
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards nanovesicle-based disease diagnostics: a rapid single-step exosome assay within one hour through in situ immunomagnetic extraction and nanophotonic label-free detection
Qinming
Zhang
a,
Hannah J.
Loghry
b,
Jingjing
Qian
a,
Michael J.
Kimber
*b,
Liang
Dong
 *ac and
Meng
Lu
*acd
*ac and
Meng
Lu
*acd
aDepartment of Electrical and Computer Engineering, Iowa State University, Ames, Iowa 50011, USA. E-mail: ldong@iastate.edu; menglu@iastate.edu
bDepartment of Biomedical Sciences, Iowa State University, Ames, Iowa 50011, USA. E-mail: michaelk@iastate.edu
cMicroelectronics Research Centre, Iowa State University, Ames, Iowa 50011, USA
dDepartment of Mechanical Engineering, Iowa State University, Ames, Iowa 50011, USA
First published on 13th July 2021
Abstract
Exosomes have been considered as high-quality biomarkers for disease diagnosis, as they are secreted by cells into extracellular environments as nanovesicles with rich and unique molecular information, and can be isolated and enriched from clinical samples. However, most existing exosome assays, to date, require time-consuming isolation and purification procedures; the detection specificity and sensitivity are also in need of improvement for the realization of exosome-based disease diagnostics. This paper reports a unique exosome assay technology that enables completing both magnetic nanoparticle (MNP)-based exosome extraction and high-sensitivity photonic crystal (PC)-based label-free exosome detection in a single miniature vessel within one hour, while providing an improved sensitivity and selectivity. High specificity of the assay to membrane antigens is realized by functionalizing both the MNPs and the PC with specific antibodies. A low limit of detection on the order of 107 exosome particles per milliliter (volume) is achieved because the conjugated MNP–exosome nanocomplexes offer a larger index change on the PC surface, compared to the exosomes alone without using MNPs. Briefly, the single-step exosome assay involves (i) forming specific MNP–exosome nanocomplexes to enrich exosomes from complex samples directly on the PC surface at the bottom of the vessel, with a >500 enrichment factor, and (ii) subsequently, performing in situ quantification of the nanocomplexes using the PC biosensor. The present exosome assay method is validated in analyzing multiple membrane proteins of exosomes derived from murine macrophage cells with high selectivity and sensitivity, while requiring only about one hour. This assay technology will provide great potential for exosome-based disease diagnostics.
Introduction
Extracellular vesicles (EVs), such as exosomes, are nanoscale membrane-bound structures secreted by living cells that carry molecular traits associated with their parent cells.1,2 EVs have been widely studied as disease biomarkers since they have been identified in various types of body fluids, such as blood, urine, and saliva.3–8 Compared with conventional small molecule, protein, and nucleic acid biomarkers, EV biomarkers are information-rich, with a group of cargoes including membrane proteins, exosomal proteins, mRNA fragments, and miRNAs.9 Importantly, EVs can be isolated and enriched from complex samples to eliminate interfering molecules and thus assure the biomarker quality.10 Extraction and detection of EVs are two required assay steps to process EV samples for disease diagnosis and biomedical research. Several EV extraction approaches have been developed based on the target EVs' biochemical and physical characteristics. For example, differential ultracentrifugation, size exclusion chromatography and polymer-based precipitations are the most common approaches to isolate EVs from clinical samples.11–15 These methods are time-consuming and lack the ability to distinguish EVs from particles of similar size. Recently, microfluidic technologies, such as deterministic lateral displacement and acoustic separations, have been successfully demonstrated but with limited throughput.16,17 Alternatively, immunomagnetic methods using functionalized magnetic micro- and nanoparticles can enable efficient, fast, and specific extractions of EVs.18–27 Following an extraction process, the EV samples need to be eluted and detected using an EV sensing approach. For example, nanoparticle tracking analysis can be used to obtain EVs' size and phenotype.28–30 Enzyme-linked immunosorbent assays can quantify EVs based on their membrane protein constituents.31 Label-free EV detection assays have also been adopted using surface plasmonic resonances, photonic crystals (PCs), nuclear magnetic resonance, and electrochemical biosensors.32–35This paper reports a single-step EV assay capable of performing both high-efficiency extraction and high-sensitivity and selectivity detection of multiple membrane proteins on the surface of exosomes in a single vessel. The whole process requires only about one hour from collecting the EVs from cell culture media using magnetic nanoparticles (MNPs) to quantifying the conjugated MNP–EV nanocomplexes using a photonic crystal (PC)-based label-free biosensor (Fig. 1(a)). The MNP-based exosome isolation provides an enrichment factor of 523 times. Because of the high-refractive-index MNPs used, the assay offers an improved sensitivity and a low limit of detection (LOD). In addition, the specificity of the assay is enhanced, due to the functionalization of both the MNPs and the biosensor using antibodies specific to the target EVs.
 | ||
| Fig. 1 Schematic illustration of the single-step in situ EV assay. (a) The EV assay involves mixing 50 nm-sized Fe2O3 MNPs and macrophage cell-derived EVs in a culture medium, conjugating the MNPs and EVs, collecting the formed MNP–EV nanocomplexes using a magnet, washing and re-suspending in a buffer solution, and quantifying the MNP–EVs using a PC-based biosensor located at the bottom of the sample chamber. All these steps were performed in a miniature test vessel. (b) Schematic showing the immobilization of the conjugated MNP–EVs on the surface of a PC biosensor. (c) Optical setup for measuring reflection from the PC sensor tube. The light source, beam splitter, and collection fiber were placed on a kinematic mount to facilitate the alignment. The inset shows the bottom of the vessel containing the PC biosensor. (d) SEM image of captured MNP–EVs on the surface of the PC biosensor. The inset shows the TEM image of an MNP–EV nanocomplex with two MNPs conjugated to an EV particle. (e) Schematic showing the detection of a spectral change in reflectance corresponding to the immobilization of the EVs, MNPs and MNP–EVs on the sensor surface. | ||
Results and discussion
Preparation of the label-free PC biosensor
The PC biosensor consisted of a one-dimensional (1D) grating and a thin-film coating of titanium oxide (TiO2), as shown in Fig. 1(b). Illuminated by broadband incident light, the PC grating can generate a resonant reflection whose peak reflection wavelength (λr) is sensitive to the refractive index or mass density changes occurring on the surface of the PC biosensor.36 The PC grating was fabricated using the low-cost replica molding approach, as described in the Methods and materials section.37–40 To functionalize the PC biosensor using EV-specific antibodies, the PC surface was treated using a polyvinylamine (PVAm) layer and a subsequent bifunctional linker, glutaraldehyde (GA). The scanning electron microscopy (SEM) image in Fig. 1(d) shows the surface of a fabricated PC grating with immobilized MNP–EVs. The transmission electron microscopy (TEM) image (the inset of Fig. 1(d)) shows one of the MNP–EVs. To measure the binding of EVs to the antibodies, the reflection spectra of the PC biosensor were recorded using the setup shown in Fig. 1(c), where the PC sensor was illuminated using a broadband light source, and its reflection was analyzed using a spectrometer. The resonant wavelength, λr, was calculated by fitting the peak in the reflection spectrum and the shift of the resonant wavelength (Δλr) was used as the sensor output. For example, when EV–MNPs are captured, the PC's resonant reflection shifts to a longer wavelength by Δλ. Because the MNPs have a relatively high refractive index (nMNP = 2.42), the detection of MNP–EVs exhibits a larger Δλr than the detection of EVs, as schematically illustrated in Fig. 1(e).Extraction of EVs using MNPs
Magnetic microbeads have been widely used for the separation of nucleic acids, proteins, and EVs from clinical samples.19–27 Although the microbeads are fast and efficient to be collected, the high material loss of microscale beads significantly quenches the resonance of the PC when the beads are immobilized. In contrast, the MNPs, whose diameter is much smaller than the operation wavelength, can be used to enhance the PC output without deteriorating the resonance mode. As shown in Fig. 2(a), the carboxylic acid modified MNPs were coated using anti-CD63 antibodies via the EDC and sulfo-NHS coupling reaction. The details of the MNP functionalization procedure are explained in the Methods and materials section. To evaluate the MNPs' extraction performance, the CD63 antibody-coated MNPs were mixed with a cell culture that contained EVs. The concentrations of EVs and MNPs used were 4 × 106 EVs per mL and 1.2 × 1012 particles per mL, respectively. Fig. 2(b) summarizes the major steps to add MNPs, and separate and wash the MNP–EVs from the macrophage cultures. The EVs and MNPs were incubated in a 10 mL culture medium for 30 min at room temperature (23 °C). Following the EV–MNP conjugation, the MNPs were attracted to the bottom of the sample tube using a magnet for 20 min, the supernatant was removed, and the MNPs were subsequently resuspended in phosphate buffered saline (PBS). The MNP–EV collection and resuspension process was repeated three times to remove interfering molecules and the samples were diluted in PBS to a final volume of 50 μL. Fig. 2(c) shows the nanoparticle tracking analysis (NTA) of EVs and MNPs before and after MNP conjugation, indicating the separated and bonded MNPs with EVs. The black curve exhibits two separated peaks, corresponding to ∼97 nm EVs and ∼54 nm bare MNPs, respectively. Because of lacking the CD63 antibody coating, the bare MNPs were not bound to the EVs, as shown in Fig. 2(d). In contrast, the MNPs with the CD63 antibody coating can form the MNP–EV nanocomplexes, which corresponds to the 151 nm peak in the red NTA curve in Fig. 2(c). To compare the size distributions before and after the MNP conjugation, the concentration of the MNP–EV nanocomplexes shown in the red curve in Fig. 2(c) was diluted by a factor of 1000 before the NTA analysis. The TEM images of MNP–EV nanocomplexes are summarized in Fig. 2(e)–(g), showing the isolated MNP–EV pairs, and the aggregated complexes. The MNP-based EV extraction increased the concentration of MNP–EV nanocomplexes (120–250 nm) from ~3.55 × 109 particles per mL to ∼1.75 × 1012 particles per mL, representing an enrichment factor of 493 times. To estimate the EV capture efficiency, we compared the EV concentrations in the cell culture medium before and after the MNP extraction. After the incubation of EV–MNPs and collection of MNPs using the magnet, the supernatant was sampled and measured using a nanoparticle analyzer (NanoSight). The concentration of the nanoparticles under 200 nm decreased by 42.3%, which represented an approximately 40% EV capture efficiency.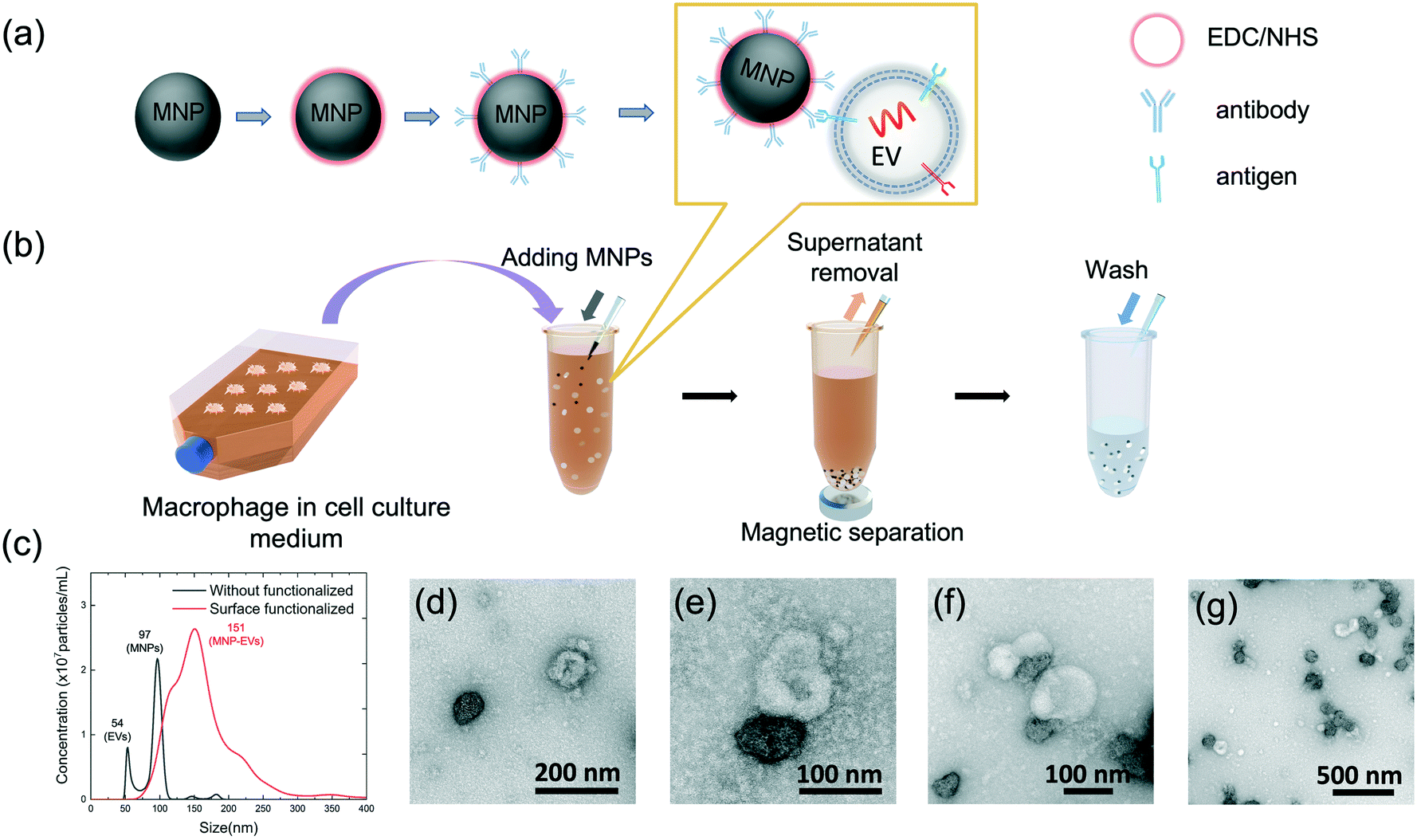 | ||
| Fig. 2 Extraction of EVs using MNPs. (a) Carboxylic acid modified MNPs are coated using anti-CD63 antibodies via the EDC/sulfo-NHS coupling reaction. The target EVs can bind to the MNPs via the EV's CD63 antigens. (b) Schematic of the MNP–EV extraction from macrophage cell cultures. The anti-CD63 coated MNPs were incubated with the macrophage cell culture to form MNP–EV conjugates. Then, the MNP–EVs were collected and washed using a buffer solution. (c) Size distributions of unconjugated MNPs and EV nanoparticles (black), and the MNP–EV nanocomplexes (red). (d) TEM image of a dispersed EV and bare MNP without the anti-CD63 coating. (e)–(g) TEM images of the MNP–EV nanocomplexes formed after the incubation of MNPs in a macrophage culture. | ||
Kinetic measurement of MNP–EV bindings using the PC biosensor
The label-free detection of MNP–EV nanocomplexes was performed using the PC biosensors as shown in Fig. 3(a). Here, three membrane antigens, CD9, CD63, and CD81, were chosen as the binding targets.41 These membrane markers have been used to distinguish macrophage-derived EV subtypes.42 Three different PC biosensors were functionalized using the anti-CD63, anti-CD9, and anti-CD81 capture antibodies via the PVAm/GA coupling reaction. After the coating of these antibodies, the non-specific bindings on the sensor surfaces were blocked using a bovine serum albumin (BSA)/GA composite.43 By minimizing the absorptions of the MNPs without EVs and other interfering molecules in the cell culture, the use of this new BSA/GA blocker made it possible to integrate the EV extraction and detection steps with the PC biosensor. The MNP–EV and control samples were pipetted onto the sensor surfaces (at t = 5 min) and allowed to bind to the antibodies. During the tests, the reflection spectra of the biosensors were recorded and the Δλr values were calculated. Fig. 3(b) shows the Δλr as a function of time when the EVs were immobilized onto the sensor surfaces via the CD9, CD63, and CD81 antibodies, respectively. The sensor outputs for the EV samples stabilized after 35 min. To show the stability of the assay, the kinetic PC sensor outputs were measured for 120 min. After the biosensor outputs were fully stabilized (at t = ∼120 min), all unbound materials were washed away using PBS to obtain the endpoint reading. For the positive control, the GA-coated biosensor was used to capture unspecific EV–MNP nanocomplexes. The positive control exhibited an output of Δλr= 7.21 nm. In contrast, the MNP sample (1 × 1011 MNPs per mL) incubated in pure DMEM containing 10% FBS was used as the negative control sample, exhibiting the lowest Δλr of 0.28 nm. The end-point values of ΔλCD9, ΔλCD63, ΔλCD81, Δλ−ctrl, and Δλ+ctrl are compared in Fig. 3(c). The absorption of EV–MNPs via the CD9 and anti-CD9 antibody binding generated the largest ΔλCD9 of 4.65 nm, which corresponds to the relatively high concentration of CD9 on the EV membrane protein. In contrast, the binding associated with the CD81 antigen resulted in the lowest Δλr of 1.97 nm. It can be seen that CD9 and CD63 were expressed at higher concentrations than CD81, which agrees well with the results previously reported by Kowal et al.41 (Kowal et al., 2016). All measured MNP–EV signals fell in the range of the positive and negative references Δλr = 7.06 nm and 0.28 nm. | ||
| Fig. 3 Label-free detection of MNP–EVs using the PC biosensor. (a) Schematic of the label-free MNP–EV assay using PC biosensors functionalized using three different antibodies. (b) Kinetic response of Δλr as a function of time. The Δλr values were measured every 5 sec during binding of MNP–EVs to the PC biosensors coated with CD9, CD63, and CD81 antibodies, respectively. (c) Column plot of endpoint Δλr values after the washing of unbound MNP–EVs. The error bars represent the standard deviations of the Δλr calculated based on the results obtained from nine independent tests. | ||
MNP-enhanced quantitative analysis of EVs
The conjugation of MNPs to EVs can enhance the sensitivity of the assay owing to the high refractive index of Fe2O3 MNPs. To demonstrate this using the MNPs as a signal amplifier, we compared the responses of the PC biosensor for three different test sets, including set #1: detecting EVs without using MNPs; set #2: tagging MNPs to the EVs immobilized on the PC biosensor; and set #3: measuring MNP–EV nanocomplexes. The dose–response curves for the three sample sets are shown in Fig. 4(a) by plotting the measured Δλr as a function of EV concentrations. For test sets #1 and #2, the EVs were purified using ultracentrifugation to 2.45 × 1012 EVs per mL and then diluted in PBS to six consecutive concentrations ranging from 4 × 106 to 1.25 × 1011 EVs per mL. The PC biosensors were functionalized using the anti-CD63 antibodies and blocked using the BSA/GA blocker. The PC-based detection of EVs without MNPs was performed following the steps reported in our previous work (Wang et al., 201834). The blue dose–response curve in Fig. 4(a) represents the dose–response curve of the direct detection of EVs based on the membrane CD63 antigen. For test set #2, the CD63 antibody-coated MNPs (2 × 1011 MNPs per mL) were introduced to the sample wells as a tagging material. After incubation for 20 min, unbound particles were washed away using PBS, and the MNP amplified Δλr value was calculated for each EV concentration. The red dose–response curve in Fig. 4(a) demonstrates an increase in Δλr after the MNPs were conjugated to the adsorbed EVs on the sensor surfaces. At the EV concentrations of 2 × 109 and 2 × 1011 EVs per mL, the sensor outputs were amplified by a factor of 16.8 and 3.6, respectively.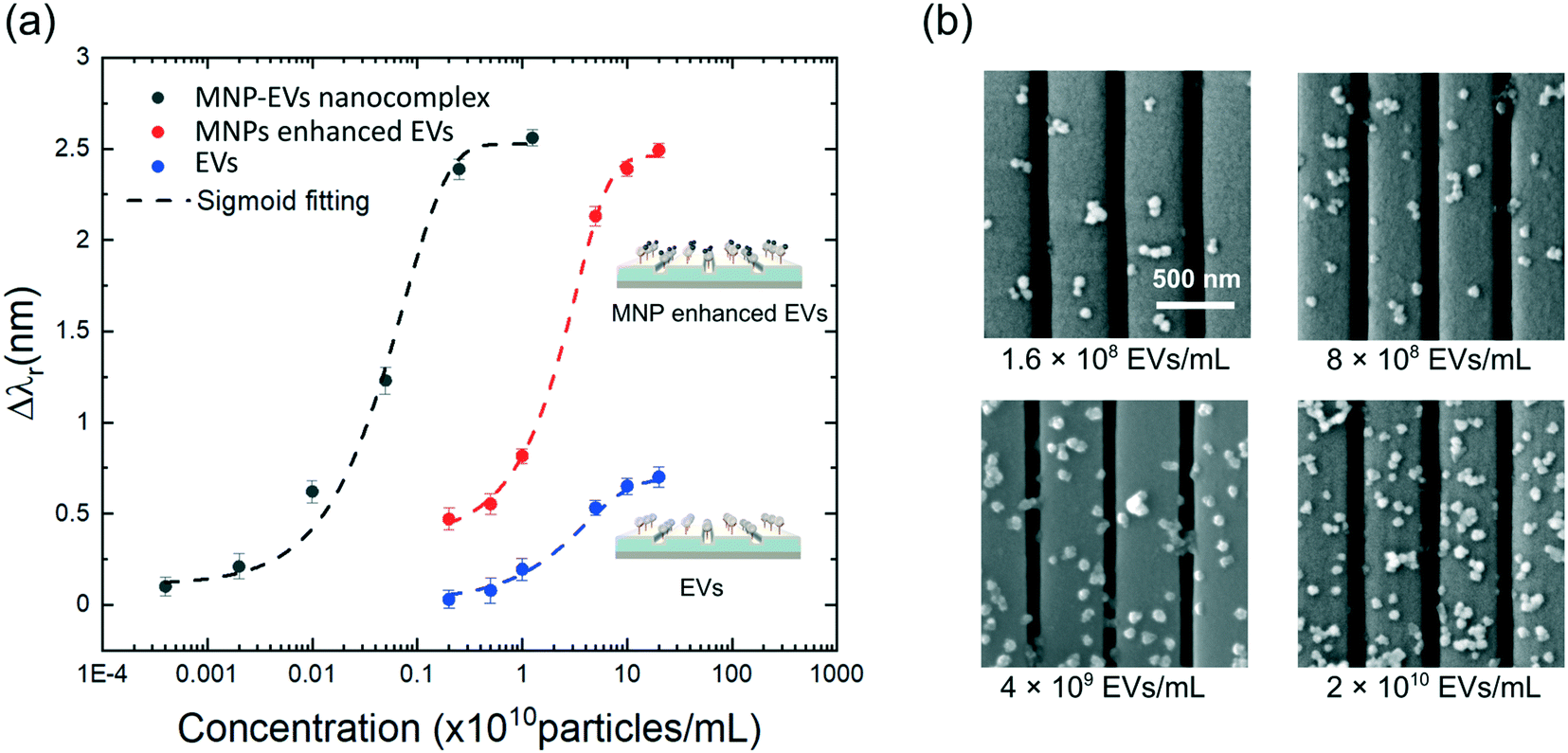 | ||
| Fig. 4 Quantitative EV detections of the PC biosensor. (a) Dose–response curves for the detection of EVs with MNPs (red), without MNPs (blue), and MNP–EV nanocomplexes (black). Measured data points were fitted using the sigmoid function. (b) SEM images of the MNP–EVs immobilized on the PCs for the EV concentrations of 1.6 × 108, 8 × 108, 4 × 109, and 2 × 1010 EVs per mL, respectively. | ||
Test set #3 took advantage of both the MNP-based signal enhancement and EV extraction functions to improve the assay sensitivity at the same time. The EV samples at the concentrations of 1.6 × 108, 8 × 108, 4 × 109, and 2 × 1010 EVs per mL were mixed with the CD63–MNPs (2 × 1011 particles per mL). After the incubation, the MNP–EVs were washed and re-suspended in 20 μL PBS. The six samples of MNP–EV nanocomplexes were measured using the PC biosensors with the CD63 antibody coating and BSA/GA blocker. The black dose–response curve in Fig. 4(a) summarizes the testing results and the SEM images in Fig. 4(b) illustrate the density of immobilized MNP–EVs on PC gratings. To calculate the LODs, the dose–response curves were fitted with the sigmoid function. The LODs were found by calculating the concentration that corresponds to the sensor output noise level of Δλr = 0.05 nm. Without the MNPs, the PC biosensor exhibited a LOD of 2.18 × 109 EVs per mL. With the enrichment using MNPs, the LOD was reduced to 3.44 × 107 EVs per mL, which represents an improvement of 63.4 times.
Extraction-and-detection analysis of EVs
The use of MNPs can further simplify the EV detection assay by combining both extraction and detection steps in one sample tube. The extraction-and-detection assay was performed in a 2 mL sample tube with the PC sensor attached to its bottom (Fig. 5(a)). The sensor surface was functionalized using the anti-CD63 antibody ligand and subsequently blocked using the BSA/GA blocker. The culture medium was collected from the macrophage cell culture flask and filtered using a 0.22 μm filter to remove the cells and debris. Cell culture media at a concentration of 1.69 × 1010, 8.44 × 1010 and 4.22 × 1011 particles per mL were used in the test. For the single-step analysis, the filtered culture medium containing EVs was mixed with CD63 antibody-coated MNPs (2 × 1011 particles per mL) in the tube above the sensor surface. After 20 min conjugation, a magnet was placed at the sidewall of the tube to collect the MNP–EV particles for another 20 min. The supernatant (∼1.9 mL) was then removed from the tube, and the MNP–EV particles remained inside the tube. After resuspension in PBS (20 μL), the enriched MNP–EVs were allowed to bind to the sensor surface for 15 min before the final wash step. During the assay, the tube was fixed on the detection setup to collect reflection spectra from the PC sensors in real time. It took only about one hour to complete the collection of the EVs from the cell culture media to the quantification of the target membrane proteins on the surface of the collected EVs. The kinetic detections of the EV samples at three different concentrations and negative control samples are shown by the red, orange, yellow, blue, and black curves, respectively (Fig. 5(b)). The red, orange and yellow curves correspond to the samples with both EVs and MNPs, which shows Δλr = 4.32 nm, 3.64 nm and 2.79 nm, respectively, compared with the starting point and ending point, while the black curve represent the assay without MNPs and EVs. The black curve exhibited the lowest Δλr = 1.59 nm caused by DMEM. As a reference, the MNP sample in the culture medium without EVs was also measured as the blue curve in Fig. 5(b), which shows Δλr = 1.85 nm due to the DMEM and MNP remnants. The integrated extraction and detection assay can significantly simplify EV analysis and reduce the total assay time. | ||
| Fig. 5 In situ EV assay performing both extraction and detection in a single test tube. (a) In situ EV analysis. The PC biosensor was attached to a bottomless sample tube. The sample tube was placed on a reflection measurement setup. The photos in the bottom illustrate the incubation of EVs and MNPs, collection of MNP–EVs using a magnet, removal of culture medium, and resuspension of MNP–EVs in PBS. (b) Kinetic detection of the MNP–EV, EV, and reference samples in the sample tube. The entire assays were carried out in the sample tube with the PC attached to its bottom. The sensor outputs were measured every 5 sec during the incubation, washing, resuspension, and binding steps. | ||
Most existing EV assay studies are focused on enhancing the EV extraction efficiency or improving the detection sensitivity. The latest developments towards rapid EV assays are summarized in Table 1, where microfluidics has enabled EV extraction and detection. For example, Lee et al.44 developed a paper microfluidic device to enrich and detect EVs on paper with an assay time of 2 h and a LOD of ~106 EVs per mL. Wang et al.34 demonstrated a microfluidic biosensor to detect EVs with an assay time of >5.5 h and a LOD of 2.18 × 109 EVs per mL. Liang et al.45 utilized a double-filtration method for EV isolation and ELISA for EV quantification with a total assay time of >4 h. Profoundly, our single-step EV analysis enables both the extraction and detection steps in a sample tube, reduces the total assay time to about 1 h, and provides a considerably low LOD on the order of 107 EVs per mL, while eliminating the need for any microfluidic control components and accessories, such as channels and syringe pumps.
| Method | EV isolation approach | EV detection approach | LOD | Assay time | Ref. |
|---|---|---|---|---|---|
| Microfluidic chip | Immunomagnetic enrichment | ELISA | 1–2 AU μL−1 | 1.5 h | 24 |
| Microfluidic photonic crystal biosensor | Differential centrifugation | PC resonance | 2.18 × 109 EVs per mL | >5.5 h | 34 |
| Paper microfluidics | Streptavidin agarose resin paper | On-paper ELISA | 106–107 EVs per mL | <2 h | 44 |
| Double-filtration microfluidic device | Double filtration | On-chip ELISA | 109 EVs per mL | >4 h | 45 |
| Quantum dot-based sensitive detection | Immunomagnetic enrichment | Electrochemical sensor | 105 EVs per mL | ∼2 h | 46 |
| Alternating current electrohydrodynamic induced nanoshearing | Alternating current electrohydrodynamic flow | Tunable alternating current electrohydrodynamic | 2.76 × 106 EVs per mL | ∼3 h | 47 |
| Gold-loaded nanoporous ferric oxide nanozymes | Immunomagnetic enrichment | Electrochemical sensor | 103 EVs per mL | ∼4 h | 48 |
| Two-in-one EV assay | Immunomagnetic nanoparticles | MNP-enhanced label-free photonic crystal sensor | 3 × 107 EVs per mL | <1 h | This work |
Conclusions
The present work has demonstrated a rapid in situ EV analysis assay capable of accomplishing both rapid extraction and selective detection of EVs in a single vessel. The immunomagnetic extraction enriched EVs from a macrophage cell culture by a factor of 523 within 40 min, while the label-free PC biosensor analyzed three membrane proteins (CD9, CD63, and CD81) carried by the EVs. When the MNPs were conjugated to the target EVs, the MNPs functioned as a signal tag to enhance the sensor output. For the detection of CD63 antibodies, the single-step assay platform improved the resonance wavelength shift by up to 16.8 times and the LOD by about two orders of magnitude, compared to those obtained using the PC biosensor alone without MNPs. The entire EV assay, including incubation, enrichment, purification, and detection, required less than one hour.While a 1D PC grating biosensor was incorporated in this work, it is possible to employ other label-free biosensors, such as plasmonic resonance sensors and microarray platforms, to analyze EVs in clinical samples. It should be noted that most heterogeneous immunoassays suffer from a slow response due to the mass transfer limit.11–14 Our future work therefore will be focused on using MNPs to further reduce the time required for driving EVs from complex clinical samples towards the sensing surface, thus making it more practical for rapid point-of-care EV analysis in disease diagnostics.
Methods and materials
Fabrication and surface functionalization of PC biosensors
The 1D grating structure was produced by the replica molding approach using a 555.5 nm-period silicon grating (LightSmyth Technologies) as the mold and optical adhesive (NOA 86, Norland Products) on a plastic sheet. The replicated grating was coated with a 100 nm TiO2 film using an electron beam evaporator. The fabricated PC sensors were cut into 12.7 mm-diameter disks and attached to glass sample tubes using a UV epoxy. To coat the fabricated PC sensor with selective ligands, the PC sensor was functionalized using a two-step process. The first step was to treat the TiO2 surface using polyvinylamine (PVAm, BASF), which provided amine functional groups. Then, a bi-functional linker, glutaraldehyde (GA, Sigma Aldrich), was used to activate the sensor for the attachment of specific antibodies. The capture antibodies, anti-CD63, anti-CD9, and anti-CD81 (BioLegend), were dispensed onto the GA-coated PC sensor at 0.5 mg mL−1 as the EV-specific ligands. GA/BSA was used to block the unspecific binding. The antibody-coated PC sensors can be soaked in PBS and stored at 4 °C for future use.Macrophage cell culture
The EVs used in this study were secreted by murine macrophage cells (J774.1 cell line, ATCC). Murine macrophages were cultured in Dulbecco's modified Eagle medium (DMEM) containing 10% fetal bovine serum (Sigma-Aldrich) at 37 °C with 5% CO2. The culture medium with FBS was stored at 4 °C for more than 15 days before use. The EVs were ready for extraction when macrophages reached 90% confluency after the cells were cultured for approximately three days. Then, the culture media were filtered using 0.22 μm filters to remove the debris. The filtered media can be mixed with MNPs for the MNP-based EV enrichment.Preparation of EV samples
The EVs were also extracted using a conventional ultracentrifugation process as a reference. To extract EVs from the cell culture medium, cells and debris were removed through a 0.22 μm filter and were then collected into an ultracentrifuge tube. The sample was centrifuged at 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 90 min. Then, the pellet was resuspended in PBS, and the sample was transferred into a 1.5 mL Beckman ultracentrifuge tube. The sample was centrifuged again at 55000 rpm for an additional 2 hours, and the pellet was resuspended in PBS. All centrifugations were completed at 4 °C. The purified EVs were stored in a −80 °C freezer. The concentrations and size distributions of EVs and EV–MNP complexes were measured by nanoparticle tracking analysis (NanoSight LM10, Malvern Instruments Inc.).
000g for 90 min. Then, the pellet was resuspended in PBS, and the sample was transferred into a 1.5 mL Beckman ultracentrifuge tube. The sample was centrifuged again at 55000 rpm for an additional 2 hours, and the pellet was resuspended in PBS. All centrifugations were completed at 4 °C. The purified EVs were stored in a −80 °C freezer. The concentrations and size distributions of EVs and EV–MNP complexes were measured by nanoparticle tracking analysis (NanoSight LM10, Malvern Instruments Inc.).
Surface functionalization of MNPs
The 50 nm diameter MNPs (SC0050, Ocean NanoTech) were carboxylic acid modified and functionalized using CD63 antibodies (anti-mouse CD63 antibodies, BioLegend). Before coating with the CD63 antibodies, the MNPs were activated using an EDC/sulfo-NHS covalent coupling procedure by mixing 0.2 mL of the MNPs (10 mg mL−1), 0.2 mL activation buffer, and 20 μL EDC solution (20 mg mL−1) for 15 min at room temperature (Peterson et al., 201439). Then, 0.3 mL CD63 antibodies (1 mg mL−1 in activation buffer) was added into the MNP solution, and the mixture was incubated for 2.5 h at room temperature. After the CD63 antibody conjugation, the MNP surface was blocked using 0.1 mL quenching buffer (Ocean NanoTech) for 30 min (Fig. 2(a)). To collect the CD63-coated MNPs, the mixture was placed on a magnetic separator for 20 min (Fig. 2(b)), the supernatant was removed and the MNPs were resuspended in 200 μL PBS solution. The processed MNP wash process was repeated three times and the resuspended MNPs in PBS were stored at 4 °C for future use.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the United States National Science Foundation under Grant No. ECCS 17-11839 and ECCS 16-53673. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation. The authors thank the Nano Fabrication Centre at the University of Minnesota for the support for device fabrication.References
- C. Théry, L. Zitvogel and S. Amigorena, Nat. Rev. Immunol., 2002, 2, 569–579 CrossRef PubMed.
- G. Raposo and W. Stoorvogel, J. Cell Biol., 2013, 200, 373–383 CrossRef CAS.
- Y. Zhang, Y. Liu, H. Liu and W. H. Tang, Cell Biosci., 2019, 9, 1–18 CrossRef.
- H. Zhou, T. Pisitkun, A. Aponte, P. S. T. Yuen, J. D. Hoffert, H. Yasuda, X. Hu, L. Chawla, R.-F. Shen, M. A. Knepper and R. A. Star, Kidney Int., 2006, 70, 1847–1857 CrossRef CAS.
- H. Sonoda, N. Yokota-Ikeda, S. Oshikawa, Y. Kanno, K. Yoshinaga, K. Uchida, Y. Ueda, K. Kimiya, S. Uezono, A. Ueda, K. Ito and M. Ikeda, Am. J. Physiol., 2009, 297, F1006–F1016 CAS.
- T. An, S. Qin, Y. Xu, Y. Tang, Y. Huang, B. Situ, J. M. Inal and L. Zheng, J. Extracell. Vesicles, 2015, 4, 27522 CrossRef.
- Q. Cai, B. He, A. Weiberg, A. H. Buck and H. Jin, PLoS Pathog., 2019, 15, e1008090 CrossRef CAS.
- I. O. Sun and L. O. Lerman, Diagnostics, 2020, 10, 311 CrossRef CAS.
- E. Cocucci, G. Racchetti and J. Meldolesi, Trends Cell Biol., 2009, 19, 43–51 CrossRef CAS.
- A. Yekula, K. Muralidharan, K. M. Kang, L. Wang, L. Balaj and B. S. Carter, Methods, 2020, 177, 58–66 CrossRef CAS PubMed.
- M. A. Livshits, E. Khomyakova, E. G. Evtushenko, V. N. Lazarev, N. A. Kulemin, S. E. Semina, E. V. Generozov and V. M. Govorun, Sci. Rep., 2015, 5, 1–14 Search PubMed.
- M. A. Rider, S. N. Hurwitz and D. G. Meckes, Sci. Rep., 2016, 6, 23978 CrossRef CAS.
- P. G. K. Patel, M. A. Khan, H. Zubair, S. K. Srivastava, M. Khushman, S. Singh and A. P. Singh, Sci. Rep., 2019, 9, 1–10 CAS.
- A. Gámez-Valero, M. Monguió-Tortajada, L. Carreras-Planella, M. Franquesa, K. Beyer and F. E. Borràs, Sci. Rep., 2016, 6, 33641 CrossRef.
- H.-K. Woo, V. Sunkara, J. Park, T.-H. Kim, J.-R. Han, C.-J. Kim, H.-I. Choi, Y.-K. Kim and Y.-K. Cho, ACS Nano, 2017, 11, 1360–1370 CrossRef CAS.
- B. H. Wunsch, J. T. Smith, S. M. Gifford, C. Wang, M. Brink, R. L. Bruce, R. H. Austin, G. Stolovitzky and Y. Astier, Nat. Nanotechnol., 2016, 11, 936–940 CrossRef CAS PubMed.
- M. Wu, Y. Ouyang, Z. Wang, R. Zhang, P.-H. Huang, C. Chen, H. Li, P. Li, D. Quinn, M. Dao, S. Suresh, Y. Sadovsky and T. J. Huang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10584–10589 CrossRef CAS.
- J. Chen, Y. Xu, Y. Lu and W. Xing, Anal. Chem., 2018, 90, 14207–14215 CrossRef CAS.
- H. Shao, J. Chung, K. Lee, L. Balaj, C. Min, B. S. Carter, F. H. Hochberg, X. O. Breakefield, H. Lee and R. Weissleder, Nat. Commun., 2015, 6, 1–9 Search PubMed.
- A. Clayton, J. Court, H. Navabi, M. Adams, M. D. Mason, J. A. Hobot, G. R. Newman and B. Jasani, J. Immunol. Methods, 2001, 247, 163–174 CrossRef CAS.
- (a) S. Jeong, J. Park, D. Pathania, C. M. Castro, R. Weissleder and H. Lee, ACS Nano, 2016, 10, 1802–1809 CrossRef CAS.
- Z. Zhao, Y. Yang, Y. Zeng and M. He, Lab Chip, 2016, 16, 489–496 RSC.
- J. Park, H.-Y. Lin, J. P. Assaker, S. Jeong, C.-H. Huang, A. Kurdi, K. Lee, K. Fraser, C. Min, S. Eskandari, S. Routray, B. Tannous, R. Abdi, L. Riella, A. Chandraker, C. M. Castro, R. Weissleder, H. Lee and J. R. Azzi, ACS Nano, 2017, 11, 11041–11046 CrossRef CAS.
- W. Chen, H. Li, W. Su and J. Qin, Biomicrofluidics, 2019, 13, 054113 CrossRef PubMed.
- Y. Wan, G. Cheng, X. Liu, S.-J. Hao, M. Nisic, C.-D. Zhu, Y.-Q. Xia, W.-Q. Li, Z.-G. Wang, W.-L. Zhang, S. J. Rice, A. Sebastian, I. Albert, C. P. Belani and S.-Y. Zheng, Nat. Biomed. Eng., 2017, 1, 1–11 CrossRef.
- F. Z. Farhana, M. Umer, A. Saeed, A. S. Pannu, M. Shahbazi, A. Jabur, H. J. Nam, K. Ostrikov, P. Sonar, S. H. Firoz and M. J. Shiddiky, ACS Appl. Nano Mater., 2021, 4, 1175–1186 CrossRef CAS.
- K. Boriachek, M. K. Masud, C. Palma, H.-P. Phan, Y. Yamauchi, M. S. Hossain, N.-T. Nguyen, C. Salomon and M. J. Shiddiky, Anal. Chem., 2019, 91, 3827–3834 CrossRef CAS PubMed.
- R. A. Dragovic, C. Gardiner, A. S. Brooks, D. S. Tannetta, D. J. P. Ferguson, P. Hole, B. Carr, C. W. G. Redman, A. L. Harris, P. J. Dobson, P. Harrison and I. L. Sargent, Nanomed.: Nanotechnol., Biol. Med., 2011, 7, 780–788 CrossRef CAS PubMed.
- T. Soares Martins, J. Catita, I. Martins Rosa, O. A. B. da Cruz e Silva and A. G. Henriques, PLoS One, 2018, 13, e0198820 CrossRef.
- W. Oosthuyzen, N. E. Sime, J. R. Ivy, E. J. Turtle, J. M. Street, J. Pound, L. E. Bath, D. J. Webb, C. D. Gregory, M. A. Bailey and J. W. Dear, J. Physiol., 2013, 591, 5833–5842 CrossRef CAS.
- R. M. Lequin, Clin. Chem., 2005, 51, 2415–2418 CrossRef CAS.
- H. Im, K. Yang, H. Lee and C. M. Castro, Methods Mol. Biol., 2017, 133–141 CrossRef CAS.
- H. Im, H. Shao, Y. I. Park, V. M. Peterson, C. M. Castro, R. Weissleder and H. Lee, Nat. Biotechnol., 2014, 32, 490–495 CrossRef CAS PubMed.
- Y. Wang, W. Yuan, M. Kimber, M. Lu and L. Dong, ACS Sens., 2018, 3, 1616–1621 CrossRef CAS.
- H. Shao, J. Chung, L. Balaj, A. Charest, D. D. Bigner, B. S. Carter, F. H. Hochberg, X. O. Breakefield, R. Weissleder and H. Lee, Nat. Med., 2012, 18, 1835–1840 CrossRef CAS.
- S. Fan and J. D. Joannopoulos, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 235112 CrossRef.
- I. D. Block, L. L. Chan and B. T. Cunningham, Microelectron. Eng., 2007, 84, 603–608 CrossRef CAS.
- S. M. Shamah and B. T. Cunningham, Analyst, 2011, 136, 1090 RSC.
- R. D. Peterson, B. T. Cunningham and J. E. Andrade, Biosens. Bioelectron., 2014, 56, 320–327 CrossRef CAS PubMed.
- D. Gallegos, K. D. Long, H. Yu, P. P. Clark, Y. Lin, S. George, P. Nath and B. T. Cunningham, Lab Chip, 2013, 13, 2124 RSC.
- J. Kowal, G. Arras, M. Colombo, M. Jouve, J. P. Morath, B. Primdal-Bengtson, F. Dingli, D. Loew, M. Tkach and C. Théry, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E968–E977 CrossRef CAS.
- Y. Wang, Q. Zhang, W. Yuan, Y. Wang, H. J. Loghry, Z. Zhao, M. J. Kimber, L. Dong and M. Lu, Lab Chip, 2021, 21, 196–204 RSC.
- J. Sabaté del Río, O. Y. Henry, P. Jolly and D. E. Ingber, Nat. Nanotechnol., 2019, 14, 1143–1149 CrossRef.
- J. Lee, H. Kim, Y. Heo, Y. K. Yoo, S. I. Han, C. Kim, D. Hur, H. Kim, J. Y. Kang and J. H. Lee, Analyst, 2020, 145, 157–164 RSC.
- L.-G. Liang, M.-Q. Kong, S. Zhou, Y.-F. Sheng, P. Wang, T. Yu, F. Inci, W. P. Kuo, L.-J. Li, U. Demirci and S. Q. Wang, Sci. Rep., 2017, 7, 1–10 CrossRef PubMed.
- K. Boriachek, M. N. Islam, V. Gopalan, A. K. Lam, N.-T. Nguyen and M. J. Shiddiky, Analyst, 2017, 142, 2211–2219 RSC.
- R. Vaidyanathan, M. Naghibosadat, S. Rauf, D. Korbie, L. G. Carrascosa, M. J. Shiddiky and M. Trau, Anal. Chem., 2014, 86, 11125–11132 CrossRef CAS PubMed.
- M. K. Masud, S. Yadav, M. N. Islam, N.-T. Nguyen, C. Salomon, R. Kline, H. R. Alamri, Z. A. Alothman, Y. Yamauchi, M. S. Hossain and M. J. Shiddiky, Anal. Chem., 2017, 89, 11005–11013 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |