen, M. Heuer, C. Selhuber-Unkel, W. Peukert and J. K. Keppler, Eur. Polym. J., 2019, 120, 109211 CrossRef CAS.
- Y. Wang, X. Wang, G. Wang and X. Wang, J. Am. Oil Chem. Soc., 2020 DOI:10.1002/aocs.12393.
- A. Mishra, R. Bansal, S. Sreenivasan, R. Dash, S. Joshi, R. Singh, A. S. Rathore and G. Goel, J. Chem. Inf. Model., 2020, 60, 3304–3314 CrossRef CAS.
- E. Mari, C. Ricci, S. Pieraccini, F. Spinozzi, P. Mariani and M. G. Ortore, Life, 2020, 10, 60 CrossRef CAS.
- G. Ge, Y. Han, J. Zheng, M. Zhao and W. Sun, Food Chem., 2020, 309, 125614 CrossRef CAS.
- E. Dachmann, V. Nobis, U. Kulozik and J. Dombrowski, Food Hydrocolloids, 2020, 107, 105981 CrossRef CAS.
- H. Wu and T. W. Randolph, J. Pharm. Sci., 2020, 109, 1473–1482 CrossRef CAS.
- M. Wolz, E. Mersch and U. Kulozik, Food Hydrocolloids, 2016, 56, 396–404 CrossRef CAS.
- R. Brogna, H. Oldenhof, H. Sieme, C. Figueiredo, T. Kerrinnes and W. F. Wolkers, PLoS One, 2020, 15, e0234502 CrossRef CAS.
- Z. Zhao, K. Engholm-Keller, M. M. Poojary, S. G. Boelt, A. Rogowska-Wrzesinska, L. H. Skibsted, M. J. Davies and M. N. Lund, J. Agric. Food Chem., 2020, 68, 6701–6714 CrossRef CAS.
- T. Kaffe-Abramovich and R. Unger, Folding Des., 1998, 3, 389–399 CrossRef CAS.
- T. Sumi and K. Koga, Sci. Rep., 2019, 9, 5186 CrossRef.
- C. N. Pace, B. A. Shirley, M. McNutt and K. Gajiwala, FASEB J., 1996, 10, 75–83 CrossRef CAS.
- R. Jaenicke, Biochemistry, 1991, 30, 3147–3161 CrossRef CAS.
- R. Jaenicke and R. Rudolph, in Protein structure: a practical approach, ed. T. E. Creighton, IRL Press, Oxford, UK, 1989, pp. 191–223 Search PubMed.
- K. A. Dill, Biochemistry, 1990, 29, 7133–7155 CrossRef CAS.
- F. Franks, R. H. M. Hatley and H. Friedman, Biophys. Chem., 1988, 31, 307–315 CrossRef CAS.
- J. M. Andrews and C. J. Roberts, J. Phys. Chem. B, 2007, 111, 7897–7913 CrossRef CAS.
- C. J. Roberts, J. Phys. Chem. B, 2003, 107, 1194–1207 CrossRef CAS.
- P. L. Privalov, Crit. Rev. Biochem. Mol. Biol., 1990, 25, 281–306 CrossRef CAS.
- P. L. Privalov and S. J. Gill, Advances in protein chemistry, Elsevier, 1988, vol. 39, pp. 191–234 Search PubMed.
- R. L. Remmele, S. D. Bhat, D. H. Phan and W. R. Gombotz, Biochemistry, 1999, 38, 5241–5247 CrossRef CAS.
- A. I. Azuaga, C. M. Dobson, P. L. Mateo and F. Conejero-Lara, Eur. J. Biochem., 2002, 269, 4121–4133 CrossRef CAS.
- A. M. Tsai, J. H. van Zanten and M. J. Betenbaugh, Biotechnol. Bioeng., 1998, 59, 273–280 CrossRef CAS.
- B. Chen, T. Arakawa, C. F. Morris, W. C. Kenney, C. M. Wells and C. G. Pitt, Pharm. Res., 1994, 11, 1581–1587 CrossRef CAS.
- B. Luan, B. Shan, C. Baiz, A. Tokmakoff and D. P. Raleigh, Biochemistry, 2013, 52, 2402–2409 CrossRef CAS.
- S. C. Meliga, W. Farrugia, P. A. Ramsland and R. J. Falconer, J. Phys. Chem. B, 2013, 117, 490–494 CrossRef CAS.
- R. Wälchli, P.-J. Vermeire, J. Massant and P. Arosio, J. Pharm. Sci., 2020, 109, 595–602 CrossRef.
- R. Esfandiary, A. Parupudi, J. Casas-Finet, D. Gadre and H. Sathish, J. Pharm. Sci., 2015, 104, 577–586 CrossRef CAS.
- R. Esfandiary, D. B. Hayes, A. Parupudi, J. Casas-finet, S. Bai, H. S. Samra, A. U. Shah and H. A. Sathish, J. Pharm. Sci., 2013, 102, 62–72 CrossRef CAS.
- Y. Wang, A. Lomakin, R. F. Latypov, J. P. Laubach, T. Hideshima, P. G. Richardson, N. C. Munshi, K. C. Anderson and G. B. Benedek, J. Chem. Phys., 2013, 139, 121904 CrossRef.
- P. Atkins and J. De Paula, Elements of physical chemistry, Macmillan, 2009 Search PubMed.
- A. Oliva, J. B. Fariña and M. Llabrés, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1022, 206–212 CrossRef CAS.
- Z. Sahin, Y. K. Demir and V. Kayser, Eur. J. Pharm. Sci., 2016, 86, 115–124 CrossRef CAS.
- N. Chakroun, D. Hilton, S. S. Ahmad, G. W. Platt and P. A. Dalby, Mol. Pharmaceutics, 2016, 13, 307–319 CrossRef CAS.
- J. M. Andrews and C. J. Roberts, Biochemistry, 2007, 46, 7558–7571 CrossRef CAS.
- A. Fazeli, M. Haji-Abdolvahab, S. A. Shojaosadati, H. Schellekens, K. Khalifeh, A. A. Moosavi-Movahedi and M. R. Fazeli, AAPS PharmSciTech, 2014, 15, 1619–1629 CrossRef CAS.
- W. Wang and C. J. Roberts, AAPS J., 2013, 15, 840–851 CrossRef CAS.
- D. Ramakrishna, M. D. Prasad and A. K. Bhuyan, Arch. Biochem. Biophys., 2012, 528, 67–71 CrossRef CAS.
- A. Saluja, V. Sadineni, A. Mungikar, V. Nashine, A. Kroetsch, C. Dahlheim and V. M. Rao, Pharm. Res., 2014, 31, 1575–1587 CrossRef CAS.
- F. Bickel, E. M. Herold, A. Signes, S. Romeijn, W. Jiskoot and H. Kiefer, Eur. J. Pharm. Biopharm., 2016, 107, 310–320 CrossRef CAS.
- D. Reichert, S. Gröger and C. Hackel, Biopolymers, 2017, 107, 39–45 CrossRef CAS.
- M.-T. Schermeyer, A. K. Wöll, B. Kokke, M. Eppink and J. Hubbuch, mAbs, 2017, 9, 1169–1185 CrossRef CAS.
- V. Filipe, B. Kükrer, A. Hawe and W. Jiskoot, J. Pharm. Sci., 2012, 101, 2327–2339 CrossRef CAS.
- G. V. Barnett, W. Qi, S. Amin, E. Neil Lewis and C. J. Roberts, Biophys. Chem., 2015, 207, 21–29 CrossRef CAS.
- L. Zhang, L. Yu, J. Zhang-Van Enk, G. Huang and J. Zhang, J. Pharm. Sci., 2017, 106, 3287–3292 CrossRef CAS.
- C. Calero-Rubio, R. Ghosh, A. Saluja and C. J. Roberts, J. Pharm. Sci., 2018, 107, 1269–1281 CrossRef CAS.
- E. Sahin, W. F. Weiss, A. M. Kroetsch, K. R. King, R. K. Kessler, T. K. Das and C. J. Roberts, J. Pharm. Sci., 2012, 101, 1678–1687 CrossRef CAS.
- T. Iwura, J. Fukuda, K. Yamazaki and F. Arisaka, J. Biochem., 2014, 156, 345–352 CrossRef CAS.
- L. Nielsen, R. Khurana, A. Coats, S. Frokjaer, J. Brange, S. Vyas, V. N. Uversky and A. L. Fink, Biochemistry, 2001, 40, 6036–6046 CrossRef CAS.
- R. Pearlman and Y. J. Wang, Formulation, characterization, and stability of protein drugs, Springer Science & Business Media, 1996, vol. 9 Search PubMed.
- M. W. Townsend and P. P. DeLuca, J. Pharm. Sci., 1990, 79, 1083–1086 CrossRef CAS.
- S. Krishnan, E. Y. Chi, J. N. Webb, B. S. Chang, D. Shan, M. Goldenberg, M. C. Manning, T. W. Randolph and J. F. Carpenter, Biochemistry, 2002, 41, 6422–6431 CrossRef CAS.
- A. Fatouros, T. Österberg and M. Mikaelsson, Int. J. Pharm., 1997, 155, 121–131 CrossRef CAS.
- K. Takano, K. Tsuchimori, Y. Yamagata and K. Yutani, Biochemistry, 2000, 39, 12375–12381 CrossRef CAS.
- M. Kutsch, P. Hortmann, C. Herrmann, S. Weibels and H. Weingärtner, Biointerphases, 2016, 11, 19008 CrossRef.
- S. Wang, G. Wu, X. Zhang, Z. Tian, N. Zhang, T. Hu, W. Dai and F. Qian, Eur. J. Pharm. Biopharm., 2017, 114, 263–277 CrossRef CAS.
- W. G. Lilyestrom, S. Yadav, S. J. Shire and T. M. Scherer, J. Phys. Chem. B, 2013, 117, 6373–6384 CrossRef CAS.
- A. K. Buell, P. Hung, X. Salvatella, M. E. Welland, C. M. Dobson and T. P. J. Knowles, Biophys. J., 2013, 104, 1116–1126 CrossRef CAS.
- K. Baumgartner, S. Großhans, J. Schütz, S. Suhm and J. Hubbuch, J. Pharm. Biomed. Anal., 2016, 128, 216–225 CrossRef CAS.
- P. Arosio, B. Jaquet, H. Wu and M. Morbidelli, Biophys. Chem., 2012, 168–169, 19–27 CrossRef CAS.
- S. Campioni, B. Mannini, J. P. López-Alonso, I. N. Shalova, A. Penco, E. Mulvihill, D. V. Laurents, A. Relini and F. Chiti, J. Mol. Biol., 2012, 424, 132–149 CrossRef CAS.
- B. C. Cunningham, M. G. Mulkerrin and J. A. Wells, Science, 1991, 253, 545–548 CrossRef CAS.
- S. N. Timasheff, Adv. Protein Chem., 1998, 51, 355–432 CrossRef CAS.
- M. G. Cacace, E. M. Landau and J. J. Ramsden, Q. Rev. Biophys., 1997, 30, 241–277 CrossRef CAS.
- J. C. Lee and S. N. Timasheff, J. Biol. Chem., 1981, 256, 7193–7201 CrossRef CAS.
- L. Nicoud, M. Sozo, P. Arosio, A. Yates, E. Norrant and M. Morbidelli, J. Phys. Chem. B, 2014, 118, 11921–11930 CrossRef CAS.
- A. M. Tsai, J. H. van Zanten and M. J. Betenbaugh, Biotechnol. Bioeng., 1998, 59, 281–285 CrossRef CAS.
- T. Arakawa and S. N. Timasheff, Biochemistry, 1984, 23, 5912–5923 CrossRef CAS.
- R. A. Curtis, J. Ulrich, A. Montaser, J. M. Prausnitz and H. W. Blanch, Biotechnol. Bioeng., 2002, 79, 367–380 CrossRef CAS.
- B.-L. Chen, X. Wu, S. J. Babuka and M. Hora, J. Pharm. Sci., 1999, 88, 881–888 CrossRef CAS.
- M. Z. Zhang, J. Wen, T. Arakawa and S. J. Prestrelski, Pharm. Res., 1995, 12, 1447–1452 CrossRef CAS.
- D. S. Maclean, Q. Qian and C. R. Middaugh, J. Pharm. Sci., 2002, 91, 2220–2229 CrossRef CAS.
- I. K. Ventouri, D. B. A. Malheiro, R. L. C. Voeten, S. Kok, M. Honing, G. W. Somsen and R. Haselberg, Anal. Chem., 2020, 92, 4292–4300 CrossRef CAS.
- S. K. Senthil, P. K. Murugan, S. Selvam, J. P. Chandhana, T. G. S. Babu and E. Kandasamy, Mater. Today: Proc., 2020, 33, 2328–2330 CAS.
- K. Uesugi, H. Ogi, M. Fukushima, M. So, H. Yagi, Y. Goto and M. Hirao, Jpn. J. Appl. Phys., 2013, 52, 07HE10 CrossRef.
- L. Brückl, T. Schröder, S. Scheler, R. Hahn and C. Sonderegger, J. Pharm. Sci., 2016, 105, 1810–1818 CrossRef.
- S. A. McBride, C. F. Tilger, S. P. Sanford, P. M. Tessier and A. H. Hirsa, J. Phys. Chem. B, 2015, 119, 10426–10433 CrossRef CAS.
- S. Kiese, A. Papppenberger, W. Friess and H.-C. Mahler, J. Pharm. Sci., 2008, 97, 4347–4366 CrossRef CAS.
- G. Bai, J. S. Bee, J. G. Biddlecombe, Q. Chen and W. T. Leach, Int. J. Pharm., 2012, 423, 264–280 CrossRef CAS.
- A. S. Sediq, R. B. van Duijvenvoorde, W. Jiskoot and M. R. Nejadnik, J. Pharm. Sci., 2016, 105, 519–529 CrossRef CAS.
- A. V. Gandhi, M. R. Pothecary, D. L. Bain and J. F. Carpenter, J. Pharm. Sci., 2017, 106, 2178–2186 CrossRef CAS.
- Y. Yoshimura, Y. Lin, H. Yagi, Y.-H. Lee, H. Kitayama, K. Sakurai, M. So, H. Ogi, H. Naiki and Y. Goto, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 14446–14451 CrossRef CAS.
- C. Zhou, W. Qi, E. N. Lewis, T. W. Randolph and J. F. Carpenter, J. Pharm. Sci., 2016, 105, 2302–2309 CrossRef CAS.
- V. Kumar, V. K. Sharma and D. S. Kalonia, Int. J. Pharm., 2009, 366, 88–98 CrossRef CAS.
- M. Agarkhed, C. O’Dell, M.-C. Hsieh, J. Zhang, J. Goldstein and A. Srivastava, AAPS PharmSciTech, 2013, 14, 1–9 CrossRef CAS.
- C. Du, G. Barnett, A. Borwankar, A. Lewandowski, N. Singh, S. Ghose, M. Borys and Z. J. Li, Eur. J. Pharm. Biopharm., 2018, 127, 37–43 CrossRef CAS.
- S. R. Singh, J. Zhang, C. O’Dell, M.-C. Hsieh, J. Goldstein, J. Liu and A. Srivastava, AAPS PharmSciTech, 2012, 13, 422–430 CrossRef CAS.
- A. Sreedhara, J. Yin, M. Joyce, K. Lau, A. T. Wecksler, G. Deperalta, L. Yi, Y. John Wang, B. Kabakoff and R. S. K. Kishore, Eur. J. Pharm. Biopharm., 2016, 100, 38–46 CrossRef CAS.
- C. Arzeni, O. E. Pérez and A. M. R. Pilosof, Food Hydrocolloids, 2012, 29, 308–316 CrossRef CAS.
- L. G. Stepanskiy, J. Theor. Biol., 2012, 298, 77–81 CrossRef CAS.
- H. Kiminami, A. B. Krueger, Y. Abe, K. Yoshino and J. F. Carpenter, J. Pharm. Sci., 2017, 106, 1001–1007 CrossRef CAS.
- R. Lumry and H. Eyring, J. Phys. Chem., 1954, 58, 110–120 CrossRef CAS.
- A. Singla, R. Bansal, V. Joshi and A. S. Rathore, AAPS J., 2016, 18, 689–702 CrossRef CAS.
- A. H. Pekar and B. H. Frank, Biochemistry, 1972, 11, 4013–4016 CrossRef CAS.
- D. N. Brems, L. A. Alter, M. J. Beckage, R. E. Chance, R. D. DiMarchi, L. K. Green, H. B. Long, A. H. Pekar, J. E. Shields and B. H. Frank, Protein Eng., 1992, 5, 527–533 CrossRef CAS.
- J. R. Alford, B. S. Kendrick, J. F. Carpenter and T. W. Randolph, J. Pharm. Sci., 2008, 97, 3005–3021 CrossRef CAS.
- S. W. Raso, J. Abel, J. M. Barnes, K. M. Maloney, G. Pipes, M. J. Treuheit, J. King and D. N. Brems, Protein Sci., 2005, 14, 2246–2257 CrossRef CAS.
- B. S. Kendrick, J. F. Carpenter, J. L. Cleland and T. W. Randolph, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 14142–14146 CrossRef CAS.
- E. Y. Chi, S. Krishnan, T. W. Randolph and J. F. Carpenter, Pharm. Res., 2003, 20, 1325–1336 CrossRef CAS.
- R. Krishnamurthy and M. C. Manning, Curr. Pharm. Biotechnol., 2002, 3, 361–371 CAS.
- S. Hermeling, H. Schellekens, C. Maas, M. F. B. G. Gebbink, D. J. A. Crommelin and W. Jiskoot, J. Pharm. Sci., 2006, 95, 1084–1096 CrossRef CAS.
- Y. K. Al-Hilaly, L. Biasetti, B. J. F. Blakeman, S. J. Pollack, S. Zibaee, A. Abdul-Sada, J. R. Thorpe, W.-F. Xue and L. C. Serpell, Sci. Rep., 2016, 6, 39171 CrossRef CAS.
- A. K. Tyagi, T. W. Randolph, A. Dong, K. M. Maloney, C. Hitscherich Jr and J. F. Carpenter, J. Pharm. Sci., 2009, 98, 94–104 CrossRef CAS.
- E. Y. Chi, J. Weickmann, J. F. Carpenter, M. C. Manning and T. W. Randolph, J. Pharm. Sci., 2005, 94, 256–274 CrossRef CAS.
- H. G. Barth, C. Jackson and B. E. Boyes, Anal. Chem., 1994, 66, 595–620 CrossRef.
- P. Hong, S. Koza and E. S. P. Bouvier, J. Liq. Chromatogr. Relat. Technol., 2012, 35, 2923–2950 CrossRef CAS.
- J. S. Philo, Analysis of Aggregates and Particles in Protein Pharmaceuticals, John Wiley & Sons, Ltd, 2012, pp. 303–333 Search PubMed.
- T. Wang, S. B. Joshi, O. S. Kumru, S. Telikepalli, C. R. Middaugh and D. B. Volkin, in Biophysics for Therapeutic Protein Development, ed. L. O. Narhi, Springer New York, New York, NY, 2013, pp. 205–243 Search PubMed.
- J. Janča, TrAC, Trends Anal. Chem., 1983, 2, 278–281 CrossRef.
- H.-C. Mahler, W. Friess, U. Grauschopf and S. Kiese, J. Pharm. Sci., 2009, 98, 2909–2934 CrossRef CAS.
- A. P. Minton, Anal. Biochem., 2016, 501, 4–22 CrossRef CAS.
- D.-M. Smilgies and E. Folta-Stogniew, J. Appl. Crystallogr., 2015, 48, 1604–1606 CrossRef CAS.
- J. Stetefeld, S. A. McKenna and T. R. Patel, Biophys. Rev., 2016, 8, 409–427 CrossRef CAS.
- S. Amin, G. V. Barnett, J. A. Pathak, C. J. Roberts and P. S. Sarangapani, Curr. Opin. Colloid Interface Sci., 2014, 19, 438–449 CrossRef CAS.
- P. Garidel, A. Herre and W. Kliche, Analysis of Aggregates and Particles in Protein Pharmaceuticals, John Wiley & Sons, Ltd, 2012, pp. 269–302 Search PubMed.
- J. J. Sung, N. N. Pardeshi, A. M. Mulder, S. K. Mulligan, J. Quispe, K. On, B. Carragher, C. S. Potter, J. F. Carpenter and A. Schneemann, J. Pharm. Sci., 2015, 104, 750–759 CrossRef CAS.
- T. O. Pleshakova, N. S. Bukharina, A. I. Archakov and Y. D. Ivanov, Int. J. Mol. Sci., 2018, 19, 1142 CrossRef.
- Y.-L. Chiang, Y.-C. Chang, I.-C. Chiang, H.-M. Mak, I.-S. Hwang and Y.-L. Shih, PLoS One, 2015, 10, e0142506 CrossRef.
- H. Fu, G. Grimsley, J. M. Scholtz and C. N. Pace, Protein Sci., 2010, 19, 1044–1052 CrossRef CAS.
- I. B. Durowoju, K. S. Bhandal, J. Hu, B. Carpick and M. Kirkitadze, J. Visualized Exp., 2017, 121, e55262 Search PubMed.
- C. M. Johnson, Arch. Biochem. Biophys., 2013, 531, 100–109 CrossRef CAS.
- D. Prozeller, S. Morsbach and K. Landfester, Nanoscale, 2019, 11, 19265–19273 RSC.
- M. De, C.-C. You, S. Srivastava and V. M. Rotello, J. Am. Chem. Soc., 2007, 129, 10747–10753 CrossRef CAS.
- K. Rajarathnam and J. Rösgen, Biochim. Biophys. Acta, 2014, 1838, 69–77 CrossRef CAS.
- M. Winiewska, E. Bugajska and J. Poznański, PLoS One, 2017, 12, e0173260 CrossRef.
- R. Esfandiary and C. R. Middaugh, Analysis of Aggregates and Particles in Protein Pharmaceuticals, John Wiley & Sons, Ltd, 2012, pp. 169–200 Search PubMed.
- S. Patke, S. Srinivasan, R. Maheshwari, S. K. Srivastava, J. J. Aguilera, W. Colón and R. S. Kane, PLoS One, 2013, 8, e64974 CrossRef CAS.
- L. H. Garcia-Rubio, C. A. Lopez-Menacho and S. Grossman, Chem. Eng. Commun., 1993, 122, 85–101 CrossRef CAS.
- S. Siddhanta, I. Barman and C. Narayana, Soft Matter, 2015, 11, 7241–7249 RSC.
- V. Joshi, T. Shivach, N. Yadav and A. S. Rathore, Anal. Chem., 2014, 86, 11606–11613 CrossRef CAS.
- H. Zhang, X. Zheng, R. T. K. Kwok, J. Wang, N. L. C. Leung, L. Shi, J. Z. Sun, Z. Tang, J. W. Y. Lam, A. Qin and B. Z. Tang, Nat. Commun., 2018, 9, 4961 CrossRef.
- S. Benjwal, S. Verma, K.-H. Röhm and O. Gursky, Protein Sci., 2006, 15, 635–639 CrossRef CAS.
- A. Micsonai, F. Wien, L. Kernya, Y.-H. Lee, Y. Goto, M. Réfrégiers and J. Kardos, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3095–E3103 CrossRef CAS.
- B. Shivu, S. Seshadri, J. Li, K. A. Oberg, V. N. Uversky and A. L. Fink, Biochemistry, 2013, 52, 5176–5183 CrossRef CAS.
- E. Calabrò and S. Magazù, Electromagn. Biol. Med., 2020, 39, 129–138 CrossRef.
- M. van de Weert and L. Jørgensen, Analysis of Aggregates and Particles in Protein Pharmaceuticals, John Wiley & Sons, Ltd, 2012, pp. 227–248 Search PubMed.
- M. B. Taraban, R. A. DePaz, B. Lobo and Y. B. Yu, Anal. Chem., 2017, 89, 5494–5502 CrossRef CAS.
- P. C. A. van der Wel, Solid State Nucl. Magn. Reson., 2017, 88, 1–14 CrossRef CAS.
- R. Linser, Solid State Nucl. Magn. Reson., 2017, 87, 45–53 CrossRef CAS.
- O. S. Makin and L. C. Serpell, FEBS J., 2005, 272, 5950–5961 CrossRef CAS.
- M. S. Hunter, D. P. DePonte, D. A. Shapiro, R. A. Kirian, X. Wang, D. Starodub, S. Marchesini, U. Weierstall, R. B. Doak, J. C. H. Spence and P. Fromme, Biophys. J., 2011, 100, 198–206 CrossRef CAS.
- L. Giehm, D. I. Svergun, D. E. Otzen and B. Vestergaard, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3246–3251 CrossRef CAS.
- C. L. P. Oliveira and J. S. Pedersen, Amyloid Fibrils and Prefibrillar Aggregates, John Wiley & Sons, Ltd, 2013, pp. 85–102 Search PubMed.
- J. Porath and P. Flodin, Nature, 1959, 183, 1657–1659 CrossRef CAS.
- H. G. Barth, B. E. Boyes and C. Jackson, Anal. Chem., 1996, 68, 445–466 CrossRef CAS.
- D. M. Walsh, D. M. Hartley, Y. Kusumoto, Y. Fezoui, M. M. Condron, A. Lomakin, G. B. Benedek, D. J. Selkoe and D. B. Teplow, J. Biol. Chem., 1999, 274, 25945–25952 CrossRef CAS.
- D. M. Walsh, A. Lomakin, G. B. Benedek, M. M. Condron and D. B. Teplow, J. Biol. Chem., 1997, 272, 22364–22372 CrossRef CAS.
- M. B. Podlisny, D. M. Walsh, P. Amarante, B. L. Ostaszewski, E. R. Stimson, J. E. Maggio, D. B. Teplow and D. J. Selkoe, Biochemistry, 1998, 37, 3602–3611 CrossRef CAS.
- W. Wang, I. Perovic, J. Chittuluru, A. Kaganovich, L. T. T. Nguyen, J. Liao, J. R. Auclair, D. Johnson, A. Landeru, A. K. Simorellis, S. Ju, M. R. Cookson, F. J. Asturias, J. N. Agar, B. N. Webb, C. Kang, D. Ringe, G. A. Petsko, T. C. Pochapsky and Q. Q. Hoang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 17797–17802 CrossRef CAS.
- A. Rekas, R. B. Knott, A. Sokolova, K. J. Barnham, K. A. Perez, C. L. Masters, S. C. Drew, R. Cappai, C. C. Curtain and C. L. L. Pham, Eur. Biophys. J., 2010, 39, 1407–1419 CrossRef CAS.
- K. Štulík, V. Pacáková and M. Tichá, J. Biochem. Biophys. Methods, 2003, 56, 1–13 CrossRef.
- J. Lebowitz, M. S. Lewis and P. Schuck, Protein Sci., 2002, 11, 2067–2079 CrossRef CAS.
- J. L. Cole, J. W. Lary, T. P. Moody and T. M. Laue, Methods Cell Biol., 2008, 84, 143–179 CAS.
- P. Schuck, Biophys. Rev., 2013, 5, 159–171 CrossRef CAS.
- S. E. Harding and A. J. Rowe, Biochem. Soc. Trans., 2010, 38, 901–907 CrossRef CAS.
- J. P. Gabrielson, M. L. Brader, A. H. Pekar, K. B. Mathis, G. Winter, J. F. Carpenter and T. W. Randolph, J. Pharm. Sci., 2007, 96, 268–279 CrossRef CAS.
- T. Arakawa, D. Ejima, T. Li and J. S. Philo, J. Pharm. Sci., 2010, 99, 1674–1692 CrossRef CAS.
- J. P. Gabrielson, T. W. Randolph, B. S. Kendrick and M. R. Stoner, Anal. Biochem., 2007, 361, 24–30 CrossRef CAS.
- J. P. Gabrielson, K. K. Arthur, B. S. Kendrick, T. W. Randolph and M. R. Stoner, J. Pharm. Sci., 2009, 98, 50–62 CrossRef CAS.
- S. Cao, J. Pollastrini and Y. Jiang, Curr. Pharm. Biotechnol., 2009, 10, 382–390 CAS.
- K.-G. Wahlund, J. Chromatogr. A, 2013, 1287, 97–112 CrossRef CAS.
- W. Fraunhofer and G. Winter, Eur. J. Pharm. Biopharm., 2004, 58, 369–383 CrossRef CAS.
- H. Zhang and D. Lyden, Nat. Protoc., 2019, 14, 1027–1053 CrossRef CAS.
- J. Pollastrini, T. M. Dillon, P. Bondarenko and R. Y.-T. Chou, Anal. Biochem., 2011, 414, 88–98 CrossRef CAS.
- P. Y. Lee, J. Costumbrado, C.-Y. Hsu and Y. H. Kim, J. Visualized Exp., 2012, 62, e3923, DOI:10.3791/3923.
- A. B. Nowakowski, W. J. Wobig and D. H. Petering, Metallomics, 2014, 6, 1068–1078 RSC.
- D. A. Fancy and T. Kodadek, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 6020–6024 CrossRef CAS.
- A. M. Tsai, J. H. van Zanten and M. J. Betenbaugh, Biotechnol. Bioeng., 1998, 59, 273–280 CrossRef CAS.
- A. Ramzi, M. Sutter, W. E. Hennink and W. Jiskoot, J. Pharm. Sci., 2006, 95, 1703–1711 CrossRef CAS.
- M. Y. Lobanov, N. S. Bogatyreva and O. V. Galzitskaya, Mol. Biol., 2008, 42, 623–628 CrossRef CAS.
- C. Wang, X. Zhong, D. B. Ruffner, A. Stutt, L. A. Philips, M. D. Ward and D. G. Grier, J. Pharm. Sci., 2016, 105, 1074–1085 CrossRef CAS.
- P. A. Hassan, S. Rana and G. Verma, Langmuir, 2015, 31, 3–12 CrossRef CAS.
- A. Lomakin, D. S. Chung, G. B. Benedek, D. A. Kirschner and D. B. Teplow, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 1125–1129 CrossRef CAS.
- A. Lomakin and D. B. Teplow, Methods Mol. Biol., 2012, 849, 69–83 CrossRef CAS.
- S. Watanabe and E. M. Jorgensen, Methods Cell Biol., 2012, 111, 283–306 CAS.
- V. L. Anderson and W. W. Webb, BMC Biotechnol., 2011, 11, 125 CrossRef CAS.
- C. Goldsbury, U. Baxa, M. N. Simon, A. C. Steven, A. Engel, J. S. Wall, U. Aebi and S. A. Müller, J. Struct. Biol., 2011, 173, 1–13 CrossRef CAS.
- H. A. Bladen, M. U. Nylen and G. G. Glenner, J. Ultrastruct. Res., 1966, 14, 449–459 CrossRef CAS.
- L. Nielsen, S. Frokjaer, J. F. Carpenter and J. Brange, J. Pharm. Sci., 2001, 90, 29–37 CrossRef CAS.
- B. Michen, C. Geers, D. Vanhecke, C. Endes, B. Rothen-Rutishauser, S. Balog and A. Petri-Fink, Sci. Rep., 2015, 5, 9793 CrossRef CAS.
- E. Callaway, Nature, 2015, 525, 172–174 CrossRef CAS.
- J. A. Rodriguez, M. I. Ivanova, M. R. Sawaya, D. Cascio, F. E. Reyes, D. Shi, S. Sangwan, E. L. Guenther, L. M. Johnson, M. Zhang, L. Jiang, M. A. Arbing, B. L. Nannenga, J. Hattne, J. Whitelegge, A. S. Brewster, M. Messerschmidt, S. Boutet, N. K. Sauter, T. Gonen and D. S. Eisenberg, Nature, 2015, 525, 486–490 CrossRef CAS.
- K. J. Vargas, N. Schrod, T. Davis, R. Fernandez-Busnadiego, Y. V. Taguchi, U. Laugks, V. Lucic and S. S. Chandra, Cell Rep., 2017, 18, 161–173 CrossRef CAS.
- A. Ponce, S. Mejía-Rosales and M. José-Yacamán, Methods Mol. Biol., 2012, 906, 453–471 CAS.
- H. Li, F. Rahimi, S. Sinha, P. Maiti, G. Bitan and K. Murakami, Encyclopedia of Analytical Chemistry, American Cancer Society, 2009 Search PubMed.
- A. K. Paravastu, R. D. Leapman, W.-M. Yau and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18349–18354 CrossRef CAS.
- A. Sen, U. Baxa, M. N. Simon, J. S. Wall, R. Sabate, S. J. Saupe and A. C. Steven, J. Biol. Chem., 2007, 282, 5545–5550 CrossRef CAS.
- A. T. Petkova, R. D. Leapman, Z. Guo, W.-M. Yau, M. P. Mattson and R. Tycko, Science, 2005, 307, 262–265 CrossRef CAS.
- B. Chen, K. R. Thurber, F. Shewmaker, R. B. Wickner and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14339–14344 CrossRef CAS.
- C. Wasmer, A. Lange, H. Van Melckebeke, A. B. Siemer, R. Riek and B. H. Meier, Science, 2008, 319, 1523–1526 CrossRef CAS.
- R. W. Hepler, K. M. Grimm, D. D. Nahas, R. Breese, E. C. Dodson, P. Acton, P. M. Keller, M. Yeager, H. Wang, P. Shughrue, G. Kinney and J. G. Joyce, Biochemistry, 2006, 45, 15157–15167 CrossRef CAS.
- W. B. J. Stine, S. W. Snyder, U. S. Ladror, W. S. Wade, M. F. Miller, T. J. Perun, T. F. Holzman and G. A. Krafft, J. Protein Chem., 1996, 15, 193–203 CrossRef CAS.
- S. Grudzielanek, A. Velkova, A. Shukla, V. Smirnovas, M. Tatarek-Nossol, H. Rehage, A. Kapurniotu and R. Winter, J. Mol. Biol., 2007, 370, 372–384 CrossRef CAS.
- Y. Chen, J. Cai, Q. Xu and Z. W. Chen, Mol. Immunol., 2004, 41, 1247–1252 CrossRef CAS.
- C. Harnagea, M. Vallières, C. P. Pfeffer, D. Wu, B. R. Olsen, A. Pignolet, F. Légaré and A. Gruverman, Biophys. J., 2010, 98, 3070–3077 CrossRef CAS.
- M. Raspanti, M. Reguzzoni, M. Protasoni and D. Martini, Biomacromolecules, 2011, 12, 4344–4347 CrossRef CAS.
- X. Zheng, H. Pan, Z. Wang and H. Chen, J. Microsc., 2011, 241, 162–170 CrossRef CAS.
- Y. Chen, Y. Wu and J. Cai, Biochem. Biophys. Res. Commun., 2007, 361, 391–397 CrossRef CAS.
- I. Greving, M. Cai, F. Vollrath and H. C. Schniepp, Biomacromolecules, 2012, 13, 676–682 CrossRef CAS.
- M. E. van Raaij, I. M. J. Segers-Nolten and V. Subramaniam, Biophys. J., 2006, 91, L96–L98 CrossRef CAS.
- T. Kowalewski and D. M. Holtzman, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3688–3693 CrossRef CAS.
- G. Bruylants, J. Wouters and C. Michaux, Curr. Med. Chem., 2005, 12, 2011–2020 CrossRef CAS.
- R. O’Brien and I. Haq, Biocalorimetry 2, John Wiley & Sons, Ltd, 2005, pp. 1–34 Search PubMed.
- M. J. Cliff and J. E. Ladbury, J. Mol. Recognit., 2003, 16, 383–391 CrossRef CAS.
- R. Zhao, M. So, H. Maat, N. Ray, F. Arisaka, Y. Goto, J. Carver and D. Hall, Biophys. Rev., 2016, 8, 445–471 CrossRef CAS.
- L. H. Lucas, B. A. Ersoy, L. A. Kueltzo, S. B. Joshi, D. T. Brandau, N. Thyagarajapuram, L. J. Peek and C. R. Middaugh, Protein Sci., 2006, 15, 2228–2243 CrossRef CAS.
- S. V. Thakkar, K. M. Allegre, S. B. Joshi, D. B. Volkin and C. R. Middaugh, J. Pharm. Sci., 2012, 101, 3051–3061 CrossRef CAS.
- G. Rao, V. Iyer, M. P. Kosloski, D. S. Pisal, E. Shin, C. R. Middaugh and S. V. Balu-Iyer, J. Pharm. Sci., 2010, 99, 1697–1706 CrossRef CAS.
- C. R. Middaugh, J. M. Kehoe, M. B. Prystowsky, B. Gerber-Jenson, J. C. Jenson and G. W. Litman, Immunochemistry, 1978, 15, 171–187 CrossRef CAS.
- W. Wang, Y. J. Wang and D. Q. Wang, Int. J. Pharm., 2008, 347, 31–38 CrossRef CAS.
- G. A. Eberlein, P. R. Stratton and Y. J. Wang, PDA J. Pharm. Sci. Technol., 1994, 48, 224–230 CAS.
- C. Lendel, B. Bolognesi, A. Wahlström, C. M. Dobson and A. Gräslund, Biochemistry, 2010, 49, 1358–1360 CrossRef CAS.
- Y. S. Kim, T. W. Randolph, M. C. Manning, F. J. Stevens and J. F. Carpenter, J. Biol. Chem., 2003, 278, 10842–10850 CrossRef CAS.
- P. Frid, S. V. Anisimov and N. Popovic, Brain Res. Rev., 2007, 53, 135–160 CrossRef CAS.
- M. Groenning, J. Chem. Biol., 2010, 3, 1–18 CrossRef.
- H. Naiki, K. Higuchi, M. Hosokawa and T. Takeda, Anal. Biochem., 1989, 177, 244–249 CrossRef CAS.
- A. Hawe, M. Sutter and W. Jiskoot, Pharm. Res., 2008, 25, 1487–1499 CrossRef CAS.
- M. R. H. Krebs, E. H. C. Bromley and A. M. Donald, J. Struct. Biol., 2005, 149, 30–37 CrossRef CAS.
- M. Biancalana, K. Makabe, A. Koide and S. Koide, J. Mol. Biol., 2009, 385, 1052–1063 CrossRef CAS.
- V. I. Stsiapura, A. A. Maskevich, V. A. Kuzmitsky, K. K. Turoverov and I. M. Kuznetsova, J. Phys. Chem. A, 2007, 111, 4829–4835 CrossRef CAS.
- V. Foderà, F. Librizzi, M. Groenning, M. van de Weert and M. Leone, J. Phys. Chem. B, 2008, 112, 3853–3858 CrossRef.
- A. Bothra, A. Bhattacharyya, C. Mukhopadhyay, K. Bhattacharyya and S. Roy, J. Biomol. Struct. Dyn., 1998, 15, 959–966 CrossRef CAS.
- B. Ranjbar and P. Gill, Chem. Biol. Drug Des., 2009, 74, 101–120 CrossRef CAS.
- N. J. Greenfield, Nat. Protoc., 2006, 1, 2876–2890 CrossRef CAS.
- S. Frokjaer and D. E. Otzen, Nat. Rev. Drug Discovery, 2005, 4, 298–306 CrossRef CAS.
- C. Perez-Iratxeta and M. A. Andrade-Navarro, BMC Struct. Biol., 2008, 8, 25 CrossRef.
- L. Whitmore and B. A. Wallace, Nucleic Acids Res., 2004, 32, 668–673 CrossRef.
- X. Li-Blatter and J. Seelig, J. Phys. Chem. B, 2019, 123, 10181–10191 CrossRef CAS.
- S. Ma, X. Cao, M. Mak, A. Sadik, C. Walkner, T. B. Freedman, I. K. Lednev, R. K. Dukor and L. A. Nafie, J. Am. Chem. Soc., 2007, 129, 12364–12365 CrossRef CAS.
- P. I. Haris and F. Severcan, J. Mol. Catal. B: Enzym., 1999, 7, 207–221 CrossRef CAS.
- B. W. Caughey, A. Dong, K. S. Bhat, D. Ernst, S. F. Hayes and W. S. Caughey, Biochemistry, 1991, 30, 7672–7680 CrossRef CAS.
- H. Li, R. Lantz and D. Du, Molecules, 2019, 24, 186 CrossRef.
- P. Kheddo, M. J. Cliff, S. Uddin, C. F. van der Walle and A. P. Golovanov, mAbs, 2016, 8, 1245–1258 CrossRef CAS.
- N. L. Fawzi, J. Ying, D. A. Torchia and G. M. Clore, J. Am. Chem. Soc., 2010, 132, 9948–9951 CrossRef CAS.
- K.-P. Wu and J. Baum, J. Am. Chem. Soc., 2010, 132, 5546–5547 CrossRef CAS.
- T. Muraoka, K. Adachi, M. Ui, S. Kawasaki, N. Sadhukhan, H. Obara, H. Tochio, M. Shirakawa and K. Kinbara, Angew. Chem., Int. Ed., 2013, 52, 2430–2434 CrossRef CAS.
- J. Cavanagh, W. J. Fairbrother, A. G. Palmer III, M. Rance and N. J. Skelton, Protein NMR Spectroscopy, Academic Press, Burlington, 2nd edn, 2007 Search PubMed.
- R. Rajan, Y. Suzuki and K. Matsumura, Macromol. Biosci., 2018, 18, 1800016 CrossRef.
- J. T. Pedersen and N. H. H. Heegaard, Anal. Chem., 2013, 85, 4215–4227 CrossRef CAS.
- M. Ahmed, J. Davis, D. Aucoin, T. Sato, S. Ahuja, S. Aimoto, J. I. Elliott, W. E. Van Nostrand and S. O. Smith, Nat. Struct. Mol. Biol., 2010, 17, 561–567 CrossRef CAS.
- S. Chimon, M. A. Shaibat, C. R. Jones, D. C. Calero, B. Aizezi and Y. Ishii, Nat. Struct. Mol. Biol., 2007, 14, 1157–1164 CrossRef CAS.
- H. Heise, M. S. Celej, S. Becker, D. Riedel, A. Pelah, A. Kumar, T. M. Jovin and M. Baldus, J. Mol. Biol., 2008, 380, 444–450 CrossRef CAS.
- A. K. Paravastu, I. Qahwash, R. D. Leapman, S. C. Meredith and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 7443–7448 CrossRef CAS.
- V. Prasanna, B. Gopal, M. R. Murthy, D. V. Santi and P. Balaram, Proteins, 1999, 34, 356–368 CrossRef CAS.
- K. Hinds, J. J. Koh, L. Joss, F. Liu, M. Baudys and S. W. Kim, Bioconjugate Chem., 2000, 11, 195–201 CrossRef CAS.
- L. G. Czaplewski, J. McKeating, C. J. Craven, L. D. Higgins, V. Appay, A. Brown, T. Dudgeon, L. A. Howard, T. Meyers, J. Owen, S. R. Palan, P. Tan, G. Wilson, N. R. Woods, C. M. Heyworth, B. I. Lord, D. Brotherton, R. Christison, S. Craig, S. Cribbes, R. M. Edwards, S. J. Evans, R. Gilbert, P. Morgan, E. Randle, N. Schofield, P. G. Varley, J. Fisher, J. P. Waltho and M. G. Hunter, J. Biol. Chem., 1999, 274, 16077–16084 CrossRef CAS.
- K. Y. Fang, S. A. Lieblich and D. A. Tirrell, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 264–267 CrossRef CAS.
- S. A. Lieblich, K. Y. Fang, J. K. B. Cahn, J. Rawson, J. LeBon, H. T. Ku and D. A. Tirrell, J. Am. Chem. Soc., 2017, 139, 8384–8387 CrossRef CAS.
- D. Samuel, T. K. Kumar, G. Ganesh, G. Jayaraman, P. W. Yang, M. M. Chang, V. D. Trivedi, S. L. Wang, K. C. Hwang, D. K. Chang and C. Yu, Protein Sci., 2000, 9, 344–352 CrossRef CAS.
- M. Kudou, K. Shiraki, S. Fujiwara, T. Imanaka and M. Takagi, Eur. J. Biochem., 2003, 270, 4547–4554 CrossRef CAS.
- E. De Bernardez Clark, D. Hevehan, S. Szela and J. Maachupalli-Reddy, Biotechnol. Prog., 1998, 14, 47–54 CrossRef CAS.
- R. Rudolph and H. Lilie, FASEB J., 1996, 10, 49–56 CrossRef CAS.
- P. Alam, K. Siddiqi, S. K. Chturvedi and R. H. Khan, Int. J. Biol. Macromol., 2017, 103, 208–219 CrossRef CAS.
- S. Prabhudesai, S. Sinha, A. Attar, A. Kotagiri, A. G. Fitzmaurice, R. Lakshmanan, M. I. Ivanova, J. A. Loo, F.-G. Klärner, T. Schrader, M. Stahl, G. Bitan and J. M. Bronstein, Neurotherapeutics, 2012, 9, 464–476 CrossRef CAS.
- S. Sinha, D. H. J. Lopes, Z. Du, E. S. Pang, A. Shanmugam, A. Lomakin, P. Talbiersky, A. Tennstaedt, K. McDaniel, R. Bakshi, P.-Y. Kuo, M. Ehrmann, G. B. Benedek, J. A. Loo, F.-G. Klärner, T. Schrader, C. Wang and G. Bitan, J. Am. Chem. Soc., 2011, 133, 16958–16969 CrossRef CAS.
- A. Gaeta and R. C. Hider, Br. J. Pharmacol., 2005, 146, 1041–1059 CrossRef CAS.
- M. Di Vaira, C. Bazzicalupi, P. Orioli, L. Messori, B. Bruni and P. Zatta, Inorg. Chem., 2004, 43, 3795–3797 CrossRef CAS.
- C. Cabaleiro-Lago, I. Lynch, K. A. Dawson and S. Linse, Langmuir, 2010, 26, 3453–3461 CrossRef CAS.
- A. Bellova, E. Bystrenova, M. Koneracka, P. Kopcansky, F. Valle, N. Tomasovicova, M. Timko, J. Bagelova, F. Biscarini and Z. Gazova, Nanotechnology, 2010, 21, 65103 CrossRef.
- S. Il Yoo, M. Yang, J. R. Brender, V. Subramanian, K. Sun, N. E. Joo, S.-H. Jeong, A. Ramamoorthy and N. A. Kotov, Angew. Chem., Int. Ed., 2011, 50, 5110–5115 CrossRef.
- H. A. Popiel, T. Takeuchi, J. R. Burke, W. J. Strittmatter, T. Toda, K. Wada and Y. Nagai, Neurotherapeutics, 2013, 10, 440–446 CrossRef CAS.
- K. Rajasekhar, S. N. Suresh, R. Manjithaya and T. Govindaraju, Sci. Rep., 2015, 5, 8139 CrossRef CAS.
- J. A. Loureiro, R. Crespo, H. Börner, P. M. Martins, F. A. Rocha, M. Coelho, M. C. Pereira and S. Rocha, J. Mater. Chem. B, 2014, 2, 2259–2264 RSC.
- E.-N. Lee, H.-J. Cho, C.-H. Lee, D. Lee, K. C. Chung and S. R. Paik, Biochemistry, 2004, 43, 3704–3715 CrossRef CAS.
- A. A. Valiente-Gabioud, M. C. Miotto, M. E. Chesta, V. Lombardo, A. Binolfi and C. O. Fernández, Acc. Chem. Res., 2016, 49, 801–808 CrossRef CAS.
- P. W. Yang, T. K. Kumar, G. Jayaraman and C. Yu, Biochem. Mol. Biol. Int., 1996, 38, 393–399 CAS.
- V. Srinivas and D. Balasubramanian, Langmuir, 1995, 11, 2830–2833 CrossRef CAS.
- U. Brinkmann, J. Buchner and I. Pastan, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 3075–3079 CrossRef CAS.
- R. Sakamoto, S. Nishikori and K. Shiraki, Biotechnol. Prog., 2004, 20, 1128–1133 CrossRef CAS.
- J. Buchner and R. Rudolph, Biotechnology, 1991, 9, 157–162 CAS.
- A. Sgarbossa, Int. J. Mol. Sci., 2012, 13, 17121–17137 CrossRef CAS.
- J. Folch, M. Ettcheto, D. Petrov, S. Abad, I. Pedrós, M. Marin, J. Olloquequi and A. Camins, Neurologia, 2018, 33, 47–58 CrossRef CAS.
- J. M. Ringman, S. A. Frautschy, G. M. Cole, D. L. Masterman and J. L. Cummings, Curr. Alzheimer Res., 2005, 2, 131–136 CrossRef CAS.
- M. de la Luz Cádiz-Gurrea, S. Fernández-Arroyo and A. Segura-Carretero, Int. J. Mol. Sci., 2014, 15, 20382–20402 CrossRef.
- J. W. Betts, A. S. Sharili, L. M. Phee and D. W. Wareham, J. Nat. Prod., 2015, 78, 2145–2148 CrossRef CAS.
- N. Bitu Pinto, B. da Silva Alexandre, K. R. T. Neves, A. H. Silva, L. K. A. M. Leal and G. S. B. Viana, J. Evidence-Based Complementary Altern. Med., 2015, 2015, 161092 Search PubMed.
- F. Yang, G. P. Lim, A. N. Begum, O. J. Ubeda, M. R. Simmons, S. S. Ambegaokar, P. P. Chen, R. Kayed, C. G. Glabe, S. A. Frautschy and G. M. Cole, J. Biol. Chem., 2005, 280, 5892–5901 CrossRef CAS.
- K. Ono and M. Yamada, Neurobiol. Dis., 2007, 25, 446–454 CrossRef CAS.
- S. M. Yatin, S. Varadarajan and D. A. Butterfield, J. Alzheimer's Dis., 2000, 2, 123–131 CAS.
- Z. Liu, T. Zhou, A. C. Ziegler, P. Dimitrion and L. Zuo, Oxid. Med. Cell. Longevity, 2017, 2017, 2525967 Search PubMed.
- K. Brieger, S. Schiavone, F. J. J. Miller and K.-H. Krause, Swiss Med. Wkly., 2012, 142, w13659 CAS.
- V. Lobo, A. Patil, A. Phatak and N. Chandra, Pharmacogn. Rev., 2010, 4, 118–126 CrossRef CAS.
- M. E. Goldberg, N. Expert-Bezançon, L. Vuillard and T. Rabilloud, Folding Des., 1996, 1, 21–27 CrossRef CAS.
- L. Vuillard, T. Rabilloud and M. E. Goldberg, Eur. J. Biochem., 1998, 256, 128–135 CrossRef CAS.
- K. Wangkanont, K. T. Forest and L. L. Kiessling, Protein Expression Purif., 2015, 115, 19–25 CrossRef CAS.
- N. Expert-Bezançon, T. Rabilloud, L. Vuillard and M. E. Goldberg, Biophys. Chem., 2003, 100, 469–479 CrossRef.
- M. Hagihara, A. Takei, T. Ishii, F. Hayashi, K. Kubota, K. Wakamatsu and N. Nameki, FEBS Open Bio, 2012, 2, 20–25 CrossRef.
- S. Choudhary, N. Kishore and R. V. Hosur, Sci. Rep., 2015, 5, 17599 CrossRef CAS.
- M. Dasgupta and N. Kishore, PLoS One, 2017, 12, e0172208 CrossRef.
- A. Venkatraman, E. Murugan, S. J. Lin, G. S. L. Peh, L. Rajamani and J. S. Mehta, Sci. Rep., 2020, 10, 4011 CrossRef CAS.
- K. Tsumoto, M. Umetsu, I. Kumagai, D. Ejima, J. S. Philo and T. Arakawa, Biotechnol. Prog., 2004, 20, 1301–1308 CrossRef CAS.
- S. Choudhary, S. N. Save, N. Kishore and R. V. Hosur, PLoS One, 2016, 11, e0166487 CrossRef.
- V. L. N. Ngoungoure, J. Schluesener, P. F. Moundipa and H. Schluesener, Mol. Nutr. Food Res., 2015, 59, 8–20 CrossRef CAS.
- S. Ghosh, S. Banerjee and P. C. Sil, Food Chem. Toxicol., 2015, 83, 111–124 CrossRef CAS.
- N. Ahsan, S. Mishra, M. K. Jain, A. Surolia and S. Gupta, Sci. Rep., 2015, 5, 9862 CrossRef CAS.
- Y. Song, E. G. Moore, Y. Guo and J. S. Moore, J. Am. Chem. Soc., 2017, 139, 4298–4301 CrossRef CAS.
- A. J. Doig, Curr. Opin. Drug Discovery Dev., 2007, 10, 533–539 CAS.
- C. Adessi and C. Soto, Curr. Med. Chem., 2002, 9, 963–978 CrossRef CAS.
- P. Alam, S. K. Chaturvedi, M. K. Siddiqi, R. K. Rajpoot, M. R. Ajmal, M. Zaman and R. H. Khan, Sci. Rep., 2016, 6, 26759 CrossRef CAS.
- N. V. Katre, Adv. Drug Delivery Rev., 1993, 10, 91–114 CrossRef CAS.
- F. Kreppel and S. Kochanek, Mol. Ther., 2008, 16, 16–29 CrossRef CAS.
- F. M. Veronese and G. Pasut, Drug Discovery Today, 2005, 10, 1451–1458 CrossRef CAS.
- A. Kolate, D. Baradia, S. Patil, I. Vhora, G. Kore and A. Misra, J. Controlled Release, 2014, 192, 67–81 CrossRef CAS.
- A. Bunker, Phys. Procedia, 2012, 34, 24–33 CrossRef.
- J. D. Belsey, M. Geraint and T. A. Dixon, Int. J. Clin. Pract., 2010, 64, 944–955 CrossRef CAS.
- J. A. Di Palma, J. R. Smith and M. vB Cleveland, Am. J. Gastroenterol., 2002, 97, 1776–1779 CrossRef CAS.
- S.-L. Chen, R.-H. Fu, S.-F. Liao, S.-P. Liu, S.-Z. Lin and Y.-C. Wang, Cell Transplant., 2018, 27, 275–284 Search PubMed.
- D. S. Pisal, M. P. Kosloski and S. V. Balu-Iyer, J. Pharm. Sci., 2010, 99, 2557–2575 CrossRef CAS.
- J. K. Dozier and M. D. Distefano, Int. J. Mol. Sci., 2015, 16, 25831–25864 CrossRef CAS.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS.
- N. Kameta, T. Matsuzawa, K. Yaoi and M. Masuda, RSC Adv., 2016, 6, 36744–36750 RSC.
- K. Ikeda, T. Okada, S.-I. Sawada, K. Akiyoshi and K. Matsuzaki, FEBS Lett., 2006, 580, 6587–6595 CrossRef CAS.
- S. Sawada, Y. Sasaki, Y. Nomura and K. Akiyoshi, Colloid Polym. Sci., 2011, 289, 685–691 CrossRef CAS.
- Y. Nomura, M. Ikeda, N. Yamaguchi, Y. Aoyama and K. Akiyoshi, FEBS Lett., 2003, 553, 271–276 CrossRef CAS.
- T. Furuki and M. Sakurai, Cryobiol. Cryotechnol., 2016, 62, 47–51 Search PubMed.
- D. K. Hincha and A. Thalhammer, Biochem. Soc. Trans., 2012, 40, 1000–1003 CrossRef CAS.
- K. Goyal, L. J. Walton and A. Tunnacliffe, Biochem. J., 2005, 388, 151–157 CrossRef CAS.
- T. Furuki and M. Sakurai, Biochim. Biophys. Acta, Proteins Proteomics, 2016, 1864, 1237–1243 CrossRef CAS.
- R. Rajan and K. Matsumura, J. Mater. Chem. B, 2015, 3, 5683–5689 RSC.
- R. Rajan and K. Matsumura, Sci. Rep., 2017, 7, 45777 CrossRef CAS.
- R. J. Mancini, J. Lee and H. D. Maynard, J. Am. Chem. Soc., 2012, 134, 8474–8479 CrossRef CAS.
- J. Lee, E.-W. Lin, U. Y. Lau, J. L. Hedrick, E. Bat and H. D. Maynard, Biomacromolecules, 2013, 14, 2561–2569 CrossRef CAS.
- H. Sun, J. Liu, S. Li, L. Zhou, J. Wang, L. Liu, F. Lv, Q. Gu, B. Hu, Y. Ma and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 5988–5993 CrossRef CAS.
- R. Chai, C. Xing, J. Qi, Y. Fan, H. Yuan, R. Niu, Y. Zhan and J. Xu, Adv. Funct. Mater., 2016, 26, 9026–9031 CrossRef CAS.
- L. Breydo, B. Newland, H. Zhang, A. Rosser, C. Werner, V. N. Uversky and W. Wang, Biochem. Biophys. Res. Commun., 2016, 469, 830–835 CrossRef CAS.
- K. Akiyoshi, S. Deguchi, N. Moriguchi, S. Yamaguchi and J. Sunamoto, Macromolecules, 1993, 26, 3062–3068 CrossRef CAS.
- T. Nishikawa, K. Akiyoshi and J. Sunamoto, Macromolecules, 1994, 27, 7654–7659 CrossRef CAS.
- T. Nishikawa, K. Akiyoshi and J. Sunamoto, J. Am. Chem. Soc., 1996, 118, 6110–6115 CrossRef CAS.
- K. Akiyoshi, Y. Sasaki and J. Sunamoto, Bioconjugate Chem., 1999, 10, 321–324 CrossRef CAS.
- Y. Nomura, Y. Sasaki, M. Takagi, T. Narita, Y. Aoyama and K. Akiyoshi, Biomacromolecules, 2005, 6, 447–452 CrossRef CAS.
- T. Nochi, Y. Yuki, H. Takahashi, S. Sawada, M. Mejima, T. Kohda, N. Harada, I. G. Kong, A. Sato, N. Kataoka, D. Tokuhara, S. Kurokawa, Y. Takahashi, H. Tsukada, S. Kozaki, K. Akiyoshi and H. Kiyono, Nat. Mater., 2010, 9, 572–578 CrossRef CAS.
- Y. Liu, S. Chakrabortee, R. Li, Y. Zheng and A. Tunnacliffe, FEBS Lett., 2011, 585, 630–634 CrossRef CAS.
- T. Furuki, Y. Takahashi, R. Hatanaka, T. Kikawada, T. Furuta and M. Sakurai, J. Phys. Chem. B, 2020, 124, 2747–2759 CrossRef CAS.
- A. V. Popova, M. Hundertmark, R. Seckler and D. K. Hincha, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 1879–1887 CrossRef CAS.
- M. A. S. Artur, J. Rienstra, T. J. Dennis, J. M. Farrant, W. Ligterink and H. Hilhorst, Front. Plant Sci., 2019, 10, 1–15 CrossRef.
- D. Tolleter, M. Jaquinod, C. Mangavel, C. Passirani, P. Saulnier, S. Manon, E. Teyssier, N. Payet, M.-H. Avelange-Macherel and D. Macherel, Plant Cell, 2007, 19, 1580–1589 CrossRef CAS.
- M. Hundertmark and D. K. Hincha, BMC Genomics, 2008, 9, 1–22 CrossRef.
- S. Li, N. Chakraborty, A. Borcar, M. A. Menze, M. Toner and S. C. Hand, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 20859–20864 CrossRef CAS.
- D. Kovacs, B. Agoston and P. Tompa, Plant Signaling Behav., 2008, 3, 710–713 CrossRef.
- A. Banerjee and A. Roychoudhury, Plant Growth Regul., 2016, 79, 1–17 CrossRef CAS.
- S. E. Kudaibergenov, Polyampholytes: synthesis, characterization and application, Springer Science & Business Media, 2012 Search PubMed.
- K. Matsumura, R. Rajan, S. Ahmed and M. Jain, Biopolymers for Medical Applications, CRC Press, Boca Raton, FL, 2016, pp. 165–182 Search PubMed.
- K. M. Zurick and M. Bernards, J. Appl. Polym. Sci., 2014, 131, 40069 CrossRef.
- H. Kitano, T. Mori, Y. Takeuchi, S. Tada, M. Gemmei-Ide, Y. Yokoyama and M. Tanaka, Macromol. Biosci., 2005, 5, 314–321 CrossRef CAS.
- H. Kitano, T. Kondo, T. Kamada, S. Iwanaga, M. Nakamura and K. Ohno, Colloids Surf., B, 2011, 88, 455–462 CrossRef CAS.
- A. J. Keefe and S. Jiang, Nat. Chem., 2011, 4, 59–63 CrossRef.
- N. Sharma, R. Rajan, S. Makhaik and K. Matsumura, ACS Omega, 2019, 4, 12186–12193 CrossRef CAS.
- D. Zhao, R. Rajan and K. Matsumura, ACS Appl. Mater. Interfaces, 2019, 11, 39459–39469 CrossRef CAS.
- K. Matsumura and S.-H. Hyon, Biomaterials, 2009, 30, 4842–4849 CrossRef CAS.
- P. Zhang, F. Sun, C. Tsao, S. Liu, P. Jain, A. Sinclair, H.-C. Hung, T. Bai, K. Wu and S. Jiang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12046–12051 CrossRef CAS.
- J. H. Moon and Kenry, Conjugated Polymers for Biological and Biomedical Applications, John Wiley & Sons, Ltd, 2018, pp. 215–241 Search PubMed.
- T. Congdon, R. Notman and M. I. Gibson, Biomacromolecules, 2013, 14, 1578–1586 CrossRef CAS.
- D. E. Mitchell, A. E. R. Fayter, R. C. Deller, M. Hasan, J. Gutierrez-Marcos and M. I. Gibson, Mater. Horiz., 2019, 6, 364–368 RSC.
- N. K. Jain and I. Roy, Protein Sci., 2009, 18, 24–36 CAS.
- A. A. Sofronova, V. A. Izumrudov, V. I. Muronetz and P. I. Semenyuk, Polymer, 2017, 108, 281–287 CrossRef CAS.
- K. Debnath, N. Pradhan, B. K. Singh, N. R. Jana and N. R. Jana, ACS Appl. Mater. Interfaces, 2017, 9, 24126–24139 CrossRef CAS.
- J. Madeira do O, F. Mastrotto, N. Francini, S. Allen, C. F. van der Walle, S. Stolnik and G. Mantovani, J. Mater. Chem. B, 2018, 6, 1044–1054 RSC.
- Y. J. Wang and G. Yu, J. Polym. Sci., Part B: Polym. Phys., 2019, 57, 1557–1558 CrossRef CAS.
- J. Liu and B. Liu, Conjugated Polymers for Biological and Biomedical Applications, John Wiley & Sons, Ltd, 2018, pp. 1–33 Search PubMed.
- C. Xie and K. Pu, Conjugated Polymers for Biological and Biomedical Applications, John Wiley & Sons, Ltd, 2018, pp. 111–133 Search PubMed.
- X. Liu, W. Huang and Q. Fan, Conjugated Polymers for Biological and Biomedical Applications, John Wiley & Sons, Ltd, 2018, pp. 171–213 Search PubMed.
- H. Sun, F. Lv, L. Liu and S. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 22973–22978 CrossRef CAS.
- J. Honda, H. Andou, T. Mannen and S. Sugimoto, in Bioseparation Engineering, ed. I. Endo, T. Nagamune, S. Katoh and T. Yonemoto, Elsevier, 2000, vol. 16, pp. 101–105 Search PubMed.
- M. P. Murphy and H. LeVine 3rd, J. Alzheimer's Dis., 2010, 19, 311–323 Search PubMed.
- R. W. Carrell and B. Gooptu, Curr. Opin. Struct. Biol., 1998, 8, 799–809 CrossRef CAS.
- P. J. Thomas, B. H. Qu and P. L. Pedersen, Trends Biochem. Sci., 1995, 20, 456–459 CrossRef CAS.
- C. Soto, J. Mol. Med., 1999, 77, 412–418 CrossRef CAS.
- J. W. Kelly, Curr. Opin. Struct. Biol., 1998, 8, 101–106 CrossRef CAS.
- G. Zettlmeissl, R. Rudolph and R. Jaenicke, Biochemistry, 1979, 18, 5567–5571 CrossRef CAS.
- T. Kiefhaber, R. Rudolph, H.-H. Kohler and J. Buchner, Bio/Technology, 1991, 9, 825–829 CAS.
- D. L. Hevehan and E. De Bernardez Clark, Biotechnol. Bioeng., 1997, 54, 221–230 CrossRef CAS.
- G. Taylor, M. Hoare, D. R. Gray and F. A. O. Marston, Bio/Technology, 1986, 4, 553–557 CAS.
- H. H. Wong, B. K. O’Neill and A. P. Middelberg, Bioseparation, 1996, 6, 361–372 CAS.
- C. Tin Lee, G. Morreale and A. P. J. Middelberg, Biotechnol. Bioeng., 2004, 85, 103–113 CrossRef.
- N. K. Puri, E. Crivelli, M. Cardamone, R. Fiddes, J. Bertolini, B. Ninham and M. R. Brandon, Biochem. J., 1992, 285(Pt 3), 871–879 CrossRef CAS.
- R. H. Khan, K. B. Rao, A. N. Eshwari, S. M. Totey and A. K. Panda, Biotechnol. Prog., 1998, 14, 722–728 CrossRef CAS.
- K. Tsumoto, M. Umetsu, I. Kumagai, D. Ejima and T. Arakawa, Biochem. Biophys. Res. Commun., 2003, 312, 1383–1386 CrossRef CAS.
- S. S. Mahmoud, S. Wang, M. M. Moloney and H. R. Habibi, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 1998, 120, 657–663 CrossRef CAS.
- E. D. B. Clark, Curr. Opin. Biotechnol., 1998, 9, 157–163 CrossRef.
- M. Yasuda, Y. Murakami, A. Sowa, H. Ogino and H. Ishikawa, Biotechnol. Prog., 1998, 14, 601–606 CrossRef CAS.
- M. Umetsu, K. Tsumoto, M. Hara, K. Ashish, S. Goda, T. Adschiri and I. Kumagai, J. Biol. Chem., 2003, 278, 8979–8987 CrossRef CAS.
- S. Machida, S. Ogawa, S. Xiaohua, T. Takaha, K. Fujii and K. Hayashi, FEBS Lett., 2000, 486, 131–135 CrossRef CAS.
- M. M. Altamirano, R. Golbik, R. Zahn, A. M. Buckle and A. R. Fersht, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 3576–3578 CrossRef CAS.
- N. S. Preston, D. J. Baker, S. P. Bottomley and M. G. Gore, Biochim. Biophys. Acta, 1999, 1426, 99–109 CrossRef CAS.
- K. Tsumoto, M. Umetsu, H. Yamada, T. Ito, S. Misawa and I. Kumagai, Protein Eng., 2003, 16, 535–541 CrossRef CAS.
- T. Mannen, S. Yamaguchi, J. Honda, S. Sugimoto and T. Nagamune, J. Biosci. Bioeng., 2001, 91, 403–408 CrossRef CAS.
- N. Karuppiah and A. Sharma, Biochem. Biophys. Res. Commun., 1995, 211, 60–66 CrossRef CAS.
- A. Cooper, J. Am. Chem. Soc., 1992, 114, 9208–9209 CrossRef CAS.
- S.-C. Lin, K.-L. Lin, H.-C. Chiu and S. Lin, Biotechnol. Bioeng., 2000, 67, 505–512 CrossRef CAS.
- Y.-J. Chen, L.-W. Huang, H.-C. Chiu and S.-C. Lin, Enzyme Microb. Technol., 2003, 32, 120–130 CrossRef CAS.
- I. Roy and M. N. Gupta, Protein Eng., 2003, 16, 1153–1157 CrossRef CAS.
- Z. Huang, C. Ni, X. Zhou, Y. Liu, Y. Tan, J. Xiao, W. Feng, X. Li and S. Yang, Biotechnol. Prog., 2009, 25, 1387–1395 CrossRef CAS.
- K. Mondal, S. Raghava, B. Barua, R. Varadarajan and M. N. Gupta, Langmuir, 2007, 23, 70–75 CrossRef CAS.
- S. M. Doyle and S. Wickner, Trends Biochem. Sci., 2009, 34, 40–48 CrossRef CAS.
- M. Nakamoto, T. Nonaka, K. J. Shea, Y. Miura and Y. Hoshino, J. Am. Chem. Soc., 2016, 138, 4282–4285 CrossRef CAS.
- C. T. Lee, A. M. Buswell and A. P. J. Middelberg, Chem. Eng. Sci., 2002, 57, 1679–1684 CrossRef CAS.
- H. Hohenblum, K. Vorauer-Uhl, H. Katinger and D. Mattanovich, J. Biotechnol., 2004, 109, 3–11 CrossRef CAS.
- S. Katoh, Y. Sezai, T. Yamaguchi, Y. Katoh, H. Yagi and D. Nohara, Process Biochem., 1999, 35, 297–300 CrossRef CAS.
- D. B. Wetlaufer and Y. Xie, Protein Sci., 1995, 4, 1535–1543 CrossRef CAS.
- M. Cardamone, N. K. Puri and M. R. Brandon, Biochemistry, 1995, 34, 5773–5794 CrossRef CAS.
- Y. Maeda, H. Yamada, T. Ueda and T. Imoto, Protein Eng., Des. Sel., 1996, 9, 461–465 CrossRef CAS.
- I. Kurucz, J. A. Titus, C. R. Jost and D. M. Segal, Mol. Immunol., 1995, 32, 1443–1452 CrossRef CAS.
- J. Maachupalli-Reddy, B. D. Kelley and E. De Bernardez Clark, Biotechnol. Prog., 1997, 13, 144–150 CrossRef CAS.
- F.-H. Ma, Y. An, J. Wang, Y. Song, Y. Liu and L. Shi, ACS Nano, 2017, 11, 10549–10557 CrossRef CAS.
- B. M. Gorovits and P. M. Horowitz, Biochemistry, 1998, 37, 6132–6135 CrossRef CAS.
- R. J. St John, J. F. Carpenter and T. W. Randolph, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 13029–13033 CrossRef.
- D. Foguel, C. R. Robinson, P. C. de Sousa Jr, J. L. Silva and A. S. Robinson, Biotechnol. Bioeng., 1999, 63, 552–558 CrossRef CAS.
- B. G. Lefebvre, M. J. Gage and A. S. Robinson, Biotechnol. Prog., 2004, 20, 623–629 CrossRef CAS.
- B. E. Schoner, K. S. Bramlett, H. Guo and T. P. Burris, Mol. Genet. Metab., 2005, 85, 318–322 CrossRef CAS.
- R. Schlegl, G. Iberer, C. Machold, R. Necina and A. Jungbauer, J. Chromatogr. A, 2003, 1009, 119–132 CrossRef CAS.
- R. Schlegl, R. Necina and A. Jungbauer, Chem. Eng. Technol., 2005, 28, 1375–1386 CrossRef CAS.
- B.-J. Park, C.-H. Lee and Y.-M. Koo, Korean J. Chem. Eng., 2005, 22, 425–432 CrossRef CAS.
- T. H. Cho, S. J. Ahn and E. K. Lee, Bioseparation, 2001, 10, 189–196 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |