
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmplified spontaneous emission from oligo(p-phenylenevinylene) derivatives†
Masashi
Mamada
 *abc,
Hajime
Nakanotani
ab and
Chihaya
Adachi
*abcd
*abc,
Hajime
Nakanotani
ab and
Chihaya
Adachi
*abcd
aCenter for Organic Photonics and Electronics Research (OPERA), Kyushu University, Fukuoka 819-0395, Japan. E-mail: mamada@opera.kyushu-u.ac.jp; adachi@cstf.kyushu-u.ac.jp
bJST, ERATO, Adachi Molecular Exciton Engineering Project c/o Center for Organic Photonics and Electronics Research (OPERA), Kyushu University, Nishi, Fukuoka 819-0395, Japan
cAcademia-Industry Molecular Systems for Devices Research and Education Center (AIMS), Kyushu University, Nishi, Fukuoka 819-0395, Japan
dInternational Institute for Carbon Neutral Energy Research (WPI-I2CNER), Kyushu University, Nishi, Fukuoka 819-0395, Japan
First published on 30th April 2021
Abstract
For an improved understanding of molecular design for efficient laser materials, we report structure–property relationships in a series of oligo(p-phenylenevinylene) derivatives. The π-extended phenylenevinylene systems have been studied less frequently than the well-known 1,4-distyrylbenzene framework, despite easy syntheses and great prospects for optoelectronic devices. Here, light amplification behaviours, with amplified spontaneous emission thresholds as low as 7.2 μJ cm−2, were characterized for oligo(p-phenylenevinylene) crystals. Substitution with cyano groups significantly changes the crystal packing and emission properties, while also improves the lasing behaviour.
Introduction
Organic lasers have had a long history since the first demonstration of the dye laser in 1966.1 Research and development has been extended to solid-state lasers, with the potential of innovative optoelectronic applications.2–4 Although organic semiconductor laser devices that enable light amplification under electrical pumping have been studied for several decades, they have not been realized until very recently.5,6The gain media for organic solid-state lasers can be classified into two main classes: amorphous films of small molecules and polymers, and organic crystals.7–14 Amorphous films are compatible with organic light-emitting diode (OLED) structures; therefore, they are important for the development of organic semiconductor laser diodes (OSLDs).5 Single crystals have several advantages, such as higher mobilities, light outcoupling, and better stabilities based on long-range molecular order. They can also be used in light-emitting field-effect transistors (LEFETs).6 For both classes, the molecular structures of the laser materials involve common basic π-electronic systems, such as simple oligo-aromatics and oligomeric phenylenevinylenes.15–24 Therefore, fundamental knowledge of the relationship between basic structure and properties is necessary to develop a wide variety of laser materials.
Stilbene is an important moiety that is used in numerous solid-state laser materials. Stilbene derivatives with carbazole rings form amorphous films with excellent laser properties,25–28 which were key to the success of quasi-continuous-wave lasers and electrically pumped lasers.5,29,30 In addition, poly(p-phenylenevinylene) (PPV) is a notable backbone for deriving excellent laser materials.31–33 Meanwhile, high-quality single crystals can easily be made from 1,4-distyrylbenzene (DSB/2PV/P3V2, 1a in Fig. 1).34–36 Thus, there have been many derivatives based on 1a that have several substituents, such as cyano and alkoxy groups, which control crystal-packing structures.11,14 In particular, there are many reports on compounds having cyano groups on the vinyl groups (1b and 1c).37–53 Both compounds exhibit polymorphism with blue and green emission. The blue phase of 1b and 1c has a herringbone packing structure similar to 1a, which results in similar emission maxima in the blue region. These are also known to have lasing capabilities. On the other hand, the green phases have strong π-stacking structures that exhibit excimer-like emission. Therefore, they have high mobilities in single-crystal LEFETs but exhibit no light amplification. The relationship between laser properties and crystal-packing structures has been well-documented in review papers.12,14
 | ||
| Fig. 1 Chemical structures of phenylenevinylene small molecules. | ||
Compared with P3V2 derivatives, there is a very limited number of π-extended molecules except for PPV-based polymers.19,36,54–56 Because the net-gain cross-section for lasers is a function of the stimulated emission cross-section (σem) and the population inversion density (ΔN), a large transition dipole moment in a molecule can reduce the threshold energy for light amplification. A longer π-conjugation length increases the oscillator strength of an electronic transition, which is proportional to the square of the transition dipole moment. Therefore, extended π-structures are an effective strategy to improve laser properties. Nevertheless, the π-conjugation length of 1a is relatively short compared with those of excellent amorphous-type laser materials.20
In this work, we investigated several oligo(p-phenylene-vinylene) derivatives (Fig. 1). The laser properties are expected to be improved for the P5V2 and P4V3 series relative to those of the P3V2 derivatives since 3a had a better amplified spontaneous emission (ASE) threshold than 1a.36 However, it is more difficult to predict packing structures that greatly affect laser properties. Although linear oligo-aromatics generally have a herringbone packing because of the low C/H ratio,57 cyano-substituted compounds should have more complex intermolecular interactions that can form various arrangements, including π-stacking structures. Here, their crystal structures and photophysical properties are presented.
Results and discussion
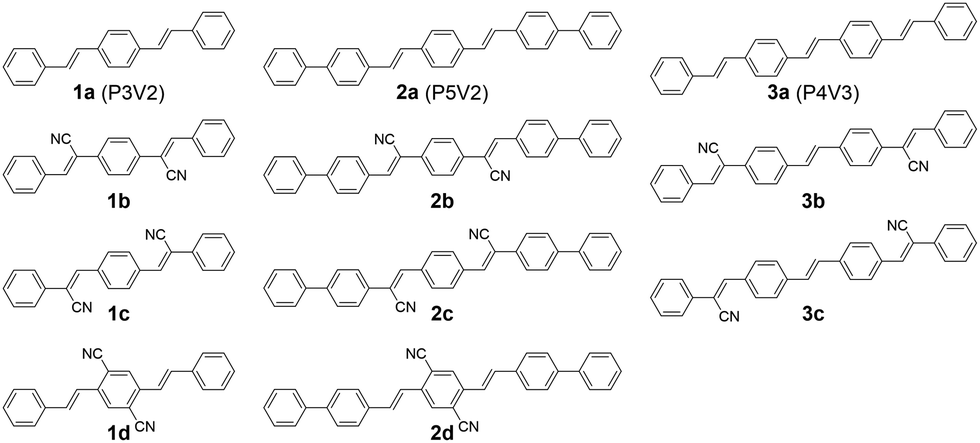
The compounds 1d, 2a, 2d and 3a were synthesized with the Wittig–Horner reaction, and 1b, 1c, 2b, 2c, 3b and 3c were synthesized by the Knoevenagel reaction. Both reactions produced good yields (Methods S1 in the ESI†).58 The compounds were purified by vacuum sublimation and their crystals were grown via sublimation under a nitrogen flow. The sublimation temperatures were 300–350 °C for the P5V2 series and 270–300 °C for the P4V3 series.The absorption and photoluminescence (PL) spectra for these compounds in chloroform solutions and in the crystalline form are shown in Fig. 2. In general, because of extended π-conjugation, the P5V2 and P4V3 series spectra were at longer wavelengths than those of the P3V2 series. Although the absorption maxima of 1b, 2b, and 3b in solution appear at shorter wavelengths relative to those of 1a, 2a and 3a, respectively, the onset of absorption is in the order a < b < c < d. Hence, the optical energy gap (Eoptg) decreases in order from a to d, which agrees well with the calculations (Methods S2 and Tables S1–S3 in the ESI†). The Stokes shifts for 1b, 1c, 2b, and 2c are larger than those for 1d and 2d, as indicated by the calculations (Table S3, ESI†), and the 0–0 peaks for b and c have a gentle slope relative to those for a and d. These results might be related to the high degree of rotational motion of the vinyl groups that were substituted with cyano groups in b and c. Such compounds are known to exhibit aggregation-induced emission enhancement (AIEE) and crystallization-induced emission enhancement (CIEE).59 Thus, the PL quantum yields (ΦPL) in solution are very low for b and c, although 2c was slightly emissive (Table 1). The radiative rate constant (kr) values for 1a, 2a, and 3a in solution are very high at 7.3 × 108, 1.1 × 109, and 7.7 × 108 s−1, respectively, which were consistent with the conjugation lengths and the calculated oscillator strengths.
 | ||
| Fig. 2 Ultraviolet-visible absorption (dashed lines) and emission (solid lines) spectra for (a) 1a–1d in chloroform, (b) crystalline 1a–1d, (c) 2a–2d in chloroform, (d) crystalline 2a–2d, (e) 3a–3c in chloroform, and (f) crystalline 3a–3c. | ||
| Compound | Condition | λ abs [nm] | λ PL [nm] | Φ PL [—] | τ [ns] | k r/108 [s−1] | E ASEth [μJ cm−2] | λ ASE [nm] |
|---|---|---|---|---|---|---|---|---|
| a Absolute PL quantum yield evaluated using an integrating sphere. b Taking from the literature (ref. 36). c Could not be determined because of the measurement limit. | ||||||||
| 1a | Chloroform | 355, 377 | 392, 414 | 0.83 | 1.1 | 7.3 | — | — |
| Crystalb | — | 452, 472 | 0.85 | 2.9 | 2.9 | 18 ± 6 | 466 | |
| 1b | Chloroform | 348 | 426, 449 | <0.01 | —c | — | — | — |
| Crystal (blue) | — | 476 | 0.65 | 1.6, 6.1 | 3.2 | 18 | 465 | |
| Crystal (green) | — | 526 | 0.36 | 6.1, 14 | 0.36 | No ASE | No ASE | |
| 1c | Chloroform | 368 | 426, 448 | <0.01 | —c | — | — | — |
| Crystal (blue) | — | 477 | 0.55 | 1.6, 8.3 | 3.2 | — | — | |
| Crystal (green) | — | 524 | 0.72 | 14, 24 | 0.43 | No ASE | No ASE | |
| 1d | Chloroform | 363, 395 | 428, 442 | 0.55 | 1.7 | 3.3 | — | — |
| Crystal | — | 511 | 0.64 | 18 | 0.36 | No ASE | No ASE | |
| 2a | Chloroform | 377 | 417, 441 | 0.93 | 0.82 | 11.3 | — | — |
| Crystal | — | 485, 508 | 0.58 | 1.5, 3.0 | 2.4 | 18 ± 2 | 515 | |
| 2b | Chloroform | 371 | 463 | 0.01 | —c | — | — | — |
| Crystal | — | 514 | 0.67 | 1.4, 2.6 | 3.6 | 7.4 ± 2 | 513 | |
| 2c | Chloroform | 385 | 457, 484 | 0.16 | 0.45 | 3.5 | — | — |
| Crystal | — | 510, 528 | 0.71 | 2.1, 3.7 | 2.4 | 12 and 8.5 | 504 and 531 | |
| 2d | Chloroform | 399 | 453, 469 | 0.73 | 1.0 | 7.0 | — | — |
| Crystal | — | 531 | 0.55 | 9.2 | 0.60 | No ASE | No ASE | |
| 3a | Chloroform | 383, 409 | 427, 452 | 0.79 | 1.0 | 7.7 | — | — |
| Crystal | — | 495, 517 | 0.65 | 2.4 | 2.7 | 11 ± 4 | 522 | |
| 3b | Chloroform | 376 | 445, 465 | 0.03 | —c | — | — | — |
| Crystal | — | 493 | 0.63 | 0.7, 1.8 | 3.7 | 7.2 ± 2 | 495 | |
| 3c | Chloroform | 393 | 450, 478 | 0.02 | —c | — | — | — |
| Crystal | — | 592 | 0.30 | 7.7, 21 | 0.17 | No ASE | No ASE | |
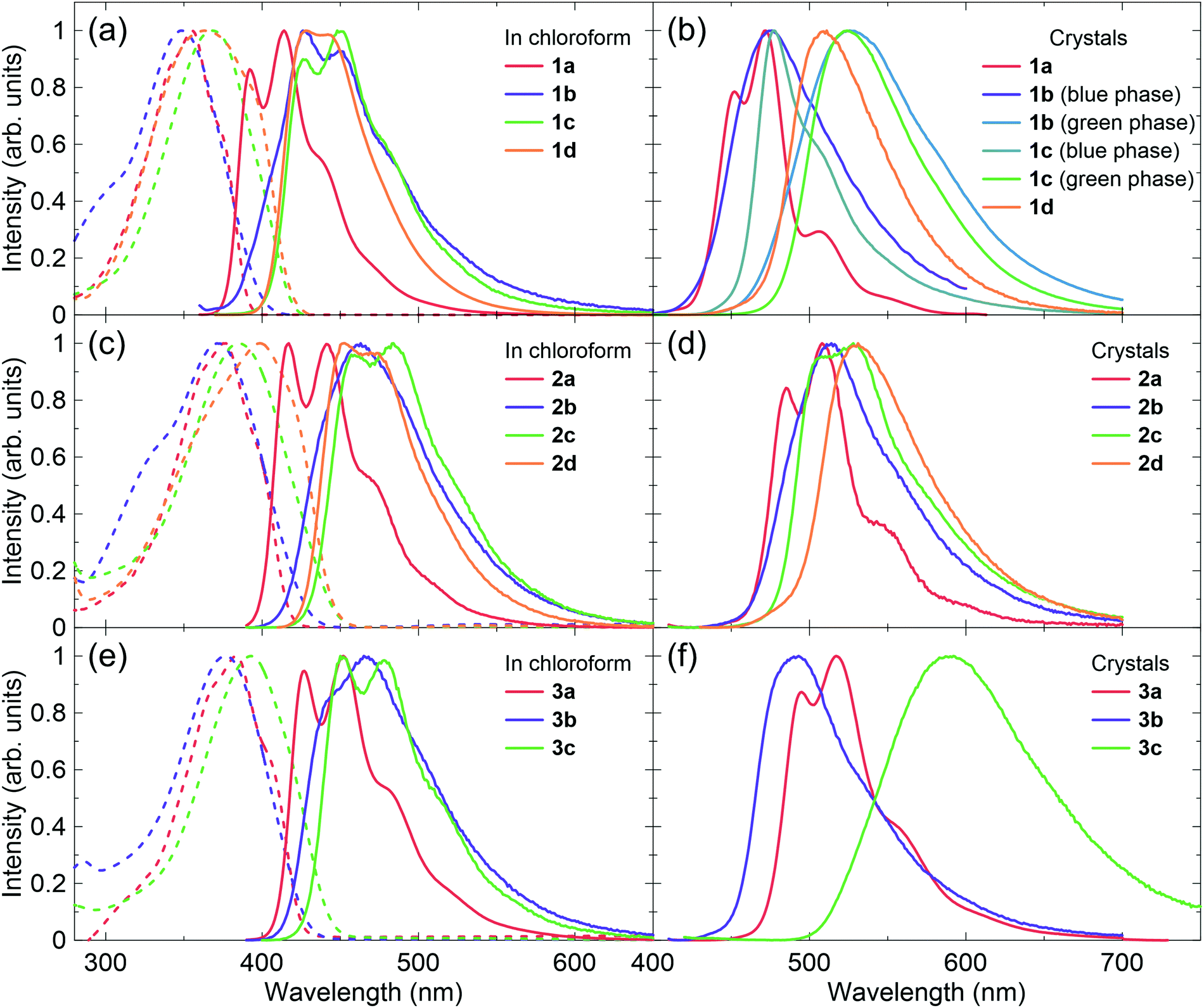
The photophysical properties in the crystal are distinctively different from those in solution because the molecular geometries are fixed in the solid state and the intermolecular interactions greatly affect their electronic structures. Single-crystal X-ray structure analysis is thus the best way to understand the relationship between solid-state optoelectronic properties and structures. Crystal structures have been reported for 1a (CCDC 1181381,34 and 92199835), the 1b blue phase (CCDC 1247728,37 799014,38 892284,39 and 1473570,40 note that these structures are substantially the same), the 1b green phase (CCDC 892287,39 and 147357140), the 1c blue phase (CCDC 815708),46 the 1c green phase polymorph 1 (CCDC 860278),48 and the 1c green phase polymorph 2 (CCDC 1542079).52 The crystal structure of 2a was reported,54 but no CCDC number is available. Hence, we solved the crystal structures for 1d, 2b, 2c, 2d, 3a, 3b, and 3c (Fig. 3 and Tables S4–S10, ESI†). Judging from their emission spectra, the crystals for these compounds are unlikely to exhibit polymorphism. The emission properties of the crystals are also summarized in Table 1. Significant differences in the crystal structures of these compounds occur in the packing motifs, that is, herringbone or π-stacking. Note that crystals of these compounds are often referred to as H- and J-aggregates for the specific arrangement of two molecules in the stacking direction. However, the molecules have three-dimensional interactions, complicating the dipole couplings. The excitation spectra for these crystals (Fig. S1, ESI†) indicate that the classification is difficult because 1a, which is often described as a H-aggregate with herringbone packing,12 might exhibit a red-shifted absorption in the crystal compared with that in solution. Therefore, we avoid mentioning H- and J-aggregates for the new compounds. Further discussion should be reported in the future, with the combination of theoretical calculations and full experimental data, including optical spectra and crystal structures.
 | ||
| Fig. 3 Crystal structures of (a) 1d, (b) 2b, (c) 2c, (d) 2d, (e) 3a, (f) 3b, (g) 3c. | ||
The compounds without cyano groups (1a, 2a, and 3a) had typical herringbone packing with nearly flat stilbene structures and similar kr values (>2 × 108 s−1). Compounds 1b and 1c, both with polymorphisms, have blue-emissive herringbone packing and green-emissive π-stacking structures. The emission wavelengths of the blue phases are similar to those of 1a, although 1b and 1c have twisted conformations. The large bathochromic shifts of the emission with broad spectral shapes observed for the π-stacking molecules are attributed to excimer-like features. The cyano-vinylene groups are overlapped with phenyl groups in the π-stacking, which enables intermolecular charge transfer (CT). For 1d, the 1,4-dicyanobenzene apparently inhibited herringbone packing. Thus, the crystal packing is relatively similar to the green phases of 1b and 1c, with strong π-stacking. The emission properties are also similar.
Compounds 2b and 2c have herringbone packing, but did not form π-stacking structures. This can be explained by the non-planar biphenyl rings, which prevent interactions between the cyano-vinylene groups and the phenyl rings, while still facilitating herringbone packing between the biphenyl rings. The emission wavelengths of 2b and 2c are not significantly red-shifted from those of 2a. The values of ΦPL and kr are high and are expected to exhibit ASE. Similar to 1d, the 1,4-dicyanobenzene structure in 2d hinders the herringbone packing, while biphenyl rings also limit π-stacking. As a result, 2d has a hybrid structure of herringbone and π-stacking. The excimer features are not strong because the 1,4-dicyanobenzene rings are mainly overlapped between themselves, which results in a small bathochromic shift compared with 2a–2c. However, the emission decay lifetime is relatively long and kr was less than 1 × 108 s−1.
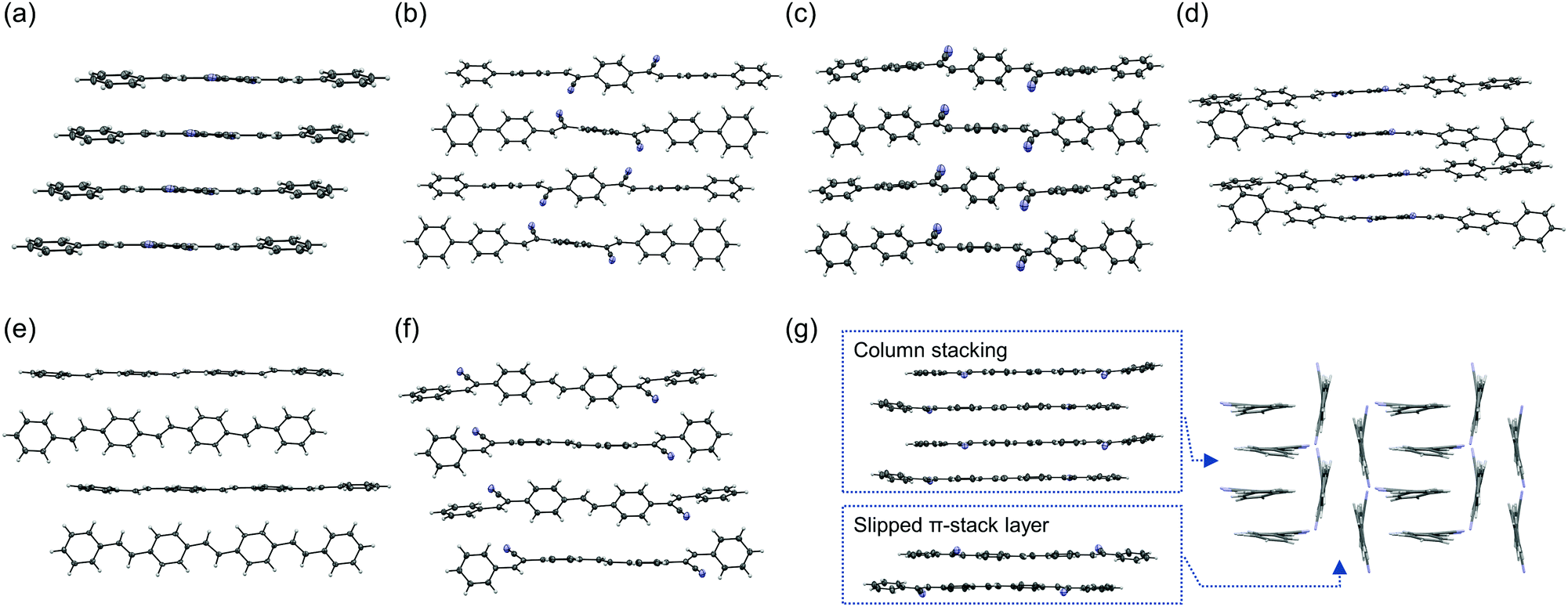
Interestingly, compounds 3b and 3c had very different features. The 3b emission was blue-shifted from that of 3a, but the 3c emission was largely red-shifted. The 3c crystal included two types of π-stacking: single column stacking and two molecular layers with slipped π-stacking between the columns. The broad orange emission implies extensive excimer formation, probably in the molecules forming the column stacking. The ΦPL of the 3b crystal is relatively high and that of 3c is moderate, although the fluorescence in solution was very weak for both compounds. Thus, these compounds are considered to be CIEE materials, such as 1b and 1c. The AIEE characteristics were also examined in mixed tetrahydrofuran (THF)/water solvents (Fig. 4). The suspension in solutions with higher water content includes various aggregated states, which may be different from the bulk crystals. Compound 3b had AIEE characteristics, although the ΦPL even in 90% water was lower than that in the crystals. The 3c spectra were clearly shifted with increasing water content up to 70%. However, the ΦPL were almost constant. Therefore, the highly ordered molecular arrangements are expected to be efficiently emissive. The values of kr for the 3b crystal was slightly higher than that of the blue-emissive 1b crystal. This is attributed to the larger oscillator strength. The value of kr for the 3c crystal was expected to be low because of its relatively long fluorescence lifetime based on excimer formation.
 | ||
| Fig. 4 Emission spectra of (a) 3b and (b) 3c in x%-water/(100 − x)%-tetrahydrofuran (THF) mixed solvent. The concentration was fixed at 3.0 × 10−5 M. (c) Water fraction dependence of the ΦPL, and the ΦPL in the crystal. | ||
Fig. 5, 6 and Fig. S2 (ESI†) show ASE characteristics for these crystals. The blue-emissive crystals of 1b and 1c are reported to exhibit ASE with the extremely high thresholds of 1.8 × 104 μJ cm−2 and 360 μJ cm−2, respectively.14 The thresholds are often increased in the low-quality crystals. In general, sublimation under a nitrogen flow forms better-quality crystals compared to solution-based techniques. The oligo(p-phenylenevinylene) derivatives tend to form large single crystals. In many cases, thin platelet crystals with thicknesses ranging from several hundred nanometres to several micrometres are favourable for ASE measurements and LEFET fabrications. Fortunately, large enough crystals of blue-emissive 1b could be obtained, although the larger green-emissive crystals were the main products. An ASE threshold as low as 18 μJ cm−2 can be observed for the blue-phase crystal (Fig. S2, ESI† and Table 1). Meanwhile, the 1c blue-emissive crystals were very small, which hindered the determination of a reliable ASE threshold with our apparatus. Note that the green-emissive crystals with π-stacking structures did not exhibit ASE for 1b and 1c up to 200 μJ cm−2.
 | ||
| Fig. 5 Output photoluminescence (PL) intensity and full width at half maximum (FWHM) values from edges of the crystals of (a) 2a, (b) 2b, (d) 3a, (e) 3b. The stimulated cross-section spectra and PL spectra above the amplified spontaneous emission (ASE) threshold for (c) 2a and 2b, and (f) 3a and 3b. | ||
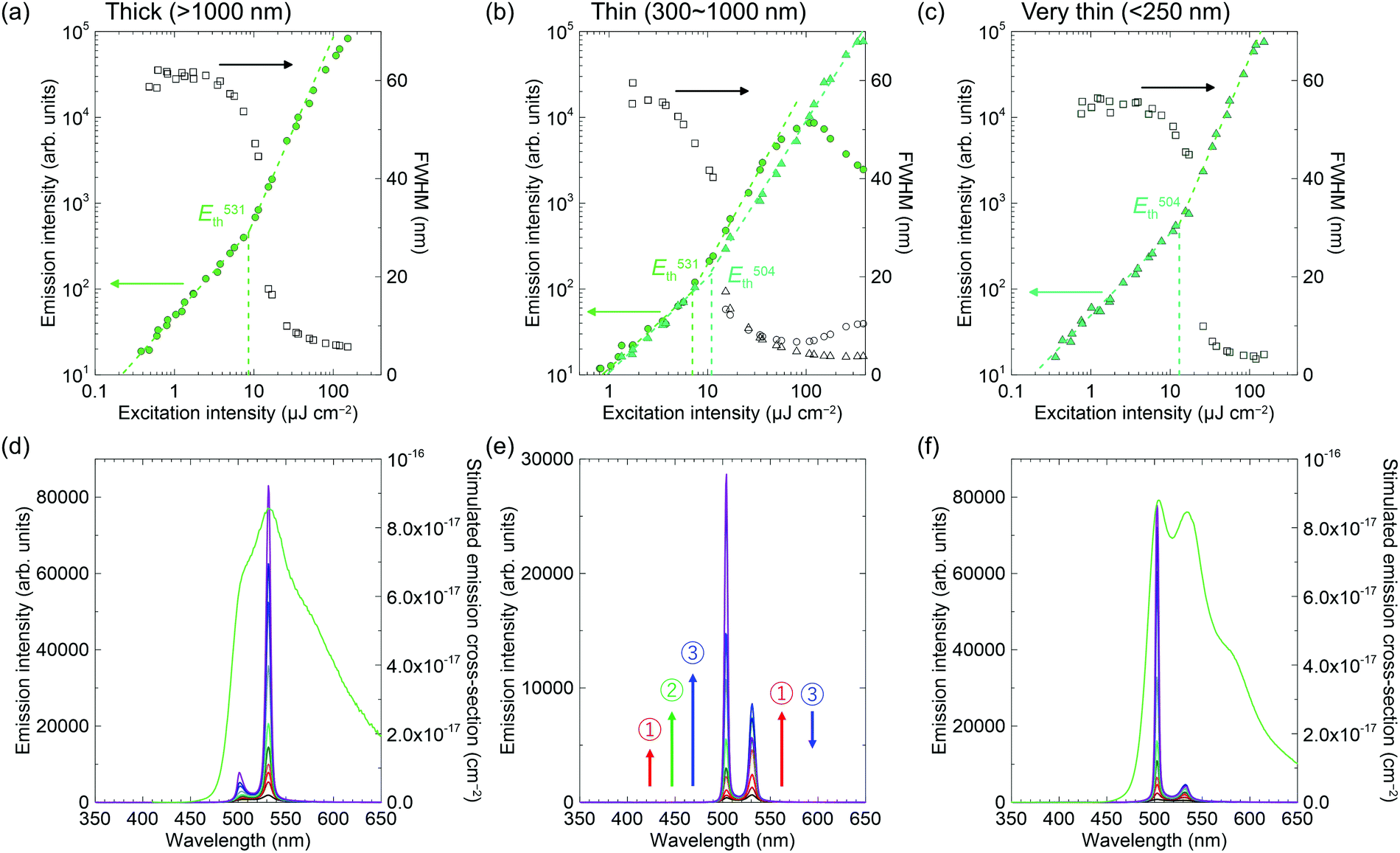 | ||
| Fig. 6 Output photoluminescence (PL) intensity and full width at half maximum (FWHM) values and PL spectra at different excitation energies from the edges of 2c crystals. Amplified spontaneous emission (ASE) behaviour for (a and d) thick crystals (>1000 nm), (d and e) thin crystals (300–1000 nm), and (c and f) very thin crystals (<250 nm). | ||
The ASE for 2a and 2b were observed at ca. 515 nm, which is nearly the same as the peaks of stimulated emission cross-section spectra (Fig. 5). The ASE thresholds of 2a and 2b were 18 μJ cm−2 and 7.4 μJ cm−2, respectively. Similarly, the thresholds for 3a and 3b were 11 μJ cm−2 and 7.2 μJ cm−2, respectively. The introduction of electron-withdrawing cyano groups into the compounds with donor units resulted in red-shifted emission, a decrease in kr, and an increase in the ASE thresholds because of increased intramolecular CT character.27 However, we have demonstrated improved ASE thresholds for cyano-substituted compounds without strong donor–acceptor interactions. These results indicated that cyano-substitution paves the way for improving the laser properties of p-phenylenevinylene derivatives, including polymers based on PPV.
The ASE of 2c has two peaks that are apparently different from the others (Fig. 6), but more similar to that of methyl-substituted P3V2, 1,4-bis(4-methylstyryl)benzene.60 The results seem to correlate with crystal thicknesses. The thicker crystals (>1000 nm) had ASE mainly at the lower-energy sub-band at 531 nm (referred to the 0–2 transition, according to ref. 60). In contrast, the thinner crystals (∼250 nm) had the ASE at the higher-energy sub-band at 504 nm (referred to the 0–1 transition, according to ref. 60). In addition, the two ASE peaks evolved simultaneously in the moderately thick crystals (300–1000 nm), where the peak at 531 nm first increased (number 1 in Fig. 6e), then the peak at 504 nm increased (number 2 in Fig. 6e), while the 531 nm peak then decreased (number 3 in Fig. 6e). Note that the out-of-plane X-ray diffractions for these crystals excluded polymorphism (Fig. S3, ESI†). The dual-wavelength ASE has been explained by re-absorption by molecules in the ground state or in excited states.61–63 The experimental results for 2c, including the thickness dependence and the different energy behaviours of the two ASE peaks, suggest the same mechanism as that reported previously for the two-transition ASE origin. Because the optical amplifications at 504 nm and 531 nm are competitive processes, the 531 nm intensity decreased with increasing 504 nm intensity in Fig. 6e when the net gain at 504 nm became larger than that at 531 nm with intense excitation. The best thresholds were 12 μJ cm−2 and 8.5 μJ cm−2 for the 504 nm and 531 nm transitions, respectively.
As expected, the other compounds (1d, 2d, and 3c) with π-stacking structures did not exhibit ASE. These results also verified that solid-state interactions are very important when developing highly efficient laser materials.
Finally, charge transport properties for 2a were evaluated and summarized in Table S11 (ESI†) since the reported mobilities were relatively low (μh = 1.6 × 10−4 cm2 V−1 s−1) compared with those of 1a (μh = 0.12 cm2 V−1 s−1 and μe = 1.3 × 10−2 cm2 V−1 s−1) and 3a (μh = 0.12 cm2 V−1 s−1 and μe = 0.11 cm2 V−1 s−1).36,54 This is certainly attributed to the different types of active media and device configurations: thin-film vs. single-crystal and gold source–drain (S–D) electrodes vs. asymmetric gold–calcium S–D electrodes. The single-crystal transistors normally exhibited better performances because of their perfect molecular arrangements that were free of grain boundaries and had minimal defects.64 The asymmetric electrodes with different work-functions (gold: 4.5–5.1 eV and calcium: 2.9–3.0 eV) ease the injection of both hole and electron carriers.65 Therefore, we fabricated 2a-based single-crystal transistors using gold–calcium electrodes. Both p- and n-type characteristics were clearly observed with saturated mobilities of μh = 0.05 ± 0.01 cm2 V−1 s−1 and μe = 0.25 ± 0.06 cm2 V−1 s−1 for three devices (Fig. 7). Although the hole mobility was slightly lower than those in 1a and 3a, it was significantly improved compared to those in the thin-film devices. In addition, high electron mobility was observed owing to effective carrier injection from the calcium electrode. These high mobilities suggest considerable potential of p-phenylenevinylene derivatives for electrically pumped lasers. Unfortunately, the devices of the cyano-derivatives such as 2d and 3b did not show improved performances. In addition, the 3c crystals are needles, which differ from the thin-platelets of the other compounds; this makes device fabrication difficult.
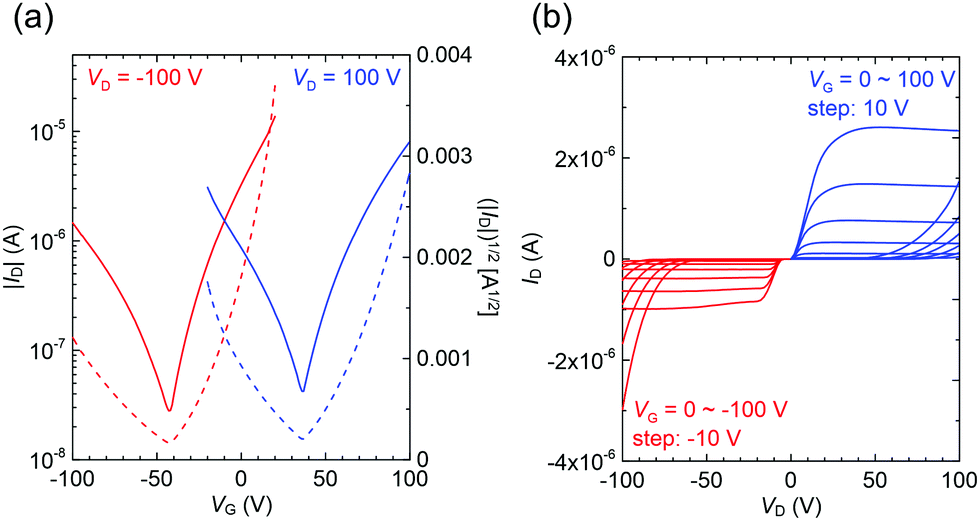 | ||
| Fig. 7 (a) Transfer and (b) output characteristics of a single-crystal field-effect transistor (FET) for 2b with gold–calcium source–drain electrodes. The channel length and width are ca. 100 μm and ca. 300 μm, respectively. | ||
Conclusions
In this study, we developed several π-extended stilbene derivatives to increase the understanding of structure–property relationships. The crystal structures were fully investigated and classified according to herringbone and π-stacking structures. The kr values were strongly related to the packing motifs, and high values up to 3.7 × 108 s−1 were achieved for compounds with herringbone packing. Since the biphenyl groups prevent π-stacking, it would be useful to design the laser materials. On the other hand, it is more difficult to predict the crystal packing for the compounds with cyano groups on the vinyl groups. Compounds with π-stacking structures did not exhibit ASE, while clear ASE peaks were observed for those with herringbone packing. The ASE thresholds were mostly lower for the π-extended compounds and were as low as 7.2 μJ cm−2 with cyano substitutions. These results give a systematic insight into light amplification in the single crystals.Experimental section
General
Commercially available materials were used as received from the suppliers for the synthesis (Table S12, ESI†). Details of instruments and physical measurements are given in Table S13 (ESI†).Materials synthesis and characterization
The synthesis procedures and characterization data of materials used in this study are described in the ESI† (Methods S1) and NMR spectra are given in Data S1.Quantum calculations
Details of the calculations are described in the ESI† (Methods S2 and Tables S1–S3).X-Ray structure analysis
Single crystals suitable for X-ray analysis were grown by slow sublimation under nitrogen flow. X-Ray diffraction data were collected on a Rigaku Saturn724 diffractometer with Mo-Kα radiation (λ = 0.71075 Å) at 123 K for 2b, 3b and 3c, a Rigaku R-AXIS RAPID diffractometer with Cu-Kα radiation (λ = 1.54187 Å) at 183 K for 2c, and a Rigaku AFC HyPix-6000 diffractometer with Mo-Kα radiation at 100 K for 1d, 3a, and 2d. All non-hydrogen atoms were refined anisotropically. The positions of all hydrogen atoms were calculated geometrically and refined as a riding model. CCDC 2034947–2034953.†ASE measurements
The thin flexible crystals were grown by sublimation under nitrogen flow. The crystal samples were placed on the cleaned quartz substrate. ASE properties were characterized by optically pumping with a randomly polarized nitrogen gas laser (KEN2020, Usho Optical Systems Co., Ltd) at an excitation wavelength of 337 nm with a 0.8 ns pulse (operating frequency of 10 Hz). The input laser beam was focused into a stripe with dimensions of ca. 0.5 cm × 0.1 cm using a cylindrical lens. Neutral density filters were used to adjust excitation intensity. ASE measurements were performed under a nitrogen atmosphere to prevent degradation. Output light emission from the edge of the sample was collected into an optical fiber connected to a spectrometer (Hamamatsu Photonics PMA-11). ASE thresholds were identified from the plot of output versus input intensity. The thresholds of absorbed energy density were not calculated because absorbance of a single crystal sample is difficult to measure. Reproducibility was confirmed by measuring at least five different samples.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Ms K. Kusuhara and Ms N. Nakamura for the characterization of materials, and Mr T. Matsumoto for the measurements of X-ray single crystal analysis. This work was financially supported by JST ERATO Grant Number JPMJER1305, JSPS KAKENHI Grant Number 19H02790 and 20K21227, The Murata Science Foundation and the JSPS Core-to-Core Program.Notes and references
- F. P. Schäfer, W. Schmidt and J. Volze, Appl. Phys. Lett., 1966, 9, 306–309 CrossRef.
- N. Tessler, G. Denton and R. Friend, Nature, 1996, 382, 695–697 CrossRef CAS.
- F. Hide, M. A. DiazGarcia, B. J. Schwartz, M. R. Andersson, Q. B. Pei and A. J. Heeger, Science, 1996, 273, 1833–1836 CrossRef CAS.
- M. D. McGehee and A. J. Heeger, Adv. Mater., 2000, 12, 1655–1668 CrossRef CAS.
- A. S. D. Sandanayaka, T. Matsushima, F. Bencheikh, S. Terakawa, W. J. Potscavage, C. Qin, T. Fujihara, K. Goushi, J.-C. Ribierre and C. Adachi, Appl. Phys. Express, 2019, 12, 061010 CrossRef CAS.
- T. Kanagasekaran, H. Shimotani, K. Kasai, S. Onuki, R. D. Kavthe, R. Kumashiro, N. Hiroshiba, T. Jin, N. Asao and K. Tanigaki, Phys. Opt., arXiv:1903.08869, 2019, https://arxiv.org/abs/1903.08869.
- I. D. W. Samuel and G. A. Turnbull, Chem. Rev., 2007, 107, 1272–1295 CrossRef CAS PubMed.
- S. Chénais and S. Forget, Polym. Int., 2012, 61, 390–406 CrossRef.
- A. J. C. Kuehne and M. C. Gather, Chem. Rev., 2016, 116, 12823–12864 CrossRef CAS PubMed.
- C. Grivas and M. Pollnau, Laser Photonics Rev., 2012, 6, 419–462 CrossRef.
- Y. Jiang, Y.-Y. Liu, X. Liu, H. Lin, K. Gao, W.-Y. Lai and W. Huang, Chem. Soc. Rev., 2020, 49, 5885–5944 RSC.
- J. Gierschner and S. Y. Park, J. Mater. Chem. C, 2013, 1, 5818–5832 RSC.
- H.-H. Fang, J. Yang, J. Feng, T. Yamao, S. Hotta and H.-B. Sun, Laser Photonics Rev., 2014, 8, 687–715 CrossRef.
- J. Gierschner, S. Varghese and S. Y. Park, Adv. Opt. Mater., 2016, 4, 348–364 CrossRef CAS.
- R. Hibino, M. Nagawa, S. Hotta, M. Ichikawa, T. Koyama and Y. Taniguchi, Adv. Mater., 2002, 14, 119–122 CrossRef CAS.
- G. Kreiza, P. Baronas, E. Radiunas, P. Adomėnas, O. Adomėnienė, K. Kazlauskas, J.-C. Ribierre, C. Adachi and S. Juršėnas, Adv. Opt. Mater., 2017, 5, 1600823 CrossRef.
- P. Baronas, G. Kreiza, M. Mamada, S. Maedera, P. Adomėnas, O. Adomėnienė, K. Kazlauskas, C. Adachi and S. Juršėnas, Adv. Opt. Mater., 2019, 7, 1901670 Search PubMed.
- T. Oyamada, C. H. Chang, T. C. Chao, F. C. Fang, C. C. Wu, K. T. Wong, H. Sasabe and C. Adachi, J. Phys. Chem. C, 2007, 111, 108–115 CrossRef CAS.
- H. Nakanotani, M. Saito, H. Nakamura and C. Adachi, Appl. Phys. Lett., 2009, 95, 103307 CrossRef.
- M. Morales-Vidal, P. G. Boj, J. M. Villalvilla, J. A. Quintana, Q. Yan, N.-T. Lin, X. Zhu, N. Ruangsupapichat, J. Casado, H. Tsuji, E. Nakamura and M. A. Diaz-Garcia, Nat. Commun., 2015, 6, 8458 CrossRef CAS PubMed.
- H. Nakanotani, S. Akiyama, D. Ohnishi, M. Moriwake, M. Yahiro, T. Yoshihara, S. Tobita and C. Adachi, Adv. Funct. Mater., 2007, 17, 2328–2335 CrossRef CAS.
- W. Zhang, Y. Yan, J. Gu, J. Yao and Y. S. Zhao, Angew. Chem., Int. Ed., 2015, 54, 7125–7129 CrossRef CAS PubMed.
- V. T. N. Mai, A. Shukla, M. Mamada, S. Maedera, P. E. Shaw, J. Sobus, I. Allison, C. Adachi, E. B. Namdas and S.-C. Lo, ACS Photonics, 2018, 5, 4447–4455 CrossRef CAS.
- B. K. Yap, R. Xia, M. Campoy-Quiles, P. N. Stavrinou and D. D. C. Bradley, Nat. Mater., 2008, 7, 376–380 CrossRef CAS PubMed.
- T. Aimono, Y. Kawamura, K. Goushi, H. Yamamoto, H. Sasabe and C. Adachi, Appl. Phys. Lett., 2005, 86, 071110 CrossRef.
- H. Nakanotani, C. Adachi, S. Watanabe and R. Katoh, Appl. Phys. Lett., 2007, 90, 231109 CrossRef.
- M. Mamada, T. Fukunaga, F. Bencheikh, A. S. D. Sandanayaka and C. Adachi, Adv. Funct. Mater., 2018, 28, 1802130 CrossRef.
- Y. Oyama, M. Mamada, A. Shukla, E. G. Moore, S.-C. Lo, E. B. Namdas and C. Adachi, ACS Mater. Lett., 2020, 2, 161–167 CrossRef CAS.
- A. S. D. Sandanayaka, K. Yoshida, M. Inoue, C. Qin, K. Goushi, J.-C. Ribierre, T. Matsushima and C. Adachi, Adv. Opt. Mater., 2016, 4, 834–839 CrossRef CAS.
- A. S. D. Sandanayaka, T. Matsushima, F. Bencheikh, K. Yoshida, M. Inoue, T. Fujihara, K. Goushi, J.-C. Ribierre and C. Adachi, Sci. Adv., 2017, 3, e1602570 CrossRef PubMed.
- T. W. Lee, O. O. Park, D. H. Choi, H. N. Cho and Y. C. Kim, Appl. Phys. Lett., 2002, 81, 424–426 CrossRef CAS.
- A. Rose, Z. G. Zhu, C. F. Madigan, T. M. Swager and V. Bulovic, Nature, 2005, 434, 876–879 CrossRef CAS PubMed.
- E. B. Namdas, M. Tong, P. Ledochowitsch, S. R. Mednick, J. D. Yuen, D. Moses and A. J. Heeger, Adv. Mater., 2009, 21, 799–802 CrossRef CAS.
- C. C. Wu, M. C. DeLong, Z. V. Vardeny and J. P. Ferraris, Synth. Met., 2003, 137, 939–941 CrossRef CAS.
- S. Varghese, S. K. Park, S. Casado, R. C. Fischer, R. Resel, B. Milian-Medina, R. Wannemacher, S. Y. Park and J. Gierschner, J. Phys. Chem. Lett., 2013, 4, 1597–1602 CrossRef CAS PubMed.
- H. Nakanotani, M. Saito, H. Nakamura and C. Adachi, Appl. Phys. Lett., 2009, 95, 033308 CrossRef.
- G. P. Bartholomew, G. C. Bazan, X. Bu and R. J. Lachicotte, Chem. Mater., 2000, 12, 1422–1430 CrossRef CAS.
- S.-J. Yoon and S. Y. Park, J. Mater. Chem., 2011, 21, 8338–8346 RSC.
- Y. Xu, K. Wang, Y. Zhang, Z. Xie, B. Zou and Y. Ma, J. Mater. Chem. C, 2016, 4, 1257–1262 RSC.
- Y. Xu, Z. Xie, H. Zhang, F. Shen and Y. Ma, CrystEngComm, 2016, 18, 6824–6829 RSC.
- B. Lu, Y. Zhang, X. Yang, K. Wang, B. Zou and D. Yan, J. Mater. Chem. C, 2018, 6, 9660–9666 RSC.
- J. Shi, L. E. A. Suarez, S.-J. Yoon, S. Varghese, C. Serpa, S. Y. Park, L. Lüer, D. Roca-Sanjuán, B. Milián-Medina and J. Gierschner, J. Phys. Chem. C, 2017, 121, 23166–23183 CrossRef CAS.
- J. Han, J. You, X. Li, P. Duan and M. Liu, Adv. Mater., 2017, 29, 1606503 CrossRef PubMed.
- J. S. Ramírez-Pradilla, C. Blanco-Tirado and M. Y. Combariza, ACS Appl. Mater. Interfaces, 2019, 11, 10975–10987 CrossRef PubMed.
- T. Zhanga, G. Zhu, L. Lin, J. Mu, B. Ai, Y. Li and S. Zhuo, Org. Electron., 2019, 68, 264–270 CrossRef.
- Y. Xu, H. Zhang, F. Li, F. Shen, H. Wang, X. Li, Y. Yu and Y. Ma, J. Mater. Chem., 2012, 22, 1592–1597 RSC.
- X. Li, Y. Xu, F. Li and Y. Ma, Org. Electron., 2012, 13, 762–766 CrossRef CAS.
- C.-W. Chang, C. J. Bhongale, C.-S. Lee, W.-K. Huang, C.-S. Hsu and E. W.-G. Diau, J. Phys. Chem. C, 2012, 116, 15146–15154 CrossRef CAS.
- J. Deng, J. Tang, Y. Xu, L. Liu, Y. Wang, Z. Xiec and Y. Ma, Phys. Chem. Chem. Phys., 2015, 17, 3421–3425 RSC.
- J. Deng, Y. Xu, L. Liu, C. Feng, J. Tang, Y. Gao, Y. Wang, B. Yang, P. Lu, W. Yanga and Y. Ma, Chem. Commun., 2016, 52, 2370–2373 RSC.
- C. Wang, Y. Li, Q. Xu and L. Luo, Opt. Mater., 2017, 72, 710–716 CrossRef CAS.
- J. Shi, S.-J. Yoon, L. Viani, S. Y. Park, B. Milián-Medina and J. Gierschner, Adv. Opt. Mater., 2017, 5, 1700340 CrossRef.
- S. Hayashi, R. Hirai, S. Yamamoto and T. Koizumi, Chem. Lett., 2018, 47, 1003–1005 CrossRef CAS.
- Y.-X. Li, J.-X. Qiu, J.-L. Miao, Z.-W. Zhang and G.-X. Sun, J. Phys. Chem. C, 2015, 119, 2388–2398 CAS.
- S. E. Estrada, C. Ochoa-Puentes and C. A. Sierra, J. Mol. Struct., 2017, 1133, 448–457 CrossRef CAS.
- S. Jeon, J. P. Lee and J.-M. Kim, J. Mater. Chem. C, 2015, 3, 2732–2736 RSC.
- G. R. Desiraju and A. Gavezzotti, Acta Crystallogr., Sect. B: Struct. Sci., 1989, 45, 473–482 CrossRef.
- K. Shoji, J. Nishida, D. Kumaki, S. Tokitoab and Y. Yamashita, J. Mater. Chem., 2010, 20, 6472–6478 RSC.
- M. Martínez-Abadía, R. Giménez and M. Blanca Ros, Adv. Mater., 2017, 30, 1704161 CrossRef PubMed.
- R. Kabe, H. Nakanotani, T. Sakanoue, M. Yahiro and C. Adachi, Adv. Mater., 2009, 21, 4034–4038 CrossRef CAS.
- D. Fichou, S. Delysse and J.-M. Nunzi, Adv. Mater., 1997, 9, 1178–1181 CrossRef CAS.
- M. Polo, A. Camposeo, S. Tavazzi, L. Raimondo, P. Spearman, A. Papagni, R. Cingolani and D. Pisignano, Appl. Phys. Lett., 2008, 92, 083311 CrossRef.
- M. Mamada, R. Komatsu and C. Adachi, ACS Appl. Mater. Interfaces, 2020, 12, 28383–28391 CrossRef CAS PubMed.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- T. Yasuda, T. Goto, K. Fujita and T. Tsutsuia, Appl. Phys. Lett., 2004, 85, 2098–2100 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, characterization, computational, photophysical, electrochemical and thin-film transistor data. CCDC 2034947–2034953. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ma00756k |
| This journal is © The Royal Society of Chemistry 2021 |