
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultrafine multi-metallic carbide nanocrystals encased in a carbon matrix as durable electrocatalysts towards effective alkaline hydrogen evolution reaction†
Zhao
Wei‡
a,
Jingman
Pang‡
b,
Yuan
Ji
cd,
Xing
Yang
a,
Yundan
Liu
*cd,
Long
Ren
e and
Yun
Zhang
 *a
*a
aSchool of Physics and Opto-Electronic Technology, Collaborative Innovation Center of Rare-Earth Optical Functional Materials and Devices Development, Baoji University of Arts and Sciences, Baoji, Shaanxi, 721016, P. R. China. E-mail: zhangyun_xtu@163.com
bDepartment of Chemistry and Chemical Engineering, Baoji University of Arts and Science, Baoji 721016, China
cHunan Key Laboratory of Micro-Nano Energy Materials and Devices, Xiangtan University, Hunan 411105, P. R. China. E-mail: liuyd@xtu.edu.cn
dLaboratory for Quantum Engineering and Micro-Nano Energy Technology and Faculty of Materials and Optoelectronic Physics, Xiangtan University, Hunan 411105, P. R. China
eInstitute for Superconducting and Electronic Materials (ISEM), Australian Institute for Innovative Materials (AIIM), University of Wollongong, Wollongong, NSW 2500, Australia
First published on 16th November 2020
Abstract
Durable electrolytic hydrogen production at high-current densities (>500 mA cm−2) is required as the basis of industrial scale hydrogen evolution reaction (HER). Electrocatalysts, with the purpose of accelerating HER, however, are far from meeting the requirements of high activity, cost effectiveness, and long-term stability, especially in alkaline media. Herein, a series of ultrafine bi- or tri-metallic Mo-based carbides embedded in a graphitic carbon matrix are synthesized by a direct solid reaction between metal molybdate and dicyandiamide (DCA). The components of the proposed multi-metallic carbides can be controlled in a facile manner by precisely configuring the molybdates with different metal cations, enabling the design of electrocatalysts with optimized active sites for HER. With the carbon layer protecting against corrosion, the as-prepared series of (NixCo1−x)6Mo6C2 carbides presents effective HER performance, in which the optimized stoichiometric sample ((Ni0.2Co0.8)6Mo6C2) requires an overpotential of 100 mV to drive 10 mA cm−2 current density for HER. With this electrocatalyst, a durable current density of 1000 mA cm−2 can be achieved by applying an overpotential of 1.84 V towards HER, which is even lower than the required potential of the Pt/C commercial electrocatalyst (2.12 V) for the same current density.
1. Introduction
To meet the increasing global energy demand and decelerate the growing pollutant emissions, exploring green energy sources to replace fossil fuels has become a major challenge. As a renewable clean energy with high calorific value,1 hydrogen energy has received extensive attention from academia and industry. At present, hydrogen production methods mainly include biological hydrogen production, fossil fuel hydrogen production, photocatalytic decomposition hydrogen production, and water electrolysis hydrogen production.2 Electrolysis of water is one promising production method for high-efficiency hydrogen evolution reaction (HER).3 Typically, electrolysis requires electrocatalysts to participate in the reaction to lower the kinetic energy barrier, thereby reducing the applied driving potential and increasing the reaction rate.4,5 Although platinum-based precious metals and their compounds exhibit excellent electrocatalytic HER performance, their high price constraints limit their development and application on an industrial scale. In addition, the typical route for industrial electrolytic hydrogen production is based in alkaline media, which, however, usually requires higher overpotentials than those required for acidic electrolytes due to the relatively slow kinetics caused by an additional water dissociation step.6 Therefore, it is highly demanded to develop highly efficient, durable and cost-effective electrocatalysts for industrial scale HER under alkaline conditions.In recent years, intense efforts have been made in developing effective, long-term durable and earth-abundant metal-based substitutes including carbides,7–12 borides,13 phosphides,14–17 oxides,18–20 nitrides,21–24 chalcogenides25–28 and carbon-based materials.9,29 Among these candidate materials, transition metal carbides (TMCs) have several advantages, such as high electronic conductivity, noble-metal-like electronic structure for catalysis, broad pH applicability and chemical stability.30–37 As a representative among various TMCs, molybdenum carbides (e.g. Mo2C and MoC) have been reported to be some of the most active non-precious metal-based electrocatalysts for HER, due to their similar-band electronic configuration to those of Pt-group catalysts.38,39 Subsequently, it was found that the introduction of other transition metal elements into the transition metal carbides, using the synergistic effect between different transition metals, can optimize the Gibbs free energy and M–H bond energy of hydrogen adsorption40–42 and this type of interfacial structure in the material also accelerates the process of water splitting and active hydrogen bonding, all of which contribute to the enhanced hydrogen evolution catalytic performance.43 Though polymetallic carbides theoretically have many advantages for achieving improvement in intrinsic catalytic activity for electrocatalysis, few carbides with more than two metal ions for alkaline electrocatalytic HER have been reported. Thus, it is highly demanded to develop high-quality polymetallic carbides for practical hydrogen production.
In this work, we developed a general route to “synthesize” a series of ultrafine Mo-based HER-active bi-/tri-metallic “carbides” as electrocatalysts by “direct” carbonization of multi-metal “molybdate” precursors. The unique framework crystal structure of molybdates in the form of AMoO4 offers the capability to construct multi-metal compounds, and the multi-metal components can be adjusted to achieve regulation of the electronic configuration and thereby the catalytic activity. Following this strategy, various bimetallic and trimetallic Mo-based carbides, Fe3Mo3C, Co6Mo6C2, Mo6Ni6C, and (NixCo1−x)6Mo6C2, were synthesized. As typical developed examples for HER, a series of ultrafine (NixCo1−x)6Mo6C2 nanocrystals with a small size of 4 nm was grown by using dicyandiamide (DCA) as a carbon source, resulting in a nano-architectural carbide uniformly embedded in a conductive carbonaceous matrix with high chemical and mechanical stability. With the carbon layer protecting against corrosion and accelerating interfacial charge transfer, the as-prepared series of (NixCo1−x)6Mo6C2 carbides presents effective HER performance, in which the optimized stoichiometric sample (Ni0.2Co0.8)6Mo6C2 requires an overpotential of 100 mV to drive 10 mA cm−2 current density for alkaline HER. Using such an electrocatalyst, a durable current density of 1000 mA cm−2 can be achieved by applying an overpotential of 1.84 V towards HER, which is even lower than the potential required by the Pt/C commercial electrocatalyst (2.12 V) for the same current density, demonstrating its good potential for industrial HER application.
2. Experimental
2.1. Materials
Ammonium heptamolybdate tetrahydrate ((NH4)6Mo7O24·4H2O), cobalt nitrate hexahydrate (Co(NO3)2·6H2O), nickel nitrate hexahydrate (Ni(NO3)2·6H2O), and dicyandiamide (DCA) were purchased from Aladdin Chemical Reagent Co. Ltd. Nafion solution (5 wt%) was purchased from Alfa Aesar. Commercial platinum on graphitized carbon (20 wt% Pt/C) was purchased from Aldrich. All reagents were used as received without further purification.2.2. Synthesis of ultrafine (NixCo1−x)6Mo6C2 nanocrystals
Aqueous solutions containing Co(NO3)2·6H2O, Ni(NO3)2·6H2O and (NH4)6Mo7O24·4H2O in a particular ratio were mixed and stirred in a water bath at 80 °C for 4 h. At the end of the reaction, the resulting light purple NixCo1−xMoO4 precursor was collected by centrifuging at 3000 rpm and washed several times. Finally, the sample was dried at 80 °C overnight in a vacuum oven.(NixCo1−x)6Mo6C2 samples were synthesized by high-temperature carbonization. In a typical procedure, 0.5 g of dicyandiamide (DCA) was firstly dissolved in 5 mL of H2O at 100 °C under magnetic stirring to form a transparent solution. Then, 50 mg of NixCo1−xMoO4 was added to the above hot solution, which was then stirred and boiled until all water evaporated. A dry powder was obtained. Afterwards, the powder was placed in an alumina boat, heated in a pipe furnace at 500 °C for 30 min under a N2 atmosphere with a heating rate of 2 °C min−1, and then further heated at 900 °C for 6 h with a heating rate of 5 °C min−1.
2.3. Synthesis of Co6Mo6C2, DCA-Co and DCA-Mo
Co6Mo6C2 was prepared according to the same method as that of (NixCo1−x)6Mo6C2 except that the Ni element was removed from the precursor. Similarly, DCA-Co or DCA-Mo was prepared according to the same method as that of (NixCo1−x)6Mo6C2 except that the precursor was replaced by Co(NO3)2·6H2O or (NH4)6Mo7O24·4H2O.2.4. Characterization
The powder X-ray diffraction (XRD) patterns were recorded using an X-ray diffractometer using Cu Kα (Dandong TD-3500, 30 kV and 20 mA). The morphology and microstructures were characterized using scanning electron microscopy (SEM, Hitachi, SU-8010) and high-resolution transmission electron microscopy (TEM/HRTEM, JEM-2100). The surface chemical compositions were studied using X-ray photoelectron spectroscopy (XPS, ESCA Lab MKII).2.5. Preparation of the working electrodes
The catalyst (5 mg) was dispersed in a mixture of water (0.75 mL) and ethanol (0.2 mL) containing 5% Nafion solution (50 μL) under ultrasonic irradiation for ca. 1 h until a homogeneous ink was formed. Then, 5 μL of the ink containing 25 μg of the catalyst was transferred onto an L-shaped glassy carbon electrode of 3 mm diameter, yielding a catalyst level of 0.35 mg cm−2. Similarly, 15 μL of the ink containing 75 μg of the catalyst was transferred onto a glassy carbon disk electrode of 5 mm diameter, yielding a catalyst level of 0.38 mg cm−2. The electrode with the catalyst was dried at 50 °C and was used as the working electrode for further electrochemical measurements.2.6. Electrochemical measurement
The electrochemical performance was tested by using an AUTOLAB PGSTAT302N (Methrohm) via a three-electrode system at room temperature. The reference and counter electrodes were the standard Hg/HgO electrode and a carbon rod, respectively. The L-shaped glassy carbon electrode (d = 3 mm) and disk glassy carbon electrode (d = 5 mm) were used as the working electrodes for HER catalysis. Linear sweep voltammetry (LSV) measurements were performed in 1 M KOH. Before HER tests, the electrolyte was deoxygenated with N2 for at least 30 minutes to completely remove oxygen. Electrochemical impedance spectroscopy (EIS) was performed from the frequency of 0.001 Hz to 10 kHz at the open circuit potential or at other potentials. Long-term stability tests were executed using the chronopotentiometric method at the current densities of 25, 125 and 225 mA cm−2 for 48 h and cyclic voltammograms (CVs) recorded at a scan rate of 50 mV s−1 for 2000 times. Furthermore, considering the demands of practical application, chronopotentiometric curves were recorded at large current densities of 1000 mA cm−2 for over 48 h. The polarization curves were replotted as overpotential (η) vs. log current density (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) j), and the Tafel slope (b) was obtained according to the Tafel equation (η = a + blog(j)). Potentials were referenced to RHE according to the equation: ERHE = 0.098 V + 0.059 × pH + EHg/HgO. Cyclic voltammogram curves were obtained at various scan rates. Electrochemically active surface areas (ECSAs) of the catalysts were determined by electrochemical capacitance measurements with potential ranging from −0.025 to 0.175 V (vs. RHE) at the scan rates of 10, 20, 30, 50, 70 and 100 mV s−1 to get the electrochemical capacitance. The measured data were plotted as a function of scan rate, and a linear fitting determined the specific capacitance.
j), and the Tafel slope (b) was obtained according to the Tafel equation (η = a + blog(j)). Potentials were referenced to RHE according to the equation: ERHE = 0.098 V + 0.059 × pH + EHg/HgO. Cyclic voltammogram curves were obtained at various scan rates. Electrochemically active surface areas (ECSAs) of the catalysts were determined by electrochemical capacitance measurements with potential ranging from −0.025 to 0.175 V (vs. RHE) at the scan rates of 10, 20, 30, 50, 70 and 100 mV s−1 to get the electrochemical capacitance. The measured data were plotted as a function of scan rate, and a linear fitting determined the specific capacitance.
3. Results and discussion
As schematically illustrated in Fig. 1a, Am(MoO4)n (A = Fe3+, m = 2, and n = 3; or A = Co2+, Ni2+, and (NixCo1−x)2+, and m = n = 1), which is a precursor for the preparation of polymetallic compounds, possesses a special molybdate (tetrahedron) crystal structure. The AMoO4 precursor was mixed with DCA by using a solution evaporation method, and then a series of single-phase multi-metal Mo-based carbides (Fe3Mo3C, Mo6Ni6C, Co6Mo6C2, and (NixCo1−x)6Mo6C2) was obtained through a two step high-temperature carbonization. The chemical compositions and crystal properties of the as-prepared samples were characterized using XRD. As shown in Fig. 1b and Fig. S1 (ESI†), different cationic Mo-based bimetallic carbides were successfully synthesized, corresponding to Fe3Mo3C (JCPDS No. 47-1191), Mo6Ni6C (JCPDS No. 65-4436) and Co6Mo6C2 (JCPDS No. 80-0339), respectively. Specifically, as shown in the XRD pattern of (Ni0.2Co0.8)6Mo6C2, five distinct characteristic peaks at 35.31°, 39.86°, 42.39°, 46.37° and 72.37° were indexed to the (331), (422), (511), (440) and (822) facets of cubic Co6Mo6C2 (Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (227), JCPDS No. 80-0339), respectively, which confirmed the presence of the tri-metallic carbide (NixCo1−x)6Mo6C2 with a relatively low Ni content maintaining a cubic Co6Mo6C2 structure. Namely, the addition of Ni does not destroy the structure of the carbides, forming a single-phase multi-metal alloy carbide structure. Meanwhile, the XRD results of DCA-Co, DCA-Mo and Co6Mo6C2 indicating the peaks of metallic cobalt (JCPDS No. 15-0806), molybdenum carbide and cubic Co6Mo6C2, respectively, are also presented for comparison.
m (227), JCPDS No. 80-0339), respectively, which confirmed the presence of the tri-metallic carbide (NixCo1−x)6Mo6C2 with a relatively low Ni content maintaining a cubic Co6Mo6C2 structure. Namely, the addition of Ni does not destroy the structure of the carbides, forming a single-phase multi-metal alloy carbide structure. Meanwhile, the XRD results of DCA-Co, DCA-Mo and Co6Mo6C2 indicating the peaks of metallic cobalt (JCPDS No. 15-0806), molybdenum carbide and cubic Co6Mo6C2, respectively, are also presented for comparison.
 | ||
| Fig. 1 (a) Illustration of the preparation of the (NixCo1−x)6Mo6C2 catalyst. DCA: dicyandiamide; AMoO4: different cationic molybdates; the support with a gold colour indicates the N-doped carbon layers as the support materials. The blue central core represents the (NixCo1−x)6Mo6C2 nanoparticle. (b) XRD patterns of DCA-Co, DCA-Mo, Co6Mo6C2 and (Ni0.2Co0.8)6Mo6C2. | ||
The addition of DCA is regarded to not only act as the carbon source in the final high-temperature carbonization step for crystallization of the carbides, but also as the partitioning and reducing agent for the deoxygenation of the metal molybdate precursors in the first thermal-treatment step, which facilitates the anchoring of intermediate precursors on the carbon-based matrix and the following crystallization without agglomeration.
In view of Ni- and Co-based compounds being active for HER, for the purpose of developing effective electrocatalysts, the following characterization and discussion take the as-prepared (NixCo1−x)6Mo6C2 as examples. The optimized (NixCo1−x)6Mo6C2 catalyst for HER with a particular ratio of Ni and Co could be identified. The microstructure and morphology of (Ni0.2Co0.8)6Mo6C2 as a representative of the (NixCo1−x)6Mo6C2 series were characterized using scanning electron microscopy (SEM) and transmission electron microscopy (TEM).
As shown in the representative SEM images (Fig. 2a and b), (Ni0.2Co0.8)6Mo6C2 nanoparticles are uniformly anchored onto a porous carbon matrix. The relevant X-ray energy-dispersive spectroscopy (EDS) elemental mappings (Fig. 2c–g) illustrate that Co, Mo, Ni, C and N elements as the components of (Ni0.2Co0.8)6Mo6C2 nanoparticles are uniformly distributed. The quantified results also confirmed the ratios of the metal elements, which were consistent with the initial amounts used in the reaction (Fig. S2, ESI†). The TEM image (Fig. 2h) shows that the ultra-small (Ni0.2Co0.8)6Mo6C2 nanoparticles with an average diameter of ca. 4.0 nm are evenly embedded into the N-doped carbon matrix.44,45 The successive lattice fringe with a plane distance of 0.22 nm shown in the high-resolution TEM (HRTEM) image (Fig. 2i) of these particles, which is consistent with the (511) lattice plane of Co6Mo6C2, further confirmed the successful growth of single-phase multi-metal carbides. In addition, these nanoparticles are well enveloped by few-layer graphitic carbon shells (with a typical lattice spacing of 0.36 nm similar to that of graphitic carbon) exhibiting high mechanical and chemical stability. The uniform dispersion of the (Ni0.2Co0.8)6Mo6C2 nanocrystals with an ultra-small size can be ascribed to the confining effect of the N-doped carbon originating from the carbonization of DCA during the reaction and crystallization of the carbides.
 | ||
| Fig. 2 (a and b) SEM images of (Ni0.2Co0.8)6Mo6C2; (c–g) elemental mapping of Co, Mo, Ni, C and N in (Ni0.2Co0.8)6Mo6C2; (h) TEM image of (Ni0.2Co0.8)6Mo6C2; (i) HRTEM image of (Ni0.2Co0.8)6Mo6C2. | ||
To reveal the chemical state and bonding configuration of the as-prepared (Ni0.2Co0.8)6Mo6C2, X-ray photoelectron spectroscopy (XPS) analysis was carried out, as shown in Fig. 3. The full survey spectrum of (Ni0.2Co0.8)6Mo6C2 confirmed the existence of peaks in the regions of Ni 2p, Co 2p, Mo 3d, O 1s, N 1s, and C 1s (Fig. 3a). For the Co 2p XPS spectra (Fig. 3b), the binding energy peaks of Co 2p3/2 and Co 2p1/2 appear at 781.2 eV and 796.5 eV, respectively. The binding energy peaks at 786.2 eV and 803.4 eV are their corresponding satellite structures (S1 and S2), respectively. In addition, slightly weaker metal Co (Co0) peaks (779.5 eV and 794.7 eV) of (Ni0.2Co0.8)6Mo6C2 could be found, which suggests the formation of cobalt molybdenum bimetallic carbides.46 The spectrum of Mo 3d (Fig. 3c) implies four valence states (0, 2+, 4+, and 6+) for the catalyst. After a curve deconvolution, the doublets of 232.5 eV/232.1 eV and 235.5 eV/234.9 eV can be attributed to 3d3/2/3d5/2 of Mo4+ and 3d3/2/3d5/2 of Mo6+, which stem from the MoO2 and MoO3 species on the (Ni0.2Co0.8)6Mo6C2 nanoframe surface.47 The peak located at 228.8 eV for Mo 3d5/2 can be ascribed to the Mo2+ species, which is assigned to Mo–C bonding.48 The peak located at a binding energy of 227.7 eV is assigned to metallic Mo (Mo0).49 The Mo0 species is derived from the metal (Mo)–metal bond existing in Mo-based carbides, which has already been proved by previous reports.50 For the Ni 2p spectrum (Fig. 3d), the peaks located at 856.1 eV and 871.4 eV with satellite peaks can be assigned to Ni2+ 2p3/2 and Ni2+ 2p1/2 from (Ni0.2Co0.8)6Mo6C2,51 respectively. The details of the C 1s XPS spectra are shown in Fig. 3e. The main peak at 284.2 eV corresponds to the sp2 bonded graphite-like carbon, and the peak at 285.2 eV corresponding to sp3 bonded carbon was also observed. It should be noted that there is only a tiny peak at 288.7 eV, which is ascribed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/C–OH groups, indicating that most oxygen containing functional groups of DCA are eliminated during the high temperature treatment and a significant increase in structure ordering of the carbon matrix has been achieved.52 The N 1s spectrum (Fig. 3f) confirmed the N-doping in the carbon matrix including the existence of pyridinic N (397.9 eV) and pyrrolic N (400.2 eV).53
O/C–OH groups, indicating that most oxygen containing functional groups of DCA are eliminated during the high temperature treatment and a significant increase in structure ordering of the carbon matrix has been achieved.52 The N 1s spectrum (Fig. 3f) confirmed the N-doping in the carbon matrix including the existence of pyridinic N (397.9 eV) and pyrrolic N (400.2 eV).53
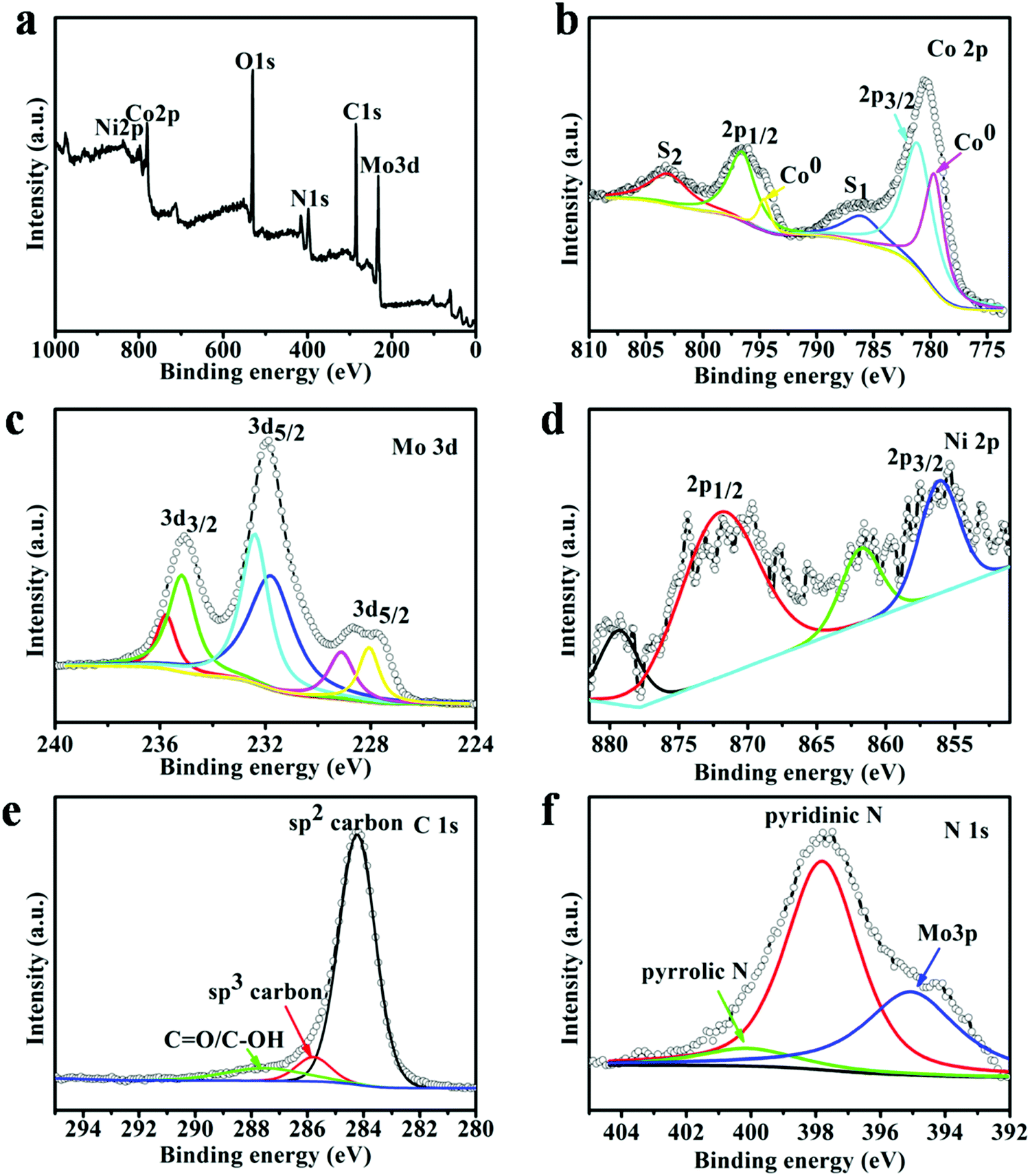 | ||
| Fig. 3 (a) XPS spectrum of (Ni0.2Co0.8)6Mo6C2, high resolution XPS spectra and the fitted data of Co 2p (b), Mo 3d (c), Ni 2p (d), C 1s (e) and N 1s (f) of (Ni0.2Co0.8)6Mo6C2. | ||
The electrocatalytic activity of the (NixCo1−x)6Mo6C2 series for HER was systematically evaluated using a standard three-electrode system in 1 M KOH electrolyte. For comparison, the performance of counterparts including DCA-Co, DCA-Mo, Co6Mo6C2 and a commercial 20% Pt/C catalyst was also studied in parallel. The polarization curves in Fig. 4a and Fig. S3 (ESI†) reveal that the Ni/Co/Mo ternary carbides (NixCo1−x)6Mo6C2 possess better electrocatalytic activities towards the HER in comparison to their binary and mono metal carbide counterparts, and (Ni0.2Co0.8)6Mo6C2 exhibits the highest activity among the (NixCo1−x)6Mo6C2 series. To achieve the current density of 10 mA cm−2, as shown in the inset of Fig. 4a, (Ni0.2Co0.8)6Mo6C2 needs an overpotential of 100 mV, which is smaller than those of Co6Mo6C2 (152 mV), DCA-Mo (174 mV) and DCA-Co (245 mV).
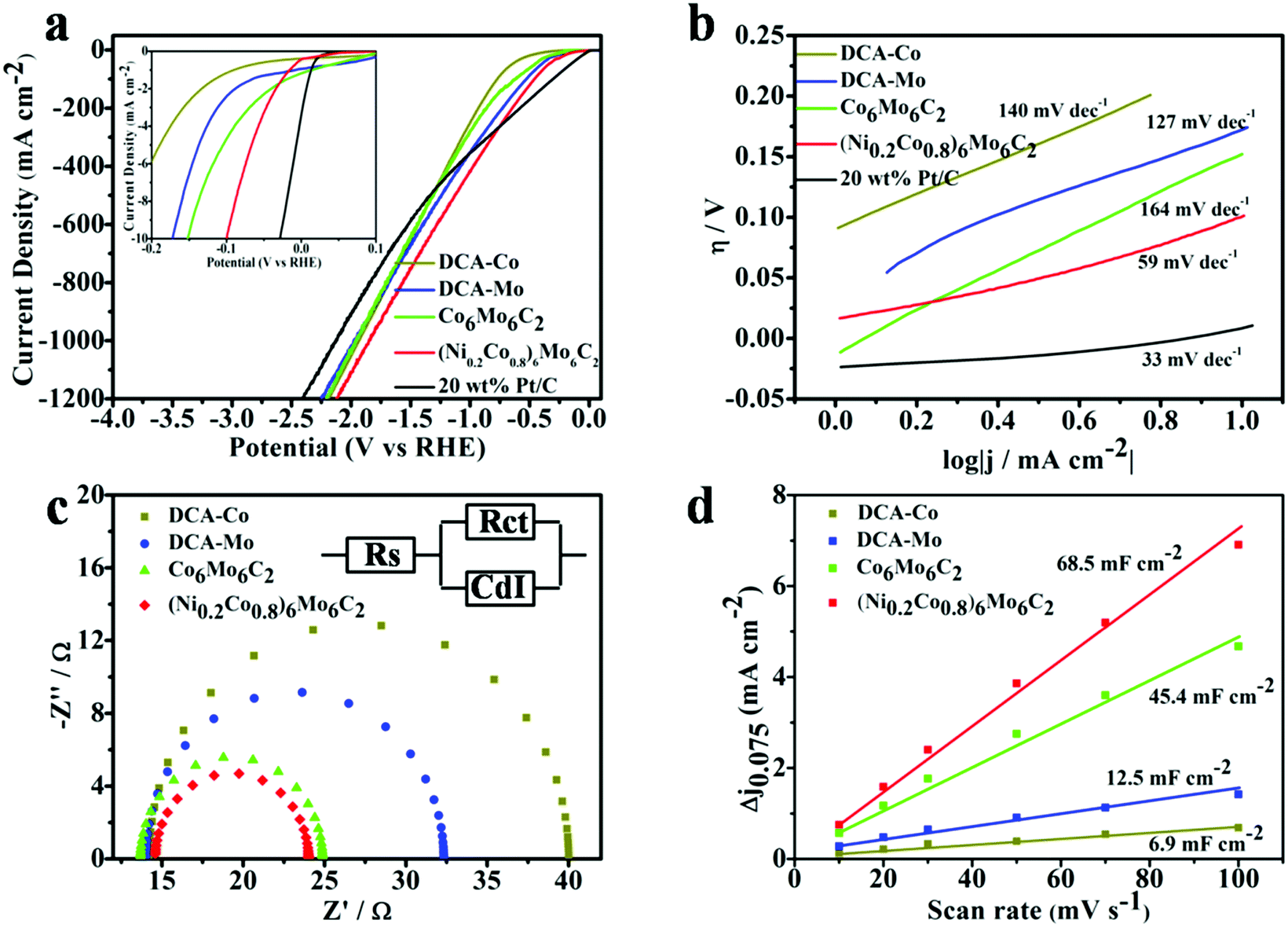 | ||
| Fig. 4 (a) Polarization curves of (Ni0.2Co0.8)6Mo6C2, Co6Mo6C2, DCA-Mo, DCA-Co and Pt/C on a glassy carbon disk electrode in 1 M KOH. Inset: Polarization curves of the same catalyst on an L-shaped glassy carbon electrode in the range of 0–10 mA cm−2 current density; (b) Tafel plots of (Ni0.2Co0.8)6Mo6C2, Co6Mo6C2, DCA-Mo, DCA-Co and Pt/C in 1 M KOH; (c) Nyquist plots of electrochemical impedance spectroscopy (EIS) of (Ni0.2Co0.8)6Mo6C2, Co6Mo6C2, DCA-Mo and DCA-Co in 1 M KOH; (d) the capacitive current at 0.075 V as a function of scan rate for (Ni0.2Co0.8)6Mo6C2, Co6Mo6C2, DCA-Mo and DCA-Co in 1 M KOH. | ||
In view of practical hydrogen production at an industrial scale, a current density with a higher order of magnitude is usually required for electrolysis operation. Therefore, to probe the potential of the as-prepared ternary carbides for practical applications, the HER activities at a higher current (such as 1000 mA cm−2) of these samples were also evaluated. The overpotential required for the current density of 1000 mA cm−2 using the (Ni0.2Co0.8)6Mo6C2 electrocatalyst is only 1.84 V, which is superior to other carbide counterparts in this work and even better than the performance of the commercial Pt/C (20% Pt) catalyst (2.12 V required for 1000 mA cm−2). It should be noted that although the activity of commercial Pt/C at a low potential (29 mV required for 10 mA cm−2) is better than that of (Ni0.2Co0.8)6Mo6C2, (Ni0.2Co0.8)6Mo6C2 can drive a higher current density when applying a potential over 745 mV. These results suggest that (Ni0.2Co0.8)6Mo6C2 has great potential for practical HER applications.
The Tafel plots were derived from the HER polarization curves by deriving the logarithm of the current density, according to the following equation: η = a + blogj, where η is the overpotential (η10), b is the Tafel slope, and j is the current density. The equation is usually used to evaluate the reaction kinetics in the electrocatalytic hydrogen generation process.54 Hydronium ion reduction consists of three steps: the Volmer, Heyrvosky and Tafel steps, respectively.
Electrochemical hydrogen adsorption (Volmer step)
| H2O + M + e− → M–H* + OH− |
| M–H* + H2O + e− → M + OH− + H2 |
| 2M–H* → 2M + H2 |
The Tafel slope is regarded as an intrinsic parameter to reveal the rate determining step in the kinetics of HER. Fig. 4b demonstrates that the Tafel slope of the (Ni0.2Co0.8)6Mo6C2 catalyst is about 59 mV dec−1, while the Tafel slopes of DCA-Co, DCA-Mo and Co6Mo6C2 are 140, 127 and 164 mV dec−1, respectively. Thus, the Volmer–Heyrovsky mechanism is presumed to dominate the HER process when using (Ni0.2Co0.8)6Mo6C2 as the catalyst, and the hydrogen generation rate could be increased rapidly by applying a higher overpotential, resulting in high HER activity with high voltages applied.57,58 To better understand the role of the interface reaction and electrode kinetics for HER, EIS analysis was further employed.59 As the Nyquist plots show in Fig. 4c, the charge transfer resistance Rct of DCA-Co, DCA-Mo, Co6Mo6C2, and (Ni0.2Co0.8)6Mo6C2 is Rct = 25.8 Ω, 18.0 Ω, 11.2 Ω, and 9.42 Ω, respectively. Compared with the other catalysts, (Ni0.2Co0.8)6Mo6C2 displays a much lower Rct, indicating that the addition of Co and especially Ni in the multi-metal carbides significantly enhances the transfer rate of carriers and the faradaic process.60 It is generally believed that the electric double layer capacitance (Cdl) is directly proportional to the electrochemically active surface area (ECSA) during the electrocatalytic process. When there are more effective active components on the catalyst surface, the larger the ECSA, the better the corresponding catalytic activity, so the electric double layer capacitance can often be used to determine the catalyst activity.61 Thus, the CVs of these samples were recorded in the potential range not containing the redox states at different scan rates (10, 20, 30, 50, 70 and 100 mV s−1) in 1 M KOH. As shown in Fig. 4d and Fig. S4 (ESI†), one can clearly observe that the Cdl of (Ni0.2Co0.8)6Mo6C2, Co6Mo6C2, DCA-Mo and DCA-Co is 68.5, 45.4, 12.5 and 6.9 mF cm−2, respectively, manifesting that the Cdl of (Ni0.2Co0.8)6Mo6C2 is the highest, which implies that the multi-metal synergy helps the carbides in exposing more accessible electrochemically active sites.
The catalytic stability is another critical criterion for determining practical HER capability. As shown in Fig. 5a, the stability of the as-prepared (Ni0.2Co0.8)6Mo6C2 was evaluated by a cycling electrochemical test in the form of cyclic voltammetry (CV) sweeps between 0.02 V –and −0.25 V vs. RHE at a scan rate of 50 mV s−1 in 1 M KOH. The negligible change between the polarization curves obtained before and after 2000 continuous CV tests demonstrates the superior stability of (Ni0.2Co0.8)6Mo6C2 in electrochemical processes. The HER durability at a high current density of (Ni0.2Co0.8)6Mo6C2 was also studied by carrying out long-term chronopotentiometry. Only a slight catalytic current depression can be observed after the long-term electrolysis operated at different constant potentials (160 mV, 470 mV, and 650 mV) over 48 hours (Fig. 5b), illustrating the remarkable stability of such ternary carbide catalysts in the alkaline HER process. In comparison, the electrolysis current density driven by the commercial Pt/C catalyst at 650 mV constant potential attenuates by 40% after 15 hours. In addition, considering the demands of practical application, chronopotentiometric curves were recorded for the (Ni0.2Co0.8)6Mo6C2 catalyst by driving a long-term high current density of 1000 mA cm−2. As shown in Fig. 5c, the potential required for maintaining 1000 mA cm−2 remains reasonably stable and lasts over 48 h, suggesting the great potential of the as-prepared (Ni0.2Co0.8)6Mo6C2 for commercial HER utilization. The TEM and XPS characterization results (Fig. S6 and S7, ESI†) of the catalyst after the long-time constant-current HER process demonstrate that the structure and composition of (Ni0.2Co0.8)6Mo6C2 are not obviously changed, showing the good stability of such materials.
 | ||
| Fig. 5 (a) Polarization curves of (Ni0.2Co0.8)6Mo6C2 initially and after 2000 cycles in 1 M KOH; (b) chronoamperometric responses of (Ni0.2Co0.8)6Mo6C2 and Pt/C at different static over-potentials for 48 h; (c) chronopotentiometric curves at a constant current density of 1000 mA cm−2 for 48 h. | ||
Based on the experimental results obtained in this work, it is clear that the synergistic effect between different transition metals in the carbides can optimize the Gibbs free energy and bonding energy of hydrogen adsorption, which then affects the process of water splitting and HER performance. Compared to DCA-Co and DCA-Mo, the Co6Mo6C2 carbides obtained by mixing Co and Mo exhibit improved HER activity. By including Ni in the crystal structure of Co6Mo6C2, the electrocatalytic activity of the multi-metal carbides is further enhanced. The active sites for HER in (Ni0.2Co0.8)6Mo6C2 are significantly increased in relation to the single metal carbides, which can be confirmed by the analysis of the electrochemically active surface areas of different samples. The superior HER activity and durability of (Ni0.2Co0.8)6Mo6C2 in alkaline media can be concluded as due to the synergistic effects of the multi-metal coordinated carbides and the carbon-based architectures. In detail, the following aspects could be outlined: (1) first of all, the small size of (Ni0.2Co0.8)6Mo6C2 ensures a large amount of exposed active sites; (2) double metal cations (Co and Ni) introduce multi-metal synergy to assist fast water dissociation to yield a sufficient amount of protons to accelerate the hydrogen production; (3) the carbon matrix and protective layer derived from DCA offer good chemical and mechanical stability, as well as high interfacial conductivity for the electrocatalysts.
4. Conclusions
In summary, we reported a simple and novel strategy to synthesize multi-metal carbide nanoparticles covered by few-layer N-doped graphitic carbon shells, which were in situ prepared by a two-step method from NixCo1−xMoO4 and DCA in a N2 atmosphere. This (NixCo1−x)6Mo6C2 series of materials, especially (Ni0.2Co0.8)6Mo6C2, shows good HER performance and high long-term stability in alkaline solution even at a large current density. The superior HER activity can be attributed to the cooperative/synergistic effects of the nanosized multi-metallic carbides (NixCo1−x)6Mo6C2 and the carbon-based protective support. The novelty of this work is not only the fundamental understanding and scientific discovery of new nanomaterials, but also their readiness for large-scale production and industrial application.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 11704007), Baoji University of Arts and Sciences Key Research (Grant No. ZK16070), the Provincial Natural Science Foundation of Hunan (No. 2019JJ50602), and the Scientific Research Fund of Hunan Provincial Education Department (No. 18C0105).References
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS.
- X. X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- X. F. Lu, L. Yu and X. W. Lou, Sci. Adv., 2019, 5, eaav6009 CrossRef CAS.
- W. F. Chen, J. T. Muckerman and E. Fujita, Chem. Commun., 2013, 49, 8896–8909 RSC.
- J. Li and D. Zheng, Adv. Sci., 2017, 4, 1600380 CrossRef.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. B. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef.
- J. S. Li, Y. Wang, C. H. Liu, S. L. Li, Y. G. Wang, L. Z. Dong, Z. H. Dai, Y. F. Li and Y. Q. Lan, Nat. Commun., 2016, 7, 11204–11211 CrossRef CAS.
- R. G. Ma, Y. Zhou, Y. F. Chen, P. X. Li, Q. Liu and J. C. Wang, Angew. Chem., Int. Ed., 2015, 54, 14723–14727 CrossRef CAS.
- Y. P. Liu, G. T. Yu, G. D. Li, Y. H. Sun, T. Asefa, W. Chen and X. X. Zou, Angew. Chem., Int. Ed., 2015, 54, 10752–10757 CrossRef CAS.
- Y. Huang, Q. F. Gong, X. N. Song, K. Feng, K. Q. Nie, F. P. Zhao, Y. Y. Wang, M. Zeng, J. Zhong and Y. G. Li, ACS Nano, 2016, 10, 11337–11343 CrossRef CAS.
- M. Qamar, A. Adam, B. Merzougui, A. Helal, O. Abdulhamid and M. N. Siddiqui, J. Mater. Chem. A, 2016, 4, 16225–16232 RSC.
- F. X. Ma, H. B. Wu, B. Y. Xia, C. Y. Xu and X. W. Lou, Angew. Chem., Int. Ed., 2015, 54, 15395–15399 CrossRef CAS.
- H. Vrubel and X. Hu, Angew. Chem., Int. Ed., 2012, 51, 12703e6 CrossRef.
- J. Q. Tian, Q. Liu, A. M. Asiri and X. P. Sun, J. Am. Chem. Soc., 2014, 136, 7587–7590 CrossRef CAS.
- L. Jiao, Y. X. Zhou and H. L. Jiang, Chem. Sci., 2016, 7, 1690–1695 RSC.
- Y. Zhang, H. J. Zhang, Y. Y. Feng, L. Liu and Y. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 26684–26690 CrossRef CAS.
- H. J. Yan, Y. Q. Jiao, A. P. Wu, C. G. Tian, X. M. Zhang, M. Wang, Z. Y. Ren and H. G. Fu, Chem. Commun., 2016, 52, 9530–9533 RSC.
- Z. H. Xiao, Y. Wang, Y. C. Huang, Z. X. Wei, C. L. Dong and J. M. Ma, Energy Environ. Sci., 2013, 10(12), 2563–2569 RSC.
- J. Yin, P. P. Zhou, L. An, L. Huang, C. W. Shao, J. Wang, H. Y. Liu and P. X. Xi, Nanoscale, 2016, 8, 1390 RSC.
- H. H. Shi, H. F. Liang, F. W. Ming and Z. C. Wang, Angew. Chem., Int. Ed., 2017, 56, 573 CrossRef CAS.
- H. J. Yan, C. G. Tian, L. Wang, A. P. Wu, M. C. Meng, L. Zhao and H. G. Fu, Angew. Chem., Int. Ed., 2015, 54, 6325–6329 CrossRef CAS.
- Y. Zhao, K. Kamiya, K. Hashimoto and S. Nakanishi, J. Am. Chem. Soc., 2015, 137, 110–113 CrossRef CAS.
- J. F. Xie, S. Li, X. D. Zhang, J. J. Zhang, R. X. Wang, H. Zhang, B. C. Pan and Y. Xie, Chem. Sci., 2014, 5, 4615–4620 RSC.
- W. F. Chen, J. M. Schneider, K. Sasaki, C. H. Wang, J. Schneider, S. Iyer, Y. M. Zhu, J. T. Muckerman and E. Fujita, ChemSusChem, 2014, 7, 2414–2418 CrossRef CAS.
- L. L. Feng, G. T. Yu, Y. Y. Wu, G. D. Li, H. Li, Y. Sun, H. Asefa, T. W. Chen and X. X. Zou, J. Am. Chem. Soc., 2015, 137, 14023–14026 CrossRef CAS.
- T. Y. Wang, L. Liu, Z. W. Zhu, P. Papakonstantinou, J. B. Hu, H. Y. Liu and M. X. Li, Energy Environ. Sci., 2013, 6, 625–633 RSC.
- K. Zhang, H. J. Kim, J. T. Lee, G. W. Chang, X. J. Shi, W. J. Kim, M. Ma, K. J. Kong, J. M. Choi, M. S. Song and J. H. Park, ChemSusChem, 2014, 7, 2489–2495 CrossRef CAS.
- S. F. Zhuo, Y. Xu, W. W. Zhao, J. Zhang and B. Zhang, Angew. Chem., Int. Ed., 2013, 52, 8602–8606 CrossRef CAS.
- H. Vrubel and X. I. Hu, Angew. Chem., Int. Ed., 2012, 51, 12703–12706 CrossRef CAS.
- Y. Liu, T. G. Kelly, J. G. Chen and W. E. Mustain, ACS Catal., 2013, 3, 1184–1194 CrossRef CAS.
- M. Miao, J. Pan, T. He, Y. Yan, B. Y. Xia and X. Wang, Chem. – Eur. J., 2017, 23, 10947–10961 CrossRef CAS.
- Y. C. Kimmel, X. Xu, W. Yu, X. Yang and J. G. Chen, ACS Catal., 2014, 4, 1558–1562 CrossRef CAS.
- D. Vikraman, S. Hussain, K. Karuppasamy, A. Feroze, A. Kathalingam and A. Sanmugam, Appl. Catal., B, 2020, 264, 118531 CrossRef.
- S. Hussain, I. Rabani, D. Vikraman, A. Feroze and K. Karuppasamy, ACS Sustainable Chem. Eng., 2020, 32, 12248–12259 CrossRef.
- D. Vikraman, K. Akbar, S. Hussain, G. Yoo, J. Y. Jang and S. H. Chun, Nano Energy, 2017, 35, 101–114 CrossRef CAS.
- D. Vikraman, H. G. Abbas, S. Hussain, K. Akbar and K. Karuppasamy, ACS Sustainable Chem. Eng., 2019, 7, 458–469 CrossRef CAS.
- S. Hussain, I. Rabani, D. Vikraman, A. Feroze, M. Ali, Y. S. Seo, H. S. Kim and S. H. Chun, Nanomaterials, 2020, 10, 1597 CrossRef CAS.
- C. Wan, Y. N. Regmi and B. M. Leonard, Angew. Chem., Int. Ed., 2014, 53, 6407–6410 CrossRef CAS.
- H. Ang, H. T. Tan, Z. M. Luo, Y. Zhang, Y. Y. Guo, G. Guo, H. Zhang and Q. Yan, Small, 2015, 11, 6278–6284 CrossRef CAS.
- G. Wu, C. M. Johnston and N. H. Mack, J. Mater. Chem., 2011, 21, 11392–11405 RSC.
- S. Bukola, B. Merzougui and A. Akinpelu, Electrochim. Acta, 2016, 190, 1113–1123 CrossRef CAS.
- Y. Zhao, K. Kamiya and K. Hashimoto, Angew. Chem., Int. Ed., 2013, 52, 13638–13641 CrossRef CAS.
- D. V. Esposito, S. T. Hunt and Y. C. Kimmel, J. Am. Chem. Soc., 2012, 134, 3025–3033 CrossRef CAS.
- Y. P. Liu, G. T. Yu, G. D. Li, Y. H. Sun, T. Asefa, W. Chen and X. X. Zou, Angew. Chem., Int. Ed., 2015, 54, 10752–10757 CrossRef CAS.
- Y. T. Xu, X. F. Xiao, Z. M. Ye, S. L. Zhao, R. A. Shen, C. T. He, J. P. Zhang, Y. D. Li and X. M. Chen, J. Am. Chem. Soc., 2017, 139, 5285–5288 CrossRef CAS.
- X. Ma, H. Meng, M. Cai and P. K. Shen, J. Am. Chem. Soc., 2012, 134, 1954–1957 CrossRef CAS.
- L. F. Pan, Y. H. Li, S. Yang, P. F. Liu, M. Q. Yu and H. G. Yang, Chem. Commun., 2014, 50, 13135–13137 RSC.
- C. Shi, A. Zhang, X. Li, S. Zhang, A. Zhu, Y. Ma and C. Au, Appl. Catal., A, 2012, 431, 164–170 CrossRef.
- Y. Liu, G. Yu, G. D. Li, Y. Sun, T. Asefa, W. Chen and X. Zou, Angew. Chem., Int. Ed., 2015, 54, 10752–10757 CrossRef CAS.
- M. Y. Zu, P. F. Liu, C. Wang, Y. Wang, L. R. Zheng, B. Zhang, H. Zhao and H. G. Yang, ACS Energy Lett., 2018, 3, 78–84 CrossRef CAS.
- H. Liu, Q. He, H. Jiang, Y. Lin, Y. Zhang, M. Habib, S. Chen and L. Song, ACS Nano, 2017, 11, 11574–11583 CrossRef CAS.
- C. He, S. P. Jiang and P. K. Shen, Sci. Rep., 2013, 3, 2144 CrossRef.
- Y. Zheng, Y. Jiao, L. H. Li, T. Xing, Y. Chen, M. Jaroniec and S. Z. Qiao, ACS Nano, 2014, 8, 5290–5296 CrossRef CAS.
- Y. Y. Chen, Y. Zhang, W. J. Jiang, X. Zhang, Z. H. Dai, L. J. Wan and J. S. Hu, ACS Nano, 2016, 10, 8851–8860 CrossRef CAS.
- M. G. de Chialvo and A. C. Chialvo, J. Electrochem. Soc., 2000, 147, 1619–1622 CrossRef CAS.
- B. E. Conway and L. J. Bai, J. Electroanal. Chem. Interfacial Electrochem., 1986, 198, 149–175 CrossRef CAS.
- B. E. Conway and B. V. Tilak, Advances in catalysis, Academic Press, 1992, vol. 38, pp. 1–147 Search PubMed.
- S. A. Vilekar, I. Fishtik and R. Datta, J. Electrochem. Soc., 2010, 157, B1040–B1050 CrossRef CAS.
- R. G. Ma, Y. Zhou, Y. F. Chen, P. X. Li, Q. Liu and J. C. Wang, Angew. Chem., Int. Ed., 2015, 54, 14723–14727 CrossRef CAS.
- Y. Yin, J. C. Han, Y. M. Zhang, X. H. Zhang, P. Xu, Q. Yuan, L. Samad, X. J. Wang, Y. Wang, Z. H. Zhang, P. Zhang, X. Z. Cao, B. Song and S. Jin, J. Am. Chem. Soc., 2016, 138, 7965–7972 CrossRef CAS.
- J. F. Xie, S. Li, X. D. Zhang, J. J. Zhang, R. X. Wang, H. Zhang, B. C. Pan and Y. Xie, Chem. Sci., 2014, 5, 4615 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00571a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |