
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAutomated optimization of endoderm differentiation on chip†
Jessi Carolina
Ardila Riveros‡
a,
Anna Karolina
Blöchinger‡
b,
Scott
Atwell‡
 a,
Michel
Moussus
a,
Nina
Compera
a,
Omid
Rajabnia
c,
Tihomir
Georgiev
a,
Heiko
Lickert
bdef and
Matthias
Meier
*ae
a,
Michel
Moussus
a,
Nina
Compera
a,
Omid
Rajabnia
c,
Tihomir
Georgiev
a,
Heiko
Lickert
bdef and
Matthias
Meier
*ae
aHelmholtz Pioneer Campus, Helmholtz Zentrum München, Munich, Germany. E-mail: matthias.meier@helmholtz-muenchen.de
bInstitute of Diabetes and Regeneration Research, Helmholtz Zentrum München, D-85764 Neuherberg, Germany
cLaboratory for MEMS Application, IMTEK-Department of Microsystems Engineering, University of Freiburg, Germany
dTUM School of Medicine, Technical University of Munich, Munich, Germany
eGerman Center for Diabetes Research (DZD), D-85764 Neuherberg, Germany
fInstitute of Stem Cell Research, Helmholtz Zentrum München, D-85764 Neuherberg, Germany
First published on 9th November 2021
Abstract
Human induced pluripotent stem cells (hiPSCs) can serve as an unlimited source to rebuild organotypic tissues in vitro. Successful engineering of functional cell types and complex organ structures outside the human body requires knowledge of the chemical, temporal, and spatial microenvironment of their in vivo counterparts. Despite an increased understanding of mouse and human embryonic development, screening approaches are still required for the optimization of stem cell differentiation protocols to gain more functional mature cell types. The liver, lung, pancreas, and digestive tract originate from the endoderm germ layer. Optimization and specification of the earliest differentiation step, which is the definitive endoderm (DE), is of central importance for generating cell types of these organs because off-target cell types will propagate during month-long cultivation steps and reduce yields. Here, we developed a microfluidic large-scale integration (mLSI) chip platform for combined automated three-dimensional (3D) cell culturing and high-throughput imaging to investigate anterior/posterior patterns occurring during hiPSC differentiation into DE cells. Integration of 3D cell cultures with a diameter of 150 μm was achieved using a U-shaped pneumatic membrane valve, which was geometrically optimized and fluidically characterized. Upon parallelization of 32 fluidically individually addressable cell culture unit cells with a total of 128 3D cell cultures, complex and long-term DE differentiation protocols could be automated. Real-time bright-field imaging was used to analyze cell growth during DE differentiation, and immunofluorescence imaging on optically cleared 3D cell cultures was used to determine the DE differentiation yield. By systematically alternating transforming growth factor β (TGF-β) and WNT signaling agonist concentrations and temporal stimulation, we showed that even under similar DE differentiation yields, there were patterning differences in the 3D cell cultures, indicating possible differentiation differences between established DE protocols. The automated mLSI chip platform with the general analytical workflow for 3D stem cell cultures offers the optimization of in vitro generation of various cell types for cell replacement therapies.
Introduction
The research progress of three-dimensional (3D) cell culture systems has opened new possibilities for refining in vitro cellular test systems for fundamental research and industry screening applications. While two-dimensional (2D) cell cultures offer simplicity, reproducibility, and low costs for high numerical repeats for cellular experiments, 3D cell cultures additionally include cell–cell and cell–extracellular matrix interactions. In such tissue like microenvironments spontaneous cell patterns could be firstly observed during mouse embryonic-stem-cell-derived retinal epithelium formation.1 Since then various pluripotent stem cells, progenitors, and/or differentiated cells have been used to generate self-organized cell patterns with tissue-like structures. Thus, with 3D cell cultures it becomes possible to recapitulate aspects of native tissue architecture and function in so-called organoids.2,3 The value of organoid systems is particularly visible by achievements in the early development of the human embryonic phase. The definitive endoderm (DE) is one of the three primary germ layers, which together with the mesoderm and ectoderm give rise to all adult organs. DE cells are first specified through the process of gastrulation followed by gastrointestinal tract formation. Therefore, the DE cells pattern along the embryo anterior–posterior and dorso–ventral axes and form the primitive gut tube, with division into fore-, mid- and hindgut domains; leading to the formation of endoderm-derived organs, including the thymus, thyroid, lungs, liver, and pancreas.4,5 The recapitulation of endodermal development including mechanisms for axis pattern signals are pivotal for generating in vitro endodermal derived cell types for cell replacement therapies.6Several chemical induction protocols have been described to differentiate hiPSCs into definitive endoderm cells by activating TGF-β and WNT signaling pathways7–9 with activin A (AA) and CHIR-99021 (CHIR), respectively. Concentrations and stimulation time of both biologicals during DE differentiation vary slightly between the reported protocols. Despite this observation the efficiency of DE induction, quantified by the expression of pan-endoderm marker (e.g. FOXA2, or SOX17), is still high for all protocols. However, stem cell differentiation is a deterministic process, and small transcriptional and presumably pattern changes in the early stages will propagate and define further downstream organ specific progenitor cell types. Consequently, off-target cell types in the early differentiation stages reduce the yield of targeted cell types, such as β-cells or hepatocytes, in the later stages.10 In fact, isolation of specific DE cells has been already exploited to enrich for more functional β-cells,11 which argues that DE cell type differences exist. Optimization of the differentiation protocol is therefore a central operation in all current stem cell replacement therapy approaches. Cell culture automation and parallelization technology can help to reduce manually introduced variation and increase cost efficiency in the multi-chemical screening of cell differentiation protocols.
It has been recognized that microfluidics technology enables the formation, handling, chemical screening, and analysis of 3D cell cultures. Numerous microfluidic platforms have been designed to integrate 3D cell cultures, and have been reviewed in detail.12,13 However, fully automated higher-throughput microfluidic systems for 3D cell culture and analysis are rare, particularly for the handling of human 3D stem cell cultures. Microfluidic large-scale integration chip technology (mLSI) has set standards for automated 2D cell culture systems.14 The integration of multiple pneumatic membrane valves (PMVs) on mLSI chips allowed the automation of complex fluid logics and programs, which improved microenvironmental control and increased the parallelization of cell culture on the chip.14–16 Extension of the mLSI technologies upon integration of 3D cell cultures is challenging because of their relatively large size, which can range from 50 μm to several millimeters in diameter.17 Channel networks and PMVs on mLSI chips have been optimized on the scale of tens of microns.18 Upscaling of microfluidic channel networks and PMVs for mLSI has been reported by exploiting milling19 or additive production technologies for polydimethylsiloxane (PDMS) casting molds.20,21 However, general operation units and elements for forming and handling 3D cell cultures are missing for mLSI. Deflecting membranes used for PMVs have previously been exploited to compartmentalize cell culture chambers22 and to protect cells from direct shear flow. Uniform 3D cell cultures have been formed with a U-shaped valve system; however, integration on an mLSI chip of this grouping unit element has not been demonstrated.23,24 One additional demand for newly developed mLSI chip platforms for 3D cell culture systems is their compatibility with high-content imaging25 and other analytical workflows to interrogate molecular and cellular information as, for example, detection of cell patterns during differentiation.
In this study, we developed an mLSI chip for culturing human 3D stem cell cultures in parallel. For the formation, patterning and long-term stability of 3D cell cultures, we optimized the size and closing pressures of deflecting membrane valves in a U-shaped form. The viability of the 3D cell cultures was tested in relation to the feeding rates. Automation of the 3D cell culturing process allowed us to screen various stem cell endoderm differentiation protocols. The efficiency of DE differentiation protocols was evaluated by real-time bright-field and immunofluorescence imaging of the transcription factors SOX17 and FOXA2 at the endpoints of optically cleared 3D cell cultures. Upon using CER1 as specific anterior marker, pattern formation was evaluated. By systematically varying the two chemical DE induction components for the TGF-β and WNT signaling pathways, that is, AA and CHIR, in time and concentration, we observed DE efficiency and pattern changes in pluripotent stem cell-derived 3D DE cultures.
Experimental procedures
3D microfluidic chip design and fabrication
A microfluidic chip was fabricated using standard soft lithography methods.26 Briefly, the control and flow-layer channel networks were designed using the AutoCAD software (AutoDesk, 2019). The control layer mold containing channels of 50 μm height was printed on a silicon wafer using a negative photoresist (SU-8 3025; MicroChem). The flow-layer mold was produced via a two-step printing process. In the first step, 40 μm half-rounded channels were printed with a reflowable positive photoresist (AZ40XT; MicroChemicals). In the second step, cell culture chambers with a height of 150 μm were printed using a negative photoresist (SU-8 3050; MicroChem). All molds were printed with a micropattern generator (μPG101; Heidelberg Instruments) and then coated with CYTOP™ (CTL-809 M; AGC Chemicals) to prevent the adhesion of the PDMS. Pre-polymer and cross-linking reagents (Sylgard™ 184 silicone elastomer kit, 001003072799) were mixed in a ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 20:1, respectively, at 1500 rpm for 6 min to fabricate flow and control layers. The PDMS mixes were poured onto the respective molds. To remove trapped air, the flow layer was placed in vacuum for 15–20 min. The control layer was spin-coated with PDMS at 500 rpm for 15 s (acceleration 500 ms), followed by spinning at 1700 rpm for 30 s (acceleration 1200 ms). Both layers were half-cured at 80 °C for 16 min. After punching the corresponding inlets, the flow layer was aligned to the top of the control layer. The assembly was then further bonded at 80 °C for 1 h. Next, the remaining inlet ports that tunnel both layers were punched. Finally, the PDMS chip was irreversibly sealed with a glass slide through oxygen plasma bonding (20 W at 0.9 mbar for 25 s).
:1 and 20:1, respectively, at 1500 rpm for 6 min to fabricate flow and control layers. The PDMS mixes were poured onto the respective molds. To remove trapped air, the flow layer was placed in vacuum for 15–20 min. The control layer was spin-coated with PDMS at 500 rpm for 15 s (acceleration 500 ms), followed by spinning at 1700 rpm for 30 s (acceleration 1200 ms). Both layers were half-cured at 80 °C for 16 min. After punching the corresponding inlets, the flow layer was aligned to the top of the control layer. The assembly was then further bonded at 80 °C for 1 h. Next, the remaining inlet ports that tunnel both layers were punched. Finally, the PDMS chip was irreversibly sealed with a glass slide through oxygen plasma bonding (20 W at 0.9 mbar for 25 s).
Cell culture chip operation
The control lines of the chips were directly connected by Tygon tubings (ND 100-80; Proliquid) to the solenoid valves (LMV155RHY-5A-Q; SMC) operated by MATLAB (MathWorks®, version R2019a). The control line for the catching valve was operated using a manual pressure regulator. Optimal valve closing pressures for the catching valve ranged from 10–15 psi and were determined for each chip. Light-proof pressured bottles containing reagents were directly connected to the eight media inlets (Fig. 1a) with Tygon tubings. All reagent bottles were set under a fluidic forward pressure of 50 mbar. The chips were placed in a microscope stage top incubator (STX; Tokai Hit®) to provide a constant culture environment of 37 °C and 5% CO2 in a humidified atmosphere.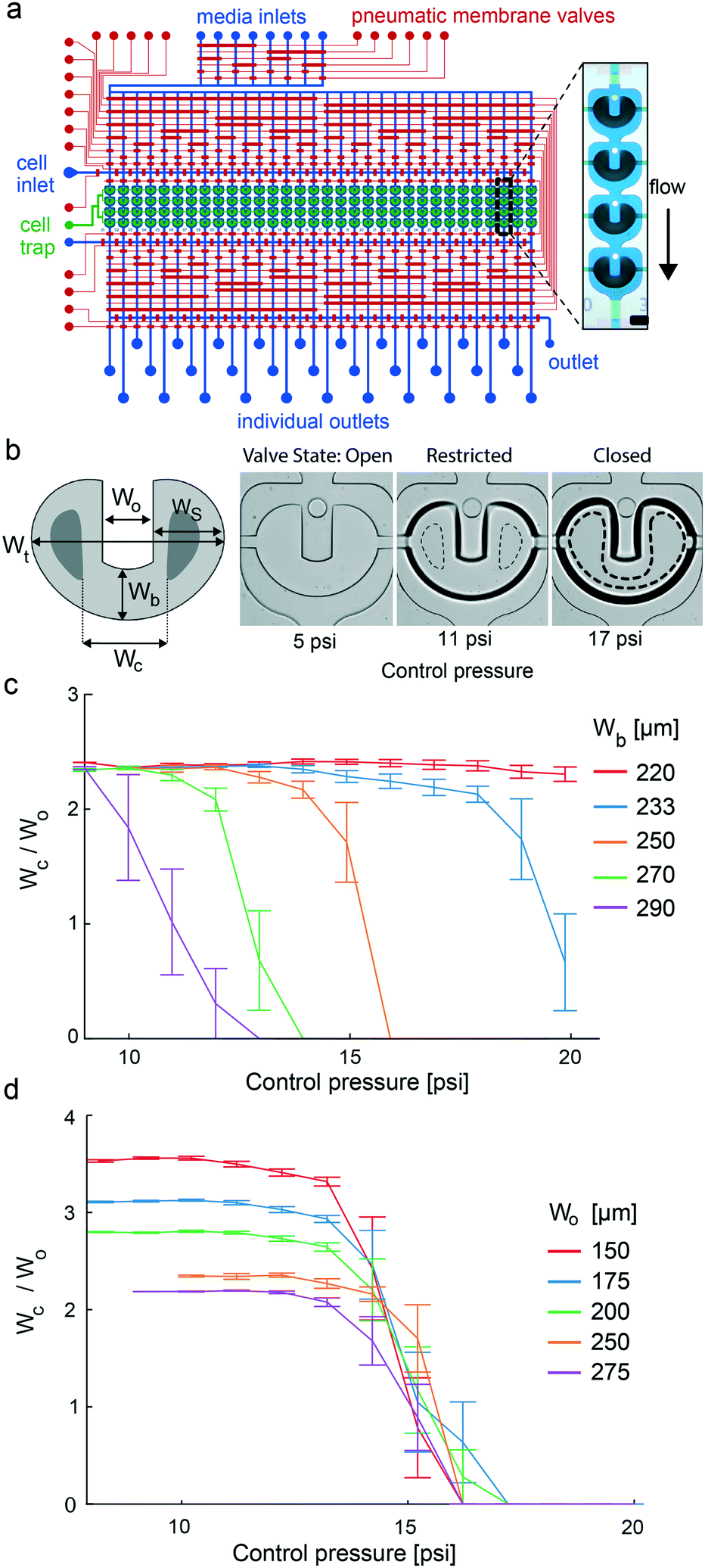 | ||
| Fig. 1 Microfluidic large-scale integration chip platform for hiPSC culturing and differentiation. (a) Layout of the two-layered mLSI chip platform. Microchannels within the pneumatic control and flow layer are depicted in red and blue, respectively. Zoom-in view: the pneumatic valve structures used for cell catching within a cell unit are enlarged in green. Round pillars on top of the catching structures prevent the loss of the 3D cell cultures during long-term culturing processes. (b) Schematic of a PMV for cell catching with parameterized design and with indicated length parameters. Bright-field images on the right are representative of the U-shaped valve in its three actuation states. The valve is designed in a push-up configuration, where the dashed lines within the valve area indicate the touching points between the PDMS membrane and the top of the flow chamber. (c) The bottom width of the U-shaped valve (Wb) defines the closing behavior from the open to restricted and fully closed states. All valves exhibited a constant opening width Wo of 250 μm. (d) The open width Wo of the valve had no influence on closing behavior. | ||
Prior to cell seeding on chip, the cell culture chambers were coated with 10% Pluronic® F-127 (Cat# 9003-11-6; Sigma-Aldrich) in phosphate-buffered saline (PBS) overnight to inhibit cell adhesion to the PDMS. The cell culture chambers were incubated in maintenance medium for at least 1 h. For chip seeding, adherent cells were harvested at 80% confluence with Accutase cell dissociation reagent (Cat# A1110501; Gibco) and resuspended at a density of 1 × 106 cells per ml in maintenance medium with 10 μM ROCK inhibitor (Y) (Y-27632, Cat# sc-281642A; Santa Cruz Biotechnology). The single-cell suspension was flushed into the chip via the cell inlet (Fig. 1a) and uniformly distributed in all cell culture chambers (Fig. S1†). Cells located outside the cell culture chamber and the catching valve area were removed by a brief rinse with the maintenance medium containing Y. For recovery, cells were allowed to adapt to the new microenvironment for a 4 h settling period without any fluid perfusion. Before starting endoderm induction, cells were fed for the first 24 h with the maintenance medium containing Y, and for the next 24 h without Y. If not stated otherwise, the medium was renewed every 4 h.
Cell culture
Human iPSCs (HMGUi001)27 were maintained as adherent monolayers on conventional cell culture plates coated with Geltrex (Cat# A1413302; Life Technologies). To promote self-renewal and avoid spontaneous differentiation, hiPSCs were fed daily with StemMACS™ iPS-Brew XF maintenance medium (Cat# 130-104-368; Miltenyi Biotec). The cells were incubated in a humidified atmosphere at 37 °C and 5% CO2. After reaching 70–80% confluence, the cells were passaged with 0.5 mM EDTA (Cat# A4892; AppliChem) in PBS. To enhance cell viability after splitting, the maintenance medium was supplemented with 10 μM Y for the following 24 h. Mycoplasma-free cell culture was regularly confirmed using a MycoAlert™ Plus mycoplasma detection kit (Cat# LT07-703; Lonza).Definitive endoderm differentiation
In all chip experiments, DE differentiation was induced 48 h after cell seeding (D0). The differentiation protocol (P1) was performed according to a previously published method,28,29 where the basal medium (Table S1†) was supplemented with 100 ng ml−1 activin A (AA) (Cat# 120-14-300; Peprotech) and 5 μM CHIR-99021 (CHIR), a GSK3β inhibitor (Cat# 24804-0004; Tebu-bio), on the first day of differentiation (D1). P1 basal medium supplemented with 100 ng ml−1 AA and 0.3 μM CHIR was added on the second day (D2), and P1 basal medium with 100 ng ml−1 AA was added on the third day (D3) of differentiation. Variations in the differentiation protocol (P2–4 and C1–6) are described in detail in Tables S2–S4.†Immunocytochemistry on chip
At the end of on-chip differentiation induction, 3D cell cultures were fixed in 4% paraformaldehyde for 1 h and cleared with 8% (w/v) sodium dodecylsulfate (SDS) in 0.1 M PBS (pH 7.5) for 2 h (protocol adapted30). After clearing, the 3D cell cultures were permeabilized with 0.2% Triton X-100 and 100 mM glycine in distilled H2O for 2 h and blocked in a blocking solution containing 3% donkey serum (CAT# P30-0101; PAN Biotech), 10% fetal calf serum (CAT# 35-079-CV; Corning Media Tech), 0.1% Tween-20, and 0.1% bovine serum albumin (BSA, CAT# A3311; Sigma Aldrich) in PBS. Three-dimensional cell cultures were incubated overnight with primary antibodies diluted in the blocking solution followed by 1 h washing with PBS. The 3D cell cultures were then incubated with secondary antibodies diluted in the blocking solution for at least 24 h. Nuclei were counterstained with DAPI. Fluorescently stained 3D cell cultures were thoroughly perfused with PBS and flushed with the X-CLARITY™ mounting solution (Cat# 13101; Biolab). All steps were programmed on a chip and performed at room temperature. Detailed information about the primary and secondary antibodies used for the assessment of pluripotency and DE differentiation efficiency are listed in Table S5.†Flow cytometry analysis
For quantitative analysis of cellular marker expression by flow cytometry (FACS), 3D cell cultures were dissociated using Accutase solution. Single cells were retrieved from the chip via 32 individual outlets (Fig. 1a). To evaluate pluripotency and DE stage commitment, single cells were stained for the protein expression of SSEA-4/TRA-1-60 or FOXA2/SOX17, respectively. Corresponding isotype and unstained controls were included according to the manufacturer's instructions. FACS analysis was performed using a BD FACSAria™ III cell.Image acquisition and analysis
Bright-field images were acquired using a Zeiss Axio Observer Z1 inverted microscope with an AxioCam MRm camera. Image processing was performed using customized ImageJ31 and MATLAB macros. Immunofluorescence images were acquired using a Zeiss LSM 880 Airyscan inverted confocal microscope. Z-stacks of entire 3D cell cultures were taken in the corresponding channels (DAPI, 488, 555, and 647) with a 20× objective lens. The middle sections of the images were extracted as TIFF files. Nuclei were detected and segmented with the Stardist32 library from ImageJ. A custom Python script was then used to calculate the mean fluorescence intensity for the different transcription factors within nuclei areas of 100 px2, which converted to 6.25 μm2.Statistical analysis
Error bars represent the mean standard deviation, unless otherwise mentioned. The box and whisker plots show the maximum point range as indicated.Results
Microfluidic large-scale integration for 3D cell culture
For parallelization of stem cell differentiation protocols with 3D cell cultures, we engineered a microfluidic large-scale integration chip platform. The fluidic design of the mLSI chip is illustrated in Fig. 1a. The platform integrates the process steps to (i) uniformly generate 3D cell cultures from a single-cell suspension; (ii) enable long-term culturing under changing microenvironmental conditions; (iii) automatize analytical workflows, including tissue clearing and immunocytochemistry; (iv) perform high-resolution imaging of 3D cell cultures; and (v) retrieve cell samples from the individual screened conditions for downstream off-chip analysis. The core function of the chip was 32 fluidically individually addressable microchambers. Each microchamber was segmented into four compartments, each containing an independent U-shaped PMV. One cell culture chamber exhibited a volume of 0.18 μL. Channels for the fluid routing had a height of 40 μm for the proper function of the PMVs. Fluid routing was enabled by including a multiplexer upstream and downstream of the cell culture chambers. All PMVs were designed in a push-up configuration, where the bottom PDMS sheet layer was 150 μm, allowing real-time bright-field and fluorescence imaging with high numerical aperture objectives.U-shaped valves have been previously developed to facilitate the formation of 3D cell cultures from single-cell suspensions. The functional principle of the U-shaped PMV is described by its three actuation states, which are the resting, half-actuated, and fully actuated states (see Fig. 1b). In the resting state, fluids pass through the cell culture chamber without a mechanical barrier. Upon increasing the control pressure of the U-shaped valve, the PDMS membrane deflects into the cell culture chamber. The geometric design difference between the side and bottom areas of the U-shaped valve leads to the closure of its side areas, whereas the bottom area of the valve is still open but restricted. As a result, the flow is focused through a U-shaped valve, and cells are trapped within the U-shaped area. Further, an increase in the actuation pressure leads to the full closure of all U-shaped elements. Consequently, the laminar flow surrounds the valve area, and the cells in the valve area self-aggregate into a 3D arrangement. Although previous publications investigated geometric design rules to balance the sides and bottom areas of the U-shaped valve to enable the three different operational valve states,23 further optimization is required for integration onto an mLSI chip. This is explained by the fact that the operational state transitions of the U-shaped valve have to be within the pressure range of the PMVs used for fluid routing, which is approximately 1.5 bar. To achieve this, we parameterized the U-shaped valve design using the length scales, namely, the total width (Wt), side area width (Ws), opening width (Wo), bottom width (Wb), and closing width (Wc) (schematic Fig. 1b). Ws governs the actuation pressure for the transition from the open to the restricted state, whereas Wb governs the transition from the restricted to the closed state of the U-shaped valve. We decided to keep the total width of the valve and the diameter of the cell culture chamber constant at 950 μm and 1.2 mm, respectively. This ensured that the surrounding flow profiles for all U-shaped valves were equal. Further changes in Ws have relatively little influence on the actuation pressure for reaching the restricted valve state as long as Ws/Wo is large. Notably, Wo influences the initial diameter of 3D cell cultures as the width of the U-shaped valve increases, and more cells can be trapped. For parameterization of the transition between the restricted and closed valve states, we defined Wc, which is the inner distance between the touching points of the PDMS membrane on the cell culture chamber sealing on the right and left U-shaped arms at a given pressure. Wc is dependent on the actuation pressure and is zero for fully closed U-shaped valves. The optimal U-shaped valve design for the integration of the mLSI chip platform was then determined by generating an mLSI chip with the same design as shown in Fig. 1a but with 20 geometrically different U-shaped valves. Table S6† summarizes all valve types tested. A representative example of a U-shaped valve actuated with three different pressures is shown in Fig. 1b. The closing behavior of the five U-shaped valves with different Wb values is plotted in relation to the actuation pressure in Fig. 1c. As expected, the reduction in Wb led to an increase in the closing pressure of the U-shaped valve. A U-shaped valve with a Wb of 250 μm exhibited a closing pressure of 14 psi, which was equal to that of the PMVs used for fluid routing in the inlet and outlet flow channel networks. Furthermore, this valve geometry exhibited a 5 psi pressure difference between the transition from the open/restricted to the restricted/closed state, which is sufficient for robust manufacturing. Interestingly, Wo had no effect on the closing behavior of the different U-shaped valves (Fig. 1d). Consequently, manufacturing chips for different aggregate sizes can be achieved by changing the cell-trapping volume by adapting Wo. All cell culture experiments were performed with an mLSI chip containing 128 U-shaped valves with 250 μm Wb and Wo parameters. A flow stream analysis around the geometrically optimized U-shaped valve in its three states at a typical flow rate of 1 μL min−1 is given in Fig. S2.†
Parallel culturing of hiPSC-derived 3D cell cultures on an mLSI chip
To obtain 128 homogenous 3D cell cultures, the cell culture chambers were filled with a high cell density suspension of hiPSCs from the cell inlet port. The U-shaped valves were set in a restricted valve state during the seeding process. In the next step, the U-shaped valves were set to the closed state, and cells outside the trap region were rinsed out through a common outlet port. The process lasted for less than 2 min. Although trapping cells out of the flow stream is possible with the U-shaped valve, it requires more time and leads to less homogenous filling because hiPSCs start to self-aggregate and clog the channel network. After seeding the entire chip, we followed the morphological changes in the 128 3D cell cultures in the U-shaped valve area by bright-field microscopy. Three-dimensional cell cultures were formed within the next 12 h (Fig. 2a). The mean occupied cell area decreased within the first 12 h after cell seeding, whereas the mean circularity increased (Fig. 2b). After 12 h of chip culturing, the mean 3D culture area increased owing to cell proliferation. After 24 h, the mean diameter of the 128 hiPSC-derived 3D cultures formed with the U-shaped valve approach on one mLSI chip was 145 ± 30 μm (Fig. 2c). The homogenous 3D cell culture formation process is reproducible as shown by measuring the chip-to-chip variability of the 3D cell culture diameter between five chips cultured under the same conditions (Fig. 2d). To quantify the pluripotency of the cultures after 48 h, we dissociated them and retrieved the single-cell suspension for FACS analysis. The pluripotency markers TRA-1-60 and SSEA-4 were co-expressed in 97.4% of the cells. Similar results were found by on-chip immunofluorescence analysis of the 3D cell cultures co-expressing pluripotency transcription factors NANOG and OCT4 (Fig. S3†). Notably, for long-term stability of the 3D cell culture, it was essential to coat the chip platform with 10% Pluronic for 12 h. Without surface coating, hiPSCs started to adhere and formed over time mixed 2D/3D cell cultures. Further, we included a micropillar at the entry of the U-shaped valve, which led to a 41% increase in the probability of maintaining the 3D cell culture at a position compared to U-shaped valves without pillars used over a culture period of 1 week (Fig. S4†). Pulse feeding with fresh media was initiated after 4 h of cell seeding. A media feeding pulse lasted 45 s per chamber to fully exchange the cell culture chamber volume. For long-term culturing, the repetition cycle for cell feeding could be between 2 and 8 h without any observable changes in cell morphology. Extending the time gap between feeding pulses over 12 h led to the disaggregation of the 3D cell culture. | ||
| Fig. 2 On-chip formation of 3D cell cultures from a hiPSC single cell suspension. (a) Representative bright-field images of a pneumatic U-shaped valve after cell seeding. The single hiPSCs started to aggregate within the first 2 h, formed a compact aggregate after 12 h and proliferated over 48 h. Scale bar denotes 500 μm. (b) 3D orthogonal representative of fluorescence images from one cleared 3D aggregate stained for the nucleus with DAPI. Scale bar denotes 50 μm. (c) Time trajectory of the mean 3D hiPSC culture area (dark gray) and circularity (light gray) after seeding. (d and e) Distribution of 3D hiPSC culture diameters obtained on a single and on multiple chips. Error bars represent SD (n = 5, SD = 25.63, SEM = 11.46). | ||
On-chip differentiation of hiPSCs into definitive endoderm
In the next step, we aimed to show that the mLSI chip platform can be used to automatize chemical induction protocols for stem cell differentiation. For this, we differentiated 3D hiPSC-derived cultures into DE by activating the TGF-β/nodal and WNT signaling pathways with AA and CHIR. While cells were exposed to AA at a constant concentration for 72 h, CHIR was added at a decreasing concentration within the first 48 h. The precise differentiation protocol is shown in Fig. 3a with representative brightfield images of one aggregate undergoing the DE differentiation in Fig. 3b. For the on-chip experiment, we seeded hiPSCs according to the previously described method and started the differentiation protocol 48 h later. All cell culture media were exchanged at each step every 4 h. At the end of the DE differentiation protocol, the 3D cell cultures were fixed, optically cleared, and stained with cell stage-specific antibodies using an automated fluid program. Fig. 3c shows representative confocal immunofluorescence images of a 3D cell culture stained for FOXA2 and SOX17. Co-expression of the two transcription factors is indicative of a DE cell type. The efficiency of on-chip differentiation was evaluated by measuring the fluorescence intensity of FOXA2 and SOX17 markers within the nuclei area (Fig. 3c) of confocal sections distributed along the Z-axis of the 3D cell cultures. Cells were considered positive when the fluorescence intensity of the DE marker antibodies was two times higher than the background signal obtained from primary antibody control samples. The image analysis revealed that 89.4% of the cells within the 3D cultures were double-positive for the expression of FOXA2 and SOX17 markers, which was comparable to control differentiation experiments performed off chip (Fig. 3d top panel).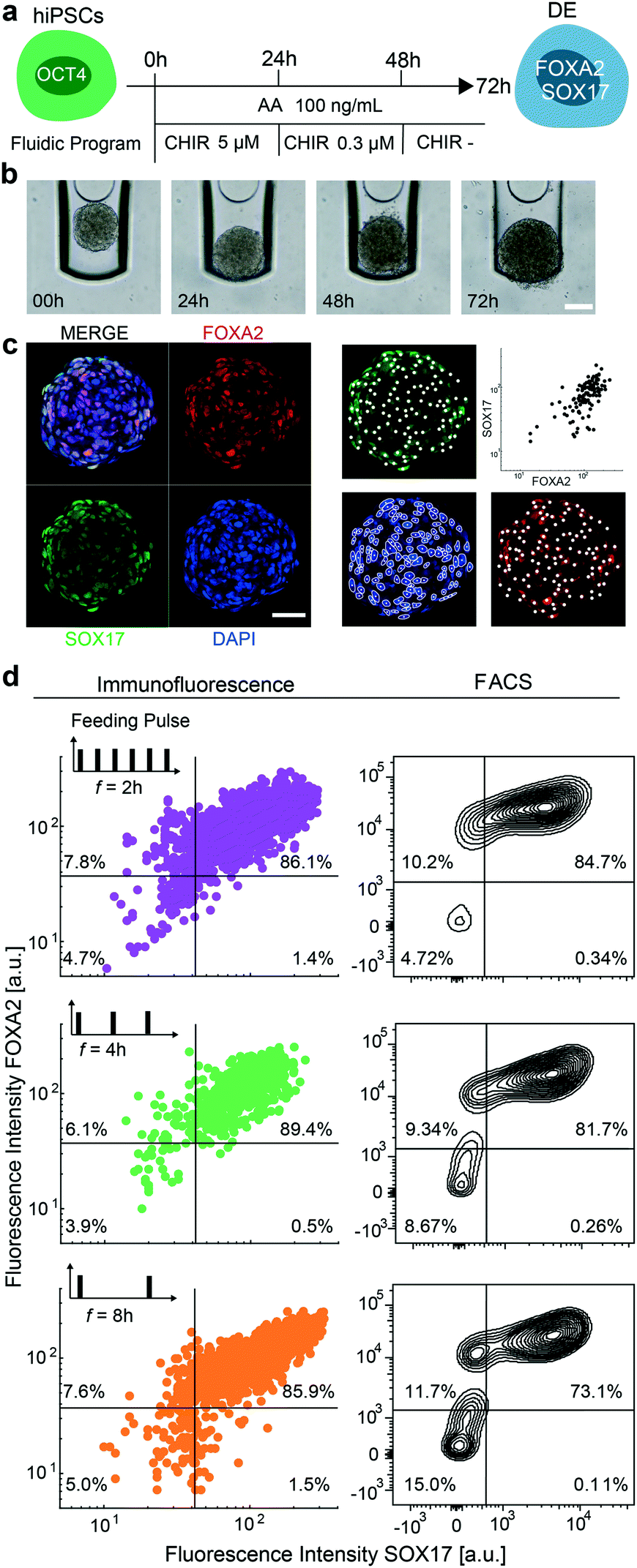 | ||
| Fig. 3 On-chip differentiation of 3D hiPSC cultures into DE under changing feeding frequency. (a) Chemical program used to differentiate hiPSCs into the DE stage. (b) Brightfield images of one representative hiPSC 3D aggregate undergoing DE differentiation. Scale bar denotes 100 μm. (c) Representative fluorescence signal intensities of FOXA2 (red), SOX17 (green), and DAPI (blue) in single cells of the 3D cell cultures after 3 days of DE differentiation. Scale bar denotes 50 μm. Nuclei are segmented from the DAPI signal using Stardist and the mean fluorescence intensities are extracted from a 100 pix2 area at the center of mass of each nuclei. (d) Fluorescence signal intensity of DE markers in cells of 3D cell cultures differentiated into DE within 3 days with feeding frequencies of 2, 4, or 8 h acquired from immunofluorescence microscopy (left) and FACS analysis (right). FACS data was acquired from the chip by dissociating the 3D cell cultures and retrieving the cells. | ||
One of the main differences between standard cell culture formats and cell cultures integrated on an mLSI chip platform is the need for a higher cell culture media exchange rate. Under static conditions, a single 3D cell culture on chip has access to 0.18 μL of media, which is only 50 to 100 times the volume of its own size. To determine whether the media exchange rate affects DE differentiation, we repeated the on-chip DE differentiation experiment with media exchange rates of 2, 4, and 8 h over the entire period of the experiment. Concomitantly, we challenged our image analysis by comparing the results with the FACS data. For this, 24 3D cell cultures differentiated under the same conditions were retrieved and pooled as single cells at the end of the differentiation protocol and then subjected to FACS analysis. DE differentiation efficiency performed with 2 and 8 h media exchange rates led to similar results (Fig. 3c) as with a media exchange rate of 4 h. All 3D cell cultures exhibited 85% of cells co-expressing FOXA2 and SOX17. The FACS analysis matched these results for the 2 and 4 h media exchange rates but indicated a lower DE state (73%) for the differentiation performed with a media exchange rate of 8 h. One possible explanation for this finding is that cells under these conditions are more stressed, and the relatively long FACS workflow, including cell dissociation and retrieval, in the still life state from the chip led to a decreased number of double-positive cells.
Despite the fact that we obtained high differentiation efficiencies for the DE state, we asked if the chemical composition of the differentiation cocktail was biased by the material of the mLSI platform. Although small hydrophobic compounds are generally desired for cell manufacturing purposes to reduce the variability of recombinant proteins,33 they have to be tested on microfluidics devices due to possible partition behavior in bulk material. Compounds with logP values larger than 1.5 have been shown to partition into the PDMS. CHIR had a predicted logP value of 4.92.34 A biological analogue for CHIR activity is the protein WNT3A, which is only soluble in water. In the following we repeated the DE differentiation with WNT3A and CHIR on the same chip experiment to compare the differentiation efficiencies and 3D structure. Indeed, we did not observe any significant differences for DE induction induced by CHIR and WNT3A (Fig. S5†) on chip based on the DE marker expression. The same was observed for an off-chip experiment within standard 2D cell culture format (data omitted). With this we can exclude that the DE differentiation is biased by partition of CHIR into the PDMS. Taken together, we verified the functionality of the mLSI chip platform for the differentiation of hiPSCs in a parallel fashion.
Screening DE differentiation protocols on chip
In the next step, we aimed to test the differentiation efficiency of four protocols (P1–4) to generate the DE cell type. Two DE differentiation protocols are currently applied to generate stem cell-derived cells. Both protocols, named P1 and P3, included AA and CHIR to induce DE cell differentiation; however, the concentrations of CHIR differed. Further, the use of an inhibitor of Rho-associated protein kinase (ROCK) is currently debated. ROCK inhibitors enhance cell adhesion, viability, and differentiation,33 and at the same time inhibit apoptosis, which was frequently observed in chip conditions, predominately during the first day of differentiation. Upon including ROCK (Y) to P1 and P3, we generated the differentiation protocols P2 and P4. The differentiation protocols are summarized in Fig. 4a. | ||
| Fig. 4 On-chip screening of differentiation protocols for the DE cell type. (a) Chemical composition and temporal resolution of four screened differentiation protocols for DE used during the first differentiation step of stem cell-derived β-like cells. (b) Changes in the mean diameter of the 3D cell cultures during DE differentiation indicate a clear difference between the four differentiation protocols. Error bars represent SD. (c) Representative immunofluorescence images of on-chip DE-differentiated 3D cell cultures stained for FOXA2 (red), SOX17 (green), OCT4 (gray), and DAPI (blue). Scale bars denote 50 μm. (d) Quantitative single-cell image analysis of the 3D cell cultures to resolve the percentage of double-positive FOXA2+/SOX17+-expressing cells after 3 days of differentiation. | ||
Chip run was performed as previously described. Bright-field images taken before the start of the differentiation protocols and at 48 h after seeding showed a homogeneous size distribution of the 3D cell cultures (inset Fig. 4b). Size differences in the 3D cell culture emerged during DE differentiation. Under P1 and P2, the size of the 3D cell cultures was relatively constant during differentiation, whereas 3D cultures under P3 and P4 started to grow from day 1 and were up to 40% larger in diameter at the end of differentiation. Immunofluorescence (IF) images of cleared 3D cell cultures differentiated with protocols P1 and P2 exhibited only DE cells, whereas 3D cell cultures differentiated with protocols P3 and P4 showed approximately 90% and 96% of cells positive for DE markers, respectively. Using IF images, we identified spatial segregation of FOXA2/SOX17 positive and negative cells in 3D cell cultures under P3 and P4. Cells in the center of the 3D cell cultures maintained pluripotency as confirmed by OCT4 expression. These cells were surrounded by a layer of double-positive FOXA2 and SOX17 expressing cells. On the outside, only SOX17 expressing cells were found. This spatial distribution was not observed in the 3D cell cultures differentiated under P1 and P2 (Fig. 4c). Pluripotent cells are proliferative, supporting the observation that 3D cell cultures with OCT4 expressing cells are larger in diameter. The addition of Y reduced the OCT4 expression in P4 compared to P3, whereas between P1 and P2 no obvious effect on DE differentiation could be observed.
The four tested differentiation protocols showed variations in their differentiation efficiencies towards DE with changes of up to 10%. Differences in that magnitude have been observed between chip-to-chip and off-chip differentiation experiments. This variability is not circumvented upon microfluidic integration and is most likely explained by cell stage differences when starting the differentiation. Growth differences and spatial patterns introduced by the different differentiation protocols, however, cannot be explained by experimental variations because the starting points on the same chip were identical for all 3D cell cultures.
Concentration gradient screening for mesendoderm segregation and definitive endoderm patterning on chip
In the last step, we investigated the concentration gradients of AA and CHIR for mesendoderm segregation and endoderm patterning on the chip. In humans, the endoderm and mesendoderm arise from the primitive streak (PS). In dependence of highest concentrations of TGF-β/WNT pathway activators, most PS cells in the anterior region will generate DE, whereas the posterior PS cells will form mesoderm upon lower concentrations. The anterior DE gives rise to liver and pancreatic cell types.35,36In vitro protocols induced this transition by CHIR (WNT pathway) and AA (TGF-β pathway) in a balanced manner. A high concentration of AA during differentiation leads to the progression towards DE, whereas the absence of AA leads to mesoderm induction.37 The relative concentration of AA/CHIR required to stimulate germ layer progression varies throughout the literature. Therefore, we investigated different concentrations of AA and CHIR on a single chip to determine the concentration that maximizes the specification of different cell types during DE patterning. Here, we screened six differentiation protocols (Fig. 5a). Chip experiments were performed as described above. | ||
| Fig. 5 Chemical concentration screening to investigate anterior DE and mesoderm differentiation. (a) Schematic of the six chemical differentiation protocols (C1–6) used to induce anterior DE and mesoderm on chip. (b) Changes in the normalized mean areas of the 3D cultures during differentiation with different chemical protocols. (c) Representative IF images of 3D cell cultures stained for the mesoderm marker Brachyury-T (red), DE marker SOX17 (green), and anterior DE marker CER1 (magenta). Nuclei were visualized by DAPI staining. Scale bars denote 50 μm. (d) Fraction of cells within the 3D cell cultures with marker fluorescence intensity 2σ over the background for Brachyury-T, SOX17, and CER1. Error bars represent SD. (e) Normalized changes in protein marker expression levels of cells from the 3D cultures differentiated with the chemical programs C1–6. The dashed line denotes the fluorescent background signals intensity of the protein marker. | ||
Bright-field images of the differentiation progression of each hiPSC-derived 3D cell culture revealed growth differences between the different applied conditions (Fig. 5b). Protocols C1 and C6 with the highest and lowest AA concentrations led to a stronger decrease in the mean 3D cell culture area during the first day of differentiation, whereas protocols C2–C5 with intermediate AA concentrations resulted in only small decreases. All 3D cell cultures were stained for SOX17, CER1 (an anterior-endoderm marker), and Brachyury-T (a mesoderm marker) by immunofluorescence. The highest levels of SOX17 were found in the 3D cultures differentiated under high concentrations of AA. Under conditions C1 to C3, more than 70% of cells in every 3D cell culture were SOX17-positive, whereas under conditions C4 to C6, less than 20% of cells per 3D cell culture were SOX17-positive. The anterior domain marker CER1 was strongly induced within the C1 protocol with 65% of positive cells per 3D cell culture. Notably, minimal reduction of AA but maintained CHIR concentration, as shown in C2, led to a strong reduction of CER1 positive cells, representing less than 10% per 3D cell culture. However, C6 cells only expressed the early mesodermal marker Brachyury-T. The absence of AA and high concentration of CHIR led to the expression of Brachyury-T in 66% of the cells. This finding corroborates the inhibitory effect of AA on the expression of Brachyury-T. Fig. 5c shows representative immunofluorescence images of 3D cell cultures from protocols C1, C3, and C6. Representative images of the other differentiation protocols are shown in Fig. S6.† In addition to the changes in marker expression, we observed morphological differences between the 3D cell cultures. 3D cell cultures under high AA (C1 and C2) or high CHIR concentration (C6) maintained a compact non-organized aggregate shape, whereas cultures with intermediate concentrations of AA and CHIR (C3–C5) showed epithelium-like structures and lumen formation, suggesting that these cultures were committed to off-target cell types. Taken together, marker and morphological profiling on the chip can help to optimize the generation of more homogeneous DE cells.
Discussion
Three-dimensional human pluripotent stem cell cultures are the starting materials for developing cell replacement therapies. To obtain endodermal-specific cell types, including β-cells for diabetes transplantation, we have to closely recapitulate the embryonic endodermal signaling mechanisms under in vitro conditions. To establish the minimal chemical media composition, concentration, stimulation times, and cell pattern information for differentiation protocols with 3D cell cultures requiring as long as 30–60 days, new cell culture technologies for parallel screening are required.In this study, we developed an mLSI chip platform for the formation, long-term culturing, and chemical screening of 3D human pluripotent stem cell cultures. The general cell unit design with the closing pressure optimized U-shaped valve will allow the scaling up of 32 individual addressable conditions on successor mLSI platforms. The U-shaped valve-assisted formation process of 3D cell cultures led to equally homogenous aggregate sizes of 145 ± 30 μm. The low size variation is comparable to the aggregate formation process achieved on hanging drop or microcavity chip platforms. On average, 1 million cells were required to fill the 128 U-shaped valve structures, which were approximately the same number used for filling 128 2D cell culture chambers on an mLSI chip. In contrast to the integration of 2D hiPSCs on mLSI chips, the 3D cell culture mLSI chip does not require covalent surface coatings for long-term culturing,38 which strongly simplifies the chip production process and increases the robustness of the cell culture maintenance. Although the size of the cell culture chamber is fixed, it was possible to culture 3D hiPSC cultures up to 5 days before their size exceeded the U-shape valve footprint. For differentiation experiments, however, culture times can last more than two weeks since proliferation and differentiation of cells showed antagonistic behavior. While progenitor cells remain proliferative, proliferation in more mature cell types is arrested and the cell cycle is permanently exited.39 Miniaturization of the cell culture volume comes with the need for constant renewal of the cell culture media. Although flow logic on the mLSI chips allowed the feeding of independent cell cultures sequentially, feeding rates between 2 and 8 h had no influence on the variability and pluripotency of hiPSCs.
With the differentiation of hiPSCs into DE, we proved that it is possible to automate stem cell differentiation protocols using the mLSI chip technology. On the mLSI chip, DE differentiation efficiencies of over 80% were obtained based on the presence of double-positive cells expressing both SOX17 and FOXA2 markers. This differentiation efficiency is comparable to the results observed in standard well plate 2D and 3D culture systems.7–9,40 The PDMS material of the chip platform did not reduce the efficiency owing to chemical absorption, which was observed upon exchange of the hydrophobic CHIR with the hydrophilic WNT3A protein. In small volumes, the accumulation of endogenous signals is sufficient to drive stem cell fate,41 and constant flow conditions are known to negatively impact stem cell viability. In addition, medium exchange rates between 2 and 8 h had no significant influence on the DE differentiation efficiency, which suggests that the time interval is sufficient to maintain possible secreted pro-survival factors for the cell cultures in the chamber.
Upon comparing the two DE differentiation protocols,10,28,29 we showed the effectiveness of our platform. Homogeneous size distribution of the 3D cell cultures on the chip reduced the batch variability of the differentiation protocols and allowed systematic investigation of media composition changes during DE differentiation. Although the overall DE differentiation efficiency between the two tested base differentiation protocols was low, it was observed that changes in the cell culture medium formulation with reduced BSA (from 2% to 0.5%) and CHIR (from 5 to 3 μM) concentrations within the first 24 h of DE differentiation induced distinct phenotypes. Monolayer stem cell experiments revealed that CHIR in combination with AA led to loss of cells, presumably through a combination of cell death and substrate detachment caused by extracellular matrix remodeling.42 3D cell cultures differentiated under P1 showed fewer cells and smaller size after 24 h. However, as expected from 2D cell cultures, 3D cell cultures differentiated under P3 increased in cell number and size. One explanation for this is the BSA substitution, which has been suggested to contribute directly to stem cell survival by carrying growth hormones, lipids, and other factors.43 Concomitantly, BSA could diminish CHIR activity through binding inhibition. The finding of maintained pluripotent stem cells within the core of 3D cell cultures differentiated under P3 suggests that diminished CHIR concentration led to the growth phenotype. Concomitantly, it was observed that minor media additives had a profound influence on differentiation experiments. Prolonged addition of the Y to P2 or P4 did not affect the DE marker and growth behavior of the 3D cell cultures.
To further investigate DE differentiation, we systematically screened the dose-dependent activation of the TGF-β and WNT pathways. It is known that a high AA concentration induces stem cell development towards the endoderm via the anterior PS stage, and a reduction in AA leads to mesodermal cell development via the posterior PS stage. Both inductions require the presence of CHIR.37 Indeed, this concentration effect of DE/mesodermal development could be recapitulated on the chip. In accordance with the literature, anterior-like DE cells were obtained at AA concentrations higher than 50 μM, based on the SOX17 and CER1 expression, whereas mesodermal-like cells were obtained upon removal of AA and increased CHIR concentration, based on the Brachyury-T expression. Intermediate concentrations of AA and CHIR led to increased structural and cell-type heterogeneity within the 3D cell cultures. For example, a decrease in AA concentration from 100 to 50 μM led to the formation of complex epithelial layers within the 3D cell cultures, whereas 3D cell cultures differentiated under 100 μM AA showed homogeneous DE marker expression and no internal aggregate structures. This indicates that a combined on-chip morphological and marker expression analysis can help optimize protocol development.
In summary, the mLSI chip platform developed here is a cost-effective tool for optimizing stem cell differentiation protocols in order to obtain cell types for cell replacement therapies. Evaluation of self-organization in 3D cell cultures allow to evaluate and specify differentiation protocols for cell types arising from the definitive endoderm. The integrated workflows and unit cell operations for the formation and high-content image processing of 3D cell culture may extend the applicability of mLSI chips in the future.
Author contributions
J. C. A. R., A. K. B., S. A. performed investigation. J. C. A. R., A. K. B., S. A., M. M. analyzed the data, J. C. A. R., S. A., N. C., M. M., O. R., T. G. provided methodologies. J. C. A. R., A. K. B., S. A., H. L., M. Meier wrote the manuscript. H. L. and M. Meier supervised the work.Conflicts of interest
The authors declare not conflict of interest.Acknowledgements
This work is supported by the Helmholtz Pioneer Campus, the German Ministry of Education and Research (INDIMED-PancChip), and the ERC (Consolidator Grant Number MicroAdiPSChip – 772646). We thank Alina Platen for cell culture assistance. J. C. A. R. was supported by Doctoral fellowship from the Deutscher Akademischer Austauschdienst (DAAD).References
- M. Eiraku, N. Takata, H. Ishibashi, M. Kawada, E. Sakakura, S. Okuda, K. Sekiguchi, T. Adachi and Y. Sasai, Nature, 2011, 472, 51–56 CrossRef CAS PubMed.
- X. Yin, B. E. Mead, H. Safaee, R. Langer, J. M. Karp and O. Levy, Cell Stem Cell, 2016, 18, 25–38 CrossRef CAS PubMed.
- T. Tao, Y. Wang, W. Chen, Z. Li, W. Su, Y. Guo, P. Deng and J. Qin, Lab Chip, 2019, 19, 948–958 RSC.
- K. A. D'Amour, A. D. Agulnick, S. Eliazer, O. G. Kelly, E. Kroon and E. E. Baetge, Nat. Biotechnol., 2005, 23, 1534–1541 CrossRef PubMed.
- M. R. C. Kraus and A. Grapin-Botton, Curr. Opin. Genet. Dev., 2012, 22, 347–353 CrossRef CAS PubMed.
- M. D. Ungrin, G. Clarke, T. Yin, S. Niebrugge, M. C. Nostro, F. Sarangi, G. Wood, G. Keller and P. W. Zandstra, Biotechnol. Bioeng., 2012, 109, 853–866 CrossRef CAS PubMed.
- U. Diekmann, H. Wolling, R. Dettmer, I. Niwolik, O. Naujok and F. F. R. Buettner, Sci. Rep., 2019, 9, 1–14 CAS.
- U. Diekmann and O. Naujok, Methods Mol. Biol., 2016, 1341, 157–172 CrossRef CAS PubMed.
- F. W. Pagliuca, J. R. Millman, M. Gürtler, M. Segel, A. V. Dervort, J. H. Ryu, Q. P. Peterson, D. Greiner and D. A. Melton, Cell, 2014, 159, 428–439 CrossRef CAS PubMed.
- L. Velazco-Cruz, J. Song, K. G. Maxwell, M. M. Goedegebuure, P. Augsornworawat, N. J. Hogrebe and J. R. Millman, Stem Cell Rep., 2019, 12, 351–365 CrossRef CAS PubMed.
- P. U. Mahaddalkar, K. Scheibner, S. Pfluger, Ansarullah, M. Sterr, J. Beckenbauer, M. Irmler, J. Beckers, S. Knöbel and H. Lickert, Nat. Biotechnol., 2020, 38, 1061–1072 CrossRef CAS PubMed.
- K. Moshksayan, N. Kashaninejad, M. E. Warkiani, J. G. Lock, H. Moghadas, B. Firoozabadi, M. S. Saidi and N. T. Nguyen, Sens. Actuators, B, 2018, 263, 151–176 CrossRef CAS.
- A. Essaouiba, T. Okitsu, R. Kinoshita, R. Jellali, M. Shinohara, M. Danoy, C. Legallais, Y. Sakai and E. Leclerc, Biochem. Eng. J., 2020, 164, 107783 CrossRef CAS.
- R. Gómez-Sjöberg, A. A. Leyrat, D. M. Pirone, C. S. Chen and S. R. Quake, Anal. Chem., 2007, 79, 8557–8563 CrossRef PubMed.
- J. Zhang, X. Wei, R. Zeng, F. Xu and X. J. Li, Future Sci. OA, 2017, 3, FSO187 CrossRef CAS PubMed.
- M. Blazek, T. S. Santisteban, R. Zengerle and M. Meier, Lab Chip, 2015, 15, 726–734 RSC.
- M. Hofer and M. P. Lutolf, Nat. Rev. Mater., 2021, 6, 402–420 CrossRef CAS PubMed.
- P. M. Fordyce, C. A. Diaz-Botia, J. L. DeRisi and R. Gómez-Sjöberg, Lab Chip, 2012, 12, 4287 RSC.
- T. S. Santisteban, O. Rabajania, I. Kalinina, S. Robinson and M. Meier, Lab Chip, 2017, 18(1), 9 Search PubMed.
- J. C. McDonald and G. M. Whitesides, Acc. Chem. Res., 2002, 35, 491–499 CrossRef CAS PubMed.
- N. Compera, S. Atwell, J. Wirth, B. Wolfrum and M. Meier, Lab Chip, 2021, 21, 2986–2996 RSC.
- T. Robinson, P. Kuhn, K. Eyer and P. S. Dittrich, Biomicrofluidics, 2013, 7, 044105 CrossRef CAS PubMed.
- W. Liu, J. C. Wang and J. Wang, Lab Chip, 2015, 15, 1195–1204 RSC.
- W. Liu, J. Xu, T. Li, L. Zhao, C. Ma, S. Shen and J. Wang, Anal. Chem., 2015, 87, 9752–9760 CrossRef CAS PubMed.
- B. Schuster, M. Junkin, S. S. Kashaf, I. Romero-Calvo, K. Kirby, J. Matthews, C. R. Weber, A. Rzhetsky, K. P. White and S. Tay, Nat. Commun., 2020, 11, 5271 CrossRef CAS PubMed.
- M. A. Unger, H. P. Chou, T. Thorsen, A. Scherer and S. R. Quake, Science, 2000, 288, 113–116 CrossRef CAS PubMed.
- X. Wang, M. Sterr, I. Burtscher, S. Chen, A. Hieronimus, F. Machicao, H. Staiger, H. U. Häring, G. Lederer, T. Meitinger, F. M. Cernilogar, G. Schotta, M. Irmler, J. Beckers, M. H. de Angelis, M. Ray, C. V. E. Wright, M. Bakhti and H. Lickert, Mol. Metab., 2018, 9, 57–68 CrossRef CAS PubMed.
- Z. Zhu, Q. V. Li, K. Lee, B. P. Rosen, F. González, C. L. Soh and D. Huangfu, Cell Stem Cell, 2016, 18, 755–768 CrossRef CAS PubMed.
- Z.-D. Shi, K. Lee, D. Yang, S. Amin, N. Verma, Q. V. Li, Z. Zhu, C.-L. Soh, R. Kumar, T. Evans, S. Chen and D. Huangfu, Cell Stem Cell, 2017, 20, 675–688.e6 CrossRef CAS PubMed.
- N. Xu, A. Tamadon, Y. Liu, T. Ma, R. K. Leak, J. Chen, Y. Gao and Y. Feng, Sci. Rep., 2017, 7, 1–15 CrossRef CAS PubMed.
- M. D. Abràmoff, P. J. Magalhães and S. J. Rams, Biophotonics Int., 2004, 11, 36–42 Search PubMed.
- J. C. Caicedo, A. Goodman, K. W. Karhohs, B. A. Cimini, J. Ackerman, M. Haghighi, C. Heng, T. Becker, M. Doan, C. McQuin, M. Rohban, S. Singh and A. E. Carpenter, Nat. Methods, 2019, 16, 1247–1253 CrossRef CAS PubMed.
- A. Korostylev, P. U. Mahaddalkar, O. Keminer, K. Hadian, K. Schorpp, P. Gribbon and H. Lickert, Mol. Metab., 2017, 6, 640–650 CrossRef CAS PubMed.
- T. Sterling and J. J. Irwin, J. Chem. Inf. Model., 2015, 55, 2324–2337 CrossRef CAS PubMed.
- K. D. Tremblay and K. S. Zaret, Dev. Biol., 2005, 280, 87–99 CrossRef CAS PubMed.
- P. P. L. Tam, P.-L. Khoo, S. L. Lewis, H. Bildsoe, N. Wong, T. E. Tsang, J. M. Gad and L. Robb, Development, 2007, 134, 251–260 CrossRef CAS PubMed.
- M. Hashemitabar and E. Heidari, J. Cell. Physiol., 2019, 234, 7811–7827 CrossRef CAS PubMed.
- C. Luni, S. Giulitti, E. Serena, L. Ferrari, A. Zambon, O. Gagliano, G. G. Giobbe, F. Michielin, S. Knöbel, A. Bosio and N. Elvassore, Nat. Methods, 2016, 13, 446–452 CrossRef CAS PubMed.
- S. Ruijtenberg and S. van den Heuvel, Cell Cycle, 2016, 15, 196–212 CrossRef CAS PubMed.
- J. H. Shim, J. H. Kim, J. Han, S. Y. An, Y. J. Jang, J. Son, D. H. Woo, S. K. Kim and J. H. Kim, Cell Transplant., 2015, 24, 2155–2168 Search PubMed.
- J. Guild, A. Haque, P. Gheibi, Y. Gao, K. J. Son, E. Foster, S. Dumont and A. Revzin, Stem Cells, 2016, 34, 1501–1512 CrossRef CAS PubMed.
- Q. Li, A. P. Hutchins, Y. Chen, S. Li, Y. Shan, B. Liao, D. Zheng, X. Shi, Y. Li, W.-Y. Chan, G. Pan, S. Wei, X. Shu and D. Pei, Nat. Commun., 2017, 8, 15166 CrossRef PubMed.
- G. L. Francis, Cytotechnology, 2010, 62, 1–16 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1lc00565k |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |