
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Virus removal from semen with a pinched flow fractionation microfluidic chip†
T.
Hamacher
 *a,
J. T. W.
Berendsen
a,
J. E.
van Dongen
a,
R. M.
van der Hee
b,
J. J. L. M.
Cornelissen
b,
M. L. W. J.
Broekhuijse
cd and
L. I.
Segerink
a
*a,
J. T. W.
Berendsen
a,
J. E.
van Dongen
a,
R. M.
van der Hee
b,
J. J. L. M.
Cornelissen
b,
M. L. W. J.
Broekhuijse
cd and
L. I.
Segerink
a
aBIOS Lab on a Chip Group, MESA+ Institute for Nanotechnology & Technical Medical Centre, Max Planck – University of Twente Center for Complex Fluid Dynamics, University of Twente, P.O. Box 217, 7500 AE, Enschede, The Netherlands. E-mail: t.hamacher@utwente.nl; j.t.w.berendsen@utwente.nl; j.e.vandongen@utwente.nl
bDepartment of Molecules & Materials, MESA+ Institute, University of Twente, P.O. Box 217, 7500 AE, Enschede, The Netherlands
cCRV, Wassenaarweg 20, 6843NW, Arnhem, The Netherlands
dTopigs Norsvin, 227, 5263LT Vught, The Netherlands
First published on 12th October 2021
Abstract
Nowadays pigs are bred with artificial insemination to reduce costs and transportation. To prevent the spread of diseases, it is important to test semen samples for viruses. Screening techniques applied are enzyme-linked immunosorbent assays and/or polymerase chain reaction, which are labor-intensive and expensive methods. In contrast to the current used screening techniques, it is possible to remove viruses physically from semen. However, existing methods for virus removal techniques have a low yield of spermatozoa. Therefore, we have developed a microfluidic chip that performs size-based separation of viruses and spermatozoa in boar semen samples, thereby having the potential to reduce the risk of disease spreading in the context of artificial insemination in the veterinary industry. As the head of a spermatozoon is at least twenty times larger than a virus particle, the particle size can be used to achieve separation, resulting in a semen sample with lower viral load and of higher quality. To achieve the size separation, our microfluidic device is based on pinched-flow fractionation. A model virus, cowpea chlorotic mottle virus, was used and spiked to porcine semen samples. With the proposed microfluidic chip and the optimized flow parameters, at least 84 ± 4% of the model viruses were removed from the semen. The remaining virus contamination is caused by the model virus adhering to spermatozoa instead of the separation technique. The spermatozoa recovery was 86 ± 6%, which is an enormous improvement in yield compared to existing virus removal techniques.
Introduction
The introduction of artificial insemination (AI) in the veterinary industry was revolutionary. Besides reducing the cost of animal transportation and the use of semen from superior males for a multitude of females, the spread of diseases was minimized.1,2 Nowadays, AI is the gold standard reproduction technology; approximately 90% of pigs in Europe and North America are bred using AI.2,3Although the spread of diseases is minimized by AI, micro-organisms can still be found in semen and their presence has a major influence on the semen quality.4–6 The negative impact on sperm quality caused by viruses includes decreases in sperm motility and viability7 as well as sperm abnormalities.8,9 Furthermore, one infected boar can transfer a disease to a multitude of sows which can lead to a disease outbreak and severe economic costs.4 An example is the epidemic of classical swine fever (CSF) in the Netherlands in 1997–1998. During this epidemic, the semen of infected boars was used for AI leading to CSF outbreaks in farms, whose only contact with CSF was via the contaminated semen.10 This outbreak caused the eradication of 12 million pigs and the economic loss was estimated to be $2.3 billion.11,12 To prevent another epidemic caused by virus-infected semen, new laws and regulations strictly control the biosafety and animal health of boar centers and pig farms for semen trade and import within the European Union.13 Special agreements with selected countries outside the European Union have been made to control semen import and export.14
Currently, both enzyme-linked immunosorbent assays (ELISA) and polymerase chain reactions (PCR) are used to detect the presence of viruses in semen.6 These techniques are costly (up to €150 per test), labor intensive and time consuming (3–12 working days), since these tests are performed by external providers instead of by the AI centers themselves. Due to the long screening times, virus screening test results of porcine semen are usually not available prior to insemination, because necessary waiting time reduces semen quality.15,16
Numerous viral pathogens have been identified in animal ejaculates. Most of the viruses can be found free in the seminal plasma or in somatic cellular components and not in the spermatozoa fraction.17–20 With the implementation of a virus removal processing step during daily AI laboratory procedure, the viral load and therewith the risk of disease transmission is decreased. Since the chance of disease transmission is related to the viral load, lowering this will facilitate safer insemination with fresh semen, which is currently used for AI of sows.
To reduce the viral load, it could be interesting to separate spermatozoa from the viruses. A combination of single-layer centrifugation (SLC) and “swim-up” procedure has been proposed to remove porcine circovirus type 2 (PCV2) from semen.21 The total spermatozoa yield was 45 ± 20% and the spermatozoa motility was 72 ± 23% with respect to unprocessed spermatozoa motility of 80 ± 16%.21 This method claims to reduce more than 99% of PCV2 from the semen samples. Similarly, a double SLC procedure has been proposed to remove equine arteritis virus from equine semen.22 The mean yield of motile spermatozoa was 52 ± 16% and the virus level was reduced, but the exact virus reduction was not stated.22
The poor spermatozoa yield of the previously mentioned virus removal techniques can be improved by using a microfluidic device to separate viruses from spermatozoa. Microfluidics is a fast-emerging field dealing with the flow of liquids inside micrometer-sized channels.23 The advantage of a separation technique based on microfluidics is that microfluidic processes can be automated and standardized, resulting in less manual performed processing steps.
Many different principles to separate spermatozoa from other particles or cell types using microfluidic devices have been reviewed.24–26 Microfluidic approaches are both aimed at increasing spermatozoa motility and separation of spermatozoa from different cell types. An example of devices aimed at separation is the spiral channel proposed by Son et al., which focuses particles based on their size and in this way motile spermatozoa are separated from both immotile spermatozoa and erythrocytes.27 The spermatozoa recovery was 81% with an erythrocyte removal of 99%. Liu et al.28 and Berendsen et al.29 used pinched-flow fractionation (PFF) to remove epithelial cells and erythrocytes from spermatozoa, respectively. Liu et al. achieved a spermatozoa recovery of 41 ± 3% with a purity of 97 ± 2%,28 whereas Berendsen et al. have reached a spermatozoa recovery of 94 ± 8% with an erythrocyte removal of more than 90%.29
The size separation technique PFF is based on the sudden broadening of a channel immediately after a pinched segment.30 A sample flow and a sheath flow coincide at the pinched segment, where the particles are focused onto the sidewall in case of a higher sheath than sample flow rate. The width of the sample fluid in the pinched segment should be smaller than the bigger particles present in the sample, which are the spermatozoa in our application. By choosing the flow rates in an appropriate manner, the system will operate in the laminar flow regime, which causes the streamlines to divert into the broadened segment without crossing each other. This allows the particles to effuse according to their initial position in the pinched segment, where the larger particles follow the streamlines more to the center of the channel, whereas smaller particles follow the streamlines closer to the channel wall. This allows for a separation based on each particle's size and deformability. Due to the differences in size between spermatozoa (head: 7 μm long, 4 μm wide; length: 45 μm (ref. 31)) and viruses (10–400 nm6), the size-based separation principle PFF could, in principle, be used to separate viruses from spermatozoa. PFF is advantageous in contrast to other separation methods, such as labelling with DNA stains, of which contradictory statements have been made regarding the toxicity,32–34 or motility-based methods, which can cause spermatozoa exhaustion.35 Besides that, PFF does not seem to be harmful to the cells, since we have previously shown that the effect of PFF on the viability of boar and bull spermatozoa, is less than or similar to the current used processing techniques such as centrifugation and flow cytometry.36
To our knowledge, for the first time a microfluidic device is presented that can efficiently separate viruses from porcine spermatozoa to decrease the virus load prior AI. As a virus model cowpea chlorotic mottle viruses (CCMVs) were used, which have a similar size (28 nm) to typical viruses found in semen.37 The chip design and flow parameters were optimized to achieve high spermatozoa recovery while separating most of the viruses. With a virus cleaning step implemented in the daily processing flow of porcine semen, the biosecurity of AI with porcine semen is improved.
Materials and methods
Microfluidic chip: design and fabrication
The microfluidic chip has a typical PFF design, of which a schematic illustration is shown in Fig. 1A, consisting of two inlets, a pinched segment, a broadened segment and two outlets. The two parallel inlets, one for the sample (inlet 1) and one for the buffer (inlet 2), conjunct at the pinched segment (width (wp) 50 μm, length 100 μm). The angle of the boundary between the pinched and broadened segment is 180° and the corners of the broad segment are rounded, which prevents the adherence of air bubbles and spermatozoa, because there is almost no fluid flow in the corners. The broadened segment width (wb) is 1100 μm (design I) or 2200 μm (design II). After a length of 1500 μm in the broadened segment, the separation channel with a width of 45 μm branches off to outlet 1, whereas the broadened segment ends in outlet 2. The device height was designed to be 50 μm.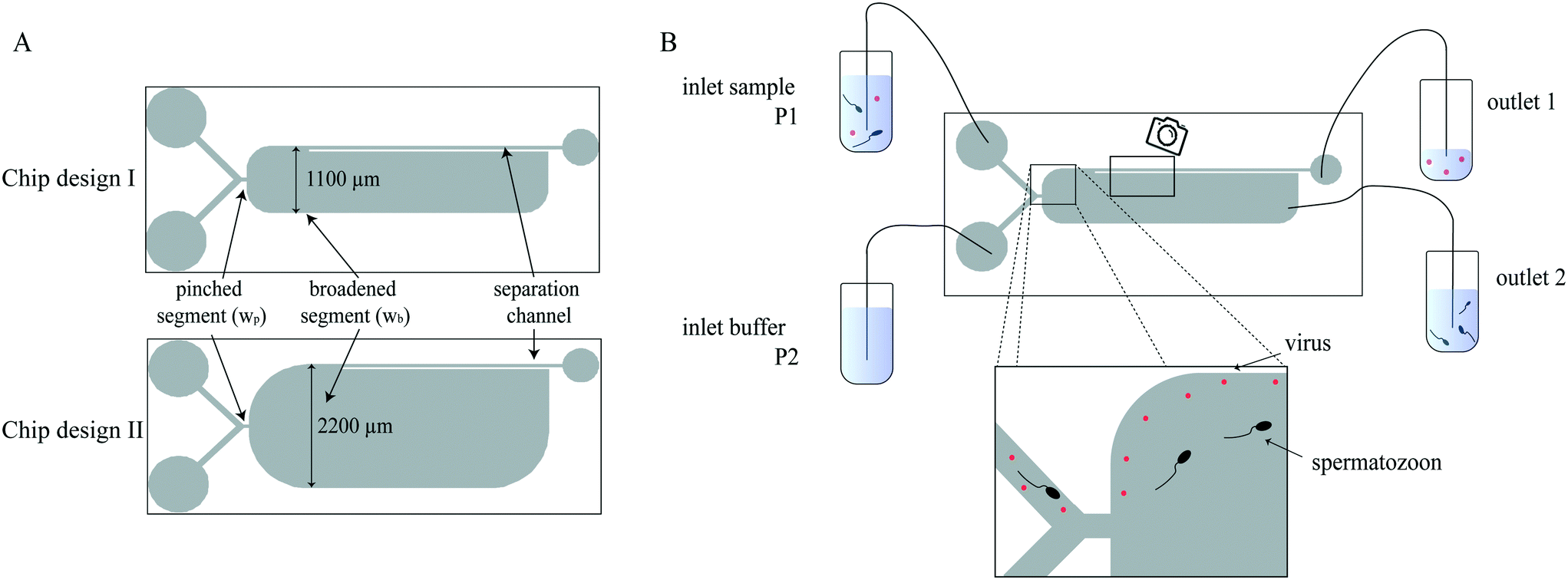 | ||
| Fig. 1 A) Schematic representation of the two chip designs. The difference between both designs is the width of the broadened segment. B) Schematic representation of the PFF set-up. The flow of the microfluidic chip is controlled by a pressure pump which is connected individually to the sample and buffer inlets. Particles are pinched and separated to follow different streamlines according to their size. The larger particles (spermatozoa) follow the streamlines further away from the channel wall and exit the chip at outlet 2. The smaller particles (viruses) are closer to the channel wall and exit the chip at outlet 1 into the waste container. | ||
The chips were designed using CleWin software (version 5.0.12, WieWeb software, Hengelo, The Netherlands) and master molds were produced using standard photolithography. The polydimethylsiloxane (PDMS) chips were fabricated in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v ratio of base versus curing agent (Sylgard 184, Dow Corning, Midland, MI, USA). PDMS was poured onto a silicon wafer, degassed, and cured at 60 °C for three hours. After curing, microfluidic inlets and outlets were punched using a Harris Uni-Core puncher (tip inner diameter (ID) 1.0 mm, Ted Pella Inc., Redding, CA, USA). The chips were bonded to glass microscope slides after activation by oxygen plasma using a plasma cleaner (model CUTE, Femto Science, Hwaseong-Si, South Korea).
:1 v/v ratio of base versus curing agent (Sylgard 184, Dow Corning, Midland, MI, USA). PDMS was poured onto a silicon wafer, degassed, and cured at 60 °C for three hours. After curing, microfluidic inlets and outlets were punched using a Harris Uni-Core puncher (tip inner diameter (ID) 1.0 mm, Ted Pella Inc., Redding, CA, USA). The chips were bonded to glass microscope slides after activation by oxygen plasma using a plasma cleaner (model CUTE, Femto Science, Hwaseong-Si, South Korea).
CCMV preparation and characterization
All chemicals described in this section were obtained from Sigma-Aldrich (Zwijndrecht, the Netherlands) unless stated otherwise. The CCMV were grown in cowpea plants. After 14 to 18 days, the virus was isolated and purified as described by Verduin38,39 and adapted by Comellas Aragonès.37 In short, the harvested leaves were homogenized with cold pH 4.8 buffer containing 0.2 M sodium acetate, 10 mM ascorbic acid and 10 mM disodium ethylenediaminetetraacetic acid (EDTA). The homogenate was squeezed through a cheesecloth and the filtrate was centrifuged (12000 × g, 10 min, 4 °C) to remove the bigger particles. The supernatant containing CCMV was precipitated with polyethylene glycol (PEG)-6000 (10% w/v) by centrifugation (12000 × g, 15 min, 4 °C). After reconstitution of the pellet in virus buffer (0.1 M sodium acetate buffer pH 5.0 containing 1 mM sodium azide and 1 mM disodium EDTA), CCMV was centrifuged for 16 h at 145000 × g at 10 °C in 37.5% cesium chloride with a Sorvall WX80 ultracentrifuge in a vertical rotor TV-1665 (Thermo Fisher Scientific, Waltham, MA, USA). This creates a gradient with a CCMV band at a density of 1.36 g L−1. After collection CCMV is dialyzed 3× with 300 ml virus buffer and stored at 4 °C.
CCMV were fluorescently labelled with Atto 647 hydroxysyccinimide (NHS) ester and Atto 488 NHS ester. Before, CCMV was dialyzed with encapsulation buffer consisting of 5 mM magnesium chloride (MgCl2) in phosphate-buffered saline (PBS) pH 7.4. The buffer was refreshed twice. The Atto-NHS dye was dissolved in oxygen free, dry DMSO and mixed in a 1:0.03 (w/w) ratio with CCMV in PBS. To remove the excess dye, CCMV was dialyzed with encapsulation buffer and refreshed twice. CCMV was stored in encapsulation buffer at 4 °C. The CCMV concentration and degree of labeling were determined by measuring the absorption of CCMV with a nanodrop system (NanoDrop One, Thermo Fisher Scientific, MA, USA). For CCMV labelled with Atto 647 NHS (CCMV-647), absorption at 260 nm and 644 nm were measured. CCMV-647 concentration was 3.3 mg ml−1, and the degree of labelling was 0.025 dyes/capsid protein. For CCMV labelled with Atto 488 NHS (CCMV-488), absorption at 260 nm and 488 nm were measured. CCMV-488 concentration was 2.4 mg ml−1, and the degree of labelling was 0.03 dyes/capsid protein. The size distribution of CCMV was measured with dynamic light scattering (DLS) using a Microtrac Nanotrac Wave W3043. Viscosity and refractive index of water and the refractive index of CCMV (1.54) were used.
Semen sample preparation
Fresh boar semen at a concentration of 20 × 106 cells per ml was obtained from a local artificial insemination center (Varkens KI Twenthe, Fleringen, The Netherlands). The semen was stored at 16 °C and used within four days. The samples were diluted with Solusem Bio+ (AIM Extender, AIM Worldwide, Vught, The Netherlands).Experimental setup
A schematic representation of the microfluidic set-up is shown in Fig. 1B. Both chip inlets were connected to a container using fused silica capillaries (Polymicro Technologies, ID 100 μm, outer diameter (OD) 360 μm, length (L) 9 cm, Molex, Surrey, UK) and Tygon tubing (ND 100-80, ID 250 μm, OD 760 μm, L 20 cm, Saint-Gobain Performance Plastics, Akron, OH, USA). The flow for the inlets was controlled with a pressure pump consisting of two Flow-EZ modules (LineUp Series, Fluigent, Le Kremlin-Bicêtre, France) with a p-cap connector (Fluigent, Le Kremlin-Bicêtre, France). The sheath pressure was 400 mbar (≈64 μl min−1), and the sample pressure was varied between 28–32 mbar (≈1.5–2.8 μl min−1). The outlet tubing was chosen such that 2–3% of the flow exited at outlet 1. Outlet 1 was connected to a container using fused silica capillaries (L = 11 cm) and Tygon tubing (L = 25 cm). Outlet 2 was connected to a container with Tygon tubing (L = 10–13 cm).Shortly before use, the chips were hydrophilized using a plasma cleaner (model CUTE, Femto Science, Hwaseong-Si, South Korea). The chips were then rinsed and incubated with poly(L-lysine)-grafted-poly(ethylene glycol) (PLL-g-PEG, SuSoS, Dübendorf, Switzerland) at a concentration of 100 μg ml−1 in deionized (DI) water for at least 15 minutes to prevent particle and cell adhesion. After coating, the chip was connected to the pressure pump. Sample and buffer solution, the latter one Solusem Bio+ for all experiments, were introduced via inlets 1 and 2, respectively. Flow was induced by applying the desired pressures to the sample and buffer inlet. At the outlets, the processed sample was collected in Eppendorf tubes.
Experimental procedure
:1 (total flow/sample flow), respectively. The ratio of outlet 1 (waste) flow/total flow was 2.4%.
For the comparison of the separation quality of chip design I with chip design II, CCMVs labeled with Atto 647 NHS ester were used. The applied sheath pressure was 400 mbar (design I: 65 μl min−1, design II: 64 μl min−1) for both designs and the sample pressures were 27 (2.5 μl min−1) and 30 mbar (2.4 μl min−1) for design I and II, respectively. The flow rate ratio for chip design I was 27 (total flow/sample flow) and for chip design II was 28 (total flow/sample flow). Outlet 1 tubing was 10 and 13 cm for chip design I and II, respectively, so that for both designs 2.8% of the total flow were collected in outlet 1.
For the other separation experiments performed with chip design II, CCMVs labeled with Atto 488 NHS ester were used. Outlet 2 tubing was 12, 12.5 or 13 cm long, so that 3.0%, 2.7%, 2.5% of the total volume were collected at outlet 1. The sheath buffer was for all experiments 400 mbar, which is equivalent to a flow rate of 64 μl min−1 and the sample pressure was 30 mbar (≈2.5 μl min−1).
For a constant outlet 1 (waste) removal rate of 2.7% with chip design II, different sample buffer pressures were applied, namely 28 mbar (2.1 μl min−1), 30 mbar (2.5 μl min−1), and 32 mbar (2.8 μl min−1). With a sheath buffer of 400 mbar (64.5 μl min−1), the flow rate ratios were 32, 27 and 24 (total flow/sample flow) for a sample pressure of 28, 30 and 32 mbar, respectively.
Separation analysis
The flow profile of CCMVs in the PFF chip was analyzed using fluorescence microscopy. Fluorescence images were taken from the separation channel and broadened segment with the EVOS microscope as shown in Fig. 1. The obtained fluorescent images were analyzed using a Matlab script (Matlab 2017b, Mathworks, Natick, MA). Exemplary images of the figures obtained with the Matlab script can be found in ESI A.† In short, the images were processed with Gaussian noise removal (low-pass Wiener filter) and the image intensity values were saturated. Then the image intensity values of a line orthogonal to the outer channel wall were plotted and the width of the fluid flow containing CCMVs was determined.The concentrations of CCMVs in the sample and in both outlets were determined with an EnSpire multimode plate reader (Pelkin Elmer, Waltham, MA, USA). Samples were pipetted in black FLUOTRAC™ 600 96-well-plates (Greiner Bio-one, Essen, Germany) and measured in the multimode plate reader in fluorescence mode (λex = 495 nM, λem = 520 nm, 50 flashes). The fluid collected in outlet 1 (≈20 μl) was diluted and 100 μl was used to measure the fluorescence intensity. The fluid collected in outlet 2 (≈500 μl) was directly used and the fluorescence intensity of 250 μl was measured. For both sample volumes, calibration lines were used to determine the CCMV concentration from the fluorescence intensity (ESI B†). The CCMV removal is defined as the percentage of CCMVs present in outlet 1 relative to the CCMVs present in both outlets. The CCMV concentration in weight was transformed to a particle concentration with CCMV weight (4.6 × 106 avogram per CCMV particle, equivalent to 7.6 × 10−21 kg per CCMV particle40). The limit of detection was 0.01 ng ml−1 (1.3 × 109 CCMVs per ml).
The spermatozoa collected in outlet 1 and 2 were manually counted using a Neubauer counting chamber. A volume of 10 μl from the fluid collected at the outlet 1 was deposited onto a Neubauer chamber. For each experiment, at least 100 spermatozoa were enumerated depending on the cell concentration. It was corrected for the difference in obtained volumes from outlet 1 and 2. The spermatozoa recovery is defined as the percentage of spermatozoa present in outlet 2 relative to the spermatozoa in both outlets.
Statistical analysis
Independent t-tests were performed to compare the separation qualities of the experiments with a constant outlet 1 (waste) removal rate of 2.7%. The separation qualities obtained for a sample pressure of 28 mbar were compared to the separation qualities of 30 and 32 mbar. The significance level of the two-tailed test was chosen to be 0.05.Results and discussion
CCMV characterization
To investigate if CCMVs stay intact in Solusem Bio+, the dilution medium for porcine semen, the size distributions of CCMVs in Solusem Bio+, and CCMVs in encapsulation buffer were measured using DLS. CCMVs were diluted in Solusem Bio+ to a concentration of 740 ng ml−1. The structure of CCMV is dependent on pH and salt concentration. The pH of Solusem Bio+ is with 7.2 neutral and similar to the pH of the encapsulation buffer (pH 7.4). The size distribution of CCMVs in Solusem Bio+ and CCMVs in encapsulation buffer with the standard deviations were 26.5 ± 9.8 nm and 26.7 ± 10.6 nm, respectively. The respective size distribution graphs obtained with DLS are shown in the ESI C.† The pH and the CCMV size distribution in Solusem Bio+ were like the values obtained in encapsulation buffer, confirming that the CCMVs stayed intact in Solusem Bio+ and were also comparable to CCMVs in virus buffer (27.7 nm).CCMVs in microfluidic chip
The flow behavior of the fluorescently labelled model virus was followed by a fluorescent microscope in the PFF chips to investigate if separation is technically feasible. The CCMVs were diluted to a concentration of 370 ng ml−1 in Solusem Bio+. The CCMV concentration was chosen to be higher than realistic virus concentrations to guarantee the detection of the fluorescence signal. The diluted CCMVs were investigated with chip design I (broadened segment width: 1100 μm) and the sample pressures were 24, 26 or 32 mbar, whereas the sheath buffer pressure was constant (400 mbar). With the fluorescent microscope, images were taken from the position where the separation channel branches off the broadened segment (Fig. 2A–C). After image processing, intensity profiles (Fig. 2D–F) orthogonal to the outer channel wall were obtained, which indicate the width of the CCMV containing fluid flow. | ||
| Fig. 2 Merged brightfield and fluorescent images (A–C) and intensity profiles (D–F) of Atto 647 labelled CCMVs in PFF chip with broadened segment width of 1100 μm (design I): sample pressure was varied (A and D: 24 mbar; B and E: 26 mbar; C and F: 32 mbar) with constant sheath buffer pressure (400 mbar). With increasing sample pressure, the fluid stream containing CCMVs broadens (FR: flow rate ratio of total flow/sample flow; red arrow: line of intensity plot). | ||
The intensity profiles show that with increasing sample pressure, the fluid flow containing CCMVs broadens. With an increase in sample pressure, the sample flow rate increases and the flow rate ratio decreases when the sheath buffer pressure is constant. The CCMV flow width for sample pressures of 24 and 26 mbar was smaller than the separation channel width of 45 μm and CCMVs exited the chip at outlet 1. When the sample flow was getting too high, the fluorescent image and intensity plot show that CCMVs exit also at outlet 2 and separation from larger particles was not provided. The flow rate ratio in the pinched segment with width wp is linearly amplified in the broadened segment with width wb by wb/wp.30 With this relationship, the fluid width in the broadened segment can be calculated and compared with the experimental results. The theoretical determined width of the CCMV fluid flow is 25 and 30 μm for 24 and 26 mbar, respectively, and therefore below the width of the separation channel of 45 μm. For a sample pressure of 32 mbar, the CCMV fluid width is 46 μm and efflues also to outlet 2, which is in accordance with the experimental results. The theoretical values can deviate slightly from experimental results, because design parameters are not perfectly translated to the chip. The position of the separation channel wall overlaps with the CCMV containing fluid for a sample pressure of 32 mbar. The width of the position of the separation channel wall was designed to be 45 μm, but the actual width of the PDMS chip can deviate slightly due to the processing steps. Furthermore, during image processing pixels were converted to distance, which can also cause some inaccuracy. Nevertheless, with the intensity profiles, it can be distinguished whether the model virus flow out of the chip via outlet 1 (waste) or outlet 2.
Separation of spermatozoa from viruses
We optimized the spermatozoa recovery while maintaining the CCMVs removal, by comparing the separation efficiencies of both chip designs and investigating the effect of the fluid removal ratios of both outlets. A wider broadened segment width improves the separation efficiency because the effluent position of the spermatozoa and viruses is farther apart from each other (ESI D†). This also corresponds to the previously mentioned linear amplification relationship of wb/wp. Therefore, chip design II, which had a broadened segment width of 2200 μm, was used for the next experiments. The optimal fluid removal ratio must be found, as this parameter also determines the separation efficiency. With a higher fluid removal ratio, it is expected that more spermatozoa exit the chip at the waste outlet, whereas with a low fluid removal ratio, the virus may also exit the chip at outlet 2. Fig. 3A presents the obtained separation quality with respect to the CCMV removal and spermatozoa recovery. For a fluid removal ratio of 3.0%, most spermatozoa are lost, whereas with a fluid removal ratio of 2.7% almost 90% of the spermatozoa are collected in outlet 2. A higher fluid removal ratio means, that the flow to outlet 1 is higher and with a fluid removal of 3.0% many spermatozoa exit the chip at outlet 1. For all experiments, more than 75% of the CCMVs were removed from the spermatozoa. The best CCMVs removal of 89% was achieved with 2.7% of flow to outlet 1. In Fig. 3B, the CCMV concentrations in the outlets are shown. The concentrations in outlet 1 were more than two order of magnitude higher compared to the concentrations obtained in outlet 2. The CCMV concentration in outlet 1 is lower than the input concentration, which therefore cannot be traced back to the separation efficiency of CCMVs. There are several explanations for this dilution. The largest impact on the dilution is that a third of the volume exiting at outlet 1 is the buffer solution. This can be seen by the CCMV flow width, which is approximately a third of the separation channel width. Additionally, approximately 20% of the viruses are lost during processing with the microfluidic chip and tubing (ESI E†). With all fluid removal ratios, most CCMVs were separated from spermatozoa, because the CCMV fluid width is smaller than the separation channel width of 45 μm (Fig. 3C). While performing the experiments, spermatozoa were fluorescently visible under the microscope, although they are not known to be autofluorescent. It has been examined, that CCMV and the dye adhere to spermatozoa (ESI F†), which justifies some of the fluorescent signal obtained from samples collected at outlet 2. | ||
| Fig. 3 Separation quality for different fluid removal ratios of chip design II A) spermatozoa recovery and CCMV removal. B) The CCMV concentration after the separation in both outlets. C) The CCMV flow width from the outer wall in the broadened segment (multiple images were taken during one experiment; ±X, in which X represents 1 SD, N ≥ 3). With all fluid removal ratios, CCMV are collected in outlet 1 (waste outlet). The best spermatozoa recovery was achieved with a fluid removal ratio of 2.7% (sheath pressure 400 mbar; sample pressure: 30 mbar; N = 1). | ||
The best separation with chip design II was obtained with a fluid removal rate of 2.7%, because this fluid removal rate achieved both the highest spermatozoa recovery and virus removal. The separation efficiency was further investigated by varying the pressure for the sample flow. The individual separation efficiencies with sample pressure of 28 mbar (1.8 μl min−1), 30 mbar (2.0 μl min−1), and 32 mbar (2.4 μl min−1) are shown in the ESI G.† When using a sample pressure below 28 mbar, the flow rate ratio was too high, and the sample flow was blocked by the sheath flow. The results of independent t-tests between 28 mbar and the other applied pressures did not report a statistical difference for the spermatozoa recovery and CCMV removal rates (ESI G†). In Fig. 4 the separation efficiencies are summarized. Spermatozoa recovery of 86 ± 6% and CCMV removal of 84 ± 4% were achieved.
 | ||
| Fig. 4 Separation quality with chip design II and 2.7% fluid removal ratio (error bars = 1 SD, sheath pressure 400 mbar; sample pressure: 28–32 mbar; N = 9). | ||
Up to date only a few microfluidic separation techniques were applied to purify spermatozoa from other types of cells such as blood cells and epithelial cells. Dean flow fractionation, which uses inertial forces in a spiral channel, was applied to separate erythrocytes and white blood cells from spermatozoa.27,41 Spermatozoa recoveries of 80% (ref. 27) and 89% (ref. 41) were achieved, while removing 99% of the erythrocytes27 and 82% of the white blood cells.41 Dean flow fractionation was further improved with a channel of a trapezoidal cross-section.42 With this device, the spermatozoa recovery was 96%, whereas epithelial cells (86%), white blood cells (approximately 95%) and erythrocytes (approximately 75%) were removed.42 The reported spermatozoa recovery with PFF and the virus removal rate are similar to the ones reported with Dean flow fractionation. Channel dimensions, particle size and flow characteristics are key design parameters for inertial microfluidics. Small deviations from optimal flow characteristics can have an impact on particle separation. Another proposed spermatozoa purification method is acoustic trapping. Spermatozoa were trapped by an acoustic standing wave, while other biological components originating from the female victim, such as free DNA, pass the acoustic field.43,44 Spermatozoa were successfully purified from a 40-fold excess of female epithelial cells over spermatozoa.44 Limitations of acoustic trapping are that it is not a continuous separation technique and an external field is required. Inertial microfluidics and acoustic trapping can also be applied to purify spermatozoa from virus containing semen. In contrast to inertial microfluidics and acoustic trapping, PFF has several benefit such as the simple design and no need for an outer field. The most important parameters of PFF are the width of the pinched segment and its ratio with the broadened segment. Key separation parameters such as flow rate ratio and fluid removal ratio can be easily optimized during testing, as previously reported by Berendsen et al.29 and shown in this study.
The reported spermatozoa recovery (86 ± 6%) is almost twice as high as the spermatozoa recovery reported by other virus separation techniques (≈45%), which have used combinations of “swim-up” and density gradient centrifugation.21,22 Nevertheless, the spermatozoa recovery is slightly lower than the spermatozoa recovery of similar PFF spermatozoa separation techniques as proposed by Berendsen et al., who have achieved a spermatozoa recovery of up to 95%.29
The asymmetrical shape of spermatozoa improves the separation. Berendsen et al. have reported that the tumbling effect influences the spermatozoa behavior in PFF; the average spermatozoa position in the broadened segment is further away from the channel wall and the distribution is broader than when considering the spermatozoa head size.29 Therefore, in PFF spermatozoa can be associated with an average particle size of 15 μm, instead of a 4 μm one. Without the tumbling effect most spermatozoa would not be separated from the virus containing fluid, since the width of the sample flow in the pinched segment is equal or lower than 4 μm. However, due to the tumbling and as evidenced by the results, spermatozoa can be purified from viruses with our set-up. Another spermatozoa characteristic is the motility and its rheotactic behavior; spermatozoa tend to swim against the flow stream with a stream velocity of 100 μm s−1.41 The flow velocity in the broadened segment of the PFF device is with approximately 9000 μm s−1 higher than the flow velocity needed for rheotaxis. Additionally, separation was performed at room temperature to prevent spermatozoa movement and subsequent spermatozoa fatigue, so both effects did not influence the separation. Moreover, it has been shown that the effect of microfluidic processing, including PFF, on the spermatozoa viability is low.36 The used chip dimensions and flow rates used in the viability study36 were similar and in the same range as the ones used in this study. There were differences in our proposed separation technique and the previously proposed one by Berendsen et al., which can cause the difference in spermatozoa recovery such as the chip design, flow rate ratio and sample composition. Berendsen et al. have used a sample which consisted mainly of erythrocytes and was spiked with spermatozoa, whereas in this study, a spermatozoa sample was spiked with smaller CCMVs and the particle density was lower. The particle density may influence the separation efficiency.
For analysis and quantification of the CCMV separation, two techniques based on the fluorescence signal were used. Fluorescence microscopy visualizes the CCMV fluid width in the broadened segment of the microfluidic chip and determines its width of the channel wall, whereas with a fluorescence imaging plate reader the concentration of the viruses after the separation was determined. Fluorescence microscopy implies that all viruses are sorted from the spermatozoa, because the width of the fluid width is below 45 μm and the viruses exit the chip at outlet 1. However, the technique determining the CCMV concentration after the separation reveals that CCMVs are also found in outlet 2. As is shown in ESI F,† both CCMV and the Atto 488 dye adhere to spermatozoa, which increase the fluorescence intensity and therewith the CCMV concentration in outlet 2. For several virus types, it has been shown that most virus particles are free in the seminal plasma instead of penetrating or attaching to spermatozoa.17–19 Taking both CCMV analysis techniques into account, the CCMV removal is at least 84 ± 4%. Similar to other virus removal techniques from semen,21,22 not a complete virus elimination was achieved. To further eliminate CCMVs from the sample, it is an option to process the sample with the PFF device multiple times. A PFF device with two cascading devices has been proposed to remove both larger particles and smaller particles from spermatozoa.45 To improve the virus removal of our application, cascading two PFF chips may increase the virus removal 87% to 98%, assuming that for every device the efficiency is the same. However, simultaneously, this would decrease the spermatozoa recovery to 81%.
PFF is based on the typical laminar flow behavior in microfluidic systems. Although the flow is laminar, particles experience Brownian motion and diffuse across streamlines. For our application, it is interesting to investigate the diffusion distance of viruses between entering the broadened segment and the separation channel. The diffusion coefficient for spherical viruses with a size of 10–400 nm ranges from 30 μm2 s−1 to 0.8 μm2 s−1.46 For one-dimensional diffusion the average distance x travelled by a particle can be calculated with  , which D the diffusion coefficient and t the time.47 Assuming the time critical for the diffusion is 0.15 s, which is the calculated time a particle needs to move between the pinched segment and the branch-off of the separation channel, the average distance is 3 μm to 0.5 μm for viruses with a size of 10 nm to 400 nm, respectively. With a separation channel width of 45 μm and a virus containing fluid width in the broadened segment of smaller than 40 μm, the virus separation is not affected.
, which D the diffusion coefficient and t the time.47 Assuming the time critical for the diffusion is 0.15 s, which is the calculated time a particle needs to move between the pinched segment and the branch-off of the separation channel, the average distance is 3 μm to 0.5 μm for viruses with a size of 10 nm to 400 nm, respectively. With a separation channel width of 45 μm and a virus containing fluid width in the broadened segment of smaller than 40 μm, the virus separation is not affected.
Due to the regulations in our laboratory, we have chosen to use CCMVs as a model virus, because viruses found in semen are potential transmitters of diseases and require higher safety regulations in the laboratory. CCMVs with a size of 28 nm belong to the small viruses, when comparing it to the size range of viruses found in semen (10–400 nm5). Examples with a comparable size to CCMVs are the foot-and-mouth disease (FMD) virus (25–30 nm (ref. 48)) and CSF virus (40–60 nm). Another important virus type, which can be present in semen, is the African swine fever (AFS) virus (200 nm (ref. 49)). With the current use of PFF, spermatozoa are removed from the liquid containing sample based on the spermatozoa size. As large viruses are more than twenty times smaller than spermatozoa, it is expected that spermatozoa can be separated from all virus types found in semen. As viruses can have a diameter of up to 400 nm, we performed PFF separation of semen spiked with 500 nm polystyrene beads. The results show that 98% of polystyrene beads were removed from semen while recovering 93% of the spermatozoa (ESI H†). The PFF chip was similar to chip design I, but the chip was made out of cyclic olefin copolymer (COC). The semen sample was spiked with 74 ng ml−1 CCMVs, which is approximately 9.5 × 109 particles per ml. The high virus concentration was used to enable the analysis based on fluorescence intensity. CCMV analysis techniques have the limitation of a fluorescence signal threshold. If the signal is lower than the threshold, the intensity and therewith the CCMV will not be detected. In outlet 2 the sample is diluted with the sheath buffer, which is needed to pinch the particles. This dilution also decreases the measured fluorescence signal. Other virus separation techniques have used virus concentration of a few 1 × 103 to 2 × 105 plaque forming units per ml.21,22 Assuming that each plaque forming unit is one virus particle, in this study high virus concentrations were used. In the future, the separation of spermatozoa from viruses can be confirmed by using samples spiked with viruses found in semen, such as the CSF and AFS virus, at realistic concentrations.
The present study is a proof-of-principle for the separation of viruses from semen used by the veterinary industry. Before implementing this in routine semen processing, many steps must be performed, such as using a more realistic sample as mentioned previously. The only necessary pretreatment step is sample dilution, which is also currently part of routine semen processing. Another important aspect of the separation is the sample throughput, as the total spermatozoa count of a boar ejaculate ranges between 75 × 109–100 × 109 cells.31 The throughput of the proposed separation techniques is only a few μl min−1, which would take too long to process a whole boar ejaculate. To minimize sample pretreatment and to increase the sample throughput, it is suggested to investigate PFF with higher spermatozoa concentrations. There is a need to increase the throughput to become of more interest for the veterinary industry. Furthermore, the higher the spermatozoa recovery the more attractive the separation technique will be, as every individual spermatozoon represents a fertilization opportunity.
Conclusion
The presented microfluidic chip based on PFF separates spermatozoa from virus spiked semen. With the optimized flow rate ratio and fluid removal fraction, a spermatozoa recovery of 86 ± 6% and removal of at least 84 ± 4% of a model virus were achieved. The spermatozoa recovery of this technique is twice as high as the spermatozoa recovery of other virus separation techniques. By removing potential viruses from porcine semen before its distribution to recipient farms, the transmittance of diseases by artificial insemination is further reduced.Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.Author contributions
T. H. carried out the experiments and wrote the manuscript. J. B. and J. D. supported the experimental design and analysis. R. H. prepared the CCMVs under the supervision of J. C. M. B. and L. S. supervised the project. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare that they have no conflict of interest.References
- R. A. Godke, M. Sansinena and C. R. Youngs, in Transgenic Animal Technology, Elsevier, 2014, pp. 581–638 Search PubMed.
- J. M. Morrell, in Artificial insemination in farm animals, 2011, vol. 1, pp. 1–14 Search PubMed.
- D. Waberski, A. Riesenbeck, M. Schulze, K. F. Weitze and L. Johnson, Theriogenology, 2019, 137, 2–7 CrossRef PubMed.
- B. Guerin and N. Pozzi, Theriogenology, 2005, 63, 556–572 CrossRef CAS PubMed.
- D. Maes, H. Nauwynck, T. Rijsselaere, B. Mateusen, P. Vyt, A. de Kruif and A. Van Soom, Theriogenology, 2008, 70, 1337–1345 CrossRef CAS PubMed.
- D. Maes, A. Van Soom, R. Appeltant, I. Arsenakis and H. Nauwynck, Theriogenology, 2016, 85, 27–38 CrossRef PubMed.
- H. Feitsma, H. J. Grooten, F. W. V. Schie and B. Colenbrander, PrOf 12thInt Congr Anim Reprod, 1992, pp. 1710–1712 Search PubMed.
- L. B. Hall Jr, J. P. Kluge, L. E. Evans and H. T. Hill, Can. J. Comp. Med., 1984, 48, 192 CAS.
- L. B. Hall Jr, J. P. Kluge, L. E. Evans, T. L. Clark and H. T. Hill, Can. J. Comp. Med., 1984, 48, 303 CAS.
- A. Stegeman, A. Elbers, H. de Smit, H. Moser, J. Smak and F. Pluimers, Vet. Microbiol., 2000, 73, 183–196 CrossRef CAS PubMed.
- A. J. De Smit, A. Bouma, C. Terpstra and J. T. Van Oirschot, Vet. Microbiol., 1999, 67, 239–249 CrossRef CAS PubMed.
- M. P. M. Meuwissen, S. H. Horst, R. B. M. Huirne and A. A. Dijkhuizen, Preventive Veterinary Medicine, 1999, 42, 249–270 CrossRef CAS PubMed.
- Council of the European Union, Council Directive 92/65/EEC, 2012.
- Council Directive 2008/73/EC of 15 July 2008 simplifying procedures of listing and publishing information in the veterinary and zootechnical fields, Official Journal of the European Union, L219, European Union, 2008.
- L. A. Johnson, J. G. Aalbers, C. M. T. Willems and W. Sybesma, J. Anim. Sci., 1981, 52, 1130–1136 CrossRef CAS PubMed.
- J. Roca, J. M. Vázquez, M. A. Gil, C. Cuello, I. Parrilla and E. A. Martínez, Reprod. Domest. Anim., 2006, 41, 43–53 CrossRef PubMed.
- J. W. Nash, L. A. Hanson and K. C. C. St, Am. J. Vet. Res., 1995, 56, 760–763 CAS.
- C. M. Gradil, R. E. Watson, R. W. Renshaw, R. O. Gilbert and E. J. Dubovi, Vet. Microbiol., 1999, 70, 21–31 CrossRef CAS PubMed.
- R. A. Burger, P. D. Nelson, K. Kelly-Quagliana and K. S. Coats, Am. J. Vet. Res., 2000, 61, 816–819 CrossRef CAS PubMed.
- J. Kim, D. U. Han, C. Choi and C. Chae, J. Virol. Methods, 2001, 98, 25–31 CrossRef CAS PubMed.
- G. Blomqvist, M. Persson, M. Wallgren, P. Wallgren and J. M. Morrell, Anim. Reprod. Sci., 2011, 126, 108–114 CrossRef PubMed.
- J. M. Morrell, P. Timoney, C. Klein, K. Shuck, J. Campos and M. Troedsson, Reprod. Domest. Anim., 2013, 48, 604–612 CrossRef CAS PubMed.
- A. Folch, Introduction to bioMEMS, CRC Press, 2016 Search PubMed.
- S. M. Knowlton, M. Sadasivam and S. Tasoglu, Trends Biotechnol., 2015, 33, 221–229 CrossRef CAS PubMed.
- R. Nosrati, P. J. Graham, B. Zhang, J. Riordon, A. Lagunov, T. G. Hannam, C. Escobedo, K. Jarvi and D. Sinton, Nat. Rev. Urol., 2017, 14, 707 CrossRef PubMed.
- G. Marzano, M. S. Chiriacò, E. Primiceri, M. E. Dell'Aquila, J. Ramalho-Santos, V. Zara, A. Ferramosca and G. Maruccio, Biotechnol. Adv., 2020, 40, 107498 CrossRef PubMed.
- J. Son, K. Murphy, R. Samuel, B. K. Gale, D. T. Carrell and J. M. Hotaling, Anal. Methods, 2015, 7, 8041–8047 RSC.
- W. Liu, W. Chen, R. Liu, Y. Ou, H. Liu, L. Xie, Y. Lu, C. Li, B. Li and J. Cheng, Biomicrofluidics, 2015, 9, 44127 CrossRef PubMed.
- J. T. W. Berendsen, J. C. T. Eijkel, A. M. Wetzels and L. I. Segerink, Microsyst. Nanoeng., 2019, 5, 24 CrossRef PubMed.
- M. Yamada, M. Nakashima and M. Seki, Anal. Chem., 2004, 76, 5465–5471 CrossRef CAS PubMed.
- S. Bonet, I. Casas, W. V. Holt and M. Yeste, Boar reproduction: fundamentals and new biotechnological trends, Springer Science & Business Media, 2013 Search PubMed.
- D. L. Garner, Theriogenology, 2009, 71, 11–21 CrossRef CAS PubMed.
- I. Parrilla, J. M. Vázquez, C. Cuello, M. A. Gil, J. Roca, D. Di Berardino and E. A. Martínez, Reproduction, 2004, 128, 615–621 CAS.
- J. M. Vazquez, I. Parrilla, J. Roca, M. A. Gil, C. Cuello, J. L. Vazquez and E. A. Martinez, Theriogenology, 2009, 71, 80–88 CrossRef CAS PubMed.
- P. Ditsayabut, W. Pongsena, N. Promsawat, K. Makbun, P. Kupittayanant, P. Janphuang and C. Wanapu, Biomed. Phys. Eng. Express, 2018, 4, 65016 CrossRef.
- T. Hamacher, J. T. W. Berendsen, S. A. Kruit, M. L. W. J. Broekhuijse and L. I. Segerink, Biomicrofluidics, 2020, 14, 44111 CrossRef CAS PubMed.
- M. Comellas-Aragonès, H. Engelkamp, V. I. Claessen, N. A. J. M. Sommerdijk, A. E. Rowan, P. C. M. Christianen, J. C. Maan, B. J. M. Verduin, J. J. L. M. Cornelissen and R. J. M. Nolte, Nat. Nanotechnol., 2007, 2, 635–639 CrossRef PubMed.
- B. J. M. Verduin, FEBS Lett., 1974, 45, 50–54 CrossRef CAS PubMed.
- B. J. M. Verduin, J. Gen. Virol., 1978, 39, 131–147 CrossRef CAS.
- J. B. Bancroft, E. Hiebert, M. W. Rees and R. Markham, Virology, 1968, 34, 224–239 CrossRef CAS PubMed.
- A. Jafek, H. Feng, D. Broberg, B. Gale, R. Samuel, K. Aston and T. Jenkins, Microfluid. Nanofluid., 2020, 24, 1–9 CrossRef.
- S. A. Vasilescu, S. Khorsandi, L. Ding, S. R. Bazaz, R. Nosrati, D. Gook and M. E. Warkiani, Sci. Rep., 2021, 11, 1–11 CrossRef PubMed.
- J. V. Norris, M. Evander, K. M. Horsman-Hall, J. Nilsson, T. Laurell and J. P. Landers, Anal. Chem., 2009, 81, 6089–6095 CrossRef PubMed.
- C. P. Clark, K. Xu, O. Scott, J. Hickey, A. C. Tsuei, K. Jackson and J. P. Landers, Forensic Sci. Int.: Genet., 2019, 41, 42–49 CrossRef CAS PubMed.
- J. T. W. Berendsen, in Microfluidic Spermatozoa Selection for Clinical Applications, 2019, pp. 83–92 Search PubMed.
- A. G. Murray and G. A. Jackson, Mar. Ecol.: Prog. Ser., 1992, 89, 103–116 CrossRef.
- H. Bruus, Physics, 2008, 18, 363 Search PubMed.
- S. Tomar, S. Mahajan and R. Kumar, in Genomics and Biotechnological Advances in Veterinary, Poultry, and Fisheries, Elsevier, 2020, pp. 435–468 Search PubMed.
- I. Galindo and C. Alonso, Viruses, 2017, 9, 103 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1lc00643f |
| This journal is © The Royal Society of Chemistry 2021 |